

Abstract
One of the most viable options to tackle the growing resistance to the antimalarial drugs such as artemisinin is to resort to synthetic drugs. The multi-target strategy involving the use of hybrid drugs has shown promise. In line with this, new hybrids of quinoline with pyrimidine have been synthesized and evaluated for their antiplasmodial activity against both CQS and CQR strains of Plasmodium falciparum. These depicted activity in nanomolar range and were found to bind to heme as well as AT rich pUC18 DNA.
Keywords: Antimalarials, 4-Aminoquinolines, Heme binding, Hybrid molecules, Drug discovery
Graphical abstract
Efficaciously synthesized new 4-aminoquinoline–pyrimidine hybrids depict antiplasmodial activity (nM) against CQR and CQS strains of Plasmidum falciparum. Representative compounds revealed binding with heme and AT rich pUC18 DNA, spectrophotometically.

Highlights
-
•
New hybrids of 4-aminoquinoline and pyrimidine were synthesized efficaciously.
-
•
nM range antiplasmodial activity.
-
•
Activity against both CQS and CQR strains of Plasmodium falciparum.
-
•
Effective binding to heme as well as AT rich pUC18 DNA.
1. Introduction
Malaria is one of the most widespread diseases besides tuberculosis and AIDS which affects more than 500 million people worldwide and results in around 1–3 million causalities every year [1]. In Africa alone, around 20% childhood deaths are due to malaria and a child dies every 30 s [2] and it is estimated that an African child has on an average 1.6–5.4 episodes of malaria fever each year. Of the four typically recognized Plasmodium species causing disease in humans, Plasmodium falciparum is most deadly to children below the age of five leading to mortality while Plasmodium vivax is most morbidity prone, and is responsible for latent infection that hampers current control and future elimination efforts [3]. The development of drug resistance for the common antimalarials such as 4-/8-aminoquinolines, 4-methanol quinolines, antifolate drugs, sesquiterpene lactones etc. (Fig. 1 ) is a rather serious issue which has stimulated considerable research efforts in the development of new drugs using different approaches [4], [5] of which the molecular hybridization approach [6], [7] is quite an attractive strategy which involves design of new chemical entities by covalent fusion of two drugs, both active compounds and/or pharmacophoric units derived from known bioactive molecules with complimentary activities and multiple pharmacological targets. The multiple target strategy led to the design of hybrid of 4-aminoquinoline with species such as triazine [8], [9], ferrocene [10], rhodanine [11], thiazolidine-4-one [12], chalcone [13], trioxane [14], isatin [15] and recently, pyrimidines [16], [17], [18] (Fig. 2 ).
Fig. 1.

Small molecule antimalarial agents.
Fig. 2.
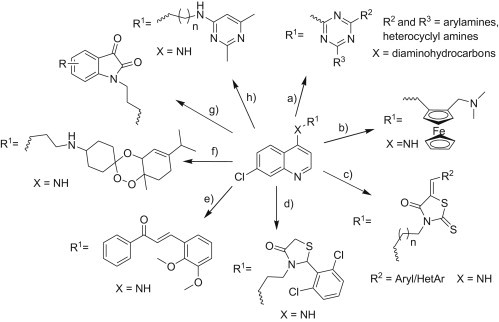
Representative designs of 4-aminoquinoline based hybrid drugs showing antimalarial activity (quinoline hybrids with (a) triazine, (b) ferrocene, (c) rhodanine, (d) thiazolidin-4-one, (e) chalcone, (f) trioxane, (g) isatin and (h) pyrimidine).
Quinoline containing drugs (chloroquine and primaquine, Fig. 1) are known to affect parasite metabolism and cause parasite death by blocking the polymerization of the toxic heme, into an insoluble and non-toxic pigment, hemozoin, resulting in cell lysis and parasite cell auto digestion [19], [20], [21]. On the other hand, pyrimidine-based compounds are well known for their wide range of promising antiviral [22], antitubercular [23], anti-AIDS [24], antinociceptive [25], antifungal [26], antitumor [27] and antimalarial activities [28] apart from their role in the nucleic acid synthesis. Thus, linking of the quinoline unit with pyrimidine might furnish conjugate hybrids that are capable of showing useful antimalarial activity.
Recently, antimalarial activities of some quinoline–pyrimidine hybrids with activities in the micromolar to nanomolar range have been reported (Fig. 3 ) [16], [17], [18], [29]. In yet another report on the evaluation of quinoline–pyrimidine, the activity (in micromolar range) was also reported for fixed combinations of the chloroquine and pyrimethamine. In all these reports, the pyrimidines were linked to the quinoline unit through 2-, 4- and 6-positions. We have employed rather conformationally flexible pyrimidine-5-carboxylates linked covalently to 4-aminoquinoline core. These novel pyrimidine carboxylate hybrids interact with the iron center of free heme within the physiological environment (pH 5.6), a key step in the accumulation of heme which is selectively toxic to the parasite. To enhance the possibility to accumulate within the digestive vacuole via weak-base trapping (the mechanism by which CQ and other quinoline antimalarials attain high concentrations inside this compartment), we developed a novel class of antimalarials using a pharmacophore hybridization approach in which the pyrimidine-5-carboxylate motif was hybridized with an iron-complexing, 4-aminoquinoline moiety through C-2 position [29]. To further elaborate the structure–activity profile, here, we present additional new 4-aminoquinoline–pyrimidine carboxylate hybrids. We also report on their antimalarial activity against both CQ sensitive (CQs Dd2) as well as CQ resistant (CQR D10) strains. Finally, the mechanism of action studies with the representative compounds has also been performed.
Fig. 3.

Quinoline–pyrimidine and quinoline–pyrimidine carboxylate hybrids.
2. Chemistry
The 4-aminoquinoline–pyrimidine-5-carboxylate hybrids were synthesized in economical way using synthetic approach outlined in Scheme 1 . The key starting compound, 3,4-dihydropyrimidin-2(1H)-one 1 was prepared through NH4Cl/TFA [30], [31] catalyzed three-component Biginelli condensation of an alkyl acetoacetate, urea and appropriate aldehyde or its formyl equivalent: 1,3-oxazinane derivative, in acetonitrile or under solvent-free reaction conditions, in some cases. Subsequent oxidation of 1 using 60% nitric acid readily furnished pyrimidinones 2 which upon chlorination with POCl3 yielded 3 [32]. The nucleophilic substitution reaction of 3 with appropriate 4-amino-7-chloroquinoline 4 which in turn was prepared from the commercially available 4,7-dichloroquinoline and diaminoalkanes [33], gave 5a–g in good yields (Table 1). Structures of 1–5 were unambiguously established on the basis of spectral (1H NMR, 13C NMR, MS, FT IR) as well as microanalytical analysis.
Scheme 1.

Reagents and conditions. (a) NH4Cl, 100 °C, 3 h, (b) 60% HNO3, 0 °C, 30 min, (c) POCl3, 105 °C, 45 min, (d) THF/MeCN, K2CO3, 70 °C, 48 h.
