

Abstract
In many cases, at the beginning of a high throughput screening experiment some information about active molecules is already available. Active compounds (such as substrate analogues, natural products and inhibitors of related proteins) are often identified in low throughput validation studies on a biochemical target. Sometimes the additional structural information is also available from crystallographic studies on protein and ligand complexes. In addition, the structural or sequence similarity of various protein targets yields a novel possibility for drug discovery. Co-crystallized compounds from homologous proteins can be used to design leads for a new target without co-crystallized ligands. In this paper we evaluate how far such an approach can be used in a real drug campaign, with severe acute respiratory syndrome (SARS) coronavirus providing an example. Our method is able to construct small molecules as plausible inhibitors solely on the basis of the set of ligands from crystallized complexes of a protein target, and other proteins from its structurally homologous family. The accuracy and sensitivity of the method are estimated here by the subsequent use of an electronic high throughput screening flexible docking algorithm. The best performing ligands are then used for a very restrictive similarity search for potential inhibitors of the SARS protease within the million compounds from the Ligand.Info small molecule meta-database. The selected molecules can be passed on for further experimental validation.
1. Introduction
Severe acute respiratory syndrome (SARS) is a life-threatening form of pneumonia characterized by high fever, nonproductive cough, chills, myalgia, lymphopenia, and progressing infiltrates in chest radiography (Neuman et al 2005). Between 2002 and 2003, an epidemic emerged that, facilitated by international air travel, spread within a few weeks from its origin in Guangdong Province, China, to many other countries. The WHO reported over 8000 SARS cases and nearly 800 deaths resulting from the infection with the SARS-associated coronavirus (SARS CoV) (WHO 2004). Studies on SARS CoV resulted in identification of protein targets for potential drugs, which included SARS CoV protease, polymerase and helicase (Kliger et al 2005).
This paper proposes a novel in silico method that predicts potent inhibitors for SARS CoV protease and captures the specific features of inhibitor molecules, as opposed to ligands that hit other targets. The method is based on experimental publicly available structural information databases and presents a good starting point for the further experimental validation. It is well known that genomic research (and especially structural biology) provide an ever increasing number of potential drug targets and allow for intensive use of structural data in various drug discovery projects. In this study we use all structural homologues of SARS CoV protease co-crystallized with various ligands in order to design novel, potential inhibitors for use in vHTS (virtual high throughput screening methods) experiments for this enzyme.
2. Computational methods
2.1. Protein structures with co-crystallized ligands
We extracted from the Protein Data Bank (PDB) database (Berman et al 2000a, 2000b) all viral cysteine protease of the trypsin fold based on the structural classification of proteins (SCOP) (Murzin et al 1995, Lo Conte et al 2002, Andreeva et al 2004). The family is a part of the trypsin-like serine protease superfamily that possesses closed barrel type fold with Greek-key duplication. The family of viral cysteine proteases encompasses three different groups of domains: (1) the 3C cysteine protease (picornain 3C) domain with three proteins: human rhinovirus type 2 (1CQQ), human hepatitis A virus (1QA7, 1HAV) and poliovirus type I (1L1N); (2) 2A cysteine proteinase with one protein human rhinovirus 2 (2HRV); and (3) coronavirus main proteinase (3Cl-pro, putative coronavirus nsp2) with four proteins: transmissible gastroenteritis virus (1LVO), transmissible gastroenteritis virus (1P9U), human coronavirus (1P9S), and SARS coronavirus (1Q2W, 1UJ1, 1UK2, 1UK3, 1UK4). The sequence similarity search was performed on all selected proteins from the PDB database in order to find homologous protein structures that are not included in the recent version of the SCOP database. The list of ligands co-crystallized with these proteins include AG7, ACE, DMS, GOL, OCS, DOX, MPD, MSE and a few peptides specifically, two peptides and a single ligand (chloroacetone) were found to be crystallized with the original protein target, i.e. SARS coronavirus protease (1UK4)—see figure 1.
Figure 1.

The peptides and chloroacetone ligand co-crystallized with the SARS coronavirus protease (1UK4).
All protein structures were aligned superimposed using 3D-Hit software (Plewczynski et al 2002, 2004). Protein chains were divided into single domains and all domains were structurally aligned in order to analyse the common binding mode for various ligands. Figure 2 presents structures superimposed on 1UK4 together with all ligands located in their active sites. From aligned structures we extracted the 1UK4 protein active site neighbourhood with ligands co-crystallized in it. We used 1UK4 as the template for a flexible docking experiment in order to adjust the conformation of ligands in the new structural neighbourhood. The target structure with structurally aligned ligands (co-crystallized with structurally homologous proteins) is presented in figure 3. The data were then selected for further analysis in order to remove some inconsistencies in PDB entries for selected ligands.
Figure 2.

The structure superposition from the protein chains from the SCOP family of viral cysteine proteases of the trypsin fold. The co-crystallized ligands located in common active site are shown as balls and sticks.
Figure 3.

