

Abstract
The T cell infiltrates that are formed in human cancers are a modifier of natural disease progression and also determine the probability of clinical response to cancer immunotherapies. Recent technological advances that enable the single-cell analysis of phenotypic and transcriptional states have revealed a vast heterogeneity of intratumoral T cell states, both within and between patients, and the observation of this heterogeneity makes it critical to understand the relationship between individual T cell states and therapy response. This Review covers our current knowledge regarding the T cell states that are present in human tumors and the role that different T cell populations have been hypothesized to play within the tumor microenvironment, with a particular focus on CD8+ T cells. Three key models that are discussed herein are: 1). The dysfunction of T cells in human cancer is associated with a change in T cell functionality rather than inactivity; 2). Antigen recognition in the tumor microenvironment is an important driver of T cell dysfunctionality and the presence of dysfunctional T cells can hence be used as a proxy for the presence of a tumor-reactive T cell compartment; 3). A less dysfunctional population of tumor-reactive T cells may be required to drive a durable response to T cell immune checkpoint blockade.
Introduction
It has long been known that the presence of T cells in cancer lesions is correlated with better patient prognosis in a number of human malignancies. As an example, it has been appreciated for over twenty years that the presence of brisk T cell infiltrates is associated with improved overall survival in human melanoma1. In subsequent work, the magnitude of intratumoral T cell infiltrates was shown to form an independent positive prognostic marker in colorectal cancer (CRC) and ovarian cancer2,3, and similar results have been obtained in several other malignancies4. However, correlation does obviously not imply causation, and the observed relationship between intratumoral T cell numbers and patient prognosis could for many years be ‘explained away’, for instance, by assuming that T cell entry into tumors was influenced by the oncogenic pathways that were activated in an individual tumor, with more benign tumors by chance being more permissive to T cell accumulation.
The direct evidence that the T cell infiltrates in human cancer should be seen as a true modifier of cancer growth came from parallel efforts to enhance tumor-specific T cell reactivity, either by infusion of T cell products expanded ex vivo from tumor-infiltrating lymphocytes5, or by antibody-mediated blockade of T cell checkpoint molecules6–8. Therapies that block the T cell checkpoint molecules cytotoxic T lymphocyte-associated antigen 4 (CTLA4) and in particular programmed cell death protein 1 (PD1) have shown a significant rate of clinical responses, and sometimes durable complete responses, in a range of tumor types, with an understandable bias - only recognized in hindsight - towards tumors that are characterized by higher amounts of DNA damage9. Blockade of the CTLA4 checkpoint is thought to predominantly induce a broadening of the tumor-specific T cell response, by abolishing the inhibitory effect of CTLA4 during T cell priming10–12. In contrast, blockade of the PD1–PD1 ligand 1 (PDL1) axis is thought to primarily boost pre-existing tumor-specific T cell responses13. In spite of this presumed difference in mode of action, both therapies ultimately rely on the activity of a, pre-existing or newly induced, tumor-resident T cell pool to achieve tumor elimination. The recent identification of high diversity in the activation and dysfunctional states of the T cells that are present in human cancer lesions therefore raises a number of crucial issues: Which cell states are associated with an ongoing tumor-specific T cell response? How do the current immunotherapies impact these different T cell states? And finally, how does the presence of individual T cell states predict response to immune checkpoint blockade (ICB)?
T cell states in human cancer
Overview of the T cell states that have been identified in human tumors
The simplest distinction between T cells is that of the CD4+ and CD8+ T cell subsets. The evidence for a role of the CD8+ T cell subset in tumor control is compelling, as for instance reflected by a series of prognostic analyses (listed in 4 and 14), the association between pre-treatment intratumoral CD8+ T cell numbers and response to PD1 blockade15, and the clinical activity of CD8+ T cell-enriched cell products in melanoma16. These observations explain the focus of most of the recent single-cell analyses, and also this Review, on the CD8+ T cell compartment. However, we feel that it is also important to briefly describe the cell states that are assumed by CD4+ T cells in the tumor microenvironment (TME), as CD4+ T cells have been shown to play a substantial role in tumor control in both preclinical models and in patient case studies (e.g. 17–19). Furthermore, prior data already revealed that distinct CD4+ T cell subsets are associated with either good or poor clinical prognosis4, suggesting that a more granular analysis of CD4+ T cell states is likely to yield further information on the role of different intratumoral CD4+ T cell pools. A brief overview of the CD4+ T cell states that have been identified in human tumors to date is provided in Box 1.
Box 1. CD4+ T cell states in human cancer.
While CD8+ T cells are considered major drivers of anti-tumor immunity, CD4+ T cells also play a prominent role in tumor control, either promoting or inhibiting anti-tumor responses74. For instance, conventional CD4+ T cells (Tconv cells) can promote tumor control through stimulation of, amongst others, CD8+ T cells, natural killer (NK) cells, and a broad range of other innate immune cell types (reviewed in 75). In addition to this function of facilitating anti-tumor immune responses, Tconv cells can exert cytotoxic functions that result in killing of human leukocyte antigen (HLA) class II-expressing tumor cells, or inhibit tumor growth through secretion of interferon γ (IFNγ) and tumor necrosis factor (TNF)76. In addition to the Tconv cell pool, a T follicular helper cell (TFH)-like population of CD4+ T cells that is characterized by expression of B cell lymphoma 6 (BCL6) and the capacity to produce high levels of CXC-chemokine ligand 13 (CXCL13) has been identified in multiple human tumor types77. Although the exact role of TFH cells in tumor immunity is unclear, these cells may contribute to the generation of tertiary lymphoid structures (TLS) at the tumor site and thereby shape intratumoral CD8+ T cell and B cell responses77,78. In contrast, tumor-resident regulatory T (Treg) cells have been shown to counteract tumor-specific immune responses by suppressing the infiltration and anti-tumor activity of, amongst others, CD8+ T cells and macrophages75. Single cell RNA-sequencing (scRNA-seq) studies have described a variety of CD4+ T cell states, including dysfunctional CD4+ T cells, naïve-like or memory CD4+ T cells, cytotoxic effector CD4+ T cells, Treg cells and TFH cells20,21,28–31,34. Notably, unlike the major CD8+ T cell states, these CD4+ T cell states do not appear to be ubiquitously present in all tumor types. Another interesting observation of single-cell sequencing as well as cytometry by time of flight (CyTOF) studies has been that Treg cells in the tumor express higher levels of tumor necrosis factor receptor superfamily member 9 (TNFRSF9; encoding 4-1BB), inducible T-cell costimulator (ICOS), and cytotoxic T lymphocyte antigen 4 (CTLA4) than Treg cells in blood or adjacent normal tissue, possibly reflective of an activated state29,79. In addition, the intratumoral Treg cell pool displays substantial diversity, for example as shown by their variable expression levels of TNFRSF9 21,29,31. Furthermore, in melanoma, both Treg cells and TFH cells displayed levels of proliferation that were comparable with those observed in dysfunctional CD8+ T cells21. By analogy with the dysfunctional CD8+ T cell pool, it may be hypothesized that this proliferative signature reflects a response of these cell pools to a local (antigen) signal and suggests that both Treg cells and TFH may play pivotal roles in the intratumoral CD4+ T cell response.
Circulating and lymph node-resident CD8+ T cells are classically subdivided according to their state of differentiation into naïve T cells, effector T cells, and subsets of memory T cells. The development of high dimensional profiling techniques such as cytometry by time of flight (CyTOF) and single-cell RNA sequencing (scRNA-seq) has enabled the field to go substantially beyond this relatively coarse profiling of CD8+ T cells based on the expression of just a few protein markers, and has over the past years been used to profile T cell infiltrates in human tumors. In three independent melanoma cohorts, the major intratumoral T cell populations that were identified based on transcriptional profiling using different scRNA-seq platforms displayed strong resemblance across the studies. In one study, ‘naïve’ CD8+ T cells, marked by expression of CC-chemokine receptor 7 (CCR7), transcription factor 7 (TCF7), lymphoid enhancer-binding factor 1 (LEF1), and L-selectin (SELL), and ‘cytotoxic’ cells, amongst others expressing perforin 1 (PRF1), granzyme A (GZMA), GZMB, and natural killer cell granule protein 7 (NKG7), were identified (Supplementary Table S1)20. Likewise, ‘naïve-like’ (marked by expression of amongst others CCR7, LEF1, interleukin 7 receptor (IL7R), and TCF7) and ‘cytotoxic effector’ (for instance characterized by CX3C chemokine receptor 1 (CX3CR1), PRF1, killer cell lectin-like receptor subfamily G member 1 (KLRG1), and fibroblast growth factor-binding protein 2 (FGFBP2)) cell states were defined in a second study21. In a third cohort, CD8+ T cell states with similar characteristics were observed, but were named differently, identifying a ‘memory’ state with expression of CCR7, IL7R, LEF1, and TCF7 (matching the naïve(-like) cells observed in the other two studies, and a ‘cytotoxic’ state defined by expression of Fcγ receptor IIIA (FCGR3A), KLRG1, PRF1, and GZMB 22. Combined protein and gene expression analyses may be required to clarify whether the first of these two populations is either composed of true naïve CD8+ T cells, memory CD8+ T cells, a stem cell-like subset of memory cells (as described by Gattinoni et al.23), or a mixture of these, as the transcriptional profiles of these subsets display many similarities. In the absence of data that conclusively settles this issue, we will here refer to this population as ‘naïve-like’. The presence of these naïve-like CD8+ T cells at tumor sites does represent somewhat of a conundrum: While cytotoxic effector cells are known for their capacity to home to peripheral tissues, naïve and (stem cell-like) memory T cells typically circulate through blood and lymphoid organs. One hypothesis may be that intratumoral naïve-like cells reside in the intratumoral lymph node-like aggregates that are referred to as tertiary lymphoid structures (TLS), but more work to substantiate this is clearly required.
