

Abstract
The pyrazolone structural motif is a critical element of drugs aimed at different biological end-points. Medicinal chemistry researches have synthesized drug-like pyrazolone candidates with several medicinal features including antimicrobial, antitumor, CNS (central nervous system) effect, anti-inflammatory activities and so on. Meanwhile, SAR (Structure-Activity Relationship) investigations have drawn attentions among medicinal chemists, along with a plenty of analogues have been derived for multiple targets. In this review, we comprehensively summarize the biological activity and SAR for pyrazolone analogues, wishing to give an overall retrospect and prospect on the pyrazolone derivatives.
Keywords: Pyrazolone, Antimicrobial, Antitumor, CNS agents, Anti-inflammatory, Derivatives
Graphical abstract
1. Introduction
Pyrazolones delegate a cluster of compounds with the nucleus of 1H-pyrazol-3-ol and pyrazolin-5-one (Fig. 1 ) which have been investigated because of their multiple features and applications. Since 1883, the synthesis of antipyrine (1) by Knorr, numerous attention has caused by the analgesic and antipyretic activities of pyrazolone analogues [1]. The discovery of these features encouraged the researches to synthesize other pyrazolone derivatives with similar behavior but with a better therapeutic action. Nowadays, multiple FDA approved drugs containing pyrazolone nucleus have been explored (Fig. 2 ), for instances, edaravone (2) has been used as free radical scavenger for the treatment of amyotrophic lateral sclerosis (ALS) [2], aminophenazone (3) with antipyretic and anti-inflammatory activities has been used in breath tests to measure the cytochrome P-450 metabolic activity in liver function evaluations [3], eltrombopag (4) has been used for the treatment of low blood platelet counts in adults with idiopathic chronic immune thrombocytopenia [4], dichloralphenazone (5) has been used for the relief of tension and vascular headaches [5], metamizole (6) has been used for perioperative pain, cancer pain, acute injury and other forms of pain and is considered as the strongest antipyretic [6]. Several investigational small molecules containing pyrazolone have been regarded as drug candidates including sulfamazone (7) [7], propyphenazone (8) [8] and nifenazone (9) [9].
Fig. 1.
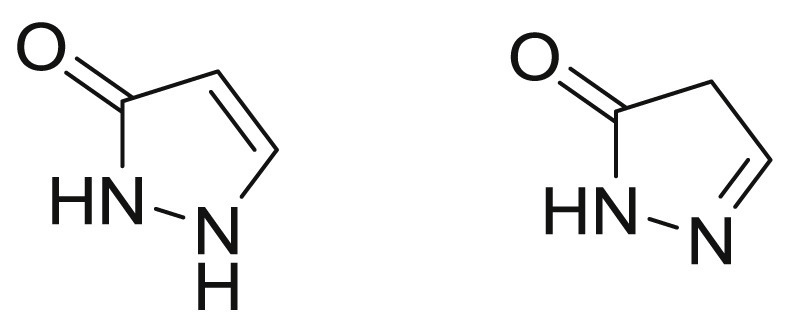
Structure of 1H-pyrazol-3-ol and pyrazolin-5-one.
Fig. 2.

FDA approved drugs containing pyrazolone nucleus and investigational pyrazolone derivatives.
Regarding pyrazolone derivatives, it is important to underline the enormous diversity of classes of synthetic pyrazolone published in existing studies. As for the pharmacology investigators, this review is a concise and critical account focusing on the properties of pyrazolones as antimicrobial, antitumor, CNS effect, anti-inflammatory, antioxidant, anti-tubercular, antiviral, lipid-lowering, antihyperglycemic agents and protein inhibitors. We summarized advances of pyrazolone derivatives as well as their SAR (10–189). Currently, the existing reviews [10,11] about pyrazolone derivatives are mainly concerning their catalytic asymmetric synthesis or coordination, the comprehensive review of the bioactivity and the SAR studies are still vacant. This review is in order to fill gaps of systematic summarization of biological activities and SAR that could be beneficial for researchers to design novel pyrazolone derivatives, in addition, docking analysis is used to explain interactions between some typical pyrazolone derivatives and potential targets to give a visual appreciation of the roles of pyrazolone group play in the bioactivity.
2. Synthetic strategies of pyrazolones
Recent advances in the synthesis of pyrazolones have been reported in the literature [12]. Condensation of hydrazines with β-ketoester compounds is the classical method for the synthesis of pyrazolones (Scheme 1 ), the catalytic conditions are usually the use of organic base like piperidine or inorganic base like NaH in the system of the boiling ethanol or methanol solvent. The nucleus of pyrazolones has been widely used to ulteriorly synthesize broad range of derivatives including spiroheterocycles and acylation productions [13], which features multiple reactive sites. The electrophilic substitution at the C-4 position of pyrazolones is an effective synthetic route for the construction of pyrazolones linked with chiral groups and 4-disubstituted pyrazolones. In addition, as an important synthon, α, β-unsaturated pyrazolones (R1: C C-R`/C N-R`/C O) can be transformed into diverse pyrazolones. When R1 is C C-R′, they can undergo 1,4-Michael addition as acceptors to generate 4-substituted pyrazole analogues.
Scheme 1.

Synthesis of pyrazolone nucleus.
3. Pharmacological profile of pyrazolones
The broad-spectrum of pharmacology properties about pyrazolone derivatives have been reported including antimicrobial, antitumor, CNS activity, anti-inflammatory, antioxidant, anti-tubercular, antiviral, lipid-lowering, antihyperglycemic and protein inhibitory activities.
3.1. Antimicrobial agents
Pyrazolone derivatives have been reported to show the significant antimicrobial activities against multiple types of bacteria and fungi. Generally, the antibacterial effect of pyrazolones is better than its antifungal effect, and the inhibitory activity on Gram positive (Gram +ve) and Gram negative (Gram −ve) strains was distinct as well.
The bacterial cell membrane is generated with a dense wall comprising several peptidoglycan and teichoic acid layers [[14], [15], [16], [17]]. In Gram +ve bacteria, the adsorption of the biocidal molecules is occurred on the lipoteichoic acid layer which is characterized by the charged nature and the ability to interact with the heteroatoms in the biocide molecules. While in the Gram −ve bacteria, the lipid layer is the target of the biocide molecules. The drug resistance of traditional antibiotics has aroused people’s anxiety about superbacteria, and the attentions giving pyrazolones will pay a light in this field.
3.1.1. Pyrazolones as antimicrobial agents and their SAR
Nasser S. A. M. Khalil [18] described Schiff base compound 10 and its acetyl glucoside derivative 11 (Fig. 3 ) which exhibited the most potent inhibitory effect against the strains of Penicillium italicum, Syncephalastrum racemosum, Aspergillus fumigatus, Bacillus subtilis, Staphylococcus aureus and Escherichia coli at the dose of 400 μg/mL. SAR could be deduced that the analogues with non-substituted on ring A exhibited the strongest activity and the acetyl glucoside group was more helpful to promote the inhibitory effect than N-glucoside. It was notable that the possible intramolecular H-bonding formed by the carbonyl group and the NH moiety improved the inhibition and the triad of permeability, solubility, and potency of the analogues were changed accordingly [19].
Fig. 3.

Pyrazolone derivatives with antimicrobial activity.
In 2007, Padmavathi and colleagues [20] synthesized multiple amino-pyrazolone, amino-isoxazolone and amino-pyrimidinone analogues, and evaluated their bioactivities. Thereinto, the amino-pyrazolone derivatives 12 and 13 (Fig. 3) showed the most pronounced antimicrobial activity (Table 1 ). SAR study elucidated that analogues with amino-hydroxypyrimidinone and N, N-dimethyliminopyrimidinedione showed weak inhibitory effect when compared with analogues modified with amino-pyrazolone and amino-isoxazolone groups. Additionally, benzyl sulfonyl analogues exhibited strongest inhibitory effect among the productions.
Table 1.
Antimicrobial activity as MICS (mg/mL) of pyrazolone derivatives against tested microorganisms.
| Com. | MIC (μg/mL) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Antibacterial |
Antifungal |
|||||||||
| Gram Positive |
Gram negative |
|||||||||
| S. aureus | B. subtilis | S. pyogenes | P. aeruginosa | E. coli | A. fumigatus | G. candidum | C. albicans | S. racemosum | A. niger | |
| 12 | 100 | 100 | – | – | – | – | – | – | – | 100 |
| 13 | 25 | 25 | – | – | – | – | – | – | – | 50 |
| 14 | 19 | 9.5 | – | 39 | 19 | 19 | 19 | 19 | 39 | – |
| 15 | 39 | 19 | – | 39 | 19 | 19 | 39 | 78 | 39 | – |
| 16 | 78 | 39 | – | 78 | 156 | 78 | 78 | 156 | 625 | – |
| 17 | 39 | 19 | – | 39 | 78 | 39 | 78 | 156 | 313 | – |
| 21 | 100 | 100 | 100 | 200 | 62.5 | – | – | 1000 | – | >1000 |
| 22 | 125 | 125 | 62.5 | 250 | 200 | – | – | >1000 | – | >1000 |
| 23 | 62.5 | 62.5 | 200 | 500 | 500 | – | – | 250 | – | >1000 |
| 24 | 250 | 250 | 200 | 62.5 | 125 | – | – | 1000 | – | 500 |
| 25 | 8.02 | – | – | 9.58 | 15.28 | – | – | 9.22 | – | 5.30 |
| 26 | 12.04 | – | – | 11.92 | 26.72 | – | – | 9.08 | – | 6.28 |
| 27 | 7.30 | – | – | 7.39 | 12.68 | – | – | 6.18 | – | 6.80 |
| 28 | 7.96 | – | – | 10.24 | 13.27 | – | – | 8.58 | – | 7.52 |
| 29 | – | 10.0 | – | – | 4.08 | – | – | – | – | 8.16 |
| 30 | – | – | – | – | – | – | – | – | – | 5.1 |
| 31 | – | ≤12.5 | ≤12.5 | ≤12.5 | >200 | – | >200 | – | – | – |
| 41 | – | – | 50 | 50 | 50 | 50 | – | 25 | – | – |
| 42 | – | – | 50 | 50 | 100 | 50 | – | 50 | – | – |
| 43 | – | – | 50 | 50 | 50 | 25 | – | 25 | – | – |
| 44 | – | – | – | – | 125 | – | – | 7.8 | – | 2.9 |
| 45 | – | – | – | – | 187.5 | – | – | 15.6 | – | 5.8 |
| 46 | – | – | – | – | 125 | – | – | 11.6 | – | 5.8 |
| 47 | – | 100 | – | – | – | – | – | – | – | – |
| 48 | – | 25 | – | – | – | – | – | – | – | – |
| 49 | – | 25 | – | – | – | – | – | – | – | – |
| 50 | 100 | 25 | – | – | – | – | – | – | – | – |
Note: not determined.
Aly and co-workers [21] synthesized antipyrine derivatives 14, 15, 16 and 17 (Fig. 3) which were regarded as the prospective inhibitor (Table 1). SAR investigation expatiated that the presence of pyrazole and the introducing of effective moieties including –COCH3, –CH3, -Cl and thiophene group on the scaffold of 4-aminoantipyrine showed significant inhibitory effect.
Hamama and teammates [22] described the antibacterial activity of several pyrazolone analogues, among which compound 18 (Fig. 3) was the most hopeful candidate against E. coli and B. subtilis with the inhibition zoom diameter values of 40 and 36 mm respectively at the concentration of 100 ppm, which was approximately twofold to the reference drug ampicillin. The initiatory SAR can be considered as the introduction of piperidine group which was helpful to improve the antibacterial effect.
Active antimicrobial compounds 19 and 20 produced by Rasapalli et al. [23] (Fig. 3) exhibited noticeable antibiofilm activity towards S. aureus strains (Table 2 ). Compound 19 suppressed biofilm formation of Staphylococcus epidermidis as well. SAR study suggested that the antibacterial scaffold can be acquired by condensation of the carbonyl compounds with the active methylene group on pyrazolone, and the 4-Cl and 2-Br substituted groups on A ring were proved to be the potential active modification for further development.
Table 2.
Antibacterial effects of compounds 19 and 20.
| Com. | Percent inhibition of overall growth or biofilm formation by 50 μM of compounds |
|||||||
|---|---|---|---|---|---|---|---|---|
|
Staphylococcus epidermidis |
S. aureus |
P. aeruginosa |
||||||
| GTC 18972 |
ATCC 35556 |
MSSA |
MRSA 1094 |
|||||
| Growth | Biofilm | Growth | Biofilm | Growth | Biofilm | Growth | Biofilm | |
| 19 | 32.348 | 20.421 | 88.071 | 88.071 | 21.951 | 86.697 | 38.003 | 21.468 |
| 20 | 54.976 | 82.624 | −6.961 | 65.846 | 8.690 | 46.471 | 53.468 | 87.936 |
Gadhave and colleagues [24] synthesized multi-fluorinated pyrazole-pyrazolone and chromone-pyrazolone compounds 21, 22, 23 and 24 (Fig. 3) which exerted favorable antibacterial effects (Table 1), whereas all the compounds exhibited poor antifungal activity profile. Regarding SAR, it can be speculated as that multi-fluorinated substitution was contributive to antibacterial while further modification was needed to improve the antifungal effect.
Narayana Rao et al. [25] designed and synthesized several pyrazolone analogues 25, 26, 27 and 28 (Fig. 3) with favorable activity (Table 1). As for SAR, the authors disclosed that the amino group on the A ring of pyrazolone markedly elevated the inhibitory effect compared with methyl, and the antimicrobial sequence of R group on the p-position of phenylhydrazine can be generally summarized as: Cl > Br > OMe > OC2H5.
In 2017, Abdel Reheim and Baker [26] synthesized multiple pyrazolone derivatives and screened the antimicrobial activity, among which compound 29 exhibited strongest antibacterial and antifungal activities, and compound 30 (Fig. 3) showed significant antifungal effect (Table 1). The results of antimicrobial test can be concluded as that the introduction of active substitutions such as pyrano, pyridazine and pyrimidine can promote the antimicrobial activity of the pyrazolone nucleus to different degrees.
Alkhaldi et al. [27] synthesized pyrazolone derivative 31 (Fig. 3) with multiple antimicrobial effects (Table 1). Concerning SAR, meta dichloro phenyl group on the benzene ring of the Schiff base was helpful to improve the antibacterial activity, and the subsequent molecular docking of the interactions of 31 within potential target glucosamine-6-phosphate synthase (Fig. 4 ) provided evidence for the antimicrobial effect of 31, the pyrazolone and the 2,4-dichloro-phenyl moieties made the different binding models between 31 and glucosamine 6-phosphate, but the inhibitory potency is significant as well.
Fig. 4.