Ligands co-crystallized with proteins from the SCOP family of viral cysteine proteases of the trypsin fold in the active site of SARS coronavirus protease (1UK4).
In the case of an inhibitor taken from 1UK4 structure the following interactions were identified. ARG188, GLN189 and GLN 192 are in close contact with ASN1 of the inhibitor with the methyl group of MET165. Such an arrangement warrants good orientation of the peptide inside the protein cavity. NH2 of the GLN189 interacts with O of the first peptide bond of the inhibitor. The OH group of SER2 of the inhibitor interacts with the carboxylate moiety of GLU166. GLU166 also forms hydrogen bond with amide oxygen of GLN5. HIS163 and LEU141 interact with the terminal side chain of the GLN5 of the protein inhibitor. The position of the HIS163 is stabilized by parallel interaction with the phenyl ring of PHE140 (see figures 4 and 5).
Figure 4.

Schematic representation of the main interactions between SARS CoV proteinase (pdb code 1UK4) amino acid residues and the original inhibitor molecule. The residues within 3.6 Å distance from the inhibitor molecule are shown.
Figure 5.

Protein inhibitor (orange) inside a cavity of the SARS proteinase (blue ribbon for the protein and yellow licorice for most important amino acids).
2.2. From peptide to small molecule lead
Although a peptide can bind strongly to a protein target and effectively block or regulate its activity, it is also prone to hydrolysis of its peptide bonds. Therefore over the last few years various research groups searched for peptide bond mimics, analogues, substitutes, or isosters in order to obtain a molecule whose shape and electrostatic potential are very similar to these of the parental peptide (see table 1 for detailed references). We generated here a set of molecules for further virtual screening by substituting a peptide bond by its mimics. Table 1 presents various linkages capable of substituting peptide bonds. Almost 40 moieties have been described in the literature and some of them can be present in various stereoisomeric forms. The substitutes for a peptide bond can be grouped into various subcategories. For example with respect to the number of atoms separating the groups connected initially by a peptide bond one can identify linkages with one, two, three and more atoms. There is also a set of peptide bond substitutes where rings are involved in binding two groups of the parental peptide. In addition there is a group of molecules where side chains of amino acids are substituted by atoms forming ring structures that substitute both peptide bonds and some side chains. The set with one-atom linkages shorter than the original peptide bond is composed of amine –NH– and ether –O– bridges (Baker et al 1992). The other set with three or more atoms separating the two groups previously joined by –CO–NH– is larger. There are linkages where additional carbonyl –CO–(–CO–CF2–CO– and –CO–CO–NH– (Thaisrivongs et al 1986, Watt 1986, Wolfe et al 1999)) or other –O– (–CO–NH–O–), –N = CH– (–NH–N = CH–CO–), –CH2– (–CH(OH)–CH2–NH–) are inserted. In the set where rings are substituting peptide bonds one can find azaphosphinane rings (Grembecka et al 2003), cyclopropane rings (Martin et al 1992), pyrrole rings (Smith et al 1994), and thiazole rings (Singh et al 2001), while in the group where ring structures substitute not only a peptide bond but also some amino acid side chains one can find para-substituted phenyl rings (Temple et al 1998, Tyndall et al 2000, Yin et al 2005) and thiazolopyridine rings (Olson et