In addition to the naïve-like and cytotoxic CD8+ T cell states, a third, substantially more heterogeneous, pool of T cells that displays features of ‘dysfunction’ or ‘exhaustion’ was observed in all three studies20–22. In line with original data in chronic viral infection models in mice, dysfunctionality of T cells in human tumors is characterized by the increased cell surface expression of inhibitory receptors such as PD1, lymphocyte activation gene 3 protein (LAG3), T cell immunoglobulin mucin receptor 3 (TIM3; encoded by HAVCR2), 2B4, CD200, and CTLA4, and a reduced capacity of the cells to carry out classical CD8+ T cell effector functions, including the capacity to produce cytokines such as tumor necrosis factor (TNF), interleukin 2 (IL-2), and interferon γ (IFNγ) under (semi-)physiological conditions (i.e. directly ex vivo or after stimulation with cognate antigen or low dose anti-CD3)24–27. Because of the transcriptional but also functional heterogeneity that is observed in this T cell pool (see below), it may be further divided into subgroups, and we will use the nomenclature pre-dysfunctional – early dysfunctional – late dysfunctional in the subsequent sections.
At present it has not been fully established whether the same or similar populations of dysfunctional CD8+ T cells are present in all human tumor types, but early data do point to considerable similarities. Specifically, in non-small-cell lung cancer (NSCLC) and hepatocellular carcinoma (HCC), a dysfunctional CD8+ T cell population (named ‘exhausted’) has been described that is characterized by high-level expression of inhibitory receptor genes such as PDCD1 (encoding PD1), LAG3, CTLA4, T cell immunoreceptor with Ig and ITIM domains (TIGIT), LAYN, and HAVCR228,29. Similarly, CD8+ T cells expressing amongst others PDCD1, LAG3, and HAVCR2 have been observed in basal cell carcinoma (BCC)30. Also in CRC, dysfunctional T cells that express a comparable set of signature genes (PDCD1, HAVCR2, LAYN) were identified, both in patients with microsatellite stable (MSS) and microsatellite instable (MSI) tumors31. In a breast cancer study investigating the immune infiltrates of patients with different breast cancer subtypes, only T cells with effector–memory or central–memory profiles, and no cells with a dysfunctional state, were distinguished32. Nevertheless, one of the two computationally defined components that explained most variation between the T cell states in this study was ‘terminal differentiation’, and this component was defined by the expression of inhibitory and costimulatory genes, including CD2, tumor necrosis factor receptor superfamily member 18 (TNFRSF18; encoding GITR), TNFRSF4 (encoding OX40), TNFRSF9 (encoding 4-1BB), CTLA4, and TIGIT 32, which are often associated with dysfunction.
Two studies in patients with NSCLC and triple negative breast cancer (TNBC) have described the presence of tissue-resident memory CD8+ T (TRM) cells expressing integrin αE (ITGAE, encoding CD103)33. Interestingly, in the lung cancer cohort, a subset of ‘tumor TRM T cells’ was found to display increased expression of dysfunctional markers such as HAVCR2, PDCD1, CTLA4, and LAYN when compared to TRM cells from adjacent healthy tissue33. The TRM CD8+ T cells reported in the TNBC tumors were similarly characterized by markers that are largely consistent with a dysfunctional profile (expression of amongst others HAVCR2, LAG3, and PDCD1)34, implying that at least part of the TRM population in these tumors share characteristics with the dysfunctional cells reported in other studies. Taken together, whereas alignment of T cell states across studies poses a challenge, both because of variation in sequencing technology and analysis strategy, the majority of the investigated tumor types contain a CD8+ T cell population that displays characteristics of dysfunction at the transcriptional level.
While similar groups of dysfunctional and also naïve-like and cytotoxic CD8+ T cells appear present in a large fraction of different tumor types, the existence of CD8+ T cell types that seem specific for certain tumor types has also been proposed, including mucosal associated invariant T (MAIT) cells in NSCLC, HCC, and CRC, intraepithelial lymphocytes (IELs) in CRC, and γδ T cells in TNBC28,29,31,34. However, further studies that profile rare intratumoral cell types with considerable depth are required to understand whether these (invariant) T cell subsets are truly restricted to these tumor types. Another striking finding of the single-cell sequencing analyses carried out to date is that the relative abundance of naïve-like, cytotoxic, and dysfunctional cells is highly variable between tumors with, for instance, dysfunctional T cell fractions ranging from 5% to 80% of the total T cell infiltrate in melanoma21,22. However, which (environmental) factors exactly drive this diversity requires further investigation. Below, we will discuss the recent advances in our understanding regarding the connection between the major CD8+ T cell states that have been observed in human cancers, and their presumed biological contribution to tumor control.
T cell dysfunctionality is a gradual, not a binary, state
Similar to the naïve, memory, and effector T cell subsets, dysfunctional T cells have frequently been viewed as a defined, well demarcated, subset of T cells. However, at least in the context of cancer, it is doubtful whether a binary classification of cells as being dysfunctional or not (or exhausted or not) is justified. In both mice and humans a remarkable phenotypic diversity is observed within the intratumoral T cell pool that displays characteristics of dysfunction25,35–38, reflected both by varying combinations and levels of inhibitory and costimulatory receptors, such as TIM3, CTLA4, CD39, 4-1BB, and also variable surface levels of PD125,35,36,38. Furthermore, single-cell transcriptome analyses of CD8+ T cells in various human cancers have identified a pre-dysfunctional cell state, characterized by an expression of inhibitory receptors that is higher than that of naïve-like and cytotoxic populations but lower than that of the dysfunctional cells. In one of the aforementioned melanoma studies21, these pre-dysfunctional cells (referred to as ‘transitional cells’) were defined by high expression of GZMK and intermediate expression of, amongst others, PDCD1 and LAG3. In the melanoma cohort of Sade-Feldman et al., a ‘lymphocyte’ population that expresses TCF7 and IL7R as well as GZMK was identified22, potentially reflecting a state similar to the GZMK-expressing pre-dysfunctional population in Li et al.21. Similar populations, marked by GZMK and ZNF683 expression and showing low to intermediate expression levels of inhibitory receptors, have also been identified in NSCLC and HCC28,29. Moreover, GZMK-expressing subsets of effector–memory T cells (further defined by CD44 expression) and memory T cells (further defined by eomesodermin homolog (EOMES) and CXC-chemokine receptor 3 (CXCR3)) were described in CRC31 and BCC30 respectively, possibly resembling the pre-dysfunctional states identified in melanoma, NSCLC, and HCC. In breast tumors, cells with a pre-dysfunctional state were not explicitly identified32,34. However, the presence of the terminal differentiation T cell component containing markers of dysfunction suggests that a range of dysfunctional states may also exist in breast cancer32. Collectively, these data support two conclusions; first, the presence of a gradient of cell states rather than discrete populations is consistent with an intratumoral differentiation process, resulting in cells that reside along a continuum of dysfunction (with T cell activation as one probable driver of this process, see below). In Box 2, we briefly summarize current knowledge on the T cell-intrinsic factors that contribute to the different levels of dysfunctionality within the CD8+ T cell compartment. Second, this research field has somewhat of an issue with nomenclature, with respect to the fact that seemingly similar cell pools are named differently across studies. In Box 3 we further address the nomenclature challenges that have appeared with the increased use of high dimensional single-cell profiling techniques, and discuss the value of creating a consensus nomenclature in this field. In the absence of such a consensus, we have aimed to align the T cell pools distinguished in the recent studies in Table 1, using their (partial) overlap in signature genes (Figure 1).
Box 2. Cell intrinsic factors involved in the development of the dysfunctional CD8+ T cell state.
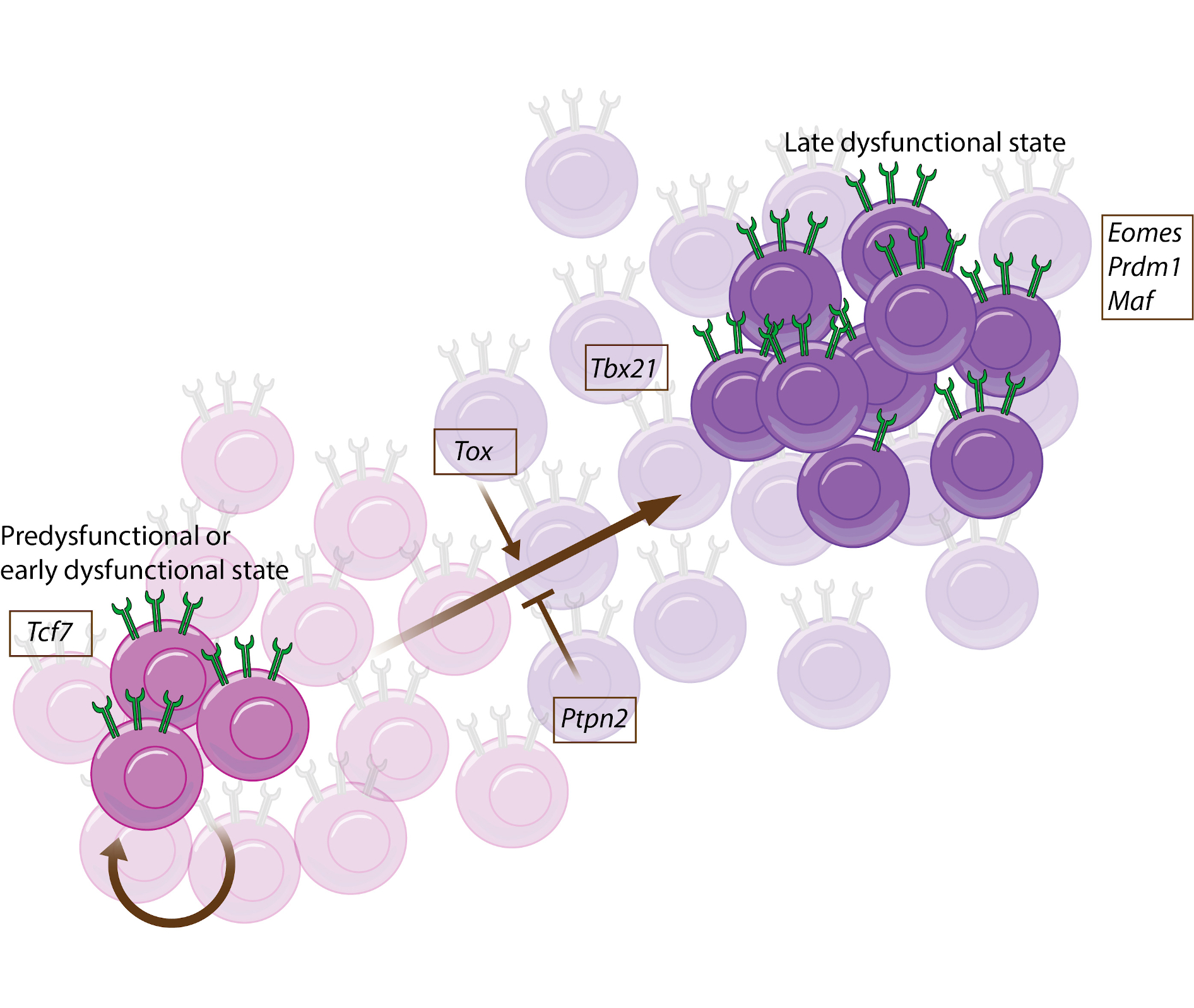
CD8+ T cell dysfunctionality appears to be tightly regulated by a variety of transcription factors (TFs), including eomesodermin homolog (EOMES), T-box expressed in T cells (T-BET; encoded by TBX21), B lymphocyte-induced maturation protein 1 (BLIMP1, encoded by PR domain zinc finger protein 1 (PRDM1)), MAF, TOX, and TCF1 (encoded by TCF7)44,62,63,80–83. Interestingly, the expression levels of many of these TFs are interdependent in mouse studies, with, for example, deletion of Eomes leading to upregulation of TCF1 and downregulation of TOX levels81. Similarly, Tox expression positively correlates with Prdm1 expression83, while Maf is negatively correlated with Prdm1 82, suggesting that dysfunctionality is controlled by a complex regulatory network. Interestingly, Tox deletion leads to changes in the chromatin accessibility of genes including Pdcd1, Entpd1 (less accessible), Tcf7 and Il7r (more accessible), suggesting that transcriptional regulation of dysfunctionality by TOX might (in part) be due to epigenetic imprinting83. In early studies of CD8+ T cell dysfunctionality in mouse models of chronic viral infection, T-BET and EOMES were identified as major regulators and have been used to distinguish CD8+ T cells that exist at different stages along the dysfunctional gradient84. More recent data from mouse studies of viral infection and cancer suggest that the pre-dysfunctional cells that are required for persistence of both the pre-dysfunctional and (late) dysfunctional pools are more strictly defined by the expression of TCF144,62,65. This raises the question of whether T-BET and EOMES expression might distinguish between early and late dysfunctional cells. TCF1 appears to be an important regulator of anti-tumor immunity, as deletion of Tcf7 abolishes tumor control both in the untreated setting and upon immune checkpoint blockade (ICB)63. Besides the anti-tumor role of the TCF1-positive self-renewing population, a recent paper has shown that the conversion of a TCF1-positive pre- or early dysfunctional state to a more dysfunctional state also contributes to the response to ICB, as tumor growth was substantially reduced upon anti-programmed cell death protein 1 (PD1) therapy when the phosphatase protein tyrosine phosphatase non-receptor type 2 (Ptpn2) was deleted, thereby converting CD8+ T cells towards (late) dysfunctionality69.
