

Abstract
Severe acute respiratory syndrome (SARS) coronavirus (CoV) spread from China to more than 30 countries, causing severe outbreaks of atypical pneumonia and over 800 deaths worldwide. CoV primarily infects the upper respiratory and gastrointestinal tract; however, SARS-CoV has a unique pathogenesis because it infects both the upper and lower respiratory tracts and leads to human respiratory diseases. SARS-CoV genome has shown containing 14 open reading frames (ORFs) and 8 of them encode novel proteins. Previous reports show that overexpression of ORF-3a, ORF-3b and ORF-7a induce apoptosis. In this report, we demonstrate that overexpression of ORF-6 also induces apoptosis and that Caspase-3 inhibitor and JNK inhibitor block ORF-6 induced apoptosis. Importantly, the protein level of ER chaperon protein, GRP94, was up-regulated when ORF-6 was overexpressed. All these data suggest that ORF-6 induces apoptosis via Caspase-3 mediated, ER stress and JNK-dependent pathways.
Keywords: ORF-6, Severe acute respiratory syndrome coronavirus, Endoplasmic reticulum, Caspase, Apoptosis, c-Jun N-terminal kinase, ER stress
1. Introduction
In 2003, Hong Kong experienced an outbreak of SARS. The outbreak incurred huge economical and social losses. SARS mainly presents as pneumonia-like symptoms and the lungs are pathologically the most affected organs. Immunohistochemistry and in situ hybridization of organs from SARS patients who died revealed that the virus was found not only in the lungs and intestines, but also in the liver, distal convoluted renal tubules, sweat glands, parathyroid, pituitary glands, pancreas, adrenal gland, and cerebrum. The etiological agent for this disease is a novel member of the coronavirus (SARS-CoV) family with limited sequence homology to other coronaviruses [1], [2], [3], [4], [5], [6], [7], [8]. Coronaviruses are enveloped, plus-stranded RNA viruses that lead to respiratory diseases such as the avian coronavirus, Infectious Bronchitis Virus (IBV). SARS-CoV encodes 23 putative proteins including four typical structural proteins: the spike (S), nucleocapsid (N), membrane (M), and envelope (E) proteins [4], [7]. These four proteins contribute to generating the host's immune response, as has been observed in many other transmissible viruses, such as gastroenteritis coronavirus, bronchitis virus, porcine respiratory coronavirus, and mouse hepatitis virus. In addition to the structural proteins, replicase 1a ORF, replicase 1b ORF and eight novel ORFs have been identified.
Although the sequence of the SARS-CoV has now been characterized, the pathogenesis of the virus, the host's response to the viral infection, and the mechanism of the disease remain largely elusive. Viruses are known to utilize their own proteins to influence host cells. Apoptosis is a physiological mechanism to control cell numbers during development and to respond to foreign infections, including bacterial and viral infections [9]. Viruses have evolved strategies either to inhibit or stimulate host cell apoptosis, depending on particular virus–host interactions. Many viruses encode either pro-apoptotic or anti-apoptotic proteins, such as BRLF-A and LMP-1 of the Epstein–Barr virus, Crm of the poxvirus, p35 of the baculovirus, and E1A which can specifically inhibit or delay apoptotic pathways, resulting in increased virus production [10], [11]. Apoptosis in later stages of infection may also be advantageous in facilitating virus dissemination and limiting the host's inflammatory response. Apoptosis usually shows typical morphological changes such as cell blebbing nuclei condensation and DNA fragmentation and involves the activation of caspases, which cleave a variety of cellular substrates. Caspases are synthesized as precursors and activated upon induction by apoptotic signals. Mitochondria play a crucial role in the activation of caspases, leading to the activation of caspase-9 [12]. The Bcl-2 family proteins play an important role in regulating cytochrome c releases, while pro-apoptotic members such as Bax, Bak, Bid and Bik promote cytochrome c release [13].
