

Abstract
Coumarins have received a considerable attention in the last three decades as a lead structures for the discovery of orally bioavailable non-peptidic antiviral agents. A lot of structurally diverse coumarins analogues were found to display remarkable array of affinity with the different molecular targets for antiviral agents and slight modifications around the central motif result in pronounced changes in its antiviral spectrum. This manuscript thoroughly reviews the design, discovery and structure–activity relationship studies of the coumarin analogues as antiviral agents focusing mainly on lead optimization and its development into clinical candidates.
Keywords: Coumarin, Antiviral, Anti-HIV, Anti-HCV, Anti-influenza, Anti-herpes
Graphical abstract

Highlights
-
•
Coumarins are promising chemotype against the viruses.
-
•
Coumarins emerged as an orally bioavailable non-peptidic antiviral agents.
-
•
This review summarizes the design, discovery, and SAR of coumarin analogues.
-
•
Future perspectives of coumarins as possible clinical candidates are also discussed.
1. Introduction
Viruses are causative agents for several epidemic fatal diseases such as human immunodeficiency virus (HIV), hepatitis B and C viruses (HBV and HCV, respectively), coronaviruses (Middle east respiratory Syndrome, MERS; severe acute respiratory syndrome, SARS), influenza (seasonal, pandemic), smallpox, viral haemorrhagic fevers (Ebola), dengue, and chikungunya etc. Each of these diseases has caused global societal and economic impact related to unexpected illnesses and deaths, as well as troubling day-to-day normal life activities. Recent emergence of newer pandemics e.g. H1N1 influenza, Ebola, and Zika virus etc. are also a major threat to public health [1], [2]. All these emerging viruses have more or less RNA genomes and as a result are capable of rapid mutation and resistance to the clinically available antiviral drugs. Thus threat of resistance poses major challenges in clinical management of these viral infections [3]. However, the field of antiviral drug discovery has witnessed a steady flow of exciting developments since the advent of nucleoside analogue acyclovir in 1960 [4]. Subsequently, more than 60 antiviral drugs of diverse chemical classes have been approved by the FDA, mainly for the management of HIV, herpes, the hepatitis B and C, and influenza A and B viruses and still many molecules are in various stages of clinical trials. Despite of these progress in the development of antiviral agents in recent years, there is still a pressing need for newer drugs acting through different mechanism to combat the problem of viral resistance as viruses are constantly evolving and have been developing new ways to evade the drugs [5]. Moreover, all of these render the search of antiviral therapy indistinct since it is difficult to identify unique biochemical features of viruses that may be appropriate for selective attack without harming the host cells [6].
Coumarins (benzo-α-pyrone) are considered as a privileged structure for designing novel agents having high affinity and specificity to different molecular targets for antiviral agents [7]. Because, coumarins are endowed with a unique characteristic pharmacophore of planar aromatic nucleus connected with a hydrogen bond acceptor; lactone group as a facilitator of protein-ligand binding [8]. Coumarins have gained momentous attention in the last three decades as a lead structures for the discovery of orally bioavailable non-peptidic antiviral agents. Several of natural, semisynthetic, synthetic lead molecules bearing coumarin scaffold have been discovered in the recent past and are in various phase of drug development [9]. Although some erstwhile reviews have already been published, but all of them have specifically focused on the anti-HIV potential of coumarins derivatives [10], [11]. The present review provides a comprehensive account of therapeutic potential of coumarins against the different viral diseases and summarizes detailed structure-activity relationship (SAR) and recent advances with emphasis on their molecular mechanisms.
2. Molecular targets for coumarin derivatives
Recent years have observed an outstanding advancement in the discovery and development of antiviral agents, fuelled by advances in virology which in turn have provided new prospects for therapeutic intervention. A number of virus-specific proteins or processes have been identified as targets for chemotherapeutic intervention [12]. Most of the antiviral drugs block intracellular events affecting the synthesis and dynamics of viral proteins and nucleic acids and amongst these, viral polymerases constitute the major target for many antiviral drugs [13]. Viral replication cycle generally consists of virus adsorption, virus-cell fusion, reverse transcription, integration, translation, proteolytic cleavage, glycosylation, assembly, or release. All these steps could be envisaged as targets for chemotherapeutic intervention [14]. In addition to these virus-specific events, there are a number of host enzymes and processes that are innately involved with viral DNA, RNA, and glycoprotein syntheses. Also, these processes [i.e., inosine 5′-monophosphate (IMP) dehydrogenase for RNA viruses, S-adenosylhomocysteine (SAH) hydrolase for Ebola, orotidine 5′-monophosphate decarboxylase, CTP synthetase, glycosylation pathways, etc.] may be considered as targets for antiviral agents. The other strategies involve HIV reverse transcriptase, herpes viral DNA polymerase, HIV adsorption inhibitors as vaginal microbicides in the prevention of AIDS, inhibition of HIV-cell fusion through their interaction with the viral gp41, HIV nucleocapsid p7 zinc finger-binding, HIV integrase inhibition, viral (HIV, HSV, CMV, HCV, etc.) protease inhibition, HIV-1 Vpr inhibition, picorna viral capsid binders, influenza A virus uncoating inhibition, HIV Tat and Rev antagonism, HIV and HBV glycosylation inhibition, and influenza A and B virus neuraminidase inhibition [15], [16].
Different classes of coumarin compounds have been found to act through a diverse set of above-mentioned mechanisms. The molecular targets, mechanisms of action, antiviral activity spectra, and clinical applications of different classes of coumarin compounds along with chemical structures for representative prototype compounds are schematically reviewed in Table 1 and Fig. 1 .
Table 1.
Coumarin derivatives as antiviral drugs or on their way to clinics.
| Chemical classes | Prototype compounds | Mechanisms of action | Viruses | Clinical status |
|---|---|---|---|---|
| 4-Hydroxycoumarins (4-HC) |
Warfarine analogues: 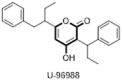
|
HIV-1 and 2 protease inhibitor | Human immunodeficiency virus type 1 (HIV-1) | 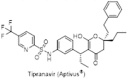 |
Tetramer of 4-HC: 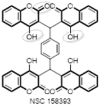
|
HIV-1 integrase, HIV-1 reverse transcriptase (RT) and Protease inhibitor | HIV-1 | – | |
| Pyranocoumarins |
Khellactone: 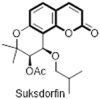
|
Inhibition of production of double-stranded viral DNA from the single-stranded DNA intermediate | HIV-1, and multi-RT inhibitor resistant HIV strain. | – |
Calanolide: 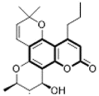
|
Non-nucleoside reverse transcriptase inhibitor (NNRTI) | AZT-resistant G-9106 and pyridinone-resistant A17 HIV strain | (+)-calanolide A (Sarawak MediChem Pharmaceuticals) | |
| Furanocoumarin | 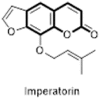 |
Inhibit the Sp1-related genes (D1 expression) thereby arresting the cells at the G(1) phase of cell cycle | HIV | – |
 |
Inhibition of viral RNP of influenza viruses and Tat-mediated transcription by Akt pathway of HIV | Oseltamivir-resistant influenza A and B viruses and HIV | – | |
 |
Inhibition of both viral DNA synthesis and virion production | Kaposi's sarcoma-associated herpesvirus (KSHV) | – | |
| 3-phenylcoumarins | 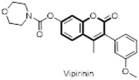 |
Arrest activity of HIV-1 viral protein R (Vpr) activity | HIV | – |
| 4-Phenylcoumarins | 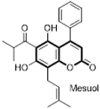 |
Interference of NF-κB and Tat functions | HIV | – |
| Coumarin-benzimidazole Conjugate | 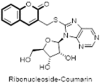 |
– | Hepatitis C virus (HCV) | – |
| Anilinocoumarin | 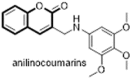 |
Induction of interferon-α (IFN)-mediated antiviral responses | HCV | – |
| 7-Hydroxycoumarins analogues | 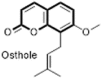 |
DNA polymerase inhibition | Bovine viral diarrhoea virus (BVDV), Respiratory syncytial virus (RSV) and HCV | – |
| Coumestans |  |
NS5B RNA-dependent RNA polymerase (RdRp) inhibition | HCV | – |
| Toddacoumaquinone | 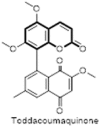 |
– | Herpes simplex virus type 1 and 2 (HSV-1 & 2), HIV-1 | – |
Fig. 1.

Different classes of coumarins effective against various viral diseases.
3. Coumarin analogues as anti-HIV agents
Recently numerous classes of coumarin compounds have been evaluated for inhibitory effects against HIV replication, and some of them have been found to inhibit different stages in the HIV replication cycle excellently.
3.1. 4-Hydroxycoumarin analogues: an alternative to pepetidomimetics
Previously, more or less all the clinically available HIV protease inhibitors are pepetidomimetics. Although these peptide-based inhibitors have got the edge over other inhibitors because of their very high binding affinity, but generally lacking the bioavailability requirements essential for cellular and oral activity and metabolic instability. This led to the search for inhibitors containing non-peptidic templates that might offer superior biopharmaceutical properties. 4-hydroxycoumarins (4-HCs) as non-peptidic HIV protease inhibitors have received considerable attention in the 90s, owing to their low molecular weight, high oral bioavailability, low clearance and more importantly, these are already being in use as therapeutic agents in human. Warfarin (1) is a 4-HC analogue, which is clinically available drug used in the prevention of thrombosis and thromboembolism. It acts by inhibiting vitamin K-dependent conversion of prothrombin into an active form thrombin, which is a serine protease enzyme catalysing many other coagulation-related reactions [17]. Moreover, several studies demonstrate that the protease is also an essential viral enzyme and an attractive target for anti-HIV drugs [18]. Prompted by these correlations, Bourinbaiar and his colleagues pioneered the study of testing the effects of the oral anticoagulant, warfarin, on viral replications and spread. Remarkably, they found that the low doses of warfarin (10−9 to 10−8 M) effectively inhibited the 50% HIV infections [19]. This discovery of 4-HC as a lead compound having promising anti-protease activity pave the way for the development of a new class of non-peptidic oral anti-viral agents. In pursuit of this goal, the Upjohn laboratory scientists Thaisrivongs et al. evaluated some similar coumarin derivatives as potential HIV-protease inhibitors using fluorescence based high-throughput screening. Surprisingly, an another coumarin based anticoagulant phenprocoumon (2) emerged as a novel lead template possessing weak HIV protease inhibitory activity (Ki = 1 μM) with antiviral ED50 of 100–300 μM but having superior pharmacokinetics (Fig. 2 ). Protein crystallography of 2 in complex with HIV-1 protease was successfully carried out in order to get a clear insight of interactions and lead optimization. This crystal structure revealed that the 4-HC scaffold binds in a mode very similar to that of peptide inhibitors at the catalytic site, with the C-4 hydroxyl forming two hydrogen-bonds with aspartic residues (Asp25A and Asp25B) and lactone group forming two hydrogen bonds with isoleucine residues (Ile50A and Ile50B) (Fig. 3 ). This study led to the postulation of 4-HC as an essential pharmacophoric element which formed the basis of ensuing structure based drug design of more active analogues.
Fig. 2.

