

Abstract
Survival has improved in patients with cystic fibrosis (CF), in part because of aggressive antimicrobial management. Two multidrug-resistant environmental bacteria, the Burkholderia cepacia group and nontuberculous mycobacteria, have emerged. Improving genomic and proteomic technologies are allowing better identification of bacteria and fungi found in the CF lung and detection of viral agents that may be associated with pulmonary exacerbations. Anaerobic bacteria and Streptococcus angionsus group organisms may play a role in chronic CF lung infections. The diversity of organisms declines perhaps as a result of aggressive antimicrobial therapy, and an apex predator, Pseudomonas aeruginosa, may emerge in many patients with CF.
Keywords: Infection, Cystic fibrosis, Diagnostic microbiology, Update
Key points
-
•
Cystic fibrosis is the most important genetic disease in Caucasians. Patients with this disease die prematurely primarily as a result of chronic lung infection. Staphylococcus aureus and mucoid Pseudomonas aeruginosa continue to be the key pulmonary pathogens.
-
•
Survival has improved in patients with CF in part because of aggressive antimicrobial management. An unintended consequence of this therapy has been the emergence of 2 multidrug-resistant environmental bacteria: the Burkholderia cepacia group and nontuberculous mycobacteria.
-
•
Burkholderia cenocepacia, a species within the Burkholderia cepacia complex, is associated with high mortality and is a contraindication for lung transplantation. The key nontuberculous mycobacterial pathogen, Mycobacterium abscessus, is not so virulent and is not a lung transplantation contraindication. Both present an infection control challenge, because they can be spread from person to person.
-
•
Improving genomic and proteomic technologies are allowing better identification of bacteria and fungi found in the CF lung and to detect viral agents that may be associated with pulmonary exacerbations. Chronic rhinovirus infections are of particular interest.
-
•
Microbiome studies have identified 2 groups of bacteria that may play a role in chronic CF lung infections: anaerobic bacteria and Streptococcus angionsus group organisms. Microbiome studies also show that as the diversity of organisms decline, perhaps as a result of aggressive antimicrobial therapy, an apex predator, etc., Pseudomonas aeruginosa, may emerge in many patients with CF.
Introduction, epidemiology, and clinical presentation
Cystic fibrosis (CF) is the most common autosomal-recessive genetic disease that occurs in non-Hispanic Caucasians populations, although other racial groups may have this disease as well.1 Affected individuals have mutation in the CF transmembrane conductance gene (CFTR), a membrane protein involved in sodium and chloride transport in epithelial cells.2 The resulting dysregulation in electrolyte transport leads to depletion in airway surface liquid on bronchial epithelial cell surfaces. As a result, patients with CF have thick, dry, tenacious mucus, which impairs mucociliary clearance of particulates, especially bacteria and fungal conidia, from the airways. This environment is ideal for the growth of a limited number of organisms, primarily those that thrive in natural environments such as water. This thickened mucus provides an ideal niche for the establishment of chronic infection. It is this chronic infection that results in the premature death that in seen in CF.3
More than 1800 CFTR mutations have been associated with CF.4 The most common mutation is F508del, which is found in ∼85% of people in the United States; approximately 47% are homozygous for this mutated gene.4 Further carrier rate for mutated CFTR genes is estimated to range from 1/25 for non-Hispanic Caucasians to 1/61 for African Americans to 1/94 for Asian Americans.1 CF is seen most frequently in North America, Northern Europe, Australia, New Zealand, Brazil, and Argentina. It is estimated that 1 in 3500 live births result in clinical disease.4
Currently life expectancy in US patients with CF is approximately 38 years, significantly less than that of the general population.4 Cardiopulmonary failure secondary to chronic lung disease is responsible for 85% of premature deaths in CF. The airways of patients with CF become infected in infancy. This situation begins periods of chronic infection and lung inflammation with accompanying cough, which is a lifelong reality in patients with CF. A hallmark of chronic infection and airway inflammation is periods of pulmonary exacerbations. Pulmonary exacerbations are characterized by worsening symptoms, including increased cough and sputum production, hemoptysis, shortness of breath, increased respiratory rate, loss of appetite, weight loss, increased neutrophil counts, and declining pulmonary function.5 The events that trigger these pulmonary exacerbations are not clearly understood, although viral infections and perhaps changes in the microbiome may be important.6, 7 Exacerbations are characterized by the recruitment of neutrophils, cytokine release, and high level of neutrophil-derived elastases in the bronchi and bronchioles, causing significant lung disease.8 Antimicrobial therapy has been shown to be effective in treating exacerbations symptomatically.9 However, over time, lung function deteriorates and becomes so low, that it is no longer compatible with life (Fig. 1 ). Only lung transplantation can successfully reverse this disease course.8
Fig. 1.

Lung function by age group, 2011. FEV1, forced expiratory volume in 1 second.
(From Cystic Fibrosis Foundation. Cystic Fibrosis Foundation patient registry 2011 annual data report. 2012.)
Microbiology, diagnostic considerations, and susceptibility testing of agents of chronic infection
Over the past 4 decades, our understanding of the complex nature of chronic lung infections has greatly expanded. Over the past 3 decades, there has been more than a doubling in life expectancy in the population with CF.4 Three factors have been central to this improvement:
-
a.
More effective antimicrobial therapy and treatment strategies, with early eradication of Pseudomonas aeruginosa being a key strategy
-
b.
Improvement in airway clearance techniques
-
c.
Improvements in infection control techniques to prevent the spread of organisms highly virulent to patients with CF, especially Burkholderia cenocepacia 2, 9
With the use of broader-spectrum antimicrobials, we are seeing a plethora of emerging highly resistant bacteria and fungi in the CF airways. Our understanding of the role of these organisms in chronic infection and inflammation is poorly delineated (Box 1 ). Over the past decades, new technologies (Fig. 2 ) have been developed and applied to this understanding. These technologies include nucleic acid amplification techniques (NAATs) for direct organism detection, including multiplex NAAT for viruses; the use of DNA sequence analysis for organism identification; molecularly based epidemiologic techniques, including pulsed field gel electrophoresis (PFGE), multilocus sequence type, whole genome sequencing; and matrix-assisted laser desorption ionization–time of flight mass spectroscopy (MALDI-TOF MS).
Box 1. Pathogenic potential of commonly recovered organisms from chronic CF airway infections or pulmonary exacerbations.
-
•Known
-
○Pseudomonas aeruginosa
-
○Staphylococcus aureus
-
▪Methicillin resistant
-
▪Small colony variant
-
▪
-
○Burkholderia multivorans
-
○Burkholderia cenocepacia
-
○Burkholderia dolosa
-
○Aspergillus spp
-
○Scedosporium spp
-
○Mycobacterium abscessus
-
○Influenza virus
-
○Respiratory syncytial virus
-
○
-
•Possible/likely
-
○Haemophilus influenzae
-
○Mycobacterium avium complex
-
○Anaerobic bacteria especially Prevotella spp
-
○Streptococcus anginosus group
-
○Respiratory viruses other than influenza virus and respiratory syncytial virus
-
○
-
•Unknown
-
○Nocardia spp
-
○Pandoraea spp
-
○Other members of the Burkholderia cepacia complex
-
○Inquilinus spp
-
○Trichosporon spp
-
○
-
•Unlikely
-
○Stenotrophomonas maltophilia
-
○Achromobacter spp
-
○Ralstonia spp
-
○Burkholderia gladioli
-
○Streptococcus pneumoniae
-
○Candida albicans
-
○
Fig. 2.

CF lung infection diagnostics 2014.
Perhaps the most exciting development in the application of new technologies is the use of direct sequencing technique to study the CF microbiome.5, 10, 11, 12, 13 These studies hold the promise for new understanding of the disease process, with accompanying improved treatment strategies.
Staphylococcus aureus
Before the antimicrobial era, few children with CF survived past the age of 2 years. The children died primarily from failure to thrive, coupled with bacterial pneumonia. The organism found in the lungs at autopsy in these children was almost always Staphylococcus aureus.14 With the advent of antistaphylococcal antimicrobial therapy, mortality caused by S aureus decreased substantially, but it continues to be the predominant CF pathogen from birth to adolescence, with 80% of children infected in their preadolescence and early adolescence.4 Improvements in antistaphylococcal therapy, especially the use of oral agents such as oral cephalosporins and trimethoprim-sulfamethoxazole (TMP-SMX), are likely to have contributed to the improved life expectancy seen in patients with CF.3, 15 However this effective antistaphylococcal therapy is not without its costs.