Apart from mitochondria, endoplasmic reticulum (ER) also plays an important role in apoptosis. Functions of the ER are affected by various intra-cellular and extra-cellular stimuli, so called ER stress, which includes inhibition of glycosylation, reduction of disulfide bonds, calcium depletion from the ER lumen, impairment of protein transport to the Golgi, and expression of mutated proteins in the ER. Under ER stress, unfolded proteins accumulate in the lumen of the ER, which eventually induces conflicting cellular activities; survival and apoptosis. When misfolded proteins accumulate in the ER, cells activate a self-protective mechanism, termed the ER-stress response, to survive ER-stress conditions. The ER-stress responses of eukaryotic cells consist of three different mechanisms: translational attenuation to reduce accumulation of misfolded proteins [14], transcriptional activation of gene encoding ER-resident chaperones [15] and ER-associated degradation (ERAD) [16]. The ER-stress response is initiated by three types of ER membrane receptors, ATF6, IRE1 and PERK. If the adaptive responses are not sufficient to relieve cells from ER stress, cells undergo apoptosis to destroy the ER-stress-damaged cells.
Eight SARS-CoV-encoded proteins have been shown to induce apoptosis [17]. These data suggest that apoptosis may indeed play an important role in helping with virus dissemination in vivo, minimizing inflammatory reaction and evasion of the host's defense mechanisms. Importantly, pathological studies have revealed diffuse alveolar damage as the most notable feature in patients who died of SARS. It is reasonable to believe that apoptosis occurs in the alveolar epithelia cells in SARS patients. Three accessory proteins of SARS-CoV have been shown to induce apoptosis, ORF-3a (also known as U274, SARS X1, or ORF-3), ORF-3b (also known as ORF-4) and ORF-7a (also known as U122, SARS X4, or ORF-8) [18], [19]. ORF-3a protein is ER and Golgi apparatus localized protein. It has been shown to upregulate expression of all three subunits (Aα, Bβ and γ) of fibrinogen in lung epithelial cells and induce chromatin condensation and DNA fragmentation [20], [21]. ORF-3b is predominantly localized in the nucleolus and partially in the mitochondria. ORF-3b also induces G0/G1 arrest and apoptosis in transfected cells [22], [23]. ORF-7a is an ER-localized protein. It has been shown to have a signal peptide at the N-terminus and a typical ER retrieval motif at the C-terminus [24]. Moreover, overexpression of ORF-7a induces apoptosis via the caspase-dependent pathway [19]. However, the signaling pathway for ORF-3a, ORF-3b and ORF-7a induced apoptosis remains elusive.
This study reports the characterization of another SARS-CoV group-specific gene product encoded by ORF-6 (also known as X3, ORF-7). ORF-6 (nucleotide 27,074–27,265 in Tor2 genome sequence [4]) contains 63 amino acids and has no significant sequence homology to other proteins. It has been shown to accelerate replication of a related mouse virus and interact with nonstructural protein (nsp8), which may be involved in virus replication [25], [26]. Recently, ORF-6 has been identified as an ER/Golgi membrane localized protein [27]. Here, we show that first; overexpression of ORF-6 induces apoptosis. Second, caspase-3 was activated in the present of ORF-6. Third, apoptosis induced by ORF-6 and ORF-7a was blocked by caspase-3 inhibitor, z-DEVD, and JNK(c-Jun N-terminal kinase) inhibitor. Finally, ORF-6 and ORF-7a up-regulates ER chaperone protein, 94 kDa glucose-regulated protein (GRP94), protein levels. All these data suggest that in addition to ORF-3a, ORF-3b and ORF-7a, ORF-6 is a new player involved in SARS-CoV-induced apoptosis and both ORF-6 and ORF-7a share a similar pathway to induce apoptosis.
2. Materials and methods
2.1. Cell culture
Human embryonic kidney cells HEK293T, Cercopithecus aethiops kidney cells COS-7 and C. aethiops kidney cells Vero E6 were grown in Dulbecco's modified minimal essential medium (DMEM), supplemented with 10% heat-inactivated fetal bovine serum and 2 mM glutamine, 100 U/ml of penicillin and 100 μg/ml of streptomycin in a humidified incubator with 5% (v/v) carbon dioxide at 37 °C.
