

Abstract
Acetaldehyde is a highly reactive, DNA damaging metabolite, produced upon alcohol consumption1. Impaired acetaldehyde detoxification is common in the Asian population, and is associated with alcohol related cancers1,2. Cellular protection against acetaldehyde-induced damage is provided by DNA crosslink repair; when impaired this causes Fanconi anaemia (FA), a disease resulting in failed blood production and cancer predisposition3,4. Strikingly, combined inactivation of acetaldehyde detoxification and the FA pathway induces mutation, accelerates malignancies and causes the rapid attrition of blood stem cells5–7. A key question concerns the nature of DNA damage caused by acetaldehyde, and how this is repaired. Here we generate acetaldehyde-induced DNA interstrand crosslinks (AA-ICLs) and determine their repair mechanism in Xenopus egg extract. We discover that two replication-coupled pathways repair these lesions. The first is the FA pathway, that operates using excision, analogous to the mechanism used for chemotherapeutic crosslinks caused by cisplatin. Yet, this AA-ICL repair results in elevated mutation frequency and altered mutational spectrum. The second repair modality requires replication fork convergence but unexpectedly does not involve DNA incisions, instead the acetaldehyde-crosslink itself is broken. The Y-family DNA polymerase REV1 completes repair, culminating in a distinct mutation spectrum. This work defines how DNA interstrand crosslinks caused by an endogenous and alcohol-derived metabolite are repaired, identifying an excision-independent mechanism.
To study the repair of alcohol-induced DNA damage, we generated an acetaldehyde-crosslinked DNA substrate. Acetaldehyde reacts with guanine creating a crosslink precursor, N2-propanoguanine (PdG) (Fig. 1a)8. In a 5’-CpG sequence, PdG reacts with the N2-amine of guanine on the opposite strand to create an interstrand acetaldehyde crosslink (AA-ICL). The crosslink exists in equilibrium between three states. We synthesized a site-specific native AAnat-ICL within an oligonucleotide duplex (Extended Data Fig. 1a, b, d, Supplementary Information Fig. 1). A control reaction of PdG with deoxyinosine (dIno), lacking an N2-amine, did not crosslink, confirming AAnat -ICL site-specificity (Extended Data Fig. 1c, for gel source data see Supplementary Information Fig. 2). AAnat -ICLs were stable at physiological pH and temperature (< 10% reversal after 72 h at 37 °C) (Extended Data Fig. 1e). Elevated temperature (55 °C) or acid did however reverse AAnat-ICL, consistent with Schiff base hydrolysis and dG N2-amine protonation (Extended Data Fig. 1e, f)10. DNA crosslink repair is conserved among vertebrates and comprehensively studied in Xenopus egg extract11. To examine AAnat-ICL repair using this system, the oligonucleotide was ligated into a plasmid (pICL-AAnat). We also generated cisplatin ICL (pICL-Pt), PdG (pPdG) and unmodified (pCon) control plasmids (Extended Data Figs. 1g, h). Crosslinked vectors were stable in non-replicating Xenopus egg extract (Extended Data Fig. 1i).
Fig. 1. AA-ICL repair by Fanconi-dependent and independent mechanisms.

(a) Reaction scheme of AAnat-ICL formation. Two acetaldehydes react with deoxyguanine (dG) generating N2-deoxypropanoguanine (PdG), this reacts with a 5’-CpG guanine on the opposite strand. The AAnat-ICL exists in a three-state equilibrium. (b) Replication intermediates generated during ICL repair. (c) Plasmids were replicated in Xenopus egg extract, reaction products were resolved by native agarose gel and visualized by autoradiography. Figure 8 structures (F8), later replication/repair intermediates (RRI), open circle/supercoiled products (OC/SC, grey arrow) are indicated. Six independent experiments. (d) Scheme for the NotI ICL repair assay. Wavy lines: synthesized during repair. (e) Plasmids were replicated in extract, repair intermediates were isolated, NotI-digested, and resolved by denaturing PAGE. Accumulation of the 44 nt product (open arrow) indicates ongoing replication and repair. (*) Product probably generated from end-joining activity in some extracts. Ten independent experiments. (f) Quantification of repair based on the gels in (e), as described in methods (Supplementary Information Methods). Ten independent experiments. (g) Quantification of repair in mock or FANCD2-depleted extract. Based on gel in Extended Data Fig. 2e. Three independent experiments. (h) Scheme of the reduction of ICL-AAnat to ICL-AARED. (i) Plasmids were replicated in extract and products were resolved by native agarose gel. Three independent experiments. (j) Quantification of repair with or without p97i. Based on gel in Extended Data Fig. 3b. Three independent experiments.