Replacement of α-acetonyl with ethyl group led to weak HIV protease inhibitory activity but better pharmacokinetic.
Fig. 3.

Docked structure of phenprocoumon at the active site of HIV-1 protease (PDB ID: 1UPJ) and schematic representation of 4-HC pharmacophore.
Structure-based drug discovery (SBDD) is becoming an essential tool in assisting fast and cost-efficient lead discovery and optimization, as it aims to understand the molecular basis of a disease and utilizes the knowledge of the 3-D structure of the biological target in the process. Using the SBDD approach and X-ray crstallography, the 4-HC template was further optimised and developed into the first clinical candidate 4-hydroxy-2-pyrone derivative U-96988 (3) as non-peptidic HIV protease inhibitor which entered phase I clinical trials (Fig. 4 ). Compound 3 showed excellent HIV-1 protease inhibition (Ki = 38 nM), significant antiviral activity (IC50 = 3 μM) and improved pharmacokinetic properties and thus considered as a potential therapeutic agent for the treatment of HIV infection [20].
Fig. 4.

Structure-based design of 4-hydroxy-2-pyrone as a first clinical candidate against HIV protease.
Molecular hybridization of different pharmacophoric subunits, is an important tool for lead optimization and have provided the discovery of numerous ligands with synergistic properties. Based on these approach Thaisrivongs and co-workers (Upjohn Lab.) did an extensive research and examined the crystal structure of 4-HC analogues bound to HIV protease superimposed along with previously reported peptide-based inhibitors (4). The relative positioning of peptidic and non-peptidic compounds in HIV-protease revealed that addition of amide chain at the meta position of the phenyl ring of 4-HC could improve the binding affinity of coumarin-based inhibitors. This design strategy resulted into an exciting series of a several promising amino acid and non-amino acid residue containing 4-HCs (5–10) with improved enzyme-binding affinity. These study revealed that, in addition to the essential pharmacophore 4-HC, 5,6-dihydro-4-hydroxy-2-pyrones incorporated side chains at the C-6 position are also suitably accommodated into the S1′ and S2′ subsites of the enzyme active site. Thus, previously reported compound 3 also provided a basis for possible further modification of these series into potent 2-pyrone analogues. Consequently, pyrone compound containing L-histidyl amino acid residue (11), was found to be most potent analogue of the series having Ki = 4 nM. Further resolution of its four specific diastereomers had no significant impact on enzyme-binding affinity which clearly indicates their insignificant stereochemical preferences. Cell culture assay proved the compound as a nontoxic (TD50 = >>30 μM) with antiviral potency of IC50 value 3–6 μM, which is comparable to that previously reported for the inhibitor 3 (Fig. 5 ) [21]. Replacement of ethyl group by cyclopropyl group at C-3α position has also resulted in compounds 12–15 with improved efficacies. The general SAR of the series has been illustrated in Fig. 6 .
Fig. 5.

Structure-based design of hybrid carboxamide derivatives of 4-hydroxycoumarins.
Fig. 6.

General structure-activity relationship (SAR) of 4-hydroxycoumarins as HIV protease inhibitors.
Romines et al. at the Upjohn laboratories demonstrated a unique strategy for the enhancement secondary binding of 4-HC inhibitors to the protease enzyme. Instead of pursuing 4-hydroxy-2-pyrones they explored an alternate strategy for increasing the flexibility by replacing the benzene ring of 4-HC with saturated cycloalkyl rings i.e. cyclopentylpyranone (16) and cyclooctylpyranone (17) (Fig. 7 ). The results were surprising and cyclopropyl containing cyclooctylpyranone (18) emerged as a potential protease enzyme inhibitor having good pharmacokinetics but relatively very low activity in cell culture (IC50 = 57 μM). The size variation of cycloalkyl ring also provided some interesting information about the binding mode of conformationally-flexible cyclooctyl ring on the inhibitors [22]. Further, they also designed some meta substituted carboxamide derivatives (19 and 20) which were significantly more active than the corresponding unsubstituted cyclooctylpyranone due to its additional hydrogen bonding by the amide group [23]. Substitution of cyclooctylpyranones with a wide variety of arenesulfonamides led to analogues with markedly enhanced antiviral activity. Especially p-cyanophenyl sulphonamide derivative (U-103017) (21) was found to be potent (Ki = 0.8 nM, IC50 = 1.5 μM). Resolution via chiral HPLC yielded more potent enantiomer having a Ki = 0.6 nM and an antiviral IC50 = 1 μM. Oral bioavailability of this compound ranged from 42% (rat) to 77% (dog) with t1/2 = 6 h [24], [25]. Saturation of the 5,6-double bond in the pyrone ring led to the identification of a cycloalkyldihydropyrone derivative (22) with superb binding affinity for the HIV protease (Ki values in the 0.05 nM) and excellent antiviral activity in cell culture, with significantly less ED50 value of 0.95 μM [26]. Unfortunately, all the previous agents (U-96988 and U-103017) had failed in the early 1990s, but Pharmacia & Upjohn (now Pfizer) was again successful in generating buzz in June of 1999 when they reported the promising results of its new compound tipranavir (PNU-140690) (23), a potent orally bioavailable HIV-1 protease analogue of the 5,6-dihydro-4-hydroxy-2-pyrones sulfonamide class (Ki = 8 pM) effective against the 96 out of 107 highly resistant strains [27]. Later in the year 2000, Boehringer Ingelheim (BI) acquired the exclusive rights to tipranavir (Aptivus®) and subsequently, it was approved by the US FDA in 2005.
Fig. 7.

Conformationally flexible analogues of 4-hydroxycoumarins.
A group of scientist Tummino et al. also did a parallel research for the development of non-peptidic inhibitor. They screened the Parke-Davis compound library using a high through-put assay and identified phenoxypropyl derivatives of 4-HC (PD099560) (24) and phenylthio derivative of 4-hydroxypyrone (25) as reversible competitive inhibitors effective at micro molar level. These two derivatives were excellent lead molecules in the development of therapeutically effective HIV-1 protease inhibitors, since they are small achiral nonpeptide molecules which can be easily synthesized and possessed promising pharmacological properties [28]. Binding mode of the compounds were studied employing a Monte Carlo-based docking procedure and X-ray crystal analysis. Subsequently, several analogues were prepared to test the binding interactions and improve the overall binding affinity. Amongst all, 4,7-dihydroxy-3-(4-(2-methoxyphenyl)butyl)-2H-chromen-2-one (26) emerged as the most active compound having IC50 0.52 μM. Further optimization of pyrone derivative yielded several potent analogues (27–32) having several fold higher binding affinity among which PD-178390 (31) EC50 of 200 nM) was selected for further preclinical evaluation [29], [30], [31], [32], [33], [34]. Unfortunately, no further clinical progress of this compound has been reported, probably as a consequence of the merging of the company with Pfizer. Parke-Davis laboratories also identified a dimethyl derivative of warfarin (32) which was nine-fold more potent than the parent compound (IC50 value 1.9 μ M) but later they did not pursue warfarin analogues (Fig. 8 ) [35].
Fig. 8.

4-hydroxycoumarins and related pyrones developed by Parke-Davis.
Collaborative endeavours of 3D database searching by Wang's group at the Laboratory of Medicinal Chemistry, and the Frederick Cancer Research and Development Centre, National Cancer Institute (NCI-US), resulted in a group of eight promising non-peptide HIV-1 protease inhibitors of 4-HCs along with some other class of compounds (33–40) (Fig. 9 ). The pharmacophore query used in the search was derived directly from the X-ray crystallography structures of protease-inhibitor complexes. Results of the in-vitro HIV protease bioassay evaluations clearly suggested that, while compounds containing a single 4-HC moiety are capable of binding to HIV-1 protease, compounds such as NSC 32180 (33) (ID50 0.32 μM) and 158393 (40) (ID50 1.7 μM, IC50 11.5 μM), which contain a “dimer” or “tetramer” of 4-HC, may be superior leads for further development. Compound NSC 158393 was also found to be an inhibitor of HIV-1 integrase and HIV-1 reverse transcriptase (RT). This compound therefore represents a multi-target lead for the development of potential anti-AIDS drugs. Based on their structural studies, they also proposed a pharmacophore essential for the HIV protease activity [36]. Further optimization of lead molecule NSC 158393 into structurally simpler coumarin derivatives through fragmentation of tetramer revealed that a coumarin dimer containing an arylmethylene (41) is an essential HIV Integrase pharmacophore [37]. Indeed, it turned out that modification of central phenyl ring with more lipophilic extended aromatic systems and addition of 7-hydroxy substituents to the coumarin rings (42) significantly enhanced its potency [38].
Fig. 9.

Dimers and tetramers of 4-hydroxycoumarins as dual integrase and reverse transcriptase inhibitors.
Considering the conspicuous significance of 4-hydroxycoumarinic derivatives as HIV-1 protease inhibitor, Kirkiacharian and co-workers have synthesized some sets of 3-substituted 4-HCs (43) with the objective to investigate the influence of some modifications at the 3-position of coumarin. The results demonstrated that any hydroxyl group nearer to the 4-hydroxyl group interfere with the hydrogen bond between this group and Asp25/Asp25′ of HIV-1 protease however, distant 7-hydroxy derivatives were most active anti-HIV agents. Replacement of 3-phenyl with benzyl group indicates that flexibility did not lead to any significant change in activity [39]. Similarly, another scientist Stanchev et al. also reported some 4-HC derivatives as HIV-1 protease inhibitors. All these compounds were tested for anti-HIV-1 protease activity in MT4 cells infected by HIV-1. The highest inhibition of HIV-1 PR (25%) and highest MT4 cell survival (78%) were demonstrated by compound containing m-nitrophenyl derivative (44) (Fig. 10 ) [40].
Fig. 10.