In addition to their role in regulating dysfunctionality, many of the above-mentioned TFs are involved in effector and memory T cell formation; however, whilst TOX is critical for the development of a dysfunctional phenotype, it appears dispensable for effector and memory development in CD8+ T cells83,85,86. Interestingly, whereas TOX deficiency resulted in reduced expression of inhibitory receptors in CD8+ T cells, the capacity to produce cytokines was not rescued in TOX-deficient T cells in mouse liver tumors83, suggesting that a larger set of TFs may in concert regulate the distinct aspects of dysfunction.
The schematic shows a model for the roles of cell intrinsic factors in the development of CD8+ T cell dysfunction. The proposed associations between the expression of the TF genes Tcf7, Tox, Prdm1, Maf, Eomes, and Tbx21 and the phosphatase gene Ptpn2, and the development of CD8+ T cell dysfunction is depicted, based on their overexpression and/or deletion in mouse tumor models44,62,63,69,80–83.
Box 3. Challenges in cell state definitions and nomenclature.
With the increasing use of in-depth profiling technologies such as cytometry by time of flight (CyTOF) and single cell RNA-sequencing (scRNA-seq), cell states are now analyzed with a level of detail that goes far beyond cell type identification based on classical T cell differentiation markers. However, these emerging methods bring with them a number of challenges, both in the way that cell states are defined and named: 1). Choices made in analysis methodology can influence the detection of specific cell populations in such studies. As an example, two scRNA-seq studies in melanoma and non-small-cell lung cancer (NSCLC) have reported the existence of a T cell population marked by the expression of multiple heat shock proteins (HSPs)22,33. Since stress signatures can be induced by sample processing, stress genes (including HSPs) were filtered out during data analysis in a third study, in which - understandably - no HSP-dominated population was found21. At present, it is unclear whether this population is biologically meaningful and under which conditions inclusion or exclusion of stress genes is preferable. Similarly, additional cell type-independent gene modules - such as a proliferation signature - can influence clustering when included or excluded. 2). As some of the intratumoral T cell states appear to be closely connected to neighbouring cell populations, with for instance pre-dysfunctional cells and dysfunctional cells appearing to form a continuum rather than separate cell states, the definitions of cell states can vary by the model that is used for clustering. 3). Limited numbers of marker proteins and/or genes are often used to define cell states, raising the question of whether these sets of markers accurately define the (functional) differences between intratumoral CD8+ T cell subsets within and between studies. 4). The marker genes that are used to define cell states vary between studies, complicating direct comparisons of cell states (such as between tissue-resident memory T (TRM) T cells positive for HAVCR2 (encoding TIM3)33,34 and dysfunctional T cells) within these studies. 5). T cell populations that display strong resemblance to each other have been designated different names in separate reports, as exemplified by the memory and naïve(-like) populations (both characterized by the expression of interleukin 7 receptor (IL7R), CC-chemokine receptor 7 (CCR7), and TCF7) identified in different studies of melanoma20–22. Aligning the cell states identified by scRNA-seq of human tumors with those defined in mouse tumours using various protein and gene markers poses an additional challenge, especially because full consensus on the exact function and appropriate naming of these populations in mice is still somewhat lacking87.
In Table 1, we have aimed to align the T cell states in human tumors that have been identified across single-cell sequencing studies using the associated marker genes. Note that sequencing platforms differ in sensitivity and that various data analysis strategies were used in these studies. Comprehensive efforts to enable the comparison of datasets from different sequencing platforms are now ongoing and will help settle this issue88,89. Along similar lines, comparative studies are required to elucidate whether the TCF7-positive subset of CD8+ T cells that is responsive to immune checkpoint blockade (ICB) in mice mainly consists of pre-dysfunctional and early dysfunctional T cells, or possibly also comprises naïve-like cells. In future efforts, complementation of transcriptional data with data on protein expression90, functional properties, tumor-reactivity of the associated T cell receptor (TCR)46, transcriptional regulators, and epigenetic regulatory mechanisms (reviewed in 91 and 92) will help to establish a more robust nomenclature to describe intratumoral T cell states.
Table 1. Integration of CD8+ T cell states that display similarities based on single cell RNA-sequencing studies.
| Dataset | Sequencing method | Gene signature | Annotation | Expression of inhibitory receptors | Clonality | Transcriptionally linked to | TCR sharing with | Functional characteristics |
|---|---|---|---|---|---|---|---|---|
| Naïve-like | ||||||||
| Clarke (NSCLC) 33 | 10X Sequencing |
TCF7, CCR7, SELL |
Central memory | Low | Low | NA | NA | NA |
| Guo (NSCLC) 29 | SMART-Seq2 |
LEF1, SELL, TCF7, CCR7 |
Naïve | Low | Low* | Pre-dysfunctional**** | None | Low abundance in tumor |
| Li (melanoma) 21 | MARS-seq |
IL7R, CCR7, TCF7 |
Naïve-like | Low | Low | NA | None | NA |
| Sade-Feldman (melanoma) 22 | SMART-Seq2 |
TCF7, LEF1, SELL, IL7R, LTB |
Memory | Low | NA** | NA** | NA | Correlated with response to ICB |
| Savas (TNBC) 34 | 10X Sequencing | NA | NA | NA | NA | NA | NA | NA |
| Tirosh (melanoma) 20 | SMART-Seq2 |
CCR7, TCF7, LEF1, SELL |
Naïve | Low | Low*** | NA | NA | NA |
| Yost (BCC) 30 | 10X Sequencing |
IL7R, CCR7 |
Naïve | Low | Low | NA | Pre-dysfunctional, cytotoxic, dysfunctional | NA |
| Zhang (CRC) 31 | SMART-Seq2 | LEF1, SELL, CCR7, TCF7 | Naïve | Low | Low* | Pre-dysfunctional, cytotoxic**** | NA | Low abundance in tumor |
| Zheng (HCC) 28 | SMART-Seq2 |
LEF1, CCR7 |
Naïve | Low | Low-intermediate *** | Pre-dysfunctional, cytotoxic | None | Low abundance in tumor |
| Pre-dysfunctional | ||||||||
| Clarke (NSCLC) 33 | 10X Sequencing | NA | NA | NA | NA | NA | NA | NA |
| Guo (NSCLC) 29 | SMART-Seq2 |
GZMK, PDCD1 ZNF683, ITGAE CD28 |
Pre-exhausted | Intermediate | Intermediate | Naïve-like, cytotoxic, dysfunctional**** | Dysfunctional, cytotoxic**** | NA |
| Li (melanoma) 21 | MARS-seq | GZMK | Transitional | Intermediate | Intermediate | Dysfunctional | Dysfunctional | NA |
| Sade-Feldman (melanoma) 22 | SMART-Seq2 |
TCF7, IL7R, GZMK,, FYN |
Lymphocytes | Intermediate | NA** | NA** | NA | NA |
| Savas (TNBC) 34 | 10X Sequencing |
GZMK, KLRG1, LYAR, GZMM, TXNIP, FCRL6 |
Effector-memory***** | Intermediate | Intermediate | Dysfunctional | None | NA |
| Tirosh (melanoma) 20 | SMART-Seq2 | NA | NA | NA | NA | NA | NA | NA |
| Yost (BCC) 30 | 10X Sequencing |
EOMES, GZMK, CXCR3 |
Memory | Low | Intermediate | NA | Dysfunctional, cytotoxic, naïve-like | NA |
| Zhang (CRC) 31 | SMART-Seq2 |
GZMK, CXCR4, CXCR3, CD44 |
Effector-memory | Intermediate | Intermediate | Dysfunctional, naïve-like, cytotoxic **** | Dysfunctional, cytotoxic**** | NA |
| Zheng (HCC) 28 | SMART-Seq2 |
GZMK, PDCD1 |
Transitional | Intermediate | Low-intermediate *** | Dysfunctional, naïve-like, cytotoxic | Dysfunctional, cytotoxic | NA |
| Dysfunctional | ||||||||
| Clarke (NSCLC) 33 | 10X Sequencing |
HAVCR2, GZMB, IFNG, CXCL13, PDCD1, ITGAE |
TIM3+ IL7R- | High | High | NA | NA | Proliferative subset, potentially HSP-expressing subset |
| Guo (NSCLC) 29 | SMART-Seq2 |
LAYN, LAG3, TIGIT, PDCD1, HAVCR2, CTLA4, ITGAE |
Exhausted | High | Intermediate | Pre-dysfunctional | Pre-dysfunctional | NA |