Table 1.
In vitro antimalarial activity of compounds 5a–g against P. falciparum (CQS) D10 strain and (CQR) Dd2 strain for n = 3 (n = number of replicates).
| Compound | Structure | Yield (%) | D10 IC50 (nM)a, b | Dd2 IC50 (nM)b, c | C log Pd | CC50 (μM)e, f | SIg |
|---|---|---|---|---|---|---|---|
| 5a |  |
86 | 659 | 542 | 6.71 | 1.7 | 3.13 |
| 5b |  |
83 | 156 | 153 | 7.66 | 0.6 | 3.92 |
| 5c |  |
75 | 1461 | nd | 7.10 | 0.8 | – |
| 5d |  |
90 | 478 | 483 | 7.24 | 21.1 | 43.6 |
| 5e |  |
85 | 1926 | nd | 8.07 | 2.2 | – |
| 5f |  |
72 | 2759 | nd | 7.50 | 11.4 | – |
| 5g |  |
89h | 211 | 336 | 5.25 | 10.3 | 30.65 |
| 6ai |  |
– | 202j | 26.1k | 6.82 | 0.8 | 15.33 |
| 6bi |  |
– | 247.5j | 52.2k | 7.35 | 0.9 | 34.41 |
| 6ci |  |
– | 18.2j | 3.6k | 7.3 | 2.3 | 638 |
| CQ | 35 | 221.9 | – | – | – | ||
| ASN | – | 31.2 | – | – | – | ||
| MMV390048 | – | 17.8 |
CQ sensitive strain.
Data represents the mean of three independent experiments.
CQ resistant strain.
Calculated from Chem draw Ultra 11.0.
Determined on Madin Darby canine kidney (MDCK) cells.
50% cytotoxic concentration, as determined by measuring the cell viability with the colorimetric formazan-based MTS assay (reference drugs used: Oseltamivir carboxylate CC50/MIC > 100, Ribavirin CC50/MIC > 100, Amantadine CC50/MIC > 200 and Rimantadine CC50/MIC > 200).
Selectivity index (S.I.) is calculated as CC50/IC50 (Dd2 Strain) ratio.
Prepared by using perhydro 1,3-oxazine (1.5 equiv.) as formaldehyde equivalent (CH3CN:TFA (10:1, v/v) solution/70 °C) [31].
See Ref. [29].
3D7 strain used.
K1 strain used.
3. Results and discussion
3.1. Antimalarial activity and structure–activity relationships (SARs)
We have previously established that the 4-aminoquinoline–pyrimidine hybrids 6a–c intercepted by a diaminoalkyl spacer showed optimum potency (Table 1 ), when the flexible spacer consisted of three or four carbon atoms [29]. Further, the introduction of nitro substituent at ortho position of the phenyl ring at the C-4 of the pyrimidine core furnished the most potent compound 6c with antimalarial activity superior to the standard CQ and close to artesunate [29]. Keeping these observations in mind, we planned to further refine the activity of these persuasive 4-aminoquinoline–pyrimidine by incorporating electron withdrawing substituents at C-4 phenyl of pyrimidine core, as well as by varying the C-5 ester substituent and also by altering the basicity of diaminoalkyl spacer.
The in vitro antimalarial screening of the new synthesized compounds 5a–g revealed good to moderate activities in nM range against both the tested Dd2 (CQS) and D10 strains (CQR) of P. falciparum (Table 1). Although the tested hybrids were not as active as the standard drugs viz. CQ and ASN, interesting SARs have been drawn. Analysis of the activity of the compounds recorded in Table 1 reveals that replacing C-5 ethyl ester of the previously reported [29] compound 6a [IC50 247.5 nM (CQS); 52.2 nM (CQR)] with methyl ester 5a [IC50 659 nM (CQS); 542 nM (CQR)] led to the decrease in antimalarial activity against both the chloroquine sensitive and chloroquine resistant strains of P. falciparum. However, comparison of hybrids 5b, 5c with 6b, 6c having an identical butyl spacer showed that incorporation of isopropyl ester at C-5 of pyrimidine motif (5b and 6b) increased the antimalarial activity against the CQS strain whereas considerable decrease in activity was observed for CQR strain of P. falciparum. Also, the most potent compound 5b of the series displayed 2-fold increase in antimalarial activity than the standard CQ against CQR strain of P. falciparum. When the diaminoalkyl linker of compound 6a was replaced with relatively less basic alkoxy amino linker 5d considerable decrease in antimalarial activity was observed which in turn linked to the decreased accumulation of compound via pH trapping into the digestive vacuole. It was not unexpected since the basicity of alkyl chain linker plays crucial role in determining the antimalarial activity of this class of compounds. Furthermore, the introduction of a nitro substituent on the phenyl ring at the C-4 position of the pyrimidine core to create 5c resulted in a significant increase in anti-plasmodial activity in comparison to the p-chloro/p-fluoro substituents (5e and 5f). Moreover, the hybrid 5g lacking a C-4 substituent on the pyrimidine motif led to an increase in antimalarial activity against both the CQS as well as CQR strains of P. falciparum. However, although the antimalarial activity of 5g was superior to 5c, 5e & 5f, it was less than the corresponding C-4 phenyl substituted analogs 5b as well as 6b.
Thus, the SAR study suggested that both the substitution of the C-4 phenyl group with electron withdrawing groups and alterations in basicity of linker leads to better antimalarial activity. Unfortunately, these compounds suffer from high ClogP values which are in the range 5–8 (Table 1), which are suggestive of the fact that these possess limited aqueous solubility. However, it is not a serious limitation in view of recent advancements in formulation methods.
3.2. Cytotoxicity and antiviral activity
Compounds 5a–g were evaluated for their toxicity against various (HeLa, Vero, CRFK, HEL and MDCK) cell cultures (Table 1 & SI Table S1). Toxicity data revealed that these compounds exhibit high toxicity (low CC50) against MDCK cell cultures while CC50 values are quiet high for other cell cultures. The CC50 values for inhibition of MDCK cells summarized in Table 1 indicate that the strongest antimalarial compound 5b was mildly cytotoxic (Table 1). Further, the ratio of the cytotoxicity (CC50 in μM) and in vitro antimalarial activity (IC50 in nM for Dd2 strain) enabled the determination of selectivity index (SI) for these compounds. Compound 5d with alkoxy amino linker and compound 5g bearing C-4 unsubstituted pyrimidine motif exhibited high CC50 values and thus led to fairly safe selectivity index values (Table 1). Compound 5d having less basic alkyl spacer, displayed highest SI (43.6) whereas the most potent compound 5b exhibit relatively low SI value (3.92). Thus, the compounds depicted structure dependent SI values.