2.2. Construction of plasmids
ORF-6 and ORF-7a cDNA was kindly provided by Dr. Zhanguo Wu (Department of Biochemistry, HKUST). ORF-6 and ORF-7a were amplified by PCR and restriction sites BamHI and XhoI were introduced at 5′ and 3′ end, respectively. The fragments ORF-6 and ORF-7a were sub-cloned into mammalian expression vector GFP-N1.
2.3. Drug treatment
Vero E6 cells or COS-7 cells were pre-incubated with 50 μM of z-DEVD-fmk (Calbiochem) or 40 μM of JNK inhibitor or Dimethyl Sulfoxide (DMSO) for 30 min before transfection. For the control group of ER-stress experiments, VeroE6 cells were treated with 1 μM of Thapsigargin for 24 h.
2.4. Transfection and Western blot
Transfection was performed by using calcium chloride or Lipofectamine Transfection Reagent (Invitrogen) according to the manufacturer's instructions. For the Western blot, 24 h after transfection, cells were lysed with 300 μl cold lysis buffer [20 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM sodium orthovanadate, 1 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride] and were scrapped into 1.5 ml Eppendorf tubes. The lysates were exposed to sonication and cleaned by centrifugation at 15,000 ×g for 30 min. The supernatant was collected and the protein concentration was measured and adjusted to a concentration of 2 mg/ml by the lysis buffer. Sample buffer was added and the samples were subjected to SDS-PAGE and were transferred onto PVDF membranes (Hybond). The membranes were blotted with 5% non-fat milk for an hour and then immunoblotted by using appropriate primary and secondary antibodies. The specific protein bands were visualized by SuperSignal West Pico Luminol (PIERCE) and exposed on Fuji Medical X-ray film (Fujifilm).
2.5. Measurement of cell death
After 24 h of transfection, 1 μM Hoechst 33324 (Sigma) was applied to the cells for 10 min. Apoptotic cells were characterized by counting condensed and fragmented nuclei under a fluorescent microscope. The numbers of GFP-expressed cells and GFP-expressed apoptotic cells were counted. At least three hundred cells were counted each time and each experiment was repeated three times.
3. Results
3.1. Overexpression of ORF-6 protein induces apoptosis
In a search for SARS proteins that could induce apoptosis, the SARS protein, ORF-6 was tagged with GFP at the N-terminus and was transfected into Vero E6 and COS-7 cells. We observed that ORF-6 was able to induce apoptosis when overexpressed in Vero E6 and COS-7 cells (Fig. 1A and B). Approximately one third of the cells died when 2 μg of ORF-6 DNA was transfected into cells. The number of apoptotic cells was increased when more ORF-6 DNA was transfected into the cells (Fig. 1). The death rates were comparable to the rates caused by the overexpression of Bax, a well-known pro-apoptotic member of the Bcl-family, and ORF-7a, a SARS protein that has been shown to induce apoptosis [19]. GFP-N1 served as the negative control, as the transfection of 5 μg of GFP-N1 DNA could not induce apoptosis. These results showed that the ORF-6 protein does induce apoptosis.
Fig. 1.
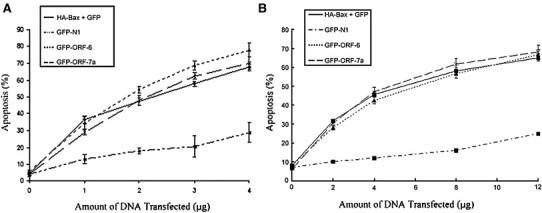
Overexpression of ORF-6-induced apoptosis in different cell lines. (A), COS-7 and (B) Vero E6 cells were transiently transfected with GFP and HA-Bax, GFP-N1, GFP-ORF-6 and GFP-ORF-7a for 24 h. The nuclei of the cells were stained by Hoechst for 15 min. The number of healthy cells was counted under fluorescence microscopy with no DNA condensation and fragmentation. The percentage of apoptotic cells was calculated by the number of healthy cells over the total number of transfected cells. Experiments were repeated three times. Standard deviations are shown.