al 1993). The largest set is the one where two atoms are separating the two groups that had been connected by a peptide bond. It is easy here to identify a subgroup with a–PO– group instead of –CO– in peptides (Bone et al 1991, Fraser et al 1992, Tsukamoto et al 1998, Collinsova and Jiracek 2000, Demange et al 2002, Grembecka et al 2003), and –SO2– (Langenhan et al 2001). There is a subgroup with a fluorine atom bound to a carbon atom (Thaisrivongs et al 1986, Fraser et al 1992, Silva et al 1996, Volonterio et al 2003, Annedi et al 2005, Xiao et al 2005) and a subgroup where a carbon–carbon double bond substitutes a C–N bond in peptide linkage (Lehman de Gaeta and Czarniecki 1989, Jaskolski et al 1991, Gardner et al 1995, Zhang et al 1998, Xiao et al 2005). There is also the set with a carbonyl group in a peptide linkage reduced to a hydroxyl group (Kempf et al 1990, Jaskolski et al 1991). Other linkages include ester –CO–O– (Amblard et al 1993), ethylene –CH2–CH2– (Amblard et al 1993), and –CH2–NH– (Jaskolski et al 1991, Guichard et al 1994, 1995) substitutes, as well as retro-inverto-peptides (Guichard et al 1994, 1995, Beglova et al 2000).
Table 1.
Various peptide bond mimics, isosteres, analogues, and substitutes have been proposed over the last 20 years. In this search for inhibitors of SARS protease we have examined various possibilities that can substitute R1–CO–NH–R2 peptide bonds.
| Substituted peptide bond | Peptide bond mimic | References |
| R1–CO–NH–R2 | R1–CH2–CH2–R2 | (Amblard et al 1993) |
| R1–CO–NH–R2 | R1–CO–O–R2 | (Amblard et al 1993) |
| R1–CO–NH–R2 | R1–CHF–S–R2 | (Annedi et al 2005) |
| R1–CO–NH–R2 | R1–CF2–NH–R2 | (Fraser et al 1992) |
| R1–CO–NH–R2 | R1–CO–CF2–R2 | (Silva et al 1996) |
| R1–CO–NH–R2 | R1–CO–CF2–CO–R2 | (Thaisrivongs et al 1986, Watt 1986, |
| Wolfe et al 1999) | ||
| R1–CO–NH–R2 | R1–NH–CH(CF3)–R2 | (Volonterio et al 2003) |
| R1–CO–NH–R2 | R1–NH–R2 | (Baker et al 1992) |
| R1–CO–NH–R2 | R1–O–R2 | (Baker et al 1992) |
| R1–CO–NH–R2 | R1–NH–N = CH–CO–R2 | (Gaertner et al 1994) |
| R1–CO–NH–R2 | R1–CO–NH–O–R2 | (Yang et al 2003) |
| R1–CO–NH–R2 | R1–CO–CO–NH–R2 | (Ocain and Rich 1992) |
| R1–CO–NH–R2 | R1–CH2-NH–R2 | (Jaskolski et al 1991, Guichard et al 1995, |
| Benkirane et al 1996) | ||
| R1–CO–NH–R2 | R1–CH(OH)–CH2–NH–R2 | (Swain et al 1990, Jaskolski et al 1991) |
| R1–CO–NH–R2 | R1–C(OH) = CH–R2 | (Jaskolski et al 1991) |
| R1–CO–NH–R2 | R1–CH(OH)–CH(OH)–R2 | (Jaskolski et al 1991) |
| R1–CO–NH–R2 | R1–CH(OH)–CH2–R2 | (Kempf et al 1990, Abdel-Meguid et al 1994) |
| R1–CO–NH–R2 | R1–CH = CH–R2 | (Lehman de Gaeta and Czarniecki 1989) |
| R1–CO–NH–R2 | R1–CMe = CMe–R2 | (Gardner et al 1995) |
| R1–CO–NH–R2 | R1–CH = C(Pro) | (Zhang et al 1998, Hart and Etzkorn 1999) |
| R1–CO–NH–R2 | R1–C(CF3) = CH–R2 | (Xiao et al 2005) |
| R1–CO–NH–R2 | R1–C(CH3) = CH–R2 | (Xiao et al 2005) |
| R1–CO–NH–R2 | R1–SO2–NH–R2 | (Langenhan et al 2001) |
| R1–CO–NH–R2 | R1–PO(OPh)–O–R2 | (Bone et al 1991, Collinsova and Jiracek 2000) |
| R1–CO–NH–R2 | R1–PO(OH)–O–R2 | (Fraser et al 1992, Tsukamoto et al 1998, |
| Collinsova and Jiracek 2000) | ||
| R1–CO–NH–R2 | R1–PO(OH)–NH–R2 | (Collinsova and Jiracek 2000, |
| Grembecka et al 2003) | ||
| R1–CO–NH–R2 | R1–PO(OEt)–NH–R2 | (Demange et al 2002) |
| R1–CO–NH–R2 | R1–PO(OH)–CH2–R2 | (Fraser et al 1992, Collinsova and Jiracek 2000, |
| Grembecka et al 2003) | ||
| R1–CO–NH–R2 | R1–PO(NH2)–CH2–R2 | (Collinsova and Jiracek 2000) |
| R1–CO–NH–R2 | R1–NH–CO–R2 (retro-inverto-peptide) | (Guichard et al 1994, Beglova et al 2000) |
| R1–CO–NH–R2 |