Chloroquine is known to elicit antiviral effects against several viruses, including human immunodeficiency virus type 1, HCoV-229E, hepatitis B virus, and herpes simplex virus type 1 [34], [35], [36], [37]. Thus, we determined in vitro antiviral activities of 5a–g against (i) herpes simplex virus-1 (HSV-1; KOS), herpes simplex virus-2 (HSV-2; G), vaccinia virus, vesicular stomatitis virus, herpes simplex virus-1 (TK_KOS ACVR) in HEL cell cultures, (ii) parainfluenza-3 virus, reovirus-1, Sindbis virus, Coxsackie virus B4, Punta Toro virus in vero cell cultures, (iii) influenza A virus (H1N1 and H3N2) and influenza B virus in MDCK cell cultures, (iv) vesicular stomatitis virus, coxsackie virus B4, respiratory syncytial virus in HeLa cell cultures, (v) cytomegalovirus using AD-169 and Davis strain in HeL cells, (vi) varicella-zoster virus (VZV) in HEL cells (SI Table S1) and (vii) feline corona virus (FIPV) and feline herpes virus activity in CRFK cell cultures (Table 2 ). The anti-viral activity of most of the compounds was not impressive except compounds 5a and 5c which exhibited relatively low EC50's only against the feline corona virus (FIPV) and feline herpes virus in CRFK cell cultures (Table 2).
Table 2.
Anti-Feline Corona Virus (FIPV) and anti-Feline Herpes Virus activity and cytotoxicity in Crandell-Rees Feline Kidney (CRFK) cell cultures.
| Compound | CC50 (μM)a | EC50 (μM)b |
|
|---|---|---|---|
| Feline corona virus (FIPV) | Feline herpes virus | ||
| 5a | 25.1 | 1.1 | >20 |
| 5b | 50.6 | >20 | >20 |
| 5c | >100 | 9.0 | >100 |
| 5d | >100 | >100 | >100 |
| 5e | >100 | >100 | >100 |
| 5f | >100 | >100 | >100 |
| 5g | >100 | >100 | >100 |
| HHA (μg/ml) | >100 | 32.5 | 2.9 |
| UDA (μg/ml) | 63.2 | 1.6 | 1.0 |
| Ganciclovir | >100 | >100 | 4.1 |
50% Cytotoxic concentration as determined by measuring the cell viability with the colorimetric formazon-based MTS assay.
50% Effective concentration or concentration producing 50% inhibition of virus-induced cytopathic effect as determined by measuring the cell viability with the colorimetric formazon-based MTS assay.
3.3. Mode of action studies
3.3.1. Heme binding studies
Quinoline antimalarials (e.g., CQ, amodiaquine and quinine) act principally by forming adducts with ferriprotoporphyrin IX, thus blocking haemozoin formation [38]. In this study, we have evaluated the mechanism of antimalarial activity of the most potent compound 5b of the series by studying its binding with heme [Fe(III)PPIX] in solution and inhibition of β-hematin formation using UV–visible spectrophotometer. The incremental addition of 5b (0–25 μM) into monomeric heme (2.4 μM, DMSO:H2O/4:6, v/v) in 0.02 M HEPES buffer (pH 7.4) showed a substantial decrease in the intensity of the Fe(III) PPIX Soret band at 402 nm with no shift in the wavelength of the absorption maximum (Fig. 4 ). The titration of monomeric heme was also performed at the Plasmodial food vacuole pH 5.6 using MES buffer instead of HEPES to ensure that the compound 5b binds with heme even at acidic pH (S1). A 1:1 stoichiometry of the most stable complex of 5b with monomeric heme at pH 7.4 and 5.6 was established from the Job's plot (SI Figure S1). The association constants (Table 3 ) were calculated by analyzing the titration curves obtained at pH 7.4 using HypSpec–a non-linear least square fitting programme [39]. The binding of CQ with heme under identical conditions was also determined in the similar manner and the results are presented in Table 3 for comparison. Table 3 shows that the association constants for the complexes formed between monomeric heme and 5b (log K 4.96) are comparable with those of standard antimalarial drug, CQ (log K 5.15). Furthermore, the decrease of apparent pH from 7.4 to 5.6 (Table 3) has little effect on the binding constants indicating binding is stronger even at acidic pH.
Fig. 4.

Titration of 5b with monomeric heme at (a) pH 7.4, (b) pH 5.6.
Table 3.
Binding constant (log K) of 5b and CQ with heme and DNA.
| Compound | Monomeric heme, log K ± σ |
μ-oxoheme, log K ± σ | CT DNA, log K | pUC18 DNA, log K | |
|---|---|---|---|---|---|
| pH 5.6 | pH 7.4 | pH 5.8 | |||
| 5ba | 4.58 ± 0.042 | 4.96 ± 0.029 | 5.72 ± 0.025 | 5.76 | 5.73 |
| CQ | 4.65 ± 0.052 | 5.15 ± 0.176 | 5.58 ± 0.006 | nd | nd |
| Stoichiometry | 1:1 | 1:1 | nd | ||
Calculated from HypSpec software.
To further establish the binding of 5b with monomeric heme, 1H NMR titrations were performed and shifts in the peaks as well as peak intensity noted. The addition of 30 mol% of heme dissolved in 40% DMSO to a solution of 5b in 40% DMSO:D2O\D2SO4 (10 μl) caused a shift in the aromatic proton signals (Fig. 5 ), indicating binding of 5b with heme but further addition of heme led to broadening of the peaks. An equimolar (3.9 μmol) solution of hemin chloride and 5b when analyzed in mass spectrometer depicted an intense molecular ion peak at 1119.3769 Da (Fig. 6a), corresponding to the molecular formula C62H62ClFeN9O6, suggesting the formation of 1:1 complex. Thus, we propose that 5b interacts with heme by replacing chloride atom of hemin chloride and coordinating the iron atom with its endocyclic quinoline nitrogen as proposed in Fig. 6b.
Fig. 5.

The 400 MHz 1H spectra of 5b upon addition of heme (a) 0 mol%, (b) 30 mol%, (c) 50 mol% (in 40% DMSO:D2O\D2SO4 (10 μl)) [Δδ for peak: a = 0.002, b = 0.013, c = 0.023, d = 0.008, e = 0.007, f = 0.007].
Fig. 6.

(a) The solution phase mass spectra of 5b (3.9 μmoles) upon addition of monomeric heme (3.9 μmoles) in 40% DMSO, (b) proposed binding of heme with 5b (for optimized structure of 5b, see Figure S2).
Similar titration of dimers of μ-oxo type (10 μM) at pH 5.8 using standard procedure [29] with increasing concentration of compound 5b (0–14 μM), resulted in decrease in intensity of broad peak at 362 nm (Fig. 7a, S1). Further, Job's plot calculations indicated a 1:1 stoichiometry for the most stable μ-oxo: 5b complex (Fig. 7b). In Table 3 the association constants of 5b (log K 5.72) are compared to that of standard CQ (log K 5.58) and also suggests that the binding of 5b is stronger with μ-oxo heme (log K 5.72) than monomeric heme (log K 4.96). Thus, the compound 5b inhibits hemozoin formation by blocking the growing face of heme resulting in the observed antimalarial activity. Furthermore, the β-hematin inhibition assay (SI Table S2) shows that there is no correlation between antimalarial activity and β-hematin inhibition and also, all the compounds inhibit β-hematin formation although less than that of standard CQ.
