

Abstract
Whole genome sequences (WGS) greatly increase our ability to precisely infer population genetic parameters, demographic processes, and selection signatures. However WGS can still be not affordable for a representative number of individuals/populations. In this context, our goal was to assess the efficiency of several SNP genotyping strategies by testing their ability to accurately estimate parameters describing neutral diversity and to detect signatures of selection. We analysed 110 WGS at 12X coverage for four different species, i.e. sheep, goats and their wild counterparts. From these data we generated 946 datasets corresponding to random panels of 1K to 5M variants, commercial SNP chips and exome capture, for sample sizes of 5 to 48 individuals. We also extracted low-coverage genome re-sequencing of 1X, 2X and 5X by randomly sub-sampling reads from the 12X re-sequencing data. Globally, 5K to 10K random variants were enough for an accurate estimation of genome diversity. Conversely, commercial panels and exome capture displayed strong ascertainment biases. Besides the characterization of neutral diversity, the detection of the signature of selection and the accurate estimation of linkage disequilibrium required high-density panels of at least 1M variants. Finally, genotype likelihoods increased the quality of variant calling from low coverage re-sequencing but proportions of incorrect genotypes remained substantial, especially for heterozygote sites. Whole genome re-sequencing coverage of at least 5X appeared to be necessary for accurate assessment of genomic variations. These results have implications for studies seeking to deploy low-density SNP collections or genome scans across genetically diverse populations/species showing similar genetic characteristics and patterns of LD decay for a wide variety of purposes.
Keywords: Whole Genome Sequencing, mammals, depth of coverage, SNP chip, population genomics, genotyping-by-sequencing
Introduction
Demographic and adaptive processes such as migration, genetic bottlenecks and selection are evolutionary forces that have influenced patterns of variation in genomes. Combined with genetic processes such as recombination they result in a non-uniform distribution of genetic variation across the genome. Since the middle of the last century (Wright, 1931; Fisher, 1958), population genetics has been providing theoretical models to infer how these processes have shaped evolution by studying genetic variations among individuals, populations or species. This has set up a conceptual framework for studying the historical role of these processes from the study of current genetic variation. Several metrics allow assessing genetic diversity and inferring historical evolutionary processes. Parameters such as heterozygosity (observed Ho or expected He), nucleotide diversity (π), allelic richness, genotypic richness, linkage disequilibrium (LD) or mutational diversity (θ) and effective population size (Ne) are widely used to infer neutral within-population diversity (Hughes, Inouye, Johnson, Underwood, & Vellend, 2008). They reflect either the number of alleles or haplotypes within a population and/or the evenness of allele or haplotype frequencies (Frankham, Ballou, & Briscoe, 2002). Otherwise, genetic measures such as fixation index (Fst) and analogues, genetic distance, e.g. (Nei’s D, D-tajima) or Bayesian ancestry account for genetic differences among populations and inferring population structure, e.g. (Nei, 1973; Weir & Cockerham, 1984; Frichot, Mathieu, Trouillon, Bouchard, & Francois, 2014). Contrary to demographic effects that similarly impact neutral loci across the genome, selection acts on specific (i.e.,‘outlier’) loci causing varying behavior (Luikart, England, Tallmon, Jordan, & Taberlet, 2003). Selection studies exploit this property by identifying outlier loci using either traditional metrics such as Fst (Luikart et al., 2003), or LD variations, e.g. XP-EHH (Sabeti et al., 2007), or specifically developed metrics, e.g. XP-CLR (Chen, Patterson, & Reich, 2010), HapFLK (Fariello, Boitard, Naya, SanCristobal, & Servin, 2013). Generally, these metrics are effective in quantifying the impacts of demographic processes on populations and predicting their evolution under various ecological scenarios, with applications in conservation biology, agronomy or medicine (Jorde, Watkins, & Bamshad, 2001; Hughes et al., 2008).
Thus, a mandatory prerequisite of evolutionary studies has been the design of panels of molecular markers representative of genome variations (Goodwin, McPherson, & McCombie, 2016). This step has always been challenging. Until the last decade, the efficiency of molecular markers was mainly limited by technical issues. Co-dominant markers such as microsatellites give access to allelic frequencies and are informative for inferring demographic processes (e.g. Di Rienzo et al., 1998; Pritchard, Seielstad, Perez-Lezaun, & Feldman, 1999) but a maximum of a few dozen markers were usually genotyped. Other markers such as Amplified Fragment Length Polymorphism (AFLP) are more representative of whole genome variations, as a few hundred can be genotyped simultaneously, but they are dominant and do not allow for the estimation of allelic frequencies. Even co-dominant markers that can be genotyped across the whole genome such as Single Nucleotide Polymorphisms (SNPs) were first revealed in limited numbers, e.g. (Holloway et al., 1999). Due to these limitations, there was a strong risk for misestimating whole genome variations and linkage disequilibrium (Jones et al., 2013), and to infer incorrect assumptions about past demographic and selection signatures.
Recent technological developments have made possible the typing of very large numbers of co-dominant markers, mostly SNPs, which have considerably increased genome coverage and lead to the development of population genomics approaches (Black, Baer, Antolin, & DuTeau, 2001; Goldstein & Weale, 2001; Jorde, Watkins, & Bamshad, 2001; Luikart, England, Tallmon, Jordan, & Taberlet, 2003). So far, genome wide genotyping using SNPs has essentially been limited to model organisms because of the need for whole genome reference data. Today, methods that overcome this challenge have been developed (Davey et al., 2011; Everett, Grau, & Seeb, 2011), and it is possible to manage whole genome data (in terms of both computation capacity and cost) on nearly any studied species (Goodwin et al., 2016). Moreover, references genomes are now increasingly produced and improved for many species (Howe et al., 2013; Dong et al., 2013; Bennetzen et al., 2012; Carneiro et al., 2014). Nevertheless, producing reference genomes and setting up population genomic studies by re-sequencing whole genomes of several individuals at sufficient coverage remains both costly and computationally time consuming (Fuentes-Pardo & Ruzzante, 2017).
In this context, several strategies have been developed in order to reduce these costs while aiming to keep reliable and representative information of genome-wide variation. One strategy is to reduce the depth of coverage of the WGS data to obtain information on the whole genome (Fuentes-Pardo & Ruzzante, 2017). A few studies have promoted the use of low to medium coverage shotgun WGS (Jansen et al., 2013; Bizon et al., 2014; Dastjerdi, Robert, & Watson, 2014; Therkildsen & Palumbi, 2017). Nevertheless there is a strong risk of losing accuracy in variant calling and individual genotyping. These problems can be overcome by sequencing key individuals or increasing the number sequenced individuals, e.g. (Y. Li, Sidore, Kang, Boehnke, & Abecasis, 2011; Pasaniuc et al., 2012; Alex Buerkle & Gompert, 2013; Han, Sinsheimer, & Novembre, 2014), or by imputing genotypes from genotype likelihoods inferred with dedicated algorithms such as ANGSD (Korneliussen, Albrechtsen, & Nielsen, 2014) or ngsTOOLS (Fumagalli, Vieira, Linderoth, & Nielsen, 2014).
A second strategy to reduce the costs is to avoid whole genome sequencing and genotype a panel of a limited number of variants. For instance, commercial DNA chips or arrays for SNP typing are already available for several species (e.g. human, cattle, sheep, chicken) and can be designed for the purpose of any species. The Restriction-site Associated DNA sequencing (RAD-seq) method reduces genome complexity by re-sequencing stretches of genomic DNA adjacent to restriction endonuclease sites (M. R. Miller, Dunham, Amores, Cresko, & Johnson, 2007; Baird et al., 2008). The RNA-seq method gives access to the transcriptome by sequencing the complementary DNA (cDNA) (Wilhelm et al., 2008; Mudge et al., 2011). Genome enrichment methods allow the extraction of targeted regions of the genome, and one main application is the exome capture used for sequencing protein-coding regions (Ng et al., 2009; Choi et al., 2009; Teer & Mullikin, 2010; Cosart et al., 2011), which can be used in population genomics studies (Mascher et al., 2013; Campbell et al., 2013). However, when using these approaches we face the key question of their ability to produce representative genotypes (Fountain, Pauli, Reid, Palsbøll, & Peery, 2016). The panel of genotyped variants should reliably represent genome variations for all studied individuals to avoid the ascertainment bias that results in the misestimating of genetic parameters (Clark, Hubisz, Bustamante, Williamson, & Nielsen, 2005). Only a few studies evaluated the accuracy of such genotyping approaches, and these have demonstrated an impact of ascertainment bias on measures of population diversity for SNP data (Nielsen, Hubisz, & Clark, 2004; Clark et al., 2005; Albrechtsen, Nielsen, & Nielsen, 2010), and for RAD-seq data (Arnold, Corbett-Detig, Hartl, & Bomblies, 2013; Fountain et al., 2016; Lowry et al., 2016). Moreover, to our knowledge, no study has evaluated empirically the impact of subsampling panels of variants compared to WGS data generated using High-Throughput Sequencing Technologies, when studying genome diversity, population genetic structure and genes under selection.
In this context, our study aimed at assessing the accuracy of low to medium coverage whole genome sequences as well as different variant subsampling methods for describing whole genome diversity. We produced 110 WGS at 12X coverage for four mammal species: sheep (Ovis aries), goat (Capra hircus) and their closely related wild species, the Asiatic mouflon (Ovis orientalis) and the Bezoar ibex (Capra aegagrus). From these WGS data we extracted lower re-sequencing coverages associated to genotype likelihoods (GLs) to evaluate the impact of sequencing depth on the assessment of whole genome diversity. We also extracted panels of genomic variants corresponding to different genome sampling strategies (i.e., exome capture, commercial SNP chips or random panels) in order to evaluate the impact of variants subsampling on the estimation of genome diversity and on the detection of a selection signature. This allowed defining appropriate sampling of genome-wide markers to describe neutral diversity and to detect selection signatures in population genomic studies.
Materials and Methods
Sampled individuals
Tissue samples were collected for 48 sheep (Ovis aries) and 30 goats (Capra hircus) widely spread across the Northern half of Morocco (North of latitude 28°) between January 2008 and March 2012 (Table S1). Tissues from the distal part of the ear were collected and placed in alcohol for one day, before transfer into silica-gel tubes until DNA extraction. Tissues from 15 Asiatic mouflon (Ovis orientalis) and 20 Bezoar ibex (Capra aegagrus) were collected in Iran, either from captive or recently hunted animals and conserved in silica-gel after one day in alcohol, or from frozen corpses or tissues archived in alcohol by the Iranian local Department of Environment and transferred in silica-gel until extraction.