Three antimicrobial resistance issues have arisen, which must be considered in the care of patients with CF. First, the use of oral TMP-SMX has been associated with the emergence of small colony variants (SCV) of S aureus.16, 17, 18 The resistance mechanism in these organisms is their ability to use thymidine obtained from the environment, bypassing the folic acid synthesis pathway, the target of TMP-SMX. Such organisms are considered to be thymidine dependent. Such organisms are not as fit as wild-type organism and grow slowly, producing small, streptococcallike colonies on most enrichment media.16 Another SCV that is seen in S aureus are strains with defect in electron transport. These strains are resistant to aminoglycosides. These mutants cannot generate sufficient proton motor force to drive transport of these large, highly charged molecules into the bacterial cytoplasm.17 SCV are less susceptible to antimicrobials, especially β-lactams, because of their slow growth rate, making them more challenging to treat and resulting in a greater decline in pulmonary function.17, 19
The second resistance problem in patients with CF is the problem of methicillin-resistant S aureus (MRSA). It is now recognized that approximately 20% to 25% of patients with CF have MRSA (Fig. 3 ).4, 20 It has also been recognized that patients with CF with MRSA have a higher mortality than those patients with CF without this organism.21 Most CF MRSA isolates can be divided into 2 molecular types based on staphylococcal chromosomal cassette (SCC) mecA type and whether they have the gene for Panton-Valentine leukocidin (pvl). Approximately 70% are SCCmecA type II pvl negative and 17% are SCCmecA type IV pvl positive; the other 13% are SCCmecA type IV pvl negative.20 SCCmecA type IV, pvl positive MRSA isolates are also referred to as community-acquired (CA)-MRSA strains. Initial studies of respiratory infections in healthy adults caused by CA-MRSA suggested that this organism caused severe, necrotizing pneumonia, with significant morbidity and mortality in excess of 50% in some series.22 We were concerned that this organism would be a problem in patients with CF as well. In a survey performed between 2005 and 2007, we found that approximately 3% of patients with CF in our institution had CA-MRSA recovered from their respiratory specimen and that approximately 1% of patients in our center were chronically infected. Further, no episodes of necrotizing pneumonia were seen in our population of patients with CF during that period.23
Fig. 3.

Germs found in the lungs of people with CF by age, 2012.
(From Cystic Fibrosis Foundation. Cystic Fibrosis Foundation patient registry 2012 annual data report. 2013.)
The third antimicrobial resistance issue we have observed is vancomycin-intermediate S aureus (VISA). The mechanism of resistance is believed to be a thickened cell wall, which prevents vancomycin from reaching the site of peptidoglycan synthesis.24 We have screened S aureus isolates for the VISA phenotype (a vancomycin minimum inhibitory concentration [MIC] of 4–8 μg/mL) for the past 5 years by the use of vancomycin E-test or the Vitek 2 susceptibility testing (Biomerieux, Durham, NC). We have tested in excess of 1500 isolates from more than 700 patients with CF (Miller M and Gilligan P, 2013, unpublished data). We have found VISA in only 1 patient. This patient had been chronically infected with MRSA for more than 10 years and had received repeated courses of vancomycin. Over time, her pulmonary function declined. With the emergence of VISA, she was treated with multiple courses of linezolid and ceftaroline. Neither was successful in eradicating the organism. Because of declining lung function, she received a double lung transplant. On her most recent bronchoscopy culture 9 months after transplant, she had positive S aureus infection culture; the organism was both vancomycin and oxacillin susceptible, but the VISA strain has not been recovered from multiple bronchoscopy specimens obtained during the posttransplant period (Miller M and Gilligan P, 2013, unpublished data).
Laboratory detection and identification of S aureus from patients with CF is straightforward. Because these patients often are infected with other organisms, especially mucoid P aeruginosa, selective media for the recovery should be used.25 Two media, mannitol salts agar (Fig. 4 ) and chromogenic S aureus agar, are effective in the recovery of SCV and wild-type strains.16, 26 SCV S aureus have typical morphology on mannitol salts agar, whereas on chromogenic media, these organisms may grow more slowly and not give typical phenotypic reactions.16, 26 Organism identification is straightforward, with catalase and latex agglutination or coagulase testing being highly accurate.27 MALDI-TOF MS works well for identification of S aureus from patients with CF, although its ability to speciate SCV has not been reported.28 Susceptibility testing is straightforward, except for the detection of VISA strains. For those isolates, the most accurate method is an MIC-based method. Because E-test MICs tend to run higher than more conventional agar or broth dilution methods, all S aureus isolates with a vancomycin MIC of 4 μg/mL or greater should be confirmed by a conventional MIC method.29
Fig. 4.

Staphylococcus aureus on mannitol salt agar.
Pseudomonas aeruginosa
The key pathogen in chronic lung disease is an unusual morphotype of P aeruginosa, referred to as mucoid. The organism is typically obtained from the patient’s environment, although patient-to-patient spread of virulent clones does occur.30 The pathogenesis of P aeruginosa is dependent on the transition of the organism from a motile, virulent, nonmucoid organism to a nonmotile, comparatively avirulent, mucoid organism, which has adapted to living in the CF airways. The transition is believed to be caused by mutations that inactivate the mucA gene, which regulates alginate production. Alginate-producing organisms grow in a biofilm within the mucous layer in bronchioles of the CF lung. These organisms may be observed as microcolonies when the mucous layer is observed microscopically.31 On agar plates, these organisms appear as mucoid colonies (Fig. 5 ). In the lung, these organisms are resistant to mechanical clearance, phagocytosis, and antimicrobial therapy.3, 31 Although they are comparatively avirulent, they induce a chronic inflammatory response, which is believed to be responsible for the lung damage that results in the patient’s demise.31 This disease process takes place over a period of years to decades.
Fig. 5.
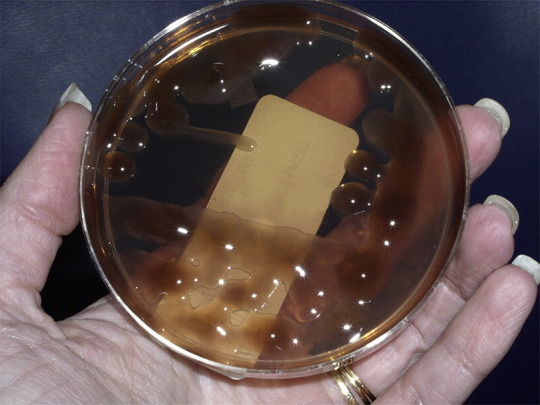
Mucoid Pseudomonas aeruginosa on MacConkey agar.
In the United States, 60% to 75% of adults are infected with this organism.4 The longer a patient can remain free of chronic infection with P aeruginosa, the longer the life expectancy is. A highly aggressively antimicrobial therapeutic approach, first used in Danish patients with CF, for eradicating early infection with P aeruginosa has been shown to lengthen the time that patients with CF remain P aeruginosa free. As a result, Danish patients with CF have perhaps the longest life expectancy of any CF populations in the world, at 42 years.9
Laboratory diagnosis of infections with this organism is easily accomplished. It is readily recovered on both nonselective agar (chocolate) or selective agar (MacConkey). Selective agar specific for P aeruginosa is not required. In addition, identification is easily accomplished. The organism is oxidase positive, mucoid, and produces green (pyoverdin) or blue-green (pyocyanin) pigments. These characteristics identify at least 90% of CF isolates.25 MALDI-TOF MS has been shown to accurately identify both mucoid and nonmucoid strains of P aeruginosa.28, 32
Susceptibility testing of P aeruginosa from patients with CF can be challenging. Antimicrobial susceptibility results using automated susceptibility systems correlate poorly with reference methods and are not recommended. Rather, disk diffusion or E-tests are recommended.33 Paradoxically, it is unlikely that conventional susceptibility testing accurately predicts in vivo response.34 The reasons for this situation are complex but can be explained in part by comparing susceptibility of P aeruginosa with antimicrobials when the organism is grown in a conventional manner or planktonically versus a biofilm mode of growth. When P aeruginosa is grown as a biofilm, it is 8 to 32 times more resistant to all classes of β-lactams than when it is grown planktonically.35 This difference can be explained in part by reduced growth rates of mucoid colonies and by the possibility that these antimicrobials cannot penetrate to the cell through the alginate layer. There is little difference between the MICs for aminoglcyosides or fluoroquinolones grown under these 2 conditions.35 Further complicating interpretation of these results is a growing body of literature that suggests that P aeruginosa may grow under anaerobic conditions behind mucous plugs or in the thickened mucus of the airway.36 Tobramycin is not active under anaerobic conditions, because of poor transport into bacteria. Aerosolized tobramycin therapy, a key strategy in early eradication of P aeruginosa,9, 31 may act only against organisms in the airway that are growing in the superficial layer of mucus. Chronic P aeruginosa infection may be established only when the organism is growing as a biofilm in the anoxic portion of the mucous layer. Further complexity in susceptibility testing is caused by the widespread use of aerosolized antimicrobials, tobramycin, colistin, and aztreonam, used to prevent the establishment of chronic P aeruginosa or to suppress the growth of P aeruginosa in those patients chronically infected.37 Because antimicrobial susceptibility is based on achievable serum drug concentrations, susceptibility testing does not accurately predict resistance in patients receiving aerosolized antimicrobials, which reach airway levels at least 10 to 25 times greater than serum levels.