3-Substituted-4-hydroxycoumarins as HIV-1 protease inhibitor.
3.2. Pyranocoumarins
3.2.1. Tricyclic pyranocoumarins: khellactone
In an effort to identify novel anti-HIV agents from natural sources, Huang et al. reported a new class of tricyclic coumarin anti-HIV suksdofin, [(3′R,4′R)-3′-acetoxy-4′-(isovaleryloxy)-3′,4′-dihydroseselin] (45) which is a pyranocoumarin derivative having two cis-oriented acyl groups at the 3′ and 4′ positions obtained from the plant of Lomatium suksdorfii (Umbelliferae) as an anti-HIV agent [41]. The structures of suksdorin and related coumarin derivative pteryxin (46) was previously established by Willetti et al. [42]. Suksdorfin was found to inhibit HIV-1 replication in H9 lymphocytes with an in vitro EC50 value of 2.26 μM and a therapeutic index value of 30.6. They observed that varying the acyl groups at 3′ and 4′ positions of pyran ring greatly affect the anti-HIV potency of unsubstituted analogue seselin (47). This led to discovery of first synthetic lead compound 3′,4′-di-O-(−)-camphanoyl-(3′R,4′R)-(+)-cis-khellactone (DCK, 48), showing extremely potent inhibitory activity (EC50) 2.56 ± 10−4 μM) against HIV-1 replication in the H9 cell line and had a remarkable therapeutic index (TI = 136719) however, its other diastereoisomers were 10000 times less active [43]. Enthused by their promising early findings, Lee et al. reported numerous DCK analogues and its thorough structure-activity relationship (SAR) which demonstrates that, 3′R,4′R configurations and planarity of the coumarin ring system are essential structural features for anti-HIV activity; alkyl/O-alkyl substituents at the 3-, 4-, and 5-positions on the coumarin are favourable for enhanced anti-HIV activity and decreased toxicity of DCK [44], [45], [46], [47], [48]. However, development of DCK-analogues as effective anti-AIDS drugs has been hindered by problems of their low water solubility hence less bioavailability and their ineffectiveness against multidrug resistant HIV strains (RTMDR). Therefore, in furtherance to the development of DCK-related clinical drug candidates (49–51) they identified hydroxymethyl derivative of DCK (49) as first moderate orally bioavailable drug (water soluble, F = 15%) which acts inhibiting the production of double-stranded viral DNA from the single-stranded DNA intermediate [49]. Moreover, ring opened DCK analogues or seco-DCK (52) which contains lesser hydrogen bond acceptor also showed improvement in ADME properties and found to be effective against RTMDR strains [50]. Isomeric replacement of coumarin with chromone resulted in DCP analogues (53) which are potent anti-HIV agents against the resistant strains RTMDR [51]. Seco-DCP (54) and extended conjugated xanthenone analogues (55) were also turned out to be promising against these untreatable resistant strains (Fig. 11 ) [52], [53].
Fig. 11.

Tricyclic pyranocoumarins: Khellactones as anti-HIV-1 agents.
3.2.2. Tetracyclic dipyranocoumarins: calanolide
The discovery and development of the anti-HIV compounds calanolide A and its analogues is an interesting account of lead identification that brought a promising drug candidate to patients with HIV while addressing the challenging issue of drug resistance. In 1992, Kashman and co-workers at the NCI laboratory discovered a new class of coumarin derivatives known as calanolide from the tropical rainforest tree, Calophyllum lanigerum as anti-HIV chemotype. Calanolides (A, B and C) (56–58) are chemically tetracyclic dipyranocoumarins having three chiral carbon centres in the scaffold at the C-10, -11, and -12 positions. Studies with purified bacterial recombinant reverse transcriptases (RT) revealed that these calanolides are HIV-1 specific RT inhibitors active against both AZT-resistant G-9106 and pyridinone-resistant A17 strain. A 5-fold reduction in activity was observed for the 12-keto analogue 59 which contains one lesser chiral centre. Thus calanolides represent a substantial departure from the known class and therefore provide a novel new anti-HIV chemotype for drug development [54]. Because of their limited availability from natural resources, the total synthesis and racemic resolution of calanolides has been carried out. After bioassay, only (+)-calanolide A (NSC-675451) accounted for the anti-HIV activity, however its (−)- isomer was inactive [55]. Related novel RT inhibitors inophyllums A (60), B (61), C, E and P (62) were isolated from the fractions of Calophyllum inophyllum Linn. (Malaysian indigenous) [56] and cordatolide A (63) and B (64) were isolated from Calophyllum cordato-oblongum (a Srilankan indigenous) [57]. Prompted by these results, a number of closely related pyranocoumarins have been reported afterwards in different Calophyllum species and their semi-/synthetic derivatives (65–68) were also designed as potent anti-HIV agents [58], [59], [60]. Currently, (+)-calanolide A (56) (Sponsor: Sarawak MediChem Pharmaceuticals) has completed phase I/II 48-subject clinical trial as combination therapy for HIV (Fig. 12 ). The oral toxicity of the drug was minimal and reported adverse effects were dizziness, nausea, oily aftertaste.
Fig. 12.

Tetracyclic dipyranocoumarins as HIV-1 reverse transcriptase inhibitors.
3.3. Furanocoumarins
In search of natural anti-AIDS agents some furanocoumarins (69–77) have been isolated from the different plants viz. Ferula sumbul, and Prangos tschimganica (Fig. 13 ) [61], [62]. Among all the furanocoumarins, imperatronin (71) emerged as a promising anti-HIV agent. Imperatronin inhibited HIV replication in H9 lymphocytes with EC50 values of <0.10 mg/ml, and it inhibited uninfected H9 cell growth with IC50 values of >100 mg/ml, calculated therapeutic index (TI) value of >1000. Further studies on its mechanism of action revealed that unlike the other coumarins, imperatronin did not inhibit the reverse transcription nor the integrase but inhibit the Sp1-related genes (D1 expression) thereby arresting the cells at the G(1) phase of cell cycle. These results highlight the potential of SP1 transcription factor as a target of natural anti-HIV compounds, such as furancoumarins that might have potential therapeutic role in the management of AIDS [63].
Fig. 13.

Natural furanocoumarins as potential anti-HIV agents.
3.4. 3/4-Aryl coumarins
A Japanese research group led by Uchiumi et al. demonstrated that naturally occurring 3-phenylcoumarins (3-PC) compounds (78,79), generally present in tea leaves suppress TPA-induced HIV promoter activity. These compounds did not show any suppressive effect on the cytomegalovirus (CMV) promoter, suggesting that they act specifically on the HIV promoter. These results suggest that 3-phenylcoumarin derivatives might be potential candidate for developing novel anti-HIV drugs targeting at HIV promoter activity [64]. Encouraged by these observations, Olmedo et al. synthesized different 3-phenylcoumarin derivatives and evaluated their anti-HIV activity. The sulfanylphenylcoumarin derivative (80) emerged as the most potent inhibitor of HIV replication acting through dual inhibition of NF-κB and unspecific Tat functions whereas, 4-hydroxy derivative (81) (umbelliferone derivative), acts through specific Tat inhibitory activity. All the synthetic coumarins were more potent than mesuol against both NF-κB and Tat molecular targets but less effective in the RV assay. Thus, the presence of prenyl or prenacyl substituents attached to coumarin nucleus could be prerequisite structural factor reinforcing the antiviral activity (Fig. 14 ) [65].
Fig. 14.

3-Phenylcoumarins as potential candidate for anti-HIV agents.
By the high throughput screening (HTS) of 6664 compounds of RIKEN Natural Products Depository, Ong et al. identified a potent 3-PC based HTS lead NPD4456 (82) which on further optimization afforded vipirinin (83) which inhibited the cell cycle arrest activity of HIV-1 viral protein R (Vpr) in yeast and Vpr-dependent viral infection of human macrophages (Fig. 15 ). Detailed SAR and minimal pharmacophore responsible for Vpr inhibitory activity has also been explored. Binding assay of Vpr mutants revealed that the hydrophobic region about residues Glu-25 and Gln-65 are potentially involved in the binding of the inhibitor. Therefore, the study provides a new insight into molecular mechanism of Vpr-inhibitor which could be pragmatic for future drug development [66].
Fig. 15.

Anti-HIV agent vipirinin acting through novel mechanism.
Marquez et al. reported two natural 4-phenylcoumarins (4-PC), mesuol (84) and isomesuol (85) obtained from tree Marila pluricostata as effective anti-HIV agents acting through a novel mechanism of TNFα-mediated HIV-1-LTR transcriptional activity. These coumarins are devoid of the reverse transcription and intregration activity as like the other coumarins. Both the isomers suppress HIV-1 replication in Jurkat T cells with IC50 ranging from 2 to 2.5 μM and complete inhibition at 15 μM however only mesuol exhibited a potent anti-HIV activity in peripheral blood mononuclear cells acutely infected with HIV-1 clone. Thus, 4-phenyl-coumarins which are acting through a novel mechanism could be a potential lead for the development of potent therapeutic agent in the management of AIDS and drug-resistant HIV strains [67]. Later scientist from the same laboratory reported two more compounds (86, 87) of same class, 4-PC as potent HIV transcription inhibitors acting mainly through interference of NF-κB and Tat functions (Fig. 16 ) [68]. Likewise, several other studies also proved that 4-PC can be considered as promising model compounds for developing novel HIV-1 inhibitors [69].
Fig. 16.

Isoprenylated coumarins as HIV-1 inhibitors.
3.5. Hybrid coumarin analogues
The importance of the 4-HC pharmacophore for antiviral activity had been depicted in many studies. Therefore, Abd Elhafez et al. reported a series of thiazolidinone (88), oxadiazoline (89) and hydrazone (90) containing coumarin derivatives as antiviral agents. Among all these, compound having p-chlorophenyl-thiazolidinone derivative had the highest activity and was able to reduce the number of plaques by 30% at a concentration of 0.12 mg/mL (MIC) having cytotoxicity CD50 value 0.02 mg/mL [70]. Bhavsar et al., have also designed some novel anti-HIV agents by the molecular hybridization of coumarin-4-acetic acid and substituted 2-amino benzothiazoles. Most of these compounds showed moderate to potent activity against wild-type HIV-1 with an EC50 ranging from >7 to <100 μg/mL. SAR demonstrate that substitution of coumarin nucleus with hydroxyl group increases the activity whereas, bulky group decreases the anti-HIV property. The most potent compound of the series was N-1,3-benzo[d]thiazol-2-yl-2-(2-oxo-2H-chromen-4-yl)acetamide (91) (EC50 ≥ 7 μg/mL) [71] (see Fig. 17 ).
Fig. 17.

Hybrid coumarin derivatives as potent anti-HIV agents.
3.6. Miscellaneous
A new class of marine compounds for potential development of clinically useful integrase inhibitors was obtained from didemnid ascidians. Chemically these are a group of DOPA-(2-amino,3-(3′,4′-dihydroxyphenyl)propionic acid)-derived pyrrole alkaloids containing coumarin nucleus, commonly known as lamellarins. These compounds are effective against HIV-1 integrase protein which is an attractive target for anti-retroviral chemotherapy. Especially, a member of this family lamellarin α 20-sulfate (92) was found to inhibit integrase protein in vitro and viral replication in cultured cells. Its site of action on the integrase protein was also mapped by testing activity against deletion mutants of integrase [72]. Aesculetin (93) (esculetin, or cichorigenin), a dihydroxy derivative of coumarin obtained from the aerial part of Artemisia capillaris was also reported as an anti-HIV agent. It inhibited HIV replication in H9 lymphocyte cells with ED50 values of 2.51 μg/mL and therapeutic index of 11.2 (Fig. 18 ) [73].
Fig. 18.