Ligand-protein interactions of compound 31 (A) and original ligand (B) with the active site of glucosamine-6-phosphate synthase (PDB ID: 2VF5) accomplished by Discovery Studio 2019.
Bhattacharjee and colleagues [28] presented pyrazolone compounds 32, 33, 34 and 35 (Fig. 3) which exhibited favorable inhibitory effect against Bacillus cereus with the MIC values of 0.78, 0.78, 0.78 and 0.39 mg/mL, respectively, and suppressed the growth of Klebsiella pneumoniae with the MIC values of 0.78, 0.39, 0.39 and 0.78 mg/mL, respectively. The preliminary SAR extracted from the results was that pyrazolone group was necessary to the antibacterial activity and the 3,4-dimethoxyl group on A ring was the potential effective substituent group.
Another research reported that pyrazolone derivatives 36, 37 and 38 (Fig. 3) were potent compounds with anti-bacterial activity and anti-fungal activity exerted to different degree (Table 3 ) [29]. In regard to SAR, it was obvious that only compounds 36, 37 and 38 exhibited anti-fungal activity among the analogues, and in the anti-bacterial evaluation, 36, 37 and 38 were efficient inhibitor of S. mutans, S. typhi and B. subtilis, respectively. The 4-Br, 3-F-4-Br and 2,4-dinitro groups on the A ring were the potential active groups for further investigation.
Table 3.
Antimicrobial zone of pyrazolone derivatives at the dosage of 100 ppm.
| Com. | Antimicrobial zone (mm) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Anti-bacterial activity |
Anti-fungal activity |
|||||||||
| B. subtilis | S. mutans | M. luteus | P. aeruginosa | S. typhi | S. paratyphi | E. coli | C. albicans | T. viride | A. niger | |
| 36 | 18.5 | 29.5 | 18.5 | 14.5 | 19.5 | 19.5 | – | 15.5 | 13.5 | – |
| 37 | 19.5 | 18 | 16.5 | 14.5 | 20.5 | 19.5 | – | 18.5 | 10.5 | – |
| 38 | 22.5 | 14.5 | 21.5 | 13.5 | 20 | 20 | – | 28.5 | 21.5 | – |
| 39 | 18.1 | – | – | 17.4 | – | – | 15.3 | – | – | 15.2 |
| 40 | 25.3 | – | – | 19.3 | – | – | 23.1 | – | – | 25.5 |
Sayed et al. [30] synthesized active pyrazolone/pyrazole derivatives 39 and 40 (Fig. 3) with antimicrobial activity explored in Table 3. With regard to SAR, compared with the inactive pyrazolone derivative which was non-substituted on the benzene ring, compound 39 exhibited moderate activity in the inhibition zone test. The pyrazole derivative 40 displayed the strongest inhibitory effect, indicating the potential modification method in the further investigation. The author held the point that the antimicrobial effect of the analogues seemed like their trend in the discrepancy in the polarity and the interaction of analogues by cellular membrane.
Inspired from the structure of cholesterol, compounds 41, 42 and 43 (Fig. 3) were synthesized and investigated for their antimicrobial activities [31]. As shown in Table 1, compounds 41 and 43 exhibited better activity than chloro-derivative 42, and the stronger antifungal activity were observed than antibacterial activity on steroidal pyrazolones 41, 42 and 43. Docking study suggested that compound 43 exhibited strongest interactions with S4 active subsite of CYP 51 of C. albicans compared with compounds 41 and 42, in which the oxygen of pyrazolone ring can effectively interact with the H-bond donor and acceptor region.
Ibrahim Ali M. Radini synthesized pyrazolone derivatives 44, 45 and 46 (Fig. 3) as antimicrobial agents (Table 1) [32]. SAR indicated that derivatives with pyrazole-1-carbothiohydrazide moiety exhibited higher inhibitory effect than derivatives containing pyrazolyl thiadiazine moiety. Additionally, the presence of free carbothiohydrazide unit enhanced the activity of compounds 44, 45 and 46 and the presence of electron-donating groups (EDGs) at the aromatic ring promoted the inhibitory effect of compound 44.
Bihani and co-workers [33] synthesized zwitterionic pyrazolone analogues 47, 48, 49 and 50 (Fig. 3) with the antimicrobial properties listed in Table 1, the moderate inhibitory effects against Pseudomonas syringae and Proteus vulgaris were also reported. SAR can be inferred that electrophilic substituted groups on the C-3 or C-4 side of benzene ring could strengthen the activity.
Oraby and teammates [34] synthesized 2,4-disubstituted phenylhydrazonopyrazolone and isoxazolone analogues as antibacterial agents. The results of antibacterial test suggested that the compounds contain the scaffold of pyrazolone like 51 and 52 (Fig. 3) exhibited weak antibacterial effect against the multiple strains. Interestingly, the inhibitory activity was promoted simultaneously when the pyrazolone moiety was replaced to isoxazolone. Furthermore, docking study indicated that cation-π interactions between isoxazolone analogues and Arg 225, which was a residue played a crucial role in the stabilization of the cofactor during the catalysis in flavin adenine dinucleotide, increased the antibacterial effect of isoxazolones. The antibacterial effect of the analogues was influenced by the substitution on C-2 of the phenyl ring, the substitutions with electron withdrawing groups (EWGs) including F and Cl atoms selectivity increased the antibacterial effect against S. aureus compared to bulky moieties such like methyl.
3.1.2. Coordination pyrazolone compounds as antimicrobial agents
Microorganisms acquire metals from the environment and use them for many essential cellular processes [35]. Metals are able to affect bacterial growth, vitality, and survival [36], and effectively removing metals using metal chelators makes bacterial cells more susceptible to a variety of antibacterial agents, causing cell lysis and loss of viability. Chelators, including EDTA (ethylenediaminetetraacetic acid) and metal complexes of chalcone and flavonoids [37,38], have attracting attentions in this field. Recently, many research groups made unremitting efforts to the synthesis and characterization of transition metal chelates of pyrazolones. Pyrazolones were likely to form several types of coordination compounds due to the several electron-rich donor centers [39]. Coordination compounds containing pyrazolone-based ligands are reported to be superior reagents in antimicrobial agents. In 2011, a research about Mn(III) mixed-ligand complexes with bis-pyrazolones and ciprofloxacin drug was published [40], the antimicrobial outcome showed that when added into the strains of microorganisms (E. coli, Serratia marcescens, S. aureus and B. subtilis), the mixed-ligand complexes which contain the moiety of bis-pyrazolones and ciprofloxacin exhibited stronger inhibitory effect compared with bis-pyrazolones, in which compounds 53 and 54 (Fig. 5 ) were the potential active antimicrobial agents. SAR can be suspected as that the chelation decreased the polarity of the central atom due to partial sharing of its positive charge with the donor moieties and possible electron delocalization over the whole chelation ring. The complex molecule promoted the hydrophobic character of the metal chelate and the liposolubility, hence favoring its permeation through the lipoid layer of the cell membrane of microorganism.
Fig. 5.

Coordination pyrazolone compounds as antimicrobial agents.
Jadeja and co-workers [41] synthesized multiple pyrazolone-based complexes and evaluated the bioactivity. The consequence indicated that compound 55 (Fig. 5) showed stronger inhibitory effect against B. subtilis than other derivatives, at the dose of 3 mM, compound 55 suppressed the growth of B. subtilis strain with the diameter of inhibition zone of 25 mm.
Schiff bases derivatives of pyrazolone intrigue attention of coordination chemists as versatile spacers due to their viable accessibilities and structural diversity. Joseph et al. [42] prepared two kinds of copper complexes derived from 4-formylpyrazolone (56 and 57, Fig. 5) and tested the antimicrobial activity against multiple strains. The result showed that both compounds 56 and 57 suppressed the growth of stains and the effect were quite comparable to the standard drugs including Levofloxacin and Moxifloxacin. 2,4-dimethyl derivative 56 was more effective than 3,4-difluorol derivative 57.
Lunagariya et al. [43] synthesized several organometallic platinum (II) analogues containing pyrazolone and determined their bioactivities. Among the analogues, compound 58 (Fig. 5) displayed the strongest antimicrobial activities against P. aeruginosa, B. subtilis, S. Aureus, S. marcescens and E. coli with the MIC values of 35 μM, 30 μM, 25 μM, 22 μM, and 32 μM, respectively. SAR research can be recapitulated that inhibitory effect was influenced by introducing different substitutions on A ring. The presence of methoxyl group at 4-position on of A ring exhibited significant antibacterial activity against Gram +ve bacteria i.e. B. substiles and S. aureus. Moreover, the nitro and bromo groups at 4-position on A ring increased antibacterial activity against B. substiles and S. marcescens. The presence of EWGs on A ring of complexes exhibited better antibacterial activity against Gram +ve and Gram − ve micro-organism than other EDGs in complexes.
Nair and co-workers [44] prepared some metal complexes with derived from pyrazolone and evaluated their antimicrobial potent. The synthetic analogues exhibited antimicrobial effects to different degrees, among which compound 59 (Fig. 5) was the most promising one with the highest zones of inhibition i.e. 10.5, 14.3 and 11.2 mm measured in P. aeruginosa, S. aureus and E. coli. SAR research suggested that the metal chelates were more toxic than the parent ligands against the same microorganism and under identical experimental conditions. The biological activity of complexes followed the order of the change of metal ion: Ni (II) > Co (II) > Zn (II) > Cu (II) > Schiff base ligand.
3.2. Antitumor agents
In the 21st century, malignant tumor is one of the leading causes of death and developing new antitumor agents with less drug resistance and side effects needs to be done as soon as possible [45]. In recent years, pyrazolone derivatives and their metal complexes have attracted the great attention of researchers for their potential antitumor activity and their less drug resistance and side effects [46]. So far, diverse pyrazolone analogues with antitumor potency have been reported.
3.2.1. Pyrazolones as antitumor agents and their SAR
3.2.1.1. VEGFR-2 target inhibitors
Vascular Endothelial Growth Factor Receptor 2 (VEGFR-2), also known as Flk-1, is mainly expressed in vascular endothelial cells and hematopoietic stem cells [47]. It is highly expressed in a variety of malignant tumors and plays a significant regulatory role in the metastasis and angiogenesis of tumor. Taking VEGFR-2 as the target, the search for VEGFR-2 inhibitors provides a good way to find some new anticancer drugs. Based on conserved active binding sites of VEGFR-2 and similar interaction conformation with ligands, several novel inhibitors of VEGFR-2 have been developed [48], in which pyrazolones play important roles as well.
In 2006, Tripathy and co-workers [49] synthesized a train of heterocyclic-substituted pyrazolones and tested inhibitory potency against VEGFR-2 kinase, among which compound 60 (Fig. 6 ) was proved to be the most potential derivative inhibiting VEGFR2 kinase with the IC50 value of 6 nM. SAR for the analogues can be summarized that replacement on the indole ring exhibited further potency increase against VEGFR-2 especially at the position of C-4 on the ring.
Fig. 6.

Pyrazolone derivatives with antitumor activity.
Inspired from existing VEGFR-2 inhibitors, the same research group [50] modified the structure 61 by introducing a 4-benzyloxy substituent to generate compound 62. Further investigation on structural modifications of the aromatic moiety of 62 revealed that compound 63 (Fig. 6) was the most promising anaplastic lymphoma kinase (ALK) inhibitor (IC50: 5 nM). As for SAR, it was demonstrated that a plenty of halogen substituent groups on the aromatic ring were favored for ALK inhibitory potency. Moreover, the authors paid attention to the modification of the heterocyclic moiety and SAR analysis was concluded that compound bearing thiazole increased inhibition of ALK.
Gu et al. [51] prepared a series of novel pyridine analogues bearing pyrazolone skeleton and evaluated the cell proliferation activities of synthetic analogues. Among all compounds, 64, 65 and 66 (Fig. 6) exhibited favorable inhibitory potential on c-Met and VEGFR-2. The most promising analogue 66 significantly inhibited the targets c-Met (IC50: 0.11 μM) and VEGFR-2 (IC50: 0.19 μM) kinases in vitro. SAR analyses suggested that substitution of pyridine amine moiety with longer polar side chains or with hydrophilic group could improve inhibitory activity.
3.2.1.2. c-Met target inhibitors
Receptor tyrosine kinase c-mesenchymal epithelial transition factor (c-Met), a vital target in antitumor therapy, plays a crucial role in the occurrence, development, metastasis and angiogenesis of tumors. In 2012, according to the theory of Structure-Based Drug Design, Norman and co-workers [52] designed and synthesized a series of class II c-Met inhibitors, among which derivative 67 (Fig. 6) was confirmed to possess excellent potency against c-Met (Ki: 1.0 nM). This privilege compound was demonstrated to show binding affinity not only with c-Met (PDB ID: 3U6H) (Fig. 7 A), but also with VEGFR-2 kinase (PDB ID: 3U6J) (Fig. 7B) with the help of X-ray analysis of cocrystals. Obviously, the pyrazolone moiety plays crucial roles in occupying active sites of binding pocket, forming the pi-alkyl with Val 1155 in c-Met and H-bond with Asp 1046 in VEGFR-2 kinase, suggesting the broad prospect of pyrazolones developing as antitumor agents in the future. This study provides a new strategy for designing more selective analogues. Therefore, Liu et al. [53] synthesized a cluster of pyrazolone-based analogues according to compound 67 and screened the most selective class II c-Met inhibitor. Among them, compound 68 (Fig. 6) was the representative active antitumor agent. SAR investigation revealed that the 6-methoxy group of quinoline in the region (A), the pyridine in the central ring (B) and 2-hydroxypropyl side chain at N-1 could promote the selectivity and activity. In addition, the polar functionality at C-5 position of the pyrazolone was important for selectivity.
Fig. 7.

Resolved pharmaceutical cocrystal of pyrazolone derivative 67 with c-Met (A, PDB ID: 3U6H) and VEGFR-2 kinase (B, PDB ID: 3U6J).
A potential compound 69 (Fig. 6) was designed and synthesized by Zhou and colleagues [54], inspired from the structure of abozantinib and foretinib as c-Met inhibitor (IC50: 2.20 nM) to show antiproliferative effect against multiple cancer cell lines, respectively. SAR investigation could be concluded that substitution of the cyclopropane-1,1-dicarboxamide framework of foretinib with the N-aryl-pyrazolone-4-imino and 2,4-imidazolinedione group sustained the potent antitumor activity. Additionally, analogues with mono/double-EDGs on the ring B were more active, especially –CF3 at the C-2 position of ring B were more effective than –CH3 or –OCH3 groups.
3.2.1.3. Other antitumor pyrazolone derivatives
Human SIRTs (Sirtuins) are relative to cell senescence, apoptosis, metabolism, proliferation and differentiation. In the past decade, they have emerged as targets for cancer chemotherapy. SIRT1, SIRT2 and SIRT3 are relatively important in the SIRTs family with extensive research. Mahajan and teammates [55] prepared a multiple of five-membered ring pyrazolone analogues as Sirtuin inhibitors. Among these products, compounds 70 and 71 (Fig. 6) showed the inhibitory activities against SIRT1 with IC50 values of 41 μM and 27 μM, respectively. In addition, compound 70 displayed the most promising inhibitory effect against SIRT3 (IC50: 6 μM). According to the pharmacophore investigations, SAR can be sum up as the presence of pyrazolone ring was important to improve the inhibitory effect on SIRT1 and SIRT3 through maintaining the balance of H-bond donor and acceptor moieties, from the active analogue 71, the introduction of bulky hydrophobic benzene group at the C-6 position of naphthalene was helpful to increase the activity on SIRT3. Moreover, the selective inhibition on SIRT2 was significantly elevated when the H-bond donor NH group on the position 1 was replaced as H-bond acceptor O.
It’s reported that pyrazolone was potential to be the starting point for generating RalA inhibitors, which were often activated in human cancer cell lines. Derivative 72 (Fig. 6), as the most promising anticancer candidate, was designed and synthesized by Zhang and colleagues [56]. The consequence for ability of compound 72 inducing apoptotic death in PANC-1 cells showed that derivative 72 suppressed RalA according to inducing the accumulation of ROS and trigger mitochondrial apoptosis in PANC-1 cells.
Markovic and colleagues [57] designed and synthesized compound 73 and its 5-phenylpyrazole derivative 74 ( Fig. 6 ) which exhibited the most promising activity (Table 4 ). QSAR study revealed that geometrical and topological are the most momentous factors for effect of the analogues, for instance, the size and shape of the molecule. Containing planar benzene group at N-1 position of pyrazolone showed superior inhibitory potency on both cells tested for it was a more favorable conformation moiety when interacting with active sites. Herein, the inhibitory effect on the target mostly depended on fragment at the C-4 position through forming H-bonds with active place.
Table 4.
Cytotoxic activity as IC50 (μM) of pyrazolone derivatives against tested cell lines.
| Com. |
In vitro cytotoxicity IC50 (μM) |
||
|---|---|---|---|
| MCF-7 | MDA-MB-361 | MDA-MB-453 | |
| 73 | – | 9.98 | 10.24 |
| 74 | – | 12.17 | 8.75 |
| 75 | 60.72 | – | – |
| 76 | 43.41 | – | – |
| 77 | 30.68 | – | – |
| 78 | 37.22 | – | – |
| 79 | 54.23 | – | – |
| 80 | 44.99 | – | – |
| 81 | 44.99 | – | – |
Pyrazolone derivatives such as 4-aminoantipyrine are well known compound used widely prophylactic of some diseases including cancer. Ghorab and colleagues [58] synthesized a variety of pyrazolone derivatives 75–81 (Fig. 6) inspired from 4-aminoantipyrine and determined the anticancer activity (Table 4). SAR study can be summarized that the most potent compound belonged to the pyrimidine derivatives especially the halogen group.
Dube and colleagues [59] synthesized a cluster of pyrazolone heterocyclic derivatives and evaluated their cytotoxic activities in vitro. The results of brine shrimp lethality assay showed that compound 82 displayed a favorable cytotoxic activity than standard cyclophosphamide. In addition, compounds 82 and 83 (Fig. 6) were found to be prominent analogues with the lethality concentration (LC50) values being 10.8 μg/mL and 14.5 μg/mL in sulforhodamine B (SRB) assay, respectively. SAR investigation implied that the dimethoxy substitution on ring A increased the antitumor activities, the group of hydrophobicity and hydrophilicity played a significant role in cytotoxic activities.
Gouda et al. [60] prepared several pyrazole analogues and tested the cytotoxicity against the Ehrlich ascites carcinoma cells (EAC). To detail, the cytotoxicity data clearly displayed that and compounds 84 and 85 exhibited favorable activities compared with 5-fluorouracil (Table 5 ). SAR can be recapitulated that introducing a hydrazopyrazol-5-one ring in position 2 to benzothiophene was important to the cytotoxic activity, furthermore, the order of antitumor activities of compound follows 86 < 87 < 88 and 84 < 89 < 90 (Fig. 6), it can be speculated that the group of COOEt at position 3 of the thiophene ring system in compounds increased the antitumor activity.
Table 5.
In vitro cytotoxicity of tested compounds (Ehrlich ascites cells apoptosis %).
| Com. | % Apoptosis |
||
|---|---|---|---|
| 100 μg/mL | 500 μg/mL | 25 μg/mL | |
| 5-Fluorouracil | 97.3% | 68% | 38.6% |
| 84 | 84% | 53.3% | 29.8% |
| 86 | 100% | 98.6% | 94% |
| 87 | 98.4% | 81% | 65% |
| 88 | 98.1% | 79% | 60% |
| 89 | 96.5% | 66.6% | 38.1% |
| 90 | 79.4% | 71% | 43% |
3.2.2. Coordination pyrazolone compounds as antitumor agents
In 2013, Vyas and colleagues [61] synthesized the new pyrazolone based complex 91 (Fig. 8 ) for the first time, which was suggested to be a potent antitumor agent against lung cancer cell lines (A549). According to evaluation of the cytotoxic activities against A549 cell lines and noncancerous rat cardiomyocytes (H9C2) cell lines in vitro, the result displayed that compound 92 (Fig. 8) possessed a superior cytotoxic property. Moreover, LDH activity of compound 92 showed the higher levels in the culture medium treated with A549 cells. It was worth mentioning that the chelation of Cu (II) with the pyrazolone and bipyridyl ligands formed the π-π* conjugation in which gave the extended planar structure resulting in the superior cytotoxic property.
Fig. 8.