Fig. 7.

(a) Titration of 5b with μ-oxo heme at pH 5.8, (b) Job plot of μ-oxo heme complex formation at pH 5.8. x = [5b]/[5b] + [heme] is the mole fraction of the 5b, A0 is the absorbance, when x = 1 and A is the absorbance at respective values of x.
3.3.2. DNA binding studies
The mechanism of many antimalarial drugs such as CQ, quinacrine and quinine relies upon the interaction with DNA presumably through ionic interactions between phosphate groups of DNA and protonated amine in addition to the interactions between aromatic nuclei of the drug with nucleotide bases [40], [41]. Therefore, the DNA binding properties of 4-aminoquinoline–pyrimidine hybrids have been evaluated using both the UV–visible spectrophotometer and fluorescence spectrophotometry in order to probe interaction of these compounds with DNA. The addition of CT-DNA (4–200 μM) to the buffered methanolic solution of 5b (30 μM) induced hyperchromic shift of 112% in absorption band at 255 nm whereas hypochromic shift of 37% in the characteristic quinoline ring absorption at 330 nm (Fig. 8 ). Also, the bathochromic shift of ∼3 nm was observed for both the absorptions. The observed hyperchromic as well as hypochromic shifts in absorption bands of 5b upon addition of DNA results from the intercalation of 5b with CT DNA as suggested in the literature [42]. The intercalative nature of interaction of compound 5b with CT DNA was additionally supported by thermal denaturation experiment. Intercalation of molecules into the double helix is known to stabilize the DNA against thermal strand separation and thus increases thermal melting temperature (T m) [43], [44]. The derivative melting curve presented in Fig. 9 shows an increase of 7.5 °C in thermal melting temperature of CT DNA upon addition of 5b which is less than that observed for the CQ (Table S3). Thus, both the UV–visible titrations and thermal denaturation experiment advocate partial intercalative nature of interactions between compound 5b and CT DNA.
Fig. 8.

Absorption spectra of 5b (30 μM) in the presence of increasing CT DNA concentration (4–200 μM); inset shows zoom between 280 and 390 nm.
Fig. 9.

Derivative melting curves of CT DNA, 5b + CT DNA and CQ + CT DNA.
Further, to visualize the effect of DNA base composition, the fluorescence titrations of 5b were performed with both GC-rich CT DNA and AT-rich pUC18 DNA in buffered methanol. Fig. 10 shows decrease in the intensity of the emission band of 5b at 380 nm, upon addition of increasing concentration of both the DNAs. A shift of 80 nm in emission band at 380 nm was observed upon addition of CT DNA but no such shift in emission band was observed for pUC18 DNA. Comparison of binding constant of 5b with CT DNA (log K 5.76) and pUC18 DNA (log K 5.73) calculated from titration data using HypSpec [39], suggest that 5b does not discriminate between GC rich DNA and AT rich DNA.
Fig. 10.

Fluorescence emission spectra (λex = 330 nm, λem = 376 nm) of 5b (17.1 μM) in buffered CH3OH upon addition of increasing concentrations of (a) CT DNA (0.5–150 μM), (b) pUC18 DNA (0.02–15 μM).
4. Conclusions
A series of potent 4-aminoquinoline–pyrimidine hybrids with antimalarial activity in nanomolar range were reported. The compound 5b exhibits lowest IC50 value within the series against both CQS and CQR strains of P. falciparum. These hybrids displayed mild toxicity against MDCK cell cultures. The antiviral activity profiles of these hybrids indicate that the compound 5a and 5c effectively inhibit feline corona virus and feline herpes virus. Further, the mechanism of observed antimalarial activity was established in terms of binding with heme as well as DNA.
5. Experimental
5.1. General
All liquid reagents were dried/purified following recommended drying agents and/or distilled over 4 Å molecular sieves. THF was dried (Na-benzophenone ketyl) under nitrogen. 1H NMR (300 MHz) and 13C (75 MHz) NMR spectra were recorded in CDCl3 on a multinuclear Jeol FT-AL-300 spectrometer with chemical shifts being reported in parts per million (δ) relative to internal tetramethylsilane (TMS, δ 0.0, 1H NMR) or chloroform (CDCl3, δ77.0, 13C NMR). Mass spectra were recorded at Department of Chemistry, Guru Nanak Dev University, Amritsar on a Bruker LC-MS MICROTOF II spectrometer. Elemental analysis was performed on FLASH EA 112 (Thermo electron Corporation) analyzer at Department of Chemistry, Guru Nanak Dev University, Amritsar and the results are quoted in %. IR spectra were recorded on Perkin Elmer FTIR-C92035 Fourier transform spectrometer in the range 400–4000 cm−1 using KBr pellets. Melting points were determined in open capillaries and are uncorrected. For monitoring the progress of a reaction and for comparison purpose, thin layer chromatography (TLC) was performed on pre-coated aluminum sheets of Merck (60F254, 0.2 mm) using an appropriate solvent system. The chromatograms were visualized under UV light. For column chromatography silica gel (60–120 mesh) was employed and eluents were ethyl acetate/hexane or ethyl acetate/methanol mixtures. The steady state fluorescence experiments were carried out on Perkin Elmer LS55 fluorescence spectrometer at ambient temperature. UV–visible spectral studies were conducted on Shimadzu 1601 PC spectrophotometer with a quartz cuvette (path length, 1 cm). The absorption spectra have been recorded between 1100 and 200 nm. The cell holder of the spectrophotometer was thermostated at 25 °C for consistency in the recordings.
5.2. General procedure for synthesis of 5a and 5b
To the stirred solution of 3 (2 mmol) and potassium carbonate (5 mmol) in dry THF (30 ml), a solution of appropriate 4-aminoquinoline 4 (1.0 mmol) in dry THF (50 ml) was added. The reaction mixture was stirred for 48 h at room temperature. The reaction mixture was filtered and THF was removed under vacuum. The residue was purified by column chromatography using MeOH/ethyl acetate as eluent to obtain corresponding 5, which was recrystallized from DCM/hexane. Using this procedure the following compounds were isolated.