Miscellaneous natural anti-HIV coumarins.
4. Coumarin analogues as anti-hepatitis virus agents
4.1. Hybrid coumarin conjugates
Viral hepatitis is a potentially serious infectious disease mainly affecting liver causing hepatocellular carcinoma and is a major cause of morbidity and mortality worldwide. Therefore, recent research efforts have been more focusing on development of antiviral therapy and vaccination strategies against these viruses. Several coumarins analogues also proved to have a substantial anti-hepatitis virus activity. Recently a new compound library of hybrid coumarin-benzimidazole derivatives connected through methylenethio linker (—SCH2 —) as anti-hepatitis C virus (HCV) agents were reported by Hwu et al. These conjugates exhibited very appealing anti-HCV activity and become promising leads in the further development of assortment of hybrid molecules. Preliminary antiviral activity against the Huh 5–2 replicon system demonstrated that compound containing 6-bromocoumarin (94) exhibit potent inhibitory effect with EC50 value of 3.4 μM. From the results of anti-HCV activity, it was anticipated that the substitution of coumarin at 6th position with lipophilic electron withdrawing atom bromine is essential for maintaining the anti-HCV activity. Glucosidation of these compounds by linking β-d-glucose peracetate have also resulted in further 4.8-fold increase in anti-HCV activity and 6-bromocoumarin glucopyranose derivative (95) emerged as a potent antiviral agent (EC50 = 4.1 μM) inhibiting HCV RNA replication by 90% and had no effect on cell proliferation. These data in the new compound library provided clues for the development of several coumarin-benzimidazole related conjugates as anti-HCV agents [74]. Subsequent modifications of benzimidazole nucleus into heterobicycles; imidazopyridine, purine, benzoxazole, and benzothiazole were also carried out to establish the structure-activity relationship (SAR) on the basis of new compounds library. Most promising compounds were imidazopyridine-coumarin (96), purine-coumarin (97), and benzoxazole-coumarin (98), which inhibited HCV replication at an EC50 of 6.8, 2.0, and 12 μM, respectively [75]. However, the benzothiazolyl-coumarin analogue (99) was least effective against the HCV. These encouraging results of purine-coumarin conjugates pave the way for the development of corresponding ribonucleoside-coumarin analogues. Seven among the 26 new compounds were found to inhibit HCV subgenomic replicon replication in the Huh 5–2 cell line. Amongst these purine analogues, 8-(Coumarin-3′-yl)methylthio-9-(β-D-ribofuranos-1″-yl)-purine (100) showed potent activity with EC50 value of 2.0 μM [76]. In another series, the benzimidazole ring was replaced by imidazole ring to further explore the SAR of imidazole–coumarin conjugates (101). It was observed that unsubstituted imidazole (i.e. hydrogen atom at the N1 position) and Cl, F, Br, Me, and OMe substitution at the coumarin nucleus resulted into compounds having significant activity [77]. Removal of methylenethio linker between the benzimidazole and coumarin afforded directly hinged coumarin-benzimidazole derivatives as potent anti-HCV agents. The dimethylimidazole-coumarin (102) and methylimidazole-benzocoumarin (103) exhibited promising EC50 values of as low as 3.0 and 5.5 μM, respectively. Further incorporation of a d-ribofuranose afforded corresponding nucleoside (104) with slight abatement in activity [78]. Thus, all these detailed studies underline the significant potential of coumarin-benzimidazole conjugates as a lead compound for the development of novel therapeutic agents for the treatment of HCV (Fig. 19 ).
Fig. 19.

Coumarin-benzimidazole conjugates linked directly or through linker.
Recently Manvar et al., 2016 have synthesized imidazo[1,2-a]pyridine-4-hydroxy-2H-coumarins (Fig. 20 ) as potential NS5B inhibitors. The ability of titled compounds to interact with protein was investigated by in silico screening against non-structural 5B (NS5B) viral protein. The hybrid coumarin derivative 105 interact with allosteric site of NS5B protein and exhibited a docked confirmation which was in good conformity with the reference ligand [79].
Fig. 20.

Imidazopyridine based coumarin NS5B inhibitors.
Using the same methylcoumarin pharmacophore, Peng and co-workers designed some novel anilinocoumarin derivatives as agents against HCV. It was observed that substituents bearing a strong electron-donating motif on the phenyl ring were crucial for anti-HCV activity and trimethoxyanilino derivative (106) emerged as potent antiviral agent with EC50 value of 12 μM (SI 10). The compound was found to act through a novel molecular mechanism by the induction of interferon-α (IFN)-mediated antiviral responses. Thus anilinocoumarin derivatives could be a lead compound in anti-HCV therapy for circumventing drug resistance due to HCV virus mutations (Fig. 21 ) [80].
Fig. 21.

Novel anilinocoumarin derivatives as anti-HCV agents.
4.2. 7-Hydroxy coumarin derivatives
A series of Mannich bases (108) of 7-hydroxycoumarins (osthole analogues, 107) were synthesized and evaluated as anti-flaviviridae in particular anti-HCV by Mazzei et al. An interesting pattern of SAR was observed among the different viruses. The 7-propyloxy derivatives were found to be active against bovine viral diarrhoea virus (BVDV) and 7-benzoyl derivatives were active against respiratory syncytial virus (RSV) whereas 7-hydroxy derivatives were ineffective against flaviviridae group of virus. Molecular docking studies revealed that DNA polymerase is potentially involved in the inhibition of HCV. Some unsymmetrical methylene derivatives 109–111 (namely coumarins bridged to chromones or indoles) were also reported as moderately active antiviral agents [81]. Another 7-hydroxycoumarins natural compound collinin (112), which is a geranyloxycoumarin isolated from the root bark of Zanthoxylum schinifolium (Rutaceae) showed promising anti-hepatitis B virus (HBV) replication activities (IC50 = 17.1 μgmL−1) associated with inhibition of the DNA-synthesis step of virus life-cycles (Fig. 22 ) [82].
Fig. 22.

7-Hydroxycoumarin derivatives as anti-HCV and anti-HBV agents.
4.3. Coumarin phytoestrogens
Basu et al. identified and characterized coumestans, an isoflavonoid class of phytoestrogens as a novel class of anti-NS5B agents. The hepatitis C virus (HCV) NS5B is essential for viral RNA replication and is therefore a key target HCV inhibitor. A naturally occurring coumestan, wedelolactone (113) exhibited NS5B RNA-dependent RNA polymerase (RdRp) inhibition in micromolar range, which prompted them to synthesise another four different synthetic analogues (114–117). LQB34 (115) was found to be most effective NS5B inhibitor with IC50 value of 18.5 μM (∼2–20 fold active). They further concluded that the anti-NS5B efficacy of coumestans are inversely correlated with their hydrophobicity. Molecular docking studies revealed that, modification of benzofuran and coumarin core along with peripheral hydroxyl and methoxy groups will be valuable affording compounds which might be ideal inhibitors accommodating the NS5B binding pocket (Fig. 23 ) [83].
Fig. 23.

Coumestans analogues as novel potent NS5B inhibitors.
5. Coumarin analogues as anti-influenza agents
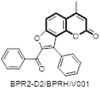
An angular furanocoumarin such as angelicin (118) was found to be safer pharmacophoric element for the anti-influneza activity. However, its linear analogue psoralen (69) is a highly phototoxic isomer due to its ability to cross-link deoxyribonucleic acid (DNA). A novel angelicin derivative 119 as potent influenza A (H1N1) virus inhibitor was identified by using a cell-based high-throughput screening of around twenty thousand compounds. The structural novelty of the hit molecule encouraged them to further pursue the lead-optimization of this class through SAR exploration. Thiophenoyl substituted angellicin (120) emerged as the optimised lead exhibiting 64-fold enhanced activity compared to hit. The compound was found to be broad spectrum anti-influenzal agent effective against both influenza A (H3N2) and influenza B strains same as drug zanamivir. Further plasmid-based reverse genetics using luciferase assay yielded key insight into the molecular mechanism of angellicin derivatives which point toward viral ribonucleoprotein (RNP) as a probable molecular target for these agents [84]. Later benzoyl substituted angelicin (121) (BPR2-D2) also emerged as a novel inhibitor against influenza virus exhibiting excellent antiviral efficacy against the oseltamivir-resistant virus (EC50 = 0.021–0.040 μM). BPR2-D2 also exhibited a broad antiviral spectrum against various strains of influenza A and influenza B viruses. The compound acts by targeting viral RNPs that are responsible for viral RNA synthesis [85]. This same molecule (BPR2-D2) was later established as an effective anti-HIV agent (BPRHIV001) which inhibits Tat-Mediated Transcription through Akt pathway (EC50 = 1.3 nM) [86]. Thus this molecule has the potential to become a promising lead compound for the development of a novel therapeutic agent effective against both HIV-1 and influenza infections (Fig. 24 ).
Fig. 24.

Angelicin derivatives as dual anti-influenza and anti-HIV agents.
6. Coumarin analogues as anti-herpes simplex agents
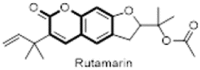
Over the years, many natural plant based compounds have been reported as potential antiviral agents effective against the Herpes simplex viruses (Fig. 25 ). In the year 1995, Ishikawa et al. isolated a unique coumarin-naphthoquinone dimer called toddacoumaquinone (122) from Toddalia asiatica (Rutaceae) and evaluated its antiviral activities against Herpes simplex virus type 1 (HSV-1), Herpes simplex virus type 2 (HSV-2) and human immunodeficiency virus type 1 (HIV-1). The compound was found to be weakly active against HSV (EC50 = 10 μg/mL) but was ineffective against HIV [87]. Xu and colleagues have also demonstrated that inhibition of human herpesvirus 8 (HHV-8) replication could be achieved by the use of (+) rutamarin (123) which is a natural furanocoumarin obtained from the plant Ruta graveolens L (common rue). In this study, a total of 7200 compounds were virtually screened for their topoisomerase II inhibition properties and subsequently tested for antiviral activities. Out of these, natural product (+) rutamarin, exhibited significant inhibition of both viral DNA synthesis and virion production, with relatively low cytotoxicity at a concentration of 20 μM. Precisely, (+) rutamarin exhibited excellent efficiency in blocking Kaposi's sarcoma-associated herpesvirus (KSHV) lytic DNA replication (EC50 = 1.62 μM) which is a proven etiological cause of Kaposi's sarcoma (KS) and other AIDS-associated malignancies. Thus, (+) rutamarin could be a potent clinical candidate for the treatment of human diseases associated with KSHV infection [88]. The sesquiterpene coumarin, kellerin (124) isolated from the gum resin of Ferula asafoetida significantly reduced the viral titre of the herpes virus type 1 (HSV-1) DNA viral strains KOS at concentrations of 10, 5 and 2.5 μg/mL with an inhibition rate of 98 ± 5.2%, 80% and 65%, respectively. Kellerin was effective in EC50 as 38 μg/mL [89].
Fig. 25.