| Li (melanoma) 21 | MARS-seq |
LAG3, PDCD1, CXCL13, TIGIT |
Dysfunctional | High | High | Pre-dysfunctional | Pre-dysfunctional | Proliferative subset, indicative of tumor-reactivity |
| Sade-Feldman (melanoma) 22 | SMART-Seq2 |
LAG3, PDCD1, HAVCR2, ENTPD1, CTLA4 |
Exhausted | High | NA** | NA** | NA | Proliferative subset, HSP-expressing subset, anti-correlated with response to ICB |
| Savas (TNBC) 34 | 10X Sequencing |
ITGAE, HAVCR2, PDCD1, TIGIT, CTLA4, LAG3 |
TRM | High | High | Pre-dysfunctional/cytotoxic | None | Proliferative subset |
| Tirosh (melanoma) 20 | SMART-Seq2 |
PDCD1, HAVCR2, TIGIT, LAG3, CTLA4 |
Exhausted | High | High | NA | NA | Proliferative subset |
| Yost (BCC) 30 | 10X Sequencing |
LAG3, HAVCR2, PDCD1, GZMB, ENTPD1, ITGAE |
Exhausted | High | High | NA | Pre-dysfunctional, naïve-like | Proliferative subset |
| Zhang (CRC) 31 | SMART-Seq2 |
LAYN, HAVCR2, CXCL13, PDCD1, IFNG, ITGAE |
Exhausted | High | High | Pre-dysfunctional | Pre-dysfunctional | Proliferative subset |
| Zheng (HCC) 28 | SMART-Seq2 | LAYN, PDCD1, HAVCR2, CTLA4 | Exhausted | High | Intermediate -high | Pre-dysfunctional | Pre-dysfunctional, Cytotoxic | NA |
| Cytotoxic | ||||||||
| Clarke (NSCLC)33 | 10X Sequencing | NA | NA | NA | NA | NA | NA | NA |
| Guo (NSCLC) 29 | SMART-Seq2 |
CX3CR1, PRF1, GZMA,, GZMB |
Effector | Low | High* | Pre-dysfunctional**** | Pre-dysfunctional**** | Low abundance in tumor |
| Li (melanoma) 21 | MARS-seq |
GZMH, GNLY, FGFBP2, CX3CR1 |
Cytotoxic | Low | Intermediate | None | None | NA |
| Sade-Feldman (melanoma) 22 | SMART-Seq2 |
KLRG1, PRF1, GZMB, FCGR3A |
Cytotoxic | Intermediate | NA** | NA** | NA | NA |
| Savas (TNBC) 34 | 10X Sequencing |
GZMK, KLRG1, LYAR, GZMM, TXNIP, FCRL6 |
Effector-memory***** | Intermediate | Intermediate | Dysfunctional | None | NA |
| Tirosh (melanoma) 20 | SMART-Seq2 |
PRF1, GZMB, NKG7, GZMA |
Cytotoxic | Intermediate | Low*** | NA | NA | NA |
| Yost (BCC) 30 | 10X Sequencing |
FGFBP2, KLRD1 |
Effector-memory | Low | Intermediate | NA | Pre-dysfunctional, naïve-like | NA |
| Zhang (CRC) 31 | SMART-Seq2 |
CX3CR1, KLRG1, FCGR3A, FGFBP2, PRF1, GZMH |
Temra | Low-intermediate | High* | Pre-dysfunctional, naïve-like**** | Pre-dysfunctional**** | Low abundance in tumor |
| Zheng (HCC) 28 | SMART-Seq2 |
CX3CR1, FCGR3A, FGFBP2 |
Effector-memory | Low | Low-intermediate*** | Pre-dysfunctional, naïve-like | Pre-dysfunctional, dysfunctional | Low abundance in tumor |
The CD8+ T cell states identified in the single cell RNA-sequencing (scRNA-seq) studies listed in Supplementary Table S1 were aligned based on their associated marker genes20–22,28–31,33,34. This Table forms the basis for the model in Figure 1 and contains cell states that were identified in treatment-naïve, on-treatment and post-treatment tumors.
The majority of cells originate from blood and/or normal tissue.
On the cell states that were defined by the original clustering and that were used in this Review, no clonality and trajectory analyses were performed.
The assumption is made that the pool of ‘low exhausted cells’ or ‘non-exhausted cells’ used for clonality analyses in these studies contain naïve-like, cytotoxic, and possibly pre-dysfunctional T cells.
As these analyses are based on T cell receptors (TCRs) from tumor, normal tissue, and blood, they do not provide direct evidence for TCR sharing at the tumor site.
Based on combined expression of granzyme K (GZMK) and killer cell lectin-like receptor subfamily G member 1 (KLRG1), the effector-memory population may contain both pre-dysfunctional and cytotoxic cells. BCC, basal cell carcinoma; CCR7, CC-chemokine receptor 7; CRC, colorectal cancer; CTLA4, cytotoxic T lymphocyte associated antigen 4; CX3CR1, CX3C chemokine receptor 1; CXCL13, CXC-chemokine ligand 13; CXCR, CXC-chemokine receptor; ENTPD1, ectonucleoside triphosphate diphosphohydrolase 1; EOMES, eomesodermin homolog; FCGR3A, Fcy receptor IIIA; FCRL6, Fc receptor-like 6; FGFBP2, fibroblast growth factor-binding protein 2; GNLY, granulysin; HCC, hepatocellular carcinoma; HSP, heat shock protein; ICB, immune checkpoint blockade; IFNG, interferon-Y; IL7R, interleukin-7 receptor; ITGAE, integrin aE; LAG3, lymphocyte activation gene 3; LEF1, lymphoid enhancer-binding factor 1; LTB, lymphotoxin-P; NA, not applicable; NSCLC, non-small-cell lung cancer; PDCD1, programmed cell death protein 1; PRF1, perforin 1; TCF7, transcription factor 7; TEMRA, recently activated effector memory or effector T; TIGIT, T-cell immunoreceptor with Ig and ITIM domains; TIM3, T-cell immunoglobulin mucin receptor 3; TNBC, triple-negative breast cancer; TRM, tissue-resident memory CD8+ T; TXNIP, thioredoxin-interacting protein.
Figure 1. Model of intratumoral CD8+ T cell states.

This schematic depicts a model describing the characteristics of, and possible connections between, the major CD8+ T cell states in human tumors, as based on data from20–22,28–31,33,34. Observations from the different studies that support this model are listed in Supplementary Table S1. In brief, the naïve-like cells described in non-small-cell lung cancer (NSCLC)29, hepatocellular carcinoma (HCC)28, colorectal cancer (CRC)31, basal cell cancer (BCC)30, and melanoma21 show a strong resemblance to the (central-)memory populations described by Sade-Feldman et al. and Clarke et al.22,33. Based on the expression of granzyme K (GZMK), intermediate expression of inhibitory molecules, and relatively low clonality, the T lymphocyte population described in Sade-Feldman et al.22 and the effector–memory population described in Zhang et al.31 were considered similar to the pre-dysfunctional cell states observed in melanoma21, HCC28, and NSCLC29. While additional research is required to determine their extent of overlap, the tissue resident memory T (TRM) population described in triple-negative breast cancer (TNBC)34 and the HAVCR2+ TRM cells in NSCLC33 are here aligned with the dysfunctional state described in all other studies. The cell state definitions from Azizi et al.32 could not be integrated into the model presented here and the effector–memory subset reported by Savas et al. may be composed of a mixture of pre-dysfunctional and cytotoxic cells, based on the combined expression of GZMK and killer cell lectin-like receptor subfamily G member 1 (KLRG1)34. In this model, we propose that the development of (pre-)dysfunctional cell states is predominantly driven by tumor-specific cues such as tumor antigen recognition and/or tumor-specific environmental factors (here referred to as tumor microenvironment (TME)-induced differentiation). Cytotoxic cell states are also encountered in healthy tissues28,29,31, indicating that the underlying differentiation process is not strictly tumor-specific (here referred to as TME-independent differentiation). Cytotoxic effector T cells are depicted as a population that is most likely developmentally distinct from the cells along the (pre-)dysfunctional axis, but additional research is required to clarify whether cytotoxic effector cells indeed originate from a distinct pool of cells, or whether they are connected to the (pre-)dysfunctional axis in some situations (as depicted by the dashed two-way arrow). Note that both trajectory and T cell receptor (TCR) sharing analyses indicate that the pre-dysfunctional and dysfunctional cells form a continuum of cell states, rather than well-demarcated populations. The line graph shows approximate levels of proliferation, CXC-chemokine ligand 13 (CXCL13) expression, and the expression of inhibitory receptors by pre-dysfunctional, early dysfunctional, and late dysfunctional CD8+ T cells. CCR7, CC-chemokine receptor 7; CX3CR1, CX3C chemokine receptor 1; CTLA4, cytotoxic lymphocyte-associated antigen 4; FCGR3A, Fcγ receptor IIIA; IL7R, interleukin 7 receptor; LAG3, lymphocyte activation gene 3; PDCD1, programmed cell death 1; PRF1, perforin 1; TCF7, transcription factor 7.
