

Abstract
Chromosomes have a complex three-dimensional (3D) architecture comprising A/B compartments, topologically associating domains and promoter–enhancer interactions. At all these levels, the 3D genome has functional consequences for gene transcription and therefore for cellular identity. The development and activation of lymphocytes involves strict control of gene expression by transcription factors (TFs) operating in a three-dimensionally organized chromatin landscape. As lymphocytes are indispensable for tissue homeostasis and pathogen defense, and aberrant lymphocyte activity is involved in a wide range of human morbidities, acquiring an in-depth understanding of the molecular mechanisms that control lymphocyte identity is highly relevant. Here we review current knowledge of the interplay between 3D genome organization and transcriptional control during B and T lymphocyte development and antigen-dependent activation, placing special emphasis on the role of TFs.
Keywords: 3D genome folding, chromosome conformation, lymphocyte differentiation, transcription factors, T-cell differentiation, B-cell differentiation
Introduction
The development and activation of immune cells has long served as a paradigm for studying transcriptional control during cell differentiation. Three-dimensional (3D) folding of the genome is implicated in the control of gene expression and cell fate, including for cells of the immune system. Our immune system is indispensable for host defense and tissue repair, for it eliminates pathogens, clears malignant cells and maintains tissue homeostasis. In vertebrates, the immune system is composed of a plethora of cell types with different and often highly specialized functions. Together and through their interplay with various other types of cells (e.g. stromal or epithelial cells), immune cells orchestrate two complementary types of responses: immediate and broadly effective responses (‘innate immunity’) as well as antigen-specific, long-lasting responses that generate immunological memory (‘adaptive immunity’). B and T Lymphocytes are central in mounting effective adaptive immune responses, and lymphocyte development and activity are governed by strict transcriptional control of cell lineage progression, cell identity and function. Importantly, impaired transcriptional control in lymphocytes can lead to aberrant immune cell development or activity, as seen in a wide variety of disorders that includes (hematological) cancers, immunodeficiencies, asthma and autoimmune diseases. Here we summarize our current knowledge of the molecular mechanisms that regulate gene expression in immune cells, with focus on the interplay between 3D genome organization and transcriptional control in lymphocyte development and activation.
Transcriptional regulation occurs in 3D
Cell-type or cell-state-specific gene expression is critically regulated by transcription factors (TFs), a class of sequence-specific DNA-binding proteins [1]. TFs regulate transcription by binding to gene regulatory elements (REs) in promoters (the region preceding the transcription start site, or TSS) and often distantly located REs called enhancers. They recruit cofactors (including coactivators, corepressors) to modify the local chromatin landscape and facilitate or impede the recruitment of the RNA polymerase 2 (RNAPol2) basal transcription machinery [1, 2]. Conversely, the chromatin state strongly influences DNA binding of TFs, for the positioning and post-translational modifications of the nucleosomes around which genomic DNA is wrapped dictate DNA accessibility for TFs [3]. A subset of TFs has the capacity to bind ‘inaccessible’ chromatin and these are often referred to as ‘pioneer’ factors [4]. Many lineage-determining TFs appear to have such pioneering capacities [5].
For our own convenience, we think of our genome as a one-dimensional string of chromatin on which genes and REs are linearly separated. However, chromosomes display a complex 3D architecture, which has been intensively studied the past 20 years and has functional consequences for almost all DNA-templated processes, including transcriptional regulation [6]. Early studies of how enhancers regulate spatiotemporal expression of their target loci over large distances led to proposed models in which 3D folding allows spatial and specific proximity, regardless of linear distance, between promoter–enhancer pairs [7–9]. Technologies to document 3D genome architecture and its dynamics have evolved rapidly over the past years. They now enable researchers to look beyond the properties and folding of individual gene loci, i.e. at gene clusters, but also at megabase-sized chromosomes. The high-throughput chromosome conformation capture (3C) assay and its subsequent 4C, 5C and Hi-C protocols have been particularly groundbreaking [10]. Their use has resulted in a multi-layered model of genome conformation that indeed goes beyond local interactions between genes and their respective enhancers, with strong links to transcriptional activity at every layer (Figure 1). 3D-folded genomes, as chromosomes, segregate into two major compartments based on chromatin state. Highly accessible regions are marked by histone modifications associated with regulatory activity (e.g. histone-3 lysine-4 dimethylation, H3K4me2; histone-3 lysine-27 acetylation, H3K27ac) and group together, whereas regions of low accessibility are marked by repressive histone marks (e.g. H3K9me3 or H3K27me3) and cluster separately [11]. These compartments are arbitrarily referred to as the A (‘active’) and B (‘inactive’) compartments, respectively. As expected from their different chromatin landscapes, the average gene expression levels are substantially higher in the A compartment [12]. At a smaller scale (~200 to 2000 kilobases), genomic regions are organized into domains of high self-interaction frequencies, named topologically associating domains [13–15]. TADs insulate genomic regions, which is thought to facilitate the frequency and specificity of intra-TAD promoter–RE interactions. Indeed, genes and REs mapped within the same TAD are more likely to find each other in 3D than when located in different TADs [16] (Figure 1).
Figure 1.
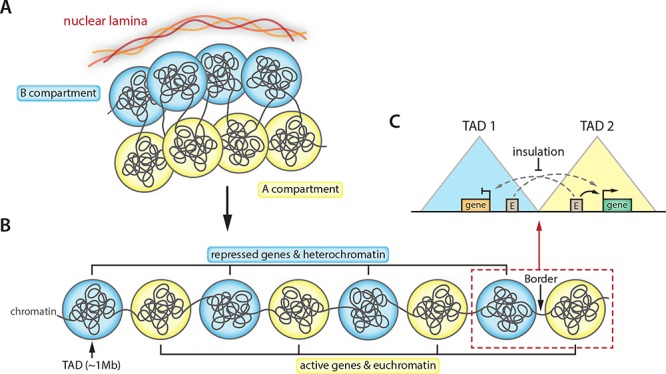
Key features of 3D genome organization. (A) Spatial chromosome folding in the nucleus separates the genome into two compartments: the A compartment that mainly consists of active genes and euchromatin (yellow spheres), and the B compartment that is enriched for inactive genes and heterochromatin (blue spheres). While regions of the A compartment localize to the nuclear interior, B compartment domains reside near the nuclear lamina. (B) A/B compartments contain megabase-sized self-interacting regions called topologically associating domains (TADs, depicted here as individual spheres). TADs tend to have a uniform compartment association and are separated by border regions. (C) TADs are considered functional units of chromosome organization in which enhancers (shown as boxes marked with ‘E’) interact with genes or other REs to control transcription. TAD border insulation is thought to restrict the search space of enhancers and promoters and to prevent unwanted regulatory contacts to be formed.