Different plant based coumarin compounds effective against herpes viruses.
Compounds having 3-hydroxycoumarin derivatives were also studied as antiviral agents but the results were somewhat not encouraging. This might be due to its non-conformity to the 4-HC pharmacophore. Way back in 1968 Giannella et al. reported some 2,4-dioxo-3-hydroxyiminochromane derivatives (125) showing good antiviral activity however, these compounds developed a high degree of cytotoxicity [90]. In order to lower their toxicity, the active molecule was modified while preserving the cyclic α-dicarbonyl structure and prepared several Mannich bases 4-N,N-dialkylaminomethyl-3-hydroxycoumarins (126). Preliminary examination of these compounds did not show any appreciable antiviral activity (Fig. 26 ) [91]. However, the 4-bromobenzylidene derivative of bis(4-hydroxycoumarin) (127) were found to exert some inhibitory activity against HSV-1 (KOS), HSV-2 (G), vaccinia virus and HSV-1 TK-KOS (ACVr) in the range of 9–12 μM [92].
Fig. 26.

Hydroxycoumarin derivatives possessing inhibitory activity against HSV.
7. Coumarins conjugates as inhibiting agents for chikungunya virus
In continuation of studies on hybrid coumarin-benzimidazole and their appealing anti-HCV activity, Hwu et al. investigated a number of new hybrid compounds of uracil–coumarin–arenes as potential inhibiting agents for chikungunya virus (CHIKV) (Fig. 27 ). A set of doubly (uracil–coumarin) and triply conjugates (uracil–coumarin–arenes) were evaluated to establish their SAR. The extension of the doubly conjugated uracil–coumarins to triply conjugated uracil–coumarin–arenes by use of the –SO2– linker was vital to their anti-CHIKV activity. Bezouracil derivatives (128) had better selectivity indexes as compared to uracil (129) or thymine [93].
Fig. 27.

Uracil containing coumarin derivatives as chikungunya virus inhibitors.
Pohjala et al. screen around 356 natural compounds and clinically approved drugs against the chikungunya virus (CHIKV) replicon cell line and Semliki Forest virus (SFV) an alpha virus in 96-well format. One of the coumarin anti-SFV screening hits exhibited excellent antiviral IC50 values in the low micromolar range. Coumarin 30 (130) was found to exhibit an IC50 value of 0.4 μM against SFV and a selectivity index of 308 and also found to be the most potent inhibitor of CHIKV-Rluc as well (IC50 value of 6.4 μM). Thenoyl derivative (131) was also effective against CHIKV (Fig. 28 ) [94].
Fig. 28.