5.2.1. Methyl 2-(3-((7-chloroquinolin-4-yl)amino)propylamino)-4-methyl-6-phenylpyrimidine-5-carboxylate (5a)
White solid. Rf: 0.47 (4% MeOH/ethyl acetate). Yield: 86%. m.p.105 °C. IR (KBr): ν max 770, 1267, 1709, 2928, 3427 cm− 1. 1H NMR (300 MHz, CDCl3, 25 °C): δ 2.00 (q, J = 6.3 Hz, 2H, CH2), 2.49 (s, 3H, C6-CH3), 3.46 (m, 2H, CH2), 3.58 (s, 3H, ester-CH3), 3.68 (q, J = 6.6 Hz, 2H, CH2), 5.57 (br, 1H, NH), 6.41 (d, 1H, ArH), 6.44 (br, 1H, NH), 7.39–7.54 (m, 7H, ArH), 7.91 (s, 1H, ArH), 8.48 (d, 1H, ArH). 13C NMR (75 MHz, CDCl3, 25 °C): δ 14.8, 20.6, 30.0, 31.6, 43.8, 90.6, 92.3, 112.7, 116.8, 119.5, 120.2, 121.5, 126.5, 141.3, 143.6, 153.5, 159.1, 160.8. Anal. Calcd. for C25H24N5O2Cl: C, 65.00; H, 5.24; N, 15.10; Found: C, 65.14; H, 5.19; N, 14.99. MS: m/z 462 [M+].
5.2.2. i-Propyl 2-(4-((7-chloroquinolin-4-yl)amino)butylamino)-4-methyl-6-phenylpyrimidine-5-carboxylate (5b)
White solid. Rf: 0.41 (4% MeOH/ethyl acetate). Yield: 83%. m.p. 140 °C. IR (KBr): ν max 1368, 1724, 2993, 3473 cm− 1. 1H NMR (400 MHz, CDCl3, 25 °C): δ 1.0 (d, J = 5.6 Hz, 6H, 2 × ester-CH3), 1.78 (m, 4H, CH2), 2.46 (s, 3H, C6-CH3), 3.33 (m, 2H, CH2), 3.50 (m, 2H, CH2), 4.99 (m, 1H, ester-CH), 5.44 (br, 1H, NH), 5.67 (br, 1H, NH), 6.34 (d, J = 5.3 Hz, 1H, ArH), 7.27–7.63 (m, 7H, ArH), 7.93 (d, J = 1.4 Hz, 1H, ArH), 8.46 (d, J = 5.3 Hz, 1H, ArH). 13C NMR (75 MHz, CDCl3, 25 °C): δ 21.3, 22.9, 25.8, 27.4, 40.7, 42.9, 68.9, 98.9, 116.0, 117.0, 121.3, 125.3, 129.5, 135.0, 138.9, 148.4, 150.0, 151.3, 161.2, 165.8, 166.9, 168.3. Anal. Calcd. for C28H30N5O2Cl: C, 66.72; H, 6.00; N, 13.89; Found: C, 66.50; H, 5.88; N, 13.65. MS: m/z 503.2 [M+].
5.3. General procedure for the synthesis of compound 5c–g
To the stirred solution of appropriate 4-aminoquinoline 4 in dry acetonitrile (50 ml) mixture of 3 (in a 1:2 molar ratio) and potassium carbonate in dry acetonitrile was added. The reaction mixture was refluxed for 24 h and then filtered. Acetonitrile was removed under vacuum and the residue was purified by column chromatography using MeOH/ethyl acetate as eluent to give 5 which is recrystallized from DCM/hexane.
5.3.1. Ethyl 2-(4-((7-chloroquinolin-4-yl)amino)butylamino)-4-methyl-6-(2-nitrophenyl)pyrimidine-5-carboxylate (5c)
Yellow solid. Rf: 0.28 (4% MeOH/ethyl acetate). Yield: 75%. m.p. 72 °C. IR (KBr): ν max 769, 1550, 1355, 1720, 2930, 3365 cm− 1. 1H NMR (400 MHz, CDCl3, 25 °C): δ 0.87 (t, J = 7.1 Hz, 3H, ester-CH3), 1.76 (m, 4H, CH2), 2.57 (s, 3H, C6-CH3), 3.27 (m, 2H, CH2), 3.47 (m, 2H, CH2), 3.96 (q, J = 7.0 Hz, 2H, ester-CH2), 6.07 (br, 1H, NH), 6.26 (d, J = 4.8 Hz, 1H, ArH), 6.33 (br, 1H, NH), 7.22–7.86 (m, 6H, ArH), 8.11 (s, 1H, ArH), 8.32 (d, J = 5.4 Hz, 1H, ArH). 13C NMR (100 MHz, CDCl3, 25 °C): δ 12.5, 22.1, 24.4, 28.6, 39.7, 42.2, 59.7, 97.4, 115.6, 121.3, 123.2, 124.5, 125.0, 128.2, 128.6, 131.9, 134.7, 145.2, 148.3, 150.1, 159.9, 165.3, 176.3. Anal. Calcd. for C27H27N6O4Cl: C, 60.62; H, 5.09; N, 15.7; Found: C, 60.34; H, 5.01; N, 15.58. MS: m/z 534.2 [M+].
5.3.2. Ethyl 2-(3-((7-chloroquinolin-4-yl)amino)propoxy)-4-methyl-6-phenylpyrimidine-5-carboxylate (5d)
White solid. Rf: 0.38 (4% MeOH/ethyl acetate). Yield: 90%. m.p.110 °C. IR (KBr): ν max 1255, 1775, 2969, 3530 cm− 1. 1H NMR (300 MHz, CDCl3, 25 °C): δ 1.05 (t, J = 7.2 Hz, 3H, ester-CH3), 2.43 (m, 2H, CH2), 2.59 (s, 3H, C6-CH3), 3.56 (m, 2H, CH2), 4.13 (t, J = 7.2 Hz, 2H, CH2), 4.68 (q, J = 6.0 Hz, 2H, ester-CH2), 5.70 (br, 1H, NH), 6.41 (d, J = 5.4 Hz, 1H, ArH), 7.20–7.76 (m, 7H, ArH), 7.93 (d, J = 2.1 Hz, 1H, ArH), 8.49 (d, J = 5.4 Hz, 1H, ArH). 13C NMR (75 MHz, CDCl3, 25 °C): δ = 13.5, 22.8, 27.7, 41.2, 61.7, 66.4, 121.3, 125.3, 130.2, 151.9. Anal. Calcd. for C26H25N4O3Cl: C, 65.47; H, 5.28; N, 11.75; Found: C, 65.12; H, 4.99; N, 11.89. MS: m/z 476.1 [M+].
5.3.3. Ethyl 4-(4-chlorophenyl)-2-(4-((7-chloroquinolin-4-yl)amino)butylamino)-6-methylpyrimidine-5-carboxylate (5e)
Yellow solid. Rf: 0.54 (4% MeOH/ethyl acetate). Yield: 85%. m.p. 135 °C. IR (KBr): ν max 1065, 1720, 2930, 3489 cm− 1. 1H NMR (300 MHz, CDCl3, 25 °C): δ 1.03 (t, J = 6.9 Hz, 3H, ester-CH3), 1.86 (m, 4H, CH2), 2.46 (s, 3H, C6-CH3), 3.40 (m, 2H, CH2), 3.59 (m, 2H, CH2), 4.07 (q, J = 6.0 Hz, 2H, ester-CH2), 5.50 (br, 1H, NH), 5.78 (br, 1H, NH), 6.36 (d, J = 5.7 Hz, 1H, ArH), 7.25–7.37 (m, 5H, ArH), 7.48 (d, J = 8.4 Hz, 1H, ArH), 7.96 (d, J = 7.8 Hz, 1H, ArH), 8.42 (d, J = 5.7 Hz, 1H, ArH). 13C NMR (75 MHz, CDCl3, 25 °C): δ 16.5, 28.8, 30.2, 32.5, 64.1, 101.8, 128.3, 131.3, 132.2, 143.2, 152.4, 161.6, 167.7. Anal. Calcd. for C27H27N5O2Cl2: C, 61.84; H, 5.19; N, 13.35; Found: C, 61.57; H, 5.05; N, 13.12. MS: m/z 523.1 [M+].