There are 2 additional problems that confront laboratorians when determining antimicrobial susceptibility of P aeruginosa. Technically, susceptibility testing of mucoid P aeruginosa is poorly reproducible and operator dependent.38 In the late stages of chronic P aeruginosa infection, after many rounds of intravenous and aerosolized antimicrobial therapy, the organism may become resistant to all classes of antimicrobial.34 One of the strategies was to perform multicombinational bactericidal testing (MCBT).39 In this method, 2-drug, 3-drug, and 4-drug combinations at serum achievable concentrations are placed in a microtiter well, inoculated with a bacteria, incubated overnight, and then subcultured to determine which combinations were bactericidal. The idea behind this method was to give a rationale for choosing antimicrobial combinations that would be used to treat these infections. However, when a randomized, double-blind, controlled clinical trial was performed to compare MCBT with conventional susceptibility, outcomes were similar for the 2 groups.40 Although the study was technically well performed, it was flawed because the patients studied were infected with organisms that had some level of susceptibility to antimicrobials and would not have needed MCBT. Based on this study, MCBT has been abandoned by the CF community.
Burkholderia cepacia complex and related organisms
In the early 1980s, a significant new pathogen, then called Pseudomonas cepacia, was recognized in lung infections in patients with CF cared for at 3 different major CF centers.41, 42, 43 In approximately 20% of patients with CF, the organism caused a fatal illness called the cepacia syndrome. In this syndrome, the patients had respiratory infections, with hallmarks of rapid decline in pulmonary function and frequent bacteremia, a highly unusual finding in patients with CF.41, 42 Subsequently, P cepacia was reclassified as belonging to the genus Burkholderia.44 Vandamme and colleagues45 showed that the organisms identified as Burkholderia cepacia represented multiple species of bacteria, which they called genomovars. The B cepacia complex is now recognized to consistent of 18 different species.46, 47 The species within this complex that is believed to be primarily responsible for the cepacia syndrome is B cenocepacia, although both B multivorans and B dolosa have also been associated with it.46 B cenocepacia or B multivorans are recovered from 80% of the B cepacia complex infections seen in patients with CF.46 The molecular epidemiology of B cenocepacia is reasonably well understood. Three different B cenocepacia clones, designated ET12,42 PHDC,43 and Midwest,44 were associated with the initial description of B cepacia as a pathogen in patients with CF.46 All 3 of these clones can be spread from person to person, although many patients have unique clones, which they likely acquired from their environment.48 It has been shown that the ET-12 strain has been spread from Toronto to Europe and that it is the predominant strain in Canada.46, 49 The organism has an unusual appendage called a cable pilin, which is believed to be responsible for its high transmissibility.50
Because B cepacia and B multivorans were associated with accelerated lung function decline and high mortality and were often panresistant to antimicrobials, lung transplantation was believed to be a life-saving therapy in those patients. Early molecular epidemiology studies using PFGE showed that patients infected with B cepacia complex after transplant had the same highly antimicrobial-resistant strain of B cepacia complex before transplant.51 It was subsequently observed that patients with CF chronically infected with B cenocepacia who received lung transplant had a higher mortality at years 1, 3, and 5 years after transplant than those who were infected with other bacteria.52, 53 Early in the posttransplant period, some patients infected with B cenocepacia had a sepsis with positive blood cultures. Some of the transplants were further complicated by wound dehiscence. Posttransplant patients with B cenocepacia sepsis had a grim prognosis, with few survivors. Bronchiolitis obliterans was an additional problem seen in this patient population.52 Other studies also showed increased risk of death after transplant in patients with CF infected before transplant with B cenocepacia.53, 54 As result, most centers no longer transplant patients with CF who are infected with B cenocepacia.
One of the difficulties in recovering B cepacia from respiratory specimens of patients with CF is that the organism grows slowly compared with mucoid P aeruginosa. Consequently, the growth of B cepacia may be obscured on most media. Three different groups developed isolation media for the recovery of B cepacia complex organism. B cepacia selective agar (BCSA)55 was superior for B cepacia recovery compared with P cepacia agar56 and oxidation-fermentation polymyxin-bacitracin-lactose agar.57 All 3 media contain polymixin as a selective agent. Not only were B cepacia complex organisms isolated on these media from CF respiratory specimens but also a large group of environmental glucose nonfermenters, which were similar to B cepacia, were also recovered. These organisms were often phenotypically difficult to separate from B cepacia using commercially available kits, resulting in misidentification of non-B cepacia isolates as B cepacia complex ones.58, 59 Alternatively, some isolates were relatively inert biochemically, making them difficult to identify using commercially available phenotypic methods.58, 59
Misidentification of non-B cepacia organisms as B cepacia has grim consequences for the patient with CF. In addition to no longer being candidates for lung transplantation in most centers, the patients are completely segregated from other children and adults with CF. They are likely to have a different clinic day, must always be on isolation when hospitalized, meaning no trips to the playroom to interact with other children, may no longer attend any CF sponsored social function or meetings, or participate in camps with other patients with CF.60 The result is social isolation for these individuals and potential exclusion from a life-saving therapy. These draconian measures have been successful in blunting the spread of this organism among patients with CF.60 Perhaps just as importantly, misidentifying B cepacia complex organism as some other species or genera results in failure to isolate patients with a potentially deadly organism, which can be spread to both patients with CF and those who do not have CF.49, 61
Burkholderia gladioli was the first organism recovered on B cepacia–selective media and recognized to be misidentified as B cepacia using phenotypic means.59, 62 By the early part of this century, it was recognized that 16s ribosomal RNA (rRNA) sequencing was superior to phenotypic methods for differentiating B cepacia complex organisms from other genera growing on B cepacia–selective media such as B gladioli.63 Our experience with 16s rRNA sequencing over the past 6 years is shown in Table 1 . With the exception of the unusual P aeruginosa phenotypes identified by sequencing, the rest of the organisms identified were recovered on BCSA, the B cepacia selective medium that we use. Many more non-B cepacia complex organisms requiring identification grow on BCSA. Almost all the organisms listed in Table 1 are recovered primarily from patients with CF. Most of them, including Ralstonia, Chyseobacterium, Inquilinus, and Pandoraea, are infrequently recovered and are believed to be of limited clinical importance. One group is reported in our laboratory as gram-negative rods, not Burkholderia, Ralstonia, or Pandoraea. Included in this group are Bordetella spp, including Bordetella broncoseptica, Comamonas, Herbaspirillium, Elizabethkingia, and Cupriavidus. These organisms have not been associated with pulmonary exacerbation in patients with CF.
Table 1.
Organisms identified by 16S rRNA sequencing from patients with CF at UNC Hospitals from Jan 2008 to Nov 2013
| Achromobacter | 67 |
| Burkholderia cepacia complexa | 64 |
| Burkholderia gladioli | 63 |
| Chryseobacterium | 38 |
| Gram-negative rods not Burkholderia cepacia complex, Ralstonia or Pandoraeab | 35 |
| Ralstonia spp | 32 |
| Pandoraea | 12 |
| Pseudomonas aeruginosa | 10 |
| Pseudomonas not aeruginosa | 10 |
| Acinetobacter spp | 4 |
| Inquilinus | 2 |
Isolates speciated by recA sequencing at Burkholderia cepacia Reference Laboratory and Repository, University of Michigan.
Includes isolates of Bordetella bronchiseptica, Comamonas, Herbaspirillium, Elizabethkingia, and Cupriavidus.
Although there are few published data suggesting a role for B gladioli in patients with CF,64 the exception seems to be in lung transplant recipients, who have decreased survival compared with those transplant recipients who do not have Burkholderia.53
Both Stenotrophomonas and Achromobacter spp are recovered with frequency in patients with CF, especially older patients who have received many rounds of antipseudomonal antimicrobials. These species are the subject of the next section of this article.
