

Abstract
Indole represents one of the most important privileged scaffolds in drug discovery. Indole derivatives have the unique property of mimicking the structure of peptides and to bind reversibly to enzymes, which provide tremendous opportunities to discover novel drugs with different modes of action. There are seven indole-containing commercial drugs in the Top-200 Best Selling Drugs by US Retail Sales in 2012. There are also an amazing number of approved indole-containing drugs in the market as well as compounds currently going through different clinical phases or registration statuses. This review focused on the recent development of indole derivatives as antiviral agents with the following objectives: 1) To present one of the most comprehensive listings of indole antiviral agents, drugs on market or compounds in clinical trials; 2) To focus on recent developments of indole compounds (including natural products) and their antiviral activities, summarize the structure property, hoping to inspire new and even more creative approaches; 3) To offer perspectives on how indole scaffolds as a privileged structure might be exploited in the future.
Keywords: Indole, Antiviral activity, Entry and fusion inhibitor, Reverse transcriptase inhibitor, Integrase inhibitor, Protease inhibitor, Polymerase inhibitor, Natural product
Graphical abstract
The recent developments of indole compounds for antiviral drug discovery were comprehensively reviewed.
Highlights
-
•
The recent development of indole derivatives as antiviral agents was reviewed.
-
•
A comprehensive list of indole antiviral agents on market or in clinical trials was provided.
-
•
The future of indole-based antiviral agents was prospected.
List of abbreviations
- IAA
indole-3-acetic acid
- 5-HT
5-hydroxytryptamine
- I3C
indole-3-carbinol
- DIM
3,3′-diindolylmethane
- WHO
world health organization
- ED
erectile dysfunction
- OD
once-daily
- BPH
benign prostatic hyperplasia
- HIV
human immunodeficiency virus
- HCV
hepatitis C virus
- HSV
herpes simplex virus
- VSV
vesivular stomatitis virus
- H1N1
influenza virus A
- CV
cyanovinyl
- IE
immediate-early
- SARS
severe acute respiratory syndrome
- RSV
Respiratory Syncytial Virus
- HAART
highly active antiretroviral therapy
- MERS
middle east respiratory syndrome
- EVD
ebola virus disease
- EFVR
efavirenz-resistant
- HDAC
histone deacetylase
- ADME
absorption, distribution, metabolism, and excretion
- RBV
ribavirin
- SVR
sustained viral response
- NHR
N-terminal heptad repeat
- DKA
diketo acid
- RT
reverse transcriptase
- RTIs
reverse-transcriptase inhibitors
- NNRTIs
non-nucleoside reverse transcriptase inhibitors
- IAS
indolylarylsulfone
- INIs
integrase inhibitors
- PPIs
protein–protein interactions
- BVDV
Bovine viral diarrhea virus
- HRV
Human rhinovirus
- FDV
Fosdevirine
1. Introduction
Indole derivatives occur widely in natural products, existing in different kinds of plants, animals and marine organisms [1]. The indole core is a near-ubiquitous component of biologically active natural products. For example, indole-3-acetic acid (IAA) 1 (Fig. 1 ), one of the most common naturally-occurrings, is a plant hormone of the auxin class [2]; tryptophan 2, an essential amino acid, participates in many essential biological processes [3]; serotonin or 5-hydroxytryptamine (5-HT) 3, biochemically derived from tryptophan, is a neurotransmitter and is found in all bilateral animals [4]; melatonin 4, is a hormone found in animals, plants, and microbes, in which animals use the variation in duration of melatonin production each day as a seasonal clock [5]. The indole core is also well known as one of the most important scaffolds for drug discovery, and it has been a major focus of research for generations [6]. Biological studies of indole-3-carbinol (I3C) 5, and 3,3′-diindolylmethane (DIM) 6, also a natural product derived from the digestion of I3C which is found at relatively high levels in cruciferous vegetables such as broccoli, Brussels sprouts, cabbage and kale, have been the subjects of on-going research due to their interesting anticarcinogenic, antioxidant, and antiatherogenic effects [7], [8], [9], [10]; Ajmalicine 7 (also known as δ-yohimbine or raubasine), an indole alkaloid found naturally in various plants, is an antihypertensive drug used in the treatment of high blood pressure [11]. It acts as a α1-adrenergic receptor antagonist with preferential actions over α2-adrenergic receptors, underlying its hypotensive rather than hypertensive effects [12]. Reserpine 8, an indole alkaloid, is used to treat high blood pressure and severe agitation in patients with mental disorders [13]. Vinblastine 9, is used to treat several types of cancer, including Hodgkin's disease, Kaposi's sarcoma, non-Hodgkin's lymphoma, and cancer of the breast or testicles [14].
Fig. 1.
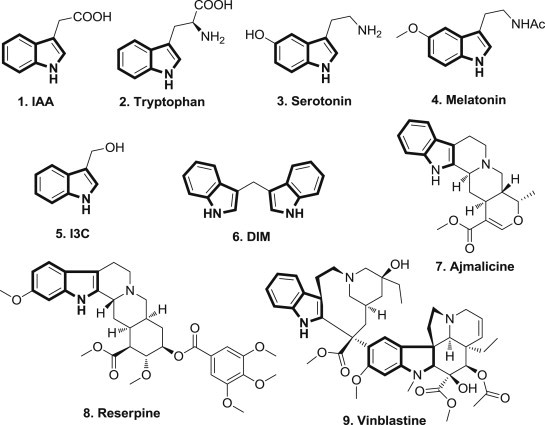
Structures of indole-containing natural products and drugs.
Indole represents one of the most important structural motifs in drug discovery, and it is described as one of the “privileged scaffolds”, a term first introduced by Evans and co-workers to define scaffolds which are capable of serving as ligand for a diverse array of receptors [15], [16], [17]. Indole derivatives have the unique property of mimicking the structure of peptides and to bind reversibly to enzymes [18], [19], [20], [21], which provide tremendous opportunities to discover novel drugs with different modes of action. There are seven indole-containing commercial drugs in the Top-200 Best Selling Drugs by US Retail Sales in 2012 [22]. This is highlighted by Cialis, an approved drug for the treatment of men's erectile dysfunction (ED), the signs and symptoms of benign prostatic hyperplasia (BPH), and both ED and the signs and symptoms of BPH [23], [24]. There are also an amazing number of approved indole-containing drugs in the market as well as compounds currently going through different clinical phases or registration statuses.
Viral diseases are extremely widespread infections. Some familiar viral diseases include common cold, influenza, chickenpox, herpes, gastroenteritis (stomach flu), human immunodeficiency virus (HIV/AIDS), hepatitis. Viral diseases can lead to serious, and potentially life-threatening complications, it is estimated that viral infections are responsible for more than 60% of the illnesses occurring in developed countries. In 2003, the severe acute respiratory syndrome (SARS) epidemic originated from southern China took the lives of nearly 800 people worldwide. Middle East Respiratory Syndrome (MERS) is a viral respiratory illness first reported in Saudi Arabia in 2012. It is caused by a corona virus called MERS-CoV. As of June, 2014, the World Health Organization (WHO) reported 699 cases of human infection with MERS, including at least 209 deaths. The most recent outbreak of Ebola virus disease (EVD) in West Africa in 2014 since its first appearance in 1976 has prompted WHO to declare international public health emergency. As of October, 2014, WHO reported that the current outbreak of Ebola has infected more than 7470 people and killed more than 3431 (Updated October 3, 2014). Antiviral drugs play an important role in fast-spreading epidemics; however almost all antivirals are subject to drug resistance as the pathogens mutate over time, becoming less susceptible to the treatment. Despite recent approvals of new antivirals in HIV and HCV therapeutic areas, there remain important unmet medical needs to improve upon the current therapy as well as those there exists no treatment.
Owing to the vast number of indole-containing molecules in the literature, this review focused primarily on antiviral agents, as limited reports were found in this area [25], [26]. This review serves as a comprehensive overview of currently published indole antiviral agents with the following objectives: 1) To present one of the most comprehensive listings of indole antiviral agents, drugs on market or compounds in clinical trials; 2) To focus on recent developments of indole compounds (including natural products) and their antiviral activities, summarize the structure property, hoping to inspire new and even more creative approaches; 3) To offer perspectives on how indole scaffolds as a privileged structure might be exploited in the future.
2. Indole antiviral agents: drugs on market or compounds in clinical trials
Indole scaffold is widely used in antiviral research. Examples of marketed indole-containing antiviral drugs include Arbidol and Delavirdine. Meanwhile, a number of indole derivatives are actively undergoing different phases of clinical evaluation, such as Atevirdine, GSK2248761 (IDX-12899), Golotimod, Panobinostat (LBH589), BILB 1941, BMS-791325, MK-8742 and Enfuvirtide (Fig. 2 ).
Fig. 2.
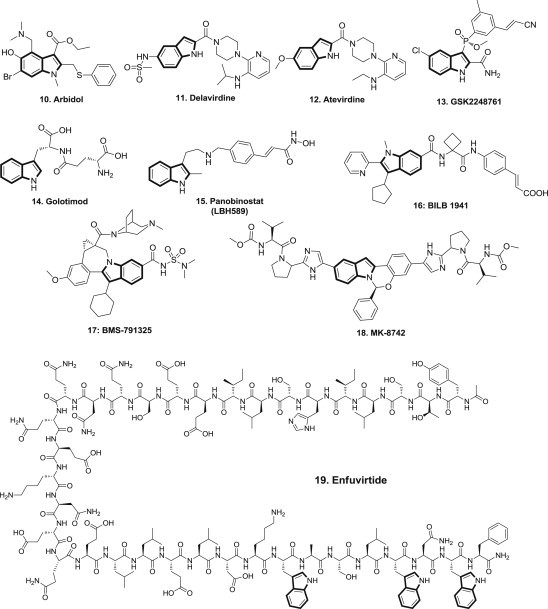
Structures of indole-containing commercial and on-study antiviral drugs.
Arbidol 10 (Umifenovir, Fig. 2) represents one of the most highly functionalized indole-containing drugs. Arbidol is a Russian developed broad spectrum antiviral which was widely used in Russia and China since 1990s. It is used for the treatment and prophylactic prevention of influenza A and B virus, respiratory syncytial virus, and SARS. It is Arbidol demonstrated both immunomodulating and anti-influenza effects, specifically against influenza groups A and B, and SARS [27], [28]. It prevents contact and entry of the virus into cells by inhibiting the fusion of viral lipid membranes with cell membranes. It has an interferon-inducing action, stimulates humoral and cell-mediated immunity, helps the phagocytic action of macrophages, and heightens the body's ability to fight infection [28]. It decreases the frequency of complications associated with viral infections, and lessens the effects of chronic bacterial illnesses. In additional, arbidol also showed in vitro and in vivo activities against a panel of human respiratory viruses including influenza A virus (FLU-A, A/PR/8/34H1N1), Respiratory Syncytial Virus (RSV), Human rhinovirus (HRV) 14, coxsackie virus B3 (CVB3), adenovirus type 7 (AdV-7) and HCV [29], [30], [31].
Delavirdine 11 (Rescriptor, Fig. 2) is a first generation non-nucleoside reverse transcriptase inhibitor (NNRTI) marketed by ViiV Healthcare. It was approved by FDA in 1997 for the treatment of human immunodeficiency virus type 1 (HIV-1). It is used as part of highly active antiretroviral therapy (HAART) [32]. Since then, better NNRTI such as efavirenz, and second generation NNRTIs such as etravirine and rilpivirine have been approved (http://aidsinfo.nih.gov/education-materials/fact-sheets/21/58/fda-approved-hiv-medicines). Delavirdine inhibits the CYP3A4-mediated metabolism of HIV protease inhibitors and thereby increases systemic exposure to protease inhibitors. The ability of delavirdine to enhance the pharmacokinetic profiles of protease inhibitors may permit the use of simplified administration regimens [33].
Atevirdine 12 (U-87201E, Fig. 2) is a new non-nucleoside (heteroarylpiperazine) reverse transcriptase inhibitor that has been studied for the treatment of HIV [34]. Atevirdine mesylate has been shown to have significant anti-HIV RT activity in vitro, it inhibits HIV-1 replication in infected peripheral blood leukocyte cultures at a 50% inhibitory concentration of 1 nM and a concentration which is cytotoxic to 50% of cells of 100 μM and also inhibits completely the formation of syncytia in human T-cell leukemia virus type III-infected MT-2 cells at 2 μM. Phase I study of atevirdine alone failed to demonstrate significant antiretroviral activity [35].
GSK2248761 (Fosdevirine, FDV, formerly IDX-12899) 13 (Fig. 2) is a novel, potent, selective, an once-daily (OD), next-generation nonnucleoside reverse-transcriptase inhibitor (NNRTI) with low nanomolar activity in vitro [36], [37]. GSK2248761 shows good activity against a broad range of HIV-1 strains, including efavirenz-resistant (EFVR) clinical isolates [38], [39]. GSK2248761 at 100–800 mg OD for 7 days was well tolerated, demonstrated potent antiviral activity in treatment-naive HIV-infected subjects, and had favorable PK and resistance profiles. The development of GSK2248761A was placed on clinical hold by the US Food and Drug Administration pending further evaluation of 5 reports of seizures in other studies of GSK2248761 in treatment-experienced patients [40].
Golotimod 14 (SCV-07, Fig. 2), an orally bioavailable synthetic peptide containing the amino acids d-glutamine and l-tryptophan connected by a gamma-glutamyl linkage with potential immunostimulating, antimicrobial and antineoplastic activities [41]. In 2010, SciClone Pharmaceuticals Inc. reported the results of phase 2b clinical trial of SCV-07 for the treatment of hepatitis C (HCV). The study evaluated the safety and immunomodulatory effects of SCV-07 as a monotherapy and in combination with ribavirin in relapsed HCV patients. The clinical data demonstrated SCV-07 to be safe and well-tolerated at both administered doses. Results showed a clear biological signal from SCV-07 but did not meet the study's primary efficacy endpoint of a 2 log reduction in viral load from baseline level. A phase 2 study is still on-going with SCV-07 in attenuating oral mucositis in subjects with head and neck cancer; however, no further study is listed for HCV [42].