Multiple forces appear to play a key role in the formation of A versus B compartments, TADs and promoter–RE interactions (for recent reviews, see [9, 17–19]). Prominent amongst these is loop extrusion, in which the ring-shaped cohesin complex extrudes chromatin to form loops until it is blocked by the DNA-binding protein CCCTC-binding factor (CTCF). The process of transcription has been implicated in shaping genome topology at various levels [17, 19]. TFs have also been directly implicated in shaping various aspects of 3D genome folding, including the establishment and maintenance of intra-TAD promoter–enhancer interactions [18, 20, 21]. Overall, 3D genome architecture displays cell-type or cell-state-specific features [22, 23], suggesting that genome conformation is linked to specific gene expression programs. Indeed, changes in 3D genome folding appear dynamically coupled to changes in chromatin landscape, but also TF-DNA binding and hence gene expression [24]. However, it should be noted that the causal relationships between all these aspects of genome function and control—particularly for 3D genome organization and transcription—are highly complex and only partially understood (reviewed in [17]). While inhibition of transcription can affect genome conformation, TADs and compartments can be established or maintained in the absence of transcription or RNAPol2 [17, 25, 26]. Nevertheless, perturbation of genome topology is able to induce altered gene expression [27–31], although the extent of this appears dependent on cell state. Surprisingly, global depletion of CTCF or cohesin—resulting in a loss of nearly all TADs—does not necessarily cause widespread transcriptional misregulation under steady-state conditions [29–32]. On the other hand, various developmentally regulated loci (e.g. Sox9, genes of the Hoxd cluster, IHH) show major transcriptional defects upon perturbation of their respective TAD(s) [27, 33, 34]. Interestingly, post-mitotic macrophages lacking RAD21 (an essential component of the cohesin complex) do not display severe transcriptional defects until they are stimulated with endotoxin [35], illustrating that 3D genome architecture may be particularly important to rapidly modify gene expression programs.
Immune cell identity and function relies on accurate transcriptional control
A cell’s phenotype is determined by its gene expression program. Thus, substantial transcriptional changes are required for immune cells to adapt to instructive cues in their environment (e.g. during differentiation or activation). In mammals, most immune cells are derived from bone marrow resident hematopoietic stem cells that differentiate guided by signals emanating from the tissue microenvironment [36]. As hematopoietic stem cells differentiate and mature, a first major lineage bifurcation into either common myeloid progenitors (CMPs) or common lymphoid progenitors (CLPs) occurs. In addition to thrombocytes (for coagulation) and erythrocytes (for oxygen transport), CMPs give rise to a variety of innate immune cells, including mast cells, neutrophils, basophils, eosinophils, monocytes and dendritic cells. CLPs give rise to the B and T lymphocytes of the adaptive immune system, as well as innate lymphoid cells including natural killer cells [36–38]. With such a diversity of cell types originating from a common progenitor, tight transcriptional control is critical to ensure a correct qualitative and quantitative cellular output as well as stable lineage fidelity.
Developing B and T lymphocytes undergo a rigid, stepwise differentiation process involving combinatorial TF action that is intimately connected to the lineage-specific, ordered gene rearrangements occurring at their antigen-receptor loci (Figure 2A). B and T cells have the unique capacity to specifically recognize foreign antigens based on surface expression of the B-cell receptor (BCR) and T-cell receptor (TCR), respectively. Whereas the BCR is composed of identical pairs of immunoglobulin (Ig) heavy (H) chains and light (L) chains (the latter of the κ or λ subtype), TCRs are dimers that consists of either an α and β chain or a γ and δ chain. The antigen-specific variable domains of the Ig/TCR chains are encoded by arrays of various gene segments—so called variable (V), diversity (D) and joining (J) segments—that are genetically recombined in a largely random fashion. Both in B cells and in T cells, the gene rearrangement events are initiated through the activity of the recombination activating gene (RAG1/RAG2) proteins [39]. Because of the very large number of unique possible combinations of this ‘VDJ-recombination’ process (often estimated at >1011 [40]) and additional processes for nucleotide sequence diversification (e.g. somatic hypermutation) in B cells, mammals are able to generate BCRs and TCRs against virtually any antigen [39]. Once mature and checked for autoreactivity, immune cells are highly receptive to external signals for their rapid activation or further specification toward effector cells, which are actively involved in pathogen destruction or tissue repair [41–43]. Upon antigen recognition by the BCR and appropriate T-cell costimulation in germinal centers, B cells either further differentiate into plasma cells that produce pathogen-neutralizing antibodies (secreted forms of the BCR) or into long-lasting circulating memory B cells [44]. When activated by antigen-presenting cells, T cells fulfill a wide range of essential immune functions, ranging from the destruction of virus-infected or malignant cells (by cytotoxic CD8+ T cells) to orchestrating tailor-made local immune responses through the production of cytokines (by helper CD4+ T cells).
Figure 2.
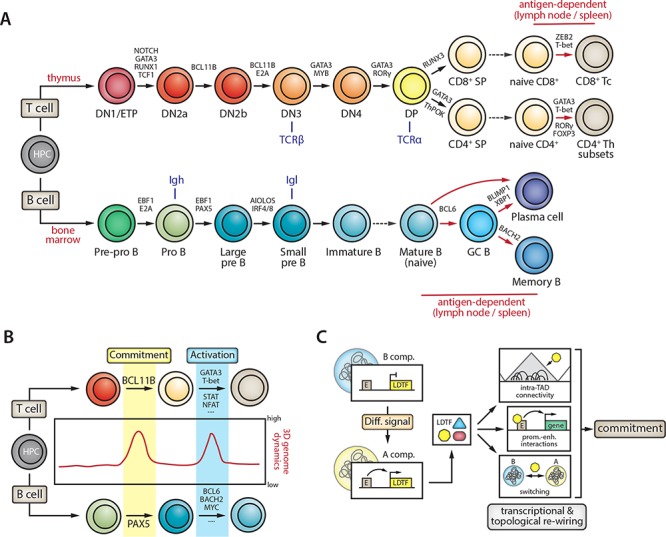
Transcription factors drive 3D genome dynamics during lymphocyte differentiation and activation. (A) Differentiation stages of T (top; for the most common αβ subset) or B (bottom) lymphocytes as they develop from hematopoietic progenitor cells (HPCs) into mature antigen-receptor-expressing cells that migrate to peripheral tissues (i.e. lymph nodes and spleen) for their antigen-dependent activation. Selected key transcription factors (TFs) critical for the various cell state transitions are indicated. Timing of TCR and immunoglobulin (Ig) locus recombination is indicated in blue. Dashed dark gray arrows denote migration of differentiated cells into the periphery; red arrows are used for antigen-dependent differentiation steps. Additional abbreviations: DN, double-negative; DP, double-positive; SP, single-positive; ETP, early thymic precursor; Th, T helper cell; Tc, cytotoxic T cell; GC, germinal center. (B) Analyses of topological genome dynamics during T-cell or B-cell lineage commitment and subsequent activation of mature lymphocytes have shown that the most extensive rewiring of 3D genome organization occurs right after the appearance of the commitment TFs BCL11b (for T cells) or PAX5 (for B cells) and after antigen-dependent activation. Several TFs critical for T-cell or B-cell activation have also been implicated in reorganizing lymphocyte genome conformation (e.g. GATA3, MYC). (C) Schematic representation of the interplay between genome topology, the activation of lineage-determining TFs (LDTF) and the subsequent action of these LDTFs. In the example, the locus encoding an LDTF (e.g. BCL11B in early T-cell development) is switched from the repressive B compartment to the transcription-competent A compartment under the influence of differentiation (Diff.) signals. As the LDTF gene is activated in the A compartment, TF proteins are produced that initiate a transcriptional and topological rewiring of the lymphocyte precursor that will eventually result in stable lineage commitment. LDTFs operate at different levels of 3D genome organization, including modifications to intra-TAD connectivity, promoter–enhancer (prom.-enh.) interactions and A/B compartment switching.