DNA extraction and re-sequencing
DNA extraction was done at Parco Tecnologico Padano (Lodi, Italy) using the Puregene Tissue Kit from Qiagen® following the manufacturer’s instructions. Then, 500ng of genomic DNA were sheared to a 150-700 bp range using the Covaris® E210 instrument. Sheared DNA was used for Illumina® library preparation by a semi-automatized protocol. Briefly, end repair, A tailing and Illumina® compatible adaptors (BiooScientific) ligation were performed using the SPRIWorks Library Preparation System and SPRI TE instrument (Beckmann Coulter), according to the manufacturer protocol. A 300-600 bp size selection was applied in order to recover most of the fragments. DNA fragments were amplified by 12 cycles PCR using Platinum Pfx Taq Polymerase Kit (Life Technologies®) and Illumina® adapter-specific primers. Libraries were purified with 0.8x AMPure XP beads (Beckmann Coulter). After library profile analysis by Agilent 2100 Bioanalyzer (Agilent Technologies®) and qPCR quantification, the libraries were sequenced using 100 bp length read chemistry in paired-end flow cell on the Illumina® HiSeq2000.
Read mapping, SNP calling and filtering
Illumina paired-end reads of Ovis were mapped on the sheep reference genome (OAR v3.1, GenBank assembly GCA_000317765.1, Jiang et al., 2014) and those of Capra on the goat reference genome (CHIR v1.0, GenBank assembly GCA_000317765.1, Dong et al., 2013) using BWA mem (H. Li & Durbin, 2009). 99.4% (± 0.1%), 99.3% (± 0.2%), 98.9% (± 0.1%) and 98.8% (± 0.4%) of the reads were mapped on the reference assembly for sheep, mouflon, goats and bezoar, respectively. The BAM files produced were then sorted using Picard SortSam and improved using Picard MarkDuplicates (http://picard.sourceforge.net), GATK RealignerTargetCreator, GATK IndelRealigner (DePristo et al., 2011) and Samtools calmd (H. Li et al., 2009).
As described by Benjelloun et al. (Benjelloun et al., 2015), variant sites were initially called using three different algorithms: Samtools mpileup (H. Li et al., 2009), GATK UnifiedGenotyper (McKenna et al., 2010) and Freebayes (Garrison & Marth, 2012). Variants were called for each group independently: sheep, mouflon, goat, and Bezoar ibex. Note that a larger dataset than that used in this study was used for variant discovery in domestic groups (160 sheep and 161 goats from Morocco; for European Nucleotide Archive ID, see Table S2). Then we ran two successive rounds of filtering variant sites. Filtering stage 1 merged together calls from the three algorithms, whilst filtering out the lowest-confidence calls. A variant site passed if it was called by at least two different calling algorithms with variant phred-scaled quality > 30. An alternate allele at a site passed if it was called by any one of the calling algorithms, and the genotype count > 0. Filtering stage 2 used Variant Quality Score Recalibration by GATK. First, we generated a training set of the highest-confidence variant sites where (i) the site is called by all three variant callers with variant phred-scaled quality > 100; (ii) the site is biallelic; (iii) the minor allele count is at least 3, counting only samples with genotype phred-scaled quality > 30. The training set was used to build a Gaussian model using the tool GATK VariantRecalibrator using the following variant annotations from UnifiedGenotyper: QD, HaplotypeScore, MQRankSum, ReadPosRankSum, FS, DP, Inbreeding Coefficient. The Gaussian model was applied to the full data set, generating a VQSLOD (log odds ratio of being a true variant). Sites were filtered out if VQSLOD < cutoff value. The cutoff value was set for each population by the following: Minimum VQSLOD = {the median value of VQSLOD for training set variants} - 3 * {the median absolute deviation VQSLOD of training set variants}. Measures of the transition/transversion ratio of SNPs suggest that this chosen cut-off criterion gives the best balance between selectivity and sensitivity.
Genotypes were improved and phased by Beagle 4 (Browning & Browning, 2013), and then filtered out where the genotype probability calculated by Beagle is less than 0.95. The genotype call sets generated at this stage constituted the WGS datasets used for within-population analyses. For cross-populations comparisons and validation of the identified WGS surrogates that were performed in each genus (i.e. Capra and Ovis), we generated a set of filtered variant sites per genus by merging the positions of filtered bi-allelic SNPs called in the different groups. For each sample, genotypes were called at each SNP position using GATK UnifiedGenotyper using the option GENOTYPE_GIVEN_ALLELES. Genotypes were improved and phased by Beagle 4 (Browning & Browning, 2013), and then filtered out where the genotype probability calculated by Beagle is less than 0.95.
Quality control of WGS data
To further assess the quality of the filtered WGS datasets, a subset of the sequenced individuals were genotyped using commercial SNP Chips by Laboratorio Genetica e Servizi (Cremona, Italy). 29 sheep and 8 Asiatic mouflon were genotyped with the Illumina® ovine 50K SNPs BeadChip, and 27 goats and 8 Bezoar ibex with the Illumina® caprine 50K SNPs BeadChip. In order to establish the concordance between WGS and chip data the coordinates of the SNPs on the chips were obtained by mapping the probes used for chip design onto the corresponding reference genome (OAR v3.1 or CHIR v1.0) using BWA aln and BWA samse (Li & Durbin, 2009). The raw data in Plink format (Purcell et al., 2007) were updated for SNP coordinates and were filtered for each group by applying the following inclusion criteria: SNPs in a known chromosome (from our mapping); minor allele frequency (MAF) > 0.02, genotype call rate (SNPs) > 0.95, genotype call rate (Animals) > 0.95 and identity-by-state (Animals) < 0.95. The filtered datasets were converted to harmonize the reference alleles with the reference genomes using a script based on the programs PlinkSeq v 0.08 (http://atgu.mgh.harvard.edu/plinkseq/index.shtml) and Plink v 1.07 (Purcell et al., 2007) which was necessary for the quality control of the re-sequencing data. After removing the positions corresponding to short indels and tri-allelic variants, which are incorrectly genotyped by the BeadChips, the number of SNPs both genotyped with the Chip and by whole genome sequencing was 47,122 for sheep, 49,467 for goats, 37,779 for Asiatic mouflon and 41,751 SNPs for Bezoar ibex. The comparison of the ovine and caprine 50K BeadChips genotyping data with the WGS data was performed. The average (± s.d.) genotype concordance between the ovine/caprine 50K BeadChips and the WGS was 99.9% (± 0.1%) in sheep, 99.7% (± 0.0%) in goats, 99.7% (± 0.1%) in Asiatic mouflon and 98.5% (± 0.3%) in Bezoar ibex.
Setting up datasets of variants
From the individuals sequenced, we defined different groups depending on the question addressed. First, to evaluate the impact of sampling panels of variants and reducing the WGS coverage on the estimation of genetic parameters we designed four groups corresponding to 30 sheep, 30 goats, 14 Asiatic mouflon and 18 Bezoar ibex. In order to assess the effect of individual sample size, each analysis was performed for the whole groups and for two random subsets corresponding approximately to one third and two thirds of the total (i.e. respectively 10 and 20 sheep and goats, 5 and 10 Asiatic mouflon and 8 and 13 Bezoar ibex). Second, for detecting a signal of selection associated to the RXFP2 locus (Kijas et al., 2012) and related to the presence/absence of horns, we had to consider additional sheep to constitute 2 contrasted groups of 15 horned and 15 polled individuals (Figure 1; Table S1).
Figure 1. Flow-chart describing sampling random and non-random panels of variants and individuals.
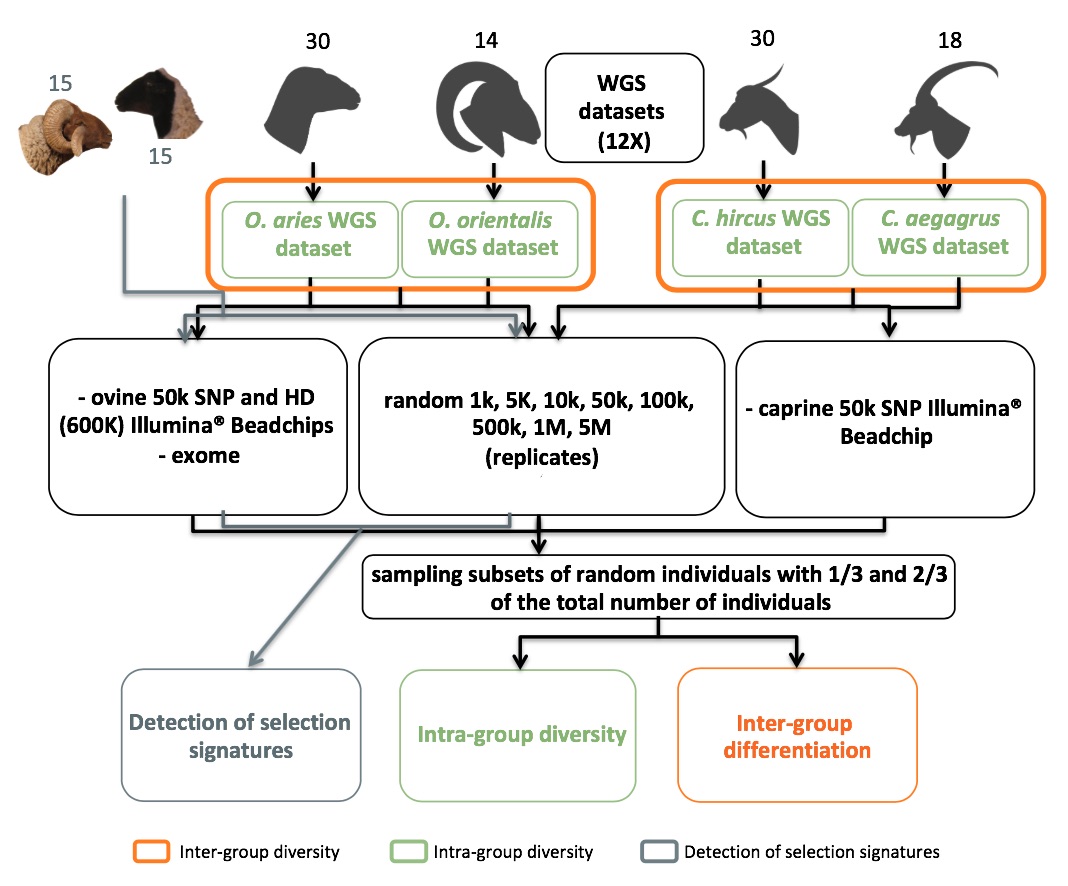
Whole genome sequences are denoted by WGS.