Cisplatin ICLs are repaired by the replication-dependent FA pathway involving ICL unhooking by endonucleases, translesion synthesis (TLS) to bypass the adduct, and homologous recombination (HR) to resolve double strand breaks (DSBs) (Fig. 1b and Extended Data Fig. 2a)12–14. Replication of pICL-Pt in Xenopus egg extract generates a temporal pattern of repair intermediates, starting with converged forks (‘figure 8’ structure), then low-mobility products that include HR intermediates (termed replication/repair intermediates (RRI)) and resolved nicked and supercoiled products (OC and SC, respectively) (Figs. 1b, c)14. Since the structures of cisplatin and acetaldehyde crosslinks differ substantially (Extended Data Fig. 1j), we asked whether they were repaired by similar mechanisms. We replicated pICL-AAnat and pICL-Pt in Xenopus egg extract, along with non-crosslinked controls, and separated the products by electrophoresis (Fig. 1c). OC/SC products accumulated rapidly for pCon and pPdG, indicating no or little impediment to replication. Replication of pICL-AAnat resulted in RRI products similar to pICL-Pt, but earlier accumulation of OC/SC products, suggesting some pICL-AAnat was repaired quickly.
We developed an assay ('NotI assay') to determine if OC/SC products observed in Fig. 1c were indeed AAnat-ICL repair products. Repair intermediates were NotI digested, 3’ end-labelled, and resolved by denaturing PAGE (Fig. 1d). Before DNA replication (t=0), this resulted in fragments of 88 nt (2 x 44 nt, crosslinked), 44 nt (low-level background of non-crosslinked plasmids), and the unresolved vector backbone. For pICL-AAnat and pICL-Pt the 44 nt fragment increased over time, confirming ICL repair (Fig. 1e). We quantified repair products and found greater accumulation for pICL-AAnat compared with pICL-Pt (~30% cf. ~20% at 180 min) and importantly, a faster rate of repair (e.g.11% cf. 1% at 50 min) (Fig. 1f and Extended Data Fig. 1k, l). These data indicate that a proportion of pICL-AAnat was processed similar to pICL-Pt, yet pICL-AAnat was additionally repaired by a second, faster mechanism.