Panobinostat (LBH589) 15 (Fig. 2) is an experimental drug developed by Novartis as a non-selective histone deacetylase inhibitor (HDAC inhibitor) for treatment of Multiple Myeloma (Phase III) and Acute Myeloid Leukemia (Phase II). The various HDAC inhibitors displayed significant potency differences in stimulating HIV-1 expression from the latently infected cell lines, and panobinostat was significantly more potent than all other HDAC inhibitors and induced virus production even at the very low concentration range (8–31 nM), proof was obtained that panobinostat induce virus production in latently infected primary cells at therapeutic concentrations, and it is currently being used in a Phase I/II clinical trial that aims at curing AIDS in patients on highly active antiretroviral therapy (HAART). In this technique, panobinostat is used to drive the HIV DNA out of the patient's DNA, in the expectation that the patient's immune system in combination with HAART will destroy it, this is the first proof of a viral “kick” leading to consistent plasma release of viral particles [43], [44].
BILB-1941 16 (Fig. 2), the first thumb pocket 1 NS5B inhibitor that demonstrated antiviral activity in patients chronically infected with genotype 1 HCV. Based on the discovery of allosteric (thumb pocket 1) non-nucleoside inhibitors of HCV NS5B polymerase that inhibit replication in replicon systems, and the identification of a metabolic liability common to many previously reported inhibitors in this series, a sparse matrix of indole-based inhibitors were generated that provided a collection of inhibitors satisfying potency criteria and displaying improved in vitro ADME profiles, and compound 16 provided the most optimal balance between antiviral potency and a consistent cross-species PK profile, it was selected for development followed by a clinical study in HCV-infected patients [45].
BMS-791325 17 (Fig. 2), a cyclopropyl-fused indolobenzazepine HCV NS5B RNA-dependent polymerase inhibitor, it inhibited cellar replication of HCV subgenomic replicons representing genotypes 1a and 1b at EC50 of 3 nM and 6 nM, respectively, it also exhibited notably enhanced pharmacokinetic profiles with improved solubility and membrane permeability, it was found to perform distinguishing antiviral, safety, and pharmacokinetic properties that resulted in its selection for clinical evaluation, Phase III studies are currently ongoing [46].
MK-8742 18 (Fig. 2), a tetracyclic indole-based NS5A inhibitor, which is currently in phase 2b clinical trials as part of an all-oral, interferon-free regimen for the treatment of HCV infection [47]. As NS5A protein plays a critical role in the replication of HCV and its inhibitors have shown impressive in vitro potency profiles in HCV replicon assays, making NS5A inhibitors attractive components for inclusion in all oral combination regimens. MK-8742, is a second generation NS5A inhibitor. In combination with MK-5172, an NS3/4A protease inhibitor, this candidate drug exhibited improvements in the genetic barrier while maintaining potency, yielding amazing results in terms of efficacy (90–100%), tolerability and safety, Phase II clinical trials are underway. In an interim analysis of treatment-naïve, non-cirrhotic patients administered a 12-week regimen of MK-5172/MK-8742, with and without ribavirin (RBV), a sustained viral response (SVR) was observed in 98 percent (42/43) of patients administered MK-5172/MK-8742 alone and 94 percent (75/80) in those administered MK-5172/MK-8742 plus RBV. This kind of interferon-free therapies is expected to lead the HCV treatment if the high cost is overcome [48], [49].
Enfuvirtide (T-20; brand name: Fuzeon) 19 (Fig. 2), the peptide anti-HIV drug targeting gp41N-terminal heptad repeat (NHR), was approved by the U.S. FDA in 2003 as the first HIV fusion/entry inhibitor for treatment of HIV/AIDS patients who fail to respond to the current antiretroviral drugs. However, because T20 lacks the pocket-binding domain, it exhibits low anti-HIV-1 activity and short half-life [50].
TMC647055 25 (Fig. 3 ), a nonzwitterionic 17-membered macrocyclic indole, has yielded potent and selective finger-loop inhibitors of the hepatitis C virus (HCV) NS5B polymerase [51]. Lead optimization from 20 to 25 in conjunction with in vivo evaluation in rats identified this compound showing nanomolar potency (EC50 = 77 nM) in HCV replicon cells, limited toxicity and off–target activities, and encouraging preclinical pharmacokinetic profiles characterized by high liver distribution, and it is currently being evaluated in phase II clinical trials in combination with simeprevir [52].
Fig. 3.
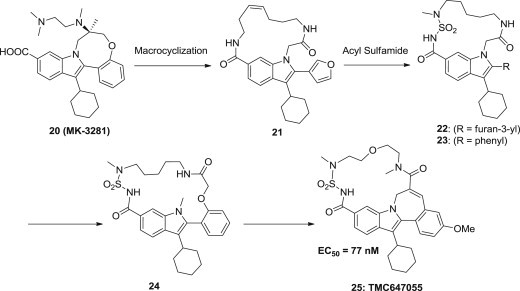
Optimization route of macrocyclic indole from MK-3281 to TMC647055.
3. Discovery of novel indole derivatives and their antiviral activity
The general idea of modern antiviral drug design is to identify viral proteins, or parts of proteins, that can be disabled. Generally speaking, these “targets” should be as unlike any proteins or parts of proteins in humans as possible, to reduce the likelihood of side effects. The targets should also be common across many strains of a virus, or even among different species of virus in the same family, that means a single drug will have broad effectiveness. Once targets are identified, candidate drugs can be selected, either from drugs already known to have appropriate effects, or by actually designing the candidate at the molecular level with a computer-aided design program.
3.1. Indole derivatives as entry and fusion inhibitor
As mentioned in the introduction, Arbidol acts as an inhibitor of virus entry and membrane fusion, and it is a broad-spectrum antiviral agent that inhibits acute and chronic HCV infection. Using Arbidol as a lead compound, Grazia Sellitto and co-workers reported the synthesis and evaluation of a series of ethyl 1H-indole-3-carboxylate derivatives 26–29 (Fig. 4 ) [28]. Compounds 26, 27 and 28 exhibited strong anti-HCV effects, while compound 27, 28 and 29 showed higher selectivity indices of inhibition on entry and replication. Of all the synthetic initial hits, compound 28 was the most potent candidate. This revealed that the elimination of the phenylsulfonyl moiety preserved the anti-HCV activity, and the removal of the 6-bromo and 5-hydroxy groups from the indole ring of Arbidol did not have an influence on its anti-HCV efficacy. This implies that the indole core rather than these groups might be the antiviral pharmacophore.
Fig. 4.
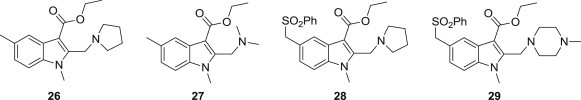
Structures of Arbidol derivatives.
BMS-378806 (32) is reported as a prototype of novel HIV attachment inhibitors that block the gp120 and CD4 interaction, the first step of HIV-1 entry into cells (Fig. 5 ). The initial screen hit was an indole analog 30, which interferes with the interaction of the HIV surface protein gp120 with the host cell receptor CD4, and the 4-fluoro derivative 31 exhibited markedly enhanced potency and was bioavailable in the rat, dog, and cynomolgus monkey when administered orally as a solution formulation. However, aqueous suspensions of 31 were poorly bioavailable, indicative of dissolution-limited absorption. The 7-azaindole derivative 32, BMS-378806, exhibited improved pharmaceutical properties while retaining the HIV-1 inhibitory profile of compound 31 [53], [54], [55].
Fig. 5.
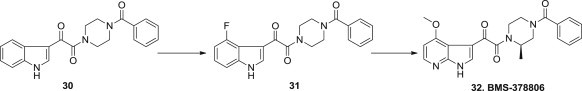
Optimization route for BMS-378806.
Seung Joo Cho and coworkers employed in silico methods such as molecular docking, MD simulations and three-dimensional quantitative structure-activity relationship (3D-QSAR), to guide the molecular design of new indole derivatives (Fig. 6 ). A data set of 68 indole-based inhibitors along with their biological activities was collected from the literatures [56], [57], [58]. In this approach, lowest energy conformation of the most potent compound 34 derived from systematic conformational search was considered as template for sketching the rest of the molecules. This study can also contribute to the better understanding of binding pose between these inhibitors and gp120 glycoprotein, and provide structural information affecting activity of these inhibitors and improve the understanding of ligand–receptor interactions, resulting in some useful and rational suggestions for further design of novel inhibitors for HIV/AIDS therapy [59].
Fig. 6.
Indole-containing inhibitors employed in 3D QSAR study.
As virus-cell fusion is the primary means by which the human immunodeficiency virus-1 (HIV) delivers its genetic material into the human T-cell host, fusion is mediated in large part by the viral glycoprotein 41 (gp41), and the hydrophobic pocket in the HIV-1 gp41 N-terminal heptad repeat (NHR) domain plays an important role in viral fusion and entry into the host cell, and serves as an attractive target for development of HIV-1 fusion/entry inhibitors. Robert C. Rizzo and co-workers reviewed current approaches to study the interactions between inhibitors and gp41, putting an emphasis on atomic-level computer modeling methods (Fig. 7 ). Compounds 35–38, having anti-fusion activity, were reported to interact in a specific manner with the binding site formed by the most conserved residues [50], [60]. For example, compound 35, discovered by virtual screening as a fusion inhibitor targeting gp41 [61], had high geometric overlap between its carboxylic acid and the position of Asp121 inside the binding pocket. Compound 36, discovered by a structure-based approach, exhibited inhibition constant of 2.1 μM and IC50 of 1.1 μM for cell–cell fusion inhibition [61]. 37 with a long hydrophobic interface showed submicromolar binding, fusion inhibition and blocking viral replication. The molecular simulations showed that 37 was able to adopt a structure mimicking the hydrophobic contacts of the D-peptide PIE7 [62]. 38 exhibited effective activity against HIV-1 Env-mediated cell–cell fusion with IC50 value of 1.3 μM, and bound in the hydrophobic pocket of gp41 to make a favorable interaction with Lys574 [63].
Fig. 7.
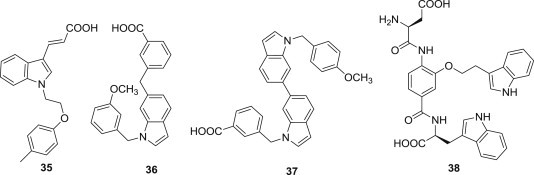
Indole-containing structures with gp41-binding activity.
3.2. Indoles as reverse transcriptase inhibitors
Reverse-transcriptase inhibitors (RTIs) are a class of antiretroviral drugs used to treat HIV infection or AIDS, and in some cases hepatitis B. RTIs inhibit the activity of reverse transcriptase, a viral DNA polymerase that is required for replication of HIV and other retroviruses. What's summarized below is the recent progress of discovery and design of indole derivatives as RTIs.
3.2.1. Indolylarylsulfones (IAS) as reverse transcriptase inhibitors
Compound 39 (L-737,126) (Fig. 8 ), an indolylarylsulfone (IAS) derivative endowed with high selectivity and potency against HIV-1 WT and Y181C mutant, developed by Merck in 1992 as a novel inhibitor of HIV reverse transcriptase. It is useful in the prevention or treatment of HIV infection and AIDS [64], [65]. The following development of indolylarylsulfones (IASs) NNRTIs was based on L-737,126 as the reference compound, there are mainly four structural modification sites, including the benzene group of SO2Ph, the sulfonyl, the amide and the substituent(s) of indole ring.
Fig. 8.
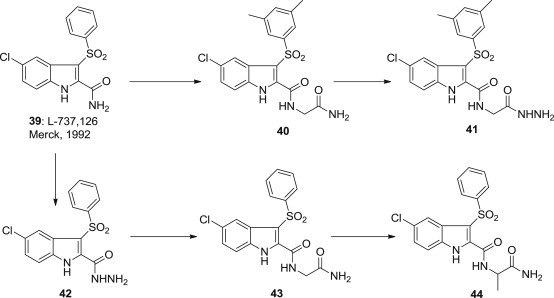
Optimization route of Sulfonylindolecarboxamide as NNRTI.
Romano Silvestri et al. [66] reported a series of indolylarylsulfone derivative 40–44 (Fig. 8) as analogues of L-737,126. These compounds were obtained by introducing two methyl groups at positions 3 and 5 of the benzenesulfonyl moiety 40–41 and coupling the glycinamide/alaninamide units to its carboxyamide function 42–44. The cell-based assays showed that these compounds exhibited higher potency against HIV-1 wild type and NNRTI-resistant mutants than the parent indole derivative 39.
Based on the previous work, Giuseppe La Regina and coworkers synthesized a series of novel IAS derivatives bearing nitrogen-containing substituents at the indole-2-carboxamide linked through a CH2/CH2CH2 spacer (Fig. 9 ) [67]. Most of these IASs 46 were proved to be potent inhibitors of the HIV-1 WT (NL4−3 strain) in MT-4 cells at low nanomolar concentrations and weakly cytotoxic. Several compounds were also identified as potent inhibitors of the mutant HIV-1 strains.
Fig. 9.
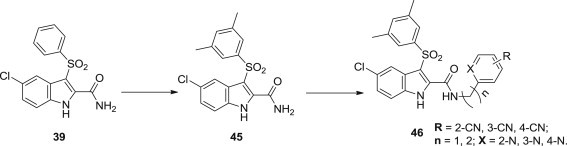
Optimization route of novel IAS derivatives as NNRTIs.
Zhijian Zhao and coworkers from Merck reported the design, synthesis, and biological evaluation of novel 3-indole sulfonamides as potent NNRTIs with balanced profiles against common HIV RT mutants K103N and Y181C (Fig. 10 ) [68]. Introduction of a pyrrolidine sulfonamide at the 3-position of the indole ring of lead compound 39 led to analog 47 with excellent wild-type HIV RT potency and comparatively weak K103N and Y181C RT activities. Compounds 48–50 with variation in the indole 2-substituent showed improved activities against wild-type and some mutants, such as K103N and Y181C.
Fig. 10.