3.2. Overexpression of ORF-6-induced apoptosis is caspase-3 dependent
ORF-7a induces apoptosis via a caspase-3 dependent pathway [19]. To determine if cell death induced by ORF-6 is also caspase-3 dependent, a caspase-3-specific inhibitor, z-DEVD, was used to block the caspase-3 activation in the cells. In the absence of z-DEVD, approximately 60% of cells underwent apoptosis when ORF-6 and ORF-7a were transiently transfected into the Vero E6 cells; however, when ORF-6 or ORF-7a were overexpressed in z-DEVD pre-treated cells, the percentage of apoptotic cells was significantly decreased to approximately 20% (Fig. 2A). In parallel, Caspase-3 activities were also monitored when the Vero E6 cells were transfected with ORF-6. The activity of Caspase-3 can be detected by the amount of the 17 kD active form of Caspase-3 in cells by using Caspase-3 specific antibodies. Results in Fig. 2B showed that both ORF-6 and ORF-7a induced Caspase-3 activation, as the 17 kD active form of Caspase-3 was detected in both. Cells without any treatment served as a negative control and cells treated with Staurosporine (STS) served as a positive control. Similar results were obtained when using 293T or COS-7 cells (data not shown). Our observation suggests that overexpression of ORF-6 induced apoptosis via a Caspase-3-dependent pathway.
Fig. 2.
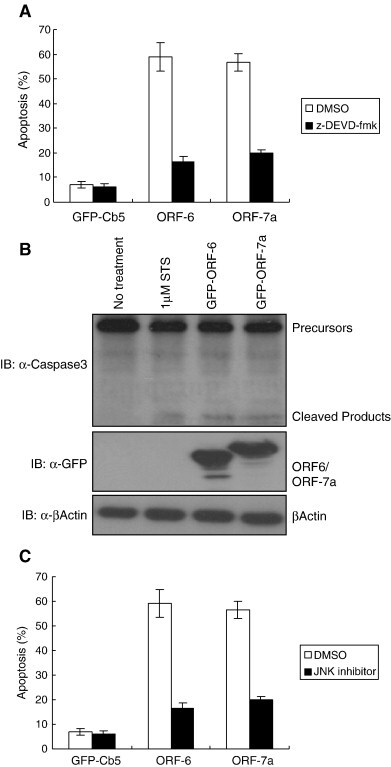
Overexpression of ORF-6 induced apoptosis is Caspase-3 and JNK-dependent. (A) Caspase-3 inhibitor (z-DEVD) blocks ORF-6-induced apoptosis. Vero E6 cells were incubated with either Dimethyl Sulfoxide (DMSO) or 50 μM z-DEVD-fmk for 30 min before they were transiently transfected with 3 μg of GFP-Cb5, GFP-ORF-6 and GFP-ORF-7a for 24 h. The nuclei of the cells were stained by Hoechst for 15 min. The number of healthy cells was counted under fluorescence microscopy with no DNA condensation and fragmentation. The percentage of apoptotic cells was calculated by the number of healthy cells over the total number of transfected cells. Experiments were repeated three times and the standard deviations are shown. (B) Overexpression of ORF-6- and ORF-7a induces Caspase-3 activation. Vero E6 cells were transiently transfected with GFP-ORF-6 and GFP-ORF-7a for 24 h. Cell lysates were normalized to 2 μg/ μl by lysis buffer and subjected to Western Blot with α-GFP, α-Caspase-3 and α-Actin. For positive controls, cells were treated with 1 μM of Staurosporine (STS) for 8 h. Cells without any treatment were served as negative control. (C) JNK inhibitor blocked ORF-6 induced apoptosis. Vero E6 cells were incubated with either Dimethyl Sulfoxide (DMSO) or 40 μM JNK inhibitor for 30 min before they were transiently transfected with 3 μg of GFP-Cb5, GFP-ORF-6 and GFP-ORF-7a for 24 h. Cell counts were done as mentioned in B.