Coordination pyrazolone compounds as antitumor agents.
Zhao and colleagues [62] prepared a cluster of novel mixed-ligand dibutyltin (IV) complex pyrazolone, all of the synthesized complexes were screened against two human cancer cell lines (KB and HeLa) in vitro and utilized MTT assay to evaluate antitumor activity. The results suggested that all complexes display a favorable antiproliferative activity against KB and HeLa cell lines with the IC50 values ranging from 0.29 to 0.54 μM for KB and 0.05–0.31 μM for HeLa. Compound 93 (Fig. 8) exhibited the most potent activity (IC50: 0.05 μM) against HeLa cell. SAR study implied that acetyl from acylpyrazolone and para-fluorobenzoate ligand may play a considerable role for increasing the antitumor activities.
Three kinds of pyrazolone complexes 94–96 (Fig. 8) were synthesized by Bakr and co-workers [63]. The MTT assay outcomes implied that HL exhibited the better cytotoxic activity against HePG2 and PC3 cell lines than 5-Fluorouracil with IC50 values being 5.69 and 6.80 μg/mL, respectively (Table 6 ). For metal complexes, the order of cytotoxicity was found to be 95 > 94 > 96.
Table 6.
In vitro cytotoxic activity of compounds 94–96 towards the cell lines.
| Com. | In vitro cytotoxicity IC50(μg/mL) |
|||
|---|---|---|---|---|
| HePG2 | HCT-116 | PC3 | MCF-7 | |
| 5-Fu | 7.9 | 5.2 | 8.3 | 5.5 |
| 94 | 15.31 | 16.93 | 17.30 | 27.47 |
| 95 | 10.73 | 14.65 | 11.45 | 12.96 |
| 96 | 28.30 | 20.59 | 20.40 | 35.62 |
Zhang and colleagues [64] synthesized two new transition metal complexes with 4-acylpyrazolone derivative as potential antitumor agents. According to the results of antitumor determination, the complex 97 (Fig. 8) was proved to exhibit higher cytotoxicity activity with IC50 values being 1.9 μg/mL for human esophageal cancer cells (Eca-109) and 1.2 μg/mL for cervical cancer cells (HeLa). The study suggested that the complex 97 might be an effective agent for tumor.
3.3. CNS agents
Because of the intricate pathogenesis and devastating effects, CNS disease is still a challenge for researchers to find novel precursors as the therapy [65]. In view of the experiment of the development of edaravone, several pyrazolones have been taken into account to be the CNS agents, existing literatures mainly focus on the anticonvulsant, antidepressant, anti-amyotrophic lateral sclerosis, anti-Alzheimer’s activities and inhibitory effect against other CNS related targets. Herein, we summarized the bioactivity and SAR for the synthetic derivatives, wishing to give evidence for pyrazolones to develop as CNS clinical agents.
3.3.1. Anticonvulsant agents
Epilepsy is a chronic neurological disorder syndrome identified by the spontaneous recurrence of seizures which can disrupt periods of more or less normal electroencephalographic (EEG) activity and behavior [66]. Currently the anticonvulsant drugs are chiefly derived from benzodiazepines or GABA (γ-aminobutyric acid) analogues, pyrazolone derivatives are likely to provide a novel orientation for drug design. Eldebss et al. [67] synthesized pyrazolone-based heterocycle compounds as monoamine oxidase (MAO) inhibitors and evaluated the tryptamine seizure potentiation in rats. The results manifested that compounds 97 and 98 (Fig. 9) were the most promising candidates (Table 7 ).
Fig. 9.

Pyrazolones as CNS agents.
Table 7.
Bioactivity of compounds 97 and 98.
| Com. | Ki (nM) MAO-A | Ki (nM) MAO-B | SIa | MAO-A IC50 (nM) | MAO-B IC50 (nM) | ED50 (μM) in vivo | |
|---|---|---|---|---|---|---|---|
| Deprenyl | – | – | – | 3.90 × 10−6 | 3.00 × 10−8 | 0.30 | |
| 97 | 0.014 | 121.34 | 8667.143 | 2.34 × 10−6 | 13.23 × 10−8 | 19.23 | |
| 98 | 0.346 | 765.87 | 2213.497 | 7.99 × 10−6 | 45.43 × 10−8 | 47.54 | |
SI (selectivity index) = Ki (MAO-B)/Ki (MAO-A) ratio.
Viveka et al. [68] prepared pyrazolone analogues 99 and 100 (Fig. 9 ) with favorable anticonvulsant activities. In the maximal electroshock (MES) test, compound 99 performed the best protective potency with the protection ratio of 79.76% and no significant toxicity was determined at the dosage of 25 mg/kg, while in the analgesic test it showed weak effect. Compound 100 exhibited moderate protection on the reaction time of mice. SAR study demonstrated that the existing of pyrazolone moiety elevates the anticonvulsant potency. As for the analgesic effect di-Cl substituent group on the heterocyclic systems promoted the activity, whereas slightly less activity was determined for the analogues with di-F substituents.
A cluster of pyrazolone analogues were synthesized and assessed for the anticonvulsant and antidepressant activities by Abdel-Aziz and colleagues [69]. Compounds 101, 102 and 103 (Fig. 9) displayed the most potent protective potency against pentylenetetrazole (PTZ)-induced clonic seizures in mice at a dosage level of 20 mg/kg with the protective ratio for 74.5, 78.7 and 74%, respectively, which was close to phenobarbital sodium (30 mg/kg) and better than phenytoin sodium (30 mg/kg). Preliminary SAR study can be concluded that the existence of cricoid pyrazolone group increased the anticonvulsant effect compared with the linear chain-hydrazide analogues, and thiophene acyl moiety was the most potent substituted group, while linear chain-hydrazide analogues performed better than pyrazolones in the antidepressant determination.
3.3.2. Antidepressant agents
Merugumolu and co-workers [70] prepared multiple pyrazolone derivatives and estimated the anti-depressant effect in vivo. The consequence demonstrated that the most promising derivatives 104 and 105 (Fig. 9) possessed remarkable anti-depressant activity in forced swimming test and tail suspension test (Table 8 ), with the relative values in the same magnitude of imipramine. As for SAR, the substituted groups on the 3,4-position of the ring of aromatic hydrazine exhibited better activity than other substituted groups.
Table 8.
Antidepressant effect of compounds 104 and 105.
| Com. | Forced swimming test |
Tail suspension test |
||
|---|---|---|---|---|
| Duration of immobility (s) | Change in immobility (%) | Duration of immobility (s) | Change in immobility (%) | |
| Control | 203.3 | – | 185.8 | – |
| Imipramine | 94.0 | −53.8 | 109.0 | −41.3 |
| 104 | 97.7 | −51.9 | 114.3 | −38.4 |
| 105 | 129.5 | −36.3 | 111.0 | −40.2 |
3.3.3. Anti-amyotrophic lateral sclerosis agents
The pyrazolone skeleton is an effective group characterized in a cell-based high throughput screening assay targeting mutant Cu/Zn superoxide dismutase 1 (SOD1) induced toxicity and aggregation as a marker for amyotrophic lateral sclerosis (ALS), which is an orphan neurodegenerative disease so for without a cure with the incidence of 1–2 per 100000 per year and 2–5 years from diagnosis to death [71,72].
A cluster of pyrazolone analogues were screened and evaluated for the anti-ALS activity from FDA approved drugs and biochemical reagents by Radhia and co-workers [73], as the most potent skeleton, the arylsulfanyl pyrazolones showed 100% efficacy compared with the positive control, radicicol (80% efficacy) in the test of G93A-SOD1 expression in PC12 cells model. CMB-003299 (106, Fig. 9) was the typical compound to decrease the mutant SOD1 aggregation with the ED50 value of 400 nM, and the weak cytotoxic effect was detected with the LD50 > 100 μM.
In the sequent research, a series of ether analogues with more metabolically stability were synthesized [74]. The most effective derivative 107 (CMB-087229) (Fig. 9) showed superior potency and in vitro pharmacological and pharmacokinetic features including protection against the mutant SOD1-induced cytotoxicity (ED50: 67 nM), good performance in the evaluation of potassium channel (10 μM), protection of primary cortical neurons, Caco-2 permeability, rat liver microsomes and cytochrome P450 isozymes. Moreover, in vivo for its efficacy in an ALS mouse model, pharmacokinetic profile and brain penetration were investigated as well, compound 107 (1.0, 10, and 20 mg/kg) dose-dependently extended the survival of SOD1 G93A mice at the dose of 300 mg/kg. In addition, compound 107 exhibited favorable blood-brain barrier penetration effect. SAR research revealed that the size and electronics were imperative features at the meta-positions of the derivatives, the potency of analogues reduced in the sequence: Cl > CF3 > F > Br > Ph.
3.3.4. Anti-Alzheimer’s agents
Based on the structure of leading compound edaravone, Tok and co-workers [75] synthesized a cluster of analogues and investigated the potential anti-Alzheimer activity. Among the analogues, compounds 108 and 109 (Fig. 9) exerted most potent inhibitory effects, with the detail data listed in Table 9 . For the instance of the most potent AChE inhibiting agent 108 and MAO-B inhibiting agent 109, the initial SAR could be summarized as that the variation of substituent groups strongly affected the inhibitory potency of analogues. Basic nitrogen atom in substituent groups of 108 significantly promoted the AChE inhibitory activity. In addition, incorporation of EWG increased the MAO-B inhibitory activity of 109. It can be seen from the docking analysis that compound 108 was prepared with similar size with donepezil (Fig. 10 ), and the room of cyclopentanone ring can be occupied by pyrazolone ring, providing the evidence of the activity and the idea to capture Alzheimer disease.
Table 9.
Inhibition ratio of 108 and 109 on Alzheimer’s enzymes.
| Com. | AChE IC50 (nM) | BChE inhibition % (10−3 M) | MAO-A inhibition % | MAO-B IC50 (nM) |
|---|---|---|---|---|
| Donepezil | 29 | 94.1 | – | – |
| 108 | 57 | 96.3 | 42.3 | – |
| 109 | – | 35.3 | 38.1 | 49 |
Fig. 10.

Ligand-protein interactions of compound 108 (blue) and donepezil (green) with AChE (PDB ID: 4EY7) accomplished by Discovery Studio 2019. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.3.5. Other CNS target agents
A major inhibitory neurotransmitter in the mammalian brain is GABA that delivers primarily through the GABAA receptors [76]. GABAA receptors are considered to be heteropentameric transmembrane glycoprotein spanning both extracellular and intracellular regions [77]. Inspired from the pyrazolone-based precursors 110–112 (Fig. 9), Hintermann and co-workers [78] synthesized several analogues as the ligands for the benzodiazepine binding site of the GABAA receptor. Among them, compound 113 (Fig. 9) was the typical compound with the affinity of IC50 values of α1 GABAA receptor, α2 GABAA receptor, α1 GABAA BZ receptor, α2 GABAA BZ receptor for 49, 271, 46 and 271 nM, respectively, however, the weak activity of compound 113 was observed in anxiety models in vivo, which was suspected due to the high level of clearance (in vitro: CLint rat: 128.3 μL/min*mg). SAR study suggested that the 2-Cl-benzyl substituent group was initially maintained for the purpose to investigate the influence of core modifications, variations of substituent groups on the aromatic pyrazolone moiety leading to the mixed results. The corresponding analogues were very weak agonists at α2 and some behaved as antagonists at α1.
Glycogen synthase kinase 3 (GSK-3), a regulator of glycogen metabolism, has been well known to be involved in a variety of intracellular signaling, and a lot of GSK-3 inhibitors has been used for the treatment of CNS disorders including Alzheimer’s disease (AD) [79], Parkinson’s disease [80], stroke, traumatic brain injury, and bipolar disorders [81]. Pyrazolone derivatives were chronicled to show activity on this target as well [82], the most promising compound 114 (Fig. 9) inhibited GSK-3 with IC50 value of 34 nM. Further investigation demonstrated that this compound possessed good kinase selectivity, and protective effects in oxidative stress mode in neuronal cell and locomotor hyperactivity in C57BL/6J mice were observed. SAR investigation indicated that non-substitution on the A ring was advantageous for the activity because only compound 114 exhibited the pronounced inhibitory effect among the synthetic compounds. Interestingly, the known GSK-3 inhibitors i.e. bisindol-maleimides (115) [83] and hymenialdisine (116) [84] (Fig. 9) contain the moieties structurally similar to the pyrazolone scaffold, which indicated the reasonability of using pyrazolone for the design of the GSK-3 inhibitor.
According to the structure of (S)-α-aminoadipic acid, which was an activating agent of important functional excitatory amino acid receptor mGlu2 and mGlu6 subtypes, compounds 117 and 118 (Fig. 9) were synthesized by Zimmermann et al. [85] thought some reactions. However, no significant agonist or antagonist activity from test compounds at the mGlu1a, mGlu2, mGlu4a, or mGlu6 subtypes were observed at the dose of 1 mM, the inappropriate pK a of analogues was considered as the potential reason of obstruct interaction of substituted groups with appropriate sites at the receptors.
3.4. Anti-inflammatory agents
More than 100 years of synthetic work have resulted in three privileged pyrazolones derivatives with anti-inflammatory potency, namely antipyrine, propyphenazone and metamizole which are still widely used [86]. The structure characteristic that methyl groups attach to the ring nitrogen atoms to enhance the activity has been retained from antipyrine to newly reported analogues. Based on the structure of marketed anti-inflammatory drugs including antipyrine and aminophenazone, several pyrazolone derivatives have been designed and synthesized as anti-inflammatory agents.
With the thorough understand of inflammatory reactions, using single mice or rat model to evaluate the analgesic effect gradually fall short of demand to investigate the activity, more and more targets are involved into the determination including COX-1, COX-2, 5-LOX, TNF-α, etc. Pyrazolones with anti-inflammatory effects tend to inhibited the activities of COXs and LOXs, which catalyze the conversion of arachidonic acid into prostaglandins or leukotrienes, meanwhile, phosphatase inhibitory of pyrazolones have also been disclosed [87], indicating the potential of pyrazolone motif in this field. The structural similarity between pyrazolone and COX-2 selective inhibitor celecoxib also provides possibility to develop them into potential clinical candidates [[88], [89], [90], [91]]. The gastrointestinal side effects existing in the usage of COX-1 inhibitors and the cardiovascular side effects in COX-2 inhibitors suggest researchers to consider the inhibitory potency of anti-inflammatory agents on both COX-1 and COX-2, the story of the development of imrecoxib and the conception of moderate selectivity for COX-2 enzyme have been reported as references [19]. It can be seen from the binding affinity models of celecoxib and pyrazolone derivative 131 with COX-1 and COX-2 (Fig. 12) that pyrazolone group maintain the similar pi-alkyl effect with electron group of alkyl groups in the pocket residues compared to pyrrazole ring because of its property similar to aromatic ring. The carbonyl group in pyrrazole is a favorable hydrogen acceptor which is feasible to form potential H-bond with alkaline amino acids like arginine, lysine and histidine, meanwhile, the amino group is a hydrogen donor easy to form hydrogen bond with residues like glutamine (Fig. 12 D).
Fig. 12.

Ligand-protein interactions of celecoxib (A) and pyrazolone derivative 131 (B) with the active site of COX-1 (PDB ID: 3KK6), celecoxib (C) and pyrazolone derivative 131 (D) with the active site of COX-2 (PDB ID: SLN1) accomplished by Discovery Studio 2019.
A cluster of pyrazolone and amino pyrimidine derivatives 119–124 (Fig. 11) were synthesized by Antre and co-workers [92], with obvious anti-inflammatory potency in carrageenan-induced rat paw edema model (Table 10 ). In regard to SAR, in contrast to the substitution of chlorine and nitro, the anti-inflammatory effects of these analogues were significantly promoted when the electron-donating groups of C-6 aminopyrimidine benzene ring were hydroxyl, methoxy and dimethylamine.
Fig. 11.