Dysfunctional T cells are functionally diverse
The transcriptional diversity that is observed within the dysfunctional T cell pool is accompanied by diversity in functional capacity. First, a combination of transcriptomic and proteomic approaches have shown that dysfunctional CD8+ T cells (including the comparable HAVCR2-expressing TRM cell population identified in NSCLC and TNBC) contain a highly proliferative subpopulation of cells21,22,25,33,34. In melanoma, this proliferative subpopulation of dysfunctional cells is characterized by lower expression of inhibitory receptors than their non-proliferative counterparts (although still higher than the intermediate expression seen on pre-dysfunctional cells; Figure 1)21. This is consistent with a model in which CD8+ T cells retain proliferative capacity during their transition from the pre-dysfunctional to an early dysfunctional state, but lose this capacity at the stage of more profound, ‘late’, dysfunction, either because of an intrinsic block or because their high inhibitory receptor expression suppresses T cell activation. During the progression towards late dysfunctionality, classical CD8+ T cell effector functions, such as the capacity to produce IL-2, TNF, and IFNγ, are also reduced, even though the expression of some of the genes encoding these secreted factors, and also other T cell effector function-associated genes such as PRF1 and GZMB, remains high25. This observation shows that transcriptional characteristics do not always directly translate into functional capacities, emphasizing that we should be careful in assigning functionality solely on the basis of transcriptomic analyses. In addition, this observation calls for a greater effort to understand translational control in T cells39,40.Interestingly, contrary to their reduced capacity to produce classical CD8+ T cell effector cytokines, CD8+ T cells acquire the capacity to express CXC-chemokine ligand 13 (CXCL13) mRNA20,21 and secrete CXCL13 protein25 when progressing along the (pre-)dysfunctional axis. CXCL13 is a well-established B-cell attractant, and the surprising observation that late dysfunctional CD8+ T cells constitutively produce this molecule suggests that this cell population may be one of the drivers of the formation of the TLS that are observed in a substantial fraction of human cancers. While the exact function of TLS in the TME has not been fully established, TLS are associated with clinical benefit, and the presence of antigen presenting cells (APCs) in TLS suggests a potential role for these structures in T cell activation at the tumor site (discussed in 41 and 42). Notably, the observed proliferative capacity of early dysfunctional CD8+ T cells, which in fact appears higher than that of any other CD8+ T cell subset present in the TME21, and also the acquisition of CXCL13 production capacity by late dysfunctional CD8+ T cells, provides clear evidence that dysfunctional CD8+ T cells in the human TME should not be considered inert, but rather as T cells that have assumed a novel function. In addition, the different functional characteristics of early and late dysfunctional cells strongly suggests that even within the dysfunctional compartment, CD8+ T cells play distinct roles in tumor-immunity.
Relationship between the dysfunctional state and other intratumoral T cell states
To understand the developmental relationships between the intratumoral T cell states that are observed in tumor tissues, two types of data are currently used. First, an overlap in transcriptional profiles, and in particular the occurrence of cells with transcriptional states that lie in between those of different cell groups, can be used to infer developmental relatedness. To support such analyses, algorithms have been developed that model cell state differentiation trajectories based on transcriptional relatedness (as reviewed in 43), with the caveat that it has not been well validated whether these trajectory models generally provide a correct description of the true biological cell differentiation paths. Second, as a more direct test of cellular kinship, overlap of T cell receptor (TCR) repertoires between cell populations with different states, known as TCR sharing, can be used to infer differentiation pathways32; yet this approach is only of use when analyzing clonally expanded T cell populations and cannot provide any information on the directionality of the differentiation pathways.
Trajectory analyses support the previously discussed observation that in a number of tumor types, the pre-dysfunctional and dysfunctional CD8+ T cell populations are transcriptionally related21,28,29,31. In addition, the overlap in TCR repertoire of these two cell states is higher than that observed between other T cell states21,28. Based on the increased expression of proteins associated with prolonged T cell activation in the dysfunctional T cell pool, these data are most consistent with a model in which pre-dysfunctional CD8+ T cells differentiate into early and late dysfunctional CD8+ T cells. Furthermore, direct evidence in favor of such a model comes from a mouse study that shows that CD8+ cells with a pre-dysfunctional profile, characterized by Lag3 and Tigit expression but low levels of Pdcd1 and Havcr2, could give rise to dysfunctional cells, but not vice versa44.
At present, less clarity exists with respect to the question of whether cytotoxic T cells form a developmentally distinct intratumoral CD8+ T cell population or whether they are connected to the cells that collectively form the (pre-)dysfunctional axis. In the Li et al. melanoma data set, intratumoral cytotoxic cells displayed only a minimal transcriptional overlap with either the pre-dysfunctional or the dysfunctional CD8+ T cell pool. Furthermore, the overlap between the TCR repertoire of the cytotoxic T cell pool and T cells along the (pre-)dysfunctional axis was limited21. While the number of TCRs assessed in that study was small, minimal clonotype sharing was also observed between the cytotoxic and dysfunctional populations in a larger dataset in BCC30. In contrast, cytotoxic cells did share TCRs with the GZMK-expressing memory CD8+ T cell population in this study30. This would be in line with a bifurcation model, as was proposed based on NSCLC single-cell sequencing data, in which both the cytotoxic and the dysfunctional T cell populations are connected to the pre-dysfunctional state29. A similar model has been suggested in CRC, in which cytotoxic (named recently activated effector memory or effector T cells (TEMRA) in the study) and dysfunctional T cells were transcriptionally linked to the pre-dysfunctional state31. However, it is important to note that both of these bifurcation models were built using the transcriptional overlap between T cells that were either located inside the tumor, or in adjacent normal tissue or blood. As the cytotoxic T cell pool in these data sets is primarily composed of cells obtained from normal tissue and blood, whereas the dysfunctional T cell pool is largely composed of cells obtained from tumor tissue, it is difficult to know whether these models specifically capture intratumoral differentiation dynamics29,31. In addition, the TCR clonotypes that were shared between intratumoral pre-dysfunctional and intratumoral dysfunctional T cells in CRC were mutually exclusive with the clonotypes shared between intratumoral pre-dysfunctional cells and cytotoxic cells derived from blood31. These models therefore likely reflect, at least in part, the imprint of the environmental context on T cell differentiation programs, rather than a branched differentiation process that occurs inside the tumor.
Other trajectory analyses in melanoma, breast cancer, and HCC have proposed a continuous trajectory between all intratumoral cell states22,28,32,34. Although rare, shared TCRs were observed between dysfunctional and cytotoxic T cells in HCC28. In contrast, the TCR repertoires of transcriptionally connected dysfunctional and non-dysfunctional populations in TNBC were not overlapping, incidentally suggesting that transcriptional overlap does not necessarily imply a connection by descent34. Taken together, these studies provide solid support for a model in which pre-dysfunctional and dysfunctional T cells are developmentally related, with evidence for progression of T cells from a pre-dysfunctional state to an early and then late dysfunctional state. Regarding the presence or absence of a developmental connection between the cytotoxic pool and the (pre-)dysfunctional axis, the available evidence is more ambiguous. While our interpretation of the current data is that, at least in melanoma, cytotoxic T cells do not originate from the same T cell pool as the cells that make up the dysfunctional population, additional data are required to conclusively settle this point (Figure 1).
Dysfunction as indicator of tumor-reactivity
Both during their initial activation in secondary lymphoid organs and whilst present at the site of infection or tumor growth, T cells receive numerous signals that have been shown to influence cell state and function45. Therefore, the observed heterogeneity in CD8+ T cell states in human tumors is highly suggestive of substantial variation in the signals that have been received by individual T cells, either in the recent past or their more distant (developmental) history, and below we aim to describe a potential mechanistic basis for formation of some of the T cell states that have been observed.
Recent studies have provided strong evidence that only a proportion of the T cells that reside within the TME are able to recognize antigens on surrounding tumor cells46,47. The ‘bystander’ T cell pool that has been identified in these studies has been shown to include T cells reactive against antigens derived from viruses such as Epstein Barr virus (EBV) and influenza and that were presumably attracted by chemokines such as CXCL9 and CXCL10, irrespective of the relevant antigen being present at such tumor sites47–50. In addition, it may be speculated that the bystander T cell pool could contain T cells reactive against tumor antigen–human leukocyte antigen (HLA) combinations that were lost over time; although direct experimental evidence for such ‘memories of the past’ is currently lacking.
To help to distinguish presumed bystander cells from tumor-specific T cells, a number of properties that have previously been associated with (tumor) antigen recognition may be utilized. Representing a proxy for antigen-driven T cell expansion, T cell clonality has long been used as a marker of tumor-reactivity, and increased levels of clonality in tumor tissue are associated with response to anti-PD1 therapy15. In line with these data, the most highly expanded clones in melanomas have been shown to be frequently tumor-reactive46,51. Analysis of TCR repertoires in single-cell sequencing data sets has revealed that recurrent TCRs are most abundant in the cytotoxic and dysfunctional T cell populations21,29–31,34, providing an argument to further study tumor-reactivity in these populations. However, expanded T cell clones that do not recognize tumor tissue are also present in the TME and, vice versa, small T cell clones can exhibit tumor-reactivity46. Thus, clonality is an imperfect measure for tumor recognition, emphasizing the need for alternative markers to define tumor-reactivity of intratumoral T cells.
As first established in mouse models of chronic infection, continuous antigen encounter is considered to be one of the major drivers of T cell dysfunction (Figure 2A)27. Furthermore, the effect of recognition of tumor antigens on intratumoral CD8+ T cell states has also been addressed by adoptive transfer experiments in mice. Specifically, transfer of tumor-specific CD8+ T cells into tumor-bearing mice was shown to drive the development of a dysfunctional phenotype that was characterized by increased expression of PD1, LAG3, 2B4, and TIM3, while intratumoral T cells that carried an irrelevant TCR did not display these hallmarks of dysfunction52. Likewise, in human melanoma, CD8+ T cells specific for the MART-1 tumor antigen have been shown to display a dysfunctional profile, with profound expression of PDCD1, LAG3, and CTLA4, and cell surface expression of PD124,53. Furthermore, the strength of the dysfunctional signature in a cohort of melanoma tumors (defined by the expression of a set of dysfunction-associated genes across the tumor-resident CD8+ T cell pool) was associated with the presence of a tumor-reactive T cell population, while the strength of a cytotoxic signature was negatively correlated21. As further evidence that tumor-reactivity is associated with expression of markers of dysfunction in human cancers, tumor-reactivity has been shown to be enriched in T cell populations that were expanded in vitro from CD8+ T cells with high levels of PD1, TIM3 or LAG325,54,55. Other cell surface markers that have recently been associated with tumor-reactivity are CD39 and CD103, with expression of these markers being detected on tumor antigen-specific, but not virus-specific T cells47,56. The identification of CD39 and CD103 as markers for tumor-reactivity in human tumors is consistent with data obtained in a mouse sarcoma model, in which tumor-specific T cells defined by major histocompatibility complex (MHC) tetramer staining displayed increased levels of PD1 and CD39 when compared with bystander T cells35,57. By the same token, CD39-positive CD8+ T cells from mouse melanoma tumors were more potent in eliminating tumors than their CD39-negative counterparts44. In contrast to these in vivo mouse studies, a fourth study showed that in mouse colon carcinomas, most of the in vitro tumor killing capacity in the presence of anti-PD1 was contained within the CD39-negative rather than the CD39-positive CD8+ T cell pool22. However, whether the absence of tumor killing by the CD39-positive T cell population was in this case due to a lack of tumor-reactive TCRs or a reflection of a functional limitation of these cells still needs to be addressed.