One of the problems with 16s rRNA sequence identification is that it cannot accurately differentiate the members of the B cepacia complex. Because there seem to be differences in virulence among the different species within the complex, accurate speciation is important.46 Three species are recognized as being pathogenic in patients with CF and being able to be spread from person to person: B cenocepacia, B multivorans, and B dolosa.46 Other species seen with some degree of frequency in our patient population include B vietnamienesis and B cepacia. In our experience, the other 13 species within the complex are rarely if ever isolated. Accurate speciation of isolates of the B cepacia complex can be accomplished by sequencing of the recA gene.65
The requirement to use 2 sequencing steps to accurately identify isolates belonging to the B cepacia complex is time consuming and expensive, requiring days to obtain an accurate answer. As a result, alternative identification methods have been sought. The most promising of these is MALDI-TOF MS. We66 and others28, 65, 67 have shown that MALDI-TOF accurately identifies organisms to the B cepacia complex and can differentiate them readily from other genera of organisms that grow on BCSA, most importantly B gladioli. Preliminary studies suggest that the Bruker system more accurately identifies B cenocepacia isolates than the Vitek system.65, 67 Both systems accurately speciate B multivorans and B vietnamienesis. The Bruker system misidentifies B contaminans primarily as B cepacia but may also misidentify it as B cenocepacia or B multivorans. With database improvements, it is likely that the 2 major CF pathogens, B cenocepacia and B multivorans, will be accurately identified by both systems.
B cepacia complex is one of the most antimicrobial-resistant group of organisms. These organisms are intrinsically resistant to both colistin and aminoglycosides.68 Clinical and Laboratory Standards Institute susceptibility breakpoints exist for ceftazidime, ticarcillin-clavulanic acid, TMP-SMX, meropenem. minocycline, and fluoroquinolones.69 Although a large percentage of isolates may be initially susceptible to these agents, resistance develops over time, and many strains become resistant to all these antimicrobial agents.68 Further complicating treatment is the lack of evidence that any specific antimicrobial treatment regimen is effective in treating B cepacia complex pulmonary exacerbations.70
Stenotrophomonas and Achromobacter
Both Stenotrophomonas and Achromobacter are glucose nonfermenting rods, which are found more frequently in patients with CF than are B cepacia complex organisms. Stenotrophomonas is found in approximately 10% to 20% of patients with CF, whereas Achromobacter is found in between 5% and 10%.4 The incidence of these organisms increases with age and likely reflects patients who have had multiple rounds of antipseudomonal antimicrobial therapy.
The clinical significance of both of these organisms has been debated. One study suggested that patients who had precipitating antibodies to Achromobacter antigens have a more rapid decline in lung function.71 Other studies do not support the notion that Achromobacter is pathogenic.72, 73 The story is similar with Stenotrophmonas. Patients may be more likely to have poor pulmonary function and may require lung transplantation. What is not clear is whether this organism represents a marker of severe lung disease or is the cause of this disease.74, 75
Three different approaches have been used to identify these 2 organism: conventional phenotypic methods, 16s rRNA sequencing, and MALDI-TOF MS. Phenotypic methods work reasonably well for these organisms, although we have greater confidence in the use of the VITEK 2 for Stenotrophomonas maltophilia compared with Achromobacter spp.76 MADLI-TOF MS accurately identifies both species.66, 67
Both organisms are resistant to aminoglycosides and may develop resistance to colistin.77 In addition, Stenotrophomonas produces a metallo-β-lactamase, which confers resistance to carbapenems.78 The organism may be initially susceptible to levofloxacin and TMP-SMX but can develop resistance over time.78 The initial susceptibility of Achromobacter is similar to that of B cepacia complex, with susceptibility to ceftazidime, meropenem, fluoroquinolones, and TMP-SMX being common.79 However, drug resistance can develop over time and panresistant organisms can be seen.80
Mycobacterium
Over the past 25 years, mycobacteria are being recognized as an increasingly important agent of chronic lung infection in patients with CF.81 Although we have been culturing patients with CF for Mycobacterium for more than 25 years in our institution, we have never recovered Mycobacterium tuberculosis from them.82 The mycobacterial species most frequently associated with these infections are the nontuberculous mycobacteria, M avium complex and M abscessus complex.82, 83 There is an increasing body of evidence that both M avium complex and M abscessus complex can cause chronic infection in patients with CF and may be responsible for decline in pulmonary function, although it is not as dramatic as B cepacia or P aeruginosa.81, 84, 85 In addition, the patient population infected with M avium complex seems to be older and does not have as severe disease as those infected with the M abscessus group, who seem to be younger and to have more severe lung disease.85 Recent studies using whole genome sequencing have suggested that the M abscessus group show the potential for spread from person to person in CF centers, making it another target for infection prevention.86
Attempting to recover nontuberculous mycobacteria from a CF respiratory specimen is challenging for the laboratory. Approximately 50% to 70% of specimens contain P aeruginosa, which is resistant to 0.25% n-acetyl cysteine-1% sodium hydroxide decontamination, the standard method used in the culture respiratory specimens for mycobacteria. Contamination rates between 35% and 70% may be seen using this decontamination method in CF respiratory specimens. When a second step is added, 2.5% oxalic acid, the contamination rate is reduced to 3% to 5%, although low numbers of M avium complex may not be recovered.87, 88 Chlorohexidine alone has been shown to be an effective means of decontaminating CF respiratory specimens for the recovery of nontuberculous mycobacteria.89
A useful observation concerning the recovery of M abscessus group organisms is the observation that this group of organisms can be recovered on medium that is routinely used for bacterial isolation, specifically BCSA. M abscessus group clinical isolates were recovered on BCSA from 65% to 75% of infected individuals, whereas the organism was recovered from 85% using mycobacterial-specific culture methods. Twenty-five percent of patients with CF with M abscessus complex had it first recovered on routine culture. In addition, incubation of BCSA plates for 14 days greatly enhanced recovery compared with 5-day incubation.90 As a result, we routinely examine BCSA plates for the presence of rapidly growing Mycobacterium.
Phenotypic identification of nontuberculous mycobacteria is challenging and not particularly accurate. As a result, we have used 16S rRNA sequence analysis for identification of these organisms. A 16S rRNA sequence works well for identification of M avium complex, but M abscessus complex isolates cannot be distinguished from M chelonae.91 As a result, a second target is needed. Both rpoB and hsp65 have been used for this purpose, with both being useful for differentiating M abscessus complex from M chelonae.91 Whole genome sequencing has shown that in 1 center, M abscessus subspecies massilense was predominant and there was evidence of person-to-person spread.86 Whole genome sequencing of M abscessus complex isolates from more than 100 patients with CF in our center showed M abscessus subspecies abscessus to be predominant (Grogono, Floto, Gilligan unpublished).
Antimicrobial resistance is a major problem in nontuberculous mycobacteria treatment. Anti–M tuberculosis therapy is ineffective against these organisms. Further complicating treatment is the lack of randomized, controlled trials of antimicrobial treatment against this group of organisms.92 Susceptibility testing using broth dilution is helpful in guiding antimicrobial choice, because susceptibility is not predictable.81 Broth MIC susceptibility tests of macrolides against the M abscessus group should be incubated for 14 days to detect inducible macrolide resistance. Clarithromycin seems to be a more potent inducer of this resistance than azithromycin, making azithromycin central to the treatment of M abscessus infections.93
A potential life-saving therapy for patients with CF infected with severe M abscessus is lung transplantation. However, based on anecdotal experience and case reports, M abscessus complex infection has been a contraindication for this life-saving procedure.81 This strategy seems logical, given some of the similarities between B cepacia and M abscessus. Both organisms are environment organisms, which are highly drug resistant, making them difficult to treat, able to be spread from person to person, and associated with wound dehiscence after transplant. A recent single-center study with 13 patients with CF with M abscessus receiving double lung transplant showed the same survival rate as the control population, suggesting that this organism is not a contraindication to lung transplantation.94
Fungi
Fungi have long been recognized as having a role in CF lung disease. In particular, 2 fungi, Aspergillus fumigatus and Scedosporium apiospermum, have been shown to be long-term colonizers in patients with CF.95 Both organisms are most frequently associated with allergic bronchopulmonary disease, characterized by subacute clinical disease with eosinophilia, increased IgE levels, skin test reactivity in the case of Aspergillus, presence of serum IgE specific for the Aspergillus when it is the cause, and chest imaging changes that are not attributable to bacterial agents.95 In addition, post–lung transplant infections are well described for both organisms, although colonization with these organisms is not a contraindication for lung transplantation.96, 97 Candida spp are found in as many as 70% of patients with CF, especially when selective media are used. Because patients with CF receive antibacterials from an early age, the presence of these organisms colonizing the airways is not surprising. The clinical significance of Candida in patients with CF is not clearly understood, but most clinicians discount these organisms as not significant.98
NAAT allows the detection of Pneumocystis jirovecii in the airway of patients with CF. In a prospective study, 12.5% of adults with CF were found to have Pneumocystis jirovecii in sputum. Lung function in these patients was better than in those without Pneumocystis jirovecii. These patients were more likely to have mild disease and be free of P aeruginosa.99
A fungal agent that has recently emerged in patients with CF is Trichosporon. With the increasing use of antifungal agents to treat disease associated with Aspergillus and Scedosporium, Trichosporon, which is resistant to the antifungals amphotericin and the echinocandins, may find a niche in the CF airway.100 Fungemia caused by Trichosporon has been reported in 2 patients with CF, 1 of whom was a double lung transplant recipient.101, 102 Without using a selective fungal medium, we found it in 0.2% of our patients with CF,103 and these patients tend to have more rapid decline in lung function. Whether this situation is directly attributable to Trichosporon infections is unknown.