For each group of individuals, a 12X WGS dataset was composed of all the SNPs called (see 'Read mapping, SNP calling and filtering' section) and used for within population analyses. Note that for cross-populations comparisons we only kept the variants found polymorphic in both groups considered, in order to prevent from any possible biased calling or filtering error. Then, variant panels were extracted from the 12X WGS datasets. Random panels were extracted using GATK SelectVariants (McKenna et al., 2010) consisting in 5 independent replicates for each of the 8 following numbers of variants: 1K, 5K, 10K, 50K, 100K, 500K, 1M and 5M. We also created non-random panels simulating the data obtained with commercial BeadChips or through exome capture. BeadChip data were obtained by calling variants at the Illumina® 50K Ovine or Caprine BeadChip SNP coordinates. We successfully extracted 42,117 variants for sheep, 47,245 variants for goats, 26,141 variants for Asiatic mouflon and 33,951 variants for Bezoar ibex. The combined datasets used for cross-population analyses included 30,870 variants for Ovis and 38,641 variants for Capra. In sheep, the High Density BeadChip genotyping was simulated by calling WGS variants at the coordinates of the Illumina® ovine HD BeadChip. This gave 601,456 variants for sheep and of 444,169 variants for Asiatic mouflon. The combined dataset had 419,041 variants.
We simulated an exome capture only for Ovis because of the annotation of the goat genome was insufficiently advanced. The exome annotation was obtained from the sheep genome annotation that was available in ENSEMBL database by the time of analysis (25th September 2013) (ftp://ftp.ensembl.org/pub/pre/) and corresponded to 224,871 exons in 45,972 genes. The number of variants from these regions extracted from 12X WGS was 278,568 for sheep and 155,236 for Asiatic mouflon. The 93,409 variants polymorphic in both groups constituted the combined dataset. Thus, for the identification of potential surrogates for the WGS, the genotypes produced for the different variant panels and the different groups of individuals constituted a total of 946 datasets of which 516 were used for estimating within-group genetic diversity and 430 for cross-populations comparisons (Figure 1). Furthermore, to validate the ability of some SNP panels to represent genome variations in other independent populations, a set of random 10K variants defined in sheep was extracted from WGS of Asiatic mouflon. The same approach was followed to test a random 10K markers defined in goats on bezoars.
Moreover, we simulated a ddRad-seq experiment with the Ovis genome. We located potential restriction site for the Sbfl (5’ CCTGCA|GG 3’) and MspI (5’ C|CGG 3’) enzymes using the 'seqkit locate' command (Shen, Le, Li, & Hu, 2016) on the Oar_v3.1 genomic sequence. Outputs were then processed using a custom R script in order to select DNA fragments harbouring cut on each end by a different enzyme and within the range of 250-350 bp and 250-500 bp. This resulted in two bed files corresponding to the positions on the ovis genomes of the potential ddRadseq fragments. The number of fragments was 8,895 for the range 250-350 bp and 18,979 for the range 250-500 bp. This experiment resulted in datasets of 45,485 and 118,395 variants in sheep and 30,241 and 79,134 variants in Asiatic mouflons respectively for the range 250-350 bp and 250-500 bp.
Extracting low-coverage re-sequencing data
The 12X WGS data were subsampled to simulate the output of a sequencing experiment with fewer reads were generated. For each of the 30 sheep and the 30 goats groups, three sub-sampled WGS datasets were generated comprising (i) 15 million, (ii) 30 million, (iii) 75 million paired reads, corresponding approximately to a 1X, 2X, and 5X sequencing coverage of the genome, respectively. Paired reads were randomly chosen from the full sequencing data using Picard Downsample, in such a way that all reads had an equal probability of being chosen, including duplicate or unaligned ones. Next, Picard MarkDuplicates was used to tag reads that appeared as duplicates in the sub-sampled datasets.
Variant calling and filtering were done using two different approaches. The first one was based on the approach used for 12X coverage datasets. Thus, for each variant of the list generated for the 12X WGS, genotypes were called using GATK UnifiedGenotyper with the option GENOTYPE_GIVEN_ALLELES. Genotypes were improved and phased with Beagle 4, and filtered at the individual level when the genotype probability was less than 0.95. Average depths (± standard deviation) obtained for the initially called variants were: 1.61±0.01; 2.35±0.03 and 5.07±0.09 in sheep and 1.62±0.01; 2.37±0.02 and 5.12±0.07 in goats respectively for the targeted coverages 1X, 2X and 5X.
The second approach, more suitable for low coverage data, was based on genotype likelihoods (GLs). For each sequencing depth we used ANGSD v0.921 (Korneliussen et al., 2014) to call variants using the GATK likelihood model implemented in ANGSD. We applied a significance probability threshold of 1.0 × 10−6 to call variants. Then, genotypes were improved and phased with Beagle 4.
The variants called using genotype likelihoods that were present in the 12X coverage datasets were quantified. Then, for each of the simulated coverages, the genotype at each variant position was compared to that obtained for the 12X coverage and classified as matching, un-matching or missing, for homozygotes and heterozygotes separately. Additionally, the individual observed heterozygosity was inferred for each coverage using the whole sets of the identified variants and used to estimate (i) Pearson correlation, (ii) Spearman correlation with 12X inferences. Slope values (b) were estimated for each depth and calling/filtering approaches by setting the intercept to 0. Furthermore, Site Frequency Spectra (SFS) were drawn and compared between the datasets implemented using GLs and the 12X datasets.
Description of genome diversity
Genetic diversity within groups
Using Vcftools (Danecek et al., 2011) we estimated the observed heterozygosity (Ho) and inbreeding coefficient (F) with polymorphic autosomal bi-allelic SNPs, and the nucleotide diversity (π) by taking the averaged nucleotide diversity over all autosomal variants. Furthermore, we estimated site frequency spectra (SFS) directly from vcf files using the R script vcf2sfs developed by Liu et al. (Liu, Ferchaud, Grønkjaer, Nygaard, & Hansen, 2018). Correlations between inferred spectra from different sequencing depth were made using Pearson’s rank correlations (r). Otherwise, pairwise SNPs linkage disequilibrium (r2) was also estimated with Vcftools on all bi-allelic non-rare variants (SNPs and indels with MAF>=0.05) for 5 segments of 2Mbp selected on 5 chromosomes (physical positions between 1 Mbp and 3 Mbp on chromosomes 5, 10, 15, 20 and 25). The extent of the linkage disequilibrium was assessed by the physical distance corresponding to r 2 = 0.15 (r20.15), i.e. the average distance between 2 markers with a coefficient of determination r2=0.15.
Genetic differentiation and structure
The genetic structure and differentiation was measured between domestics and wilds, as representative of population having diverged about 10,000 years ago (i.e., at the time of domestication). The averaged Fst (Weir & Cockerham, 1984) was estimated for bi-allelic variants with Vcftools. Additionally, genetic structure was investigated through the Bayesian clustering approach sNMF (Frichot, Mathieu, Trouillon, Bouchard, & Francois, 2014) using bi-allelic variants. This method was specifically developed to estimate individual admixture coefficients on large genomic datasets.
Detection of a selection signature
We targeted the genomic region surrounding the Relaxin/insulin-like family peptide receptor 2 gene (RXFP2; Chr 10: 29,454,677 – 29,502,617bp), which already showed a signature of selection related to polledness in sheep (Kijas et al., 2012; Dominik, Henshall, & Hayes, 2012) and also in wild bighorn sheep (Kardos et al., 2015). We extracted variants between positions 20 Mb and 40 Mb on chromosome 10 for 15 horned and 15 polled sheep and searched for selective sweeps in this region using two methods: (i) A standard Fst test by estimating genome-wide single nucleotide Fst (Weir & Cockerham, 1984) and choosing as outliers the top 0.1% values of Fst, and (ii) XP-CLR (Chen, Patterson, & Reich, 2010). For the later method, we estimated a constant recombination rate for this region based on the random 1M variants dataset, using the PAIRWISE program of LDhat v2.2 (Auton & McVean, 2007) with recommended parameters. XP-CLR scores were calculated for each grid point placed along the segment considered with a spacing of 5Kb. A maximum of 300 bi-allelic variants was considered in a sliding window of 0.5cM around the grid point and we down-weighted contributions of highly correlated SNPs (r2>0.99).
Results
Variant calling was done using 12X coverage whole genome sequencing data for each species. It allowed the discovery of 29.96, 29.04, 21.71 and 17.32 million polymorphic variants for sheep, Asiatic mouflon, goats and Bezoar ibex, respectively (see Table 1), which correspond mostly to SNPs but also to small indels (i.e., 6% to 10% of the variants). We created both non-random (i.e., exome and SNP BeadChips) and random variant panels across a wide range of densities by subsampling SNPs from this WGS data, and assessed the potential of each panel to accurately represent genome diversity in domestic and wild animals. Lastly, low-coverage re-sequencing data were generated by randomly sampling various fixed percentages of reads from the raw 12X re-sequencing data (see 'Material and Methods' section) and genotypes were compared to the 12X WGS variants.