Figure 2. Model for the development of CD8+ T cell dysfunction and the effect of PD1 blockade.

a) Potential drivers of dysfunction are the suboptimal priming of CD8+ T cells (I)61 and continuous T cell receptor (TCR) triggering (II)27 in combination with environmental factors, which may be cytokines such as tumor growth factor β (TGFβ)73 and interleukin 10 (IL-10)25, but also metabolic conditions such as hypoxia (III) at the tumor site. T cell priming is here depicted to occur in the lymph node, but could potentially also occur (in tertiary lymphoid structures (TLS)) at the tumor site. b) Proposed effects of anti-programmed cell death protein 1 (PD1) therapy on the CD8+ T cell states in and outside of the tumor microenvironment (TME). Mouse model studies have shown that anti-PD1 induces proliferation (denoted by the circular arrow) and conversion of pre- (and/or early) dysfunctional CD8+ T cells towards a more late dysfunctional phenotype (1) and that durable tumor control may require the proliferative capacity of a pre-dysfunctional or early dysfunctional cell population that, contrary to the late dysfunctional population, expresses transcription factor 7 (TCF7), which encodes TCF144,62. Note that it is unclear whether this TCF1-positive population contains naïve-like T cells as well. PD1 blockade may also directly increase the effector function of pre- or early dysfunctional cells (2), as well as the effector function of (late) dysfunctional cells (3). It remains to be established whether tumor regression upon PD1 blockade primarily occurs through the activity of a reactivated pre-dysfunctional or early dysfunctional cell pool, or through the activity of late dysfunctional cells that may be formed as their progeny69. A recent report in human basal cell carcinoma (BCC) has provided evidence for the possible replacement of the TCR repertoire of the dysfunctional T cell pool upon anti-PD1 treatment (4)30. This could be due to an influx of new cells (although the relevance of the systemic T cell compartment to the anti-tumor effects of anti-PD1 has been debated62,67), or expansion of lowly abundant pre-existing intratumoral clones. Finally, it remains a possibility that new T cell states (either of cells that newly infiltrated the tumor, or of pre-existing intratumoral cells) might develop upon anti-PD1 treatment; although no strong evidence in favor of such a model has thus far been obtained in mouse models or human samples (5). In this model, programmed cell death protein 1 (PD1) and PD1 ligand 1 (PDL1) are only depicted on cells in cases where the interaction is blocked by anti-PD1 therapy and are left out on other cells for clarity. Note that the model depicted here is based on the assumption that tumor-reactivity is enriched in the CD8+ T cells that reside along the (pre-)dysfunctional axis, while the cytotoxic effector T cell pool (shown in Figure 1) is enriched for bystander CD8+ T cells.
TCR triggering can, besides upregulation of the expression of inhibitory receptors, result in T cell proliferation and in increased expression of markers of T cell activation such as 4-1BB. Thus, the increased proliferative gene expression signature in the (early) dysfunctional population relative to the cytotoxic cell pool provides evidence for antigen encounter by dysfunctional T cells at the tumor site21. Equally, dysfunctional T cells as well as tumor TRM cells displaying characteristics of dysfunction show high expression of TNFRSF9 (encoding 4-1BB)20,21,29 and TNFRSF18 (encoding GITR)33, consistent with ongoing antigen stimulation. Of note, to distinguish T cell activation from T cell dysfunction, approaches are being developed to computationally uncouple the dysfunctional and activation gene modules20,58, an effort that is complicated by the fact that T cell activation is a driver of dysfunction.
In summary, the above studies provide substantial evidence that tumor-reactivity is enriched within the dysfunctional CD8+ T cell compartment, while the intratumoral cytotoxic population is likely to contain a higher fraction of bystander cells; the main characteristics of T cells that carry a tumor-specific TCR are depicted in Figure 3. Yet, it is important to recognize that these characteristics cannot be used to define tumor-reactive and bystander T cells with 100% precision. Indeed, tumor-specific T cells in mouse models, including T cells that recognize the same tumor antigen, can differ substantially in phenotype, for instance showing variable expression levels of CD39, PD1, CTLA4, and LAG322,35,59 and, as mentioned above, CD39-negative CD8+ T cells have been found to be tumor-reactive22. This phenotypic diversity amongst tumor-specific T cells may in part be due to differences in antigen affinity60, but the observation of substantial cell state diversity within T cell clones suggests that additional factors also contribute21,29,32,35,59. Such factors could include both soluble and cell surface ligands that are unevenly distributed throughout the tumor (Figure 2A). In addition, intratumoral T cell states may be influenced by cues that T cells have already received prior to tumor entry. One such molecularly well-defined example is the absence of CD4+ T cell help during CD8+ T cell activation as a driver of T cell dysfunction that influences the state that CD8+ T cells adopt later in their lifespan (reviewed in 61).
Figure 3. Hallmarks of intratumoral tumor-reactive CD8+ T cells.

This schematic depicts protein markers and functional properties that are enriched in tumor-reactive CD8+ T cells (i.e. T cells that express a tumor-reactive T cell receptor (TCR), irrespective of their functional capacity) relative to bystander CD8+ T cells at the tumor site. Note that none of these characteristics by themselves identify tumor-reactive CD8+ T cells with absolute precision and that for some of these markers (4-1BB, GITR, and CXC-chemokine ligand 13 (CXCL13)), the evidence is less well established. LAG3, lymphocyte activation gene 3 protein; PD1, programmed cell death protein 1; TIM3, T-cell immunoglobulin mucin receptor 3.
In future work, an improvement in our capacity to identify tumor-reactive T cells at the tumor site should come from the combined use of multiple parameters, such as inhibitory receptor expression and proliferative signature, thereby providing a likelihood score of tumor-reactivity for individual cells. In addition, the integration of information on the cell states of sister cells that express the same TCR should further add to our capacity to identify tumor-reactive T cells solely on the basis of phenotypic and transcriptional data.
Dysfunctional T cell states in ICB
The evidence for an enrichment of tumor-reactivity within CD8+ T cells along the (pre-)dysfunctional axis raises two key issues. First, owing to their expression of PD1 and CTLA4, these cells form potential direct targets for anti-PD1 and anti-CTLA4 therapies, and it is therefore important to understand the capacity of cells along the (pre-)dysfunctional axis to be reactivated by these therapies. Second, regardless of their reactivation potential, tumor infiltration by dysfunctional cells may be reflective of a tumor-reactive T cell response, and it is therefore of interest to understand whether the presence of dysfunctional T cells can be used as a potential biomarker in ICB.
The effect of ICB on distinct CD8+ T cell states
In view of the diversity in cell states even within the CD8+ T cell population that expresses immune checkpoint molecules such as PD1 and CTLA4, an understanding of the effect of immune checkpoint therapies on T cells with different levels of dysfunctionality is of importance. In Figure 2B, we have outlined the proposed effects of anti-PD1 therapy on the CD8+ T cell compartment, as further discussed below.
In a mouse model of HCC, tumor antigen-specific T cells were shown to display a dysfunctional profile (as reflected by high PD1 and LAG3 expression) only days after tumor induction and this dysfunctionality further increased during tumor progression, as reflected by the acquisition of 2B4 and TIM3 expression52. Whereas cytokine production and cytotoxicity could be restored by anti-PD1 or anti-PDL1 therapy in dysfunctional T cells at early time points after tumor establishment, the same effector functions failed to be restored in dysfunctional T cells targeted at later time points during tumor progression. With the caveat that human tumor development can take years, and cell states from mice early after tumor induction may therefore not be fully reflective of the T cell states that are identified in human cancer, these data suggest that pre-dysfunctional or early dysfunctional cells in human cancers may be most amenable to re-invigoration by ICB. Additional mouse studies likewise provide support for a model in which a subpopulation of less dysfunctional cells in tumors may be critical for a durable response to ICB44,62,63. Despite the use of diverse markers to characterize this subpopulation, these cells share the expression of TCF7, encoding the TCF1 transcription factor in mice (also named TCF7 in human), and are characterized by undetectable63 or low level expression44,62 of inhibitory receptors such as PD1 and TIM3, when compared with their TCF1-negative dysfunctional counterparts. Furthermore, this intratumoral CD8+ T cell pool in mice shares similarities with the previously defined CXCR5 and TCF1-expressing T cell population displaying low levels of dysfunction that appears important in sustaining the T cell response during chronic viral infection and for response to anti-PD1 therapy64–66.The observation that the TCF1-positive population in viral infection models and tumor models is capable of self-renewal but also yields the TCF1-negative population that displays higher levels of PD1, TIM3, and CTLA4 is consistent with a progenitor or stem cell-like function44,62–66. Notably, while TCF1-positive cells were critical for tumor control upon single agent anti-PD1 therapy or anti-PD1 and anti-CTLA4 combination therapy, ICB also reduced tumor growth in mice in which TCF1-expressing T cells were depleted, suggesting that the late dysfunctional compartment may also be a direct target of ICB62. Of note, it remains to be established whether ICB primarily reinvigorates a tumor-resident T cell population or also acts through mobilization of T cells from outside the tumor, as conflicting observations regarding the relevance of the systemic immune cell compartment for response to therapy have been made in different mouse studies62,67.