5.3.4. Ethyl 4-(4-fluorophenyl)-2-(4-((7-chloroquinolin-4-yl)amino)butylamino)-6-methylpyrimidine-5-carboxylate (5f)
Yellow solid. Rf: 0.34 (4% MeOH/ethyl acetate). Yield: 72%. m.p. 112 °C. IR (KBr): ν max 1156, 1682, 1333, 2928, 3395 cm− 1. 1H NMR (300 MHz, CDCl3, 25 °C): δ 1.02 (t, J = 7.2 Hz, 3H, ester-CH3), 1.83 (m, 4H, CH2), 2.46 (s, 3H, C6-CH3), 3.38 (m, 2H, CH2), 3.49 (m, 2H, CH2), 4.08 (q, J = 6.0 Hz, 2H, ester-CH2), 5.40 (br, 1H, NH), 5.74 (br, 1H, NH), 6.37 (d, J = 5.4 Hz, 1H, ArH), 7.04–7.55 (m, 6H, ArH), 7.95 (d, J = 2.1 Hz, 1H, ArH), 8.47 (d, J = 5.7 Hz, 1H, ArH). 13C NMR (75 MHz, CDCl3, 25 °C): δ 18.5, 29.6, 31.2, 63.5, 99.8, 126.3, 133.3, 145.2, 153.4, 160.4, 171.7. Anal. Calcd. for C27H27N5O2ClF: C, 63.84; H, 5.36; N, 13.79; Found: C, 63.76; H, 5.23; N, 13.88. MS: m/z 507.1 [M+].
5.3.5. Ethyl 2-(3-((7-chloroquinolin-4-yl)amino)propylamino)-4-methylpyrimidine-5-carboxylate (5g)
White solid. Rf: 0.38 (4% MeOH/ethyl acetate). Yield: 89%. m.p. 143 °C. IR (KBr): ν max 1097, 1702, 2973, 3365 cm− 1. 1H NMR (300 MHz, CDCl3, 25 °C): δ 1.36 (t, J = 7.1 Hz, 3H, ester-CH3), 1.84 (m, 4H, CH2), 2.64 (s, 3H, C6-CH3), 3.40 (d, J = 5.4 Hz, 2H, CH2), 3.58 (m, 2H, CH2), 4.31 (q, J = 7.2 Hz, 2H, ester-CH2), 5.68 (br, 2H, NH), 6.40 (d, J = 5.7 Hz, 1H, ArH), 7.33 (d, J = 2.1 Hz, 1H, ArH), 7.84 (d, J = 6.0 Hz, 1H, ArH), 7.95 (d, J = 2.1 Hz, 1H, ArH), 8.45 (d, J = 6.0 Hz, 1H, ArH), 8.70 (s, 1H, ArH). 13C NMR (75 MHz, CDCl3, 25 °C): δ 14.3, 40.8, 60.5, 99.1, 125.3, 128.8, 149.0, 151.9. Anal. Calcd. for C21H24N5O2Cl: C, 60.94; H, 5.84; N, 16.92; Found: C, 61.10; H, 5.75; N, 16.79. MS: m/z 413.1 [M+].
6. Material and methods
6.1. In vitro antimalarial activity assay
The test samples were tested in triplicate on one or two separate occasions against chloroquine sensitive (CQS) strain of P. falciparum (D10). Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified method of Trager and Jensen [45]. Quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method described by Makler et al. [46]. The test samples were prepared to a 20 mg/ml stock solution in 100% DMSO and sonicated to enhance solubility. Samples were tested as a suspension if not completely dissolved. Stock solutions were stored at −20 °C. Further dilutions were prepared on the day of the experiment. Chloroquine (CQ) was used as the reference drug in all experiments. Test samples were initially tested at three concentrations (10 μg/ml, 5 μg/ml and 2.5 μg/ml) to determine the starting concentration for the full dose–response assay. CQ was tested at three concentrations namely 30 ng/ml, 15 ng/ml and 7.5 ng/ml. A full dose–response was performed for all compounds to determine the concentration inhibiting 50% of parasite growth (IC50-value). Test samples were tested at a starting concentration of 10 μg/ml, which was then serially diluted 2-fold in complete medium to give 10 concentrations; with the lowest concentration being 0.02 μg/ml. The same dilution technique was used for all samples. CQ was tested at a starting concentration of 1000 ng/ml. Several compounds were tested at a starting concentration of 1000 ng/ml. The highest concentration of solvent to which the parasites were exposed to had no measurable effect on the parasite viability (data not shown). The IC50-values were obtained using a non-linear dose–response curve fitting analysis via Graph Pad Prism v.4.0 software.
6.2. Cytotoxicity and antiviral activity assay
Cytotoxicity was determined by exposing different concentrations of samples to Vero, HEL, HeLa and MDCK cells [29]. The antiviral assays were based on inhibition of virus-induced cytopathicity in HEL [herpes simplex virus type 1 (HSV-1), HSV-2 (G), vaccinia virus, and vesicular stomatitis virus], Vero (parainfluenza-3, reovirus-1, Coxsackie B4, and Punta Toro virus), HeLa (vesicular stomatitis virus, Coxsackie virus B4, and respiratory syncytial virus) and MDCK (influenza A (H1N1; H3N2) and B virus) cell cultures. Confluent cell cultures in microtiter 96-well plates were inoculated with 100 cell culture inhibitory dose-50 (CCID50) of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) in the presence of varying concentrations of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds [29].
Acknowledgments
We gratefully acknowledge financial assistance from CSIR, New Delhi (project 01(2364)/10/EMR-II) and UGC, New Delhi for Special Assistance Programme (SAP). H.K. thanks CSIR, New Delhi for senior research fellowship. JB thanks KU Leuven for financial support (GOA 10/14).
Footnotes
Supplementary material associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ejmech.2013.05.046.
Appendix A. Supplementary material
The following is the supplementary data related to this article:
References
- 1.Feachem R.G.A., Phillips A.A., Hwang J., Cotter C., Wielgosz B., Greenwood B.M., Sabot O., Rodriguez M.H., Abeyasinghe R.R., Ghebreyesus T.A., Snow R.W. Shrinking the malaria map: progress and prospects. Lancet. 2010;376:1566–1578. doi: 10.1016/S0140-6736(10)61270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO, 2010, http://www.who.int/malaria/world_malaria_report_2010/worldmalariareport2010.pdf.