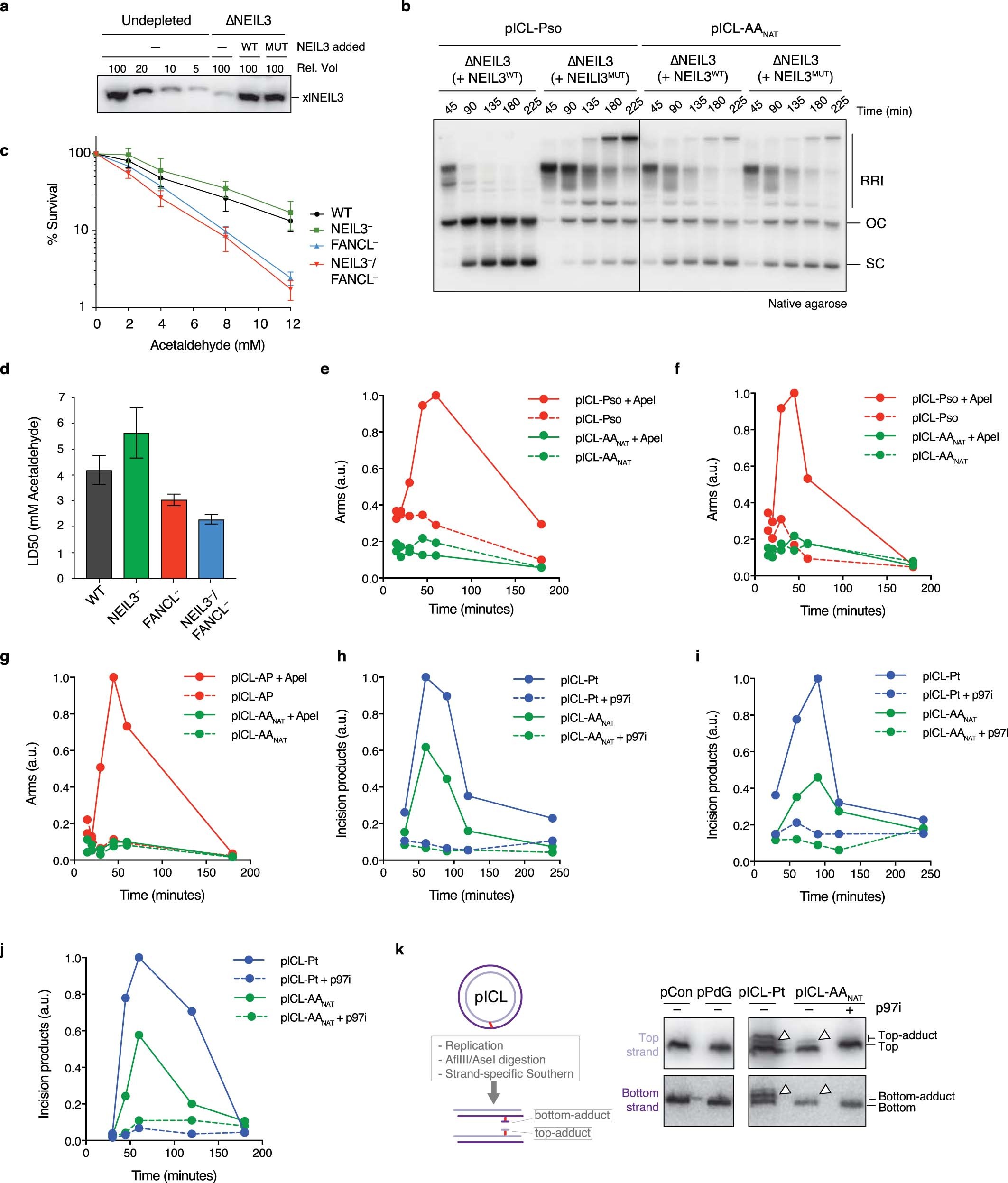
Cisplatin ICLs are repaired by the FA pathway (Extended Data Fig. 2a), we therefore examined if this also repaired AAnat-ICLs. In egg extract, pICL-Pt and pICL-AAnat stimulated monoubiquitination of FANCD2, the activation step of the FA pathway (Extended Data Fig. 2b). FANCD2-depletion causes a defect in cisplatin ICL unhooking, which results in the persistence of nascent leading strands one nucleotide before the ICL (-1 position, Extended Data Fig. 2a)12. To visualize this, we replicated plasmids in mock and FANCD2-depleted extract, digested intermediates, and separated products on a sequencing gel (Extended Data Fig. 2,d). For both crosslinks, FANCD2-depletion caused persisting -1 products (Extended Data Fig. 2d, white arrow), and fewer extension products, indicating repair of pICL-Pt and pICL-AAnat involves the FA pathway. We questioned whether the second route of AAnat-ICL repair required the FA pathway. In NotI assays, FANCD2-depleted extracts did not support pICL-Pt repair, yet pICL-AAnat was still partially repaired (Fig. 1g, Extended Data Fig. 2e-h). FA-dependent pICL-Pt repair requires CMG helicase unloading by the p97 segragase15. Consistently, a p97 segregase inhibitor (referred to as p97i) halted pICL-Pt repair (Extended Data Fig. 2i-m). In contrast, p97i only blocked a proportion of AAnat-ICL repair, while the faster route of repair was unaffected by this treatment (Extended Data Fig. 2i-m). Together, these results indicate that pICL-AAnat repair proceeds through an FA-dependent and -independent mechanism.