Table 1. Population genomics statistics from WGS data for wild and Moroccan domestic small ruminants.
| Species/Populations | Ovis | Capra | ||
|---|---|---|---|---|
| sheep | Asiatic mouflon | Goats | Bezoar ibex | |
| Number of individuals (n) | 30 | 14 | 30 | 18 |
| Number of variants | 43,478,084 | 29,274,713 | 31,775,474 | 17,449,771 |
| Number of polymorphic variants | 29,958,788 | 29,039,121 | 21,709,831 | 17,321,976 |
| Short indels | 2,805,416 | 2,713,334 | 2,139,714 | 1,344,653 |
| Variants with > 2 alleles | 817,859 | 265,998 | 219,706 | 109,520 |
| Heterozygosity (Ho) | 0.222 ± 0.026 | 0.223 ± 0.032 | 0.189 ± 0.018 | 0.194 ± 0.025 |
| Inbreeding coefficient (F) | 0.061 ± 0.108 | 0.186 ± 0.118 | 0.056 ± 0.092 | 0.182 ± 0.106 |
| Linkage disequilibrium r20.15 (Kb) | 10.22 | 4.52 | 8.70 | 6.69 |
| Nucleotide diversity (π) | 0.165 | 0.273 | 0.137 | 0.237 |
Description of genome diversity
We assessed the effect of variant subsampling on estimating genetic diversity by comparing the observed heterozygosity (Ho), inbreeding coefficient (F), nucleotide diversity (π) and Linkage disequilibrium (r2 0.15) for both the WGS dataset and variant panels (see Table 1). Even at low-densities, random panels returned diversity metrics similar to those derived from WGS. Accurate estimates were obtained with all random panels of 5K or more markers for inbreeding (F), nucleotide diversity (π), and site frequency spectra (SFS) (Figures 2, 3, S1, S2, S3, S4, S5, S6) and with random panels of 10K or more markers for observed heterozygosity (Ho) (Figure S7). Furthermore, estimates from the random panel of 10K markers defined in sheep and applied on Asiatic mouflon approximate WGS inferences in this population in terms of diversity parameters (Figures 4, S8) and site frequency spectra for which Pearson’s rank correlations with WGS=0.95 (Figure S9). Similar results were obtained for the random panel of 10K SNP defined in goats and used to infer genetic statistics in bezoar for which Pearson’s rank correlations of SFS with WGS=0.97 (Figures S10, S11, S12). On the contrary, non-random panels of variants generated strongly biased estimations. The ovine 50K SNP and HD BeadChips and the caprine 50K SNP BeadChip from Illumina® showed considerable ascertainment biases by overestimating the diversity in all groups and datasets (Figures 2-5, S8, S10). For example, in the 30 sheep there was an overestimation of 129%, 108% and 194% for π and 61%, 47% and 102% for Ho for these three panels, respectively. This ascertainment bias did not affect the estimation of the inbreeding coefficient (F) (Figures 5, S11). Whatever the bias, the ranking of individual Ho and F were not affected by the panel of variants used (Figure 5), but for π, the wilds even appeared less diverse than the domestics while WGS data showed the opposite (Figure 4, S10). Similarly, SFS inferred from SNP chips were quite different than those inferred from 12X WGS data (Pearson’s rank correlations ranging from -0.377 for the caprine beadchip in goats to 0.339 for the HD beadchip in sheep), except for the Asiatic mouflon (correlations of 0.706 for the 50K beadchip and 0.807 for the HD array) (Figures 3, S4, S5, S6). The dataset simulating exome capture underestimated π and Ho (e.g., underestimation of 20% and 6% for π and 8% and 5% for Ho in sheep and mouflon, respectively). The underestimation of the inbreeding coefficient (F) was higher in domestic but not in wild animals. Inversely, site frequency spectra were highly correlated to 12X WGS inferences (r~0.99 for different groups and population sizes). Otherwise, we did not detect any sample size effect on π inference.
Figure 2. Nucleotide diversity (π) in sheep calculated from WGS data and from random and non-random panels of variants.

Nucleotide diversity (π) was estimated for each replicate of the different numbers of variants of the random panels and for each non-random panel. Sample sizes varied for each estimate from 10 to 30 individuals.
Random panels are denoted by their number of variants (from 1K to 5M) and non-random panels by: 50K.Chip (Illumina® ovine 50K SNP Beadchip), HD.Chip (Illumina® ovine HD Beadchip) exome (exome capture simulation), WGS (all variants extracted from whole genome sequences). For each panel of variants the sample sizes are from left to right: 10 (red), 20 (green) and 30 (yellow) individuals.
Figure 3. Site frequency spectra (SFS) in sheep inferred from WGS data and from random and non-random panels of variants.

Site frequency spectra were estimated using different random and non-random panels for 30 sheep.
Random panels are denoted by their number of variants (from 1K to 5M) and non-random panels by: 50K.Beadchip (Illumina® ovine 50K SNP Beadchip), HD.Beadchip (Illumina® ovine HD Beadchip) exome (exome capture simulation), WGS (all variants extracted from whole genome sequences). Pearson correlation coefficients with the WGS inferences are shown for each panel.
Figure 4. Nucleotide diversity (π) estimated in two Ovis groups with random and commonly used panels of variants.

Plot of Nucleotide diversity (π) estimated with a random set of 10K variants sampled in sheep data (10K), and with Illumina® ovine 50K SNP Beadchip (50K.Chip), Illumina® ovine HD Beadchip (HD.Chip), and variants extracted from whole genome sequences (WGS).
Figure 5. Estimates of individual inbreeding coefficient (F) and observed heterozygosity (Ho) from different panels of variants compared to WGS data estimates in sheep.

Plot of individual inbreeding coefficient (F; top) and observed Heterozygosity (Ho; bottom) estimated with variants extracted from whole genome sequences (WGS) versus inferences with Illumina® ovine 50K SNP Beadchip (50K.Chip), Illumina® ovine HD Beadchip (HD.Chip), and 1 set of 10K variants defined in Moroccan sheep (random 10K). The red lines represent the relationship for which the estimates of the different panels are identical to the ones of WGS inferences.
At least 1M random markers in sheep and 500K random markers in the other groups were necessary to have an estimation of LD (r20.15) similar to that obtained with the WGS dataset. Smaller random panels and non-random panels biased this estimation (Figures S13, S14). Exome capture especially biased the LD estimation in sheep (but not in Asiatic mouflon with 10 individuals and more). Moreover, in all groups, decreasing the number of individuals increased r20.15. In particular, Asiatic mouflon had an r20.15 of 4.52Kb for 14 individuals and of 79.4Kb for 5 individuals (Figure S14).
We also assessed the influence of the variant panels on two methods describing the genetic differentiation of wild versus domestic populations. First, we estimated the Weir & Cockerham (Weir & Cockerham, 1984) differentiation index (Fst), which was rather high between wild and domestic animals (Fst = 0.105 in Ovis and Fst = 0.087 in Capra from WGS data; Figures 6, S15). Independently of the number of variants used, there was a strong sampling effect due to the individuals selected for estimating Fst. For a given set of individuals the number of random variants did not influence greatly the mean Fst values compared to that obtained with WGS data. The smallest random panels (from 1K to 50K) increased the variance in Fst estimates among marker-set replicates for a given set of individuals (Figures 6, S15). The caprine 50K SNP BeadChip Illumina® overestimated Fst values by 28% on average (Figure 6) and the ovine 50K and HD SNP BeadChips, and the exome capture slightly underestimated the Fst (2 to 13%). However, all non-random panels kept the ranking found with the WGS datasets for Fst estimated with different sets of individuals (r always > 0.98). Except for the caprine 50K BeadChip, the effect of the subsampling strategy on the Fst estimation was lower than that of the sample size. Second, we used the clustering method implemented in sNMF (Frichot et al., 2014) to estimate individual ancestry coefficients. The estimations depended neither on the number of markers used nor on the number of individuals in the sample. For the most likely number of clusters (K=2 for Ovis and Capra from the sNMF cross-validation values (Frichot et al., 2014)), all variant panels led to similar results (Figure S16).
Figure 6. Fixation index (Fst) between Moroccan goats and Bezoar ibex for different panels of variants and different samples of individuals.

The fixation index Fst (Weir & Cockerham, 1984) was estimated for each random panel for the 5 independent replicates, and for each non-random dataset for each sample size. Random panels are denoted by their number of variants (from 1K to 5M) and non-random panels by: 50K.Chip (Illumina® caprine 50K SNP Beadchip), WGS (all variants extracted from whole genome sequences). For each panel of variants the sample sizes are from left to right: 18 (red), 33 (green) and 48 (yellow) individuals.
Finally, we assessed the effect of the panel of SNPs used on the ability to detect a signature of selection. By contrasting 15 horned and 15 polled sheep, a standard Fst test and the XP-CLR method (Chen et al., 2010) applied on the WGS dataset allowed detection of the signal of selection previously reported on the Relaxin/Insulin-Like Family Peptide Receptor 2 gene RXFP2 (Kijas et al., 2012). This signal could also be clearly detected with random panels of 100K markers and more, with the ovine 50K and HD BeadChips and with the exome capture. (Figure 7, Figure S17). A slight signal was also seen when using the random 50K SNP panel (Figure S17). However, the intensity of the signal decreased progressively with the density of markers. Therewith, another non-previously reported sweep was detected with the XP-CLR method only for the WGS dataset with random panels of 5M and 1M variants. This signal was located in the region of the Neurobeachin NBEA and Mab21-like 1 MAB21L1 genes on chromosome 10 (positions 26,007,917-26,592,574 and 26,231,353-26,232,432 on OAR v3.1, respectively).
Figure 7. XP-CLR scores calculated along the 20M-40M bp segment on chromosome 10 in a horned-polled Moroccan sheep comparison for different sets of variants.

The two peaks of XP-CLR scores showed in the WGS data plot are located respectively in the two genes NBEA (chr 10: 26,007,917 - 26,592,574) and MAB21L1 (chr 10: 26,231,353 - 26,232,432) and in the RXFP2 gene (chr 10: 29,454,677 - 29,502,617 bp). The horizontal dashed line represents a XP-CLR score of 15 to represent a scale among the different plots.
Difference between random and non-random panels
One major difference in the design of random panels of variants and the BeadChips relies on the distribution of variants across the genome. Figure S18 illustrates this in showing the distributions of the physical distances between adjacent variants in various panels for sheep and goats. The random 50K variants as well as the random 500K variants and the HD ovine BeadChip showed a similar L-shaped curve indicating that variants were evenly distributed across the genomes. On the other hand, as it might be predicted, the caprine 50K BeadChip displayed an almost complete lack of SNPs separated by less than around 30Kb, while for the ovine 50K BeadChip the lack of SNPs in these categories is less drastic, at most around a half of the expected distribution for the shorter distances. The exome capture simulation displayed a very high occurrence of distances lower than 200 bp and a quasi absence of distance larger than 10kb, which might be expected (Figures S18, S19).
Otherwise, our simulations of RAD-seq have revealed that the datasets resulting from these experiments make it possible to obtain the same results of SFS as with random datasets of equivalent marker densities (Pearson’s rank correlations of SFS with WGS>0.999; Figures S20, S21).