Isolation of Aspergillus, Scedosporium, and Trichosporon can all be accomplished with media used for bacterial isolation. Because of its highly selective nature, BCSA is particularly useful for the isolation of fungi. Conventional techniques are sufficient for identification of these organisms. Susceptibility testing is not recommended.
Respiratory viruses
For many years, viral agents have been considered to play a prime role in the initial steps of establishing chronic lung infections.15 However, almost all the data to support this theory were based on serologic data, with few culture data to support the theory, in part because of frequent contamination of viral respiratory culture, especially in patients infected with P aeruginosa.15 With the advent of NAAT testing for viruses, a clearer picture of the role of viruses will likely emerge. It is now well recognized that acute exacerbations are associated with both influenza and respiratory syncytial virus.104, 105, 106 One of the most interesting observations was the high rate of detection of rhinovirus, approximately 30% when a multiplex polymerase chain reaction was used.107, 108 Rhinovirus tends to persist in the CF airway, and viral loads increase during pulmonary exacerbations.108 Using a CF airway epithelial model, it has been shown that rhinovirus superinfection of P aeruginosa–infected cells causes a release of planktonic bacteria. This release of planktonic bacteria during viral infection may be an important trigger of CF pulmonary exacerbations.6
Diagnosis of CF respiratory viral infections has been revolutionized by the use of NAAT. NAAT diagnostics approved by the US Food and Drug Administration are available for influenza A/B only, the combination of influenza A/B and respiratory syncytial virus (RSV), and multiplex NAAT for adenovirus, coronavirus, influenza viruses A/B, metapneumovirus, parainfluenza virus 1-4, RSV, and rhinovirus.109, 110, 111 Because there have been no evaluations comparing these different methods specifically in patients with CF, the reliability of these methods is unknown in this patient population. Given the unusual nature of the matrix in which the virus may be found, specific evaluation of NAAT performance in these specimens would be useful. A meta-analysis112 for influenza antigen detection assays suggested that these methods lack sensitivity in patients with low viral loads. Because they have not been evaluated in patients with CF, they cannot be recommended for that patient population.
Microbiome considerations
Microbiome analysis is changing our way of thinking about chronic infections. In patients with CF, we have learned that other organisms beyond those that we have already discussed may play an important role in CF lung disease. Organisms typically believed as belonging to the normal microflora of the oropharynx, Prevotella, Veillonella, and the Streptococcus anginosus group, are all found in respiratory tract of patients with CF.5, 13 It is speculated that these organisms might upregulate or downregulate virulence genes in pathogens such as P aeruginosa to obtain essential nutrients through the activity of these pathogens.12 Another observation that has arisen as a result of microbiome studies is that microbial communities are remarkably stable, even in the face of repeated rounds of antimicrobial therapy.11, 13 In addition, as chronic P aeruginosa infection is established, the diversity of the microbial community declines.10 Is this a result of P aeruginosa being the apex predator in the CF airway microbiome eliminating competitors and adapting itself by downregulating virulence genes to allow long-term survival of its human host? Over the next decade, we should get closer to this answer.
References
- 1.ACOG Committee Opinion No. 486: update on carrier screening for cystic fibrosis. Obstet Gynecol. 2011;117:1028–1031. doi: 10.1097/AOG.0b013e31821922c2. [DOI] [PubMed] [Google Scholar]
- 2.Goss C.H., Ratjen F. Update in cystic fibrosis 2012. Am J Respir Crit Care Med. 2013;187:915–919. doi: 10.1164/rccm.201301-0184UP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donaldson S.H., Wolgang M.C., Gilligan P.G. Cystic fibrosis. In: Mandell G.L., Bennett J.E., Dolin R., editors. Principles and practices of infectious diseases. 7th edition. Churchill Livingstone; New York: 2006. pp. 947–955. [Google Scholar]
- 4.Cystic Fibrosis Foundation. Cystic Fibrosis Foundation patient registry 2012 annual data report. 2013.
- 5.Goss C.H., Burns J.L. Exacerbations in cystic fibrosis. 1: epidemiology and pathogenesis. Thorax. 2007;62:360–367. doi: 10.1136/thx.2006.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sibley C.D., Grinwis M.E., Field T.R. Culture enriched molecular profiling of the cystic fibrosis airway microbiome. PLoS One. 2011;6:e22702. doi: 10.1371/journal.pone.0022702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chattoraj S.S., Ganesan S., Jones A.M. Rhinovirus infection liberates planktonic bacteria from biofilm and increases chemokine responses in cystic fibrosis airway epithelial cells. Thorax. 2011;66:333–339. doi: 10.1136/thx.2010.151431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flume P.A., Mogayzel P.J., Jr., Robinson K.A. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med. 2009;180:802–808. doi: 10.1164/rccm.200812-1845PP. [DOI] [PubMed] [Google Scholar]
- 9.Hansen C.R., Pressler T., Hoiby N. Early aggressive eradication therapy for intermittent Pseudomonas aeruginosa airway colonization in cystic fibrosis patients: 15 years experience. J Cyst Fibros. 2008;7:523–530. doi: 10.1016/j.jcf.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 10.Klepac-Ceraj V., Lemon K.P., Martin T.R. Relationship between cystic fibrosis respiratory tract bacterial communities and age, genotype, antibiotics and Pseudomonas aeruginosa. Environ Microbiol. 2010;12:1293–1303. doi: 10.1111/j.1462-2920.2010.02173.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhao J., Schloss P.D., Kalikin L.M. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci U S A. 2012;109:5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sibley C.D., Parkins M.D., Rabin H.R. The relevance of the polymicrobial nature of airway infection in the acute and chronic management of patients with cystic fibrosis. Curr Opin Investig Drugs. 2009;10:787–794. [PubMed] [Google Scholar]
- 13.Fodor A.A., Klem E.R., Gilpin D.F. The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS One. 2012;7:e45001. doi: 10.1371/journal.pone.0045001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson D.H. Therapy and prognosis of fibrocystic disease of the pancreas. Pediatrics. 1949;3:406–417. [PubMed] [Google Scholar]
- 15.Gilligan P., Kiska D.L., Appleman M.D. ASM Press; Washington, DC: 2006. Cumitech 43: cystic fibrosis microbiology. [Google Scholar]
- 16.Gilligan P.H., Gage P.A., Welch D.F. Prevalence of thymidine-dependent Staphylococcus aureus in patients with cystic fibrosis. J Clin Microbiol. 1987;25:1258–1261. doi: 10.1128/jcm.25.7.1258-1261.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Proctor R.A., von Eiff C., Kahl B.C. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol. 2006;4:295–305. doi: 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- 18.Wolter D.J., Emerson J.C., McNamara S. Staphylococcus aureus small-colony variants are independently associated with worse lung disease in children with cystic fibrosis. Clin Infect Dis. 2013;57:384–391. doi: 10.1093/cid/cit270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia L.G., Lemaire S., Kahl B.C. Antibiotic activity against small-colony variants of Staphylococcus aureus: review of in vitro, animal and clinical data. J Antimicrob Chemother. 2013;68:1455–1464. doi: 10.1093/jac/dkt072. [DOI] [PubMed] [Google Scholar]
- 20.Champion E.A., Miller M.B., Popowitch E.B. Antimicrobial susceptibility and molecular typing of MRSA in cystic fibrosis. Pediatr Pulmonol. 2014;49:230–237. doi: 10.1002/ppul.22815. [DOI] [PubMed] [Google Scholar]
- 21.Dasenbrook E.C., Checkley W., Merlo C.A. Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA. 2010;303:2386–2392. doi: 10.1001/jama.2010.791. [DOI] [PubMed] [Google Scholar]
- 22.David M.Z., Daum R.S. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev. 2010;23:616–687. doi: 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodrich J.S., Sutton-Shields T.N., Kerr A. Prevalence of community-associated methicillin-resistant Staphylococcus aureus in patients with cystic fibrosis. J Clin Microbiol. 2009;47:1231–1233. doi: 10.1128/JCM.00255-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui L., Iwamoto A., Lian J.Q. Novel mechanism of antibiotic resistance originating in vancomycin-intermediate Staphylococcus aureus. Antimicrob Agents Chemother. 2006;50:428–438. doi: 10.1128/AAC.50.2.428-438.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Report of the UK Cystic Fibrosis Trust Microbiology Laboratory Standards Working Group: Laboratory standards for processing microbiological samples form people with cystic fibrosis. Cystic Fibrosis Trust 2010.