3.3. JNK inhibitor blocks ORF-6 and ORF-7a-induced apoptosis
One of the possible mechanisms for SARS protein-induced cell death is via the JNK pathway. Previous studies have shown that JNK is phosphorylated in SARS-CoV-infected Vero E6 cells and JNK inhibitor (SP600125) can block SARS-CoV-infected Vero E6 cells-induced apoptosis. We investigated whether JNK inhibitor could block ORF-6- and ORF-7a-induced apoptosis in Vero E6 cells. Interestingly, apoptosis induced by ORF-6 and ORF-7a was blocked by the JNK inhibitor. The blocking efficiency was similar to that of z-DEVD as only approximately 10% of cells underwent apoptosis (Fig. 2C). JNK inhibitor also was able to block ORF-6 and ORF-7a in 293T and COS-7 cells (data not shown). These results suggest that the JNK inhibitor is able to block overexpression of ORF-6- or ORF-7a-induced apoptosis.
3.4. ORF-6 and ORF-7a induce ER stress
A recent report showed that ORF-6 is localized in ER/Golgi membrane [27] which is consistent with our immunostaining result of ORF-6 and ORF-7a in COS-7 cells (data not shown). Since both ORF-6 and ORF-7a are ER-localized, we suspected that they induce apoptosis through the ER-stress pathway. GRP94 is an ER-resident molecular chaperone protein which the expression level is increased upon ER-stress [28]. We compared the protein level of GRP94 in order to check whether ER stress occurred in cells transfected with ORF-6 and ORF-7a. Results in Fig. 3a showed that in cells transfected with either ORF-6 or ORF-7a, the GRP94 protein level is increased compared to cells without transfection; however, the protein level is lower than that of the cells treated with 1 μM Thapsigargin, a chemical specific for induction of ER stress. In parallel, we conducted dose dependent experiments by transfecting different amounts of ORF-6 and ORF-7a into cells. Consistently, the endogenous GRP94 protein level was increased when the amount of ORF-6 or ORF-7 increased (Fig. 3B and C). Our observation suggests that overexpression of ORF-6 and ORF-7a could induce apoptosis via ER-stress pathway.
Fig. 3.
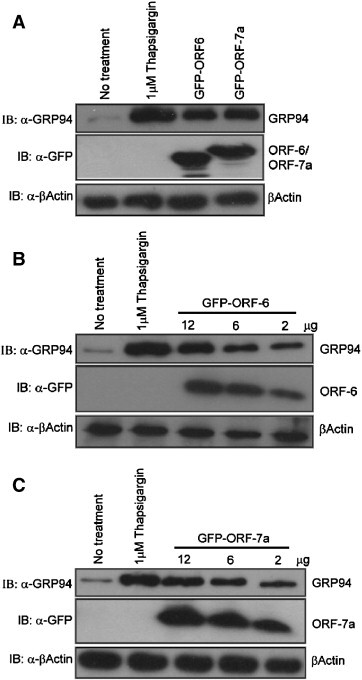
ORF-6 and ORF-7a induces ER stress. (A) Overexpression of ORF-6 and ORF-7a increase endogenous GRP94 protein level. Vero E6 cells were transiently transfected with 6 μg of GFP-ORF-6 and GFP-ORF-7a for 24 h. Cell lysates were normalized to 1 μg/μl by lysis buffer and subjected to Western blot with α-GFP, α-GRP94 and α-Actin. For positive controls, cells were treated with 1 μM of Thapsigargin for 24 h. Cells without treatment served as negative control. (B, C), ORF-6 and ORF-7a increase endogenous GRP94 protein level in a dose dependent manner. Vero E6 cells were transiently transfected with 2 μg, 6 μg and 12 μg of GFP-ORF-6 or GFP-ORF-7a for 24 h. Cell lysates were normalized to 1 μg/μl by lysis buffer and subjected to Western blot with α-GFP, α-GRP94 and α-Actin. For positive controls, cells were treated with 1 μM of Thapsigargin for 24 h. Cells without treatment served as negative control.