|
(Grembecka et al 2003) |
Table 1.
(Continued.)














2.3. Evaluation of modified ligands by a flexible docking procedure and structural similarity search over the Ligand.Info one-million-compound database
First we performed the docking procedure in order to evaluate the modification of inhibitors by replacing the peptide bond by its mimic. Electronic high throughput screening (eHiTS) is an exhaustive flexible docking method that systematically covers significant part of the conformational and positional search space, producing highly accurate docking poses at a speed practical for virtual high throughput screening (Zsoldos et al 2003). The ligand docking problem is divided here into two subproblems: pose/conformation search and scoring function. The customizable scoring function of eHiTS combines a local surface point contact evaluation with traditional empirical and statistical approaches. The search algorithm is fast, in many cases capable of finding the correct binding pose and conformation of the ligand in the virtual screening phase.
Then we performed the structural similarity search in order to select potential inhibitor molecules from a large collection of chemical compounds using the Ligand.Info database. The Ligand.Info system is designed for fast, sensitive, virtual high throughput screening of small molecule databases (von Grotthuss et al 2003). The search algorithms are based on two-dimensional structure similarity. For each molecule a vector of two-dimensional indices and their occurrence counts are calculated. The index represents a unique combination of three connected atoms. Each atom is described by six parameters: atom symbol, charge, valence, type of bond, and number of connected hydrogen and non-hydrogen atoms. In this study the modified Tanimoto coefficient (MTC) was used as the similarity metric. MTC takes values between 0 and 1, where zero means that there are no identical indices in either molecule, and one means that both molecules are composed of identical sets of indices. The system developed enables a search for similar compounds in the large (approximately a million commercially available compounds) Ligand.Info meta-database (von Grotthuss et al 2004). The database contains various publicly available sets of small molecules such as Harvard's ChemBank, which encompasses bioactive compounds and approved by FDA drugs (Strausberg and Schreiber 2003), ChemPDB ligands marked as ‘Hetero Atoms' in Protein Data Bank (PDB) files (Berman et al 2002, Boutselakis et al 2003), KEGG ligand molecules that are found in the KEGG pathways (Goto et al 2002), the Open National Cancer Institute database (Voigt et al 2001), and a few others.
3. Results and discussion
We evaluated to what extent one can utilize the structural information contained in the PDB database in the real drug discovery campaign using as an example the main protease of severe acute respiratory syndrome (SARS) coronavirus. Using the methods described we generate potential inhibitors solely on the basis of the set of ligands from crystallized complexes of a protein target and other proteins from its structurally homologous family. Two sets of compounds were built. The first one contains ligands and short peptides dissected from protein–ligand complexes of viral cysteine proteases of the trypsin fold. These ligands and peptides were then modified in order to correct the chemical structure. The second group includes ligands built from peptides of the first set with the peptide bond substituted by another linkage (see table 1) in order to obtain a molecule that potentially binds to the enzyme more strongly than the original peptide.
These two sets were then evaluated with the eHiTS flexible docking algorithm. The top rank ligands are presented in table 2 for the first data set and in table 3 for the second set. The best scoring compounds of the first set are original AG7 ligand (eHiTS docking score: −5.615), two versions of modified AG7 (with removal of some parts of the original ligand AG7; eHiTS docking scores: −5.103 and −4.803) and as the fourth/fifth scoring compounds, the original peptides of the SARS protease (1UK4, chains H and G) after improving the initial 3D structure (eHiTS docking scores: −4.795 and −4.703). The docking scores for the second group (ligands built by modification of the peptide bond) are in most cases lower than those in the case of the first group. The best scoring compounds in table 3 are modified versions of AG7 (score −5.940), where the peptide bond –CO–NH– in the middle of the parental AG7 molecule was substituted by the –PO2–CH2– moiety, original AG7 compound (score −5.615), and VAS5 peptide, in which the two residues Leu and Gln from the C termini were substituted by a thiazolopyridinone ring (modified version with score −5.465) and ligand created from the original chain G peptide from the SARS CoV target, where the peptide bond between Ser and Thr was replaced by an ether bridge (1UK4 with score −5.430).
Table 2.
Scores for selected ligands and peptides docked in the active site of the 1UK4 protein. P1 and P2 are peptides co-crystallized with 1UK4 protein (chains H and G, respectively). The P3 peptide has short sequence VNSTLQ, and AG7+NFA or VAL+NFA are pairs of ligands connected by chemical bonds. MPD is the 2-methyl-2,4-pentanediol (MPD) allosteric inhibitor.
| Ligand data | Score |
| AG7 | −5.615 |
| P1 | −4.795 |
| P2 | −4.703 |
| P3 | −4.344 |
| AG7+NFA | −3.921 |
| VAL+NFA | −3.697 |
| NFA | −3.390 |
| VAL | −3.313 |
| MPD | −2.316 |
| Chloroacetone | −1.306 |
Table 3.
Scores for ligands built on the basis of modified peptides docked in the active site of the 1UK4 protein. The AG7 M1, AG7 M2 etc are different modifications of the AG7 compound. VAL M1 is the chemically modified VAL inhibitor, whereas all modified versions of P2 peptide are marked as M1, M2, M3 up to M10.
| Ligand data | Score | |
| AG7 M1 | −5.940 |

|
| AG7 | −5.615 |

|
| VAL M1 | −5.465 |

|
| P2 M1 | −5.430 |

|
| P2 M2 | −5.412 |

|
Table 3.
(Continued.)
| AG7 M2 | −5.411 |

|
| P2 M3 | −5.398 |

|
| P2 M4 | −5.344 |

|
| P2 M5 | −5.236 |

|
| P2 M6 | −5.192 |

|
Table 3.
(Continued.)
| P2 M7 | −5.179 |

|
| P2 M8 | −5.171 |

|
| AG7 M3 | −5.151 |

|
| P2 M9 | −5.133 |
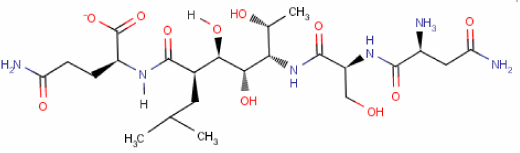
|
| P2 M10 | −5.113 |

|
Table 3.
(Continued.)
| AG7 M4 | −5.103 |

|
Next, each molecule presented in table 2 was used for screening the Ligand.Info meta-database as a query. Almost all designed potential leads did not have any close analogue in the meta-database subset. Therefore only experimental confirmation of the slowing down of the activity of SARS CoV protease by these ligands can confirm our findings, and this confirmation will require chemical synthesis. The only good seed for similarity search, i.e. 2-methyl-2,4-pentanediol (MPD) allosteric inhibitor, was observed among all the compounds from table 2. For the 21 most similar (with MTC≥0.60) analogues that were found using MPD as a query, see table 4 for details. Those compounds might be suitable as starting leads in further experimental studies, which is beyond the scope of this paper.
Table 4.
2,4-dimethyl-2-pentanol (MPD) and its 21 analogues that were found in the meta-database using MPD as a query. Molecules are sorted by similarity (modified Tanimoto coefficient, MTC). Ten most promising compounds that can be freely ordered from the National Cancer Institute (NCI) are bold.
| Ligand data | MTC | Two-dimensional structure |
| 2-methyl-2,4-pentanediol Available from NCI: Yes NSC No 8098 SMILES: CC(O)CC(C)(C)O | 1.0 |