- 3.Gamo F.-J., Sanz L.M., Vidal J., Cozar C., Alvarez E., Lavandera J.-L., Vanderwall D.E., Green D.V.S., Kumar V., Hasan S., Brown J.R., Peishoff C.E., Cardon L.R., Bustos J.F.G. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 4.Dondorp M., Nosten F., Yi P., Das D., Phyo A.P., Tarning J., Lwin K.M., Ariey F., Hanpithakpong W., Lee S.J., Ringwald P., Silamut K., Imwong M., Chotivanich K., Lim P., Herdman T., An S.S., Yeung S., Singhasivanon P., Day N.P.J., Lindegardh N., Socheat D., White N.J. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009;361:455–469. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlitzer M. Malaria chemotherapeutics, part I: history of antimalarial drug development, currently used therapeutics, and drugs in clinical development. ChemMedChem. 2007;2:944–986. doi: 10.1002/cmdc.200600240. [DOI] [PubMed] [Google Scholar]
- 6.Meunier B. Hybrid molecules with a dual mode of action: dream or reality? Acc. Chem. Res. 2008;41:69–77. doi: 10.1021/ar7000843. [DOI] [PubMed] [Google Scholar]
- 7.Muregi F.W., Ishih A. Next-generation antimalarial drugs: hybrid molecules as a new strategy in drug design. Drug Dev. Res. 2010;71:20–32. doi: 10.1002/ddr.20345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manohar S., Khan S.I., Rawat D.S. Synthesis, antimalarial activity and cytotoxicity of 4-aminoquinoline–triazine conjugates. Bioorg. Med. Chem. Lett. 2010;20:322–325. doi: 10.1016/j.bmcl.2009.10.106. [DOI] [PubMed] [Google Scholar]
- 9.Manohar S., Khan S.I., Rawat D.S. Synthesis of 4-aminoquinoline-1,2,3-triazole and 4-aminoquinoline-1,2,3-triazole-1,3,5-triazine hybrids as potential antimalarial agents. Chem. Biol. Drug Des. 2011;78:124–136. doi: 10.1111/j.1747-0285.2011.01115.x. [DOI] [PubMed] [Google Scholar]
- 10.Biot C., Glorian G., Maciejewski L.A., Brocard J.S. Synthesis and antimalarial activity in vitro and in vivo of a new ferrocene–chloroquine analogue. J. Med. Chem. 1997;40:3715–3718. doi: 10.1021/jm970401y. [DOI] [PubMed] [Google Scholar]
- 11.Chauhan K., Sharma M., Saxena J., Singh S.V., Trivedi P., Srivastava K., Puri S.K., Saxena J.K., Chaturvedi V., Chauhan P.M.S. Synthesis and biological evaluation of a new class of 4-aminoquinoline–rhodanine hybrid as potent anti-infective agents. Eur. J. Med. Chem. 2013;62:693–704. doi: 10.1016/j.ejmech.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 12.Solomon V.R., Haq W., Srivastava K., Puri S.K., Katti S.B. Synthesis and antimalarial activity of side chain modified 4-aminoquinoline derivatives. J. Med. Chem. 2007;50:394–398. doi: 10.1021/jm061002i. [DOI] [PubMed] [Google Scholar]
- 13.Guantai E.M., Ncokazi K., Egan T.J., Gut J., Rosenthal P.J., Bhampidipati R., Kopinathan A., Smith P.J., Chibale K. Enone- and chalcone–chloroquinoline hybrid analogues: in silico guided design, synthesis, antiplasmodial activity, in vitro metabolism, and mechanistic studies. J. Med. Chem. 2011;54:3637–3649. doi: 10.1021/jm200149e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cabaret O.D., Vical F.B., Robert A., Meunier B. Preparation and antimalarial activities of trioxaquines, new modular molecules with a trioxane skeleton linked to a 4-aminoquinoline. ChemBioChem. 2000;1:281–283. doi: 10.1002/1439-7633(20001117)1:4<281::AID-CBIC281>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 15.Chiyanzu I., Clarkson C., Smith P.J., Lehman J., Gut J., Rosenthal P.J., Chibale K. Design, synthesis and anti-plasmodial evaluation in vitro of new 4-aminoquinoline isatin derivatives. Bioorg. Med. Chem. 2005;13:3249–3261. doi: 10.1016/j.bmc.2005.02.037. [DOI] [PubMed] [Google Scholar]
- 16.Manohar S., Rajesh U.C., Khan S.I., Tekwani B.L., Rawat D.S. Novel 4-aminoquinoline–pyrimidine based hybrids with improved in vitro and in vivo antimalarial activity. ACS Med. Chem. Lett. 2012;3:555–559. doi: 10.1021/ml3000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pretorius S.I., Breytenbach W.J., de Kock C., Smith P.J., N’Da D.D. Synthesis, characterization and antimalarial activity of quinoline–pyrimidine hybrids. Bioorg. Med. Chem. 2012;13:3249–3261. doi: 10.1016/j.bmc.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Sharma M., Chaturvedi V., Manju Y.K., Bhatnagar S., Srivastava K., Puri S.K., Chauhan P.M. Substituted quinolinyl chalcones and quinolinyl pyrimidines as a new class of anti-infective agents. Eur. J. Med. Chem. 2009;44:2081–2091. doi: 10.1016/j.ejmech.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 19.Ettari R., Bova F., Zappala M., Grasso S., Micale N. Falcipain-2 inhibitors. Med. Res. Rev. 2010;30:136–167. doi: 10.1002/med.20163. [DOI] [PubMed] [Google Scholar]
- 20.Foley M., Tilley L. Quinoline antimalarial: mechanisms of action and resistance. Int. J. Parasitol. 1997;27:231–240. doi: 10.1016/s0020-7519(96)00152-x. [DOI] [PubMed] [Google Scholar]
- 21.Kelly J.X., Smilkstein M.J., Brun R., Sergio W., Cooper A.R., Lane K.D., Janowsky A., Johnson R.A., Dodean R.A., Winter R., Hinrichs D.J., Riscoe M.K. Discovery of dual function acridones as a new antimalarial chemotype. Nature. 2009;459:270–273. doi: 10.1038/nature07937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hossain N., Rozenski J., Clercq E.D., Herdewijn P. Synthesis and antiviral activity of the alpha-analogues of 1,5-anhydrohexitol nucleosides. J. Org. Chem. 1997;62:2442–2447. doi: 10.1021/jo961982m. [DOI] [PubMed] [Google Scholar]
- 23.Singh K., Singh K., Wan B., Franzblau S., Chibale K., Balzarini J. Facile transformation of 3,4-dihydropyrimidin-2(1H)-ones to pyrimidines in vitro evaluation as inhibitor of mycobacterium tuberculosis and modulators of cytostatic activity. Eur. J. Med. Chem. 2011;46:2290–2294. doi: 10.1016/j.ejmech.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 24.Joseph S., Burke J.M. Optimization of an anti-HIV hairpin ribozyme by in vitro selection. J. Biol. Chem. 1993;268:24515–24518. [PubMed] [Google Scholar]