- 26.Kipp F., Kahl B.C., Becker K. Evaluation of two chromogenic agar media for recovery and identification of Staphylococcus aureus small-colony variants. J Clin Microbiol. 2005;43:1956–1959. doi: 10.1128/JCM.43.4.1956-1959.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kipp F., Becker K., Peters G. Evaluation of different methods to detect methicillin resistance in small-colony variants of Staphylococcus aureus. J Clin Microbiol. 2004;42:1277–1279. doi: 10.1128/JCM.42.3.1277-1279.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desai A.P., Stanley T., Atuan M. Use of matrix assisted laser desorption ionisation-time of flight mass spectrometry in a paediatric clinical laboratory for identification of bacteria commonly isolated from cystic fibrosis patients. J Clin Pathol. 2012;65:835–838. doi: 10.1136/jclinpath-2012-200772. [DOI] [PubMed] [Google Scholar]
- 29.Howden B.P., Davies J.K., Johnson P.D. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Aloul M., Crawley J., Winstanley C. Increased morbidity associated with chronic infection by an epidemic Pseudomonas aeruginosa strain in CF patients. Thorax. 2004;59:334–336. doi: 10.1136/thx.2003.014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Folkesson A., Jelsbak L., Yang L. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol. 2012;10:841–851. doi: 10.1038/nrmicro2907. [DOI] [PubMed] [Google Scholar]
- 32.Marko D.C., Saffert R.T., Cunningham S.A. Evaluation of the Bruker Biotyper and Vitek MS matrix-assisted laser desorption ionization-time of flight mass spectrometry systems for identification of nonfermenting gram-negative bacilli isolated from cultures from cystic fibrosis patients. J Clin Microbiol. 2012;50:2034–2039. doi: 10.1128/JCM.00330-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burns J.L., Saiman L., Whittier S. Comparison of two commercial systems (Vitek and MicroScan-WalkAway) for antimicrobial susceptibility testing of Pseudomonas aeruginosa isolates from cystic fibrosis patients. Diagn Microbiol Infect Dis. 2001;39:257–260. doi: 10.1016/s0732-8893(01)00234-6. [DOI] [PubMed] [Google Scholar]
- 34.Gilligan P.H. Is there value in susceptibility testing of Pseudomonas aeruginosa causing chronic infection in patients with cystic fibrosis? Expert Rev Anti Infect Ther. 2006;4:711–715. doi: 10.1586/14787210.4.5.711. [DOI] [PubMed] [Google Scholar]
- 35.Moskowitz S.M., Foster J.M., Emerson J. Clinically feasible biofilm susceptibility assay for isolates of Pseudomonas aeruginosa from patients with cystic fibrosis. J Clin Microbiol. 2004;42:1915–1922. doi: 10.1128/JCM.42.5.1915-1922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su S., Hassett D.J. Anaerobic Pseudomonas aeruginosa and other obligately anaerobic bacterial biofilms growing in the thick airway mucus of chronically infected cystic fibrosis patients: an emerging paradigm or “old hat”? Expert Opin Ther Targets. 2012;16:859–873. doi: 10.1517/14728222.2012.708025. [DOI] [PubMed] [Google Scholar]
- 37.Lo D., VanDevanter D.R., Flume P. Aerosolized antibiotic therapy for chronic cystic fibrosis airway infections: continuous or intermittent? Respir Med. 2011;105(Suppl 2):S9–S17. doi: 10.1016/S0954-6111(11)70022-1. [DOI] [PubMed] [Google Scholar]
- 38.Foweraker J.E., Laughton C.R., Brown D.F. Phenotypic variability of Pseudomonas aeruginosa in sputa from patients with acute infective exacerbation of cystic fibrosis and its impact on the validity of antimicrobial susceptibility testing. J Antimicrob Chemother. 2005;55:921–927. doi: 10.1093/jac/dki146. [DOI] [PubMed] [Google Scholar]
- 39.Aaron S.D., Ferris W., Henry D.A. Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia. Am J Respir Crit Care Med. 2000;161:1206–1212. doi: 10.1164/ajrccm.161.4.9907147. [DOI] [PubMed] [Google Scholar]
- 40.Aaron S.D., Vandemheen K.L., Ferris W. Combination antibiotic susceptibility testing to treat exacerbations of cystic fibrosis associated with multiresistant bacteria: a randomised, double-blind, controlled clinical trial. Lancet. 2005;366:463–471. doi: 10.1016/S0140-6736(05)67060-2. [DOI] [PubMed] [Google Scholar]
- 41.Isles A., Maclusky I., Corey M. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J Pediatr. 1984;104:206–210. doi: 10.1016/s0022-3476(84)80993-2. [DOI] [PubMed] [Google Scholar]
- 42.Tablan O.C., Chorba T.L., Schidlow D.V. Pseudomonas cepacia colonization in patients with cystic fibrosis: risk factors and clinical outcome. J Pediatr. 1985;107:382–387. doi: 10.1016/s0022-3476(85)80511-4. [DOI] [PubMed] [Google Scholar]
- 43.Tablan O.C., Martone W.J., Doershuk C.F. Colonization of the respiratory tract with Pseudomonas cepacia in cystic fibrosis. Risk factors and outcomes. Chest. 1987;91:527–532. doi: 10.1378/chest.91.4.527. [DOI] [PubMed] [Google Scholar]
- 44.Yabuuchi E., Kosako Y., Oyaizu H. Proposal of Burkholderia gen. nov. and transfer of seven species of the genus Pseudomonas homology group II to the new genus, with the type species Burkholderia cepacia (Palleroni and Holmes 1981) comb. nov. Microbiol Immunol. 1992;36:1251–1275. doi: 10.1111/j.1348-0421.1992.tb02129.x. [DOI] [PubMed] [Google Scholar]
- 45.Vandamme P., Holmes B., Vancanneyt M. Occurrence of multiple genomovars of Burkholderia cepacia in cystic fibrosis patients and proposal of Burkholderia multivorans sp. nov. Int J Syst Bacteriol. 1997;47:1188–1200. doi: 10.1099/00207713-47-4-1188. [DOI] [PubMed] [Google Scholar]
- 46.Lipuma J.J. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev. 2010;23:299–323. doi: 10.1128/CMR.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peeters C., Zlosnik J.E., Spilker T. Burkholderia pseudomultivorans sp. nov., a novel Burkholderia cepacia complex species from human respiratory samples and the rhizosphere. Syst Appl Microbiol. 2013;36:483–489. doi: 10.1016/j.syapm.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 48.Heath D.G., Hohneker K., Carriker C. Six-year molecular analysis of Burkholderia cepacia complex isolates among cystic fibrosis patients at a referral center for lung transplantation. J Clin Microbiol. 2002;40:1188–1193. doi: 10.1128/JCM.40.4.1188-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holmes A., Nolan R., Taylor R. An epidemic of Burkholderia cepacia transmitted between patients with and without cystic fibrosis. J Infect Dis. 1999;179:1197–1205. doi: 10.1086/314699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun L., Jiang R.Z., Steinbach S. The emergence of a highly transmissible lineage of cbl+ Pseudomonas (Burkholderia) cepacia causing CF centre epidemics in North America and Britain. Nat Med. 1995;1:661–666. doi: 10.1038/nm0795-661. [DOI] [PubMed] [Google Scholar]
- 51.Steinbach S., Sun L., Jiang R.Z. Transmissibility of Pseudomonas cepacia infection in clinic patients and lung-transplant recipients with cystic fibrosis. N Engl J Med. 1994;331:981–987. doi: 10.1056/NEJM199410133311504. [DOI] [PubMed] [Google Scholar]
- 52.Aris R.M., Routh J.C., LiPuma J.J. Lung transplantation for cystic fibrosis patients with Burkholderia cepacia complex. Survival linked to genomovar type. Am J Respir Crit Care Med. 2001;164:2102–2106. doi: 10.1164/ajrccm.164.11.2107022. [DOI] [PubMed] [Google Scholar]
- 53.Murray S., Charbeneau J., Marshall B.C. Impact of Burkholderia infection on lung transplantation in cystic fibrosis. Am J Respir Crit Care Med. 2008;178:363–371. doi: 10.1164/rccm.200712-1834OC. [DOI] [PubMed] [Google Scholar]
- 54.Noone P.G. Lung transplant and cystic fibrosis: what's new from the UK and France? Thorax. 2008;63:668–670. doi: 10.1136/thx.2008.099622. [DOI] [PubMed] [Google Scholar]