|
| 2,4-dimethyl-2-pentanol Available from NCI: Yes NSC No 87555 SMILES: CC(C)CC(C)(C)O | 0.80 |

|
| 2-methylpentane-1,2,4-triol Available from NCI: No HET-code = MQD SMILES: CC(O)CC(C)(O)CO | 0.76 |

|
| 2-methyl-2-butanol Available from NCI: No NSC No 25498 SMILES: CCC(C)(C)O | 0.71 |

|
| 1-chloro-2-methyl-2-propanol Available from NCI: Yes NSC No 46574 SMILES: CC(C)(O)CCl | 0.71 |

|
Table 4.
(Continued.)
| 1-amino-2-methyl-2-propanol Available from NCI: Yes NSC No 17697 SMILES: CC(C)(O)CN | 0.71 |

|
| 4-amino-4-methyl-2-pentanol Available from NCI: Yes NSC No 7090 SMILES: CC(O)CC(C)(C)N | 0.70 |

|
| 2-methyl-2-pentanol Available from NCI: Yes NSC No 8686 SMILES: CCCC(C)(C)O | 0.67 |

|
| 5-amino-2-methyl-2-hexanol Available from NCI: No NSC No 163077 SMILES: CC(N)CCC(C)(C)O | 0.67 |

|
| 4-mercapto-2-methyl-2-butanol Available from NCI: Yes NSC No 207866 SMILES: CC(C)(O)CCS | 0.67 |

|
| 2,5-dimethyl-2-hexanol Available from NCI: Yes NSC No 5594 SMILES: CC(C)CCC(C)(C)O | 0.67 |

|
| 2-methyl-4-pentyn-2-ol Available from NCI: No NSC No 3515 SMILES: CC(C)(O)CC#C | 0.67 |

|
| 3-methyl-1,3-butanediol Available from NCI: Yes NSC No 62084 SMILES: CC(C)(O)CCO | 0.67 |

|
Table 4.
(Continued.)
| Heptaminol Available from NCI: No Drug approved by FDA SMILES: CC(N)CCCC(C)(C)O | 0.64 |

|
| 4-hydroxy-4-methylpentanenitrile Available from NCI: Yes NSC No 91826 SMILES: CC(C)(O)CCC#N | 0.63 |

|
| 1-(ethylamino)-2-methyl-2-propanol Available from NCI: Yes NSC No 74452 SMILES: CCNCC(C)(C)O | 0.63 |

|
| 2-methyl-2-hexanol Available from NCI: Yes NSC No 21977 SMILES: CCCCC(C)(C)O | 0.63 |

|
| 2,6-dimethyl-7-octen-2-ol Available from NCI: No HET-code = DHM SMILES: CC(CCCC(C)(C)O)C = C | 0.61 |

|
| 1-(isopentylamino)-2-methyl-2-propanol Available from NCI: Yes NSC No 128248 SMILES: CC(C)CCNCC(C)(C)O | 0.61 |

|
| 2-methyl-2-heptanol Available from NCI: Yes NSC No 898 SMILES: CCCCCC(C)(C)O | 0.60 |

|
| 4-hydroxy-4-methyl-2-pentanone Available from NCI: No NSC No 9005 SMILES: CC(= O)CC(C)(C)O | 0.60 |

|
Table 4.
(Continued.)
| 1-(hydroxy(oxido)amino)-2-methyl-2-propanol Available from NCI: Yes NSC No 17680 SMILES: CC(C)(O)C[N+]([O−]) = O | 0.60 |