- 25.Bookser B.C., Ugarkar B.G., Matelich M.C., Lemus R.H., Allan M., Tsuchiya M., Nakane M., Nagahisa A., Wiesner J.B., Erion M.D. Adenosine kinase inhibitors. Synthesis, water solubility, and antinociceptive activity of 5-phenyl-7-(5-deoxy-beta-D-ribofuranosyl) pyrrolo[2,3-d]pyrimidines substituted at C4 with glycinamides and related compounds. J. Med. Chem. 2005;48:7808–7820. doi: 10.1021/jm050394a. [DOI] [PubMed] [Google Scholar]
- 26.Hargreaves S.L., Pilkington B.L., Russell S.E., Worthington P.A. The synthesis of substituted pyridylpyrimidine fungicides using palladium catalysed cross-coupling reactions. Tetrahedron Lett. 2000;41:1653–1656. [Google Scholar]
- 27.Carlos M.G., John R.M., Charles D. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002;3:415–424. doi: 10.1016/s1470-2045(02)00788-x. [DOI] [PubMed] [Google Scholar]
- 28.Martyn D.C., Nijjar A., Celatka C.A., Mazitschek R., Cortese J.F., Tyndall E., Liu H., Fitzgerald M.M., Shea T.J., Danthi S., Clardy J. Synthesis and antiplasmodial activity of novel 2,4-diaminopyrimidines. Bioorg. Med. Chem. Lett. 2010;20:228–231. doi: 10.1016/j.bmcl.2009.10.133. [DOI] [PubMed] [Google Scholar]
- 29.Singh K., Kaur H., Chibale K., Balzarini J., Little S., Bharatam P.V. 2-Aminopyrimidine based 4-aminoquinoline anti-plasmodial agents. Synthesis, biological activity, structure–activity relationship and mode of action studies. Eur. J. Med. Chem. 2012;52:82–97. doi: 10.1016/j.ejmech.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaabani A., Bazgir A., Teimouri F. Ammonium chloride-catalyzed one-pot synthesis of 3,4-dihydropyrimidin-2-(1H)-ones under solvent-free conditions. Tetrahedron Lett. 2003;44:857–859. [Google Scholar]
- 31.Singh K., Singh J., Deb P.K., Singh H. An expedient protocol of the Biginelli dihydropyrimidine synthesis using carbonyl equivalents. Tetrahedron. 1999;55:12873–12880. [Google Scholar]
- 32.Puchala A., Belaj F., Bergman J., Kappe C.O. On the reaction of 3,4-dihydropyrimidones with nitric acid. Preparation and X-ray structure analysis of a stable nitrolic acid. J. Heterocycl. Chem. 2001;38:1345–1352. [Google Scholar]
- 33.Natarajan J.K., Alumasa J.N., Yearick K., Ekoue-Kovi K.A., Casabianca L.B., de Dios A.C., Wolf C., Roepe P.D. 4-N-, 4-S-, and 4-O-Chloroquine analogues: influence of side chain length and quinolyl nitrogen pKa on activity vs chloroquine resistant malaria. J. Med. Chem. 2008;51:3466–3479. doi: 10.1021/jm701478a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kouroumalis E.A., Koskinas J. Treatment of chronic active hepatitis B (CAH B) with chloroquine: a preliminary report. Ann. Acad. Med. 1986;15:149–152. [PubMed] [Google Scholar]
- 35.Kono M.K., Tatsumi A.M., Imai K., Saito T., Kuriyama, Shirasawa H. Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: involvement of p38 MAPK and ERK. Antiviral Res. 2008;77:150–152. doi: 10.1016/j.antiviral.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai W.P., Nara P.L., Kung H.F., Oroszlan S. Inhibition of human immunodeficiency virus infectivity by chloroquine. AIDS Res. Hum. Retroviruses. 1990;6:481–489. doi: 10.1089/aid.1990.6.481. [DOI] [PubMed] [Google Scholar]
- 37.Singh A.K., Sidhu G.S., Friedman R.M., Maheshwari R.K. Mechanism of enhancement of the antiviral action of interferon against herpes simplex virus-1 by chloroquine. J. Interferon Cytokine Res. 1996;16:725–731. doi: 10.1089/jir.1996.16.725. [DOI] [PubMed] [Google Scholar]
- 38.Dorn A., Stoffel R., Matile H., Bubendorf A., Ridley R. Malarial haemozoin/beta-haematin supports haem polymerization in the absence of protein. Nature. 1995;374:269–271. doi: 10.1038/374269a0. [DOI] [PubMed] [Google Scholar]
- 39.Gans P., Sabatini A., Vacca A. Investigation of equilibria in solution. Determination of equilibrium constants with the hyperquad suite of programmes. Talanta. 1996;43:1739–1753. doi: 10.1016/0039-9140(96)01958-3. [DOI] [PubMed] [Google Scholar]
- 40.Meshnick S.R. Chloroquine as intercalator: a hypothesis revived. Parasitol. Today. 1990;6:77–79. doi: 10.1016/0169-4758(90)90215-p. [DOI] [PubMed] [Google Scholar]
- 41.Wilson W.D., Jones R.L. Intercalating drugs: DNA binding and molecular pharmacology. Adv. Pharmacol. Chemother. 1981;18:177–222. doi: 10.1016/s1054-3589(08)60255-0. [DOI] [PubMed] [Google Scholar]
- 42.Kumari N., Maurya B.K., Koiri R.K., Trigun S.K., Saripella S., Coogan M.P., Mishra L. Cytotoxic activity, cell imaging and photocleavage of DNA induced by a Pt(II) cyclophane bearing 1,2 diamino ethane as a terminal ligand. MedChemComm. 2011;2:1208–1216. [Google Scholar]
- 43.Cohen S.N., Yielding K.L. Spectrophotometric studies of the interaction of chloroquine with deoxyribonucleic acid. J. Biol. Chem. 1965;240:3123–3131. [PubMed] [Google Scholar]
- 44.Mudasir E.T., Wahyuni D.H., Tjahjono N., Yoshioka H., Inoue Spectroscopic studies on the thermodynamic and thermal denaturation of the CT-DNA binding of methylene blue. Spectrochim. Acta Part A. 2010;77:528–534. doi: 10.1016/j.saa.2010.06.032. [DOI] [PubMed] [Google Scholar]
- 45.Trager W., Jensen J.B. Human malaria parasite in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 46.Makler M.T., Ries J.M., Williams J.A., Bancroft J.E., Piper R.C., Gibbins B.L., Hinrichs D.J. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg. 1993;48:739–741. doi: 10.4269/ajtmh.1993.48.739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.