Throughout their development and activation, the exposure of immune cells to environmental cues (e.g. cytokines, metabolites, cell-cell interactions) triggers a cell-intrinsic signal transduction cascade that converges on altered expression and/or activity of DNA-binding TFs [1]. TFs in turn drive and coordinate the transcriptional changes required for immune cell-fate determination and lineage progression or for triggering specific effector programs in mature immune cells [45–47]. For example, in the thymus the membrane-bound Delta-family of ligands on epithelial cells interact with the NOTCH receptors on lymphoid progenitors. This causes specific proteolytic cleavage of the receptor, liberating the NOTCH intracellular domain that accumulates in the nucleus, where it acts as a TF and induces a T-cell gene expression program [48]. Other classic examples of how extrinsic signals control immune cell function involve signal transduction via intracellular Janus kinases (JAKs) and signal transducer and activator of transcription proteins (STATs). Activated T cells produce the interleukin-2 (IL-2) cytokine and concomitantly upregulate IL-2 receptor expression, resulting in JAK-mediated phosphorylation of STAT5, which then dimerizes and translocates to the nucleus to activate a cell proliferation gene expression program [49]. Thus, as endpoints of a signal transduction cascade, TFs convert signals from a cell’s microenvironment into a specific and spatially temporally controlled transcriptional response. These changes in the cellular transcriptome in turn lead to a modified proteome and, ultimately, cell function(s).
Topological genome dynamics and lymphocyte biology
Lymphocyte commitment meets genome topology: B cells
In mammals, lymphoid progenitors can either remain in the bone marrow, where they will differentiate toward B cells or innate lymphoid cells, or they can migrate to the thymus to initiate T-cell differentiation. Here, we discuss how early lymphocyte development is orchestrated at the transcriptional level and how this connects to functional changes in genome topology. Given the lack of systematic investigations of 3D genome organization during the development of innate lymphoid cells, we restrict ourselves to B and T lymphocytes.
Commitment of CLPs to the B-cell lineage is tightly controlled by a regulatory network formed by the combinatorial action of TFs PU.1, Ikaros, E2A, EBF1 and PAX5 [50]. EBF1 represses alternative lineage programs (e.g. for natural killer cell differentiation) and functions as a transcriptional activator of other TF-encoding genes that are crucial for B-cell development, in particular PAX5, which stably locks in the B-cell fate at the pro-B-cell stage (Figure 2A) [51–53]. Early B-cell development is accompanied by substantial modifications to 3D genome architecture. Already in 1997, Brown et al. showed that in pre-B cells the actively transcribed gene λ5 does not associate with heterochromatin-associated Ikaros foci, while its silencing in mature B cells correlates with close nuclear proximity of the locus to heterochromatin-associated Ikaros complexes. The Cd2 locus shows the opposite dynamics: it moves away from heterochromatin-associated Ikaros foci concomitant with its upregulation in mature B cells [54]. More recently, Lin et al. report hundreds of genes switching between A and B compartments when pre-pro-B cells differentiate to pro-B cells [55]. Notably, the Ebf1 locus repositions from the B compartment at the nuclear lamina to the A compartment, concomitant with its transcriptional activation in pro-B cells [55]. Other loci that shift from B to A at this early stage include Foxo1 and the Ig light chain loci, which in general correlates with increased mRNA expression. Genes that switch without transcriptional upregulation are often marked by the repressive histone modification H3K27me3, suggesting they are actively repressed independent of their nuclear sublocalization [55]. Within these compartments, TF-binding sites frequently colocalize in nuclear space—even over large distance (>1 Mb). Interestingly, two separate classes of such TF-interaction hubs have emerged [55]. One consists of (shorter-range) interactions between CTCF and cohesin-complex sites, which form through loop extrusion and represent many of the cell-type invariant structural loops in the genome [9, 17]. Another involves strong long-range interactions between B-cell TFs (e.g. E2A, PU.1) and the enhancer-binding histone acetyltransferases P300, indicating the existence of cell type-specific 3D-organized hubs of REs. These TF-mediated hubs have also been detected in other cell types, where they may safeguard cell identity or even help establish new identities by optimizing the regulation of TF target genes [18, 56]. Hence, TFs appear not only to mediate changes in gene expression and chromatin state, but also rewire the 3D genome during early B-cell development. In line with this, the binding landscape of E2A is dramatically altered as pre-pro-B cells differentiate to pro-B cells [57]. E2A colocalizes on the chromatin with other key regulators of early B-cell development, including EBF1 and FOXO1, suggesting their collaborative control of B-cell commitment [57]. Similarly, EBF1 DNA-binding is observed at approximately 30% of known PAX5-regulated genes, which is a two-fold enrichment over an unrelated control gene set [58]. We propose that the cooperative action of TFs during B-cell development allows for stricter control of 3D chromatin interactions involved in transcriptional regulation, which require specific combinations of TFs in order to be formed or maintained.
PAX5 enforces final B-cell commitment by repressing alternative lineage programs and activating a new subset of B-cell-specific genes [59]. Over 50% of PAX5 binding sites are located outside of genes or promoters [60], suggesting that PAX5 mainly uses long-range enhancer-promoter interactions to execute its function. Indeed, Hi-C analyses of Pax5−/− pro-B cells by Johanson et al. reveal 7810 differential chromatin interactions compared to WT pro-B cells; 83% of these nuclear proximities are weakened or removed in the mutant Pax5−/− cells [60]. Importantly, cDNA-mediated PAX5 re-introduction partially rescues aberrant 3D genome folding in Pax5−/− B-cell progenitors. While the loss of PAX5 has minimal effects on A–B compartmentalization and global TAD architecture [60], it appears that PAX5 mediates local intra-TAD regulatory interactions (e.g. promoter-RE contacts) to enforce B-cell lineage commitment. Importantly, the presence of PAX5 modifies 3D chromatin architecture in a seemingly direct manner that is independent of ongoing transcription: treatment of pro-B cells with α-amanitin (a potent inhibitor of RNAPol2) did not prevent PAX5 from rescuing the majority of chromatin interactions in Pax5−/− cells [60]. Johanson et al. also used Hi-C to analyze global 3D genome configuration of various B-cell subsets, including splenic B-cell populations, activated B cells and plasma cells. Intriguingly, while modifications to genome topology occur during every state transition, 3D genome dynamics peak after Pax5 induction and upon mature B-cell activation [60]. Thus, rewiring of 3D chromatin folding positively correlates with B-cell lineage commitment (i.e. the presence of PAX5) and activation in tissues (Figure 2B).