Reduction AAnat-ICL by sodium cyanoborohydride results in a single, stable form of the acetaldehyde crosslink (AARED-ICL) as confirmed by HPLC (not shown), hydrolysis resistance and MALDI (Fig. 1h, Extended Data Fig. 1f and 3a). Replication of a plasmid containing a AARED-ICL (pICL-AARED) in egg extract, yielded RRI’s resembling the FA pathway, and OC/SC products hardly accumulated (Fig. 1i). The NotI assay revealed that pICL-AARED repair was slower than pICL- AAnat and was abolished by p97i (Fig. 1j, Extended Data Fig. 3b-d). The reduced AA-ICL is therefore repaired exclusively by the FA pathway, indicating that the alternate repair route is restricted to the native AA-ICL.
To further characterize the faster second AAnat-ICL repair route we first asked if it was replication-dependent. Addition of recombinant geminin (inhibiting DNA replication) blocked all repair of both pICL-Pt and pICL-AAnat (Fig. 2a, Extended Data Fig. 4a-c). Two replication forks must converge for pICL-Pt repair, promoting CMG helicase ubiquitination and unloading. Although CMG unloading is not required for the second route of AAnat-ICL repair, we wondered whether fork convergence was. We generated an AAnat-ICL plasmid containing a LacO array that, when bound by Lac repressor (LacR), blocks the replication fork16,17. Since the AAnat-ICL is non-symmetrical we generated two versions of this plasmid, with the ICL in either orientation, with respect to the rightward fork (pICL-AA-LacO and pICL-AAreverse-LacO respectively, Fig. 2b). We replicated these plasmids, and separated the digested repair intermediates on a sequencing gel (Fig 2b and c). In the absence of LacR, both leftward and rightward leading strands arrived at the –20 position, approached the -1 position, and were extended past the lesion over time. In the presence of LacR, arrival of the leftward fork was inhibited (indicated by the absence of –1 and –20 products), showing the LacR block was functional. The rightward fork persisted at –20, suggesting a failure in repair progression. Moreover, extension products were impeded upon incubation with LacR and this was similar for both orientations of AA-ICLnat (Fig. 2c), and pICL-Pt-LacO (Extended Data Fig. 4d). This indicates that AAnat-ICL unhooking requires replication fork convergence. Consistent with this, in the presence of LacR, repair of pICL-AA-LacO and pICL-AAreverse-LacO was greatly reduced (Extended Data Fig. 4e-h). These data show that the collision of one replication fork with an AAnat-ICL is insufficient for repair. While it is plausible that the FA-independent AAnat-ICL unhooking is a consequence of two forks colliding, we believe the mechanism is more probably enzymatic, like all known ICL repair pathways.
Fig. 2. The alternate AA-ICL repair route requires DNA replication and fork convergence but no DNA excision.