4. Discussion
We report here that ORF-6 is the fourth SARS accessory protein that can induce apoptosis. Apoptosis induced by ORF-6 is Caspase-3 dependent. Moreover, we identified that ORF-6- and ORF-7a-induced apoptosis is via JNK-dependent pathway. Finally, these two ER-localized proteins induce ER stress when overexpressed in cells. By searching through the database in NCBI (National Center for Biotechnology Information), we found that there is no significant homology between ORF-6 and other known proteins. Interestingly, we found that the molecular pathway for ORF-6-induced apoptosis is similar to that of ORF-7a.
SARS-CoV encodes 23 putative proteins and 8 novel ORFs have been identified. Five of these, ORF-3a, ORF-3b, ORF-6, ORF-7a and ORF-8a, induce apoptosis when they are overexpressed in cells [18], [19], [20], [23], [29]. However, the signaling pathway responsible for ORF-3a, ORF-3b, ORF-6 and ORF-7a induced apoptosis remains elusive. It has been shown that in SARS-CoV-infected Vero E6 cells, JNK is phosphorylated and apoptosis was inhibited by both JNK and PI3K inhibitors [30]. We identified that JNK inhibitor inhibits both ORF-6- and ORF-7a-induced apoptosis; therefore, we determined that ORF-6 and ORF-7a in SARS-CoV may be responsible for inducing phosphorylation of JNK, which leads to apoptosis.
ER is an organelle that is responsible for protein synthesis; proteins are modified in the ER to form their proper tertiary structure and are then translocated to the outer cell membrane [31]. Particular proteins in an organelle require signals that retain the protein in the correct location. A tetrapeptide sequence, Lys-Asp-Glu-Leu (KDEL), is a general sequence that has been identified for ER-resident proteins [32]; however, evaluating the amino acid sequence of ORF-6 and ORF-7a, we found that neither of them do have KDEL sequences. It has been shown that ORF-7a C-terminal KRKTE is crucial for ER localization whereas ORF-6 C-terminal mutants are still localized in ER/Golgi membrane [27]. Further mapping is needed in order to determine which region is important for ORF-6 ER localization.
One of the possible causes of ORF-6 inducing ER stress is that ORF-6 blocks STAT1 translocation to the nucleus and sequesters host nuclear import factor KPNA2 into rough ER/Golgi membrane. This may cause the overloading of protein in the ER and affect its homeostasis of ER. However, it is still unclear whether Caspase-3 activation in ORF-6 and ORF-7a overexpressed cells is via Caspase-12 or not, as we failed to detect Caspase-12 cleavage products in Thapsigargin treated Vero E6 cells (positive control) and ORF-6 and ORF-7a transfected Vero E6 cells (data not shown). Also, ER stress can activate JNK through IRE1, TRAF2 and ASK1 pathways. Since both ORF-6 and ORF-7a are ER-localized proteins, we hypothesized that ER stress induced by these two proteins will lead to Caspase-3 and JNK activation.
Activation of effector Caspases, Caspases-3, -6 and -7, causes apoptosis as they cleave a variety of proteins. To date, over 200 Caspase substrates have been discovered [33]. Caspase activation for the treatment of insufficient apoptosis, such as in cancer, is challenging whereas Caspase inhibition for the treatment of excessive apoptosis, such as in neurodegeneration, appears to be easier with the use of classical small-molecule inhibitors. Preliminary experiments in animal models using non-selective Caspase inhibitors such as z-VAD(OMe)-CH2F have shown in vivo efficacy in ischemic and hypoxic brain injury, traumatic and excitotoxic brain damage. The same approach may be applied to SARS-infected cells to identify for therapy [34], [35], [36] as two SARS proteins, ORF-6 and ORF-7a, seem to activate pro-apoptotic pathways via the Caspase-3 dependent pathway.