Lymphocyte commitment meets genome topology: T cells
T-cell development takes place in the thymus, which is seeded by early thymic progenitors that represent the double negative (or ‘DN’, because of the absence of the two canonical T-cell lineage markers CD4 and CD8) stage of T-cell development. Guided by Notch signaling and the activation of the lineage-determining TF BCL11B, definitive T-cell lineage commitment is enforced. Productive TCR rearrangements allow developing T cells to progress to the double positive (DP) stage and ultimately to CD4+ or CD8+ mature naive T cells (Figure 2A) [61, 62]. BCL11B, much like the key early B-cell TF PAX5, is critical for the silencing of alternative lineage programs in DN cells [61], in line with the substantial enrichment of BCL11B binding to regions with the repressive H3K27me3 mark [63]. Bcl11b activation is dependent on the stepwise combinatorial action of NOTCH and its downstream TF RBPJκ, the NOTCH-regulated TFs GATA3 and TCF1, as well as RUNX1, which operate at both the Bcl11b promoter region and an essential T-cell-specific enhancer located 850 kb downstream [64].
Another parallel with B-cell development is that the Bcl11b locus, like Ebf1, repositions from the B compartment in multipotent hematopoietic progenitors to the A compartment during early DN T-cell differentiation. This transition, which occurs concomitantly with Bcl11b gene activation, is mediated by transcription of a non-coding RNA called thymocyte differentiation factor (ThymoD) [65]. Furthermore, in the absence of ThymoD transcription, chromatin loops between the Bcl11b promoter and its distal enhancers are not formed, resulting in reduced Bcl11b expression and developmental arrest. Isoda et al. have shown that Bcl11b nuclear localization and 3D chromatin organization by ThymoD transcription occurs through the demethylation of CpG residues that facilitates the recruitment of CTCF and cohesin. Binding of CTCF-cohesin then promotes promoter–enhancer proximity and switching of the locus toward the A compartment [65].
At a genome-wide level, early T-cell development is accompanied by frequent A–B compartment switching, involving many loci encoding key TFs such as Bcl11b (B-to-A switching) and Meis1 (encoding a TF essential for multipotent hematopoietic precursors that is silenced and subjected to A-to-B switching) [63]. TAD border numbers vary during T-cell development (between 2768 and 3765 depending on the time point), and Hu et al. observed substantial intra-TAD interaction dynamics as cells differentiate toward the DP stage [63]. Again, 3D genome dynamics peak at the onset of lineage commitment, coinciding with upregulation of Bcl11b (Figure 2B). Like the B-cell commitment factor PAX5, BCL11B itself appears directly involved in this remodeling of the 3D chromatin landscape by promoting long-range chromatin interactions for its direct target genes, thus increasing their intra-TAD connectivity and expression levels [18, 23, 63].
From these combined analyses of B-cell and T-cell development, a picture emerges that intimately links the activity of lineage-determining TFs such as BCL11B, PAX5 and EBF1 to shaping lymphocyte genome topology. Loci encoding lymphocyte commitment TFs reside in the B compartment, possibly to help prevent their promiscuous activation. Differentiation signals from the microenvironment ‘liberate’ these TF loci, activating their expression and positioning them into the A compartment. The protein products of the now actively transcribed lineage-determining TF genes start to explore the nucleus in search for their target sequences, reshaping the 3D chromatin landscape at various levels to achieve proper gene activation and repression (Figure 2C). In line with analyses from other differentiation trajectories, the reported rewiring of the 3D chromatin landscape during the stepwise development of T cells generally coincides with major changes in histone modifications, global DNA accessibility and gene expression. However, changes in genome conformation can follow (i.e. for A-to-B switching) or foreshadow (i.e. for increased intra-TAD connectivity) transcriptional dynamics [63], indicating that gene regulation and 3D genome organization are not always strictly coupled during lymphoid development.
Antigen-receptor rearrangement requires dynamic 3D chromatin folding
Antigen-receptor recombination is restricted to specific lymphocyte lineages and developmental stages (Figure 2A). This specificity is in part controlled by the regulated expression of the RAG1/RAG2 recombinase complex in non-dividing immature lymphocytes and its recruitment to only a small focal region of active chromatin at the 3′ end of each antigen-receptor locus called the ‘recombination center’ [39]. Additionally, the timing of ordered V(D)J recombination, i.e. (i) D-to-J rearrangements preceding V-to-DJ rearrangements at the Igh/TCRβ loci and (ii) Igh/TCRβ recombination occurring before Igl/TCRα recombination, can be explained by the developmentally timed non-coding transcription of VDJ gene segments (referred to as ‘germline transcription’). Transcribed gene segments are specifically accessible to the recombinase [66]. Although the molecular mechanisms underlying this ‘accessibility hypothesis’ remain somewhat unclear, it has become apparent that the recruitment of developmental-stage-specific TF combinations to VDJ gene promoters and enhancers is critical for gene segment accessibility [67].
Antigen-receptor loci can span very large genomic distances (>1 Mb) and contain hundreds of V gene segments, each of which needs to have an equal opportunity to be used for recombination. Then, how are the very distal V genes—some of which are 2.5 Mb away from the recombination center—able to interact with the recombinase? Why do these different distances of individual V genes to the recombination center not result in a preference for proximal over distal V gene usage? It turns out that the answer to this lymphocyte-specific problem lies in the chromatin conformation of antigen-receptor loci at the time of or prior to recombination. To correct for the decreasing chance of a given V gene segment to interact with the recombination center as its linear genomic distance increases, megabase-sized antigen-receptor loci undergo extensive 3D chromatin compaction or ‘locus contraction’ (Figure 3) [68]. This way, all gene segments will have a largely similar average spatial distance to the recombination center, which ensures equal opportunities for recombination and maximum antigen-receptor repertoire diversity (for a recent more in-depth review, see Johanson et al. [69]).
Figure 3.
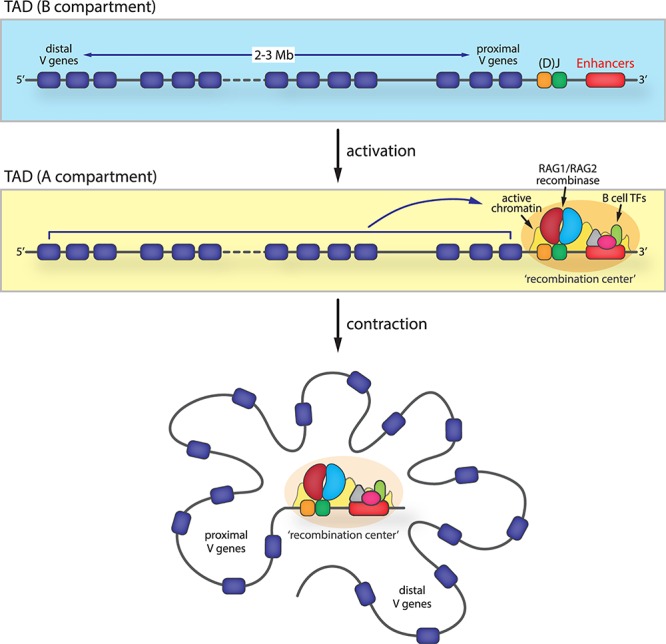
Locus contraction at antigen-receptor loci maximizes receptor diversity. Megabase-sized antigen-receptor loci (e.g. the murine immunoglobulin kappa locus shown here) are organized in TADs. Differentiation signals induce B-to-A compartment switching of the locus and trigger the establishment of a 3′-end recombination center near the (D-)J elements. At the recombination center, B-cell transcription factors (TFs) create an active chromatin environment that facilitates the recruitment of the RAG1/RAG2 V(D)J recombinase. To ensure equal average proximity of all V genes to the recombination center, antigen-receptor loci ‘contract’ to create a compact 3D structure that provides largely equal opportunities for every V gene to be recombined.