(a) Quantification of repair with or without Geminin. Based on gel in Extended Data Fig. 4a. Three independent experiments. (b) Scheme of AA-ICL LacO plasmids. (c) Plasmids were replicated in extract with or without LacR. Repair intermediates were digested and separated on a sequencing gel. Grey arrows: –1 product, open arrows: –20 product. Two independent experiments. (d) Scheme of the assays used to detect base excision (left) and nucleotide excision (right) pathways in (e) and (f), respectively. DNA fragments formed after HincII/APE1 (left box) or HincII digestion (top and right box). (e) Plasmids were replicated in extract with p97i. Repair intermediates were digested and separated by native agarose gel. Red arrows: Arms from APE1 incisions. Quantification in Extended Data Fig. 5e, replicates in Extended Data Fig. 5f, g. Four independent experiments. (f) Plasmids were replicated in extract with or without p97i. Repair intermediates were digested and separated by native agarose gel. White arrows: Arms from backbone incisions. Quantification in Extended Data Fig 5h, replicates in Extended Data Fig. 5i, j. Six independent experiments.
Whilst cisplatin crosslinks are unhooked by nucleolytic incisions, psoralen and abasic site ICLs are preferentially cleaved at a glycosidic bond by NEIL3 glycosylase12,15,18. To test a role for NEIL3 in AAnat-ICL repair, we complemented NEIL3-depleted extract with recombinant wild type or catalytically inactive NEIL3 (Extended Data Fig. 5a). Unlike psoralen-ICL plasmid (pICL-pso)15, pICL-AAnat repair intermediates were unaffected by lack of NEIL3 activity (Extended Data Fig. 5b). Furthermore, human ΔNEIL3 HAP1 cells were not acetaldehyde-hypersensitive and NEIL3 deficiency did not further sensitise a FANCL-deficient strain (Extended Data Figs. 5c, d). It is plausible another glycosylase unhooks the AAnat-ICL, so we examined the accumulation of abasic sites, the product of glycosylase cleavage. Recombinant APE1 cleaves abasic sites resulting in the generation of arm fragments upon linearization of replication intermediates. During pICL-Pso repair such arms were generated, but they did not form during AAnat-ICL repair (route 1, Figs. 2d, e, Extended Data Fig. 5e-g)15. This indicates that no abasic site is formed and that AAnat-ICL repair does not cut the N-glycosyl bond.
Next, we tested whether nucleotide excision products are formed when an AAnat-ICL is processed in Xenopus egg extract. Backbone incisions should generate arm fragments when repair intermediates are linearized, as shown for repair of pICL-Pt (route 2, Fig. 2d, f). These arms were also formed during pICL-AAnat repair, consistent with FA pathway activity. Strikingly, when p97i was added (blocking the FA repair route) no incision products formed, indicating that the second ICL-AAnat repair pathway does not create backbone incisions (Fig. 2f and Extended Data Fig. 5h-j). To further confirm this, we examined ‘adduct’ formation. In the FA pathway, incisions of one strand results in an unhooked adduct on the other (Extended Data Fig. 5k, left). Therefore, late repair intermediates of pICL-Pt contain adducts on the top or the bottom strand as either can be incised. Repair of pICL-AAnat also generated adducts, largely restricted to the top stand - possibly adducts on the bottom strand were below the detection limit or were processed in extract (Extended Data Fig. 5k, right). However, after p97-inhibition no adducts were detected, indicating they depend on the FA pathway. In conclusion, the second faster repair route for AAnat-ICLs does not involve a DNA excision step. This route must therefore operate by cutting within the crosslink itself.