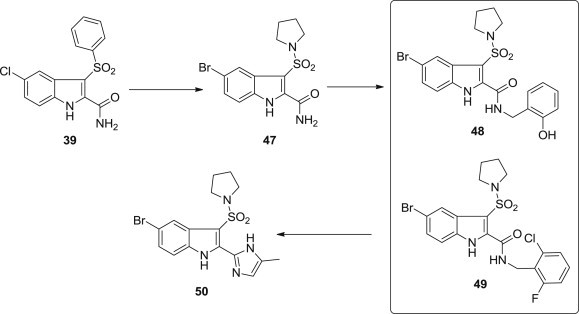
Optimization route of novel 3-indole sulfonamides as potent NNRTIs.
Rino Ragno and his coworkers performed 3D-QSAR and docking simulations to design and synthesize a series of novel derivatives of 39. N-(2-hydroxyethyl)carboxamide 51 [69] or substituted N′-carboxyhydrazide derivatives 52 [70] showed high activity against the K103N RT mutant virus (Fig. 11 ). Meanwhile, the 5-chloro-4-fluoro derivative 53 and 4,5-difluoro derivative 54 turned out to be potent inhibitors of HIV-1 WT and the NNRTI-resistant Y181C RT and K103N–Y181C RT HIV-1 strains. In particular, compound 54 was exceptionally potent against RT WT and RTs carrying the K103N, Y181I, L100I mutations.
Fig. 11.
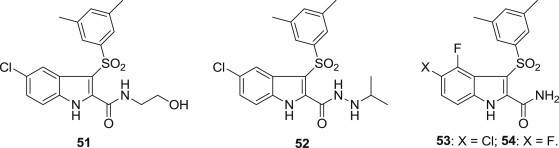
Structures of indole-containing NNRTIs.
New indolylarylsulfone derivatives 55 bearing cyclic substituents at indole-2-carboxamide linked through a methylene/ethylene spacer were designed and synthesized as novel IAS analogues by Giuseppe La Regina and his coworkers. These compounds were potent inhibitors of the WT HIV-1 replication in CEM and PBMC cells and showed inhibitory concentrations in the low nanomolar range (Fig. 12 ) [71]. The substituents introduced at positions 4 and 5 of the indole did not show a significant effect on the antiviral activity against the HIV-1 WT. 5-Bromo and 5-nitro-IASs bearing the ethylene linker were less cytostatic than the corresponding 5-chloro and 5-chloro-4-fluoro derivatives. Against the mutant L100I and K103N RT HIV-1 strains in MT-4 cells, some compounds showed antiviral potency superior to that of NVP and EFV. Further molecular docking experiments revealed that the H–bond interaction between the nitrogen atom in the carboxamide chain of IAS and Glu138B played an important role for the binding of these analogues.
Fig. 12.
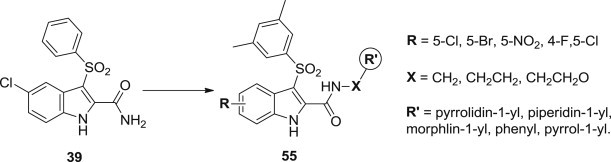
Optimization route of novel IAS derivatives as NNRTIs.
Recently, a series of new indolylarylsulfones as HIV-1 NNRTIs were synthesized and evaluated for the unexplored substitutions of the benzyl/phenylethyl group linked at the indole-2-carboxamide (Fig. 13 ) [72]. Compound 56 exhibited 535-folds, 76-folds and 18-folds higher antiviral activity against the NL4_3 HIV-1 WT strain than the references NVP, EFV and AZT, respectively. In addition, several compounds showed nanomolar potency towards the K103N HIV-1 mutant strain and were superior to EFV, while some derivatives were superior to EFV against the Y181C and L100I HIV-1 mutant strains. Very interestingly, the enantiomers 57 and 58 showed small differences of activity against the NL4-3 HIV-1 strain, but 57 turned out significantly more potent than 58 against the whole panel of mutant HIV-1 strains. The enzymatic results for 57 and 58 were in well agreement with the cellular data. The molecular simulations suggested that the difference in the observed inhibitory activities of these two isomers could be due to a kinetic rather than affinity differences. The results showed compound 57 represents a robust lead compound to develop NNRTIs with improved activity and selectivity against K103N that is the most frequently emerging HIV-1 mutation in EFV-treated patients.
Fig. 13.
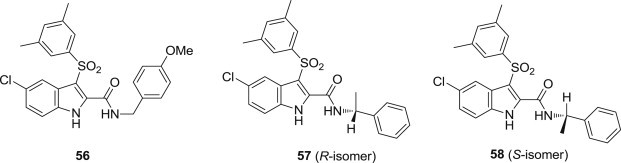
Chiral IAS derivatives as NNRTIs.
Idenix Pharmaceuticals and Merck & Co. showed a renewed interest in IASs and replaced the 3-arylsulfonyl group by either an arylphosphonyl 59 [73] or a sulfonamide group 60 [68]. Francesco Piscitelli reported new potent indolylarylsulfone (IAS) HIV-1 NNRTIs 61 obtained by coupling natural and unnatural amino acids to the 2-carboxamide and introducing different electron-withdrawing substituents at position 4 and/or 5 of the indole rings (Fig. 14 ). The new IASs inhibited the HIV-1 replication in human T-lymphocyte (CEM) cells at low/subnanomolar concentration and were weakly cytostatic [74].
Fig. 14.
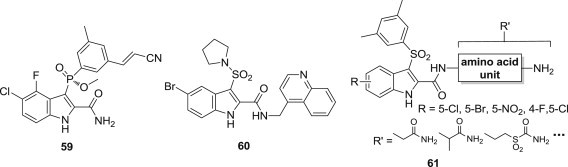
Novel IAS derivatives as NNRTIs.
Francois-Rene Alexandre and coworkers designed and synthesized a series of 3-phenylphosphinate-2-carboxamide indoles 62 as novel NNRTIs (Fig. 15 ) [75]. As the substituted bicyclic hydrogen donor scaffold could interact with K101 and F227, and the branching phosphorus tetrahedral linker would direct the substitution toward the lipophilic W229 region, chemical variation in the phosphorus linker led to the discovery of 3-phenyl-methylphosphinate-2-carboxamide, which showed excellent potency against wild-type HIV-1 and K103N and Y181C single mutants in the reverse transcriptase gene. Most importantly, in addition to the potency, the pharmacokinetic, solubility, and metabolic properties of this series compounds were also encouraging.
Fig. 15.
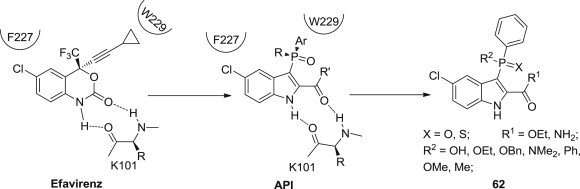
Optimization and binding of IAS derivatives as NNRTIs.
Utilizing molecular modeling, Mohammad Hassam and coworkers have designed and synthesized a series of novel cyclopropyl indole derivatives as HIV non-nucleoside reverse transcriptase inhibitors [76], and these inhibitors facilitate a double hydrogen bonding interaction to K101 and efficiently occupy the hydrophobic pockets in the regions of Y181/188 and V179. Several of these compounds, such as 63, 64 and 65 (Fig. 16 ), inhibited HIV replication as effectively as nevirapine when tested in a phenotypic assay.
Fig. 16.
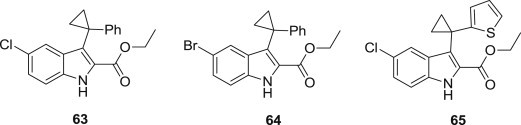
Cyclopropyl indole derivatives as NNRTIs.
3.2.2. Other indole derivatives as reverse transcriptase inhibitor
Rilpivirine 66, the most recently FDA-approved NNRTI, has an uncommon structural feature of the cyanovinyl (CV) group. For most medicinal chemists viewing the substructure, concern arises that the CV group may be sufficiently electrophilic to act as a Michael acceptor, leading to potential covalent modification of proteins, nucleic acids, or other biological entities. Although in reality unsaturated nitriles are poor Michael acceptors that require reactive organometallic nucleophiles to undergo conjugate additions, the fact is that almost no approved drugs contain a cyanovinyl group, and lack of precedent is often taken as a warning sign in drug discovery. Keeping these consideration in mind, Won-Gil Lee and coworkers carried out computer simulations to guide the molecular design of bicyclic replacements for the CVP group, which led to the discovery of compounds 67, 68 and 69 (Fig. 17 ) [77]. Against the wild-type virus, these three compounds exhibited EC50 values of 85 nM, 56 nM and 10 nM, respectively. Unfortunately, these compounds did not showed promising potency towards the mutant strains of HIV-1, including the challenging HIV-1 variant that contains K103N/Y181C double mutation in the RT enzyme.
Fig. 17.
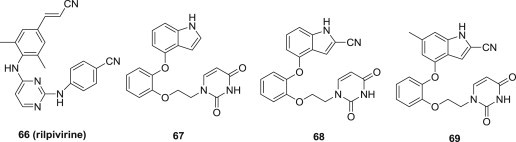
Cyanovinyl (CV) indole derivatives as NNRTIs.
Hai-Bing Zhou and coworkers described the design, synthesis and biological evaluation of indole-based trifluoropropanoates 70–74 as efficient inhibitors of reverse transcriptase (RT) of HIV-1 (Fig. 18 ). The inhibitory activities of the two enantiomers and the corresponding racemic mixture were compared. Among the non-racemic and racemic compounds, the enantiomer with the R-configuration usually showed the lowest EC50 value. For example, the R-isomer of compound 70 was identified as the most active candidate when tested in the TZM-bl cells on HIV virus type HIV–1IIIB, with an EC50 value of 19 nM, CC50 value of 210.697 μM and SI (selectivity index, CC50/EC50) value of 11,089, respectively [78].
Fig. 18.
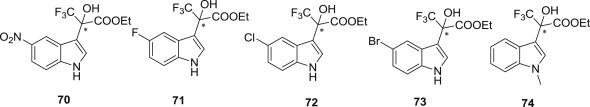
Chiral indole-based trifluoropropanoates as NNRTIs.
Based on butterfly-shaped (Wing 1--Body--Wing 2) pharmacophore requirements, Ashok Penta and coworkers have designed 50 novel 1-phenyl-2,3,4,9 -tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylic acid analogs as HIV-1 NNRTIs [79]. Molecular modeling, Lipinski rule of five parameter and toxicity parameters of designed analogs were employed to predict the rational structures (Fig. 19 ). Among the designed analogs, 75, 76 and 77 showed significant binding free energy and predicted inhibitory constant values, they have screened off for next level of study because of their predicted poor pharmacokinetic and toxicity profile.
Fig. 19.
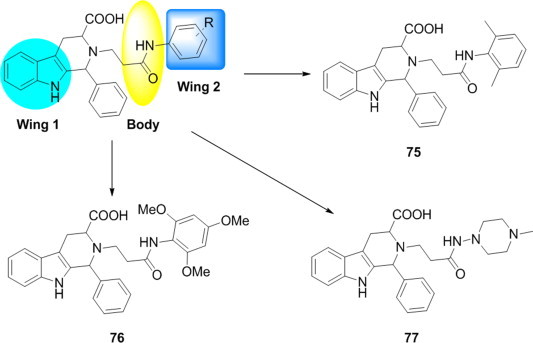
Butterfly-shaped indole compounds as NNRTIs.
3.3. Indole derivatives as integrase (IN) inhibitors
Current treatments of HIV/AIDS employ a combination of therapeutic agents that target the viral reverse transcriptase, protease enzymes and viral entry. However, the ability of HIV to rapidly evolve resistance to these agents, together with their significant side effects, requires the development of new antiviral drugs with novel mode of action. Therefore, it is an urgent task to develop novel agents that interfere with alternate stages in the viral life cycle. Integrase catalyzed the integration of the HIV genome into the cellular chromosome, an essential process for HIV replication. Because of no mammalian counterparts, integrase has become an attractive target for antiviral drug design. Furthermore, integrase uses a single active site to accommodate two different configurations of DNA substrates, which may constrain the ability of HIV to develop drug resistance to integrase inhibitors (INI). Discovery of new INI has attracted great interest from the medicinal chemists in recent years.
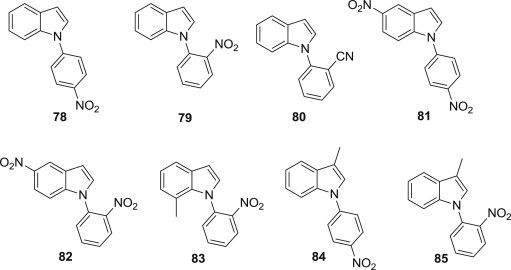
Hui Xu and coworkers designed and synthesized eight simple N-arylindoles (78–85) as HIV-1 integrase inhibitors in vitro for the first time (Fig. 20 ) [80], [81]. Among these structurally simple compounds, compound 79, 82 and 84 exhibited the highest potency against anti-HIV-1 integrase with a EC50 value of 7.88 mg/mL and a TI value of 24.61.
Fig. 20.
N-arylindoles as integrase inhibitors.
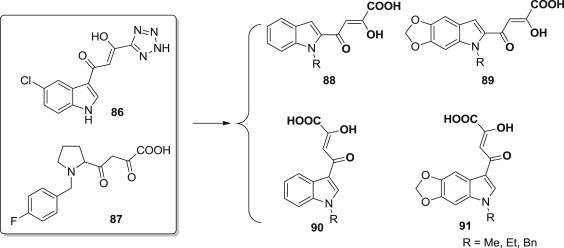
Diketo acid (DKA) derivatives 86 and 87 were independently discovered by scientists from Shionogi and Merck as a new class of HIV-1 integrase inhibitors (Fig. 21 ) [82], [83]. In order to establish a coherent structure-activity relationship among the substituted indole nucleus bearing a β-diketo acid moiety, Mario Sechi and coworkers [84] designed a series of substituted indole-based DKA derivatives 88–91, which showed anti-IN activity at low micromolar concentrations with varied selectivity against the strand transfer process. The experimental results have demonstrated that the diketo acid functionality is important for selectivity against strand transfer and that the aromatic ring plays a major role in potency. However, the selectivity for strand transfer is not sufficient for antiviral activity. Although diketo acid-containing compounds represent a novel class of compounds and are considered as a step forward in drug design in targeting IN, many analogues showed significant cytotoxicity and lack of antiviral activity. This work demonstrated once again that the physicochemical properties of a compound could potentially make a major contribution to antiviral activity in vitro.