Reliability of low-coverage re-sequencing
1X, 2X and 5X whole genome sequencing coverage were simulated by randomly sampling reads in the 12X WGS data, and used for inferring genotype likelihoods (GLs) and calling genotypes in 30 goats and 30 sheep. The 12X WGS allowed genotyping at 31,775,474 variant sites (31,735,229 at which more than 95% of individuals had genotypes called) for goats and 43,478,084 for sheep (43,105,056 at which more than 95% of individuals had genotypes called), and decreasing the coverage strongly reduced the number of variants that could be genotyped using similar algorithms for calling genotypes (missing genotypes, Table 2), while the number of variants wrongly genotyped remained rather low (mis-matching genotypes, Table 2). Heterozygous genotypes were more affected than homozygous ones. Moreover, the decreasing coverage resulted in an increasing underestimation of Ho (around 1.2, 3 and 6 times for 5X, 2X and 1X, respectively), and in a decreasing preservation of the relative ranking of Ho values among individuals (Table 2). This ranking was better preserved in sheep than in goats. Using genotype likelihoods, the numbers of called variants were substantial (from 18.3M for 1X in goats to 44.7M for 5X coverage in sheep; Table 2). However, whatever the coverage, no more than 56.8% of the variants discovered in sheep and 59.8% in goats were also in the 12X WGS dataset. The percentage of correctly genotyped heterozygotes at variant sites shared with 12X data ranged from 50% for 1X data to 88% for 5X data. These proportions ranged from 88% for 1X data to 99% for 5X data at homozygous sites shared with the 12X data. The estimations of Ho using the whole sets of variants were substantially improved using GLs. Pearson’s rank correlations with 12X data ranged from 0.88 (for 1X in goats) to 0.99 for 5X in sheep, and the Spearman correlation coefficients from 0.69 (1X in sheep) to 0.81 (obtained with 5X data in sheep; Table 2). Interestingly, inferences from 5X data from classical algorithms showed even slightly higher correlations with 12X data than 5X data from GLs (Table 2). Site frequency spectra show a relative discordance with 12X data with an overrepresentation of variants displaying allele frequencies close to 0.5 and a lower representation of rare variants (Figure S22, S23).
Table 2. Concordance between low-coverage re-sequencing and 12X coverage for homozygous and heterozygous genotypes.
| Coverage (Genotype inference) | 1x (12x-calling) | 1x (GL) | 2x (12x-calling) | 2x (GL) | 5x (12x-calling) | 5x (GL) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | Sheep | Goats | Sheep | Goats | Sheep | Goats | Sheep | Goats | Sheep | Goats | Sheep | Goats | |
| # of sites | 12,550,038 | 10,662,633 | 30,135,105 | 18,274,530 | 15,617,291 | 12,930,734 | 37,578,055 | 23,631,570 | 28,419,192 | 20,974,409 | 44,698,817 | 29,810,608 | |
| Genotypes for 100% of individuals | # of polymorphic variants | 259,177 | 249,056 | 30,134,708 | 18,274,238 | 1,783,255 | 1,737,328 | 37,577,014 | 23,630,719 | 15,847,527 | 11,296,131 | 44,691,747 | 29,805,099 |
| # of variants shared with 12x data | - | - | 17,109,335 | 10,936,768 | - | - | 20,760,524 | 13,951,043 | - | - | 24,647,380 | 17,543,844 | |
| Heterozygote genotypes | Matching 12x (%) | 0.12 ± 0.02 | 0.19 ± 0.03 | 28.6 ± 3.4 | 28.2 ± 2.9 | 6.20 ± 0.79 | 5.68 ± 0.72 | 40.6 ± 4.8 | 41.2 ± 4.1 | 74.1 ± 8.6 | 71.5 ± 7.1 | 60.0 ± 7.0 | 61.8 ± 6.1 |
| Mis-matching 12x (%) | 3.14 ± 0.33 | 4.38 ± 0.65 | 28.7 ± 3.3 | 28.1 ± 2.9 | 2.97 ± 0.30 | 3.66 ± 0.53 | 23.3 ± 2.6 | 22.9 ± 2.3 | 2.27 ± 0.24 | 2.16 ± 0.23 | 8.05 ± 0.92 | 7.80 ± 0.84 | |
| Missing (%) | 96.7 ± 11.1 | 95.4 ± 9.4 | 42.7 ± 4.9 | 43.7 ± 4.3 | 90.8 ± 10.3 | 90.7 ± 9.0 | 36.1 ± 4.1 | 36.0 ± 3.5 | 23.6 ± 2.6 | 26.4 ± 2.6 | 32.0 ± 3.7 | 30.4 ± 3.0 | |
| Homozygote genotypes | Matching 12x (%) | 34.1 ± 0.1 | 38.1 ± 0.1 | 23.6 ± 0.6 | 30.0 ± 0.4 | 49.2 ± 0.2 | 52.3 ± 0.2 | 33.5 ± 0.9 | 31.2 ± 0.6 | 87.8 ± 1.7 | 87.5 ± 1.1 | 44.9 ± 1.3 | 44.1 ± 0.9 |
| Mis-matching 12x (%) | 0.02 ± 0.02 | 0.02 ± 0.02 | 3.32 ± 0.62 | 2.45 ± 0.43 | 0.03 ± 0.01 | 0.02 ± 0.01 | 2.01 ± 0.51 | 1.50 ± 0.37 | 0.09 ± 0.10 | 0.07 ± 0.02 | 0.53 ± 0.22 | 0.39 ± 0.15 | |
| Missing (%) | 65.9 ± 2.1 | 61.9 ± 1.4 | 73.1 ± 0.9 | 76.6 ± 0.6 | 50.8 ± 1.9 | 47.7 ± 1.4 | 64.5 ± 0.8 | 67.3 ± 0.5 | 12.2 ± 0.4 | 12.5 ± 0.4 | 54.6 ± 0.7 | 55.5 ± 0.4 | |
| Correlations of Ho with 12X estimates | r (Pearson) | 0.642 | 0.173 | 0.972 | 0.877 | 0.989 | 0.507 | 0.989 | 0.953 | 0.999 | 0.989 | 0.990 | 0.980 |
| Slope (Pearson) | 0.149 | 0.176 | 0.118 | 0.128 | 0.329 | 0.281 | 0.287 | 0.305 | 0.864 | 0.818 | 0.609 | 0.628 | |
| r (Spearman) | 0.586 | 0.203 | 0.688 | 0.733 | 0.802 | 0.522 | 0.758 | 0.780 | 0.942 | 0.900 | 0.812 | 0.805 | |
For each depth, comparisons were made using for direct variant calling (12x-calling) and for variant discovery based on genotype likelihoods (GL). Ho correlations were estimated according to Pearson and Spearman to compare rankings of individuals. Slopes were estimated by forcing the intercept of the linear regression to be 0.
Discussion
A wide range of methods are used for assessing the diversity of genomes, from whole sequencing of individual genomes, e.g. (Kidd et al., 2012; Altshuler et al., 2012) to the genotyping of a panel of variants randomly chosen or specifically designed, e.g. (Kijas et al., 2012). Because the choice of the methods (e.g., commercial DNA chip, low/high coverage whole genome sequencing, random panel of SNPs) might not be straightforward depending on the goal of the study, we set up this study to test the ability and robustness of low and medium WGS coverages and a wide range of genome sampling strategies to (i) assess genome variability, (ii) infer population genetic structure and (iii) detect genome regions under selection. We applied this benchmark analyses on four different wild and domesticated groups representing different levels of diversity and linkage disequilibrium (Table 1).
Effect of sequencing coverage on the assessment of whole genome variations
Overall, the genotypes inferred from the 12X WGS were highly reliable according to the high concordance between 12X re-sequencing data and the 50K SNP BeadChips genotyping.
The extraction of 1X, 2X and 5X WGS datasets from the 12X WGS confirmed the sensitivity of population genetics inferences to the sequencing coverage without using genotype likelihoods methods (e.g. Jansen et al., 2013; Alex Buerkle & Gompert, 2013), and helped to depict the effect of reducing the coverage. As might be expected, we found that homozygote genotypes were more correctly called than heterozygote ones whatever the coverage. This is due to the fact that more reads should be mapped at a position for calling the two alleles of an heterozygote than for calling the unique allele of an homozygote. Additionally, the filtering process for variant calling induced a higher percentage of missing data for heterozygotes because it discarded any heterozygous genotype for which one allele was under or over-represented.
Thus, the decrease in WGS coverage first resulted in a decrease in variant density (increasing proportion of missing data). The density of reliable variants obtained when decreasing the coverage (> 250k for 1X and > 3M from 2X, see Table 2) would still have been sufficient to allow accurate estimation of population genetics parameters and detection of selection signatures (see below 'effect of the density of variants'). However, the trend is combined to a bias that strongly affected the estimations. This bias concerned both missing and erroneous genotyping (Figure 8), which affected mostly heterozygotes (even more when the coverage decreases) where the erroneous genotyping mostly produces homozygotes. This resulted in an underestimation of heterozygosity (Ho). However, the values obtained for the 5X coverage appeared to be just as accurate as those inferred from the 12X WGS (highly correlated values of Ho, and thus of F), for the studied species. This result is coherent with the findings of Li et al. (2011) who showed that in association studies, genotyping 3,000 individuals at 4X depth provided similar power to 30X sequencing of about 2,000 individuals. A way to overcome the concerns due to low-coverage sequencing is to analyse the data with adapted methods that use genotype likelihoods, e.g. (Korneliussen et al., 2014). Using this approach increased substantially the numbers of SNPs discovered (> 18.2M for 1X and > 23.6M for 2X, see Table 2), and in sheep the number of called variants from 5X GLs was even higher than for 12X data. It also increased the proportions of correctly genotyped heterozygote and homozygote sites (>50% of congruence with 12X data for all datasets). However, substantial numbers/proportions of polymorphic variants discovered with this approach were not shared with 12X data (ranging from 43 to 45% in sheep and from 40 to 41% in goats; Numbers of shared variants with 12X in Table 2), suggesting false positive discoveries. Yet, heterozygosity estimates using genotype likelihoods for low coverages (1X and 2X) were correlated but not fully congruent with 12X data estimates. For 5X coverage using GLs, the correlations were slightly lower than when using classical discovery and filtering algorithms (Table 2). This is explained by Site Frequency Spectra inferred from GLs datasets in comparison with 12X data (Figures S22, S23). Distributions of allele frequencies represented by these datasets suggest an over-representation of variants with allele frequencies close to 0.5 at the expense of rare variants. This fits more a probabilistic distribution as described by Fuentes-Pardo & Ruzzante (2017). This is consistent with the findings of Hendricks et al. (Hendricks et al., 2018) who reported that RAD-seq data of coverage 1X, 2X, 5X, and 10X called using genotype likelihoods led to inconsistent affiliation of four North American passerine subspecies.
Figure 8. Efficiency and accuracy of different genotyping strategies.

For each purpose, the different strategies are rated according to the accuracy of the estimates taking as a reference the WGS 12x depth inferences. Grey dots indicate that the genotyping approach allow detecting some selection signatures but could miss some further signals detected by high density panels and WGS (12x depth) data. MD chip = 50K SNP BeadChip (caprine and ovine); HD chip = 600K SNP Ovine BeadChip. Low and medium re-sequencing coverages are represented by: (i) classical variant calling and filtering denoted by 1x, 2x and 5x and (ii) variant discovery based on genotype likelihoods denoted by 1xGL, 2xGL and 5xGL.