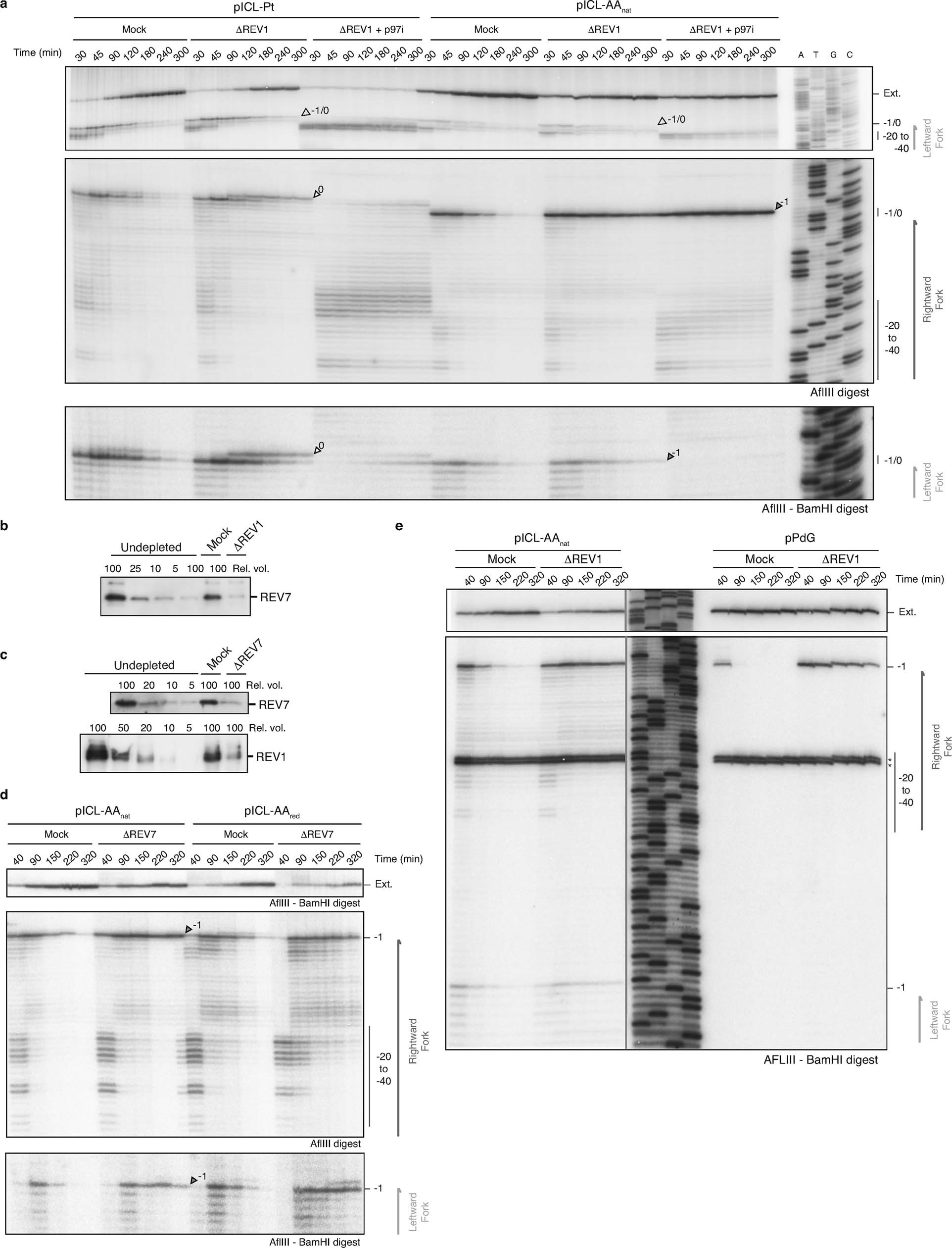
Such a repair pathway would create an adduct on one or both strands and should require TLS for nucleotide insertion opposite the adduct and extension beyond it. The TLS factor REV1 is critical for pICL-Pt repair so we tested if this also operates in AAnat-ICL repair. Plasmids were replicated in mock and REV1-depleted extracts, and digested intermediates were analyzed on a sequencing gel (Fig. 3a-c). For pICL-Pt, REV 1-depletion caused the accumulation of insertion products (0-products), and reduced extension products (Fig. 3c)19. In contrast, for pICL-AAnat, REV1-depletion caused –1 product accumulation, indicating REV 1-mediated insertion opposite the adduct (Fig. 3c and Extended Data Fig. 6a). Notably, this was also true for pICL-AARED repair, indicating different TLS mechanisms within the FA pathway. Furthermore, REV1-depletion caused more extensive leading strand stalling at the rightward than the leftward fork for AA-ICLnat (Fig. 3c). This suggests that unhooking by the second pathway may create an adduct on the bottom strand, which is bypassed by REV1. To test this, we inhibited the FA pathway using p97i and examined lesion bypass of the second pathway of AAnat-ICL repair. As expected, pICL-Pt repair in the presence of p97i caused persistent stalling at the –20 to –40 position, due to defective CMG unloading (Extended Data Fig. 6a and Semlow et al.15). Similar stalled products were observed for AAnat-ICL repair, indicating that FA pathway repair was inhibited (Extended Data Fig. 6a). However, rightward –1 products accumulated, indicating REV1-depletion prevents lesion bypass by the second pathway. In contrast, the leftward fork was extended without hindrance as no stalled products accumulated and significant extension products were formed (Extended Data Fig. 6a). Depletion of REV7 (the regulatory subunit of pol ζ encoded by FANCV), had a very similar effect on AA-ICL repair to REV1-depletion (Extended Data Fig. 6d). It also caused an insertion defect in the the FA pathway during AARED-ICL repair, and persistent stalling, especially of the rightward fork, in AAnat-ICL repair. As pol ζ is most known for its extension activity, this defect could be due to co-depletion of the REV1-polζ complex (Budzowska et al. and Extended Data Fig. 6b, c). In summary, the second pathway of AAnat-ICL repair generates an adduct on the bottom strand requiring REV1 and polζ for bypass. The top strand however is readily extended without these TLS factors.
Fig. 3. AA-ICL repair is mediated by REV1.