|
4. Conclusions
The structure-based drug discovery procedure was assessed with SARS CoV main protease serving as an example. Firstly, potential compounds were extracted from the Protein Data Bank database using sequence and structural similarity of other proteins to the selected target protein (code 1UK4). Secondly, we applied the transformation of peptide bonds in order to build ligands on the basis of co-crystallized peptides. The resulting set of compounds was ranked by docking score for the eHiTS flexible docking procedure in order to select the most promising molecules. The set of best performing compounds was then used for a similarity search over the one-million-entry Ligand.Info meta-database. The selected molecules belong to close analogues of the MPD allosteric inhibitor and can be used as starting leads for further experimental research.
MPD allosteric inhibitor (2-methyl-2,4-pentanediol) is a simple molecule with two hydroxylic groups. This molecule can have several conformations with accompanying steric effects. Using Ligand.Info we found 21 similar molecules which were modified by adding or removing a functional group, electronegative groups or atoms, and increasing or decreasing the steric effects. Some of the molecules like 2-methylpentane-1,2,4-triol, 1-amino-2-methyl-2-propanol, 4-hydroxy-4-methylpentanenitrile, 1-(hydroxy(oxido)amino)-2-methyl-2-propanol have hydrophilic character and the ability to interact with hydrophilic amino acids. Other ligands like 2,4-dimethyl-2-pentanol, 2-methyl-2-pentanol, 2,5-dimethyl-2-hexanol, 2-methyl-4-pentyn-2-ol, 2-methyl-2-hexanol, 2-methyl-2-heptanol, 2,6-dimethyl-7-octen-2-ol contain hydrophobic chains that are able to create hydrophobic interactions. All in all, these molecules may have many flexible conformations.
Modification of the peptide bond initiates new types of inhibitors. Such compounds are a mimics for peptides and can potentially replace known peptide drug types. For this instance, AG7 M1 modification interacts with SARS CoV peptidase more strongly than the original peptide (value of the score function for AG7: −5.615, and −5.940 for AG7 M1, the modified inhibitor). Replacement of the peptide bond for phosphorate ether radically changes the environment and radius of interaction. Additional oxygen atoms with two free orbitals can interact at longer distances, and for this modification symmetrically in both side of the replaced bond.
Acknowledgments
This work was supported by EC BioSapiens (LHSG-CT-2003-503265) and EC SEPSDA (SP22-CT-2004-003831) 6FP projects as well as the Polish Ministry of Education and Science (PBZ-MNiI-2/1/2005 and 2P05A00130). MvG and KE would like to thank the Foundation for Polish Science for a fellowship. Calculations of transformations of peptide bond substitutes were performed at Poznan Supercomputing and Networking Centre (PCSS) and the Interdisciplinary Computational Centre of Adam Mickiewicz University (http://ico.amu.edu.pl).
Contributor Information
Dariusz Plewczynski, Email: darman@icm.edu.pl, Interdisciplinary Centre for Mathematical and Computational Modelling, ICM, Warsaw University, Pawinskiego 5a Street, 02-106 Warsaw, Poland.
Marcin Hoffmann, BioInfoBank Institute, Limanowskiego 24A/16, 60-744 Poznan, Poland.
Marcin von Grotthuss, BioInfoBank Institute, Limanowskiego 24A/16, 60-744 Poznan, Poland.
Lukasz Knizewski, Interdisciplinary Centre for Mathematical and Computational Modelling, ICM, Warsaw University, Pawinskiego 5a Street, 02-106 Warsaw, Poland.
Leszek Rychewski, BioInfoBank Institute, Limanowskiego 24A/16, 60-744 Poznan, Poland.
Krystian Eitner, BioInfoBank Institute, Limanowskiego 24A/16, 60-744 Poznan, Poland.
Krzysztof Ginalski, Interdisciplinary Centre for Mathematical and Computational Modelling, ICM, Warsaw University, Pawinskiego 5a Street, 02-106 Warsaw, Poland.
References
- Abdel-Meguid, S S et al. An orally bioavailable HIV-1 protease inhibitor containing an imidazole-derived peptide bond replacement: crystallographic and pharmacokinetic analysis. Biochemistry 1994. 33:11671–7. 10.1021/bi00205a001 [DOI] [PubMed] [Google Scholar]
- Amblard, M et al. Synthesis and biological evaluation of cholecystokinin analogs in which the Asp–Phe–NH2 moiety has been replaced by a 3-amino-7-phenylheptanoic acid or a 3-amino-6-(phenyloxy)hexanoic acid. J. Med. Chem. 1993. 36:3021–8. 10.1021/jm00072a024 [DOI] [PubMed] [Google Scholar]
- Andreeva, A et al. SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res. 2004. 32(Database issue):D226–9. 10.1093/nar/gkh039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annedi, S C et al. Novel fluoropeptidomimetics: synthesis, stability studies and protease inhibition. Bioorg. Med. Chem. 2005. 13:2943–58. 10.1016/j.bmc.2005.02.011 [DOI] [PubMed] [Google Scholar]
- Baker, W R et al. Nonpeptide renin inhibitors employing a novel 3-aza(or oxa)-2,4-dialkyl glutaric acid moiety as a P2/P3 amide bond replacement. J. Med. Chem. 1992. 35:1722–34. 10.1021/jm00088a006 [DOI] [PubMed] [Google Scholar]