Fig. 21.
Diketo acid (DKA) derivatives as integrase inhibitors.
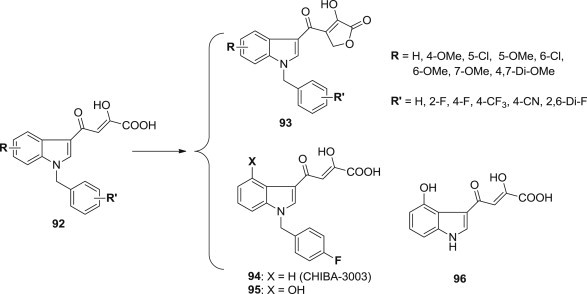
Subsequently, in order to investigate the influence of diketo acid moiety on the biological activity and cytotoxicity, Stefania Ferro and coworkers designed and synthesized a series of novel compounds 92 and its corresponding analogues, 4-[(1-benzyl-1H-indol-3-yl)- carbonyl]-3-hydroxyfuran-2(5H)-ones 93 (Fig. 22 ) [85], [86], in which 4-carbonyl-3 -hydroxy-furan-2(5H)-one moiety was introduced as a suitable replacement for the β-diketo acid motif. Even if the furanone derivatives 93 generally exhibited good potency in a micromolar range, the comparison of these derivatives with their corresponding diketo acids pointed out a diminished potency both in enzymatic and cell assays. These results suggest that the replacement of the β-diketo acid motif negatively influences the in vitro biological activity, probably due to a reduced conformational mobility of a closed system with respect to the open form.
Fig. 22.
Optimization of DKA derivatives as integrase inhibitors.
Apart from targeting the critical proteins, the manipulation of specific protein–protein interactions (PPIs) involved in the HIV life cycle could potentially result in potent drugs that lack cellular toxicity. For example, the interaction between HIV-1 integrase (IN) and the cellular protein lens epithelium-derived growth factor or transcriptional coactivator p75 (LEDGF/p75) has recently gained attention as a valuable target for a novel antiviral strategy. For example, Chimirri and his coworkers established a structure-based pharmacophore model based on the X-ray crystal structure of HIV-1 IN in complex with LEDGF/p75 IBD [87]. Then, the 3D pharmacophore model was used as a query in a virtual screening approach to filter a library of 3055 small molecules and produced compound 94 as the best hit. After optimization, compounds 95 and 96 (Fig. 22) were obtained. In agreement with the prediction, compound 95 showed a higher potency than 94, while compound 96 proved to be the most active molecule, providing a valid starting point for the discovery of new and more potent derivatives able to disrupt PPIs between IN and its intracellular cofactor LEDGF/p75.
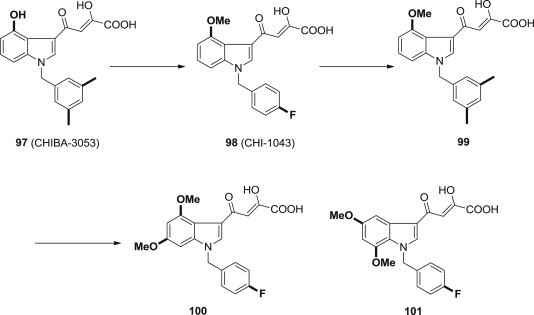
After a number of rational modifications both on the indole system and the benzyl moiety, 4-[1-(3,5-dimethylbenzyl)-4-hydroxy-1H-indol-3-yl]-2-hydroxy-4-oxobut-2-enoic acid 97 was identified as the most potent compound with an IC50 value of 3.5 μM (Fig. 23 ) [88], [89]. Methylation of the hydroxyl group afforded the first dual inhibitor of HIV-1 integration process, compound 98, which inhibits both the IN ST step (IC50 = 0.14 μM) and IN-LEDGF/p75 interaction (IC50 = 36 μM). Very interestingly, the replacement of the 4-fluorobenzyl portion at N-1 with a 3,5-dimethylbenzyl moiety negatively influenced the inhibition of IN ST step. Compound 99 drastically reduced the ST inhibitory effects with respect to compound 98 (IC50 = 5.49 μM versus IC50 = 0.14 μM). Moreover, compound 99 showed a poorer anti-HIV activity (EC50 = 4.63 μM versus EC50 = 0.59 μM) in MT4 cells and cytotoxicity at the same concentrations. The introduction of an additional methoxy group at C-5 or C-7 position increases the IN-inhibitory effects [90]. For example, the disubstituted compound 100 is more potent than the 4-monosubstituted derivative 98. Moreover, the simultaneous presence of the methoxy substituent at 5 and 7 positions afforded the most potent INSTI of this series, the 4-[1-(4-fluorobenzyl)-5,7-dimethoxy-1H-indol-3-yl]-2- hydroxy-4-oxobut-2-enoic acid 101; it proved to be active at 6 nM concentration that is comparable to raltegravir (7 nM) and better than elvitegravir (15 nM), two HIV integrase inhibitors approved by US FDA in 2007 and 2012, respectively. On the contrary, the introduction of an additional methoxy group at C-6 seems to negatively influence the activity of this class of INSTIs.
Fig. 23.
Rational modifications of DKA derivatives as integrase inhibitors.
Encouraged by these results, with the aim to identify small molecules able to inhibit both the IN strand-transfer step and IN-LEDGF/p75 interaction, Rogolino and coworkers evaluated the potent IN ST inhibitor 98 and two analogues (102 and 103) and their magnesium(II) complexes (104) for their ability to act as dual inhibitors (Fig. 24 ) [91]. Both the free ligands (102 and 103) are able to inhibit the IN-LEDGF/p75 interaction at mM values, and the metal complexes (104) exhibited IN inhibition potency in low nM or μM concentration range. Moreover, these magnesium compounds showed good antiviral activity on infected cells. This is the first data regarding the activity of metal complexes as allosteric inhibitors, and offers a promising approach to prevent viral replication and underlines the possibility to use coordination chemistry to obtain unconventional scaffold to target enzymes.
Fig. 24.
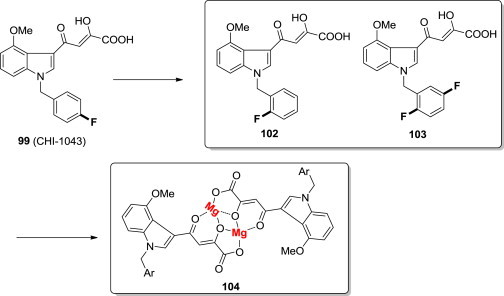
Magnesium(II) complexes of DKA derivatives as novel integrase inhibitors.
Based on the previously obtained SAR information and benzyl-indole derivatives (97, 98) as anti-HIV agents, Stefania Ferro and coworkers reported their research on the identification of new dual target small molecules able to target different steps of HIV-1 life cycle (Fig. 25 ), and obtained two series of novel anti-HIV agents (105, 106), some of which are dual inhibitors of the integration process acting both in the IN strand-transfer step and by disrupting the protein–protein interaction between IN and its cofactor LEDGF/p75, and compound 107 is the most active one [92].
Fig. 25.
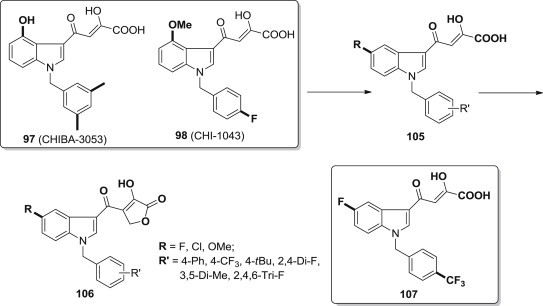
DKA derivatives as dual integrase inhibitors.
Laura De Luca and coworkers used the fragment hopping approach to design and synthesize novel non-peptidyl compounds (Fig. 26 ) that mimic the biological function of some IBD residues and in particular the LEDGF hot spot residues Ile365 and Asp366 [93]. However, these newly synthesized compounds generally were not able to produce significant inhibitory effects. The most active molecules were derivatives 109 and 111 with a percentage of inhibition of 34% and 39% at a fixed dose of 100 μM, significantly lower than that of prototype 97.
Fig. 26.
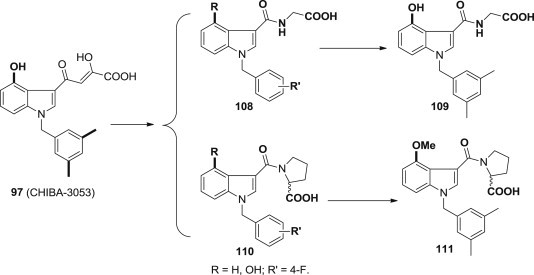
Non-peptidyl DKA derivatives as integrase inhibitors.
3.4. Indole derivatives as protease inhibitors
Protease (PR) inhibitors became available in the 1990s and have proven effective to an important mean to treat viral disease [94], considerable research has been performed to find “protease inhibitors” to attack virus at that phase of its life cycle and led to some FDA-approved protease inhibitors in clinic trail or in development [95]. For example, HIV protease plays a key role in the life cycle of the virus, its inhibition prevents maturation of the viral particles and renders them noninfectious. More than 25 years after its discovery, HIV PR remains one of the primary targets for development of novel HIV treatments. Recently, allosteric regulation of HIV PR activity has been recognized as a novel way to limit the development of drug-resistance. X-ray crystallography experiments showed that two indole-carboxylate small molecules, indole-6-carboxylic acid (112) and 3-indolepropionic acid (113) (Fig. 27 ), could be used as fragment-like starting points for the discovery of novel HIV PR inhibitors, although these two fragments did not show significant inhibition of PR up to the concentration of 1 mM. In addition, Lucia Chiummiento and coworkers reported the synthesis and antiviral activity of new non-peptidic heteroaromatic molecules 114–116 (Fig. 27) as protease inhibitors. Among them the best one 114 showed a moderate potency (IC50 = 1 μM), considering the novel skeleton and ease of preparation, this compound represented a reference structure for further modification.
Fig. 27.
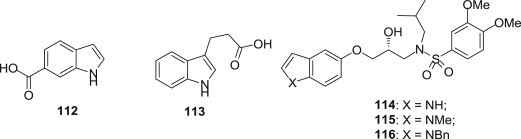
Indole-carboxylate molecules as Protease (PR) inhibitors.
The hepatitis C virus (HCV) infects approximately 170 million people worldwide. The NS3 protease has been considered as one of the most attractive targets for anti-HCV therapy because it is essential for viral replication and formation of infectious viral particles. Considerable efforts by different research groups have been directed toward development of HCV NS3 protease inhibitors [96]. For example, Nasser S. M. Ismail and coworkers used computational simulations techniques, such as molecular docking and pharmacophore modeling, to design novel indole-based derivatives as HCV NS3 protease inhibitors (Fig. 28 ). Among the compounds showing significant high simulation docking score and fit values, compounds 117 and 118 were identified as the most potent candidates with IC50 values of 15 and 13 μM, respectively [97].
Fig. 28.
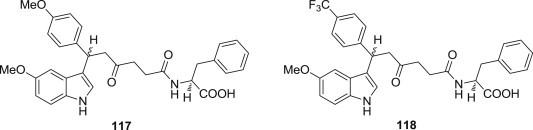
Novel indole-based derivatives as protease inhibitors.
3.5. Indole derivatives as polymerase inhibitors
In addition to NS3, the nonstructural region of the HCV genome encodes several additional enzymes that are believed to play fundamental roles in the viral life cycle. NS5B RNA-dependent RNA polymerase, one of these central enzymes in the viral replication cycle, has emerged as an especially attractive target for drug discovery efforts toward antivirals for HCV and has been described as the most drugable HCV protein. Small molecule inhibitors of this target have received much attention in the recent past as potential therapeutic agents for treatment of HCV infection, including the well-known anti-HCV drug Sovaldi. Recently, Ariamala Gopalsamy and his coworkers designed and synthesized a novel class of HCV NS5B polymerase inhibitors containing 2,3,4,9-tetrahydro-1H-carbazole and 1,2,3,4-tetrahydro-cyclopenta[b]indole scaffolds based on the lead compound 119 (Fig. 29 ) [98]. Structure-activity relationships indicated that optimization of the aromatic region showed preference for 5,8-disubstitution pattern in both the scaffolds examined while favoring the n-propyl moiety for the C-1 position. 1,2,3,4-Tetrahydro-cyclopenta[b]indole scaffold was slightly more potent than the corresponding 2,3,4,9-tetrahydro-1H-carbazole. For example, 2,3,4,9-Tetrahydro-1H-carbazole 120 and 1,2,3,4-tetrahydro-cyclopenta[b]indole 121 displayed IC50 values of 3200 nM and 550 nM against HCV NS5B enzyme, respectively.
Fig. 29.
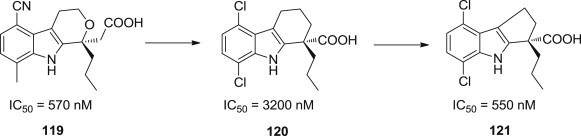
Compounds with indole scaffolds as polymerase inhibitors.
Steven Harper and coworkers reported a series of 2-(indol-1-yl)acetamides as potent allosteric inhibitors of the HCV NS5B polymerase enzyme (Fig. 30 ). Based on the lead structure of indole-N-acetamide 122, that are potent inhibitors of the NS5B enzyme and show promising activity in the replicon assay, a library-based approach toward exploration with the intracellular activity improvement was established, some derivatives (123–124) showed strong potency in the cell-based assay under routine conditions and in the presence of a high concentration of human serum, especially compound 124, it showed encouraging PK properties in both rat and dog and is clean in an extensive panel of counter screening assays [99].
Fig. 30.