(a) Scheme of products detected on the sequencing gel in (c). (b) REV1 Western blot for mock and REV1-depleted extract compared to a titration of undepleted extract. Nine independent experiments. (c) Plasmids were replicated in mock or REV1-depleted (ΔREV1) extracts, repair intermediates were digested and separated on a sequencing gel alongside a sequencing ladder. White arrows: 0 products, dark grey arrows: –1 products, light grey arrows: 0/-1 products. Three independent experiments. (d) Scheme of the primer extension assay. Late repair products are digested with AflIII and BamHI and used as template for primer extension from labelled TOP or BOT (bottom) primers. (e) Primer extension products were separated by denaturing PAGE. Four independent experiments.
To further examine adduct formation we isolated late repair products and subjected them to primer extension reactions at the ICL region using a high-fidelity polymerase (Fig. 3d). As expected, pICL-Pt repair products (forming adducts on either parental strand) caused stalled extension products on both strands (Fig. 3e). For AAnat-ICL, stalling also occurred on both strands but was more extensive on the bottom strand. Moreover, p97i treatment (blocking the FA pathway) almost entirely eliminated stalling on the top strand (Fig. 3e). These results suggest that the second repair pathway regenerates dG on the top strand, but creates a dG-adduct on the bottom strand.
Finally, we examined AAnat-ICL repair fidelity. To achieve this, we replicated plasmids in egg extract, recovered late repair products, and subjected them to high-throughput sequencing (Figs. 4a, b). Consistent with a previous report19, we confirmed pICL-Pt is repaired with a low error rate - less than 2% of the products carried a mutation at sites corresponding to crosslinked-guanines (Fig. 4b, Extended Data Fig. 7a). The most common substitution was G>T transversion. Repaired pICL-AAnat products show two differences: firstly, ~10% carry mutations at the crosslinked sites, and secondly, the mutational spectrum differs (C>G, C>A, G>C and G>T transversions) (Fig. 4b, Extended Data Fig. 7a-c). The frequency of consecutive mutations (≥ 2 nt) around the ICL was ~100-fold lower than single mutations, in agreement with a report suggesting ICLs do not drive tandem mutations caused by acetaldehyde21. AAnat-ICL repair products generated in presence of p97i (blocking the FA route) lost most mutations at position G8. This indicates that FA-dependent bypass of the top strand adduct is the predominant source of mutation at G8. Under these conditions mutations are almost entirely restricted to C7 and the spectrum is almost identical to that obtained from pPdG repair (Fig. 4b and Extended Data Fig. 7b). These data strongly suggest that the second route of AAnat-ICL repair reverses this crosslink to yield a monoadduct similar or identical to the original propanoguanine. Consistent with this model, we found that TLS past a PdG adduct is mediated by REV1 (Extended Data Fig. 6e).