7-Diethylamino coumarins effective against chikungunya virus.
8. Conclusion
The present review aims to provide an important insight into the design of newer coumarin based antiviral agents. Since the 1990s, significant researches have been devoted to the development of antiviral coumarins. A diverse class of natural, semisynthetic and synthetic coumarin analogues have been reported as promising antiviral agents effective against the broad spectrum of viruses. Among these natural coumarins remain a fascinating rich source of lead compounds and boast ingenious functionalized motifs and proven to be important precursors to many effective antiviral drugs. Several classes of coumarins which were abandoned can be further improved in terms of efficacy and selectivity for the target or achieving optimal pharmacokinetic and pharmacodynamic properties. Given all these promising results and the potential antiviral activity, coumarins could be used in future in the clinic. Moreover, this review will surely provide a new horizon for research on antiviral agents and give better understanding of role of these molecules in diseases and how therapeutic agents could be most effective.
Conflict of interest
The authors confirm that this article content has no conflicts of interest.
Acknowledgements
The authors wish to express their gratitude and appreciation to School of Chemical Science, Universiti Sains Malaysia, Penang for supporting this work. This work was funded through Research Grant No. (FRGS/203/6711462).
Contributor Information
Mohd. Zaheen Hassan, Email: drzahin@gmail.com.
Hasnah Osman, Email: ohasnah@usm.my.
Abbreviations
- HIV
Human Immunodeficiency Virus
- HBV
Hepatitis B Virus
- HCV
Hepatitis C Virus
- HSV
Herpes Simplex Virus
- CMV
Cytomegalovirus
- MERS
Middle East Respiratory Syndrome
- SARS
Severe Acute Respiratory Syndrome
- DNA
Deoxyribonucleic Acid
- RNA
Ribonucleic Acid
- IMP
Inosine 5′-Monophosphate
- CTP
Cytidine Triphosphate
- 4-HC
4-Hydroxycoumarin
- SBDD
Structure-Based Drug Discovery
- US FDA
United States Food and Drug Administration
- NCI
National Cancer Institute
- RT
Reverse Transcriptase
- DCK
3′,4′-Di-O-(−)-camphanoyl-(3′R,4′R)-(+)-cis-khellactone
- RTMDR
Multidrug Resistant HIV Strains
- AZT
Zidovudine
- 4-PC
4-Phenylcoumarin
- IC50
Half Maximal Inhibitory Concentration
- EC50
Half Maximal Effective Concentration
- BVDV
Bovine Viral Diarrhoea Virus
- RSV
Respiratory Syncytial Virus
- RNP
Ribonucleoprotein
- HHV-8
Human Herpesvirus 8
- CHIKV
Chikungunya Virus
- SFV
Semliki Forest Virus
References
- 1.Emerging Infectious Diseases: MERS-COV, Avian Influenza Remind us of the Ongoing Challenge. 2016. http://www.infectioncontroltoday.com/ (accessed 15.05.16) [Google Scholar]
- 2.Morens D.M., Fauci A.S. Emerging infectious diseases in 2012: 20 years after the institute of medicine report. MBio. 2012;3:e00494–e00512. doi: 10.1128/mBio.00494-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howard C.R., Fletcher N.F. Emerging virus diseases: can we ever expect the unexpected? Emerg. Microb. Infect. 2012;1:e46. doi: 10.1038/emi.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King D.H. History, pharmacokinetics, and pharmacology of acyclovir. J. Am. Acad. Dermatol. 1988;18:176–179. doi: 10.1016/s0190-9622(88)70022-5. [DOI] [PubMed] [Google Scholar]
- 5.Pennings P.S. HIV drug resistance: problems and perspectives. Inf. Dis. Rep. 2013;5:1–5. doi: 10.4081/idr.2013.s1.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mishra K.P., Sharma N., Diwaker D., Ganju L., Singh S.B. Plant derived antivirals: a potential source of drug development. J. Virol. Antivir. Res. 2013;2:2. [Google Scholar]
- 7.Penta S. Academic Press; 2015. Advances in Structure and Activity Relationship of Coumarin Derivatives. [Google Scholar]
- 8.Torres F.C., Brucker N., Andrade S.F., Kawano D.F., Garcia S.C., Poser G.L., Eifler-Lima V.L. New insights into the chemistry and antioxidant activity of coumarins. Curr. Top. Med. Chem. 2014;14:2600–2623. doi: 10.2174/1568026614666141203144551. [DOI] [PubMed] [Google Scholar]
- 9.Kostova I., Raleva S., Genova P., Argirova R. Structure-activity relationships of synthetic coumarins as HIV-1 inhibitors. Bioinorg. Chem. Appl. 2006;2006:68274. doi: 10.1155/BCA/2006/68274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kostova I. Coumarins as inhibitors of HIV reverse transcriptase. Curr. HIV. Res. 2006;4:347–363. doi: 10.2174/157016206777709393. [DOI] [PubMed] [Google Scholar]
- 11.Yu D., Suzuki M., Xie L., Morris-Natschke S.L., Lee K.H. Recent progress in the development of coumarin derivatives as potent anti-HIV agents. Med. Res. Rev. 2003;23:322–345. doi: 10.1002/med.10034. [DOI] [PubMed] [Google Scholar]
- 12.De Clercq E. Chemotherapy of viral infections. In: Samuel B., editor. Medical Microbiology. University of Texas Medical Branch at Galveston; Texas: 1996. [PubMed] [Google Scholar]
- 13.L. Menendez-Arias, F. Gago, Antiviral agents: structural basis of action and rational design, in: G.M. Mauricio (Ed.), Structure and Physics of Viruses: an Integrated Textbook, Springer Science, pp 599–630. [DOI] [PubMed]
- 14.Miller M.D., Hazuda D.J. New antiretroviral agents: looking beyond protease and reverse transcriptase. Curr. Opin. Microbiol. 2001;4:535–539. doi: 10.1016/s1369-5274(00)00247-2. [DOI] [PubMed] [Google Scholar]
- 15.De Clercq E. Molecular targets for antiviral agents. J. Pharm. Exp. Ther. 2001;297:1–10. [PubMed] [Google Scholar]
- 16.Ong E.B., Watanabe N., Saito A., Futamura Y., Abd El Galil K.H., Koito A., Najimudin N., Osada H. Vipirinin, a coumarin-based HIV-1 Vpr inhibitor, interacts with a hydrophobic region of VPR. J. Biol. Chem. 2011;286:14049–14056. doi: 10.1074/jbc.M110.185397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stubbs M.T., Bode W. A player of many parts: the spotlight falls on thrombins structure. Thromb. Res. 1993;69:1–58. doi: 10.1016/0049-3848(93)90002-6. [DOI] [PubMed] [Google Scholar]
- 18.Babe L.M., Craik C.S. Viral proteases: evolution of diverse structural motifs to optimize function. Cell. 1997;91:427–430. doi: 10.1016/s0092-8674(00)80426-2. [DOI] [PubMed] [Google Scholar]
- 19.Bourinbaiar A.S., Tan X., Nagorny R. Effect of the oral anticoagulant, warfarin, on HIV-1 replication and spread. AIDS. 1993;7:129–130. doi: 10.1097/00002030-199301000-00022. [DOI] [PubMed] [Google Scholar]
- 20.Thaisrivongs S., Tomich P.K., Watenpaugh K.D., Chong K.T., Howe W.J., Yang C.P., Strohbach J.W., Turner S.R., McGrath J.P., Bohanon M.J., Lynn J.C., Mulichak A.M., Spinelli P.A., Hinshaw R.R., Pagano P.J., Moon J.B., Ruwart M.J., Wilkinson K.F., Rush B.D., Dalga G.L.Z.R.J., Schwende F.J., Howard G.M., Padbury G.E., Toth L.N., Zhao Z., Koeplinger K.A., Kakuk T.J., Cole S.L., Zaya R.M., Piper R.C., Jeffrey P. Structure-based design of HIV protease inhibitors: 4-hydroxycoumarins and 4-hydroxy-2-pyronesas non-peptidic inhibitors. J. Med. Chem. 1994;37:3200–3204. doi: 10.1021/jm00046a002. [DOI] [PubMed] [Google Scholar]
- 21.Thaisrivongs S., Watenpaugh K.D., Howe W.J., Tomich P.K., Dolak L.A., Chong K.T., Tomich C.S.C., Tomasselli A.G., Turner S.R., Strohbach J.W., Mulichak A.M., Janakiraman M.N., Moon J.B., Lynn J.C., Horng M.M., Hinshaw R.R., Kimberly A., Rothrock D.J. Structure-based design of novel HIV protease inhibitors: carboxamide-containing 4-hydroxycoumarinsand 4-hydroxy-2-pyrones as potent nonpeptidic inhibitors. J. Med. Chem. 1995;38:3624–3637. doi: 10.1021/jm00018a023. [DOI] [PubMed] [Google Scholar]
- 22.Romines K.R., Watenpaugh K.D., Tomich P.K., Howe W.J., Morris J.K., Lovasz K.D., Mulichak A.M., Finze B.C., Lynn J.C., Horng M.M., Schwende F.J., Ruwart M.J., Zipp G.L., Chong K.T., Dolak L.A., Toth L.N., Howard G.M., Rush B.D., Wilkinson K.F., Possert P.L., Dalga R.J., Hinshaw R.R. Use of medium-sized cycloalkyl rings to enhance secondary binding: discovery of a new class of Human Immunodeficiency Virus (HIV) protease inhibitors. J. Med. Chem. 1995;38:1884–1891. doi: 10.1021/jm00011a008. [DOI] [PubMed] [Google Scholar]
- 23.Romines K.R., Watenpaugh K.D., Howe W.J., Tomich S.P.K., Lovasz K.D., Morris J.K., Janakiraman M.N., Lynn J.C., Horng M.M., Chong K.T., Hinshaw R.R., Dolak L.A. Structure-based design of nonpeptidic HIV protease inhibitors from a cyclooctylpyranone lead structure. J. Med. Chem. 1995;38:4463–4473. doi: 10.1021/jm00022a011. [DOI] [PubMed] [Google Scholar]
- 24.Skulnick H.I., Johnson P.D., Howe W.J., Tomich P.K., Chong K.T., Watenpaugh K.D., Janakiraman M.N., Dolak L.A., McGrath J.P., Lynn J.C., Horng M.M., Hinshaw R.R., Zipp G.L., Ruwart M.J., Schwende F.J., Zhong W.Z., Padbury G.Y., Dalga R.J., Shiou L., Rush P.L.P.B.D., Wilkinson K.F., Howard G.M., Toth L.N., Williams M.G., Kakuk T.J., Cole S.L., Zaya R.M., Lovasz K.D., Morris J.K., Romines K.R., Thaisrivongs S., Aristof P.A. Structure-based design of sulfonamide-substituted non-peptidic HIV protease inhibitors. J. Med. Chem. 1995;38:4968–4971. doi: 10.1021/jm00026a002. [DOI] [PubMed] [Google Scholar]
- 25.Skulnick H.I., Johnson P.D., Aristoff P.A., Morris J.K., Lovasz K., Howe W.J., Watenpaugh K.D., Janakiraman M.N., Anderson D.J., Reischer R.J., Schwartz T.M., Banitt L.S., Tomich P.K., Lynn J.C., Horng M.M., Chong K.T., Hinshaw R.R., Dolak L.A., Seest E.P., Schwende F.J., Rush B.D., Howard G.M., Toth L.N., Wilkinson K.R., Kakuk T.J., Johnson O.C.W., Cole O.S.L., Zaya O.R.M., Zipp O.G.L., Possert P.L., Dalga R.J., Zhong W.Z., Williams M.G., Romines K.R. Structure-based design of nonpeptidic HIV protease inhibitors: the sulfonamide-substituted cyclooctylpyranones. J. Med. Chem. 1997;40:1149–1164. doi: 10.1021/jm960441m. [DOI] [PubMed] [Google Scholar]
- 26.Romines K.R., Morris J.K., Howe W.J., Tomich P.K., Horng M.M., Chong K.T., Hinshaw R.R., Anderson D.J., Strohbach J.W., Turner S.R., Mizsak S.A. Cycloalkylpyranones and cycloalkyldihydropyrones as HIV protease inhibitors: exploring the impact of ring size on structure-activity relationships. J. Med. Chem. 1996;39:4125–4130. doi: 10.1021/jm960296c. [DOI] [PubMed] [Google Scholar]
- 27.Thaisrivongs S., Strohbach J.W. Structure-based discovery of Tipranavir disodium (PNU-140690E): a potent, orally bioavailable, nonpeptidic HIV protease inhibitor. Biopolymers. 1999;51:51–58. doi: 10.1002/(SICI)1097-0282(1999)51:1<51::AID-BIP6>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 28.Tummino P.J., Ferguson D., Hupe L., Hupe D. Competitive inhibition of HIV-1 protease by 4-hydroxy-benzopyran-2-ones and by 4-hydroxy-6-phenylpyran-2-ones. Biochem. Biophys. Res. Commun. 1994;200:1658–1664. doi: 10.1006/bbrc.1994.1642. [DOI] [PubMed] [Google Scholar]