SARS is an infectious disease that is transmissible and fatal. SARS-CoV is a previously unknown coronavirus. To date, there is no drug specifically targeting the control of SARS-CoV. The exact mechanism whereby SARS proteins induce apoptosis must be identified in order to begin to develop SARS targeted drugs.
Acknowledgments
We are grateful to Dr. Zhenguo Wu from Hong Kong University of Science and Technology for GFP-ORF6 and GFP-ORF7a constructs. We thank members of Prof. Peng Li and Dr. Yong Xie laboratories for helpful discussion. The work was supported by a grant from Research Fund for the Control of Infectious Diseases CIDHHS 03/04. SC01.
References
- 1.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Lee N., Hui D., Wu A., Chan P., Cameron P., Joynt G.M., Ahuja A., Yung M.Y., Leung C.B., To K.F., Lui S.F., Szeto C.C., Chung S., Sung J.J. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 4.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. (New York, N.Y.) [DOI] [PubMed] [Google Scholar]
- 5.Peiris J.S., Chu C.M., Cheng V.C., Chan K.S., Hung I.F., Poon L.L., Law K.I., Tang B.S., Hon T.Y., Chan C.S., Chan K.H., Ng J.S., Zheng B.J., Ng W.L., Lai R.W., Guan Y., Yuen K.Y. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poutanen S.M., Low D.E., Henry B., Finkelstein S., Rose D., Green K., Tellier R., Draker R., Adachi D., Ayers M., Chan A.K., Skowronski D.M., Salit I., Simor A.E., Slutsky A.S., Doyle P.W., Krajden M., Petric M., Brunham R.C., McGeer A.J. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- 7.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. (New York, N.Y.) [DOI] [PubMed] [Google Scholar]
- 8.Tsang K.W., Ho P.L., Ooi G.C., Yee W.K., Wang T., Chan-Yeung M., Lam W.K., Seto W.H., Yam L.Y., Cheung T.M., Wong P.C., Lam B., Ip M.S., Chan J., Yuen K.Y., Lai K.N. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1977–1985. doi: 10.1056/NEJMoa030666. [DOI] [PubMed] [Google Scholar]
- 9.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 10.Roulston A., Marcellus R.C., Branton P.E. Viruses and apoptosis. Annu. Rev. Microbiol. 1999;53:577–628. doi: 10.1146/annurev.micro.53.1.577. [DOI] [PubMed] [Google Scholar]
- 11.Teodoro J.G., Branton P.E. Regulation of apoptosis by viral gene products. J. Virol. 1997;71:1739–1746. doi: 10.1128/jvi.71.3.1739-1746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 13.Cory S., Adams J.M. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 14.Harding H.P., Zhang Y., Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 15.Gething M.J., Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 16.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 17.Tan Y.J., Lim S.G., Hong W. Regulation of cell death during infection by the severe acute respiratory syndrome coronavirus and other coronaviruses. Cell. Microbiol. 2007;9:2552–2561. doi: 10.1111/j.1462-5822.2007.01034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fielding B.C., Tan Y.J., Shuo S., Tan T.H., Ooi E.E., Lim S.G., Hong W., Goh P.Y. Characterization of a unique group-specific protein (U122) of the severe acute respiratory syndrome coronavirus. J. Virol. 2004;78:7311–7318. doi: 10.1128/JVI.78.14.7311-7318.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan Y.J., Fielding B.C., Goh P.Y., Shen S., Tan T.H., Lim S.G., Hong W. Overexpression of 7a, a protein specifically encoded by the severe acute respiratory syndrome coronavirus, induces apoptosis via a caspase-dependent pathway. J. Virol. 2004;78:14043–14047. doi: 10.1128/JVI.78.24.14043-14047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Law P.T., Wong C.H., Au T.C., Chuck C.P., Kong S.K., Chan P.K., To K.F., Lo A.W., Chan J.Y., Suen Y.K., Chan H.Y., Fung K.P., Waye M.M., Sung J.J., Lo Y.M., Tsui S.K. The 3a protein of severe acute respiratory syndrome-associated coronavirus induces apoptosis in Vero E6 cells. J. Gen. Virol. 2005;86:1921–1930. doi: 10.1099/vir.0.80813-0. [DOI] [PubMed] [Google Scholar]