Effect of the density of variants
When assessing the effect of the density of variants for various sample sizes, we generally observed a sample size effect on the estimation of summary statistics. This was observed whatever the species and the panel of variants, and the effect was especially strong when measuring population differentiation and linkage disequilibrium, even greater than the effect of variant density. Our findings disagree with Nazareno et al. (Nazareno, Bemmels, Dick, & Lohmann, 2017) who showed that accurate estimates of Fst between two Violaceae populations could be obtained with down to two individuals and about 1,500 SNPs. In our case the sample size effects reported could also be associated to the fine-scale structure of wild samples (i.e., sub-structures in Asiatic mouflon and Bezoar ibex groups that do not correspond to true populations) illustrated by high inbreeding between some individuals (Table 1) and sNMF results (Figure S16).
Many population genetics studies that infer demographic processes still rely on just a few dozens to a few hundreds of genetic markers aiming to be representative of all genome variations (Alhaddad et al., 2013; Olson, Whittaker, & Rhodes, 2013; Garza et al., 2014; Huang, Wang, Li, Wu, & Chen, 2014). We could, in fact, get a representative view of the whole genome variations by using a relatively small set of variants if they are randomly sampled across the genome (Figure 8). Low-density random panels of variants (i.e. 5K or 10K corresponding to 1 variant every ~300 or ~600Kb) gave estimates of summary statistics similar to those calculated from 12X WGS data whatever the species and its demographic history. The assessment of population structure through calculation of coefficients of ancestry was reliable whatever the panel density, while the estimations of Fst required at least 100K variants in the different populations/species. Furthermore, the estimation of LD and the detection of signatures of selection required higher variant densities: around one variant every 3 to 6Kb, which gave similar estimates to 12X WGS data with roughly one variant every 100 to 200bp.
The adequate densities of variants required for a reliable description of genomic variations depend on the pattern of LD decay across the genome. In the four studied species, those patterns represent a wide range of variation, with r 2 dropping below 0.15 within 4.5Kb in Asiatic mouflon and within more than 10Kb in sheep while excluding rare variants (Table 1). Consequently, we needed 500k to 1M variants to accurately estimate LD decay. All panels of fewer than 100K variants (~1 variant per 30kb) produced incorrect estimations of r 2 for small distances (until 50Kb depending on the panel). The same orders of magnitude of variant densities would be required in species characterized by similar patterns of LD decay such as true ungulates (Meadows, Chan, & Kijas, 2008; Wade et al., 2009; Villa-Angulo et al., 2009; Ai, Huang, & Ren, 2013; Veroneze et al., 2013; McCue et al., 2012) or even other mammals with similar genetic characteristics (e.g., Cathy Laurie et al., 2007). However, genomic patterns of LD decay depend on the demographic histories of populations, and reflect the changes in effective population sizes.
Selective sweeps, when they occur, increase LD in regions of several Kb surrounding the selected allele. This signature is then reduced by recombination, and the older the selective sweep the smaller will be the region still influenced around the selected allele (Stephens et al., 1998; Kim & Nielsen, 2004). In the case of the selective sweep that has occurred in the RXFP2 gene, the signal is still extending ~350Kb and required at least a random panel of 100K variants in order to be detected, even if the signal was also present when using the random 50K SNP panel (Figure S17). Therefore, higher density random panels would be needed to detect any weaker selective sweep (i.e. associated to lower LD).
Ascertainment bias in non-random panels
The estimation of almost all population genetics parameters was biased when using variants from commercial SNP BeadChips or exome (Figure 8). Measurements both of genome diversity and of population differentiation were affected. This might be expected knowing that SNPs included in the design of the commercial panels were intentionally chosen according to their high level of polymorphism in several breeds (mainly European industrials, Alhaddad et al., 2013). This is because these panels were designed to deploy breeding programs in connection with genomic selection and genome wide association studies, for which an accurate estimate of true population genetic diversity is irrelevant. The resulting ascertainment bias led to an overestimation of the genomic diversity. The ovine HD BeadChip suffered less from this bias compared to the 50K ovine BeadChip due to the inclusion of high, medium and low frequency SNPs (James W. Kijas et al., 2014). The exome capture data, while representing highly conserved regions, logically underestimated genetic diversity.
The biased estimation of genetic diversity and genetic differentiation would be less problematic as long as the ranking of estimated values is preserved (e.g., the most variable individuals are actually those with the highest measured diversity). For example, when estimating animal genetic resources, this will allow finding the more diverse populations/breeds. However, we showed that this ranking was inverted when comparing the diversity of wilds and domestics with the ovine and caprine SNP Beadchips, which should be used with caution when comparing well-differentiated populations. Otherwise, this inversion is explained by differences in site frequency spectra (SFS) inferred in domestics and wilds using SNP chips. In domestics, SFS of these arrays were less correlated to SFS of the WGS variants, e.g. Figure 3 for sheep and Figure S4 for Asiatic mouflons.
Several ways have been suggested to correct for such ascertainment bias already reported in humans (Nielsen et al., 2004; Clark et al., 2005; Albrechtsen, Nielsen, & Nielsen, 2010) and chicken (Malomane et al., 2018). The approach of Albrechtsen, Nielsen, & Nielsen, (Albrechtsen et al., 2010) is based on the estimation of the ascertainment scheme by modelling the underlying distribution of allele frequencies in the population using re-sequencing data. However this study cautioned there is little hope to use inferred procedures for correcting bias in populations far from the ones they studied. Another strategy suggested by Malomane et al. (Malomane et al., 2018) uses LD-based pruning to partially account for ascertainment bias. It is based on calculating LD from 50 SNPs windows, and from a pair of SNPs in LD (using variance inflation factor VIF = 1/(1-r2) threshold of 2) the SNP with lower MAF is removed, the window is shifted 5 SNPs forward and the procedure is repeated.
Distribution of variants across the genome
Besides the effects of variant density and ascertainment bias, the distribution of variants across the genome also impacts the reliability of the characterization of the genome. For similar numbers of variants, the ovine and caprine 50K BeadChips were less accurate than random panels for estimating the LD decay over short distances. This is not surprising given the underrepresentation of close adjacent SNPs (<6Kb in ovine and <30Kb in caprine BeadChips, Figure S18) and differences in site frequency spectra in these Beadchips. Moreover, the local density of BeadChip SNPs varied across the genome with some regions being far well covered than others. This explains why, like (Kijas et al., 2012), we were able to detect the signal of selection associated to the RXFP2 gene with the ovine 50K SNP BeadChip but such signal was not as clear with 50K variants random panels. The commercial BeadChip has four SNPs in a 148 Kb window centred on the RXFP2 gene, which appeared to be enough for detecting selection, while the random 50K panel used had no variant in that window. Similarly, the NBEA signal was detected by XP-CLR with 1M variants or more. This illustrates our expectation that medium and low SNPs densities are limiting for detecting less intense selective events as shown by Lowry et al. (Lowry et al., 2016) for RAD-seq data, despite the fact that the latter study has made conclusions about the approach RAD-seq in general and has been criticized by other studies (Catchen et al., 2017; McKinney, Larson, Seeb, & Seeb, 2017). Our conclusions are mainly related to the required density of markers for detecting a selection signature and not to the RAD-seq approach per se. Inversely, we found that RAD-seq experiments allowing for appropriate SNP densities would give similar results as our random datasets of similar densities (Figures S20, S21).
The distribution of variants across the genome obviously determines the ability to detect selection signatures, and high-density variant panels are required to detect selected regions. One needs variants from regions under selection to find the associated signature, which is not necessarily assumed by low and medium-density panels of variants. This is more limiting when studying populations characterised by low overall linkage disequilibrium and old or low-intensity selection signatures.
Consequences for population genomics analyses
Outcomes of this study (Figure 8) might help setting up genotyping strategies to accurately infer population genetics statistics and test hypotheses on the structure and evolution of study populations. They should be useful to study species/populations with similar genetic characteristics (i.e., ungulate species and even other mammals). When measuring population genetic diversity, we show that the studied commercial SNP panels could invert the ranking of populations. The bias induced by especially medium-density chips was also substantial when assessing inter-populations differentiation. Fst could be either overestimated (e.g. in caprine) or underestimated (e.g., in ovine). Similarly, random panels of less than 50K SNPs could lead to inaccurate estimates but the bias due to low sample size clearly exceed that due to low SNP density. Furthermore, if a commercial SNP panel was able to detect a strong selective sweep, this was related to its design (i.e. number of SNPs in the region of interest) and, as shown by random panels, medium and low SNP densities (10K SNPs and lower) are shown to be inadequate in several cases. More resolution in detecting selective sweeps can be gained when increasing the density of SNPs and the maximum is obtained with the WGS data. Medium-coverage re-sequencing (e.g. 5X) without using genotype likelihoods would be recommended for such goals.
Our study also demonstrates that low coverage re-sequencing (1X and 2X) could be improved by the use of genotype likelihoods. It could be effective to get reliable genomic information. However, the number of incorrect genotypes generated by these data as shown here should be taken into account.
Conclusion
The accuracy of panels of variants to describe genome variations depends on the distribution of these variants across the genome, according to the level of LD and its proper variability. While high to medium coverage genome sequencing produces reliable genotyping, it remains costly both in terms of money and in data management, and thus surrogates of WGS data are still needed.
For model species, commercial standardized panels are generally already available and one should know their potential biases and use them cautiously. This is particularly true if the studied populations or breeds are genetically divergent from the individuals used for designing the set of variants. Our results showed that a few thousands of markers randomly chosen across the genome provide unbiased information. Therefore, it could be valuable to include such sets of variants when designing new SNP chips or when updating existing beadChips. Especially, the strategies suggested to correct ascertainment biases could not be generalized to other populations/cases or don’t allow highly accurate inferences (e.g. LD-based pruning). In non-model species, the genotyping of individuals by SNP chips could be replaced by genotyping by sequencing approaches (RAD-seq), shown here that they fully approximate a random distribution of markers across the genome. This despite the fact that they can have sometimes some bias depending on the choice of restriction enzyme and allele dropout as reported by Arnold et al. (Arnold et al., 2013). As shown by our results, a suitable variant density should be targeted according to the aim of the study and the resources allocated. Finally, when considering Whole Genome Sequencing approaches, Genotype likelihoods are effective to increase the accuracy of low-coverage (< 5X) sequencing data in comparison with direct variant calling approaches. However, these coverages might not be fully appropriate for setting up some population genomics studies where individual correct genotypes are required. This is due to the important proportions of incorrect genotypes and the discordance of site frequency spectra with higher coverage data.