Pyrazolone derivatives with anti-inflammatory activity.
Table 10.
Result of anti-inflammatory activity of the tested derivatives.
| Com. | IC50 (μM) |
Selectivity index (SI) | Anti-inflammatory activity (% inhibition) | |
|---|---|---|---|---|
| COX-1 | COX-2 | |||
| 119 | – | – | – | 61.23 |
| 120 | – | – | – | 61.02 |
| 121 | – | – | – | 61.84 |
| 122 | – | – | – | 62.00 |
| 123 | – | – | – | 62.51 |
| 124 | – | – | – | 62.05 |
| 125 | 2.86 | 0.39 | 7.36 | 62.30 |
| 126 | – | – | – | 77.88 |
| 127 | – | – | – | 79.91 |
| 128 | – | – | – | 56.2 |
| 129 | – | – | – | 55.2 |
| 130 | 80 | 0.20 | 400 | 91.00 |
| 131 | >100 | 1.00 | >100 | 92.00 |
| 132 | 13.40 × 10−5 | 2.83 × 10−5 | 4.73 | 56.00 |
| 133 | 22.72 × 10−5 | 4.69 × 10−5 | 4.84 | 61.00 |
| 134 | 1.60 | 0.98 | 80.00 | |
| 135 | 1.57 | 0.91 | 98.00 | |
| 136 | 1.74 | 0.99 | 89.20 | |
| 137 | 4.08 | 0.77 | 5.29 | – |
| 138 | 3.81 | 0.72 | 5.29 | 72.72 |
| 139 | 3.76 | 0.66 | 5.69 | – |
| 140 | – | – | – | 72.72 |
Nadia and colleagues [93] synthesized multiple pyrazolone-pyridazine conjugates and screened their anti-inflammatory activity in vivo and in vitro. Compound 125 (Fig. 11) exhibited the strongest anti-inflammatory activity in the test of carrageenan-induced mice paw edema model and in COX-1 and COX-2 inhibition test (Table 10). It also inhibited the production of inflammatory cytokines including TNF-α and IL-6 in serum. SAR study showed that the replacement with bulky aliphatic secondary amines such as morpholine exerted good anti-inflammatory effects.
Mannich base derivatives 126 and 127 (0.03 mmol/kg) (Fig. 11) displayed the remarkable anti-inflammatory effect on the carrageenan-induced acute albino rats paw edema model with inhibitory rates of 77.88% and 79.91%, respectively, after 6 h of administration [94]. The preliminary SAR demonstrated that analogues containing EWGs in the Mannich base phenyl ring at position 4, especially sulfonic group enhanced the activity.
Abbady and co-workers synthesized some new pyridines, pyrans, and indazoles with pyrazolone ring [95]. Among the synthetic products, indazole derivatives 128 and 129 (10 mg/kg) (Fig. 11) obviously suppressed swelling of in rat hind paws induced 5 h after injection of carrageenan, with inflammatory inhibition rates of 56.20% and 55.20%, respectively, indicating excellent anti-inflammatory activity.
A series of 5-methyl-2-phenyl-1H-pyrazol-3 (2H)-one analogues were prepared and determined for their anti-inflammatory effects [96]. The outcome showed that the derivatives 130 and 131 (Fig. 11) were proved to have superior inhibitory potency against both the targets of inflammation COX-1 and COX-2 (Table 10). Furthermore, derivatives 130 and 131 also exhibited favorable anti-inflammatory effect in carrageenan induced rats paw edema model with ED50 values of 102 and 109 mg/kg in vivo, respectively. Regarding SAR, pyrazolones containing C-3 methyl group, C-4 urea substitution and carbonyl at 5-position posed an impact on COX enzymes inhibition. The substituent with urea-phenyl group at 3-phenyl ring was more active than that containing chlorine at the same position and unsubstituted urea. Moreover, thiourea derivatives imparted strengthen activity compared with those with urea and guanidine.
Ashraf et al. [97] successfully prepared two series of pyrazolone analogues and tested their anti-inflammatory effect in vivo and in vitro. The derivative 132 and the enolate133 (Fig. 11) displayed excellent inhibitory activity on COX-2 (Table 10) as well as potent edema inhibition. SAR investigation showed that these compounds with unsaturated moiety such as the allyl and aryl groups maintained stronger activities than those with alkyl groups. Thiourea analogues 132 also improved anti-inflammatory potency in the presence of the bulky hydrophobic cyclohexyl group.
El Sayed and colleagues designed and synthesized novel heterocyclic derivatives containing antipyrine and pyrazolone framework [98]. The pharmacology tests result revealed that derivatives 134–136 (Fig. 11) exhibited significant anti-inflammatory activities in different degree both in vivo and in vitro (Table 10). The SAR study indicated that the presence of the 3-benzo[d] [1,3]dioxole ring system and the N-containing heterocyclic amine attached to the methyl group at 4-position of the antipyrine structure was very essential for activity as in case of compounds 134 and 135.
A cluster of novel pyrazolone analogues containing aminosulfonyl group were synthesized and investigated for the anti-inflammatory potency [99]. The outcome showed that three compounds 137–139 (Fig. 11), as potent anti-inflammatory agents, exerted strong inhibitory effects against both COX-2 (Table 10) and 5-LOX (IC50: 0.53, 0.52 and 0.57 μM in order) with superior COX-2 SI values. Compounds 138 and 140 remarkably diminished rat paw edema induced by carrageenan with ED50 values of 0.044 and 0.079 mmol/kg, respectively, exhibiting excellent anti-inflammatory activity in vivo. In terms of SAR study, the replacement of 2-carbonyl group in the derivatives with SO2NH2 as the main active group promoted activity.
Fahmy et al. [100] tested anti-inflammatory effects of several novel O-substituted salicylamides. Among all analogues, pyrazolone derivative 141 (Fig. 11) exhibited moderate anti-inflammatory activity in carrageenan-induced edema model. The multiple bioactivity test results indicated the pyrazolone and the structurally similar group pyrazole could be the potential substitutions.
Golebiowski and co-workers synthesized some novel monocyclic pyrazolone and investigated their inhibition on the inflammatory cytokines [101]. The outcome showed that compound 142 (Fig. 11) significantly reduced the expression of TNF-α induced by lipopolysaccharide (LPS) with IC50 value of 13 nM. Furthermore, through SAR study, it can be found that benzyl amino substituents were more active than those of aminopyridine ring, and N-methyl substitution on the pyrazolone was more effective than ethyl substitution. Currently, the topic of inflammatory reaction has beyond the analgesic agents, immunoreactivity targets also involved. The author observed the interactions of p38 kinase with another potential synthesized pyrazolone derivative 143 (Fig. 13 ), in which the piperidine ring was introduced to gain the better pharmacokinetic parameters. The carbonyl group of pyrazolone ring was proved to form the H-bond with the amine group of Lys 53, revealing the importance of pyrazolone moiety of this type of inhibitor.
Fig. 13.

Resolved pharmaceutical cocrystal of pyrazolone derivative 143 with p38 kinase (PDB: 1YWR).
3.5. Antioxidant agents
Free radicals, known to be highly reactive molecules resulted via different biochemical reactions in the body. These free radicals lead the other metabolites to be oxidized and caused different diseases due to oxidative stress [102]. Pyrazolones played a role in antioxidant activity, referring to the antioxidant mechanism of edaravone, C C double bond formed by the action of tautomerism of carbonyl group on pyrazolone was considered to be important [103]. In the light of the structure motif of 4-aminoantipyrine, Schiff base derivative 144 (Fig. 14 ) was prepared and determined for the antioxidant activity [104]. The derivative 144 exerted a strong antioxidant effect with IC50 value of 31.26 μM. As a non-phenolic antioxidant agent, the antioxidant effect of compound 144 was considered to be relative to the formation of proton free radicals by the C-7 or C-11 methyl groups. The Schiff base pyrazolone analogues were also synthesized by Khan et al. [105], among the products, phenol productions 145–148 (Fig. 14) exhibited significant antioxidant effect in DPPH test with the IC50 values of 20.14, 19.12, 17.14, 15.16 μM, respectively, which was even stronger than the standard drug n-propyl gallate (IC50: 30.27 μM). SAR study revealed that double phenol group substituted analogues performed better than other moieties because of the reducibility of phenolic hydroxyl group, and 2.5-diOH substitution was stronger than 2,4-diOH.
Fig. 14.

Antioxidant of pyrazolone derivatives.
Gaffer and co-workers [106] screened the antioxidant activity of a cluster of thiazolyl-pyrazolone derivatives through 2, 2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) radical cation decolorization assay. Compounds 149 and 150 (Fig. 14) (2 mM) exhibited the strongest antioxidant potency with the inhibition of 88.6% and 85.7%, respectively. Thereinto, compound 149 exhibited better activity than standard inhibitor ascorbic acid (inhibition: 88.20%). SAR investigation was concluded as that the presence of benzene on the thiazole ring and phenylthiocarbamoyl moiety along with core pyrazolone group was benefit to the antioxidant effect. Additionally, 4-phenyl substituent group on the thiazole ring was better than the methyl substitution.
3.6. Anti-tuberculosis agents
Tuberculosis (TB) is an airborne infectious disease caused by Mycobacterium tuberculosis (MTB) and embodies one of the leading causes of death around the world [107]. The dangerous spread of TB is primarily because of its association with HIV infection and to the rapid development of multidrug-resistant (MDR) strains of MTB [108]. Pyrazolone as the precursor has been reported to show potential antitubercular activity.
Sivakumar et al. [109] prepared two series of hybridized pyrazolone analogues and determined their antitubercular effect. The most potent compounds 151 and 152 (Fig. 15 ) possessed apparent antitubercular effects (IC50 < 0.1 μg/mL). Furthermore, SAR investigation indicated that the antitubercular activity of the compounds was markedly improved in the absence of double bond in the imine side chain and the benzene ring at the end of pyrazolone moiety.
Fig. 15.

Pyrazolones with antitubercular activity.
Pethaiah et al. [110] synthesized several 2-aryl-5-methyl-2,3-dihydro-1H-3-pyrazolone analogues in water through one-pot process and investigated the antitubercular activity against MTB in vitro. The MIC value of potential compound 153 (Table 11 ) (Fig. 15) was better than clinical drugs. SAR showed that the replacement on the N-aryl ring of derivatives contributed antimycobacterial activity. Specifically, the analogues containing electron-withdrawing groups with halogens and nitro groups showed stronger activity compare with aryl rings containing electron-donating groups such as methoxide, methyl and isopropyl.
Table 11.
Antitubercular properties of the designed compounds.
| Com. | MTB MIC (μg/mL) | IC50 (μM) | Selectivity index (SI) |
|---|---|---|---|
| 151 | – | <0.37 | 633.35 |
| 152 | – | <0.44 | 630.75 |
| 153 | 0.79 | – | – |
| 154 | 4.00 | – | – |
| 155 | 4.00 | – | – |
| 156 | 4.00 | – | – |
| 157 | 3.11 | – | 26.35 |
| 158 | 1.97 | – | – |
| 159 | 0.78 | – | – |
| 160 | 0.78 | – | – |
Daniele et al. [111] prepared various pyrazole analogues and evaluated their antitubercular activities against M. tuberculosis. Compound 154 (Fig. 15) was the most potent candidate (Table 11). SAR study demonstrated that the halogens on phenyl ring and the methyl group at third position of pyrazole ring showed higher antitubercular effect than that contained alkyl in the same position.
Two series of new pyrazolone analogues were prepared and assessed for the anti-MTB activity [112]. Two analogues 155 and 156 (Fig. 15), containing the p-chlorophenyl ring, were regarded as the most promising MTB inhibitors (Table 11). Meanwhile, the presence of N-methyl-piperazine and morpholine groups remarkedly improved anti-MTB effects.
Several Mannich bases and Schiff bases of pyrazolone framework were synthesized [113]. The results showed that analogue 157 (Fig. 15) could be used as a potential antitubercular drug due to its extremely high activity (Table 11) over that of the standards ethambutol and ciprofloxacin. SAR study revealed that derivatives with acyl substituent had better anti-MTB ability than those comprising EWGs such as nitro, chlorine, and carboxyl and electron donating group including hydroxyl.
Ahsan and co-workers [114] designed some diversified pyrazolone derivatives aiming at MTB and isoniazid resistant MTB, among which compounds 158 and 159 (Fig. 15) exhibited pronounced inhibitory effect (Table 11), the outstanding inhibitory effect of 159 against isoniazid resistant MTB strain was observed as well and the compound 158 was revealed to show strong antibacterial effect simultaneously. SAR study revealed that N-aryl with electronegative substituent group showed strongest antitubercular potency, in which 4-pyridinyl group had maximum inhibition, compared to 2-chlorophenyl, 4-chlorophenyl and 4-aminophenyl group substitution. The electron donating group such as 4-methoxyphenyl, 2-methylphenyl, 3-methylphenyl and 4-methylphenyl exhibited less inhibition. In the follow up research [115], the potent compound 160 (Fig. 15) was selected from the novel synthesized analogues with excellent activity and free from cytotoxicity (>62.5 μg/mL) appeared. Better drug-likeness was obtained after adding the imine group between the oxadiazol and the phenyl ring and SAR was summarized as that EWGs produced more inhibitory effect.
3.7. Antiviral agents
Multiple viruses widely exist in the nature and threaten public health [116]. Pyrazolone has been used to generate antiviral agents, involving in anti-orthopoxvirus, anti-protease-resistant prion protein (PrP-res), anti-severe acute respiratory syndrome (SARS) and anti-buffalopox virus (BPXV) agents.
Fan et al. [117] synthesized a series of pyrimidine-pyrazolone nucleoside chimera analogues and determined the anti-orthopoxvirus activity in vitro. The consequence proved that compounds 161 and 162 (Fig. 16 ) exerted the most potent anti-orthopoxvirus effect (Table 12 ). SAR study revealed that the activity was enhanced after matching with the pyrazolone moiety.
Fig. 16.

Antiviral of pyrazolone analogues.
Table 12.
Inhibition of orthopoxvirus replication by pyrazolo-pyrimidine nucleosides.
| Com. | EC50 (μM) |
Toxicity CC50 (μM) Neutral red uptake | |||
|---|---|---|---|---|---|
| Vacciniad CPE | Vacciniad PR | Cowpox CPE | Cowpox PR | ||
| Cidofovir | 3.2 | 20 | 7.1 | 32 | >317 |
| 161 | 1.7 | 6.9 | 0.3 | 5.6 | >286 |
| 162 | 20 | 11 | 1.8 | 9.0 | >292 |
Note: CPE-reduce viral cytopathogenicity, PR-plaque formation.
In 2007, Kimata and co-workers [118] reported the synthesis and assessment of multiple PrP-res accumulation inhibitors according to the structure of edaravone, in which the most potent compound 163 (Fig. 16) displayed PrP-res inhibitory effect in the ScN2a cells with IC50 value of 3 nM, which was 130 times more active than quinacrine and more than 300-fold effective than edaravone. Furthermore, no significant SOD (superoxide dismutase)-like activity was observed, indicating potential novel mechanism of the PrP-res inhibitory effect. SAR study demonstrated that the position and class of substitutions were not directly related to the potency of suppressing the accumulation of PrP-res but substituted the nucleus with 1-cyclohexyl, 3-isopropenyl, 3-(4-nitrophenyl) and 4-benzoyl could enhance the inhibitory activity.
Srinivasan and teammates [119] synthesized several spiro-piperidinyl pyrazolone derivatives inspired from the skeleton of spiramide and assessed their antiviral properties against Buffalopox caused by BPXV on Vero cells in vitro. The results suggested that derivatives 164, 165 and 166 (Fig. 16) were the most potent analogues with electron donating groups on the aromatic ring.
Kumar and coworkers [120] synthesized multiple pyrazolone analogues as SARS-coronavirus (CoV) and Middle East Respiratory Syndrome (MERS)-CoV 3C-like protease inhibitors, among which compounds 167 and 168 (Fig. 16) exerted the strongest inhibitory effects against SARS and MERS with IC50 values of 6.4 and 5.8 μM (167), 5.8 and 7.4 μM (168), respectively. SAR study revealed that the carboxylate group is a crucial pharmacophore and its presence either at the C-2 position of ring A or moiety B is critical for the activity, meanwhile, the chlorine at C-5 position of ring A slightly decreases the activity. It can be seen from the docking studies that pyrazolone group in both compounds 167 and 168 played important roles to form H-bonds with residues Glu 166 and His 41 in SARS 3CLpro, respectively.
3.8. Lipid-lowering agents
With the change of people’s lifestyle and dietary structure, the incidence of fatty liver is increasingly high. In particular, non-alcoholic fatty liver disease (NAFLD) has seriously endangered people’s life and health [121]. However, due to the complex pathogenesis [122], it is often neglected in clinical practice. Pyrazolinone derivatives can reduce cholesterol and lipid accumulation in the body by regulating multiple indicators, such as AMPK and FXR. Therefore, it is of importance to improve the lipid-lowering activity by modifying the structure of pyrazolone. The pyrazolone derivatives are also reported to be the selective antagonists to the nonsteroidal farnesoid X receptor (FXR) [123], which maintains the bile acid homeostasis and plays crucial roles in the control of cholesterol, lipid, and glucose metabolism [124,125]. Based on the screen of the known FXR modulators, the lead compound 169 (Fig. 17 ) (IC50: 69.01 μM) was identified with the help of homogeneous time-resolved fluorescence (HTRF) assay. On the basis of 169, further modification was carried out accordingly. The most promising compound 170 (IC50: 8.96 μM) displayed antagonistic capability 10-fold and 8-fold higher than that of the control Z-guggulsterone and the original analogue 169, respectively. Compound 170 was further proved to interact with FXRαLBD with high binding affinity, and potent antagonistic activity against FXR in two cell testing platforms. In addition, compound 170 strongly inhibited the regulating activities of chenodeoxycholic acid on FXR target genes. Furthermore, compound 170 was proved to play a role in lowering the contents of triglyceride and cholesterol in human hepatoma HepG2 cells and in the cholesterol-fed C57BL/6 mice. SAR can be interpreted that changes of substituents on the 1, 3, and 4-positions of pyrazolone group had crucial influence on antagonistic effect, and appropriate structural optimizations on the above regions can substantially strengthen activity.
Fig. 17.