Hi-C analyses of developing B cells have revealed that the Ig loci are contained within TADs [70, 71]. Prior to their recombination, the Igh and Igl loci undergo a B-to-A compartment switch that positions them away from the nuclear lamina [55, 72]. Simultaneously, a marked gain in long-range interactions within the Ig-locus TADs indicates active locus contraction [55, 73]. Although the forces driving locus contraction at antigen-receptor loci are only partially understood, they appear similar to those involved in shaping chromatin conformation related to canonical transcriptional regulation. The concerted action of cell-type-specific TFs (e.g. PAX5, E2A) and general structural factors (i.e. CTCF, YY1) bound throughout the locus is critical for achieving optimal contraction and antigen-receptor diversity [67]. For example, depletion of PAX5 or CTCF during early B-cell development leads to altered long-range chromatin interactions and reduced contraction at Ig loci, resulting in impaired or proximally skewed recombination [74–76]. In this context, TFs can induce optimal locus contraction in a stepwise manner [72]. Prior to recombination, E2A is recruited to the enhancers of the Igκ locus to initiate its contraction in pro-B cells via largely random interactions with the V gene region. Subsequent differentiation into pre-B cells activates additional TFs, including IRF4, that also bind the Igκ enhancers and trigger a highly coordinated pattern of enhancer–gene interactions (‘enhancer focusing’) that strongly correlates with V gene recombination frequencies [72].
Lymphocyte activation and genome topology
After their development into mature lymphocytes, naive B and T cells enter the circulation. Once they encounter their cognate antigen and the right set of costimulatory signals, lymphocytes rapidly activate to initiate their specification into effector or memory cells. Lymphocyte activation triggers a dramatic phenotypic transformation that involves large-scale changes in gene expression accompanied by increased proliferation and a unique metabolic profile to accommodate the increased energy demands [77–79].
Regarding the role of the 3D chromatin landscape in this context, CD4+ T helper (Th) cell activation has been studied most extensively. T-cell activation requires TCR stimulation through presentation of an antigenic peptide by antigen-presenting cells (signal 1); a costimulatory signal, often via the T-cell surface protein CD28 (signal 2); and soluble factors (i.e. cytokines) released by the antigen-presenting cell or present in the microenvironment (signal 3). Together, these signals imprint Th-subtype differentiation and tissue-homing capacities onto the activated T cell [46, 80]. Depending on the specific combination of signals, CD4+ T cells will differentiate into one of the main Th subtypes: Th1 cells that express the TF T-bet and produce interferon-gamma (IFNγ) to combat intracellular pathogens; Th2 cells that express GATA3, and produce IL-4, IL-5 and IL-13 to clear parasite infections; Th17 cells that rely on the RORγ TF and produce IL-17 to respond to extracellular pathogens; or regulatory T cells (Tregs) that synthesize the TF FOXP3 and secrete anti-inflammatory cytokines such as IL-10 [80]. Important to note here is that differentiated Th cells can show considerable phenotypic plasticity, including (semi) stable intermediates (e.g. ‘Th17.1’ cells)42. Thus, environmental signals activate a specific lineage-determining TF that, together with other signal-responsive TFs (e.g. STATs), induces a cell-fate change involving widespread transcriptional remodeling.
Focused analyses of how these TFs activate the abovementioned Th signature cytokine-encoding loci have revealed that local 3D chromatin architecture plays a key role in establishing long-range promoter–enhancer communication and shaping Th-cell identity. A prime example is the activation of the clustered Il4, Il5 and Il13 genes (together often referred to as the ‘Th2 locus’) during Th2 differentiation. 3C analyses by the Flavell laboratory showed that while the Il4, Il5 and Il13 gene promoters spatially cluster in every cell type tested, the enhancer elements (organized in a locus control region or LCR) only interact with these gene promoters in T cells [81]. Surprisingly, LCR-promoter clustering also occurs—albeit somewhat weaker—in naive T cells or Th1 cells where this locus is inactive, suggesting that genome topology poises the Th2-cytokine genes for rapid activation in the presence of additional factors [81]. Remarkably, GATA3 overproduction and ionomycin-induced Nuclear factor of activated T cells (NFAT) activation induced LCR-promoter interactions even in fibroblasts [81], directly implicating these TFs in establishing promoter–enhancer communication. By contrast, interactions between the Ifng promoter and its enhancer elements are only detected in Th1 cells and depend on the presence of T-bet [82]. At the level of nuclear compartments, early microscopy studies showed that Th1 differentiation is accompanied by a progressive repositioning of Th2-associated genes toward centromeric heterochromatin [83, 84]. Together, these findings point toward a highly dynamic spatial organization of key cytokine and TF loci during CD4+ Th-cell activation.
Long-range chromatin interactions of the Il4 and Ifng genes in Th1 and Th2 cells have also been investigated in a genome-wide manner using 4C [85]. Promoter ‘interactomes’ for these two genes show substantial Th-subset specificity. Highly interacting genes also show higher subset-specific expression levels and these interaction partners are enriched for other immune-related genes. Although most interactions with the Ifng promoter can also be detected in naive T cells, many interactions become more focused (i.e. sharper, more narrow signals) upon Th1 specification, indicating a shift from promiscuous to more selective 3D genome architecture. Interestingly, 3D genome dynamics correlate with binding of STAT TFs that are activated by specific differentiation signals; the Th1-inducing cytokine IL-12 activates STAT4, while the Th2-inducing cytokine IL-4 activates STAT6. Th cells from Stat knockout mice failed to shed the promiscuous interactions with Ifng and Il4, indicating a direct role for signal-responsive TFs—alongside the TFs they themselves activate, e.g. T-bet or GATA3—in shaping lymphocyte genome topology [85]. Importantly, this work shows that TCR-mediated signaling and costimulation alone (signals ‘1’ and ‘2’, which are intact in STAT-deficient Th cells) are not sufficient to establish Th-subset-specific 3D chromatin architecture. Thus, we postulate that the canonical three signals required for T-cell activation (see above) together ensure a highly specialized TF-mediated rewiring of genome topology tailored to specific microenvironmental cues (Figure 4).
Figure 4.