Supplementary Material
Acknowledgments
This work was funded by the UE FP7 project NEXTGEN 'Next generation methods to preserve farm animal biodiversity by optimizing present and future breeding options'; grant agreement no. 244356. We thank Eric Coissac who helped in setting-up the overall approach and Bertrand Servin for the useful discussions. We are grateful to R. Hadria, M. Laghmir, L. Haounou, E. Hafiani, E. Sekkour, M. ElOuatiq, A Dadouch, A. Lberji, C. Errouidi and M. Bouali for helping in sampling in Morocco.
Footnotes
Data accessibility
The variant call sets are archived in the European Nucleotide Archive with accession numbers provided in Table S2. The accession of the sample in the Biosamples archive, and of the corresponding aligned bam file in the ENA archive are listed in Table S1.
Author contributions
The study was done within the NEXTGEN project (coordinated by P.T.). F.P. and P.T. designed and supervised the study. B.B., M.I, M.B, M.C., A.B, A.C, W.Z., H.R.R. and S.N. collected the samples. A.S. supervised the work of her research group. A.A. and S.E. produced whole-genome sequences. B.B., I.S., L.C., S.E., and F.B. contributed to bioinformatic analyses. B.B., F.B., I.S. and W.Z. did the analyses. B.B. and F.P. produced the figures and drafted the paper. I.S., F.B., F.J.A., P.T., J.K. and P.F. reviewed and amended the paper.
References
- Ai H, Huang L, Ren J. Genetic Diversity, Linkage Disequilibrium and Selection Signatures in Chinese and Western Pigs Revealed by Genome-Wide SNP Markers. Plos One. 2013;8(2) doi: 10.1371/journal.pone.0056001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrechtsen A, Nielsen FC, Nielsen R. Ascertainment Biases in SNP Chips Affect Measures of Population Divergence. Molecular Biology and Evolution. 2010;27(11):2534–2547. doi: 10.1093/molbev/msq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alex Buerkle C, Gompert Z. Population genomics based on low coverage sequencing: how low should we go? Molecular ecology. 2013;22(11):3028–3035. doi: 10.1111/mec.12105. [DOI] [PubMed] [Google Scholar]
- Alhaddad H, Khan R, Grahn RA, Gandolfi B, Mullikin JC, Cole SA, et al. Lyons LA. Extent of Linkage Disequilibrium in the Domestic Cat, Felis silvestris catus, and Its Breeds. Plos One. 2013;8(1) doi: 10.1371/journal.pone.0053537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, et al. Genomes Project, C An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold B, Corbett-Detig RB, Hartl D, Bomblies K. RADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling. Molecular Ecology. 2013;22(11):3179–3190. doi: 10.1111/mec.12276. [DOI] [PubMed] [Google Scholar]
- Auton A, McVean G. Recombination rate estimation in the presence of hotspots. Genome Research. 2007;17(8):1219–1227. doi: 10.1101/gr.6386707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Johnson EA. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. Plos One. 2008;3(10) doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjelloun B, Alberto FJ, Streeter I, Boyer F, Coissac E, Stucki S, et al. Consortium, N Characterizing neutral genomic diversity and selection signatures in indigenous populations of Moroccan goats (Capra hircus) using WGS data. Frontiers in Genetics. 2015;6:107. doi: 10.3389/fgene.2015.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Schmutz J, Wang H, Percifield R, Hawkins J, Pontaroli AC, et al. Devos KM. Reference genome sequence of the model plant Setaria. Nature Biotechnology. 2012;30(6):555–+. doi: 10.1038/nbt.2196. [DOI] [PubMed] [Google Scholar]
- Bizon C, Spiegel M, Chasse SA, Gizer IR, Li Y, Malc EP, et al. Wilhelmsen KC. Variant calling in low-coverage whole genome sequencing of a Native American population sample. Bmc Genomics. 2014;15 doi: 10.1186/1471-2164-15-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black WC, Baer CF, Antolin MF, DuTeau NM. Population genomics: Genome-wide sampling of insect populations. Annual Review of Entomology. 2001;46:441–469. doi: 10.1146/annurev.ento.46.1.441. [DOI] [PubMed] [Google Scholar]
- Browning BL, Browning SR. Improving the Accuracy and Efficiency of Identity-by-Descent Detection in Population Data. Genetics. 2013;194(2):459–+. doi: 10.1534/genetics.113.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell N, Sinagra G, Jones KL, Slavov D, Gowan K, Merlo M, et al. Taylor MRG. Whole Exome Sequencing Identifies a Troponin T Mutation Hot Spot in Familial Dilated Cardiomyopathy. Plos One. 2013;8(10) doi: 10.1371/journal.pone.0078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro M, Rubin C-J, Di Palma F, Albert FW, Alfoeldi J, Barrio AM, et al. Andersson L. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science. 2014;345(6200):1074–1079. doi: 10.1126/science.1253714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Hohenlohe PA, Bernatchez L, Funk WC, Andrews KR, Allendorf FW. Unbroken: RADseq remains a powerful tool for understanding the genetics of adaptation in natural populations. Molecular Ecology Resources. 2017;17(3):362–365. doi: 10.1111/1755-0998.12669. [DOI] [PubMed] [Google Scholar]
- Cathy C Laurie, D AN, Amy D Anderson, Bruce S Weir, Robert J Livingston, Matthew D Dean, Kimberly L Smith, Eric E Schadt, Michael W Nachman. Linkage Disequilibrium in Wild Mice. Plos Genetics. 2007 doi: 10.1371/journal.pgen.0030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Patterson N, Reich D. Population differentiation as a test for selective sweeps. Genome Research. 2010;20(3):393–402. doi: 10.1101/gr.100545.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, et al. Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15(11):1496–1502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosart T, Beja-Pereira A, Chen S, Ng SB, Shendure J, Luikart G. Exome-wide DNA capture and next generation sequencing in domestic and wild species. Bmc Genomics. 2011;12 doi: 10.1186/1471-2164-12-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. Genomes Project Anal, G The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastjerdi A, Robert C, Watson M. Low coverage sequencing of two Asian elephant (Elephas maximus) genomes. GigaScience. 2014;3:12–12. doi: 10.1186/2047-217x-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics. 2011;12(7):499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;43(5):491–+. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo A, Donnelly P, Toomajian C, Sisk B, Hill A, Petzl-Erler ML, et al. Barch DH. Heterogeneity of microsatellite mutations within and between loci, and implications for human demographic histories. Genetics. 1998;148(3):1269–1284. doi: 10.1093/genetics/148.3.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominik S, Henshall JM, Hayes BJ. A single nucleotide polymorphism on chromosome 10 is highly predictive for the polled phenotype in Australian Merino sheep. Animal Genetics. 2012;43(4):468–470. doi: 10.1111/j.1365-2052.2011.02271.x. [DOI] [PubMed] [Google Scholar]
- Dong Y, Xie M, Jiang Y, Xiao N, Du X, Zhang W, et al. Wang W. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus) Nature Biotechnology. 2013;31(2):135–141. doi: 10.1038/nbt.2478. [DOI] [PubMed] [Google Scholar]
- Everett MV, Grau ED, Seeb JE. Short reads and nonmodel species: exploring the complexities of next-generation sequence assembly and SNP discovery in the absence of a reference genome. Molecular Ecology Resources. 2011;11:93–108. doi: 10.1111/j.1755-0998.2010.02969.x. [DOI] [PubMed] [Google Scholar]
- Fariello MI, Boitard S, Naya H, SanCristobal M, Servin B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics. 2013;193(3):929–941. doi: 10.1534/genetics.112.147231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R. Polymorphism and Natural-Selection. Bulletin of the International Statistical Institute. 1958;36(3):284–289. [Google Scholar]
- Fountain ED, Pauli JN, Reid BN, Palsbøll PJ, Peery MZ. Finding the right coverage: the impact of coverage and sequence quality on single nucleotide polymorphism genotyping error rates. Molecular Ecology Resources. 2016;16(4):966–978. doi: 10.1111/1755-0998.12519. [DOI] [PubMed] [Google Scholar]
- Frankham R, Ballou JD, Briscoe DA. Introduction to Conservation Genetics. Cambridge: Cambridge University Press; 2002. [Google Scholar]
- Frichot E, Mathieu F, Trouillon T, Bouchard G, Francois O. Fast and Efficient Estimation of Individual Ancestry Coefficients. Genetics. 2014;196(4):973–+. doi: 10.1534/genetics.113.160572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Pardo AP, Ruzzante DE. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Mol Ecol. 2017;26(20):5369–5406. doi: 10.1111/mec.14264. [DOI] [PubMed] [Google Scholar]
- Fumagalli M, Vieira FG, Linderoth T, Nielsen R. ngsTools: methods for population genetics analyses from next-generation sequencing data. Bioinformatics. 2014;30(10):1486–1487. doi: 10.1093/bioinformatics/btu041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv. 2012;1207 3907v2. [Google Scholar]
- Garza JC, Gilbert-Horvath EA, Spence BC, Williams TH, Fish H, Gough SA, et al. Anderson EC. Population Structure of Steelhead in Coastal California. Transactions of the American Fisheries Society. 2014;143(1):134–152. doi: 10.1080/00028487.2013.822420. [DOI] [Google Scholar]
- Goldstein DB, Weale ME. Population genomics: Linkage disequilibrium holds the key. Current Biology. 2001;11(14):R576–R579. doi: 10.1016/s0960-9822(01)00348-7. [DOI] [PubMed] [Google Scholar]
- Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han E, Sinsheimer JS, Novembre J. Characterizing Bias in Population Genetic Inferences from Low-Coverage Sequencing Data. Molecular Biology and Evolution. 2014;31(3):723–735. doi: 10.1093/molbev/mst229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks S, Anderson EC, Antao T, Bernatchez L, Forester BR, Garner B, et al. Luikart G. Recent advances in conservation and population genomics data analysis. Evolutionary Applications. 2018;11(8):1197–1211. doi: 10.1111/eva.12659. [DOI] [Google Scholar]
- Holloway JW, Beghe B, Turner S, Hinks LJ, Day INM, Howell WM. Comparison of three methods for single nucleotide polymorphism typing for DNA bank studies: Sequence-specific oligonucleotide probe hybridisation, TaqMan liquid phase hybridisation, and microplate array diagonal gel electrophoresis (MADGE) Human Mutation. 1999;14(4):340–347. doi: 10.1002/(sici)1098-1004(199910)14:4<340::aid-humu10>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. Stemple DL. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496(7446):498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Wang H, Li L, Wu Z, Chen J. Genetic Diversity and Population Demography of the Chinese Crocodile Lizard (Shinisaurus crocodilurus) in China. Plos One. 2014;9(3) doi: 10.1371/journal.pone.0091570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AR, Inouye BD, Johnson MT, Underwood N, Vellend M. Ecological consequences of genetic diversity. Ecol Lett. 2008;11(6):609–623. doi: 10.1111/j.1461-0248.2008.01179.x. [DOI] [PubMed] [Google Scholar]