Pyrazolones as lipid-lowering agents.
NAFLD is a clinical syndrome identified by hepatic steatosis. It is closely linked to obesity, insulin resistance and dyslipidemia. AMP-activated protein kinase (AMPK) functions as an energy sensor and plays an important role in regulating lipid metabolism [126,127]. According to the structure of lead compound 171 (Fig. 17), Zhang et al. [128] synthesized multiple pyrazolone derivatives as AMPK activators to suppress lipid synthesis and reduce lipid accumulation in ob/ob mice. The most potent compound 172 directly activated the kinase domain of AMPK with an EC50 value of 2.1–0.2 μmol/L and acted as a non-selective activator of AMPK complexes, additionally, compound 172 suppressed the accumulation of triglyceride in HepG2 cells dose-dependently. The outcome demonstrated that the AMPK activators could be part of a treating method for NAFLD and related metabolic disorders.
3.9. Antihyperglycemic agents
Diabetes is considered to be relative to overindulgent life-style and thus is thought as major health concern in industrialized countries [129]. Pyrazolones have been disclosed to be antihyperglycemic through multiple targets including aldose reductase (AR), α-glucosidase and α-amylase. The antihyperglycemic activity of pyrazolone derivatives was explored by Kees et al. [130] The potent compound 173 (100 mg/kg) (Fig. 18 ) caused a 68% decrease in plasma glucose in db/db mice, moreover, it essentially normalized the level of glucose at 20 mg/kg (57% reduction) and maintained remarkable reduction at 2 mg/kg (30% decrease). SAR could be concluded that the substitution of 4-methylthio, methylsulfinyl, or ethyl to a benzyl group at C-4, in combination with trifluoromethyl at C-4 position of pyrazolone (hydroxy tautomer) formed potent antihyperglycemic agents in mice. In addition, the chemical “trapping” of four of the seven possible tautomeric forms of the heterocycle by mono- and dialkylation at the acidic hydrogens gave several additional potent analogues.
Fig. 18.

Pyrazolones as antihyperglycemic agents.
Kadam and co-workers [129] synthesized several pyrazolone derivatives as inhibitors of AR, in which compound 174 (Fig. 18) displayed the most promising inhibitory with IC50 value of 6.30 μM. In regard to SAR, it was possible to note that the hydrophobic groups like benzene ring with pyrazolone adversely influenced AR inhibition, while the introduction of carbothioamide group increased inhibitory effect. Because of the simultaneous existing of arbothioamide moiety and 3-(4I-methoxyphenyl)-1-phenyl-1H-pyrazole and 3-(4I-chlorophenyl)-1-phenyl-1H-pyrazole substituent group at first position of pyrazolone, compound 174 exerted the best activity. Docking analysis suggested that substituted groups of 174 gave the proper placement of hydrophobic groups in respective binding pockets of AR, avoiding the poor interactions because the size of substitutions was too small to occupy the binding pocket, and the pyrazolone moiety in compound 174 was considered to play a role in form H-bond with the residue Cys 298.
Eldebss and colleagues [131] prepared a series of pyrazolones acting as α-glucosidase and α-amylase inhibitors, among which compound 175 (Fig. 18) exerted the most significant inhibitory effects with IC50 values of 14.67 and 63.66 μM, respectively. In the acute diabetic mice model, compound 175 reduced serum glucose level for the ratio of 32.16%, and in subacute study the ratio was 13.34% after administration for 2 h, which was even better than the effect of pioglitazone. The docking analysis indicated that the 1,5-dimethyl group of pyrazolone was important for the interaction between compound 175 and α-glucosidase, additionally, preserving the sulphone group was momentous to the inhibitory effect.
In 2014, Shetty et al. [132] synthesized multiple pyrazolone derivatives as α-amylase inhibitors. The consequence showed that compound 176 (Fig. 18) displayed the strongest inhibitory effect with the ratio of 61.6%. The primary SAR can be suspected as that introduction of disubstituted halogen analogues promoted the inhibitory effect due to its high electronegativity, moreover, multi-fluoro substitution on A ring could be the potential active group.
3.10. Protein inhibitors
In addition to the activities mentioned above, pyrazolones have been shown to inhibit the proteins including KATP channel protein, phosphodiesterase (PDE), aromatase, divalent metal transporter 1 (DMT1), human carboxylesterase1 (hCE1) and transforming growth factor (TGF) βR1, indicating the multiple targets therapeutic action and favorable drug-likeness of pyrazolone derivatives. Drizin and co-workers [133] designed and synthesized a cluster of pyrazolone derivatives as KATP channel openers inspired from disclosed compounds (the skeleton of 177 and 178, Fig. 19 ), together with the investigation of SAR. It can be concluded that cyclopentanone in the left hand portion (A area) of the derivative was 4-fold more potent than cyclohexanone. The introduction of gem-dimethyl groups as well as incorporation of oxygen in the cyclohexanone ring in the left hand portion of the molecule elevated the potency 10-fold. In the right hand portion of the molecule, the activity was promoted 5-fold when the NH-group on the pyrazolone was replaced by oxygen atom.
Fig. 19.