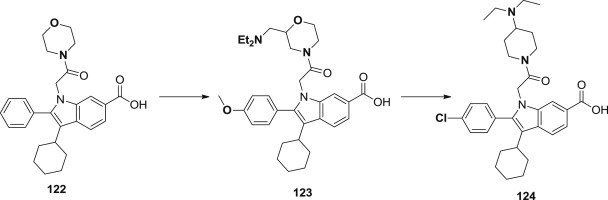
Indole-N-acetamides as polymerase inhibitors.
3.6. Indole-based natural products and their antiviral activity
During the past fifty years, natural products have served as a major source of drugs, about fifty percent of today's commercial drugs are derived from natural product. While, there are only few drugs available currently for the cure of viral diseases, including acyclovir which is modeled on a natural product parent. In order to discover novel antiviral natural products with new mode of action, many research efforts have been devoted for the discovery of new antiviral natural products, the following focus on the indole natural products and their antiviral activities, summarize the structure property, hoping to inspire new and even more creative approaches.
Sattazolin (125) (Fig. 31 ) is an indole acyloin natural product [100], it is reported to exhibit potent antiviral activity with an ID50 of 1.5 μg/mL against herpes simplex virus type 1 (HSV1) and type 2 (HSV2) [101]. Drymaritin (126), a novel indole alkaloid isolated from Drymaria diandra, is reported to exhibit anti-HIV effects in H9 lymphocytes with an EC50 value of 0.699 μg/mL and a TI of 20.6 [102]. Caulerpin (127) was isolated from the green alga Caulerpa racemosa, collected on vertical rock walls in São Pedro and São Paulo Archipelago, it is reported the antiviral activity against Bovine viral diarrhea virus (BVDV) replication, also suggest that it might be relevant to evaluate antiviral activity against HCV due to their similar characteristics [103].
Fig. 31.
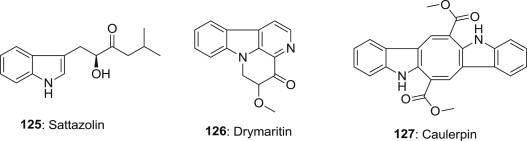
Structures of antiviral Sattazolin, Drymaritin and Caulerpin.
Minghua Chen and coworkers reported the isolation of seventeen new indole alkaloids and fourteen known analogues from an aqueous extract of the root of Isatis indigotica (Fig. 32 ), and compounds 128–130 and arvelexin (131) show antiviral activity against the influenza virus A/Hanfang/359/95 (H3N2), with IC50 values of 3.70–12.35 μM [104].
Fig. 32.
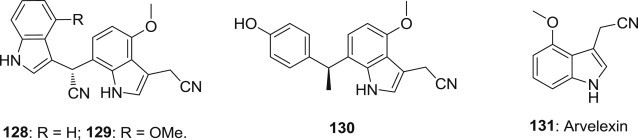
Structures of antiviral indole alkaloids from root of Isatis indigotica.
Paromita Bag and coworkers reported the isolation of an indole alkaloid 7-methoxy-1-methyl-4,9-dihydro-3H-pyrido[3,4-b]indole 132 (Harmaline, HM) from an ethnomedicinal herb Ophiorrhiza nicobarica, and it demonstrated a potent anti-HSV-1 activity against both wild type and clinical isolates of HSV-1 (Fig. 33 ). It is reported that this indole alkaloid 132 interferes with the viral immediate-early (IE) transcriptional events, and significantly reduces virus yield in mice at well-tolerated dose. Because IE complex is a critical component of herpes virus reactivation mechanism, 132 may help to prevent both the multiplication and reactivation of HSV, and provide an interesting molecular target for the development of better HSV therapy [105], [106].
Fig. 33.
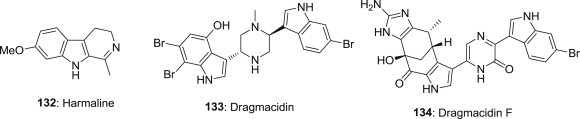
Structures of antiviral Harmaline, Dragmacidin and Dragmacidin F.
The bioactive marine natural products dragmacidins 133–134 (Fig. 33), obtained by an exhaustive set of protocols from a marine sponge of the genus Halicortex, also reported to display modest antiviral activity [107], and compound 134, dragmacidin F, containing an unprecedented carbon skeleton that is very likely derived from cyclization of a partially oxidized form of dragmacidin series derivatives, it showed in vitro antiviral activity against HSV-1 (EC50 = 95.8 μM) and HIV-1 (EC50 = 0.91 μM) [108].
The bisindole alkaloids are a class of marine natural products that show unique promise in the development of new drug leads (Fig. 34 ) [109]. Coscinamides 135–137, which contains unusual 2-ketoenamide functionality, showed partial cytoprotection activity against HIV in the NCI assay. Chondriamides 138–140 are indolic enamides and show antiviral activity against HSV II with IC50 values of about 1 μM [110].
Fig. 34.
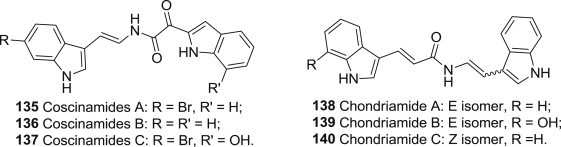
Structures of antiviral Coscinamides and Chlondriamides.
Cheng-Jian Tan and coworkers reported the isolation, structural elucidation, and anti-HIV-1 activity of three novel indole alkaloids trigonoliimines A-C (141–143) (Fig. 35 ) with unprecedented polycyclic skeletons. They were isolated from the extract of the leaves of Trigonostemon. lii Y. T. Chang. The anti-HIV-1 activity of compound 141 and 142 was tested by a microtiter syncytium formation infectivity assay with Zidovudine (EC50 = 0.02 μM, TI = 59,924) as a positive control. Trigonoliimine A showed modest anti-HIV-1 activity (EC50 0.95 μM, TI = 7.9) [111].
Fig. 35.
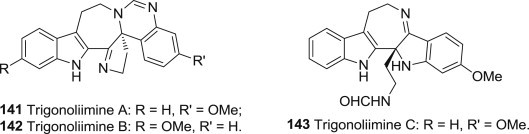
Structures of antiviral Trigonoliimines.
Zhongli Xu and coworkers independently reported the structures of pentacyclic indolocarbazoles from Streptomyces spp. (Fig. 36 ), namely the diastereomers oridamycin (144) [112] and xiamycin A (145) [113], the xiamycin B (146), and the seco-derivative indosespene (147) [114], these rare endophyte metabolites likely play an ecological role in their habitats because their antiviral activity may contribute to the antibiotic reservoir of the mangrove plants [115].
Fig. 36.
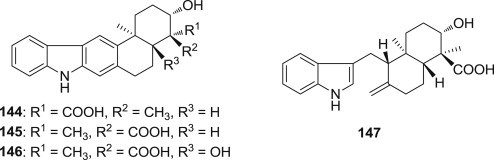
Structures of antiviral pentacyclic indolocarbazoles.
Jixing Peng and coworkers reported the isolation of three new indole alkaloids (148–150) and three known alkaloids (151–153) (Fig. 37 ) from the culture of the mangrove-derived fungus Cladosporium sp. PJX-41. All compounds were evaluated for their antiviral activities against influenza virus A (H1N1), and compounds 150 exhibited significant activities (IC50 = 85 μM) against H1N1 [116].
Fig. 37.
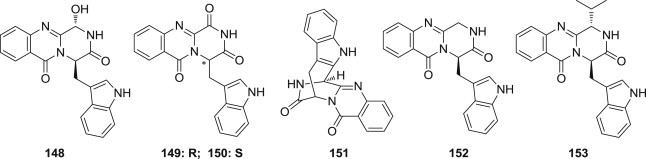
Structures of antiviral indole alkaloids from Cladosporium sp. PJX-41.
Eudistomins are β-carboline derivatives (Fig. 38 ), isolated from different kinds of ascidians (marine tunicates of the family Ascidiacea), such as Ritterella sigillinoides, Lissoclinum fragile or Pseudodistoma aureum. In recent investigations, it is reported that eudistomins containing the oxathiazepine ring (154–158, Eudistomins C, E, F, K, and L) showing the most significant antiviral activity against HSV-1, of which C and E, with a phenolic group were as active as at 0.005–0.01 μg/disk. Eudistomins C and E are also reported to possess the activities against RNA viruses such as Coxsackie A-21 virus, equine rhinovirus and against DNA viruses [117].
Fig. 38.
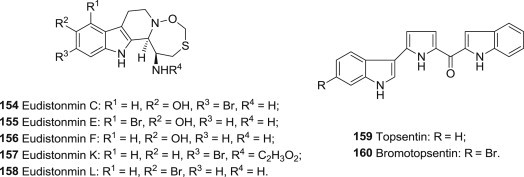
Structures of antiviral Eudistomins, Topsentin and Bromotopsentin.
What's worth noting is the indole dimer derivatives Topsentin 159 and bromotopsentin 160 (Fig. 38) isolated from the Caribbean deep-sea sponge Spongosorites ruetzleri, they exhibited antiviral activities in corona virus A-59, vitro against HSV-1 and Vesivular stomatitis virus (VSV) [117].
Compounds 161 CPI-2081a and 162 CPI-2081b (Fig. 39 ), isolated from a biologically active culture supernatant of Streptomyces sp. NCIM2081 by Sihgh et al. in 2010, as the cysteine protease inhibitor, it is found that nanomolar concentration of compounds CPI-2081a is able to inhibit papain hydrolytic activity, it also showed significantly inhibitory activity of tumor cell migration at sub cytotoxic concentration [118], [119].
Fig. 39.
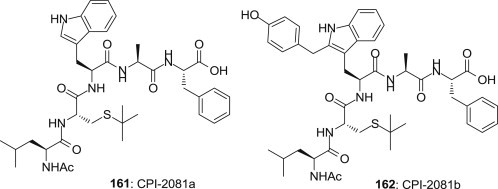
Structures of CPI-2081a and CPI-2081b.
4. Prospective
It is reported that macrocyclic indole derivatives can demonstrate favorable druglike properties [120], [121], including good solubility, increased lipophilicity, enhanced membrane penetration, improved metabolic stability, and good oral bioavailability with desirable pharmacokinetic and pharmacodynamic properties [122], [123]. Compound 25 (TMC647055), the 17-membered macrocyclic indole derivative, is currently being evaluated in phase II clinical trials. It may set up a very good example for exploited strategy of indole macrocycle as antiviral agent.
We also notice that, as an important class of marine natural products, bis and trisindole alkaloids show unique promise in the development of new drug leads [110], [124], [125]. Bis and trisindole may be treated as new drug leads due to their unique structures and functionalities. This area is well worth more scientific attention wherein lies a huge potential for development of new active compounds and exploration of various biological activities, so that they may be useful as novel lead structures in the future.
5. Conclusion
As the most abundant heterocycle in nature, indole is commonly found in biologically active natural products, pharmaceuticals and agrochemicals. There has been an increasing interest in the use of indole derivatives as bioactive molecules against different kinds of diseases. This review updates recent developments and current status of important indole derivatives in the areas of antiviral drug discovery, serves a comprehensive overview on indole antiviral agents being on the market or in clinical trials as well as currently evaluated in experimental studies, focus on recent developments of indole compounds as antiviral agents, and promote the ideas of indole macrocycle and Bis (Tris) indole as novel lead structures might be exploited in the future.
Acknowledgments
We are grateful to the financial support for this work from the National Key Technologies R&D Program (2011BAE06B05). We also thanks Dr. Vincent W.-F. Tai from Antiviral DPU GlaxoSmithKline (RTP, NC, US) for discussion and manuscript revision.
Contributor Information
Qiong Chen, Email: qchen@mail.ccnu.edu.cn.
Guang-Fu Yang, Email: gfyang@mail.ccnu.edu.cn.