TCF1-expressing PD1-positive CD8+ T cells have been observed in human NSCLC, colorectal, and melanoma tumors and were increased in tumors that were treated with ICB44,62,68. In line with the TCF1-positive cells observed in mouse tumors, this human CD8+ T cell pool shows similarities to the CXCR5 and TCF1-positive cells found in mouse models of chronic viral infection68. However, to what extent these cells are tumor-reactive has not been established. Interestingly, an association between the presence of TCF1-positive CD8+ T cells and response to anti-PD1 therapy and/or antiCTLA4 therapy has been reported in patients with melanoma22. Yet, in another melanoma cohort, the numbers of TCF1-expressing PD1-positive CD8+ T cells did not correlate with response to anti-PD1 and anti-CTLA4 combination therapy44. Nonetheless, in this study, the abundance of the TCF1- and PD1-doublepositive CD8+ T cell population was positively associated with progression free and overall survival within the patient group responding to ICB44.
Besides identifying the T cells that can respond to ICB, it will be important to address the (transcriptional) profiles of the cells that eventually account for tumor elimination upon ICB. Specifically, do the TCF1-positive pre-dysfunctional cells acquire cytotoxic capacity or is their conversion into a (late) dysfunctional cell state or a novel cell state required for anti-tumor activity? Support for a role of the late dysfunctional CD8+ T cell pool in anti-tumor immunity upon therapy comes from the observation that late dysfunctional T cells in human tumors display a more pronounced cytotoxic transcriptional profile than pre-dysfunctional and early dysfunctional cells21,25. Furthermore, a recent study in mice indicates that the conversion from a TCF1-positive pre- or early dysfunctional state to a late dysfunctional state enhances the level of tumor control69. Moreover, in human NSCLC, TRM cell clones with a dysfunctional profile (expressing HAVCR2 and other inhibitory receptors) that were present pre- and post-therapy displayed increased expression of cytotoxic genes after anti-PD1 treatment33. While transcriptional data do not allow one to draw definitive conclusions regarding functional alterations and thus should be interpreted with care, this observation would be consistent with a model in which the post-therapy dysfunctional population has an increased capacity for tumor control. Notably, in line with observations in mice59,70, these data support a model in which the effect of ICB is primarily based on inducing changes in cell states that already existed pre-therapy, rather than inducing entirely novel cell states. While the above data focus on the importance of a pre-existing intratumoral T cell population to achieve tumor control, recent work has shown that the TCR repertoire present in the dysfunctional T cell pool in BCC is altered upon anti-PD1 therapy, a phenomenon that has been named ‘clonal replacement’30. It remains to be addressed whether the newly observed TCR clonotypes originated from a small population of TCF1-positive cells already present in the tumor, or from a pool of cells outside the TME.
Taken together, the general hypothesis that arises from these data is that a durable response to anti-PD-1 therapy requires the presence of tumor-specific T cells with low levels of dysfunction. However, which states are adopted by the cells that subsequently have the capacity to achieve tumor elimination remains to be answered.
Dysfunctional T cells as a predictive marker for response to ICB
Tumor recognition by CD8+ T cells is essential for T cell-mediated tumor control and identification of the cell states that indicate the presence of a tumor-specific T cell repertoire is thus of interest. As T cell dysfunction in human tumors appears at least in part driven by tumor-reactivity, characteristics of this cell population deserve consideration as potential predictive biomarkers. In recent work, the presence of CD8+ T cells that express high levels of PD1 or PD1 and CTLA4 were reported to be predictive for response to anti-PD1 therapy in NSCLC and melanoma, respectively25,71. In an independent melanoma cohort, the pre-treatment presence of CD69 and EOMES-positive CD8+ (as well as CD4+) T cells that co-expressed T-BET, PD1 and TIGIT was associated with response to both anti-PD1 or anti-PD1 in combination with anti-CTLA472. Moreover, on-treatment biopsies of NSCLC tumors that responded to anti-PD1 treatment were shown to contain higher levels of TIM3-positive TRM cells (co-expressing CD103 and PD1) than non-responding or pre-treatment lesions33. Although it is not quite clear to what extent the reported T cell subsets overlap, collectively these studies indicate that the presence of (TRM) T cells with markers of dysfunction can be correlated with response to ICB. Of note, these data are at odds with a study that showed that the ratio of TCF1-positive, non-dysfunctional, CD8+ T cells over dysfunctional CD8+ T cells was predictive of response to PD1 and/or CTLA4 blockade in human melanoma22. A simple explanation for the discrepancy between the former studies and the latter study may be differences in the use of the cell state definitions, or the strategy to quantify the different cell fractions. When considering more conceptual explanations, in settings in which the presence or absence of tumor-reactivity is the most important variable between patients, the positive predictive value of dysfunctional T cells might potentially be most crucial. In contrast, in settings in which the presence of tumor-reactivity is a given, the frequency of TCF1-positive T cells may be of greater importance, as the differentiation state of the tumor reactive T cell pool at that point becomes a prime determinant of the capacity to achieve tumor control. Strategies that are capable of identifying tumor-reactive T cell clones with high confidence, and at the same time can assess the presence of a subpopulation within such clones that mirror the mouse TCF1-positive population required for self-renewal, would be the most optimal.
Concluding remarks
Recent studies using high-dimensional profiling techniques have led to an appreciation of the variety of states that are taken on by T cells in human tumors. While the nomenclature used to define these cells has varied, three major cell states – naïve-like, cytotoxic, and dysfunctional - have consistently been described in multiple tumor types. Notably, dysfunctional T cells do not form a homogeneous population, but rather a continuum of cell states that display increasing characteristics of dysfunction. In addition, T cells with variable levels of dysfunction differ in functional capacity, as demonstrated by the high proliferation rate of pre-dysfunctional and in particular early dysfunctional cells, and the production of CXCL13 by cells that have progressed further along the (pre-)dysfunctional axis. Antigen-recognition is a – if not the - major driver of cell state diversification amongst tumor-infiltrating CD8+ T cells, and tumor-reactive T cells appear more prone to differentiate towards a dysfunctional state than bystander cells within the same lesions. Nevertheless, T cells with the same tumor antigen-specificity can display different degrees of dysfunctionality, and the presence of tumor-reactive T cells with a low level of dysfunction may be critical for the generation of a durable anti-tumor response upon ICB. These data are compatible with a model in which T cell dysfunctionality serves as a sensitive indicator for the presence of a tumor-reactive T cell pool, with the less dysfunctional cells within this pool being required for its renewal (Figure 2). Some of the major questions that remain to be addressed are 1). Which T cell states in human cancers resemble the TCF1-positive T cell population that is required to maintain response to ICB in mice? 2). What is the identity of the effector population that is ultimately responsible for tumor killing upon ICB? And 3). How can we accurately identify and quantify those T cells in human cancer lesions that can both recognize surrounding tumor cells and have the capacity for long-term reinvigoration by ICB? Finally, the factors that drive CD8+ T cell differentiation in human tumors are presently incompletely understood, and insights into this are likely to offer new possibilities for patient stratification and therapeutic intervention.
Supplementary Material
Glossary.
Clonotype
The unique T cell receptor (TCR) sequence formed by both the TCR alpha and beta chains.
Single-cell RNA sequencing (scRNA-seq)
Gene expression profiling method that allows for unbiased transcriptome analysis of individual cells.
Tumor-specific T cell reactivity
The capacity of a T cell to recognize tumor cells, regardless of its ability to perform effector function.
Predictive biomarker
A certain measurement (e.g. T cell count or expression level of a marker gene) to make a risk estimate for the response of a patient to therapy.
Acknowledgements
We would like to thank Yaniv Lubling and Meike Logtenberg for input and insightful discussions. This work was supported by ERC AdG SENSIT to T.N.S. and the Dutch Cancer Society Bas Mulder Award to D.S.T.
Footnotes
Author contributions
A.M.v.d.L researched data for the article. A.M.v.d.L, D.S.T., and T.N.S. jointly discussed data and co-wrote the article.
Competing interests
The authors declare no competing interests.