- Beglova, N et al. Design and solution structure of functional peptide mimetics of nerve growth factor. J. Med. Chem. 2000. 43:3530–40. 10.1021/jm990441x [DOI] [PubMed] [Google Scholar]
- Benkirane, N et al. Exploration of requirements for peptidomimetic immune recognition. Antigenic and immunogenic properties of reduced peptide bond pseudopeptide analogues of a histone hexapeptide. J. Biol. Chem. 1996. 271:33218–24. 10.1074/jbc.271.52.33218 [DOI] [PubMed] [Google Scholar]
- Berman, H M et al. The Protein Data Bank and the challenge of structural genomics. Nat. Struct. Biol. 2000a. 7(Suppl.):957–9. 10.1038/80734 [DOI] [PubMed] [Google Scholar]
- Berman, H M et al. The Protein Data Bank. Nucleic Acids Res. 2000b. 28:235–42. 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H M et al. The Protein Data Bank. Acta Crystallogr. 2002. 58:899–907. 10.1107/S0907444902003451 [DOI] [PubMed] [Google Scholar]
- Bone, R et al. Crystal structures of alpha-lytic protease complexes with irreversibly bound phosphonate esters. Biochemistry 1991. 30:2263–72. 10.1021/bi00222a032 [DOI] [PubMed] [Google Scholar]
- Boutselakis, H et al. E-MSD: the European Bioinformatics Institute Macromolecular Structure Database. Nucleic Acids Res. 2003. 31:458–62. 10.1093/nar/gkg065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinsova, M; & Jiracek, J. Phosphinic acid compounds in biochemistry, biology and medicine. Curr. Med. Chem. 2000. 7:629–47. [DOI] [PubMed] [Google Scholar]
- Demange, L et al. Synthesis and evaluation of Glypsi(PO(2)R-N)Pro-containing pseudopeptides as novel inhibitors of the human cyclophilin hCyp-18. J. Med. Chem. 2002. 45:3928–33. 10.1021/jm020865i [DOI] [PubMed] [Google Scholar]
- Fraser, M E et al. Crystallographic analysis of transition-state mimics bound to penicillopepsin: phosphorus-containing peptide analogues. Biochemistry 1992. 31:5201–14. 10.1021/bi00137a016 [DOI] [PubMed] [Google Scholar]
- Gaertner, H F et al. Chemo-enzymic backbone engineering of proteins. Site-specific incorporation of synthetic peptides that mimic the 64-74 disulfide loop of granulocyte colony-stimulating factor. J. Biol. Chem. 1994. 269:7224–30. [PubMed] [Google Scholar]
- Gardner, R et al. An achiral dipeptide mimetic that promotes hairpin formation. J. Am. Chem. Soc. 1995. 117:3280–1. 10.1021/ja00116a036 [DOI] [Google Scholar]
- Goto, S et al. LIGAND: database of chemical compounds and reactions in biological pathways. Nucleic Acids Res. 2002. 30:402–4. 10.1093/nar/30.1.402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grembecka, J et al. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J. Med. Chem. 2003. 46:2641–55. 10.1021/jm030795v [DOI] [PubMed] [Google Scholar]
- Guichard, G et al. Antigenic mimicry of natural L-peptides with retro-inverso-peptidomimetics. Proc. Natl Acad. Sci. USA 1994. 91:9765–9. 10.1073/pnas.91.21.9765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichard, G et al. Efficient binding of reduced peptide bond pseudopeptides to major histocompatibility complex class I molecule. J. Biol. Chem. 1995. 270:26057–9. 10.1074/jbc.270.44.26057 [DOI] [PubMed] [Google Scholar]
- Hart, S A; & Etzkorn, F A. Cyclophilin inhibition by a (Z)-alkene cis-proline mimic. J. Org. Chem. 1999. 64:2998–9. 10.1021/jo990409a [DOI] [PubMed] [Google Scholar]
- Jaskolski, M et al. Structure at 2.5-A resolution of chemically synthesized human immunodeficiency virus type 1 protease complexed with a hydroxyethylene-based inhibitor. Biochemistry 1991. 30:1600–9. 10.1021/bi00220a023 [DOI] [PubMed] [Google Scholar]
- Kempf, D J et al. Renin inhibitors based on dipeptide analogues. Incorporation of the hydroxyethylene isostere at the P2/P3 sites. J. Med. Chem. 1990. 33:371–4. 10.1021/jm00163a059 [DOI] [PubMed] [Google Scholar]
- Kliger, Y et al. From genome to antivirals: SARS as a test tube. Drug Discov. Today 2005. 10:345–52. 10.1016/S1359-6446(04)03320-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenhan, J M et al. Evaluation of hydrogen bonding complementarity between a secondary sulfonamide and an alpha-amino acid residue. Org. Lett. 2001. 3:2559–62. 10.1021/ol016237x [DOI] [PubMed] [Google Scholar]
- Lehman de Gaeta, L; & Czarniecki, M. Synthesis of highly functionalized trans-alkene isosteres of dipeptides. J. Org. Chem. 1989. 54:4004–5. 10.1021/jo00277a053 [DOI] [Google Scholar]
- Lo Conte, L et al. SCOP database in 2002: refinements accommodate structural genomics. Nucleic Acids Res. 2002. 30:264–7. 10.1093/nar/30.1.264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, S F et al. 1,2,3-trisubstituted cyclopropanes as conformationally restricted peptide isosteres: application to the design and synthesis of novel renin inhibitors. J. Med. Chem. 1992. 35:1710–21. 10.1021/jm00088a005 [DOI] [PubMed] [Google Scholar]
- Murzin, A G et al. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995. 247:536–40. 10.1006/jmbi.1995.0159 [DOI] [PubMed] [Google Scholar]
- Neuman, B W et al. Inhibition, escape, and attenuated growth of severe acute respiratory syndrome coronavirus treated with antisense morpholino oligomers. J. Virol. 2005. 79:9665–76. 10.1128/JVI.79.15.9665-9676.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocain, T D; & Rich, D H. Alpha-keto amide inhibitors of aminopeptidases. J. Med. Chem. 1992. 35:451–6. 10.1021/jm00081a005 [DOI] [PubMed] [Google Scholar]