- 29.Lunney E.A., Hagen S.E., Domagala J.M., Humblet C., Kosinski J., Tait B.D., Warmus J.S., Wilson M., Ferguson D., Hupe D., Tummino P.J., Baldwin E., Bhat T.N., Liu B.S.J.W. Erickson, A novel nonpeptide HIV-1 protease inhibitor: elucidation of the binding mode and its application in the design of related analogs. J. Med. Chem. 1994;37:2664–2677. doi: 10.1021/jm00043a006. [DOI] [PubMed] [Google Scholar]
- 30.Prasad J.V.N.V., Para K.S., Lunney E.A., Ortwine D.F., Dunbar J.B., Jr., Ferguson D., Tummino P.J., Hupe D., Tait B.D. Novel series of achiral, low molecular weight, and potent HIV-1 protease inhibitors. J. Am. Chem. Soc. 1994;116:6989–6990. [Google Scholar]
- 31.Prasad J.V., Para K.S., Tummino P.J., Ferguson D., McQuade T.J., Lunney E.A., Rapundalo S.T., Batley B.L., Hingorani G., Domagala J.M., Gracheck S.J., Bhat T.N., Liu B., Baldwin E.T., Erickson J.W., Sawyer T.K. Nonpeptidic potent HIV-1 protease inhibitors:(4-hydroxy-6-phenyl-2-oxo-2h-pyran-3-yl)thiomethanes that span P1-P2′ subsites in a unique mode of active site binding. J. Med. Chem. 1995;38:898–905. doi: 10.1021/jm00006a007. [DOI] [PubMed] [Google Scholar]
- 32.Thaisrivongs S., Romero D.L., Tommasi R.A., Janakiraman M.N., Strohbach J.W., Turner S.R., Biles C., Morge R.R., Johnson P.D., Aristoff P.A., Tomich P.K., Lynn J.C., Horng M.M., Chong K.T., Hinshaw R.R., Howe W.J., Finzel B.C., Watenpaugh K.D. Structure-based design of HIV protease inhibitors: 5,6-dihydro-4-hydroxy-2-pyrones as effective, nonpeptidic inhibitors. J. Med. Chem. 1996;39:4630–4642. doi: 10.1021/jm960228q. [DOI] [PubMed] [Google Scholar]
- 33.Tait B.D., Hagen S., Domagala J., Ellsworth E.L., Gajda C., Hamilton H.W., Prasad J.V.N.V., Ferguson D., Graham N., Hupe D., Nouhan C., Tummino P.J., Humblet C., Lunney E.A., Pavlovsky A., Rubin J., Gracheck S.J., Baldwin E.T., Bhat T.N., Erickson J.W., Gulnik S.V., Liu B. 4-Hydroxy-5,6-dihydropyrones. 2. Potent non-peptide inhibitors of HIV protease. J. Med. Chem. 1997;40:3781–3792. doi: 10.1021/jm970615f. [DOI] [PubMed] [Google Scholar]
- 34.Prasad J.V.N.V., Boyer F.E., Domagala J.M., Ellsworth E.L., Gajda C., Hamilton H.W., Hagen S.E., Markoski L.J., Steinbaugh B.A., Tait B.D., Humblet C., Lunney E.A., Pavlovsky A., Rubin J.R., Ferguson D., Graham N., Holler T., Hupe D., Nouhan C., Tummino P.J., Urumov A., Zeikus E., Zeikus G., Gracheck S.J., Saunders J.M., VanderRoest S., Brodfuehrer J., Iyer K., Sinz M., Gulnik S.V., Erickson J.W. Nonpeptidic HIV protease inhibitors possessing excellent antiviral activities and therapeutic indices. PD 178390: a lead HIV protease inhibitor. Bioorg. Med. Chem. 1999;7:2775–2800. doi: 10.1016/s0968-0896(99)00215-1. [DOI] [PubMed] [Google Scholar]
- 35.Tummino P.J., Prasad J.V.N.V., Ferguson D., Nouhan C., Graham N., Domagala J.M., Ellsworth E., Gajda C., Hagen S.E., Lunney E.A., Para K.S., Tait B.D., Pavlovsky A., Erickson J.W., Gracheck S., McQuade T.J., Hupe D.J. Discovery and optimization of nonpeptide hiv-1 protease inhibitors. Bioorg. Med. Chem. 1996;4:1401–1410. doi: 10.1016/0968-0896(96)00134-4. [DOI] [PubMed] [Google Scholar]
- 36.Wang S., Milne G.W.A., Yan X., Posey I.J., Nicklaus M.C., Graham L., Rice W.G. Discovery of novel, non-peptide HIV-1 protease inhibitors by pharmacophore searching. J. Med. Chem. 1996;39:2047–2054. doi: 10.1021/jm950874+. [DOI] [PubMed] [Google Scholar]
- 37.Mazumder A., Wang S., Neamati N., Nicklaus M., Sunder S., Chen J., Milne G.W., Rice W.G., Burke T.R., Pommier Y., Jr. Antiretroviral agents as inhibitors of both human immunodeficiency virus type 1 integrase and protease. J. Med. Chem. 1996;39:2472–2481. doi: 10.1021/jm960074e. [DOI] [PubMed] [Google Scholar]
- 38.Zhao H., Neamati N., Hong H., Mazumder A., Wang S., Sunder S., Milne G.W.A., Pommier Y., Burke T.R., Jr. Coumarin-based inhibitors of HIV integrase. J. Med. Chem. 1997;40:242–249. doi: 10.1021/jm960450v. [DOI] [PubMed] [Google Scholar]
- 39.Kirkiacharian S., Thuy D.T., Sicsic S., Bakhchinian R., Kurkjian R., Tonnaire T. Structure-activity relationships of some 3-substituted-4-hydroxycoumarins as HIV-1 protease inhibitors. Farmaco. 2002;57:703–708. doi: 10.1016/s0014-827x(02)01264-8. [DOI] [PubMed] [Google Scholar]
- 40.Stanchev S., Jensen F., Hinkov A., Atanasov V., Genova-Kalou P., Argirova R., Manolov I. Synthesis and inhibiting activity of some 4-hydroxycoumarin derivatives on HIV-1 protease. ISRN Pharm. 2011:137637. doi: 10.5402/2011/137637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang L., Kashiwada Y., Cosentino L.M., Fan S., Lee K.H. 3′,4′-Di-o-(−)-camphanoyl-(+)-ciskhellactone and related compounds: a. new class of potent anti-HIV agents. Bioorg. Med. Chem. Lett. 1994;4:593–598. [Google Scholar]
- 42.Willette R.E., Soine T.O. Coumarins I: isolation, purification, and structure determination of pteryxin and suksdorfin. J. Pharm. S. C. 1962;51:149–156. doi: 10.1002/jps.2600510215. [DOI] [PubMed] [Google Scholar]
- 43.Huang L., Kashiwada Y., Cosentino L.M., Fan S., Chen C.H., McPhail A.T., Fujioka T., Mihashi K., Lee K.H. Anti-AIDS agents. 15. Synthesis and anti-hiv activity of dihydroseselins and related analogs. J. Med. Chem. 1994;37:3947–3955. doi: 10.1021/jm00049a014. [DOI] [PubMed] [Google Scholar]
- 44.Takeuchi Y., Xie L., Cosentino L.M., Lee K.H. Anti-AIDS agents 28. Synthesis and anti-HIV activity of methoxy substituted 3′,4′-di-O-(-)-camphanoyl-(+)-cis-khellactone (DCK) analogues. Bioorg. Med. Chem. Lett. 1997;7:2573–2578. doi: 10.1016/s0960-894x(98)00367-9. [DOI] [PubMed] [Google Scholar]
- 45.Yang Z.Y., Xia Y., Xia P., Cosentino L.M., Lee K.H. Anti-AIDS agents 31.1 synthesis and anti-HIV activity of 4-substituted 3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (DCK) thiolactone analogs. Bioorg. Med. Chem. Lett. 1998;12:1483–1486. doi: 10.1016/s0960-894x(98)00254-6. [DOI] [PubMed] [Google Scholar]
- 46.Xie L., Takeuchi Y., Cosentino L.M., Lee K.H. Anti-aids agents 33.1 Synthesis and anti-HIV activity of mono-methyl substituted 3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone (DCK) analogues. Bioorg. Med. Chem. Lett. 1998;8:2151–2156. doi: 10.1016/s0960-894x(98)00367-9. [DOI] [PubMed] [Google Scholar]
- 47.Xie L., Allaway G., Wild C., Kilgore N., Lee K.H. Anti-AIDS agents. Part 47: synthesis and anti-HIV activity of 3-substituted 3′,4′-Di-O-(S)-camphanoyl-(3′R,4′R)-(+)-cis-khellactone derivatives. Bioorg. Med. Chem. Lett. 2001;11:2291–2293. doi: 10.1016/s0960-894x(01)00437-1. [DOI] [PubMed] [Google Scholar]
- 48.Xie L., Takeuchi Y., Cosentino L.M., Lee K.H. Anti-AIDS Agents. 37. Synthesis and structure-activity relationships of(3¢R,4¢R)-(+)-cis-khellactone derivatives as novel potent anti- HIV Agents. J. Med. Chem. 1999;42:2662–2672. doi: 10.1021/jm9900624. [DOI] [PubMed] [Google Scholar]
- 49.Xie L., Yu D., Wild C., Allaway G., Turpin J., Smith P.C., Lee K.H. Anti-AIDS agents 52. synthesis and anti-hiv activity of hydroxymethyl (3′R,4′R)-3′, 4′-Di-O-(S)-camphanoyl- (+)-cis-khellactone (DCK) derivatives. J. Med. Chem. 2004;47:756–760. doi: 10.1021/jm030416y. [DOI] [PubMed] [Google Scholar]
- 50.Tang J., Qian K., Zhang B.N., Chen Y., Xia P., Yu D., Xia Y., Yang Z.Y., Chen C.H., Morris-Natschke S.L., Lee K.H. Anti-AIDS agents 82: synthesis of seco-(3′R,4′R)-3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone (DCK) derivatives as novel anti-HIV agents. Bioorg. Med. Chem. 2010;18:4363–4373. doi: 10.1016/j.bmc.2010.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu D., Chen C.H., Brossi A., Lee K.H. Anti-AIDS Agents. 60. Substituted 3′R,4′R-Di-O-(-)-camphanoyl-2′,2′-dimethyldihydropyrano[2,3-f]chromone (DCP) analogues as potent anti-HIV agents. J. Med. Chem. 2004;47:4072–4082. doi: 10.1021/jm0400505. [DOI] [PubMed] [Google Scholar]
- 52.Chen Y., Cheng M., Liu F.Q., Xia P., Qian K., Yu D., Xia Y., Yang Z.Y., Chen C.H., Morris-Natschke S.L., Lee K.H. Anti-AIDS agents 86. Synthesis and anti-HIV evaluation of 2′,3′-seco-3′-nor DCP and DCK analogues. Eur. J. Med. Chem. 2011;46:4924–4936. doi: 10.1016/j.ejmech.2011.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou T., Shi Q., Chen C.H., Huang Li, Ho P., Morris-Natschke S.L., Lee K.H. Anti-AIDS agents 85. Design, synthesis, and evaluation of 1R,2R-dicamphanoyl-3,3-dimethyldihydropyrano-[2,3-c]xanthen-7(1H)-one (DCX) derivatives as novel anti-HIV agents. Eur. J. Med. Chem. 2012;47:86–96. doi: 10.1016/j.ejmech.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kashman Y., Gustafson K.R., Fuller R.W., Cardellina J.H., McMahon J.B., Currens M.J., Buckheit R.W., Jr., Hughes S.H., Cragg G.M., Boyd M.R. The calanolides, a novel HIV-inhibitory class of coumarin derivatives from the tropical rainforest tree, Calophyllum lanigerum. J. Med. Chem. 1992;35:2735–2743. doi: 10.1021/jm00093a004. [DOI] [PubMed] [Google Scholar]
- 55.Flavin M.T., Rizzo J.D., Khilevich A., Kucherenko A., Sheinkman A.K., Vilaychack V., Lin L., Chen W., Greenwood E.M., Pengsuparp T., Pezzuto J.M., Hughes S.H., Flavin T.M., Cibulski M., Boulanger W.A., Shone R.L., Xu Z.Q. Synthesis, chromatographic resolution, and anti-human immunodeficiency virus activity of (±)-Calanolide A and its enantiomers. J. Med. Chem. 1996;39:1303–1313. doi: 10.1021/jm950797i. [DOI] [PubMed] [Google Scholar]
- 56.Patil A.D., Freyer A.J., Eggleston D.S., Haltiwanger R.C., Bean M.F., Taylor P.B., Caranfa M.J., Breen A.L., Bartus H.R., Johnson R.K., Hertzberg R.P., Westley J.W. The inophyllums, novel inhibitors of HIV-1 reverse transcriptase isolated from the malaysian tree, Calophyllum inophyllum Linn. J. Med. Chem. 1993;36:4131–4138. doi: 10.1021/jm00078a001. [DOI] [PubMed] [Google Scholar]
- 57.Dharmaratne H.R.W., Wanigasekera W.M.A.P., Mata-Greenwood E., Pezzuto J.M. Inhibition of human immunodeficiency virus type 1 reverse transcriptase activity by cordatolides isolated from Calophyllum cordato-oblongum. Planta. Med. 1998;64:460–461. doi: 10.1055/s-2006-957483. [DOI] [PubMed] [Google Scholar]
- 58.Zembower D.E., Liao S., Flavin M.T., Xu Z.Q., Stup T.L., Buckheit R.W., Jr., Khilevich A., Mar A.A., Sheinkman A.K. Structural analogues of the calanolide Anti-HIV Agents. modification of the trans-10,11-dimethyldihydropyran-12-ol ring (ring C) J. Med. Chem. 1997;40:1005–1017. doi: 10.1021/jm960355m. [DOI] [PubMed] [Google Scholar]
- 59.Ma T., Liu L., Xue H., Li L., Han C., Wang L., Chen Z., Liu G. Chemical library and structure–activity relationships of 11-demethyl-12-oxo calanolide A analogues as anti-HIV-1 agents. J. Med. Chem. 2008;51:1432–1446. doi: 10.1021/jm701405p. [DOI] [PubMed] [Google Scholar]