References
- 1.Samala S., Arigela R.K., Kant R., Kundu B. Diversity-oriented synthesis of ketoindoloquinoxalines and indolotriazoloquinoxalines from 1-(2-nitroaryl)-2-alkynylindoles. J. Org. Chem. 2014;79:2491–2500. doi: 10.1021/jo402783p. [DOI] [PubMed] [Google Scholar]
- 2.Won C., Shen X., Mashiguchi K., Zheng Z., Dai X., Cheng Y., Kasahara H., Kamiya Y., Chory J., Zhao Y. Conversion of tryptophan to indole-3-acetic acid by TRYPTOPHAN AMINOTRANSFERASES OF ARABIDOPSIS and YUCCAs in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A. 2011;108:18518–18523. doi: 10.1073/pnas.1108436108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang M.Z., Mulholland N., Beattie D., Irwin D., Gu Y.C., Chen Q., Yang G.F., Clough J. Synthesis and antifungal activity of 3-(1,3,4-oxadiazol-5-yl)-indoles and 3-(1,3,4-oxadiazol-5-yl)methyl-indoles. Eur. J. Med. Chem. 2013;63:22–32. doi: 10.1016/j.ejmech.2013.01.038. [DOI] [PubMed] [Google Scholar]
- 4.Young S.N. How to increase serotonin in the human brain without drugs. J. Psychiatry Neurosci. 2007;32:394–399. [PMC free article] [PubMed] [Google Scholar]
- 5.Diss L.B., Robinson S.D., Wu Y., Fidalgo S., Yeoman M.S., Patel B.A. Age-related changes in melatonin release in the murine distal colon. ACS Chem. Neurosci. 2013;4:879–887. doi: 10.1021/cn4000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patil S.A., Patil R., Miller D.D. Indole molecules as inhibitors of tubulin polymerization: potential new anticancer agents. Future Med. Chem. 2012;4:2085–2115. doi: 10.4155/fmc.12.141. [DOI] [PubMed] [Google Scholar]
- 7.Higdon J.V., Delage B., Williams D.E., Dashwood R.H. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol. Res. 2007;55:224–236. doi: 10.1016/j.phrs.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogan E.G. The natural chemopreventive compound indole-3-carbinol: state of the science. In Vivo. 2006;20:221–228. [PubMed] [Google Scholar]
- 9.Kim Y.S., Milner J.A. Targets for indole-3-carbinol in cancer prevention. J. Nutr. Biochem. 2005;16:65–73. doi: 10.1016/j.jnutbio.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 10.Biersack B., Schobert R. Indole compounds against breast cancer: recent developments. Curr. Drug Targets. 2012;13:1705–1719. doi: 10.2174/138945012804545551. [DOI] [PubMed] [Google Scholar]
- 11.Kurz W.G., Chatson K.B., Constabel F., Kutney J.P., Choi L.S., Kolodziejczyk P., Sleigh S.K., Stuart K.L., Worth B.R. Alkaloid Production in Catharanthus roseus cell cultures VIII. Planta Med. 1981;42:22–31. doi: 10.1055/s-2007-971541. [DOI] [PubMed] [Google Scholar]
- 12.Leon F., Habib E., Adkins J.E., Furr E.B., McCurdy C.R., Cutler S.J. Phytochemical characterization of the leaves of Mitragyna speciosa grown in U.S.A. Nat. Prod. Commun. 2009;4:907–910. [PMC free article] [PubMed] [Google Scholar]
- 13.Chen F.-E., Huang J. Reserpine: a Challenge for total synthesis of natural products. Chem. Rev. 2005;105:4671–4706. doi: 10.1021/cr050521a. [DOI] [PubMed] [Google Scholar]
- 14.Ishikawa H., Colby D.A., Boger D.L. Direct coupling of catharanthine and vindoline to provide vinblastine: total synthesis of (+)- and ent-(-)-vinblastine. J. Am. Chem. Soc. 2008;130:420–421. doi: 10.1021/ja078192m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Sa Alves F.R., Barreiro E.J., Fraga C.A. From nature to drug discovery: the indole scaffold as a ‘privileged structure’. Mini Rev. Med. Chem. 2009;9:782–793. doi: 10.2174/138955709788452649. [DOI] [PubMed] [Google Scholar]
- 16.Evans B.E., Rittle K.E., Bock M.G., DiPardo R.M., Freidinger R.M., Whitter W.L., Lundell G.F., Veber D.F., Anderson P.S., Chang R.S., Lotti V.J., Cerino D.J., Chen T.B., Kling P.J., Kunkel K.A., Springer J.P., Hirshfield J. Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 17.Welsch M.E., Snyder S.A., Stockwell B.R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaushik N.K., Kaushik N., Attri P., Kumar N., Kim C.H., Verma A.K., Choi E.H. Biomedical importance of indoles. Molecules. 2013;18:6620–6662. doi: 10.3390/molecules18066620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dolle R.E., Nelson K.H., Jr. Comprehensive survey of combinatorial library synthesis: 1998. J. Comb. Chem. 1999;1:235–282. doi: 10.1021/cc9900192. [DOI] [PubMed] [Google Scholar]
- 20.Franzen R.G. Recent advances in the preparation of heterocycles on solid support: a review of the literature. J. Comb. Chem. 2000;2:195–214. doi: 10.1021/cc000002f. [DOI] [PubMed] [Google Scholar]
- 21.Dolle R.E. Comprehensive survey of combinatorial library synthesis: 2000. J. Comb. Chem. 2001;3:477–517. doi: 10.1021/cc010049g. [DOI] [PubMed] [Google Scholar]
- 22.Vitaku E.A.I.E., Njarðarson J.T. 2013. Compiled and Produced by the Njardarson Group (The University of Arizona)http://cbc.arizona.edu/njardarson/group/top-pharmaceuticals-poster [Google Scholar]
- 23.Daugan A., Grondin P., Ruault C., Le Monnier de Gouville A.C., Coste H., Linget J.M., Kirilovsky J., Hyafil F., Labaudiniere R. The discovery of tadalafil: a novel and highly selective PDE5 inhibitor. 2: 2,3,6,7,12,12a-hexahydropyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione analogues. J. Med. Chem. 2003;46:4533–4542. doi: 10.1021/jm0300577. [DOI] [PubMed] [Google Scholar]
- 24.Daugan A., Grondin P., Ruault C., Le Monnier de Gouville A.C., Coste H., Kirilovsky J., Hyafil F., Labaudiniere R. The discovery of tadalafil: a novel and highly selective PDE5 inhibitor. 1: 5,6,11,11a-tetrahydro-1H-imidazo[1′,5′:1,6]pyrido[3,4-b]indole-1,3(2H)-dione analogues. J. Med. Chem. 2003;46:4525–4532. doi: 10.1021/jm030056e. [DOI] [PubMed] [Google Scholar]
- 25.Xu H., Lv M. Developments of indoles as anti-HIV-1 inhibitors. Curr. Pharm. Des. 2009;15:2120–2148. doi: 10.2174/138161209788489168. [DOI] [PubMed] [Google Scholar]
- 26.Olgen S. Recent development of new substituted indole and azaindole derivatives as anti-HIV agents. Mini Rev. Med. Chem. 2013;13:1700–1708. doi: 10.2174/13895575113139990075. [DOI] [PubMed] [Google Scholar]
- 27.Leneva I.A., Russell R.J., Boriskin Y.S., Hay A.J. Characteristics of arbidol-resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of arbidol. Antiviral Res. 2009;81:132–140. doi: 10.1016/j.antiviral.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 28.Boriskin Y.S., Leneva I.A., Pecheur E.I., Polyak S.J. Arbidol: a broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 2008;15:997–1005. doi: 10.2174/092986708784049658. [DOI] [PubMed] [Google Scholar]
- 29.Zhang F., Wang G. A review of non-nucleoside anti-hepatitis B virus agents. Eur. J. Med. Chem. 2014;75:267–281. doi: 10.1016/j.ejmech.2014.01.046. [DOI] [PubMed] [Google Scholar]
- 30.Pecheur E.I., Lavillette D., Alcaras F., Molle J., Boriskin Y.S., Roberts M., Cosset F.L., Polyak S.J. Biochemical mechanism of hepatitis C virus inhibition by the broad-spectrum antiviral arbidol. Biochemistry. 2007;46:6050–6059. doi: 10.1021/bi700181j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Villalain J. Membranotropic effects of arbidol, a broad anti-viral molecule, on phospholipid model membranes. J. Phys. Chem. B. 2010;114:8544–8554. doi: 10.1021/jp102619w. [DOI] [PubMed] [Google Scholar]
- 32.Romero D.L., Olmsted R.A., Poel T.J., Morge R.A., Biles C., Keiser B.J., Kopta L.A., Friis J.M., Hosley J.D., Stefanski K.J., Wishka D.G., Evans D.B., Morris J., Stehle R.G., Sharma S.K., Yagi Y., Voorman R.L., Adams W.J., Tarpley W.G., Thomas R.C. Targeting delavirdine/atevirdine resistant HIV-1: identification of (alkylamino)piperidine-containing bis(heteroaryl)piperazines as broad spectrum HIV-1 reverse transcriptase inhibitors. J. Med. Chem. 1996;39:3769–3789. doi: 10.1021/jm960158n. [DOI] [PubMed] [Google Scholar]
- 33.Mannu J., Jenardhanan P., Mathur P.P. A computational study of CYP3A4 mediated drug interaction profiles for anti-HIV drugs. J. Mol. Model. 2011;17:1847–1854. doi: 10.1007/s00894-010-0890-6. [DOI] [PubMed] [Google Scholar]
- 34.Morse G.D., Reichman R.C., Fischl M.A., Para M., Leedom J., Powderly W., Demeter L.M., Resnick L., Bassiakos Y., Timpone J., Cox S., Batts D. Concentration-targeted phase I trials of atevirdine mesylate in patients with HIV infection: dosage requirements and pharmacokinetic studies. The ACTG 187 and 199 study teams. Antiviral Res. 2000;45:47–58. doi: 10.1016/s0166-3542(99)00073-x. [DOI] [PubMed] [Google Scholar]
- 35.Been-Tiktak A.M., Vrehen H.M., Schneider M.M., van der Feltz M., Branger T., Ward P., Cox S.R., Harry J.D., Borleffs J.C. Safety, tolerance, and pharmacokinetics of atevirdine mesylate (U-87201E) in asymptomatic human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 1995;39:602–607. doi: 10.1128/AAC.39.3.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou X.J., Pietropaolo K., Damphousse D., Belanger B., Chen J., Sullivan-Bolyai J., Mayers D. Single-dose escalation and multiple-dose safety, tolerability, and pharmacokinetics of IDX899, a candidate human immunodeficiency virus type 1 nonnucleoside reverse transcriptase inhibitor, in healthy subjects. Antimicrob. Agents Chemother. 2009;53:1739–1746. doi: 10.1128/AAC.01479-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castellino S., Groseclose M.R., Sigafoos J., Wagner D., de Serres M., Polli J.W., Romach E., Myer J., Hamilton B. Central nervous system disposition and metabolism of Fosdevirine (GSK2248761), a non-nucleoside reverse transcriptase inhibitor: an LC-MS and Matrix-assisted laser desorption/ionization imaging MS investigation into central nervous system toxicity. Chem. Res. Toxicol. 2013;26:241–251. doi: 10.1021/tx3004196. [DOI] [PubMed] [Google Scholar]
- 38.Piscitelli S., Kim J., Gould E., Lou Y., White S., de Serres M., Johnson M., Zhou X.J., Pietropaolo K., Mayers D. Drug interaction profile for GSK2248761, a next generation non-nucleoside reverse transcriptase inhibitor. Br. J. Clin. Pharmacol. 2012;74:336–345. doi: 10.1111/j.1365-2125.2012.04194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Margolis D.A., Eron J.J., DeJesus E., White S., Wannamaker P., Stancil B., Johnson M. Unexpected finding of delayed-onset seizures in HIV-positive, treatment-experienced subjects in the Phase IIb evaluation of fosdevirine (GSK2248761) Antivir. Ther. 2014;19:69–78. doi: 10.3851/IMP2689. [DOI] [PubMed] [Google Scholar]
- 40.Zala C., St Clair M., Dudas K., Kim J., Lou Y., White S., Piscitelli S., Dumont E., Pietropaolo K., Zhou X.J., Mayers D. Safety and efficacy of GSK2248761, a next-generation nonnucleoside reverse transcriptase inhibitor, in treatment-naive HIV-1-infected subjects. Antimicrob. Agents Chemother. 2012;56:2570–2575. doi: 10.1128/AAC.05597-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki H., Kato K., Kumagai H. Development of an efficient enzymatic production of gamma-D-glutamyl-L-tryptophan (SCV-07), a prospective medicine for tuberculosis, with bacterial gamma-glutamyltranspeptidase. J. Biotechnol. 2004;111:291–295. doi: 10.1016/j.jbiotec.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 42.Aspinall R.J., Pockros P.J. SCV-07 (SciClone Pharmaceuticals/Verta) Curr. Opin. Investig. Drugs. 2006;7:180–185. [PubMed] [Google Scholar]