- 21.Tan Y.J. The Severe Acute Respiratory Syndrome (SARS)-coronavirus 3a protein may function as a modulator of the trafficking properties of the spike protein. Virol. J. 2005;2:5. doi: 10.1186/1743-422X-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan X., Shan Y., Yao Z., Li J., Zhao Z., Chen J., Cong Y. Mitochondrial location of severe acute respiratory syndrome coronavirus 3b protein. Mol. Cells. 2006;21:186–191. [PubMed] [Google Scholar]
- 23.Yuan X., Shan Y., Zhao Z., Chen J., Cong Y. G0/G1 arrest and apoptosis induced by SARS-CoV 3b protein in transfected cells. Virol. J. 2005;2:66. doi: 10.1186/1743-422X-2-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson C.A., Pekosz A., Lee C.A., Diamond M.S., Fremont D.H. Structure and intracellular targeting of the SARS-coronavirus Orf7a accessory protein. Structure. 2005;13:75–85. doi: 10.1016/j.str.2004.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar P., Gunalan V., Liu B., Chow V.T., Druce J., Birch C., Catton M., Fielding B.C., Tan Y.J., Lal S.K. The nonstructural protein 8 (nsp8) of the SARS coronavirus interacts with its ORF6 accessory protein. Virology. 2007;366:293–303. doi: 10.1016/j.virol.2007.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tangudu C., Olivares H., Netland J., Perlman S., Gallagher T. Severe acute respiratory syndrome coronavirus protein 6 accelerates murine coronavirus infections. J. Virol. 2007;81:1220–1229. doi: 10.1128/JVI.01515-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frieman M., Yount B., Heise M., Kopecky-Bromberg S.A., Palese P., Baric R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 2007;81:9812–9824. doi: 10.1128/JVI.01012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang K., Kaufman R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004;279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
- 29.Chen C.Y., Ping Y.H., Lee H.C., Chen K.H., Lee Y.M., Chan Y.J., Lien T.C., Jap T.S., Lin C.H., Kao L.S., Chen Y.M. Open reading frame 8a of the human severe acute respiratory syndrome coronavirus not only promotes viral replication but also induces apoptosis. J. Infect. Dis. 2007;196:405–415. doi: 10.1086/519166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizutani T., Fukushi S., Murakami M., Hirano T., Saijo M., Kurane I., Morikawa S. Tyrosine dephosphorylation of STAT3 in SARS coronavirus-infected Vero E6 cells. FEBS Lett. 2004;577:187–192. doi: 10.1016/j.febslet.2004.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grossman D., Kim P.J., Blanc-Brude O.P., Brash D.E., Tognin S., Marchisio P.C., Altieri D.C. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J. Clin. Invest. 2001;108:991–999. doi: 10.1172/JCI13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munro S., Pelham H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- 33.Fischer U., Janicke R.U., Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Endres M., Namura S., Shimizu-Sasamata M., Waeber C., Zhang L., Gomez-Isla T., Hyman B.T., Moskowitz M.A. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J. Cereb. Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Holly T.A., Drincic A., Byun Y., Nakamura S., Harris K., Klocke F.J., Cryns V.L. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J. Mol. Cell. Cardiol. 1999;31:1709–1715. doi: 10.1006/jmcc.1999.1006. [DOI] [PubMed] [Google Scholar]
- 36.Schierle G.S., Hansson O., Leist M., Nicotera P., Widner H., Brundin P. Caspase inhibition reduces apoptosis and increases survival of nigral transplants. Nat. Med. 1999;5:97–100. doi: 10.1038/4785. [DOI] [PubMed] [Google Scholar]