- 55.Henry D., Campbell M., McGimpsey C. Comparison of isolation media for recovery of Burkholderia cepacia complex from respiratory secretions of patients with cystic fibrosis. J Clin Microbiol. 1999;37:1004–1007. doi: 10.1128/jcm.37.4.1004-1007.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilligan P.H., Gage P.A., Bradshaw L.M. Isolation medium for the recovery of Pseudomonas cepacia from respiratory secretions of patients with cystic fibrosis. J Clin Microbiol. 1985;22:5–8. doi: 10.1128/jcm.22.1.5-8.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Welch D.F., Muszynski M.J., Pai C.H. Selective and differential medium for recovery of Pseudomonas cepacia from the respiratory tracts of patients with cystic fibrosis. J Clin Microbiol. 1987;25:1730–1734. doi: 10.1128/jcm.25.9.1730-1734.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kiska D.L., Kerr A., Jones M.C. Accuracy of four commercial systems for identification of Burkholderia cepacia and other gram-negative nonfermenting bacilli recovered from patients with cystic fibrosis. J Clin Microbiol. 1996;34:886–891. doi: 10.1128/jcm.34.4.886-891.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shelly D.B., Spilker T., Gracely E.J. Utility of commercial systems for identification of Burkholderia cepacia complex from cystic fibrosis sputum culture. J Clin Microbiol. 2000;38:3112–3115. doi: 10.1128/jcm.38.8.3112-3115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saiman L., Siegel J. Infection control in cystic fibrosis. Clin Microbiol Rev. 2004;17:57–71. doi: 10.1128/CMR.17.1.57-71.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bressler A.M., Kaye K.S., LiPuma J.J. Risk factors for Burkholderia cepacia complex bacteremia among intensive care unit patients without cystic fibrosis: a case-control study. Infect Control Hosp Epidemiol. 2007;28:951–958. doi: 10.1086/519177. [DOI] [PubMed] [Google Scholar]
- 62.Wilsher M.L., Kolbe J., Morris A.J. Nosocomial acquisition of Burkholderia gladioli in patients with cystic fibrosis. Am J Respir Crit Care Med. 1997;155:1436–1440. doi: 10.1164/ajrccm.155.4.9105090. [DOI] [PubMed] [Google Scholar]
- 63.Ferroni A., Sermet-Gaudelus I., Abachin E. Use of 16S rRNA gene sequencing for identification of nonfermenting gram-negative bacilli recovered from patients attending a single cystic fibrosis center. J Clin Microbiol. 2002;40:3793–3797. doi: 10.1128/JCM.40.10.3793-3797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kennedy M.P., Coakley R.D., Donaldson S.H. Burkholderia gladioli: five year experience in a cystic fibrosis and lung transplantation center. J Cyst Fibros. 2007;6:267–273. doi: 10.1016/j.jcf.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 65.Fehlberg L.C., Andrade L.H., Assis D.M. Performance of MALDI-ToF MS for species identification of Burkholderia cepacia complex clinical isolates. Diagn Microbiol Infect Dis. 2013;77:126–128. doi: 10.1016/j.diagmicrobio.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 66.Alby K., Gilligan P.H., Miller M.B. Comparison of matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry platforms for the identification of gram-negative rods from patients with cystic fibrosis. J Clin Microbiol. 2013;51:3852–3854. doi: 10.1128/JCM.01618-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Degand N., Carbonnelle E., Dauphin B. Matrix-assisted laser desorption ionization-time of flight mass spectrometry for identification of nonfermenting gram-negative bacilli isolated from cystic fibrosis patients. J Clin Microbiol. 2008;46:3361–3367. doi: 10.1128/JCM.00569-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leitao J.H., Sousa S.A., Cunha M.V. Variation of the antimicrobial susceptibility profiles of Burkholderia cepacia complex clonal isolates obtained from chronically infected cystic fibrosis patients: a five-year survey in the major Portuguese treatment center. Eur J Clin Microbiol Infect Dis. 2008;27:1101–1111. doi: 10.1007/s10096-008-0552-0. [DOI] [PubMed] [Google Scholar]
- 69.Clinical and Laboratory Institute: Performance standards for antimicrobial susceptibility testing; 23rd Informational Supplement, M100–S23. Clinical and Laboratory Standards Institute 2013.
- 70.Horsley A., Jones A.M. Antibiotic treatment for Burkholderia cepacia complex in people with cystic fibrosis experiencing a pulmonary exacerbation. Cochrane Database Syst Rev. 2012;(10) doi: 10.1002/14651858.CD009529.pub2. CD009529. [DOI] [PubMed] [Google Scholar]
- 71.Ronne Hansen C., Pressler T., Hoiby N. Chronic infection with Achromobacter xylosoxidans in cystic fibrosis patients; a retrospective case control study. J Cyst Fibros. 2006;5:245–251. doi: 10.1016/j.jcf.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 72.Raso T., Bianco O., Grosso B. Achromobacter xylosoxidans respiratory tract infections in cystic fibrosis patients. APMIS. 2008;116:837–841. doi: 10.1111/j.1600-0463.2008.00995.x. [DOI] [PubMed] [Google Scholar]
- 73.De Baets F., Schelstraete P., Van Daele S. Achromobacter xylosoxidans in cystic fibrosis: prevalence and clinical relevance. J Cyst Fibros. 2007;6:75–78. doi: 10.1016/j.jcf.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 74.Goss C.H., Mayer-Hamblett N., Aitken M.L. Association between Stenotrophomonas maltophilia and lung function in cystic fibrosis. Thorax. 2004;59:955–959. doi: 10.1136/thx.2003.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Waters V., Atenafu E.G., Lu A. Chronic Stenotrophomonas maltophilia infection and mortality or lung transplantation in cystic fibrosis patients. J Cyst Fibros. 2013;12:482–486. doi: 10.1016/j.jcf.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 76.Zbinden A., Bottger E.C., Bosshard P.P. Evaluation of the colorimetric VITEK 2 card for identification of gram-negative nonfermentative rods: comparison to 16S rRNA gene sequencing. J Clin Microbiol. 2007;45:2270–2273. doi: 10.1128/JCM.02604-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Biswas S., Dubus J.C., Reynaud-Gaubert M. Evaluation of colistin susceptibility in multidrug-resistant clinical isolates from cystic fibrosis, France. Eur J Clin Microbiol Infect Dis. 2013;32:1461–1464. doi: 10.1007/s10096-013-1898-5. [DOI] [PubMed] [Google Scholar]
- 78.Goncalves-Vidigal P., Grosse-Onnebrink J., Mellies U. Stenotrophomonas maltophilia in cystic fibrosis: improved detection by the use of selective agar and evaluation of antimicrobial resistance. J Cyst Fibros. 2011;10:422–427. doi: 10.1016/j.jcf.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 79.Wang M., Ridderberg W., Hansen C.R. Early treatment with inhaled antibiotics postpones next occurrence of Achromobacter in cystic fibrosis. J Cyst Fibros. 2013;12:638–643. doi: 10.1016/j.jcf.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 80.Amoureux L., Bador J., Siebor E. Epidemiology and resistance of Achromobacter xylosoxidans from cystic fibrosis patients in Dijon, Burgundy: First French data. J Cyst Fibros. 2013;12:170–176. doi: 10.1016/j.jcf.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 81.Leung J.M., Olivier K.N. Nontuberculous mycobacteria: the changing epidemiology and treatment challenges in cystic fibrosis. Curr Opin Pulm Med. 2013;19:662–669. doi: 10.1097/MCP.0b013e328365ab33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kilby J.M., Gilligan P.H., Yankaskas J.R. Nontuberculous mycobacteria in adult patients with cystic fibrosis. Chest. 1992;102:70–75. doi: 10.1378/chest.102.1.70. [DOI] [PubMed] [Google Scholar]