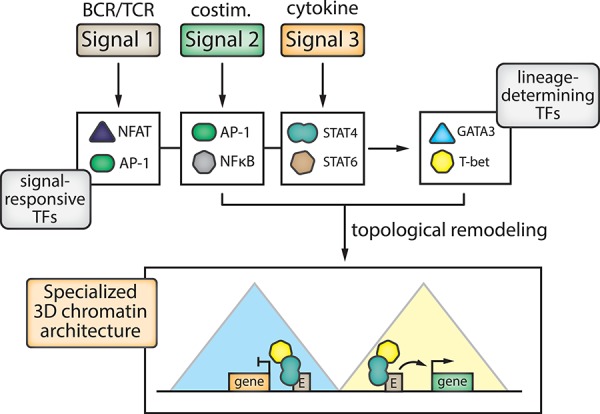
Integration of multiple signals activates a set of transcription factors that together establish the specialized 3D chromatin landscape of effector lymphocytes. Naive B and T lymphocytes require a combination of three signals (1: antigen-receptor ligation, 2: costimulation, 3: cytokines) for their full activation. These three signals will activate signal-responsive TFs (e.g. NFAT, AP-1, STAT), which will in turn activate lineage-determining TFs (e.g. GATA3, T-bet) that together initiate topological genome remodeling in a concerted manner. The resulting specialized 3D chromatin landscape that arises is tailored to the specific microenvironmental input received by the lymphocyte. Of note, complete topological remodeling by TFs appears to require all three signals. Additional abbreviations: BCR; TCR
The findings discussed above strongly suggest that the 3D regulation of gene expression is important during lymphocyte activation, at least at key effector gene loci. More recently, studies have started to investigate genome-wide chromatin conformation dynamics during the activation and effector differentiation of individual lymphocyte subtypes. Promoter capture Hi-C (PCHi-C) of 17 types of primary human blood cell showed that not only the different cell types, but also naive versus activated lymphocytes can be separated based on their specific promoter-interactomes [22]. Javierre et al. showed that only 55% of interactions are shared between resting CD4+ T cells and activated CD4+ T cells after 4 h stimulation with anti-CD3 and anti-CD28, while 22% are specific for resting and 23% for activated CD4+ T cells. Promoter–enhancer interactions positively correlate with increased gene expression [22], which was confirmed in a second PCHi-C study comparing resting and activated human peripheral blood CD4+ T cells [86]. Another study employed H3K27ac Hi-ChIP (to specifically interrogate chromatin interactions between H3K27ac+ active REs) to compare human peripheral blood CD4+ naive T cells, Th17 cells and Tregs. Likewise, interactions involving active regulatory regions are highly subtype-specific and strongly correlate to differential expression of the genes involved [87]. Enhancers involved in subset-specific interactions are enriched for known and unknown TF motifs, most notably RORγ in Th17-specific loops and FOXP3 in Treg-specific loops. Importantly, 62.2–85.8% of subset-specific loops display equal DNA accessibility across the individual T-cell subsets, suggesting that subset-specific differential gene expression programs are not only achieved through differential chromatin accessibility but also via differential TF-mediated chromatin looping [87]. In Jurkat cells, a human leukemic T-cell line carrying a competent TCR, most early responder genes such as CD69 and IL2RA, are already localized within the nuclear interior prior to activation [88]. Upon activation, 4% of the genome (approximately 5000 regions containing over 1300 genes) is either released from the lamina into the nuclear interior or vice versa. A subset of lamina-released genes also shows increased expression, including many known regulators of T-cell responses such as the cytoskeleton regulator guanylate binding protein, the proliferation-inducing cytokine IL2 and immune modulating receptors CD200 and BTLA [88]. This combination of early response genes pre-positioned in the nuclear interior together with activation-induced release from the lamina of later response genes may provide the necessary balance between rapid response capacity and prevention of promiscuous activation, which is key for maintaining immune homeostasis.
Activation of naive B cells triggers their migration to germinal centers within secondary lymphoid organs (i.e. lymph nodes or spleen). Germinal center B cells receive T-cell help, proliferate extensively and undergo full activation and maturation, eventually leaving the germinal center as active antibody-producing plasma cells or memory cells [79]. Similar to T cells, B-cell activation requires a combination of three signals and is accompanied by global changes in transcriptome and chromatin landscape. In line with this notion, murine B-cell activation triggers a marked increase in 3D genome rewiring (Figure 2B) [60, 89, 90]. More specifically, B-cell activation requires the TF MYC and involves a striking shift from long-range to short-range chromatin interactions that depend on ATP availability, resulting in weakened A/B compartmentalization and an increase in CTCF-mediated loops as well as promoter–enhancer interactions [89, 90]. These observations are in line with the global chromatin decompaction and increased nuclear size of activated B cells [89, 90]. This dramatic shift in nuclear organization reduces TF-target search times, providing activated germinal center B cells with a nuclear micro-environment optimal for the reported rapid 10-fold amplification of the B-cell transcriptome upon activation [90]. Illustrative for the multiscale rewiring of genome topology during B-cell activation is the BCL6 locus, which encodes a TF specifically expressed in and essential for activated germinal center B cells (Figure 2A) [91]. Upon activation, the BCL6 promoter rapidly engages in elevated contact frequencies with its enhancers but also the promoters of other rapid response genes. Additionally, the BCL6 promoter initiates interactions with its own 3’-UTR region upon B-cell activation, which is proposed to facilitate reloading of RNAPol2 after transcription has completed [89]. Together, these findings suggest that 3D genome remodeling during B-cell activation is required to boost and efficiently coordinate the transcription of a specialized gene expression program, enabling germinal center B cells to rapidly modify their proliferative and metabolic capacity. Although MYC plays a critical role in this process, it remains to be determined whether the TF plays a direct role in reorganizing B-cell genome topology.
Conclusions and perspectives
Recent advances have demonstrated that cell fate, gene expression and genome topology are intimately connected. Lymphocytes do not seem to be an exception, as modifications to gene expression during their development or activation strongly correlate with specific changes at various levels of 3D genome organization. However, many important questions remain unsolved. Do specific 3D genome conformations play an instructive role in lymphocyte specification or activation? For example, are the changes in nuclear positioning and local topology of the BCL11B locus a cause or consequence of its induced expression at the DN T-cell stage? Addressing this will require the controlled induction or perturbation of chromatin interactions, e.g. using CRISPR-Cas9 based mutagenesis approaches [92]. Another important topic for future studies is to elucidate the functional role of TFs in (re)shaping genome topology at various scales. While numerous studies have pointed toward a pivotal role of both signal-responsive as well as lineage-determining TFs in establishing cell-type specific chromatin interactions, we still lack a mechanistic understanding of this process. Also of interest will be to study 3D genome dynamics during the activation of innate lymphoid cells, which perform similar effector functions as T cells, but respond to different signals and with different kinetics [42].
Finally, we would like to emphasize the potential relevance of genome topology for our understanding of human disease in general. Studying disease-associated DNA variants—which are strongly enriched in distal REs [93]—or larger chromosomal abnormalities in the context of 3D chromatin architecture can pinpoint misregulated genes and identify molecular mechanisms underlying disease [94]. As protocols for studying 3D genome organization are rapidly maturing and can now deliver high-resolution data using relatively small populations of cells [95], we expect analyses of genome topology to become an integral part of studying aberrant transcriptional control in the context of immune disorders, leukemia and cancer immunosurveillance.
Key points
3D genome dynamics are intimately linked to gene expression changes and peak during lineage commitment and early after naive lymphocyte activation.
Signal-dependent (e.g. STATs) and lineage-determining (e.g. PAX5, BCL11B) transcription factors act synergistically to modify chromosome conformation at multiple levels of organization.
Anne van Schoonhoven is a PhD candidate at the Erasmus MC in Rotterdam. Her primary research interest is the molecular basis of T-cell function in health and disease.
Danny Huylebroeck is a professor at the Erasmus MC in Rotterdam and head of the Cell Biology department. His research is focused on TGFbeta/BMP signaling during embryonic development.