- Olson, G L et al. Concepts and progress in the development of peptide mimetics. J. Med. Chem. 1993. 36:3039–49. 10.1021/jm00073a001 [DOI] [PubMed] [Google Scholar]
- Plewczynski, D et al. 3D-Hit: fast structural comparison of proteins. Appl. Bioinformatics 2002. 1:223–5. [PubMed] [Google Scholar]
- Plewczynski, D et al. Comparison of proteins based on segments structural similarity. Acta Biochim. Pol. 2004. 51:161–72. [PubMed] [Google Scholar]
- Silva, A M et al. Inhibition and catalytic mechanism of HIV-1 aspartic protease. J. Mol. Biol. 1996. 255:321–46. 10.1006/jmbi.1996.0026 [DOI] [PubMed] [Google Scholar]
- Singh, Y et al. Novel cylindrical, conical, and macrocyclic peptides from the cyclooligomerization of functionalized thiazole amino acids. J. Am. Chem. Soc. 2001. 123:333–4. 10.1021/ja002666z [DOI] [PubMed] [Google Scholar]
- Smith, A B et al. Design and synthesis of peptidomimetic inhibitors of HIV-1 protease and renin. Evidence for improved transport. J. Med. Chem. 1994. 37:215–8. 10.1021/jm00028a001 [DOI] [PubMed] [Google Scholar]
- Strausberg, R L; & Schreiber, S L. From knowing to controlling: a path from genomics to drugs using small molecule probes. Science 2003. 300:294–5. 10.1126/science.1083395 [DOI] [PubMed] [Google Scholar]
- Swain, A L et al. X-ray crystallographic structure of a complex between a synthetic protease of human immunodeficiency virus 1 and a substrate-based hydroxyethylamine inhibitor. Proc. Natl Acad. Sci. USA 1990. 87:8805–9. 10.1073/pnas.87.22.8805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple, C S et al. Peptide mimics as substrates for the intestinal peptide transporter. J. Biol. Chem. 1998. 273:20–2. 10.1074/jbc.273.1.20 [DOI] [PubMed] [Google Scholar]
- Thaisrivongs, S et al. Design and synthesis of potent and specific renin inhibitors containing difluorostatine, difluorostatone, and related analogues. J. Med. Chem. 1986. 29:2080–7. 10.1021/jm00160a048 [DOI] [PubMed] [Google Scholar]
- Tsukamoto, T et al. Mechanism-based inhibition of human folylpolyglutamate synthetase: design, synthesis, and biochemical characterization of a phosphapeptide mimic of the tetrahedral intermediate. Arch. Biochem. Biophys. 1998. 355:109–18. 10.1006/abbi.1998.0703 [DOI] [PubMed] [Google Scholar]
- Tyndall, J D et al. Synthesis, stability, antiviral activity, and protease-bound structures of substrate-mimicking constrained macrocyclic inhibitors of HIV-1 protease. J. Med. Chem. 2000. 43:3495–504. 10.1021/jm000013n [DOI] [PubMed] [Google Scholar]
- Voigt, J H et al. Comparison of the NCI open database with seven large chemical structural databases. J. Chem. Inf. Comput. Sci. 2001. 41:702–12. 10.1021/ci000150t [DOI] [PubMed] [Google Scholar]
- Volonterio, A et al. Synthesis, structure and conformation of partially-modified retro- and retro-inverso psi[NHCH(CF3)]Gly peptides. Chemistry 2003. 9:4510–22. 10.1002/chem.200304881 [DOI] [PubMed] [Google Scholar]
- von Grotthuss, M et al. Ligand.Info, searching for similar small compounds using index profiles. Bioinformatics 2003. 19:1041–2. 10.1093/bioinformatics/btg117 [DOI] [PubMed] [Google Scholar]
- von Grotthuss, M et al. Ligand.Info small-molecule meta-database. Comb. Chem. High Throughput Screen 2004. 7:757–61. 10.2174/1386207043328265 [DOI] [PubMed] [Google Scholar]
- Watt, G. Design and interpretation of studies comparing individuals with and without a family history of high blood pressure. J. Hypertens. 1986. 4:1–7. 10.1097/00004872-198602000-00001 [DOI] [PubMed] [Google Scholar]
- WHO. 2004. WHO guidelines for the global surveillance of severe acute respiratory syndrome (SARS) http://www.who.int/csr/resources/publications/WHO_CDS_CSR_ARO_2004_1/en/index.html
- Wolfe, M S et al. Peptidomimetic probes and molecular modeling suggest that Alzheimer's gamma-secretase is an intramembrane-cleaving aspartyl protease. Biochemistry 1999. 38:4720–7. 10.1021/bi982562p [DOI] [PubMed] [Google Scholar]
- Xiao, J et al. Electrostatic versus steric effects in peptidomimicry: synthesis and secondary structure analysis of gramicidin S analogues with (E)-alkene peptide isosteres.. J. Am. Chem. Soc. 2005. 127:5742–3. 10.1021/ja051002s [DOI] [PubMed] [Google Scholar]
- Yang, D et al. A reverse turn structure induced by a D,L-alpha-aminoxy acid dimer. J. Am. Chem. Soc. 2003. 125:14452–7. 10.1021/ja029514j [DOI] [PubMed] [Google Scholar]
- Yin, H et al. Terephthalamide derivatives as mimetics of helical peptides: disruption of the Bcl-x(L)/Bak interaction. J. Am. Chem. Soc. 2005. 127:5463–8. 10.1021/ja0446404 [DOI] [PubMed] [Google Scholar]
- Zhang, R et al. Pseudo-A(1,3) strain as a key conformational control element in the design of poly-L-proline type II peptide mimics. J. Am. Chem. Soc. 1998. 120:3894–902. 10.1021/ja972494e [DOI] [Google Scholar]
- Zsoldos, Z et al. Software tools for structure based rational drug design. J. Mol. Struct. Theochem. 2003. 666:509–665. 10.1016/j.theochem.2003.08.105 [DOI] [Google Scholar]