- 83.Olivier K.N., Weber D.J., Lee J.H. Nontuberculous mycobacteria. II: nested-cohort study of impact on cystic fibrosis lung disease. Am J Respir Crit Care Med. 2003;167:835–840. doi: 10.1164/rccm.200207-679OC. [DOI] [PubMed] [Google Scholar]
- 84.Esther C.R., Jr., Esserman D.A., Gilligan P. Chronic Mycobacterium abscessus infection and lung function decline in cystic fibrosis. J Cyst Fibros. 2010;9:117–123. doi: 10.1016/j.jcf.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Catherinot E., Roux A.L., Vibet M.A. Mycobacterium avium and Mycobacterium abscessus complex target distinct cystic fibrosis patient subpopulations. J Cyst Fibros. 2013;12:74–80. doi: 10.1016/j.jcf.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 86.Bryant J.M., Grogono D.M., Greaves D. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet. 2013;381:1551–1560. doi: 10.1016/S0140-6736(13)60632-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Whittier S., Hopfer R.L., Knowles M.R. Improved recovery of mycobacteria from respiratory secretions of patients with cystic fibrosis. J Clin Microbiol. 1993;31:861–864. doi: 10.1128/jcm.31.4.861-864.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bange F.C., Kirschner P., Bottger E.C. Recovery of mycobacteria from patients with cystic fibrosis. J Clin Microbiol. 1999;37:3761–3763. doi: 10.1128/jcm.37.11.3761-3763.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ferroni A., Vu-Thien H., Lanotte P. Value of the chlorhexidine decontamination method for recovery of nontuberculous mycobacteria from sputum samples of patients with cystic fibrosis. J Clin Microbiol. 2006;44:2237–2239. doi: 10.1128/JCM.00285-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Esther C.R., Jr., Hoberman S., Fine J. Detection of rapidly growing mycobacteria in routine cultures of samples from patients with cystic fibrosis. J Clin Microbiol. 2011;49:1421–1425. doi: 10.1128/JCM.02379-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blauwendraat C., Dixon G.L., Hartley J.C. The use of a two-gene sequencing approach to accurately distinguish between the species within the Mycobacterium abscessus complex and Mycobacterium chelonae. Eur J Clin Microbiol Infect Dis. 2012;31:1847–1853. doi: 10.1007/s10096-011-1510-9. [DOI] [PubMed] [Google Scholar]
- 92.Waters V., Ratjen F. Antibiotic treatment for nontuberculous mycobacteria lung infection in people with cystic fibrosis. Cochrane Database Syst Rev. 2012;(12) doi: 10.1002/14651858.CD010004.pub2. CD010004. [DOI] [PubMed] [Google Scholar]
- 93.Choi G.E., Shin S.J., Won C.J. Macrolide treatment for Mycobacterium abscessus and Mycobacterium massiliense infection and inducible resistance. Am J Respir Crit Care Med. 2012;186:917–925. doi: 10.1164/rccm.201111-2005OC. [DOI] [PubMed] [Google Scholar]
- 94.Lobo L.J., Chang L.C., Esther C.R., Jr. Lung transplant outcomes in cystic fibrosis patients with pre-operative Mycobacterium abscessus respiratory infections. Clin Transplant. 2013;27:523–529. doi: 10.1111/ctr.12140. [DOI] [PubMed] [Google Scholar]
- 95.Pihet M., Carrere J., Cimon B. Occurrence and relevance of filamentous fungi in respiratory secretions of patients with cystic fibrosis–a review. Med Mycol. 2009;47:387–397. doi: 10.1080/13693780802609604. [DOI] [PubMed] [Google Scholar]
- 96.Bonvillain R.W., Valentine V.G., Lombard G. Post-operative infections in cystic fibrosis and non-cystic fibrosis patients after lung transplantation. J Heart Lung Transplant. 2007;26:890–897. doi: 10.1016/j.healun.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 97.Morio F., Horeau-Langlard D., Gay-Andrieu F. Disseminated Scedosporium/Pseudallescheria infection after double-lung transplantation in patients with cystic fibrosis. J Clin Microbiol. 2010;48:1978–1982. doi: 10.1128/JCM.01840-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Middleton P.G., Chen S.C., Meyer W. Fungal infections and treatment in cystic fibrosis. Curr Opin Pulm Med. 2013;19:670–675. doi: 10.1097/MCP.0b013e328365ab74. [DOI] [PubMed] [Google Scholar]
- 99.Hernandez-Hernandez F., Frealle E., Caneiro P. Prospective multicenter study of Pneumocystis jirovecii colonization among cystic fibrosis patients in France. J Clin Microbiol. 2012;50:4107–4110. doi: 10.1128/JCM.01974-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Miceli M.H., Diaz J.A., Lee S.A. Emerging opportunistic yeast infections. Lancet Infect Dis. 2011;11:142–151. doi: 10.1016/S1473-3099(10)70218-8. [DOI] [PubMed] [Google Scholar]
- 101.Hickey P.W., Sutton D.A., Fothergill A.W. Trichosporon mycotoxinivorans, a novel respiratory pathogen in patients with cystic fibrosis. J Clin Microbiol. 2009;47:3091–3097. doi: 10.1128/JCM.00460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hirschi S., Letscher-Bru V., Pottecher J. Disseminated Trichosporon mycotoxinivorans, Aspergillus fumigatus, and Scedosporium apiospermum coinfection after lung and liver transplantation in a cystic fibrosis patient. J Clin Microbiol. 2012;50:4168–4170. doi: 10.1128/JCM.01928-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paulson J, Kerr A, Gilligan P, et al. Trichosporon acquisition is linked to accelerated lung function decline in nontransplant cystic fibrosis patients. In: Am J Respir Crit Care Med. vol. 185. San Francisco (CA): American Thoracic Society International Meeting, poster discussion; 2012. p. A5274.
- 104.Burns J.L., Emerson J., Kuypers J. Respiratory viruses in children with cystic fibrosis: viral detection and clinical findings. Influenza Other Respir Viruses. 2012;6:218–223. doi: 10.1111/j.1750-2659.2011.00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Viviani L., Assael B.M., Kerem E. Impact of the A (H1N1) pandemic influenza (season 2009-2010) on patients with cystic fibrosis. J Cyst Fibros. 2011;10:370–376. doi: 10.1016/j.jcf.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 106.Etherington C., Naseer R., Conway S.P. The role of respiratory viruses in adult patients with cystic fibrosis receiving intravenous antibiotics for a pulmonary exacerbation. J Cyst Fibros. 2014;13(1):49–55. doi: 10.1016/j.jcf.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 107.Esther C.R., Jr., Lin F.C., Kerr A. Respiratory viruses are associated with common respiratory pathogens in cystic fibrosis. Pediatr Pulmonol. 2013 doi: 10.1002/ppul.22917. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 108.Kieninger E., Singer F., Tapparel C. High rhinovirus burden in lower airways of children with cystic fibrosis. Chest. 2013;143:782–790. doi: 10.1378/chest.12-0954. [DOI] [PubMed] [Google Scholar]
- 109.Novak-Weekley S.M., Marlowe E.M., Poulter M. Evaluation of the Cepheid Xpert Flu Assay for rapid identification and differentiation of influenza A, influenza A 2009 H1N1, and influenza B viruses. J Clin Microbiol. 2013;50:1704–1710. doi: 10.1128/JCM.06520-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Popowitch E.B., O'Neill S.S., Miller M.B. Comparison of the Biofire FilmArray RP, Genmark eSensor RVP, Luminex xTAG RVPv1, and Luminex xTAG RVP fast multiplex assays for detection of respiratory viruses. J Clin Microbiol. 2013;51:1528–1533. doi: 10.1128/JCM.03368-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Alby K., Popowitch E.B., Miller M.B. Comparative evaluation of the Nanosphere Verigene RV+ assay and the Simplexa Flu A/B & RSV kit for detection of influenza and respiratory syncytial viruses. J Clin Microbiol. 2013;51:352–353. doi: 10.1128/JCM.02504-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chartrand C., Leeflang M.M., Minion J. Accuracy of rapid influenza diagnostic tests: a meta-analysis. Ann Intern Med. 2012;156:500–511. doi: 10.7326/0003-4819-156-7-201204030-00403. [DOI] [PubMed] [Google Scholar]