- 60.Xue H., Lu X., Zheng P., Liu L., Han C., Hu J., Liu Z., Ma T., Li Y., Wang L., Chen Z., Liu G. Highly suppressing wild-type HIV-1 and Y181C mutant HIV-1 strains by 10-chloromethyl-11-demethyl-12-oxo-calanolide A with druggable profile. J. Med. Chem. 2010;53:1397–1401. doi: 10.1021/jm901653e. [DOI] [PubMed] [Google Scholar]
- 61.Zhoua P., Takaishia Y., Duana H., Chena B., Hondab G., Itohb M., Takedac Y., Kodzhimatovd O.K., Leee K.H. Coumarins and bicoumarin from Ferula sumbul: anti-HIV activity and inhibition of cytokine release. Phytochem. 2000;53:689–697. doi: 10.1016/s0031-9422(99)00554-3. [DOI] [PubMed] [Google Scholar]
- 62.Shikishima Y., Takaishi Y., Honda G., Ito M., Takeda Y., Kodzhimatov O.K., Ashurmetov O., Lee K.H. Chemical constituents of Prangos tschimganica structure elucidation and absolute configuration of coumarin and furanocoumarin derivatives with anti-HIV activity. Chem. Pharm. Bull. 2001;49:877–880. doi: 10.1248/cpb.49.877. [DOI] [PubMed] [Google Scholar]
- 63.Sancho R., Márquez N., Gomez-Gonzalo M., Calzado M.A., Bettoni G., Coiras M.T., Alcami J., Lopez-Cabrera M., Appendino G., Munoz E. Imperatorin inhibits HIV-1 replication through an Sp1-dependent pathway. J. Biol. Chem. 2004;279:37349–37359. doi: 10.1074/jbc.M401993200. [DOI] [PubMed] [Google Scholar]
- 64.Uchiumi F., Hatano T., Ito H., Yoshida T., Tanuma S. Transcriptional suppression of the HIV promoter by natural compounds. Antivir. Res. 2003;58:89–98. doi: 10.1016/s0166-3542(02)00186-9. [DOI] [PubMed] [Google Scholar]
- 65.Olmedo D., Sancho R., Bedoya L.M., Lopez-Perez J.L., Del Olmo E., Munoz E., Alcami J., Gupta M.P., San A.F. 3-Phenylcoumarins as inhibitors of HIV-1 replication. Molecules. 2012;17:9245–9257. doi: 10.3390/molecules17089245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ong E.B., Watanabe N., Saito A., Futamura Y., Abd El Galil K.H., Koito A., Najimudin N., Osada H. Vipirinin, a coumarin-based HIV-1 Vpr inhibitor, interacts with a hydrophobic region of VPR. J. Biol. Chem. 2011;286:14049–14056. doi: 10.1074/jbc.M110.185397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marquez N., Sancho R., Bedoya L.M., Alcami J., López-Perez J.L., Feliciano A.S., Fiebich B.L., Munoz E. Mesuol, a natural occurring 4-phenylcoumarin, inhibits HIV-1 replication by targeting the NF-kappa B pathway. Antivir. Res. 2005;66:137–145. doi: 10.1016/j.antiviral.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 68.Bedoya L.M., Beltrán M., Sancho R., Olmedo D.A., Sánchez-Palomino S., del Olmo E., López-Pérez J.L., Muñoz E., San A.F., Alcami J. 4-Phenylcoumarins as HIV transcription inhibitors. Bioorg. Med. Chem. Lett. 2005;15:4447–4450. doi: 10.1016/j.bmcl.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 69.Veselinović J., Veselinović A., Toropov A., Toropova A., Damnjanović I., Nikolić G. Monte carlo method based qsar modeling of coumarin derivates as potent HIV-1 integrase inhibitors and molecular docking studies of selected 4-phenyl hydroxycoumarins. Sci. J. Fac. Med. Nis. 2014;31:95–103. [Google Scholar]
- 70.Abd Elhafez O.M., El Khrisy E.E.D.A.M., Badria F., Fathy A.E.D.M. Synthesis and biological investigations of new thiazolidinone and oxadiazoline coumarin derivatives. Arch. Pharm. Res. 2003;26:686–696. doi: 10.1007/BF02976675. [DOI] [PubMed] [Google Scholar]
- 71.Bhavsar D., Trivedi J., Parekh S., Savant M., Thakrar S., Bavishi A., Radadiya A., Vala H., Lunagariya J., Parmar M., Paresh L., Loddo R., Shah A. Synthesis and in vitro anti-HIV activity of N-1,3-benzo[d]thiazol-2-yl-2-(2-oxo-2H-chromen-4-yl)acetamide derivatives using MTT method. Bioorg. Med. Chem. Lett. 2011;21:3443–3446. doi: 10.1016/j.bmcl.2011.03.105. [DOI] [PubMed] [Google Scholar]
- 72.Reddy M.V.R., Rao M.R., Rhodes D., Hansen M.S.T., Rubins K., Bushman D., Venkateswarlu Y., Faulkner D.J. Lamellarin R 20-sulfate, an inhibitor of HIV-1 integrase active against HIV-1 virus in cell culture. J. Med. Chem. 1999;42:1901–1907. doi: 10.1021/jm9806650. [DOI] [PubMed] [Google Scholar]
- 73.Wu T.S., Tsang Z.J., Wu P.L., Lin F.W., Li C.Y., Teng C.M., Lee K.H. New constituents and antiplatelet aggregation and anti-HIV principles of Artemisia capillaris. Bioorg. Med. Chem. 2001;9:77–83. doi: 10.1016/s0968-0896(00)00225-x. [DOI] [PubMed] [Google Scholar]
- 74.Hwu J.R., Singha R., Hong S.C., Chang Y.H., Das A.R., Vliegen I., De Clercq E., Neyts J. Synthesis of new benzimidazole-coumarin conjugates as anti-hepatitis C virus agents. Antivir. Res. 2008;77:157–162. doi: 10.1016/j.antiviral.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 75.Neyts J., De Clercq E., Singha R., Chang Y.H., Das A.R., Chakraborty S.K., Hong S.C., Tsay S.C., Hsu M.H., Hwu J.R. Structure-activity relationship of new anti-hepatitis C virus agents: heterobicycle-coumarin conjugates. J. Med. Chem. 2009;52:1486–1490. doi: 10.1021/jm801240d. [DOI] [PubMed] [Google Scholar]
- 76.Hwu J.R., Lin S.Y., Tsay S.C., De Clercq E., Leyssen P., Neyts J. Coumarin-purine ribofuranoside conjugates as new agents against hepatitis C virus. J. Med. Chem. 2011;54:2114–2126. doi: 10.1021/jm101337v. [DOI] [PubMed] [Google Scholar]
- 77.Tsay S.C., Lin S.Y., Huang W.C., Hsu M.H., Hwang K.C., Lin C.C., Horng J.C., Chen I.C., Hwu J.R., Shieh F.K., Leyssen P., Neyts J. Synthesis and structure-activity relationships of imidazole-coumarin conjugates against hepatitis C virus. Molecules. 2016;21:e228. doi: 10.3390/molecules21020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsay S.C., Hwu J.R., Singha R., Huang W.C., Chang Y.H., Hsu M.H., Shieh F.K., Lin C.C., Hwang K.C., Horng J.C., De Clercq E., Vliegen I., Neyts J. Coumarins hinged directly on benzimidazoles and their ribofuranosides to inhibit hepatitis C virus. Eur. J. Med. Chem. 2013;63:290–298. doi: 10.1016/j.ejmech.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 79.Manvar P., Shaikh F., Kakadiya R., Mehariya K., Khunt R., Pandey B., Shah A. Synthesis of novel imidazo[1,2-a]pyridine-4-hydroxy-2H-coumarins by Groebke-Blackburn-Bienayme multicomponent reaction as potential NS5B inhibitors. Tetrahed. 2016;72:1293–1300. [Google Scholar]
- 80.Peng H.K., Chen W.C., Lee J.C., Yang S.Y., Tzeng C.C., Lin Y.T., Yang S.C. Novel anilinocoumarin derivatives as agents against hepatitis C virus by the induction of IFN-mediated antiviral responses. Org. Biomol. Chem. 2013;11:1858–1866. doi: 10.1039/c2ob26860d. [DOI] [PubMed] [Google Scholar]
- 81.Mazzei M., Nieddu E., Miele M., Balbi A., Ferrone M., Fermeglia M., Mazzei M.T., Pricl La Colla S.P., Marongiu F., Ibba C., Loddo R. Activity of Mannich bases of 7-hydroxycoumarin against flaviviridae. Bioorg. Med. Chem. 2008;16:2591–2605. doi: 10.1016/j.bmc.2007.11.045. [DOI] [PubMed] [Google Scholar]
- 82.Chang C.T., Doong S.L., Tsai I.L., Chen I.S. Coumarins and anti-HBV constituents from Zanthoxylum schinifolium. Phytochem. 1997;45:1419–1422. [Google Scholar]
- 83.Basu N.K., Waffo A.B., Talele T.T., Basu A., Costa P.R.R., da Silva A.J.M., Sarafianos S.G., Noël F. Identification and characterization of coumestans as novel HCV NS5B polymerase inhibitors. Nucl. Acid. Res. 2008;36:1482–1496. doi: 10.1093/nar/gkm1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yeh J.Y., Coumar M.S., Horng J.T., Shiao H.Y., Kuo F.M., Lee H.L., Chen I.C., Chang C.W., Tang W.F., Tseng S.N., Chen C.J., Shih S.R., Hsu J.T., Liao C.C., Chao Y.S., Hsieh H.P. Anti-influenza drug discovery: structure-activity relationship and mechanistic insight into novel angelicin derivatives. J. Med. Chem. 2010;53:1519–1533. doi: 10.1021/jm901570x. [DOI] [PubMed] [Google Scholar]
- 85.Shih S.R., Horng J.T., Poon L.L., Chen T.C., Yeh J.Y., Hsieh H.P., Tseng S.N., Chiang C., Li W.L., Chao Y.S., Hsu J.T. BPR2-D2 targeting viral ribonucleoprotein complex-associated function inhibits oseltamivir-resistant influenza viruses. J. Antimicrob. Chemother. 2010;65:63–71. doi: 10.1093/jac/dkp393. [DOI] [PubMed] [Google Scholar]
- 86.Lin P.H., Ke Y.Y., Su C.T., Shiao H.Y., Hsieh H.P., Chao Y.K., Lee C.N., Kao C.L., Chao Y.S., Chang S.Y. Inhibition of HIV-1 Tat-mediated transcription by a coumarin derivative, BPRHIV001, through the Akt pathway. J. Virol. 2011;85:9114–91126. doi: 10.1128/JVI.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ishikawa T., Kotake K., Ishii H. Synthesis of toddacoumaquinone, a coumarin-naphthoquinone dimer, and its antiviral activities. Chem. Pharm. Bull. 1995;43:1039–1041. doi: 10.1248/cpb.43.1039. [DOI] [PubMed] [Google Scholar]
- 88.Xu B., Wang L., González-Molleda L., Wang Y., Xu J., Yuan Y. Antiviral activity of (+)-rutamarin against Kaposi's sarcoma-associated herpesvirus by inhibition of the catalytic activity of human topoisomerase II. Antimicrob. Agents. Chemother. 2014;58:563–573. doi: 10.1128/AAC.01259-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ghannadi A., Fattahian K., Shokoohinia Y., Behbahani M., Shahnoush A. Anti-viral evaluation of sesquiterpene coumarins from Ferula assafoetida against HSV-1. Iran. J. Pharm. Res. 2014;12:523–530. [PMC free article] [PubMed] [Google Scholar]
- 90.Giannella M., Gualtieri F. Studies in the field of substances with antiviral activity. Derivatives of 2,4-dioxo-3-oxyiminochromane. Farmaco. Sci. 1968;23:1104–1112. [PubMed] [Google Scholar]
- 91.Cingolani G.M., Gualtieri F., Pigini M. Researches in the field of antiviral compounds. Mannich bases of 3-hydroxycoumarin. J. Med. Chem. 1969;12:531–532. doi: 10.1021/jm00303a616. [DOI] [PubMed] [Google Scholar]
- 92.Zavrsnik D., Muratović S., Makuc D., Plavec J., Cetina M., Nagl A., Clercq E.D., Balzarini J., Mintas M. Benzylidene-bis-(4-hydroxycoumarin) and benzopyrano-coumarin derivatives: synthesis, 1H/13C-NMR conformational and X-ray crystal structure studies and in vitro antiviral activity evaluations. Molecules. 2011;16:6023–6040. doi: 10.3390/molecules16076023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hwu J.R., Kapoor M., Tsay S.C., Lin C.C., Hwang K.C., Horng J.C., Chen I.C., Shieh F.K., Leyssen P., Neyts J. Benzouracil-coumarin-arene conjugates as inhibiting agents for chikungunya virus. Antiv. Res. 2015;118:103–109. doi: 10.1016/j.antiviral.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 94.Pohjala L., Utt A., Varjak M., Lulla A., Merits A., Ahola T., Tammela P. Inhibitors of alphavirus entry and replication identified with a stable Chikungunya replicon cell line and virus-based assays. PLoS One. 2011;6:e28923. doi: 10.1371/journal.pone.0028923. [DOI] [PMC free article] [PubMed] [Google Scholar]