Pyrazolone as protein inhibitors.
Ochiai et al. [87] synthesized several 4,4-dimethylpyrazolone analogues as cyclic 30,50-nucleotide PDE 3/4-inhibitor, in which the compound 179 (Fig. 19) was demonstrated to be the most potent compound inhibiting PDE 3A and PDE 4B with the IC50 values of 0.14 and 0.15 μM, respectively. In addition, significant bronchodilatory activity was observed. SAR study demonstrated that the presence of pyrazolopyridine 7-substituent can engender potency for the inhibition of PDE 4, and the isosteric imidazopyridine substitution made for the pyrazolopyridine subunit increases the well-balanced dual of PDE 3 and PDE 4. As a member of PDEs, Trypanosoma brucei PDB1 (TbrPDEB1) was described as a crucial target for the therapy of Human African trypanosomiasis [134,135], which was a parasitic disease caused by the protozoan pathogen T. brucei. Orrling and colleagues [136] explored catechol pyrazolinones as trypanocidals inspired from the structural motif rolipram. After the scaffold merging, the premier potent compound 180 (Fig. 19) was selected with the TbrPDEB1 IC50 value of 12 μM. Furthermore, with the help of homology modeling and docking studies to guide fragment growing into the parasite-specific P-pocket, which was a unique sub-pocket that extended from the invariant glutamine (Gln 874) through the protein to the solvent in the enzyme binding site, the fragment growing compound 181 (Fig. 19) was emerged with the outstanding activities for inhibiting T. brucei rhodesiense with IC50 value of 60 nM and TbrPDEB1 with IC50 value of 49 nM. SAR investigation elucidated that pyrazolone held a conjugated π-system and additional possibility to interact with aromatic residues in the binding pocket of TbrPDEB1. Moreover, the extended alkyl chain in the analogues of 181 enabled to reach and enter the P-pocket, which displaced water from the length of the P-pocket and placed the tetrazole at the solvent-exposed exit of the pore. The tetrazole in compound 181 formed a H-bond with Tyr 845, further stabilizing the conformation. Afterwards, according to the structure of 180 and 181, Amata and co-workers [137] designed and synthesized a cluster of derivatives, while poor inhibitory effect was detected, the most promising pyrazolone compound 182 (Fig. 19) only showed inhibition for 18% at the dose of 10 μM, indicating a better understanding of the subtle structural features was necessary for an optimal enzyme inhibition.
Yi and co-workers [138] synthesized the diversified pyrazolone derivatives and screened the aromatase inhibition effect. The most active compounds were 183 and 184 (Fig. 19), showing IC50 values of 2.3 nM and 3.3 nM, respectively. The inhibition of compound 183 was even stronger than the reference letrozole (IC50: 2.8 nM). SAR analysis indicated that the pyrazolone moiety could inhibit aromatase by binding to its active site. The bioactivity result proved that the presence of substitutions in compounds 183 and 184 were good for the inhibition, as for the most promising compound 183, the cyano group was revealed to interact with the important residues Arg 115 and Met 374 of the aromatase.
From the high-throughput screening of DMT1 [139], pyrazolone hit 185 (IC50: 2.57 μM, Fig. 19) was elaborated as the structural motif. Preliminary hit-to-lead efforts gave the pyridyl analogue 186 (IC50: 1.53 μM), which imparted several undesirable features including most notably an extended conjugation motif affording an intense orange color as well as Michael acceptor features at the benzylidene α-carbon. Further investigation introduced more substitutions to modify these skeletons, and the most promising compound 187 (IC50: 0.64 μM) was distinguished because of its eminent absorption, distribution, metabolism and excretion (ADME) properties, the inhibition of CYP3A4 was significantly reduced and the rat microsome metabolic stability was markedly hoisted compared to the precursor 186. SAR of the analogues of 187 implied that EWGs on the benzene ring were slightly preferred over electron-donating ones.
Based on the existing TGFβR1 inhibitors, Guckian and colleagues [140] described a series of TGFβR1 kinase inhibitors including a pyrazolone group which allowed replacement of the ubiquitous nitrogen H-bond acceptor within the core with a carbonyl without remarkable loss in biochemical activity. The most active compound 188 (Fig. 19) inhibited the TGFβR1 kinase with the Ki value of 0.012 μM and suppressed the PAI-Luc with the IC50 value of 0.22 μM. SAR can be recapitulated that the carbonyl moiety of pyrazolone was potent to capture the H-bonding interaction with the key residue Lys 232 of TGFβR1. The A ring was the area the authors paid attention to modify, and they found that when A ring was substituted as pyridine ring could elevate the activity because it contained a basic nitrogen in the 2-position to occupy the hydrophobic pocket. As for the aromatics on A ring, substitutions on meta position was proved to be superior to ortho or para position, and this position was very sensitive to the nature of substitution with preference for small hydrophobic substituent groups without branching on the α-carbon, considering to influence of the size of substituted atom on the hydrophobic pocket, bromo group was selected for the suitable substitution for further investigation.
Asymmetric synthesis asymmetric spiro-pyrazolone derivatives have been a hotspot in recent years, while the investigations of bioactivity about these productions and the relationships between their enantioisomerism and activity are still rare. Bao et al. [141] synthesized a series of dispirotriheterocyclic scaffold derivatives and screened the inhibitory effects against hCE1. It is worth noting that the most potent compound 189 (Fig. 19), in which there were two chiral centers important for inhibitory potency, exerted the strong inhibition with IC50 value of 0.39 μM, whose activity was 20-fold stronger than its enantiomer, indicating the importance of asymmetric [3 + 2] cyclization process.
4. Conclusion and perspective
Pyrazolone core is one of the most explored precursors among diverse fused heterocycles, capable of multiple roles in different pathophysiological conditions. According to the concept of generalized bioisosteres [142,143], as a typical synthetic rather than natural motif in medicinal chemistry, pyrazolone is potential to replace multiple moieties with similar chemical structure and bioactivities including parazole, imidazolin-5-one and 2,4-dihydro-1,2,4-triazol-3-one, etc., providing an important scaffold for drug development. Being a versatile molecule with immense biological significance, pyrazolone derivatives are being developed for several pathological screening including antimicrobial, antitumor, CNS effect, anti-inflammatory, antioxidant, antitubercular, antiviral and protein inhibitors activities since long.
The development of pyrazolone derivatives can be regarded as the epitome of medicinal chemistry, starting from antipyrine, several analogues have been explored and the precursor designing methods have evolved from structural modification to Fragment Based Drug Design and high-throughput screening simultaneously. With the development of science, machine learning, virtual screening and combinatorial chemistry technologies shorten the time of filtrating the leading compounds and will play a more and more important role in drug design in the future [144,145]. Investigations for privileged scaffold from the perspective of SAR have been lasting several years [[146], [147], [148], [149], [150], [151]]. It is probable that pyrazolone scaffold has its position in the establishment of compound library due to its good drug-likeness and broad bioactive properties. Moreover, as we mentioned in this article, the pyrazolone group maintains properties like aromatic and it is feasible to form pi-alkyl effect with residues in the potential target, and the carbonyl group in pyrrazole is a favorable hydrogen acceptor which is feasible to form potential H-bond, which provide evidence for the importance of pyrazolone structural motif in medicinal chemistry.
Pan Assay Interference Compounds studies consider that some poor drug-likeness compounds bring false positive results [152,153], but pyrazolone is not on the list. Besides, the toxicity of pyrazolone derivatives is still alarming [154], because the loss and harm caused by the side effects of antipyrine and aminopyrine including leukopenia and agranulocytosis are still vivid. In this review, we have disclosed recent role of pyrazolones in the field of medicinal chemistry by discussing insights of SAR studies and binding conformations of these heterocycle compounds with a variety of targets. These intelligences are helpful to explore novel pyrazolone analogues for the challenging pathophysiological conditions nowadays.
Declaration of competing interest
None of the authors have any conflict of interest to disclose.
Acknowledgements
This work was supported by the Changjiang Scholars and Innovative Research Team in Universities, Ministry of Education of China (IRT_15R55), the 9th Group of Hundred-Talent Program of Shaanxi Province (2017), the International Science & Technology Cooperation Program of Shaanxi Province (No. 2019KWZ-001), and Natural Science Foundation of Shaanxi Province, China (Grant No. 2017JM8054).
Abbreviations
- A549
lung cancer cell lines
- ABTS
2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid)
- AChE
acetylcholine
- AD
Alzheimer’s disease
- ADME
absorption, distribution, metabolism and excretion
- ALS
amyotrophic lateral sclerosis
- ALK
anaplastic lymphoma kinase
- AMP
Adenosine monophosphate
- AMPK
AMP-activated protein kinase
- Arg
arginine
- A. niger
Aspergillus niger
- BPXV
buffalopox virus
- BZ
benzodiazepine
- Caco-2
the human coloncarcinoma cell line
- C. albicans
Candida albicans
- CLint
clearance rate
- c-Met
c-mesenchymal epithelial transition factor
- CNS
central nervous system
- CoV
coronavirus
- COX
prostaglandin-endoperoxide synthase
- CYP3A4
Cytochrome P4503A4
- DMT1
divalent metal transporter 1
- DPPH
1,1-Diphenyl-2-picrylhydrazylradical2,2-Diphenyl-1-(2,4,6-trinitrophenyl)hydrazyl
- EAC
Ehrlich ascites carcinoma cells
- EC50
half effective concentration
- ED50
median effective dose
- EDGs
electron-donating groups
- EDTA
ethylenediaminetetraacetic acid
- EWGs
electron withdrawing groups
- FDA
Food and Drug Administration
- FXR
farnesoid X receptor
- GABAA
Gamma-Amino Butyric Acid-A
- G. candidum
Geotrichum candidum
- Gln
glutamine
- Gram+ve
Gram positive
- Gram-ve
Gram negative
- GSK-3
Glycogen synthase kinase 3
- hCE1
human carboxylesterase1
- HGF
hepatocyte growth factor
- HTRF
homogeneous time-resolved fluorescence
- IC50
half maximal inhibitory concentration
- IL-6
interleukin- 6
- KATP
ATP-sensitive potassium channel
- Ki
drug concentration at which the reaction rate is half of Vmax
- LBD
ligand-binding domain
- LC50
lethality concentration
- LPS
lipopolysaccharide
- Lys
lysine
- MCF7
human tumor breast cancer cell line
- MERS
Middle East Respiratory Syndrome
- MES
maximal electroshock seizure
- Met
methionine
- mGlu
metabotropic glutamate
- MTB
Mycobacterium tuberculosis
- NAFLD
Non-alcoholic fatty liver disease
- PDB
Protein Data Bank
- PDE
phosphodiesterase
- PrP-res
protease-resistant prion protein
- P. aeruginosa
Pseudomonas aeruginosa
- PTZ
pentylenetetrazol
- MAO
monoamine oxidase
- SAR
structure-activity relationship
- SARS
severe acute respiratory syndrome
- SI
selectivity index
- SIRTs
Sirtuins
- SOD
superoxide dismutase
- SRB
sulforhodamine B
- S. pyogenes
Streptococcus pyogenes
- TbrPDEB1
trypanosoma brucei PDB1
- TGFβ
transforming growth factor β
- TNF-α
tumor necrosis factor α
- VEGFR-2
vascular endothelial growth factor receptor 2
References
- 1.Kucukguzel S.G., Senkardes S. Recent advances in bioactive pyrazoles. Eur. J. Med. Chem. 2015;97:786–815. doi: 10.1016/j.ejmech.2014.11.059. [DOI] [PubMed] [Google Scholar]
- 2.Lapchak P.A. A critical assessment of edaravone acute ischemic stroke efficacy trials: is edaravone an effective neuroprotective therapy? Expert Opin. Pharmacother. 2010;11:1753–1763. doi: 10.1517/14656566.2010.493558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meiattini F., Prencipe L., Bardelli F., Giannini G., Tarli P. The 4-hydroxybenzoate/4-aminophenazone chromogenic system used in the enzymic determination of serum cholesterol. Clin. Chem. 1978;24:2161–2165. [PubMed] [Google Scholar]
- 4.Bussel J.B., Cheng G., Saleh M.N., Psaila B., Kovaleva L., Meddeb B., Kloczko J., Hassani H., Mayer B., Stone N.L., Arning M., Provan D., Jenkins J.M. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N. Engl. J. Med. 2007;357:2237–2247. doi: 10.1056/NEJMoa073275. [DOI] [PubMed] [Google Scholar]
- 5.McHutchison J.G., Dusheiko G., Shiffman M.L., Rodriguez-Torres M., Sigal S., Bourliere M., Berg T., Gordon S.C., Campbell F.M., Theodore D., Blackman N., Jenkins J., Afdhal N.H., Grp T.P.L.S. Eltrombopag for thrombocytopenia in patients with cirrhosis associated with hepatitis C, N. Engl. J. Med. 2007;357:2227–2236. doi: 10.1056/NEJMoa073255. [DOI] [PubMed] [Google Scholar]
- 6.Freitag F.G., Cady R., DiSerio F., Elkind A., Gallagher R.M., Goldstein J., Klapper J.A., Rapoport A.M., Sadowsky C., Saper J.R., Smith T.R. Comparative study of a combination of isometheptene mucate, dichloralphenazone with acetaminophen and sumatriptan succinate in the treatment of migraine. Headache. 2001;41:391–398. doi: 10.1046/j.1526-4610.2001.111006391.x. [DOI] [PubMed] [Google Scholar]
- 7.Pecenco G.L., Apollo A., Bacciardi M. Sulphenazone in pediatric practice. Case studies, Minerva Pediatr. 1982;34:39–43. [PubMed] [Google Scholar]
- 8.Himly M., Jahn-Schmid B., Pittertschatscher K., Bohle B., Grubmayr K., Ferreira F., Ebner H., Ebner C. IgE-mediated immediate-type hypersensitivity to the pyrazolone drug propyphenazone. J. Allergy Clin. Immunol. 2003;111:882–888. doi: 10.1067/mai.2003.163. [DOI] [PubMed] [Google Scholar]
- 9.Hart F.D., Boardman P.L. Trial of nifenazone ("THYLIN") Br. Med. J. 1964;1:1553–1554. doi: 10.1136/bmj.1.5397.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casas J.S., Garcia-Tasende M.S., Sanchez A., Sordo J., Touceda A. Coordination modes of 5-pyrazolones: a solid-state overview. Coord. Chem. Rev. 2007;251:1561–1589. [Google Scholar]
- 11.Xie X., Xiang L., Peng C., Han B. Catalytic asymmetric synthesis of spiropyrazolones and their application in medicinal chemistry. Chem. Rec. 2019;19:1–28. doi: 10.1002/tcr.201800199. [DOI] [PubMed] [Google Scholar]
- 12.Hamama W.S., El-Gohary H.G., Kuhnert N., Zoorob H.H. Chemistry of pyrazolinones and their applications. Curr. Org. Chem. 2012;16:373–399. [Google Scholar]
- 13.Liu S., Bao X., Wang B. Pyrazolone: a powerful synthon for asymmetric diverse derivatizations. Chem. Commun. 2018;54:11515–11529. doi: 10.1039/c8cc06196c. [DOI] [PubMed] [Google Scholar]
- 14.Franci G., Falanga A., Galdiero S., Palomba L., Rai M., Morelli G., Galdiero M. Silver nanoparticles as potential antibacterial agents. Molecules. 2015;20:8856–8874. doi: 10.3390/molecules20058856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cukurovali A., Yilmaz I., Gur S., Kazaz C. Synthesis, antibacterial and antifungal activity of some new thiazolylhydrazone derivatives containing 3-substituted cyclobutane ring. Eur. J. Med. Chem. 2006;41:201–207. doi: 10.1016/j.ejmech.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Chaudhary A.S. A review of global initiatives to fight antibiotic resistance and recent antibiotics’ discovery. Acta Pharm. Sin. B. 2016;6:552–556. doi: 10.1016/j.apsb.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang H., Xiong M., Bi Q., Wang Y., Li C. Self-enhanced targeted delivery of a cell wall- and membrane-active antibiotics, daptomycin, against staphylococcal pneumonia. Acta Pharm. Sin. B. 2016;6:319–328. doi: 10.1016/j.apsb.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khalil N.S.A.M. A facile synthesis, structure, and antimicrobial evaluation of novel 4-arylhydrazono-5-trifluoromethyl-2,4-dihydropyrazol-3-ones, their N- and N,O-bis-beta-D-glucosides. Carbohydr. Res. 2009;344:1654–1659. doi: 10.1016/j.carres.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Guo Z. Discovery of imrecoxib. Chin. J. N. Drugs. 2012;21:223–230. [Google Scholar]
- 20.Padmavathi V., Subbaiah D.R.C.V., Mahesh K., Lakshmi T.R. Synthesis and bioassay of amino-pyrazolone, amino-isoxazolone and amino-pyrimidinone derivatives. Chem. Pharm. Bull. 2007;55:1704–1709. doi: 10.1248/cpb.55.1704. [DOI] [PubMed] [Google Scholar]
- 21.Aly H.M., Saleh N.M., Elhady H.A. Design and synthesis of some new thiophene, thienopyrimidine and thienothiadiazine derivatives of antipyrine as potential antimicrobial agents. Eur. J. Med. Chem. 2011;46:4566–4572. doi: 10.1016/j.ejmech.2011.07.035. [DOI] [PubMed] [Google Scholar]
- 22.Hamama W.S., El-Gohary H.G., Soliman M., Zoorob H.H. A versatile synthesis, PM3-semiempirical, antibacterial, and antitumor evaluation of some bioactive pyrazoles. J. Heterocycl. Chem. 2012;49:543–554. [Google Scholar]
- 23.Rasapalli S., Fan Y., Yu M., Rees C., Harris J.T., Golen J.A., Jasinski J.P., Rheingold A.L., Kwasny S.M., Opperman T.J. Detour of prenostodione synthesis towards pyrazolones for antibacterial activity. Bioorg. Med. Chem. Lett. 2013;23:3235–3238. doi: 10.1016/j.bmcl.2013.03.123. [DOI] [PubMed] [Google Scholar]
- 24.Gadhave A., Kuchekar S., Karale B. Ultrasonication-induced synthesis and antimicrobial evaluation of some multifluorinated pyrazolone derivatives. J. Chem. Neuroanat. 2013:741953–741962. [Google Scholar]