- 43.Rasmussen T.A., Schmeltz Sogaard O., Brinkmann C., Wightman F., Lewin S.R., Melchjorsen J., Dinarello C., Ostergaard L., Tolstrup M. Comparison of HDAC inhibitors in clinical development: effect on HIV production in latently infected cells and T-cell activation. Hum. Vaccin. Immunother. 2013;9:993–1001. doi: 10.4161/hv.23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prince H.M., Bishton M.J., Johnstone R.W. Panobinostat (LBH589): a potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009;5:601–612. doi: 10.2217/fon.09.36. [DOI] [PubMed] [Google Scholar]
- 45.Beaulieu P.L., Bos M., Cordingley M.G., Chabot C., Fazal G., Garneau M., Gillard J.R., Jolicoeur E., LaPlante S., McKercher G., Poirier M., Poupart M.A., Tsantrizos Y.S., Duan J., Kukolj G. Discovery of the first thumb pocket 1 NS5B polymerase inhibitor (BILB 1941) with demonstrated antiviral activity in patients chronically infected with genotype 1 hepatitis C virus (HCV) J. Med. Chem. 2012;55:7650–7666. doi: 10.1021/jm3006788. [DOI] [PubMed] [Google Scholar]
- 46.Gentles R.G., Ding M., Bender J.A., Bergstrom C.P., Grant-Young K., Hewawasam P., Hudyma T., Martin S., Nickel A., Regueiro-Ren A., Tu Y., Yang Z., Yeung K.S., Zheng X., Chao S., Sun J.H., Beno B.R., Camac D.M., Chang C.H., Gao M., Morin P.E., Sheriff S., Tredup J., Wan J., Witmer M.R., Xie D., Hanumegowda U., Knipe J., Mosure K., Santone K.S., Parker D.D., Zhuo X., Lemm J., Liu M., Pelosi L., Rigat K., Voss S., Wang Y., Wang Y.K., Colonno R.J., Gao M., Roberts S.B., Gao Q., Ng A., Meanwell N.A., Kadow J.F. Discovery and preclinical characterization of the cyclopropylindolobenzazepine BMS-791325, a potent allosteric inhibitor of the hepatitis C virus NS5B polymerase. J. Med. Chem. 2014;57:1855–1879. doi: 10.1021/jm4016894. [DOI] [PubMed] [Google Scholar]
- 47.Coburn C.A., Meinke P.T., Chang W., Fandozzi C.M., Graham D.J., Hu B., Huang Q., Kargman S., Kozlowski J., Liu R., McCauley J.A., Nomeir A.A., Soll R.M., Vacca J.P., Wang D., Wu H., Zhong B., Olsen D.B., Ludmerer S.W. Discovery of MK-8742: an HCV NS5A inhibitor with broad genotype activity. ChemMedChem. 2013;8:1930–1940. doi: 10.1002/cmdc.201300343. [DOI] [PubMed] [Google Scholar]
- 48.Nakamoto S., Kanda T., Wu S., Shirasawa H., Yokosuka O. Hepatitis C virus NS5A inhibitors and drug resistance mutations. World J. Gastroenterol. 2014;20:2902–2912. doi: 10.3748/wjg.v20.i11.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gentile I., Buonomo A.R., Zappulo E., Borgia G. Interferon-free therapies for chronic hepatitis C: toward a hepatitis C virus-free world? Expert Rev. Anti Infect. Ther. 2014;12:763–773. doi: 10.1586/14787210.2014.929497. [DOI] [PubMed] [Google Scholar]
- 50.Yu F., Lu L., Du L., Zhu X., Debnath A.K., Jiang S. Approaches for identification of HIV-1 entry inhibitors targeting gp41 pocket. Viruses. 2013;5:127–149. doi: 10.3390/v5010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Devogelaere B., Berke J.M., Vijgen L., Dehertogh P., Fransen E., Cleiren E., van der Helm L., Nyanguile O., Tahri A., Amssoms K., Lenz O., Cummings M.D., Clayton R.F., Vendeville S., Raboisson P., Simmen K.A., Fanning G.C., Lin T.I. TMC647055, a potent nonnucleoside hepatitis C virus NS5B polymerase inhibitor with cross-genotypic coverage. Antimicrob. Agents Chemother. 2012;56:4676–4684. doi: 10.1128/AAC.00245-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cummings M.D., Lin T.I., Hu L., Tahri A., McGowan D., Amssoms K., Last S., Devogelaere B., Rouan M.C., Vijgen L., Berke J.M., Dehertogh P., Fransen E., Cleiren E., van der Helm L., Fanning G., Nyanguile O., Simmen K., Van Remoortere P., Raboisson P., Vendeville S. Discovery and early development of TMC647055, a non-nucleoside inhibitor of the hepatitis C virus NS5B polymerase. J. Med. Chem. 2014;57:1880–1892. doi: 10.1021/jm401396p. [DOI] [PubMed] [Google Scholar]
- 53.Yang Z., Zadjura L., D'Arienzo C., Marino A., Santone K., Klunk L., Greene D., Lin P.F., Colonno R., Wang T., Meanwell N., Hansel S. Preclinical pharmacokinetics of a novel HIV-1 attachment inhibitor BMS-378806 and prediction of its human pharmacokinetics. Biopharm. Drug Dispos. 2005;26:387–402. doi: 10.1002/bdd.471. [DOI] [PubMed] [Google Scholar]
- 54.Lin P.F., Blair W., Wang T., Spicer T., Guo Q., Zhou N., Gong Y.F., Wang H.G., Rose R., Yamanaka G., Robinson B., Li C.B., Fridell R., Deminie C., Demers G., Yang Z., Zadjura L., Meanwell N., Colonno R. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. U. S. A. 2003;100:11013–11018. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu R.J., Tucker J.A., Pickens J., Ma Y.A., Zinevitch T., Kirichenko O., Konoplev V., Kuznetsova S., Sviridov S., Brahmachary E., Khasanov A., Mikel C., Yang Y., Liu C., Wang J., Freel S., Fisher S., Sullivan A., Zhou J., Stanfield-Oakley S., Baker B., Sailstad J., Greenberg M., Bolognesi D., Bray B., Koszalka B., Jeffs P., Jeffries C., Chucholowski A., Sexton C. Heterobiaryl human immunodeficiency virus entry inhibitors. J. Med. Chem. 2009;52:4481–4487. doi: 10.1021/jm900330x. [DOI] [PubMed] [Google Scholar]
- 56.Yeung K.S., Qiu Z., Xue Q., Fang H., Yang Z., Zadjura L., D'Arienzo C.J., Eggers B.J., Riccardi K., Shi P.Y., Gong Y.F., Browning M.R., Gao Q., Hansel S., Santone K., Lin P.F., Meanwell N.A., Kadow J.F. Inhibitors of HIV-1 attachment. Part 7: indole-7-carboxamides as potent and orally bioavailable antiviral agents. Bioorg. Med. Chem. Lett. 2013;23:198–202. doi: 10.1016/j.bmcl.2012.10.115. [DOI] [PubMed] [Google Scholar]
- 57.Yeung K.S., Qiu Z., Yin Z., Trehan A., Fang H., Pearce B., Yang Z., Zadjura L., D'Arienzo C.J., Riccardi K., Shi P.Y., Spicer T.P., Gong Y.F., Browning M.R., Hansel S., Santone K., Barker J., Coulter T., Lin P.F., Meanwell N.A., Kadow J.F. Inhibitors of HIV-1 attachment. Part 8: the effect of C7-heteroaryl substitution on the potency, and in vitro and in vivo profiles of indole-based inhibitors. Bioorg. Med. Chem. Lett. 2013;23:203–208. doi: 10.1016/j.bmcl.2012.10.117. [DOI] [PubMed] [Google Scholar]
- 58.Yeung K.S., Qiu Z., Yang Z., Zadjura L., D'Arienzo C.J., Browning M.R., Hansel S., Huang X.S., Eggers B.J., Riccardi K., Lin P.F., Meanwell N.A., Kadow J.F. Inhibitors of HIV-1 attachment. Part 9: an assessment of oral prodrug approaches to improve the plasma exposure of a tetrazole-containing derivative. Bioorg. Med. Chem. Lett. 2013;23:209–212. doi: 10.1016/j.bmcl.2012.10.125. [DOI] [PubMed] [Google Scholar]
- 59.Balupuri A., Gadhe C.G., Balasubramanian P.K., Kothandan G., Cho S.J. In silico study on indole derivatives as anti HIV-1 agents: a combined docking, molecular dynamics and 3D-QSAR study. Arch. Pharm. Res. 2014;37:1001–1015. doi: 10.1007/s12272-013-0313-1. [DOI] [PubMed] [Google Scholar]
- 60.Allen W.J., Rizzo R.C. Computer-aided approaches for targeting HIVgp41. Biol. (Basel) 2012;1:311–338. doi: 10.3390/biology1020311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou G., Wu D., Hermel E., Balogh E., Gochin M. Design, synthesis, and evaluation of indole compounds as novel inhibitors targeting Gp41. Bioorg. Med. Chem. Lett. 2010;20:1500–1503. doi: 10.1016/j.bmcl.2010.01.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou G., Wu D., Snyder B., Ptak R.G., Kaur H., Gochin M. Development of indole compounds as small molecule fusion inhibitors targeting HIV-1 glycoprotein-41. J. Med. Chem. 2011;54:7220–7231. doi: 10.1021/jm200791z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Whitby L.R., Boyle K.E., Cai L., Yu X., Gochin M., Boger D.L. Discovery of HIV fusion inhibitors targeting gp41 using a comprehensive alpha-helix mimetic library. Bioorg. Med. Chem. Lett. 2012;22:2861–2865. doi: 10.1016/j.bmcl.2012.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.S.K. Balani, T.M. Ciccarone, M.E. Goldman, W.J. Greenlee, W.S. Saari, A.D. Theoharides, J.S. Wai, T.M. Williams. Indoles as inhibitors of HIV reverse transcriptase, Patent. WO 1993005020, 1993.
- 65.Williams T.M., Ciccarone T.M., MacTough S.C., Rooney C.S., Balani S.K., Condra J.H., Emini E.A., Goldman M.E., Greenlee W.J., Kauffman L.R., O'Brien J.A., Sardana V.V., Schleif W.A., Theoharides A.D., Anderson P.S. 5-chloro-3-(phenylsulfonyl)indole-2-carboxamide: a novel, non-nucleoside inhibitor of HIV-1 reverse transcriptase. J. Med. Chem. 1993;36:1291–1294. doi: 10.1021/jm00061a022. [DOI] [PubMed] [Google Scholar]
- 66.Silvestri R., Artico M., De Martino G., La Regina G., Loddo R., La Colla M., Mura M., La Colla P. Simple, short peptide derivatives of a sulfonylindolecarboxamide (L-737,126) active in vitro against HIV-1 wild type and variants carrying non-nucleoside reverse transcriptase inhibitor resistance mutations. J. Med. Chem. 2004;47:3892–3896. doi: 10.1021/jm031147e. [DOI] [PubMed] [Google Scholar]
- 67.La Regina G., Coluccia A., Brancale A., Piscitelli F., Famiglini V., Cosconati S., Maga G., Samuele A., Gonzalez E., Clotet B., Schols D., Este J.A., Novellino E., Silvestri R. New nitrogen containing substituents at the indole-2-carboxamide yield high potent and broad spectrum indolylarylsulfone HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2012;55:6634–6638. doi: 10.1021/jm300477h. [DOI] [PubMed] [Google Scholar]
- 68.Zhao Z., Wolkenberg S.E., Sanderson P.E., Lu M., Munshi V., Moyer G., Feng M., Carella A.V., Ecto L.T., Gabryelski L.J., Lai M.T., Prasad S.G., Yan Y., McGaughey G.B., Miller M.D., Lindsley C.W., Hartman G.D., Vacca J.P., Williams T.M. Novel indole-3-sulfonamides as potent HIV non-nucleoside reverse transcriptase inhibitors (NNRTIs) Bioorg. Med. Chem. Lett. 2008;18:554–559. doi: 10.1016/j.bmcl.2007.11.085. [DOI] [PubMed] [Google Scholar]
- 69.Ragno R., Artico M., De Martino G., La Regina G., Coluccia A., Di Pasquali A., Silvestri R. Docking and 3-D QSAR studies on indolyl aryl sulfones. Binding mode exploration at the HIV-1 reverse transcriptase non-nucleoside binding site and design of highly active N-(2-hydroxyethyl)carboxamide and N-(2-hydroxyethyl)carbohydrazide derivatives. J. Med. Chem. 2005;48:213–223. doi: 10.1021/jm040854k. [DOI] [PubMed] [Google Scholar]
- 70.Ragno R., Coluccia A., La Regina G., De Martino G., Piscitelli F., Lavecchia A., Novellino E., Bergamini A., Ciaprini C., Sinistro A., Maga G., Crespan E., Artico M., Silvestri R. Design, molecular modeling, synthesis, and anti-HIV-1 activity of new indolyl aryl sulfones. Novel derivatives of the indole-2-carboxamide. J. Med. Chem. 2006;49:3172–3184. doi: 10.1021/jm0512490. [DOI] [PubMed] [Google Scholar]
- 71.La Regina G., Coluccia A., Brancale A., Piscitelli F., Gatti V., Maga G., Samuele A., Pannecouque C., Schols D., Balzarini J., Novellino E., Silvestri R. Indolylarylsulfones as HIV-1 non-nucleoside reverse transcriptase inhibitors: new cyclic substituents at indole-2-carboxamide. J. Med. Chem. 2011;54:1587–1598. doi: 10.1021/jm101614j. [DOI] [PubMed] [Google Scholar]
- 72.Famiglini V., La Regina G., Coluccia A., Pelliccia S., Brancale A., Maga G., Crespan E., Badia R., Clotet B., Este J.A., Cirilli R., Novellino E., Silvestri R. New indolylarylsulfones as highly potent and broad spectrum HIV-1 non-nucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem. 2014;80:101–111. doi: 10.1016/j.ejmech.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 73.R. Storer, F.R. Alexandre, C. Dousson, A.M. Moussa, E. Bridges, A. Stewart, J. Y. Wang, B. A. Mayes. Enantiomerically pure phosphoindoles as HIV inhibitors, Patent. WO 2008042240, 2008.