Rudi W. Hendriks is a professor at the Erasmus MC in Rotterdam and head of the research laboratory of the Pulmonary Medicine department. His research is focused on lymphocyte development and function.
Ralph Stadhouders is a principal investigator at the Erasmus MC in Rotterdam. His laboratory focuses on understanding the molecular mechanisms that control the activity of lymphocytes in health and various diseases, with a special emphasis on transcriptional control in a 3D chromatin context.
Funding
R.S. is supported by an NWO Veni Fellowship (grant No. 91617114) and an Erasmus MC Fellowship. R.W.H. and R.S. are supported by the Lung Foundation Netherlands (project 4.1.18.226).
References
- 1. Lambert SA, Jolma A, Campitelli LF, et al. The human transcription factors. Cell 2018;172:650–665. [DOI] [PubMed] [Google Scholar]
- 2. Heinz S, Romanoski CE, Benner C, et al. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 2015;16:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why. Mol Cell 2013;49:825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cirillo LA, Lin FR, Cuesta I, et al. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell 2002;9:279–289. [DOI] [PubMed] [Google Scholar]
- 5. Zaret KS, Mango SE. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr Opin Genet Dev 2016;37:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dekker J, Mirny L. The 3D genome as moderator of chromosomal communication. Cell 2016;164:1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tolhuis B, Palstra RJ, Splinter E, et al. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell 2002;10:1453–1465. [DOI] [PubMed] [Google Scholar]
- 8. Carter D, Chakalova L, Osborne CS, et al. Long-range chromatin regulatory interactions in vivo. Nat Genet 2002;32:623–626. [DOI] [PubMed] [Google Scholar]
- 9. Schoenfelder S, Fraser P. Long-range enhancer–promoter contacts in gene expression control. Nat Rev Genet 2019;20:437–455. [DOI] [PubMed] [Google Scholar]
- 10. Denker A, de Laat W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev 2016;30:1357–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simonis M, Klous P, Splinter E, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat Genet 2006;38:1348–1354. [DOI] [PubMed] [Google Scholar]
- 12. Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009;326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nora EP, Lajoie BR, Schulz EG, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012;485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sexton T, Yaffe E, Kenigsberg E, et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012;148:458–472. [DOI] [PubMed] [Google Scholar]
- 16. Gonzalez-Sandoval A, Gasser SM. On TADs and LADs: spatial control over gene expression. Trends Genet 2016;32:485–495. [DOI] [PubMed] [Google Scholar]
- 17. Rowley MJ, Corces VG. Organizational principles of 3D genome architecture. Nat Rev Genet 2018;19:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stadhouders R, Vidal E, Serra F, et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet 2018;50:238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rada-Iglesias A, Grosveld FG, Papantonis A. Forces driving the three-dimensional folding of eukaryotic genomes. Mol Syst ogy 2018;14:e8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deng W, Lee J, Wang H, et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 2012;149:1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stadhouders R, Filion GJ, Graf T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature 2019;569:345–354. [DOI] [PubMed] [Google Scholar]
- 22. Javierre BM, Burren OS, Wilder SP, et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 2016;167:1369–1384.e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krijger PH, Di Stefano B, de Wit E, et al. Cell-of-origin-specific 3D genome structure acquired during somatic cell reprogramming. Cell Stem Cell 2016;18:597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bonev B, Mendelson Cohen N, Szabo Q, et al. Multiscale 3D genome rewiring during mouse neural development. Cell 2017;171:557–572.e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hug CB, Grimaldi AG, Kruse K, et al. Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell 2017;169:216–228.e219. [DOI] [PubMed] [Google Scholar]
- 26. Du Z, Zheng H, Huang B, et al. Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 2017;547:232–235. [DOI] [PubMed] [Google Scholar]
- 27. Lupiáñez DG, Kraft K, Heinrich V, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015;161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Flavahan WA, Drier Y, Liau BB, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016;529:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nora EP, Goloborodko A, Valton A-L, et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 2017;169:930–944.e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haarhuis JHI, van der RH, Blomen VA, et al. The cohesin release factor WAPL restricts chromatin loop extension. Cell 2017;169:693–707.e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwarzer W, Abdennur N, Goloborodko A, et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017;551:51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rao SSP, Huang S-C, Glenn St Hilaire B, et al. Cohesin loss eliminates all loop domains. Cell 2017;171:305–320.e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Franke M, Ibrahim DM, Andrey G, et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016;538:265. [DOI] [PubMed] [Google Scholar]
- 34. Andrey G, Montavon T, Mascrez B, et al. A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 2013;340:1234167. [DOI] [PubMed] [Google Scholar]
- 35. Cuartero S, Weiss FD, Dharmalingam G, et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat Immunol 2018;19:932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Laurenti E, Göttgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018;553:418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cherrier DE, Serafini N, Di Santo JP. Innate lymphoid cell development: a T cell perspective. Immunity 2018;48:1091–1103. [DOI] [PubMed] [Google Scholar]
- 38. Geiger TL, Sun JC. Development and maturation of natural killer cells. Curr Opin Immunol 2016;39:82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schatz DG, Ji Y. Recombination centres and the orchestration of V(D) J recombination. Nat Rev Immunol 2011;11:251. [DOI] [PubMed] [Google Scholar]
- 40. Murugan A, Mora T, Walczak AM, et al. Statistical inference of the generation probability of T-cell receptors from sequence repertoires. Proc Natl Acad Sci 2012;109:16161–16166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kumar BV, Connors T, Farber DL. Human T cell development, localization, and function throughout life. Immunity 2018;48:202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van der Ploeg EK, Carreras Mascaro A, Huylebroeck D, et al. Group 2 innate lymphoid cells in human respiratory disorders. J Innate Immun 2019; doi: 10.1159/000496212. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. DiLillo DJ, Horikawa M, Tedder TF. B-lymphocyte effector functions in health and disease. Immunol Res 2011;49:281–292. [DOI] [PubMed] [Google Scholar]
- 44. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood 2008;112:1570–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Robinette ML, Colonna M. Immune modules shared by innate lymphoid cells and T cells. J Allergy Clin Immunol 2016;138:1243–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun 2018;87:1–15. [DOI] [PubMed] [Google Scholar]
- 47. Nutt SL, Kee BL. The transcriptional regulation of B cell lineage commitment. Immunity 2007;26:715–725. [DOI] [PubMed] [Google Scholar]
- 48. Radtke F, Robson MacDonald H, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol 2013;13:427–437. [DOI] [PubMed] [Google Scholar]
- 49. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol 2018;36:411–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Medina KL, Pongubala JMR, Reddy KL, et al. Assembling a gene regulatory network for specification of the B cell fate. Dev Cell 2004;7:607–617. [DOI] [PubMed] [Google Scholar]