- 25.Narayana Rao D.V., Raghavendra Guru Prasad A., Spoorthy Y.N., Raghunatha Rao D., Ravindranath L.K. In vitro microbiological evaluation of novel bis pyrazolones. Ann. Pharm. Fr. 2014;72:101–106. doi: 10.1016/j.pharma.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 26.Reheim M.A.M.A., Baker S.M. Synthesis, characterization and in vitro antimicrobial activity of novel fused pyrazolo[3,4-c]pyridazine, pyrazolo[3,4-d]pyrimidine, thieno[3,2-c]pyrazole and pyrazolo[3’,4’:4,5]thieno[2,3-d]pyrimidine derivatives. Chem. Cent. J. 2017;11:112–123. doi: 10.1186/s13065-017-0339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alkhaldi A.A.M., Abdelgawad M.A., Youssif B.G.M. Synthesis, antimicrobial evaluation and docking studies of new pyrazolone derivatives. Trop. J. Pharm. Res. 2018;17:2235–2241. [Google Scholar]
- 28.Bhattacharjee D., Sheet S.K., Khatua S., Biswas K., Joshi S., Myrboh B. A reusable magnetic nickel nanoparticle based catalyst for the aqueous synthesis of diverse heterocycles and their evaluation as potential anti-bacterial agent. Bioorg. Med. Chem. 2018;26:5018–5028. doi: 10.1016/j.bmc.2018.08.033. [DOI] [PubMed] [Google Scholar]
- 29.Akondi A.M., Kantam M.L., Trivedi R., Bharatam J., Vemulapalli S.P.B., Bhargava S.K., Buddana S.K., Prakasham R.S. Ce/SiO2 composite as an efficient catalyst for the multicomponent one-pot synthesis of substituted pyrazolones in aqueous media and their antimicrobial activities. J. Mol. Catal. A Chem. 2016;411:325–336. [Google Scholar]
- 30.Sayed G.H., Azab M.E., Anwer K.E., Raouf M.A., Negm N.A. Pyrazole, pyrazolone and enaminonitrile pyrazole derivatives: synthesis, characterization and potential in corrosion inhibition and antimicrobial applications. J. Mol. Liq. 2018;252:329–338. [Google Scholar]
- 31.Shamsuzzaman, Mashrai A., Ahmad A., Dar A.M., Khanam H., Danishuddin M., Khan A.U. Synthesis, evaluation and docking studies on steroidal pyrazolones as anticancer and antimicrobial agents. Med. Chem. Res. 2014;23:348–362. [Google Scholar]
- 32.Radini I.A.M. Design, synthesis, and antimicrobial evaluation of novel pyrazoles and pyrazolyl 1,3,4-thiadiazine derivatives. Molecules. 2018;23:2092–3003. doi: 10.3390/molecules23092092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bihani M., Bora P.P., Verma A.K., Baruah R., Boruah H.P.D., Bez G. PPL catalyzed four-component PASE synthesis of 5-monosubstituted barbiturates: structure and pharmacological properties. Bioorg. Med. Chem. Lett. 2015;25:5732–5736. doi: 10.1016/j.bmcl.2015.10.088. [DOI] [PubMed] [Google Scholar]
- 34.Oraby A.K., Abdellatif K.R.A., Abdelgawad M.A., Attia K.M., Dawe L.N., Georghiou P.E. 2,4-Disubstituted phenylhydrazonopyrazolone and isoxazolone derivatives as antibacterial agents: synthesis, preliminary biological evaluation and docking studies. ChemistrySelect. 2018;3:3295–3301. [Google Scholar]
- 35.Li X.-H., Lee J.-H. Antibiofihn agents: a new perspective for antimicrobial strategy. J. Microbiol. 2017;55:753–766. doi: 10.1007/s12275-017-7274-x. [DOI] [PubMed] [Google Scholar]
- 36.Gadd G.M. Metals, minerals and microbes: geomicrobiology and bioremediation. Microbiol. Sgm. 2010;156:609–643. doi: 10.1099/mic.0.037143-0. [DOI] [PubMed] [Google Scholar]
- 37.Mahapatra D.K., Bharti S.K., Asati V., Singh S.K. Perspectives of medicinally privileged chalcone based metal coordination compounds for biomedical applications. Eur. J. Med. Chem. 2019;174:142–158. doi: 10.1016/j.ejmech.2019.04.032. [DOI] [PubMed] [Google Scholar]
- 38.Selvaraj S., Krishnaswamy S., Devashya V., Sethuraman S., Krishnan U.M. Flavonoid-metal ion complexes: a novel class of therapeutic agents. Med. Res. Rev. 2014;34:677–702. doi: 10.1002/med.21301. [DOI] [PubMed] [Google Scholar]
- 39.Marchetti F., Pettinari C., Pettinari R., Cingolani A., Leonesi D., Lorenzotti A. Group 12 metal complexes of tetradentate N 2 O 2 –Schiff-base ligands incorporating pyrazole : synthesis, characterisation and reactivity toward S-donors, N-donors, copper and tin acceptors. Polyhedron. 1999;18:3041–3050. [Google Scholar]
- 40.Modi C.K., Jani D.H. Mn(III) mixed-ligand complexes with bis-pyrazolones and ciprofloxacin drug: synthesis, characterization and antibacterial activities. Appl. Organomet. Chem. 2011;25:429–436. [Google Scholar]
- 41.Jadeja R.N., Vyas K.M., Gupta V.K., Joshi R.G., Prabha C.R. Syntheses, characterization and molecular structures of calcium(II) and copper(II) complexes bearing O-2-chelate ligands: DNA binding, DNA cleavage and anti-microbial study. Polyhedron. 2012;31:767–778. [Google Scholar]
- 42.Joseph V.A., Pandya J.H., Jadeja R.N. Syntheses, crystal structure and biological evaluation of Schiff bases and copper complexes derived from 4-formylpyrazolone. J. Mol. Struct. 2015;1081:443–448. [Google Scholar]
- 43.Lunagariya M.V., Thakor K.P., Kanthecha D.N., Patel M.N. Synthesis, characterization and biological applications of substituted pyrazolone core based platinum(II) organometallic compounds. J. Organomet. Chem. 2018;854:49–63. [Google Scholar]
- 44.Nair M.S., Arish D., Johnson J. Synthesis, characterization and biological studies on some metal complexes with Schiff base ligand containing pyrazolone moiety. J. Saudi Chem. Soc. 2016;20:S591–S598. [Google Scholar]
- 45.Bray F., Jemal A., Grey N., Ferlay J., Forman D. Global cancer transitions according to the Human Development Index (2008–2030): a population-based study. Lancet Oncol. 2012;13:790–801. doi: 10.1016/S1470-2045(12)70211-5. [DOI] [PubMed] [Google Scholar]
- 46.Braña M.F., Gradillas A., Ovalles A.G., López B., Acero N., Llinares F., Mingarro D.M. Synthesis and biological activity of N,N-dialkylaminoalkyl-substituted bisindolyl and diphenyl pyrazolone derivatives. Bioorg. Med. Chem. 2006;14:9–16. doi: 10.1016/j.bmc.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 47.Masabumi S., Lena C.W. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 48.Shan Y., Wang B., Zhang J. New strategies in achieving antiangiogenic effect: multiplex inhibitors suppressing compensatory activations of RTKs. Med. Res. Rev. 2018;38:1674–1705. doi: 10.1002/med.21517. [DOI] [PubMed] [Google Scholar]
- 49.Tripathy R., Reiboldt A., Messina P.A., Iqbal M., Singh J., Bacon E.R., Angeles T.S., Yang S.X., Albom M.S., Robinson C. Structure-guided identification of novel VEGFR-2 kinase inhibitors via solution phase parallel synthesis. Bioorg. Med. Chem. Lett. 2006;16:2158–2162. doi: 10.1016/j.bmcl.2006.01.063. [DOI] [PubMed] [Google Scholar]
- 50.Tripathy R., Mchugh R.J., Ghose A.K., Ott G.R., Angeles T.S., Albom M.S., Zeck H., Aimone L.D., Mangeng C., Dorsey B.D. Pyrazolone-based anaplastic lymphoma kinase (ALK) inhibitors: control of selectivity by a benzyloxy group. Bioorg. Med. Chem. Lett. 2011;21:7261–7264. doi: 10.1016/j.bmcl.2011.10.055. [DOI] [PubMed] [Google Scholar]
- 51.Gu W., Dai Y., Hao Q., Wei S., Chen L., Zhao F., Huang W., Hai Q. Discovery of novel 2-substituted-4-(2-fluorophenoxy) pyridine derivatives possessing pyrazolone and triazole moieties as dual c-Met/VEGFR-2 receptor tyrosine kinase inhibitors. Bioorg. Chem. 2017;72:116–122. doi: 10.1016/j.bioorg.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 52.Norman M.H., Longbin L., Matthew L., Ning X., Ingrid F., D’Angelo N.D., Celia D., Karen R., Bellon S.F., Tae-Seong K. Structure-based design of novel class II c-Met inhibitors: 1. Identification of pyrazolone-based derivatives. J. Med. Chem. 2012;55:1858–1867. doi: 10.1021/jm201330u. [DOI] [PubMed] [Google Scholar]
- 53.Longbin L., Norman M.H., Matthew L., Ning X., Aaron S., Boezio A.A., Shon B., Debbie C., D’Angelo N.D., Julie G. Structure-based design of novel class II c-Met inhibitors: 2. SAR and kinase selectivity profiles of the pyrazolone series. J. Med. Chem. 2012;55:1868–1897. doi: 10.1021/jm201331s. [DOI] [PubMed] [Google Scholar]
- 54.Zhou S., Ren J., Liu M., Ren L., Liu Y., Gong P. Design, synthesis and pharmacological evaluation of 6,7-disubstituted-4-phenoxyquinoline derivatives as potential antitumor agents. Bioorg. Chem. 2014;57:30–42. doi: 10.1016/j.bioorg.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 55.Mahajan S.S., Scian M., Sripathy S., Posakony J., Lao U., Loe T.K., Leko V., Thalhofer A., Schuler A.D., Bedalov A., Simon J.A. Development of pyrazolone and isoxazol-5-one cambinol analogues as Sirtuin inhibitors. J. Med. Chem. 2014;57:3283–3294. doi: 10.1021/jm4018064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y., Wang C., Huang W., Haruehanroengra P., Peng C., Sheng J., Han B., He G. Application of organocatalysis in bioorganometallic chemistry: asymmetric synthesis of multifunctionalized spirocyclic pyrazolone-ferrocene hybrids as novel RalA inhibitors. Org. Chem. Front. 2018;5:2229–2233. [Google Scholar]
- 57.Markovic V., Eric S., Stanojkovic T., Gligorijevic N., Arandelovic S., Todorovic N., Trifunovic S., Manojlovic N., Jelic R., Joksovic M.D. Antiproliferative activity and QSAR studies of a series of new 4-aminomethylidene derivatives of some pyrazol-5-ones. Bioorg. Med. Chem. Lett. 2011;21:4416–4421. doi: 10.1016/j.bmcl.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 58.Ghorab M.M., El-Gazzar M.G., Alsaid M.S. Synthesis, characterization and anti-breast cancer activity of new 4-aminoantipyrine-based heterocycles. Int. J. Mol. Sci. 2014;15:7539–7553. doi: 10.3390/ijms15057539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dube P.N., Bule S.S., Ushir Y.V., Kumbhare M.R., Dighe P.R. Synthesis of novel 5-methyl pyrazol-3-one derivatives and their in vitro cytotoxic evaluation. Med. Chem. Res. 2015;24:1070–1076. [Google Scholar]
- 60.Gouda M.A., Eldien H.F., Girges M.M., Berghot M.A. Synthesis and antitumor evaluation of thiophene based azo dyes incorporating pyrazolone moiety. J. Saudi. Chem. Soc. 2016;20:151–157. [Google Scholar]
- 61.Vyas K.M., Jadeja R.N., Patel D., Devkar R.V., Gupta V.K. A new pyrazolone based ternary Cu(II) complex: synthesis, characterization, crystal structure, DNA binding, protein binding and anti-cancer activity towards A549 human lung carcinoma cells with a minimum cytotoxicity to non-cancerous cells. Polyhedron. 2013;65:262–274. [Google Scholar]
- 62.Zhao B., Shang X., Xu L., Zhang W., Xiang G. Novel mixed ligand di-n-butyltin(IV) complexes derived from acylpyrazolones and fluorinated benzoic acids: synthesis, characterization, cytotoxicity and the induction of apoptosis in Hela cancer cells. Eur. J. Med. Chem. 2014;76:87–97. doi: 10.1016/j.ejmech.2014.02.039. [DOI] [PubMed] [Google Scholar]
- 63.Bakr E.A., Al-Hefnawy G.B., Awad M.K., Abd-Elatty H.H., Youssef M.S. New Ni(II), Pd(II) and Pt(II) complexes coordinated to azo pyrazolone ligand with a potent anti-tumor activity: synthesis, characterization, DFT and DNA cleavage studies. Appl. Organomet. Chem. 2018;32:e4104–e4121. [Google Scholar]
- 64.Zhang Y., Li Y., Xu G.C., Li J., Luo H., Li J., Zhang L., Jia D. Synthesis, crystal structure, DNA/bovine serum albumin binding and antitumor activity of two transition metal complexes with 4-acylpyrazolone derivative. Appl. Organomet. Chem. 2019;33:e4668–e4680. [Google Scholar]
- 65.Silva A.R., Grosso C., Delerue-Matos C., Rocha J.M. Comprehensive review on the interaction between natural compounds and brain receptors: benefits and toxicity. Eur. J. Med. Chem. 2019;174:87–115. doi: 10.1016/j.ejmech.2019.04.028. [DOI] [PubMed] [Google Scholar]
- 66.Zhao Z., He X., Ma C., Wu S., Cuan Y., Sun Y., Bai Y., Huang L., Chen X., Gao T., Zheng X. Excavating anticonvulsant compounds from prescriptions of Traditional Chinese Medicine in the treatment of epilepsy. Am. J. Chin. Med. 2018:1–31. doi: 10.1142/S0192415X18500374. [DOI] [PubMed] [Google Scholar]
- 67.Eldebss T.M.A., Yi X.J., Farag A.M., Khedr A.A., Abdulla M.M., Mabkhot Y.N. Synthesis of new pyrazolone-based heterocycles as inhibitors of monoamine oxidase enzymes. J. Iran. Chem. Soc. 2018;15:1785–1800. [Google Scholar]
- 68.Viveka S., Dinesha, Shama P., Naveen S., Lokanath N.K., Nagaraja G.K. Design, synthesis, anticonvulsant and analgesic studies of new pyrazole analogues: a Knoevenagel reaction approach. RSC Adv. 2015;5:94786–94795. [Google Scholar]
- 69.Abdel-Aziz M., Abuo-Rahma G.E.-D.A., Hassan A.A. Synthesis of novel pyrazole derivatives and evaluation of their antidepressant and anticonvulsant activities. Eur. J. Med. Chem. 2009;44:3480–3487. doi: 10.1016/j.ejmech.2009.01.032. [DOI] [PubMed] [Google Scholar]
- 70.Merugumolu V.K., Chandrashekarappa R.B. Synthesis and anti-depressant evaluation of novel pyrazolone derivatives. Bangladesh J. Pharmacol. 2016;11:558–563. [Google Scholar]
- 71.Bruijn L.I., Miller T.M., Cleveland D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 72.Cronin S., Hardiman O., Traynor B.J. Ethnic variation in the incidence of ALS - a systematic review. Neurology. 2007;68:1002–1007. doi: 10.1212/01.wnl.0000258551.96893.6f. [DOI] [PubMed] [Google Scholar]
- 73.Benmohamed R., Arvanites A.C., Kim J., Ferrante R.J., Silverman R.B., Morimoto R.I., Kirsch D.R. Identification of compounds protective against G93A-SOD1 toxicity for the treatment of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:87–96. doi: 10.3109/17482968.2010.522586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen T., Benmohamed R., Kim J., Smith K., Amante D., Morimoto R.I., Kirsch D.R., Ferrante R.J., Silverman R.B. ADME-Guided design and synthesis of aryloxanyl pyrazolone derivatives to block mutant superoxide dismutase 1 (SOD1) cytotoxicity and protein aggregation: potential application for the treatment of Amyotrophic Lateral Sclerosis. J. Med. Chem. 2012;55:515–527. doi: 10.1021/jm2014277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tok F., Kocyigit-Kaymakcioglu B., Saglik B.N., Levent S., Ozkay Y., Kaplancikli Z.A. Synthesis and biological evaluation of new pyrazolone Schiff bases as monoamine oxidase and cholinesterase inhibitors. Bioorg. Chem. 2019;84:41–50. doi: 10.1016/j.bioorg.2018.11.016. [DOI] [PubMed] [Google Scholar]
- 76.Johnston G.A. GABAA receptor pharmacology. Pharmacol. Ther. 1996;69:173–198. doi: 10.1016/0163-7258(95)02043-8. [DOI] [PubMed] [Google Scholar]
- 77.Solomon V.R., Tallapragada V.J., Chebib M., Johnston G.A.R., Hanrahan J.R. GABA allosteric modulators: an overview of recent developments in non-benzodiazepine modulators. Eur. J. Med. Chem. 2019;171:434–461. doi: 10.1016/j.ejmech.2019.03.043. [DOI] [PubMed] [Google Scholar]
- 78.Hintermann S., Hurth K., Nozulak J., Tintelnot-Blomley M., Aichholz R., Blanz J., Kaupmann K., Mosbacher J. Exploring subtype selectivity and metabolic stability of a novel series of ligands for the benzodiazepine binding site of the GABA(A) receptor. Bioorg. Med. Chem. Lett. 2011;21:1523–1526. doi: 10.1016/j.bmcl.2010.12.107. [DOI] [PubMed] [Google Scholar]
- 79.Kypta R.M. GSK-3 inhibitors and their potential in the treatment of Alzheimer’s disease. Expert Opin. Ther. Pat. 2005;15:1315–1331. [Google Scholar]
- 80.Golpich M., Amini E., Hemmati F., Ibrahim N.M., Rahmani B., Mohamed Z., Raymond A.A., Dargahi L., Ghasemi R., Ahmadiani A. Glycogen synthase kinase-3 beta (GSK-3 beta) signaling: implications for Parkinson’s disease. Pharmacol. Res. 2015;97:16–26. doi: 10.1016/j.phrs.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 81.Alonso M., Martinez A. GSK-3 inhibitors: discoveries and developments. Curr. Med. Chem. 2004;11:755–763. doi: 10.2174/0929867043455738. [DOI] [PubMed] [Google Scholar]
- 82.Chen W., Gaisina I.N., Gunosewoyo H., Malekiani S.A., Hanania T., Kozikowski A.P. Structure-guided design of a highly selective glycogen synthase kinase-3 beta inhibitor: a superior neuroprotective pyrazolone showing antimania effects. ChemMedChem. 2011;6:1587–1592. doi: 10.1002/cmdc.201100231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Martinez A., Perez D.I. GSK-3 Inhibitors: a ray of hope for the treatment of Alzheimer’s disease? J. Alzheimer’s Dis. 2008;15:181–191. doi: 10.3233/jad-2008-15204. [DOI] [PubMed] [Google Scholar]
- 84.Martinez A., Alonso M., Castro A., Dorronsoro I., Gelpí J.L., Luque F.J., Pérez C., Moreno F.J. SAR and 3D-QSAR studies on thiadiazolidinone derivatives: exploration of structural requirements for glycogen synthase kinase 3 inhibitors. J. Med. Chem. 2005;48:7103–7112. doi: 10.1021/jm040895g. [DOI] [PubMed] [Google Scholar]
- 85.Zimmermann D., Janin Y.L., Brehm L., Brauner-Osborne H., Ebert B., Johansen T.N., Madsen U., Krogsgaard-Larsen P. 3-Pyrazolone analogues of the 3-isoxazolol metabotropic excitatory amino acid receptor agonist homo-AMPA. Synthesis and pharmacological testing. Eur. J. Med. Chem. 1999;34:967–976. doi: 10.1016/s0223-5234(99)00122-1. [DOI] [PubMed] [Google Scholar]
- 86.Brune K. The early history of non-opioid analgesics. Acute Pain. 1997;1:33–40. [Google Scholar]
- 87.Ochiai K., Takita S., Kojima A., Eiraku T., Ando N., Iwase K., Kishi T., Ohinata A., Yageta Y., Yasue T., Adams D.R., Kohno Y. Phosphodiesterase inhibitors. Part 4: design, synthesis and structure-activity relationships of dual PDE3/4-inhibitory fused bicyclic heteroaromatic-4,4-dimethylpyrazolones. Bioorg. Med. Chem. Lett. 2012;22:5833–5838. doi: 10.1016/j.bmcl.2012.07.088. [DOI] [PubMed] [Google Scholar]
- 88.Ahmed E.M., Kassab A.E., El-Malah A.A., Hassan M.S.A. Synthesis and biological evaluation of pyridazinone derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents. Eur. J. Med. Chem. 2019;171:25–37. doi: 10.1016/j.ejmech.2019.03.036. [DOI] [PubMed] [Google Scholar]
- 89.Lamie P.F., Ali W.A.M., Bazgier V., Rarova L. Novel N-substituted indole Schiff bases as dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase enzymes: synthesis, biological activities in vitro and docking study. Eur. J. Med. Chem. 2016;123:803–813. doi: 10.1016/j.ejmech.2016.08.013. [DOI] [PubMed] [Google Scholar]
- 90.Park C.H., Siomboing X., Yous S., Gressier B., Luyckx M., Chavatte P. Investigations of new lead structures for the design of novel cyclooxygenase-2 inhibitors. Eur. J. Med. Chem. 2002;37:461–468. doi: 10.1016/s0223-5234(02)01373-9. [DOI] [PubMed] [Google Scholar]