References
- 1.Clemente CG, et al. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77:1303–1310. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 2.Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (80-. ) 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 4.Fridman WH, Pages F, Sauts-Fridman C, Galon J. The immune contexture in human tumours: Impact on clinical outcome. Nature Reviews Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (80-. ) 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 10.Kvistborg P, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med. 2014;6 doi: 10.1126/scitranslmed.3008918. [DOI] [PubMed] [Google Scholar]
- 11.Robert L, et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res. 2014;20:2424–2432. doi: 10.1158/1078-0432.CCR-13-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cha E, et al. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci Transl Med. 2014;6 doi: 10.1126/scitranslmed.3008211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robert L, et al. Distinct immunological mechanisms of CTLA-4 and PD-1 blockade revealed by analyzing TCR usage in blood lymphocytes. Oncoimmunology. 2014;3 doi: 10.4161/onci.29244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnes TA, Amir E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. British Journal of Cancer. 2017;117:451–460. doi: 10.1038/bjc.2017.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg SA, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quezada SA, et al. Tumor-reactive CD4 + T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–650. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tran E, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (80-. ) 2014;344:641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alspach E, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574 doi: 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tirosh I, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science (80-. ) 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell. 2019;176:775–789.e18. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sade-Feldman M, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 2018;175:998–1013.e20. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gattinoni L, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baitsch L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thommen DS, et al. A transcriptionally and functionally distinct PD-1 + CD8 + T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nature Medicine. 2018;24:1–11. doi: 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by PD-L1 blockade. Proc Natl Acad Sci. 2008;105:15016–15021. doi: 10.1073/pnas.0801497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng C, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169:1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 29.Guo X, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 30.Yost KE, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25:1251–1259. doi: 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–272. doi: 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 32.Azizi E, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell. 2018;174:1293–1308.e36. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke J, et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med. 2019;216:2128–2149. doi: 10.1084/jem.20190249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Savas P, et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med. 2018;24:986–993. doi: 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 35.Fehlings M, et al. Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8+ T cells. Nat Commun. 2017;8 doi: 10.1038/s41467-017-00627-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chevrier S, et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell. 2017;169:736–749.e18. doi: 10.1016/j.cell.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giraldo NA, et al. Tumor-infiltrating and peripheral blood T-cell immunophenotypes predict early relapse in localized clear cell renal cell carcinoma. Clin Cancer Res. 2017;23:4416–4428. doi: 10.1158/1078-0432.CCR-16-2848. [DOI] [PubMed] [Google Scholar]
- 38.Wagner J, et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell. 2019;177:1330–1345.e18. doi: 10.1016/j.cell.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salerno F, Wolkers MC. T-cells require post-transcriptional regulation for accurate immune responses. Biochemical Society Transactions. 2015 doi: 10.1042/BST20150154. [DOI] [PubMed] [Google Scholar]
- 40.Salerno F, et al. Critical role of post-transcriptional regulation for IFN-γ in tumor-infiltrating T cells. Oncoimmunology. 2019;8 doi: 10.1080/2162402X.2018.1532762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dieu-Nosjean MC, et al. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev. 2016;271:260–275. doi: 10.1111/imr.12405. [DOI] [PubMed] [Google Scholar]
- 42.Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307–325. doi: 10.1038/s41568-019-0144-6. [DOI] [PubMed] [Google Scholar]
- 43.Kunz DJ, Gomes T, James KR. Immune cell dynamics unfolded by single-cell technologies. Frontiers in Immunology. 2018;9 doi: 10.3389/fimmu.2018.01435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller BC, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20:326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lanitis E, Dangaj D, Irving M, Coukos G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Annals of Oncology. 2017;28:xii18–xii32. doi: 10.1093/annonc/mdx238. [DOI] [PubMed] [Google Scholar]
- 46.Scheper W, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med. 2019;25:89–94. doi: 10.1038/s41591-018-0266-5. [DOI] [PubMed] [Google Scholar]
- 47.Simoni Y, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 48.Erkes DA, et al. Virus-Specific CD8 + T Cells Infiltrate Melanoma Lesions and Retain Function Independently of PD-1 Expression. J Immunol. 2017;198:2979–2988. doi: 10.4049/jimmunol.1601064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kvistborg P, et al. TIL therapy broadens the tumor-reactive CD8+ T cell compartment in melanoma patients. Oncoimmunology. 2012;1:409–418. doi: 10.4161/onci.18851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosato PC, et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun. 2019;10 doi: 10.1038/s41467-019-08534-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pasetto A, et al. Tumor- and Neoantigen-Reactive T-cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol Res. 2016;4:734–743. doi: 10.1158/2326-6066.CIR-16-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schietinger A, et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity. 2016;45:389–401. doi: 10.1016/j.immuni.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahmadzadeh M, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gros A, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inozume T, et al. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother. 2010;33:956–964. doi: 10.1097/CJI.0b013e3181fad2b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duhen T, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. 2018;9 doi: 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gubin MM, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singer M, et al. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell. 2016;166:1500–1511.e9. doi: 10.1016/j.cell.2016.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gubin MM, et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell. 2018;175:1014–1030.e19. doi: 10.1016/j.cell.2018.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martínez-Usatorre A, Donda A, Zehn D, Romero P. PD-1 Blockade Unleashes Effector Potential of Both High- and Low-Affinity Tumor-Infiltrating T Cells. J Immunol. 2018;201:792–803. doi: 10.4049/jimmunol.1701644. [DOI] [PubMed] [Google Scholar]
- 61.Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nature Reviews Immunology. 2018;18:635–647. doi: 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- 62.Siddiqui I, et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity. 2019;50:195–211.e10. doi: 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 63.Kurtulus S, et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1-CD8+ Tumor-Infiltrating T Cells. Immunity. 2019;50:181–194.e6. doi: 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Utzschneider DT, et al. T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity. 2016;45:415–427. doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 65.Im SJ, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu T, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol. 2016;1 doi: 10.1126/sciimmunol.aai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spitzer MH, et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell. 2017;168:487–502.e15. doi: 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brummelman J, et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J Exp Med. 2018;215:2520–2535. doi: 10.1084/jem.20180684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.LaFleur MW, et al. PTPN2 regulates the generation of exhausted CD8+ T cell subpopulations and restrains tumor immunity. Nat Immunol. 2019;20:1335–1347. doi: 10.1038/s41590-019-0480-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei SC, et al. Negative Co-stimulation Constrains T Cell Differentiation by Imposing Boundaries on Possible Cell States. Immunity. 2019;50:1084–1098.e10. doi: 10.1016/j.immuni.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Daud AI, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest. 2016;126:3447–3452. doi: 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gide TN, et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell. 2019;35:238–255.e6. doi: 10.1016/j.ccell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 73.Jiao S, et al. Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell. 2019;179:1177–1190.e13. doi: 10.1016/j.cell.2019.10.029. [DOI] [PubMed] [Google Scholar]
- 74.Kim HJ, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res. 2014;2:91–98. doi: 10.1158/2326-6066.CIR-13-0216. [DOI] [PubMed] [Google Scholar]
- 75.Ahrends T, Borst J. The opposing roles of CD4+ T cells in anti-tumour immunity. Immunology. 2018;154:582–592. doi: 10.1111/imm.12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Braumuller H, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494:361–365. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- 77.Qin L, et al. Insights into the molecular mechanisms of T follicular helper-mediated immunity and pathology. Frontiers in Immunology. 2018;9 doi: 10.3389/fimmu.2018.01884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity. 2019;50:1132–1148. doi: 10.1016/j.immuni.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lavin Y, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell. 2017;169:750–765.e17. doi: 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Giordano M, et al. Molecular profiling of CD 8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. EMBO J. 2015;34:2042–2058. doi: 10.15252/embj.201490786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li J, He Y, Hao J, Ni L, Dong C. High Levels of Eomes Promote Exhaustion of Anti-tumor CD8 + T Cells. Front Immunol. 2018;9:2981. doi: 10.3389/fimmu.2018.02981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chihara N, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature. 2018;558:454–459. doi: 10.1038/s41586-018-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scott AC, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571:270–274. doi: 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Paley MA, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science (80-. ) 2012;338:1220–1225. doi: 10.1126/science.1229620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alfei F, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571:265–269. doi: 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- 86.Khan O, et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature. 2019;571:211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blank CU, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol. 2019;19:665–674. doi: 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stuart T, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Welch JD, et al. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell. 2019;177:1873–1887.e17. doi: 10.1016/j.cell.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stoeckius M, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang C, Singer M, Anderson AC. Molecular Dissection of CD8+ T-Cell Dysfunction. Trends in Immunology. 2017;38:567–576. doi: 10.1016/j.it.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Philip M, Schietinger A. Heterogeneity and fate choice: T cell exhaustion in cancer and chronic infections. Current Opinion in Immunology. 2019;58:98–103. doi: 10.1016/j.coi.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Highlighted references
- Azizi et al., Cell (2018): Single cell transcriptome analysis of immune cells in human breast cancer demonstrating that intratumoral T cells reside along a continuum that is driven by activation and terminal differentiation. In addition, cell state diversity was shown both between and within T cell clones.
- Clarke et al., J. Exp. Med. (2019): Single cell transcriptome analysis of T cells in human lung cancer describing a PD1 and TIM3 expressing TRM cell subset that contains a highly proliferative subset and is enriched in lesions of patients responding to anti-PD1 therapy.
- Duhen et al., Nature Communications (2018): This study identifies CD103 and CD39 as markers of tumor-reactive T cells across multiple human cancer types.
- Guo et al., Nature Medicine (2018): Single cell transcriptome analysis of T cells in NSCLC, adjacent normal tissue, and blood showing the distribution of T cell states in these tissues and their relatedness based on TCR sharing between cell states in and outside the tumor. Moreover, this study provides evidence for dysfunctionality as a gradual state.
- Kurtulus et al., Immunity (2019): A mouse study that describes a PD1 low subset of T cells that respond to ICB and identifies Tcf7 expression to be required for response to anti-PD1 and anti-TIM3 combination therapy.
- Li et al., Cell (2019): Single cell transcriptome and TCR analysis in human melanoma describing a dysfunctional axis made up of cells that are transcriptionally related and show TCR sharing, as well as a separate cytotoxic population. The dysfunctional T cells identified contain a highly proliferative subset and are indicative of the presence of a tumor-reactive T cell repertoire.
- Miller et al., Nature Immunology (2019): A mouse melanoma study showing the importance of a low dysfunctional (‘progenitor exhausted’) T cell population marked by TCF1 expression and required for response to anti-PD1 therapy.
- Sade-Feldman et al., Cell (2018): Single cell transcriptome analysis in human melanoma indicating that TCF7-expressing T cells in human cancer are associated with response to ICB. Furthermore, the study shows that CD39- and TIM3-negative T cells display reactivity against mouse colon carcinoma cells in the presence of anti-PD1.
- Savas et al., Nature Medicine (2018): Single cell transcriptome analysis of T cells in two human breast cancer samples characterizing TRM cells that, amongst others, express inhibitory receptor genes such as PDCD1 and CTLA4.
- Scheper et al., Nature Medicine (2019): Unbiased analysis of the tumor-reactivity of TCRs in human cancer lesions providing formal evidence that human colorectal and ovarian cancers contain high proportions of bystander T cells.
- Siddiqui et al., Immunity (2019): A mouse study providing evidence for the role of TCF1-expressing PD1-positive CD8+ T cells in tumor control upon anti-PD1 and anti-CTLA4 combination therapy.
- Simoni et al., Nature (2018): This study demonstrates the presence of virus-specific bystander T cells in human lung and colorectal tumors. Intratumoral bystander T cells were characterized by a lack of CD39 expression, suggesting that CD39 can be used to help distinguish tumor-reactive T cells from bystander cells.
- Thommen et al., Nature Medicine (2018): This study defines a T cell subset in human NSCLC with high expression of PD1 that is enriched for tumor-reactivity, expresses markers of T cell activation, and that appears predictive for clinical response to anti-PD1 therapy.
- Tirosh et al., Science (2017): The first single cell transcriptome and TCR analysis of the TME in human melanoma, dissecting the major T cell states and tumor-specific programs.
- Zhang et al., Nature (2018): Single cell transcriptome and TCR analysis of T cells in microsatellite stable and instable colorectal tumors addressing the clonotypic relationships between intratumoral dysfunctional cells and T cells with other cell states that reside in the tumor, normal tissue, or blood.
- Zheng et al., Cell (2017): Single cell transcriptome and TCR analysis of T cells in HCC showing the distribution and connectivity between T cell states in tumor tissue, adjacent normal tissue and blood, and that also provides evidence for the presence of cell state diversity within T cell clones.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.