- Jansen S, Aigner B, Pausch H, Wysocki M, Eck S, Benet-Pages A, et al. Fries R. Assessment of the genomic variation in a cattle population by re-sequencing of key animals at low to medium coverage. Bmc Genomics. 2013;14 doi: 10.1186/1471-2164-14-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Xie M, Chen W, Talbot R, Maddox JF, Faraut T, et al. Dalrymple BP. The sheep genome illuminates biology of the rumen and lipid metabolism. Science. 2014;344(6188):1168–1173. doi: 10.1126/science.1252806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MR, Forester BR, Teufel AI, Adams RV, Anstett DN, Goodrich BA, et al. Manel S. Integrating Landscape Genomics and Spatially Explicit Approaches to Detect Loci Under Selection in Clinal Populations. Evolution. 2013;67(12):3455–3468. doi: 10.1111/evo.12237. [DOI] [PubMed] [Google Scholar]
- Jorde LB, Watkins WS, Bamshad MJ. Population genomics: a bridge from evolutionary history to genetic medicine. Human Molecular Genetics. 2001;10(20):2199–2207. doi: 10.1093/hmg/10.20.2199. [DOI] [PubMed] [Google Scholar]
- Kardos M, Luikart G, Bunch R, Dewey S, Edwards W, McWilliam S, et al. Kijas J. Whole-genome resequencing uncovers molecular signatures of natural and sexual selection in wild bighorn sheep. Molecular ecology. 2015;24(22):5616–5632. doi: 10.1111/mec.13415. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Gravel S, Byrnes J, Moreno-Estrada A, Musharoff S, Bryc K, et al. Bustamante CD. Population Genetic Inference from Personal Genome Data: Impact of Ancestry and Admixture on Human Genomic Variation. American Journal of Human Genetics. 2012;91(4):660–671. doi: 10.1016/j.ajhg.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijas JW, Lenstra JA, Hayes B, Boitard S, Neto LRP, San Cristobal M, et al. Int Sheep Genomics, C Genome-Wide Analysis of the World's Sheep Breeds Reveals High Levels of Historic Mixture and Strong Recent Selection. Plos Biology. 2012;10(2) doi: 10.1371/journal.pbio.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijas JW, Porto-Neto L, Dominik S, Reverter A, Bunch R, McCulloch R, et al. Int Sheep Genomics, C Linkage disequilibrium over short physical distances measured in sheep using a high-density SNP chip. Animal Genetics. 2014;45(5):754–757. doi: 10.1111/age.12197. [DOI] [PubMed] [Google Scholar]
- Kim Y, Nielsen R. Linkage disequilibrium as a signature of selective sweeps. Genetics. 2004;167(3):1513–1524. doi: 10.1534/genetics.103.025387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: Analysis of Next Generation Sequencing Data. Bmc Bioinformatics. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. Genome Project Data, P The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sidore C, Kang HM, Boehnke M, Abecasis GR. Low-coverage sequencing: Implications for design of complex trait association studies. Genome Research. 2011;21(6):940–951. doi: 10.1101/gr.117259.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Ferchaud AL, Grønkjaer P, Nygaard R, Hansen MM. Genomic parallelism and lack thereof in contrasting systems of three-spined sticklebacks. Mol Ecol. 2018;27(23):4725–4743. doi: 10.1111/mec.14782. [DOI] [PubMed] [Google Scholar]
- Lowry DB, Hoban S, Kelley JL, Lotterhos KE, Reed LK, Antolin MF, Storfer A. Breaking RAD: an evaluation of the utility of restriction site-associated DNA sequencing for genome scans of adaptation. Mol Ecol Resour. 2016;17(2):142–152. doi: 10.1111/1755-0998.12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart G, England PR, Tallmon D, Jordan S, Taberlet P. The power and promise of population genomics: From genotyping to genome typing. Nature Reviews Genetics. 2003;4(12):981–994. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- Malomane DK, Reimer C, Weigend S, Weigend A, Sharifi AR, Simianer H. Efficiency of different strategies to mitigate ascertainment bias when using SNP panels in diversity studies. Bmc Genomics. 2018;19(1):22. doi: 10.1186/s12864-017-4416-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden CD, Lee Y, Kreppel K, Weakley A, Cornel A, Ferguson HM, et al. Lanzaro GC. Diversity, Differentiation, and Linkage Disequilibrium: Prospects for Association Mapping in the Malaria Vector Anopheles arabiensis. G3-Genes Genomes Genetics. 2014;4(1):121–131. doi: 10.1534/g3.113.008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher M, Richmond TA, Gerhardt DJ, Himmelbach A, Clissold L, Sampath D, et al. Stein N. Barley whole exome capture: a tool for genomic research in the genus Hordeum and beyond. Plant Journal. 2013;76(3):494–505. doi: 10.1111/tpj.12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue ME, Bannasch DL, Petersen JL, Gurr J, Bailey E, Binns MM, et al. Mickelson JR. A High Density SNP Array for the Domestic Horse and Extant Perissodactyla: Utility for Association Mapping, Genetic Diversity, and Phylogeny Studies. Plos Genetics. 2012;8(1) doi: 10.1371/journal.pgen.1002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. DePristo MA. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney GJ, Larson WA, Seeb LW, Seeb JE. RADseq provides unprecedented insights into molecular ecology and evolutionary genetics: comment on Breaking RAD by Lowry et al. (2016) Molecular Ecology Resources. 2017;17(3):356–361. doi: 10.1111/1755-0998.12649. [DOI] [PubMed] [Google Scholar]
- Meadows JRS, Chan EKF, Kijas JW. Linkage disequilibrium compared between five populations of domestic sheep. Bmc Genetics. 2008;9 doi: 10.1186/1471-2156-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JM, Malenfant RM, David P, Davis CS, Poissant J, Hogg JT, et al. Coltman DW. Estimating genome-wide heterozygosity: effects of demographic history and marker type. Heredity. 2014;112(3):240–247. doi: 10.1038/hdy.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Research. 2007;17(2):240–248. doi: 10.1101/gr.5681207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudge JM, Frankish A, Fernandez-Banet J, Alioto T, Derrien T, Howald C, et al. Harrow J. The Origins, Evolution, and Functional Potential of Alternative Splicing in Vertebrates. Molecular Biology and Evolution. 2011;28(10):2949–2959. doi: 10.1093/molbev/msr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazareno AG, Bemmels JB, Dick CW, Lohmann LG. Minimum sample sizes for population genomics: an empirical study from an Amazonian plant species. Mol Ecol Resour. 2017;17(6):1136–1147. doi: 10.1111/1755-0998.12654. [DOI] [PubMed] [Google Scholar]
- Nei M. Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A. 1973;70(12):3321–3323. doi: 10.1073/pnas.70.12.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Shendure J. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272–U153. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Hubisz MJ, Clark AG. Reconstituting the frequency spectrum of ascertained single-nucleotide polymorphism data. Genetics. 2004;168(4):2373–2382. doi: 10.1534/genetics.104.031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson ZH, Whittaker DG, Rhodes OE. Translocation History and Genetic Diversity in Reintroduced Bighorn Sheep. Journal of Wildlife Management. 2013;77(8):1553–1563. doi: 10.1002/jwmg.624. [DOI] [Google Scholar]
- Palti Y, Gao G, Liu S, Kent MP, Lien S, Miller MR, et al. Moen T. The development and characterization of a 57K single nucleotide polymorphism array for rainbow trout. Molecular Ecology Resources. 2015;15(3):662–672. doi: 10.1111/1755-0998.12337. [DOI] [PubMed] [Google Scholar]
- Pasaniuc B, Rohland N, McLaren PJ, Garimella K, Zaitlen N, Li H, et al. Price AL. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nature Genetics. 2012;44(6):631–U641. doi: 10.1038/ng.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Seielstad MT, Perez-Lezaun A, Feldman MW. Population growth of human Y chromosomes: A study of Y chromosome microsatellites. Molecular Biology and Evolution. 1999;16(12):1791–1798. doi: 10.1093/oxfordjournals.molbev.a026091. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. Sham PC. PLINK: A tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, et al. Int HapMap, C Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449(7164):913–U912. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Le S, Li Y, Hu F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. Plos One. 2016;11(10):e0163962. doi: 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens JC, Reich DE, Goldstein DB, Shin HD, Smith MW, Carrington M, et al. Dean M. Dating the origin of the CCR5-Delta 32 AIDS-resistance allele by the coalescence of haplotypes. American Journal of Human Genetics. 1998;62(6):1507–1515. doi: 10.1086/301867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Human Molecular Genetics. 2010;19:R145–R151. doi: 10.1093/hmg/ddq333X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therkildsen NO, Palumbi S. Practical low-coverage genome-wide sequencing of hundreds of individually barcoded samples for population and evolutionary genomics in non-model species. Molecular Ecology Resources. 2017 doi: 10.1111/1755-0998.12593. [DOI] [PubMed] [Google Scholar]
- Veroneze R, Lopes PS, Guimaraes SEF, Silva FF, Lopes MS, Harlizius B, Knol EF. Linkage disequilibrium and haplotype block structure in six commercial pig lines. Journal of Animal Science. 2013;91(8):3493–3501. doi: 10.2527/jas.2012-6052. [DOI] [PubMed] [Google Scholar]
- Villa-Angulo R, Matukumalli LK, Gill CA, Choi J, Van Tassell CP, Grefenstette JJ. High-resolution haplotype block structure in the cattle genome. Bmc Genetics. 2009;10 doi: 10.1186/1471-2156-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade CM, Giulotto E, Sigurdsson S, Zoli M, Gnerre S, Imsland F, et al. Broad Inst Whole Genome Assembly, T Genome Sequence, Comparative Analysis, and Population Genetics of the Domestic Horse. Science. 2009;326(5954):865–867. doi: 10.1126/science.1178158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-Statistics for the Analysis of Population-Structure. Evolution. 1984;38(6):1358–1370. doi: 10.2307/2408641. [DOI] [PubMed] [Google Scholar]
- Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, et al. Bahler J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453(7199):1239–U1239. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- Wright S. Evolution in Mendelian populations. Genetics. 1931;16(2):0097–0159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.