- 91.Somakala K., Amir M. Synthesis, characterization and pharmacological evaluation of pyrazolyl urea derivatives as potential anti-inflammatory agents. Acta Pharm. Sin. B. 2017;7:230–240. doi: 10.1016/j.apsb.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Antre R.V., Cendilkumar A., Goli D., Andhale G.S., Oswal R.J. Microwave assisted synthesis of novel pyrazolone derivatives attached to a pyrimidine moiety and evaluation of their anti-inflammatory, analgesic and antipyretic activities. Saudi Pharm. J. 2011;19:233–243. doi: 10.1016/j.jsps.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Khalil N.A., Ahmed E.M., Mohamed K.O., Nissan Y.M., Zaitone S.A.-B. Synthesis and biological evaluation of new pyrazolone-pyridazine conjugates as anti-inflammatory and analgesic agents. Bioorg. Med. Chem. 2014;22:2080–2089. doi: 10.1016/j.bmc.2014.02.042. [DOI] [PubMed] [Google Scholar]
- 94.Sivakumar K.K., Rajasekaran A., Senthilkumar P., Wattamwar P.P. Conventional and microwave assisted synthesis of pyrazolone Mannich bases possessing anti-inflammatory, analgesic, ulcerogenic effect and antimicrobial properties. Bioorg. Med. Chem. Lett. 2014;24:2940–2944. doi: 10.1016/j.bmcl.2014.04.067. [DOI] [PubMed] [Google Scholar]
- 95.Abbady M.S., Youssef M.S.K. Synthesis and biological activity of some new pyridines, pyrans, and indazoles containing pyrazolone moiety. Med. Chem. Res. 2014;23:3558–3568. [Google Scholar]
- 96.Dube P.N., Bule S.S., Mokale S.N., Kumbhare M.R., Dighe P.R., Ushir Y.V. Synthesis and biologic evaluation of substituted 5-methyl-2-phenyl-1H-pyrazol-3(2H)-one derivatives as selective COX-2 Inhibitors: molecular docking study. Chem. Biol. Drug Des. 2014;84:409–419. doi: 10.1111/cbdd.12324. [DOI] [PubMed] [Google Scholar]
- 97.Moneer A.A., Mohammed K.O., El-Nassan H.B. Synthesis of novel substituted thiourea and benzimidazole derivatives containing a pyrazolone ring as anti-inflammatory agents. Chem. Biol. Drug Des. 2016;87:784–793. doi: 10.1111/cbdd.12712. [DOI] [PubMed] [Google Scholar]
- 98.El Sayed M.T., El-Sharief M.A.M.S., Zarie E.S., Morsy N.M., Elsheakh A.R., Voronkov A., Berishvili V., Hassan G.S. Design, synthesis, anti-inflammatory activity and molecular docking of potential novel antipyrine and pyrazolone analogs as cyclooxygenase enzyme (COX) inhibitors. Bioorg. Med. Chem. Lett. 2018;28:952–957. doi: 10.1016/j.bmcl.2018.01.043. [DOI] [PubMed] [Google Scholar]
- 99.Abdelgawad M.A., Labib M.B., Ali W.A.M., Kamel G., Azouz A.A., El-Nahass E.L.S. Design, synthesis, analgesic, anti-inflammatory activity of novel pyrazolones possessing aminosulfonyl pharmacophore as inhibitors of COX-2/5-LOX enzymes: histopathological and docking studies. Bioorg. Chem. 2018;78:103–114. doi: 10.1016/j.bioorg.2018.03.011. [DOI] [PubMed] [Google Scholar]
- 100.Fahmy H.H., El-Eraky W. Synthesis and evaluation of the analgesic and antiinflammatory activities of O-substituted salicylamides. Arch Pharm. Res. (Seoul) 2001;24:171–179. doi: 10.1007/BF02978252. [DOI] [PubMed] [Google Scholar]
- 101.Golebiowski A., Townes J.A., Laufersweiler M.J., Brugel T.A., Clark M.P., Clark C.M., Djung J.F., Laughlin S.K., Sabat M.P., Bookland R.G., VanRens J.C., De B., Hsieh L.C., Janusz M.J., Walter R.L., Webster M.E., Mekel M.J. The development of monocyclic pyrazolone based cytokine synthesis inhibitors. Bioorg. Med. Chem. Lett. 2005;15:2285–2289. doi: 10.1016/j.bmcl.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 102.Sun-Waterhouse D., Chen J., Chuah C., Wibisono R., Melton L.D., Laing W., Ferguson L.R., Skinner M.A. Kiwifruit-based polyphenols and related antioxidants for functional foods: kiwifruit extract-enhanced gluten-free bread. Int. J. Food Sci. Nutr. 2009;60(Suppl 7):251–264. doi: 10.1080/09637480903012355. [DOI] [PubMed] [Google Scholar]
- 103.Higashi Yukihito. Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a novel free radical scavenger, for treatment of cardiovascular diseases. Recent Pat. Cardiovasc. Drug Discov. 2006;1:85–93. doi: 10.2174/157489006775244191. [DOI] [PubMed] [Google Scholar]
- 104.Alam M.S., Lee D.-U. Synthesis, molecular structure and antioxidant activity of (E)-4- Benzylideneamino -1,5-dimethyl-2-phenyl-1H-pyrazol-3(2H)-one, a Schiff Base ligand of 4-Aminoantipyrine. J. Chem. Crystallogr. 2012;42:93–102. [Google Scholar]
- 105.Khan K.M., Khan A., Taha M., Salar U., Hameed A., Ismail N.H., Jamil W., Saad S.M., Perveen S., Kashif S.M. Synthesis of 4-amino-1,5-dimethyl-2-phenylpyrazolone derivatives and their antioxidant activity. J. Chem. Soc. Pakistan. 2015;37:802–810. [Google Scholar]
- 106.Gaffer H.E., Abdel-Fattah S., Etman H.A., Abdel-Latif E. Synthesis and antioxidant activity of some new thiazolyl-pyrazolone derivatives. J. Heterocycl. Chem. 2017;54:331–340. [Google Scholar]
- 107.Makane V.B., Krishna V.S., Krishna E.V., Shukla M., Mahizhaveni B., Misra S., Chopra S., Sriram D., Dusthackeer V.N.A., Rode H.B. Synthesis and evaluation of alpha-aminoacyl amides as antitubercular agents effective on drug resistant tuberculosis. Eur. J. Med. Chem. 2019;164:665–677. doi: 10.1016/j.ejmech.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 108.Patel H., Jadhav H., Ansari I., Pawara R., Surana S. Pyridine and nitro-phenyl linked 1,3,4-thiadiazoles as MDR-TB inhibitors. Eur. J. Med. Chem. 2019;167:1–9. doi: 10.1016/j.ejmech.2019.01.073. [DOI] [PubMed] [Google Scholar]
- 109.Krishnasamy S.K., Namasivayam V., Mathew S., Eakambaram R.S., Ibrahim I.A., Natarajan A., Palaniappan S. Design, synthesis, and characterization of some hybridized pyrazolone pharmacophore analogs against Mycobacterium tuberculosis. Arch. Pharm. 2016;349:383–397. doi: 10.1002/ardp.201600019. [DOI] [PubMed] [Google Scholar]
- 110.Gunasekaran P., Perumal S., Yogeeswari P., Sriram D. A facile four-component sequential protocol in the expedient synthesis of novel 2-aryl-5-methyl-2,3-dihydro-1H-3-pyrazolones in water and their antitubercular evaluation. Eur. J. Med. Chem. 2011;46:4530–4536. doi: 10.1016/j.ejmech.2011.07.029. [DOI] [PubMed] [Google Scholar]
- 111.Castagnolo D., De Logu A., Radi M., Bechi B., Manetti F., Magnani M., Supino S., Meleddu R., Chisu L., Botta M. Synthesis, biological evaluation and SAR study of novel pyrazole analogues as inhibitors of Mycobacterium tuberculosis. Bioorg. Med. Chem. 2008;16:8587–8591. doi: 10.1016/j.bmc.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 112.Castagnolo D., Manetti F., Radi M., Bechi B., Pagano M., De Logu A., Meleddu R., Saddi M., Botta M. Synthesis, biological evaluation, and SAR study of novel pyrazole analogues as inhibitors of Mycobacterium tuberculosis: Part 2. Synthesis of rigid pyrazolones. Bioorg. Med. Chem. 2009;17:5716–5721. doi: 10.1016/j.bmc.2009.05.058. [DOI] [PubMed] [Google Scholar]
- 113.Sivakumar K.K., Rajasekharan A., Rao R., Narasimhan B. Synthesis, SAR study and evaluation of Mannich and Schiff bases of pyrazol-5(4H)-one moiety containing 3-(Hydrazinyl)-2-phenylquinazolin-4(3H)-one. Indian J. Pharm. Sci. 2013;75:463–475. doi: 10.4103/0250-474X.119832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ahsan M.J., Samy J.G., Khalilullah H., Nomani M.S., Saraswat P., Gaur R., Singh A. Molecular properties prediction and synthesis of novel 1,3,4-oxadiazole analogues as potent antimicrobial and antitubercular agents. Bioorg. Med. Chem. Lett. 2011;21:7246–7250. doi: 10.1016/j.bmcl.2011.10.057. [DOI] [PubMed] [Google Scholar]
- 115.Ahsan M.J., Samy J.G., Jain C.B., Dutt K.R., Khalilullah H., Nomani M.S. Discovery of novel antitubercular 1,5-dimethyl-2-phenyl-4-([5-(arylamino)-1,3,4-oxadiazol-2-yl]methylamino)-1,2-dih ydro-3H-pyrazol-3-one analogues. Bioorg. Med. Chem. Lett. 2012;22:969–972. doi: 10.1016/j.bmcl.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 116.Zhao Z., Song H., Xie J., Liu T., Zhao X., Chen X., He X., Wu S., Zhang Y., Zheng X. Research progress in the biological activities of 3,4,5-trimethoxycinnamic acid (TMCA) derivatives. Eur. J. Med. Chem. 2019;173:213–227. doi: 10.1016/j.ejmech.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fan X., Zhang X., Zhou L., Keith K.A., Kern E.R., Torrence P.F. A pyrimidine-pyrazolone nucleoside chimera with potent in vitro anti-orthopoxvirus activity. Bioorg. Med. Chem. Lett. 2006;16:3224–3228. doi: 10.1016/j.bmcl.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 118.Kimata A., Nakagawa H., Ohyama R., Fukuuchi T., Ohta S., Suzuki T., Miyata N. New series of antiprion compounds: pyrazolone derivatives have the potent activity of inhibiting protease-resistant prion protein accumulation. J. Med. Chem. 2007;50:5053–5056. doi: 10.1021/jm070688r. [DOI] [PubMed] [Google Scholar]
- 119.Srinivasan R., Narayana B., Sarojini B.K., Bhanuprakash V., Raj C.G.D., Nayak P.S. Design and synthesis of novel spiro-piperidinyl pyrazolone derivatives and their potential antiviral activity. Lett. Drug Des. Discov. 2016;13:149–160. [Google Scholar]
- 120.Kumar V., Tan K.P., Wang Y.M., Lin S.W., Liang P.H. Identification, synthesis and evaluation of SARS-CoV and MERS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2016;24:3035–3042. doi: 10.1016/j.bmc.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cassidy S., Syed B.A. Nonalcoholic steatohepatitis (NASH) drugs market. Nat. Rev. Drug Discov. 2016;15:745–746. doi: 10.1038/nrd.2016.188. [DOI] [PubMed] [Google Scholar]
- 122.Neuschwander-Tetri B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 123.Huang H., Yu Y., Gao Z., Zhang Y., Li C., Xu X., Jin H., Yan W., Ma R., Zhu J., Shen X., Jiang H., Chen L., Li J. Discovery and optimization of 1,3,4-trisubstituted-pyrazolone derivatives as novel, potent, and nonsteroidal farnesoid X receptor (FXR) selective antagonists. J. Med. Chem. 2012;55:7037–7053. doi: 10.1021/jm3002718. [DOI] [PubMed] [Google Scholar]
- 124.Wang Y.D., Chen W.D., Moore D.D., Huang W. FXR: a metabolic regulator and cell protector. Cell Res. 2008;18:1087–1095. doi: 10.1038/cr.2008.289. [DOI] [PubMed] [Google Scholar]
- 125.Fiorucci S., Mencarelli A., Palladino G., Cipriani S. Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol. Sci. 2009;30:570–580. doi: 10.1016/j.tips.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 126.Hardie D.G., Schaffer B.E., Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016;26:190–201. doi: 10.1016/j.tcb.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Henao-Mejia J., Elinav E., Jin C., Hao L., Mehal W.Z., Strowig T., Thaiss C.A., Kau A.L., Eisenbarth S.C., Jurczak M.J., Camporez J.-P., Shulman G.I., Gordon J.I., Hoffman H.M., Flavell R.A. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482 doi: 10.1038/nature10809. 179-U167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang M., Xie Z., Zhang R., Chen D., Gu M., Cui S., Zhang Y., Zhang X., Yu Y., Li J., Nan F., Li J. Novel substituted pyrazolone derivatives as AMP-activated protein kinase activators to inhibit lipid synthesis and reduce lipid accumulation in ob/ob mice. Acta Pharmacol. Sin. 2018;39:1622–1632. doi: 10.1038/aps.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kadam A., Dawane B., Pawar M., Shegokar H., Patil K., Meshram R., Gacche R. Development of novel pyrazolone derivatives as inhibitors of aldose reductase: an eco-friendly one-pot synthesis, experimental screening and in silico analysis. Bioorg. Chem. 2014;53:67–74. doi: 10.1016/j.bioorg.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 130.Kees K.L., Fitzgerald J.J., Steiner K.E., Mattes J.F., Mihan B., Tosi T., Mondoro D., McCaleb M.L. New potent antihyperglycemic agents in db/db mice: synthesis and structure-activity relationship studies of (4-substituted benzyl)(trifluoromethyl)pyrazoles and -pyrazolones. J. Med. Chem. 1996;39:3920–3928. doi: 10.1021/jm960444z. [DOI] [PubMed] [Google Scholar]
- 131.Eldebss T.M.A., Farag A.M., Abdalla M.M., Khedr A.A. Synthesis of some new pyrazolone-based heterocycles containing sulphone moiety acting as alpha-glucosidase and alpha-amylase inhibitors. J. Heterocycl. Chem. 2019;56:765–780. [Google Scholar]
- 132.Shetty S., Kalluraya B., Nithinchandra, Peethambar S.K., Telkar S.B. Type II diabetes-related enzyme inhibition and molecular modeling study of a novel series of pyrazolone derivatives. Med. Chem. Res. 2014;23:2834–2846. [Google Scholar]
- 133.Drizin I., Altenbach R.J., Buckner S.A., Whiteaker K.L., Scott V.E., Darbyshire J.F., Jayanti V., Henry R.F., Coghlan M.J., Gopalakrishnan M., Carroll W.A. Structure-activity studies for a novel series of tricyclic dihydropyridopyrazolones and dihydropyridoisoxazolones as K(ATP) channel openers. Bioorg. Med. Chem. 2004;12:1895–1904. doi: 10.1016/j.bmc.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 134.Amata E., Bland N.D., Hoyt C.T., Settimo L., Campbell R.K., Pollastri M.P. Repurposing human PDE4 inhibitors for neglected tropical diseases: design, synthesis and evaluation of cilomilast analogues as Trypanosoma brucei PDEB1 inhibitors. Bioorg. Med. Chem. Lett. 2014;24:4084–4089. doi: 10.1016/j.bmcl.2014.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bouteille B., Oukem O., Bisser S., Dumas M. Treatment perspectives for human African trypanosomiasis. Fundam. Clin. Pharmacol. 2003;17:171–181. doi: 10.1046/j.1472-8206.2003.00167.x. [DOI] [PubMed] [Google Scholar]
- 136.Orrling K.M., Jansen C., Vu X.L., Balmer V., Bregy P., Shanmugham A., England P., Bailey D., Cos P., Maes L., Adams E., van den Bogaart E., Chatelain E., Ioset J.-R., van de Stolpe A., Zorg S., Veerman J., Seebeck T., Sterk G.J., de Esch I.J.P., Leurs R. Catechol pyrazolinones as trypanocidals: fragment-based design, synthesis, and pharmacological evaluation of nanomolar inhibitors of trypanosomal phosphodiesterase B1. J. Med. Chem. 2012;55:8745–8756. doi: 10.1021/jm301059b. [DOI] [PubMed] [Google Scholar]
- 137.Amata E., Bland N.D., Campbell R.K., Pollastri M.P. Evaluation of pyrrolidine and pyrazolone derivatives as inhibitors of trypanosomal phosphodiesterase B1 (TbrPDEB1) Tetrahedron Lett. 2015;56:2832–2835. doi: 10.1016/j.tetlet.2015.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yi X.J., El-Idreesy T.T., Eldebss T.M.A., Farag A.M., Abdulla M.M., Hassan S.A., Mabkhot Y.N. Synthesis, biological evaluation, and molecular docking studies of new pyrazol-3-one derivatives with aromatase inhibition activities. Chem. Biol. Drug Des. 2016;88:832–843. doi: 10.1111/cbdd.12812. [DOI] [PubMed] [Google Scholar]
- 139.Cadieux J.A., Zhang Z., Mattice M., Brownlie-Cutts A., Fu J., Ratkay L.G., Kwan R., Thompson J., Sanghara J., Zhong J., Goldberg Y.P. Synthesis and biological evaluation of substituted pyrazoles as blockers of divalent metal transporter 1 (DMT1) Bioorg. Med. Chem. Lett. 2012;22:90–95. doi: 10.1016/j.bmcl.2011.11.069. [DOI] [PubMed] [Google Scholar]
- 140.Guckian K., Carter M.B., Lin E.Y., Choi M., Sun L., Boriack-Sjodin P.A., Chuaqui C., Lane B., Cheung K., Ling L., Lee W.C. Pyrazolone based TGFbetaR1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2010;20:326–329. doi: 10.1016/j.bmcl.2009.10.108. [DOI] [PubMed] [Google Scholar]
- 141.Bao X., Wei S., Qian X., Qu J., Wang B., Zou L., Ge G. Asymmetric construction of a multi-pharmacophore-containing dispirotriheterocyclic scaffold and identification of a human carboxylesterase 1 inhibitor. Org. Lett. 2018;20:3394–3398. doi: 10.1021/acs.orglett.8b01316. [DOI] [PubMed] [Google Scholar]
- 142.Hamada Y., Kiso Y. The application of bioisosteres in drug design for novel drug discovery: focusing on acid protease inhibitors. Expert Opin. Drug Discov. 2012;7:903–922. doi: 10.1517/17460441.2012.712513. [DOI] [PubMed] [Google Scholar]
- 143.Meanwell N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 144.Schneider G., Clark D.E. Automated de novo drug design: are we nearly there yet? Angew. Chem. Int. Ed. 2019;58:10792–10803. doi: 10.1002/anie.201814681. [DOI] [PubMed] [Google Scholar]
- 145.Panteleev J., Gao H., Jia L. Recent applications of machine learning in medicinal chemistry. Bioorg. Med. Chem. Lett. 2018;28:2807–2815. doi: 10.1016/j.bmcl.2018.06.046. [DOI] [PubMed] [Google Scholar]
- 146.Wang R., Xu K., Shi W. Quinolone derivatives: potential anti-HIV agent-development and application. Arch. Der Pharm. 2019;352 doi: 10.1002/ardp.201900045. [DOI] [PubMed] [Google Scholar]
- 147.Kumari A., Singh R.K. Medicinal chemistry of indole derivatives: current to future therapeutic prospectives. Bioorg. Chem. 2019;89 doi: 10.1016/j.bioorg.2019.103021. 103021. [DOI] [PubMed] [Google Scholar]
- 148.Song Y., Chen W., Kang D., Zhang Q., Zhan P., Liu X. "Old friends in new guise": exploiting privileged structures for scaffold re-evolution/refining. Comb. Chem. High Throughput Screen. 2014;17:536–553. doi: 10.2174/1386207317666140122101631. [DOI] [PubMed] [Google Scholar]
- 149.Song Y.n., Zhan P., Liu X. Heterocycle-thioacetic acid motif: a privileged molecular scaffold with potent, broad-ranging pharmacological activities. Curr. Pharmaceut. Des. 2013;19:7141–7154. doi: 10.2174/13816128113199990505. [DOI] [PubMed] [Google Scholar]
- 150.Song Y., Zhan P., Zhang Q., Liu X. Privileged scaffolds or promiscuous binders: a glance of pyrrolo[2,1-f][1,2,4]triazines and related bridgehead nitrogen heterocycles in medicinal chemistry. Curr. Pharmaceut. Des. 2013;19:1528–1548. [PubMed] [Google Scholar]
- 151.Ju H., Zhan P., Liu X. Designing influenza polymerase acidic endonuclease inhibitors via ’privileged scaffold’ re-evolution/refining strategy. Future Med. Chem. 2019;11:265–268. doi: 10.4155/fmc-2018-0489. [DOI] [PubMed] [Google Scholar]
- 152.Guo H., Eleftheriadis N., Rohr-Udilova N., Domling A., Dekker F.J. Photoactivation provides a mechanistic explanation for pan-assay interference behaviour of 2-aminopyrroles in lipoxygenase inhibition. Eur. J. Med. Chem. 2017;139:633–643. doi: 10.1016/j.ejmech.2017.07.047. [DOI] [PubMed] [Google Scholar]
- 153.Jesus A.R., Vila-Vicosa D., Machuqueiro M., Marques A.P., Dore T.M., Rauter A.P. Targeting type 2 diabetes with C-glucosyl dihydrochalcones as selective sodium glucose co-transporter 2 (SGLT2) inhibitors: synthesis and biological evaluation. J. Med. Chem. 2017;60:568–579. doi: 10.1021/acs.jmedchem.6b01134. [DOI] [PubMed] [Google Scholar]
- 154.Jia X., Feng L., Liu Y., Zhang L. Degradation behaviors and genetic toxicity variations of pyrazolone pharmaceuticals during chlorine dioxide disinfection process. Chem. Eng. J. 2018;345:156–164. [Google Scholar]