- 74.Piscitelli F., Coluccia A., Brancale A., La Regina G., Sansone A., Giordano C., Balzarini J., Maga G., Zanoli S., Samuele A., Cirilli R., La Torre F., Lavecchia A., Novellino E., Silvestri R. Indolylarylsulfones bearing natural and unnatural amino acids. Discovery of potent inhibitors of HIV-1 non-nucleoside wild type and resistant mutant strains reverse transcriptase and coxsackie B4 virus. J. Med. Chem. 2009;52:1922–1934. doi: 10.1021/jm801470b. [DOI] [PubMed] [Google Scholar]
- 75.Alexandre F.R., Amador A., Bot S., Caillet C., Convard T., Jakubik J., Musiu C., Poddesu B., Vargiu L., Liuzzi M., Roland A., Seifer M., Standring D., Storer R., Dousson C.B. Synthesis and biological evaluation of aryl-phospho-indole as novel HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2011;54:392–395. doi: 10.1021/jm101142k. [DOI] [PubMed] [Google Scholar]
- 76.Hassam M., Basson A.E., Liotta D.C., Morris L., van Otterlo W.A., Pelly S.C. Novel Cyclopropyl-indole derivatives as HIV non-nucleoside reverse transcriptase inhibitors. ACS Med. Chem. Lett. 2012;3:470–475. doi: 10.1021/ml3000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee W.G., Gallardo-Macias R., Frey K.M., Spasov K.A., Bollini M., Anderson K.S., Jorgensen W.L. Picomolar inhibitors of HIV reverse transcriptase featuring bicyclic replacement of a cyanovinylphenyl group. J. Am. Chem. Soc. 2013;135:16705–16713. doi: 10.1021/ja408917n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Han X., Ouyang W., Liu B., Wang W., Tien P., Wu S., Zhou H.B. Enantioselective inhibition of reverse transcriptase (RT) of HIV-1 by non-racemic indole-based trifluoropropanoates developed by asymmetric catalysis using recyclable organocatalysts. Org. Biomol. Chem. 2013;11:8463–8475. doi: 10.1039/c3ob41667d. [DOI] [PubMed] [Google Scholar]
- 79.Ashok Penta S.C., Ganguly S. De novo design and in-silico studies of novel 1-phenyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole -3-carboxylic acid derivatives as HIV-1 reverse transcriptase inhibitors. Med. Chem. Res. 2014;23:3662–3670. [Google Scholar]
- 80.Xu H., Liu W.Q., Fan L.L., Chen Y., Yang L.M., Lv L., Zheng Y.T. Synthesis and HIV-1 integrase inhibition activity of some N-arylindoles. Chem. Pharm. Bull. 2008;56:720–722. doi: 10.1248/cpb.56.720. [DOI] [PubMed] [Google Scholar]
- 81.Jain S.V., Sonawane L.V., Patil R.R., Bari S.B. Pharmacophore modeling of some novel indole β-diketo acid and coumarin-based derivatives as HIV integrase inhibitors. Med. Chem. Res. 2012;21:165–173. [Google Scholar]
- 82.Goldgur Y., Craigie R., Cohen G.H., Fujiwara T., Yoshinaga T., Fujishita T., Sugimoto H., Endo T., Murai H., Davies D.R. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc. Natl. Acad. Sci. U. S. A. 1999;96:13040–13043. doi: 10.1073/pnas.96.23.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Espeseth A.S., Felock P., Wolfe A., Witmer M., Grobler J., Anthony N., Egbertson M., Melamed J.Y., Young S., Hamill T., Cole J.L., Hazuda D.J. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc. Natl. Acad. Sci. U. S. A. 2000;97:11244–11249. doi: 10.1073/pnas.200139397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sechi M., Derudas M., Dallocchio R., Dessi A., Bacchi A., Sannia L., Carta F., Palomba M., Ragab O., Chan C., Shoemaker R., Sei S., Dayam R., Neamati N. Design and synthesis of novel indole beta-diketo acid derivatives as HIV-1 integrase inhibitors. J. Med. Chem. 2004;47:5298–5310. doi: 10.1021/jm049944f. [DOI] [PubMed] [Google Scholar]
- 85.Ferro S., Barreca M.L., De Luca L., Rao A., Monforte A.M., Debyser Z., Witvrouw M., Chimirri A. New 4-[(1-benzyl-1H-indol-3-yl)carbonyl]-3-hydroxyfuran-2(5H)-ones, beta-diketo acid analogs as HIV-1 integrase inhibitors. Arch. Pharm. (Weinheim) 2007;340:292–298. doi: 10.1002/ardp.200700066. [DOI] [PubMed] [Google Scholar]
- 86.De Luca L., Barreca M.L., Ferro S., Iraci N., Michiels M., Christ F., Debyser Z., Witvrouw M., Chimirri A. A refined pharmacophore model for HIV-1 integrase inhibitors: optimization of potency in the 1H-benzylindole series. Bioorg. Med. Chem. Lett. 2008;18:2891–2895. doi: 10.1016/j.bmcl.2008.03.089. [DOI] [PubMed] [Google Scholar]
- 87.De Luca L., Barreca M.L., Ferro S., Christ F., Iraci N., Gitto R., Monforte A.M., Debyser Z., Chimirri A. Pharmacophore-based discovery of small-molecule inhibitors of protein-protein interactions between HIV-1 integrase and cellular cofactor LEDGF/p75. ChemMedChem. 2009;4:1311–1316. doi: 10.1002/cmdc.200900070. [DOI] [PubMed] [Google Scholar]
- 88.De Luca L., Gitto R., Christ F., Ferro S., De Grazia S., Morreale F., Debyser Z., Chimirri A. 4-[1-(4-Fluorobenzyl)-4-hydroxy-1H-indol-3-yl]-2-hydroxy-4-oxobut-2-enoic acid as a prototype to develop dual inhibitors of HIV-1 integration process. Antiviral Res. 2011;92:102–107. doi: 10.1016/j.antiviral.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 89.Ferro S., Grazia S.D., De Luca L., Gitto R., Faliti C.E., Debyzer Z., Chimirri A. Microwave assisted organic synthesis (MAOS) of small molecules as potential HIV-1 integrase inhibitors. Molecules. 2011;16:6858–6870. doi: 10.3390/molecules16086858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.De Luca L., De Grazia S., Ferro S., Gitto R., Christ F., Debyser Z., Chimirri A. HIV-1 integrase strand-transfer inhibitors: design, synthesis and molecular modeling investigation. Eur. J. Med. Chem. 2011;46:756–764. doi: 10.1016/j.ejmech.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 91.Rogolino D., Carcelli M., Compari C., De Luca L., Ferro S., Fisicaro E., Rispoli G., Neamati N., Debyser Z., Christ F., Chimirri A. Diketoacid chelating ligands as dual inhibitors of HIV-1 integration process. Eur. J. Med. Chem. 2014;78:425–430. doi: 10.1016/j.ejmech.2014.03.070. [DOI] [PubMed] [Google Scholar]
- 92.Ferro S., De Luca L., Lo Surdo G., Morreale F., Christ F., Debyser Z., Gitto R., Chimirri A. A new potential approach to block HIV-1 replication via protein-protein interaction and strand-transfer inhibition. Bioorg. Med. Chem. 2014;22:2269–2279. doi: 10.1016/j.bmc.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 93.De Luca L., Ferro S., Morreale F., Christ F., Debyser Z., Chimirri A., Gitto R. Fragment hopping approach directed at design of HIV IN-LEDGF/p75 interaction inhibitors. J. Enzyme Inhib. Med. Chem. 2013;28:1002–1009. doi: 10.3109/14756366.2012.703184. [DOI] [PubMed] [Google Scholar]
- 94.Anderson J., Schiffer C., Lee S.K., Swanstrom R. Viral protease inhibitors. Handb. Exp. Pharmacol. 2009;189:85–110. doi: 10.1007/978-3-540-79086-0_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Flint O.P., Noor M.A., Hruz P.W., Hylemon P.B., Yarasheski K., Kotler D.P., Parker R.A., Bellamine A. The role of protease inhibitors in the pathogenesis of HIV-associated lipodystrophy: cellular mechanisms and clinical implications. Toxicol. Pathol. 2009;37:65–77. doi: 10.1177/0192623308327119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wei H.Y., Lu C.S., Lin T.H. Exploring the P2 and P3 ligand binding features for hepatitis C virus NS3 protease using some 3D QSAR techniques. J. Mol. Graph. Model. 2008;26:1131–1144. doi: 10.1016/j.jmgm.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 97.Ismail N.S., El Dine R.S., Hattori M., Takahashi K., Ihara M. Computer based design, synthesis and biological evaluation of novel indole derivatives as HCV NS3-4A serine protease inhibitors. Bioorg. Med. Chem. 2008;16:7877–7887. doi: 10.1016/j.bmc.2008.07.084. [DOI] [PubMed] [Google Scholar]
- 98.Gopalsamy A., Shi M., Ciszewski G., Park K., Ellingboe J.W., Orlowski M., Feld B., Howe A.Y. Design and synthesis of 2,3,4,9-tetrahydro-1H-carbazole and 1,2,3,4-tetrahydro-cyclopenta[b]indole derivatives as non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA polymerase. Bioorg. Med. Chem. Lett. 2006;16:2532–2534. doi: 10.1016/j.bmcl.2006.01.105. [DOI] [PubMed] [Google Scholar]
- 99.Zhang M.Z., Wang Y., Paueksakon P., Harris R.C. Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes. 2014;63:2063–2072. doi: 10.2337/db13-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lampis G., Deidda D., Maullu C., Madeddu M.A., Pompei R., Delle Monachie F., Satta G. Sattabacins and sattazolins: new biologically active compounds with antiviral properties extracted from a Bacillus sp. J. Antibiot. (Tokyo) 1995;48:967–972. doi: 10.7164/antibiotics.48.967. [DOI] [PubMed] [Google Scholar]
- 101.Snyder K.M., Doty T.S., Heins S.P., DeSouchet A.L., Miller K.A. Asymmetric total synthesis of (+)-sattazolin. Tetrahedron Lett. 2013;54:192–194. [Google Scholar]
- 102.Hsieh P.W., Chang F.R., Lee K.H., Hwang T.L., Chang S.M., Wu Y.C. A new anti-HIV alkaloid, drymaritin, and a new C-glycoside flavonoid, diandraflavone, from Drymaria diandra. J. Nat. Prod. 2004;67:1175–1177. doi: 10.1021/np0400196. [DOI] [PubMed] [Google Scholar]
- 103.Pinto A.M.V., Leite J.P.G., Ferreira W.J., Cavalcanti D.N., Villca R.C., Giongo V., Teixeira V.L., Paixão I.C.N.D.P. Marine natural seaweed products as potential antiviral drugs against Bovine viral diarrhea virus. Br. J. Pharm. 2012;22:813–817. [Google Scholar]
- 104.Chen M., Gan L., Lin S., Wang X., Li L., Li Y., Zhu C., Wang Y., Jiang B., Jiang J., Yang Y., Shi J. Alkaloids from the root of Isatis indigotica. J. Nat. Prod. 2012;75:1167–1176. doi: 10.1021/np3002833. [DOI] [PubMed] [Google Scholar]
- 105.Bag P., Ojha D., Mukherjee H., Halder U.C., Mondal S., Biswas A., Sharon A., Van Kaer L., Chakrabarty S., Das G., Mitra D., Chattopadhyay D. A dihydro-pyrido-indole potently inhibits HSV-1 infection by interfering the viral immediate early transcriptional events. Antiviral Res. 2014;105:126–134. doi: 10.1016/j.antiviral.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 106.Karuppannan A.K., Wu K.X., Qiang J., Chu J.J., Kwang J. Natural compounds inhibiting the replication of Porcine reproductive and respiratory syndrome virus. Antiviral Res. 2012;94:188–194. doi: 10.1016/j.antiviral.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Garg N.K., Sarpong R., Stoltz B.M. The first total synthesis of dragmacidin d. J. Am. Chem. Soc. 2002;124:13179–13184. doi: 10.1021/ja027822b. [DOI] [PubMed] [Google Scholar]
- 108.Cutignano Adele, Bifulco Giuseppe, Bruno Ines, Casapullo Agostino, Gomez-Paloma Luigi, Riccio R. Dragmacidin F: a new antiviral bromoindole alkaloid from the Mediterranean sponge Halicortex sp. Tetrahedron. 2000;56:3743–3748. [Google Scholar]
- 109.Zhou X., Liu J., Yang B., Lin X., Yang X.W., Liu Y. Marine natural products with anti-HIV activities in the last decade. Curr. Med. Chem. 2013;20:953–973. [PubMed] [Google Scholar]
- 110.Gupta L., Talwar A., Chauhan P.M. Bis and tris indole alkaloids from marine organisms: new leads for drug discovery. Curr. Med. Chem. 2007;14:1789–1803. doi: 10.2174/092986707781058904. [DOI] [PubMed] [Google Scholar]
- 111.Tan C.J., Di Y.T., Wang Y.H., Zhang Y., Si Y.K., Zhang Q., Gao S., Hu X.J., Fang X., Li S.F., Hao X.J. Three new indole alkaloids from Trigonostemon lii. Org. Lett. 2010;12:2370–2373. doi: 10.1021/ol100715x. [DOI] [PubMed] [Google Scholar]
- 112.Takada K., Kajiwara H., Imamura N. Oridamycins A and B, anti-Saprolegnia parasitica indolosesquiterpenes isolated from Streptomyces sp. KS84. J. Nat. Prod. 2010;73:698–701. doi: 10.1021/np1000522. [DOI] [PubMed] [Google Scholar]
- 113.Ding L., Munch J., Goerls H., Maier A., Fiebig H.H., Lin W.H., Hertweck C. Xiamycin, a pentacyclic indolosesquiterpene with selective anti-HIV activity from a bacterial mangrove endophyte. Bioorg. Med. Chem. Lett. 2010;20:6685–6687. doi: 10.1016/j.bmcl.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 114.Ding L., Maier A., Fiebig H.H., Lin W.H., Hertweck C. A family of multicyclic indolosesquiterpenes from a bacterial endophyte. Org. Biomol. Chem. 2011;9:4029–4031. doi: 10.1039/c1ob05283g. [DOI] [PubMed] [Google Scholar]
- 115.Xu Z., Baunach M., Ding L., Hertweck C. Bacterial synthesis of diverse indole terpene alkaloids by an unparalleled cyclization sequence. Angew. Chem. Int. Ed. 2012;51:10293–10297. doi: 10.1002/anie.201204087. [DOI] [PubMed] [Google Scholar]
- 116.Peng J., Lin T., Wang W., Xin Z., Zhu T., Gu Q., Li D. Antiviral alkaloids produced by the mangrove-derived fungus Cladosporium sp. PJX-41. J. Nat. Prod. 2013;76:1133–1140. doi: 10.1021/np400200k. [DOI] [PubMed] [Google Scholar]
- 117.Gul W., Hamann M.T. Indole alkaloid marine natural products: an established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005;78:442–453. doi: 10.1016/j.lfs.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Singh J.P., Tamang S., Rajamohanan P.R., Jima N.C., Chakraborty G., Kundu G.C., Gaikwad S.M., Khan M.I. Isolation, structure, and functional elucidation of a modified pentapeptide, cysteine protease inhibitor (CPI-2081) from Streptomyces species 2081 that exhibit inhibitory effect on cancer cell migration. J. Med. Chem. 2010;53:5121–5128. doi: 10.1021/jm9014179. [DOI] [PubMed] [Google Scholar]
- 119.Heapy A.M., Patterson A.V., Smaill J.B., Jamieson S.M., Guise C.P., Sperry J., Hume P.A., Rathwell K., Brimble M.A. Synthesis and cytotoxicity of pyranonaphthoquinone natural product analogues under bioreductive conditions. Bioorg. Med. Chem. 2013;21:7971–7980. doi: 10.1016/j.bmc.2013.09.052. [DOI] [PubMed] [Google Scholar]
- 120.Yu X., Sun D. Macrocyclic drugs and synthetic methodologies toward macrocycles. Molecules. 2013;18:6230–6268. doi: 10.3390/molecules18066230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Driggers E.M., Hale S.P., Lee J., Terrett N.K. The exploration of macrocycles for drug discovery–an underexploited structural class. Nat. Rev. Drug Discov. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 122.Mallinson J., Collins I. Macrocycles in new drug discovery. Future Med. Chem. 2012;4:1409–1438. doi: 10.4155/fmc.12.93. [DOI] [PubMed] [Google Scholar]
- 123.Krahn D., Ottmann C., Kaiser M. Macrocyclic proteasome inhibitors. Curr. Med. Chem. 2011;18:5052–5060. doi: 10.2174/092986711797636063. [DOI] [PubMed] [Google Scholar]
- 124.Blunt J.W., Copp B.R., Munro M.H., Northcote P.T., Prinsep M.R. Marine natural products. Nat. Prod. Rep. 2005;22:15–61. doi: 10.1039/b415080p. [DOI] [PubMed] [Google Scholar]
- 125.Somei M., Yamada F. Simple indole alkaloids and those with a non-rearranged monoterpenoid unit. Nat. Prod. Rep. 2005;22:73–103. doi: 10.1039/b316241a. [DOI] [PubMed] [Google Scholar]