- 51. Zandi S, Mansson R, Tsapogas P, et al. Establishment of a transcription factor EBF1 is essential for B-lineage priming and network in common lymphoid progenitors. J Immunol 2008;181:3364–3372. [DOI] [PubMed] [Google Scholar]
- 52. Cobaleda C, Schebesta A, Delogu A, et al. Pax5: the guardian of B cell identity and function. Nat Immunol 2007;8:463. [DOI] [PubMed] [Google Scholar]
- 53. Tsapogas P, Zandi S, Åhsberg J, et al. IL-7 mediates Ebf-1–dependent lineage restriction in early lymphoid progenitors. Blood 2011;118:1283–1290. [DOI] [PubMed] [Google Scholar]
- 54. Brown KE, Guest SS, Smale ST, et al. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell 1997;91:845–854. [DOI] [PubMed] [Google Scholar]
- 55. Lin YC, Benner C, Mansson R, et al. Global changes in nuclear positioning of genes and intra- and inter-domain genomic interactions that orchestrate B cell fate. Nat Immunol 2012;13:1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. de Wit E, Bouwman BAM, Zhu Y, et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature 2013;501:227–231. [DOI] [PubMed] [Google Scholar]
- 57. Lin YC, Jhunjhunwala S, Benner C, et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates the B cell fate. Nat Immunol 2010;11:635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Treiber T, Mandel EM, Pott S, et al. Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription- independent poising of chromatin. Immunity 2010;32:714–725. [DOI] [PubMed] [Google Scholar]
- 59. Nutt SL, Heavey B, Rolink AG, et al. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 1999;401:556–562. [DOI] [PubMed] [Google Scholar]
- 60. Johanson TM, Lun ATL, Coughlan HD, et al. Transcription-factor-mediated supervision of global genome architecture maintains B cell identity. Nat Immunol 2018;19:1257–1264. [DOI] [PubMed] [Google Scholar]
- 61. Hosokawa H, Rothenberg EV. Cytokines, transcription factors, and the initiation of T-cell development. Cold Spring Harb Perspect Biol 2018;10:a028621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol 2014;14:529–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hu G, Cui K, Fang D, et al. Transformation of accessible chromatin and 3D nucleome underlies lineage commitment of early T cells. Immunity 2018;48:227–242.e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kueh HY, Yui MA, Ng KKH, et al. Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat Immunol 2016;17:956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Isoda T, Moore AJ, He Z, et al. Non-coding transcription instructs cohesin-dependent chromatin folding and compartmentalization to dictate enhancer-promoter communication and T cell fate. Cell 2017;171:103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bassing CH, Swat W, Alt F. The mechanism and regulation of chromosomal V(D)J recombination. Cell 2002;109:S45–S55. [DOI] [PubMed] [Google Scholar]
- 67. Ribeiro de Almeida C, Hendriks RW, Stadhouders R. Dynamic control of long-range genomic interactions at the immunoglobulin kappa light-chain locus. Adv Immunol 2015;128:183–271. [DOI] [PubMed] [Google Scholar]
- 68. Jhunjhunwala S, van Zelm MC, Peak MM, et al. Chromatin architecture and the generation of antigen receptor diversity. Cell 2009;138:435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Johanson TM, Chan WF, Keenan CR, et al. Genome organization in immune cells: unique challenges. Nat Rev Immunol 2019;19:448–456. [DOI] [PubMed] [Google Scholar]
- 70. Montefiori L, Wuerffel R, Roqueiro D, et al. Extremely long-range chromatin loops link topological domains to facilitate a diverse antibody repertoire. Cell Rep 2016;14:896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Barajas-Mora EM, Kleiman E, Xu J, et al. A B-cell-specific enhancer orchestrates nuclear architecture to generate a diverse antigen receptor repertoire. Mol Cell 2019;73:48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kosak ST, Skok JA, Medina KL, et al. Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science 2002;296:158–162. [DOI] [PubMed] [Google Scholar]
- 73. Stadhouders R, de Bruijn MJW, Rother MB, et al. Pre-B cell receptor signaling induces immunoglobulin k locus accessibility by functional redistribution of enhancer-mediated chromatin interactions. PLoS Biol 2014;12:e1001791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Medvedovic J, Ebert A, Tagoh H, et al. Flexible long-range loops in the VH gene region of the Igh locus facilitate the generation of a diverse antibody repertoire. Immunity 2013;39:229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Degner SC, Verma-Gaur J, Wong TP, et al. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc Natl Acad Sci 2011;108:9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ribeiro de Almeida C, Stadhouders R, de Bruijn MJ, et al. The DNA-binding protein CTCF limits proximal Vκ recombination and restricts κ enhancer interactions to the immunoglobulin κ light chain locus. Immunity 2011;35:501–513. [DOI] [PubMed] [Google Scholar]
- 77. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med 2015;212:1345–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Naito T, Tanaka H, Naoe Y, et al. Transcriptional control of T-cell development. Int Immunol 2011;23:661–668. [DOI] [PubMed] [Google Scholar]
- 79. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol 2015;15:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol 2010;28:445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Spilianakis CG, Flavell RA. Long-range intrachromosomal interactions in the T helper type 2 cytokine locus. Nat Immunol 2004;5:1017–1027. [DOI] [PubMed] [Google Scholar]
- 82. Sekimata M, Pérez-Melgosa M, Miller SA, et al. CCCTC-binding factor and the transcription factor T-bet orchestrate T helper 1 cell-specific structure and function at the interferon-gamma locus. Immunity 2009;31:551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Grogan JL, Mohrs M, Harmon B, et al. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity 2001;14:205–215. [DOI] [PubMed] [Google Scholar]
- 84. Hewitt SL, High FA, Reiner SL, et al. Nuclear repositioning marks the selective exclusion of lineage-inappropriate transcription factor loci during T helper cell differentiation. Eur J Immunol 2004;34:3604–3613. [DOI] [PubMed] [Google Scholar]
- 85. Hakim O, Sung M-H, Nakayamada S, et al. Spatial congregation of STAT binding directs selective nuclear architecture during T-cell functional differentiation. Genome Res 2013;23:462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Burren OS, Rubio García A, Javierre B-M, et al. Chromosome contacts in activated T cells identify autoimmune disease candidate genes. Genome Biol 2017;18:165–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mumbach MR, Satpathy AT, Boyle EA, et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet 2017;49:1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Robson MI, de las Heras JI, Czapiewski R, et al. Constrained release of lamina-associated enhancers and genes from the nuclear envelope during T-cell activation facilitates their association in chromosome compartments. Genome Res 2017;27:1126–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bunting KL, Soong TD, Singh R, et al. Multi-tiered reorganization of the genome during B cell affinity maturation anchored by a germinal center-specific locus control region. Immunity 2016;45:497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kieffer-Kwon K-R, Nimura K, Rao SSP, et al. Myc regulates chromatin Decompaction and nuclear architecture during B cell activation. Mol Cell 2017;67:566–578.e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Basso K, Dalla-Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol 2010;105:193–210. [DOI] [PubMed] [Google Scholar]
- 92. Morgan SL, Mariano NC, Bermudez A, et al. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat Commun 2017;8:15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012;337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Krijger PHL, de Laat W. Regulation of disease-associated gene expression in the 3D genome. Nat Rev Mol Cell Biol 2016;17:771–782. [DOI] [PubMed] [Google Scholar]
- 95. Díaz N, Kruse K, Erdmann T, et al. Chromatin conformation analysis of primary patient tissue using a low input Hi-C method. Nat Commun 2018;9:4938. [DOI] [PMC free article] [PubMed] [Google Scholar]