Fig. 4. AA-ICL repair causes point mutations.

(a) Scheme of the positions of DNA lesions and adducts (red) generated during repair. The FA pathway generates an adduct on the top or bottom strand, the second pathway for AA-ICL repair creates an adduct on one or both strands. The PdG monoadduct is present on the bottom strand. Mutations are generated during translesion synthesis (blue N’s and arrow). (b) Distribution and frequency of nucleotide mis-incorporation in a 15 bp region flanking the lesions. Strand specificity is lost because sample preparation involves PCR amplification, therefore, only the top sequence is indicated below the graphs. Mutation frequency is minus baseline mutations found in pCon.
In summary, we determine the repair of an important class of endogenous DNA crosslinks, caused by acetaldehyde. The central role for the FA repair pathway in removing such crosslinks agrees with the strong genetic evidence underpinning two-tier protection against this aldehyde3,6,7. However, we unexpectedly uncover a new DNA crosslink repair pathway which removes crosslinks without excision repair. This mechanism requires replication fork convergence and uniquely unhooks the ICL by cutting within the crosslink itself. Repair of AAnat-ICLs by both pathways is error prone and requires the TLS polymerases REV1 and polζ. However, this new modality of repair has an obvious advantage since it avoids the creation of DNA strand breaks or abasic sites, that can promote large-scale genome instability.
Extended Data
Extended Data Figure 1. Acetaldehyde ICLs are stable and repaired in Xenopus egg extract.

(a) Scheme for the synthesis of the precursor, 4-(R)-Aminopentane-1,2-diol. (b) Site-specific synthesis of an (R)-α-CH3-γ-OH-1,N2-Propano-2’-deoxygunosine (PdG) adduct in a DNA oligonucleotide. (c) Denaturing PAGE showing crosslink formation between dG and PdG, but not between dG and Inosine (Ino), which lacks an N2 amine. Two independent experiments. (d) Confirmation of AA-ICL formation by MALDI. The peak at 12,370.2 represents imine or pyrimidopurinone form. Two further peaks at 5979.41 and 6409.74 Da, equate to masses for the two parent oligos, consistent with the mass of the carbinolamine form after dissociation back to PdG and dG upon desorption/ionization conditions. Three independent experiments. (e) Stability of AANAT-ICL as a function of temperature and time, as determined by radiolabelling (α-32P-dCTP) and resolution by denaturing PAGE. Error Bars represent S.E.M., from three independent experiments. (f) The AANAT-ICL is susceptible to hydrolysis with aqueous acid, whereas AARED-ICL is stable. Pre-purification crosslink reactions were incubated with or without formic acid and products were resolved by denaturing PAGE. Three independent experiments. (g) Scheme depicting the type and position of the DNA lesions used in this study. Duplex DNA with or without the indicated lesion was annealed into a backbone vector to generate a circular plasmid with lesion. (h) To determine the percentage of crosslinks the ICL-containing plasmids were digested with NotI, 3’ labelled by end filling with α-32P-dCTP, and separated by denaturing PAGE. Crosslinked DNA (88 nt) will show slower mobility compared to non-crosslinked DNA (44 nt). The crosslink percentage was calculated by comparing the 88 nt with the 44 nt products. Two independent experiments. (i) ICL-AANAT and ICL-Pt are stable in Xenopus egg extract. Plasmids were incubated in a highspeed supernatant (HSS) extract. DNA was extracted and analyzed as described in (h). Three independent experiments. (j) Solution structures of a cisplatin ICL and a reduced form of an acetaldehyde ICL (pdb: 1DDP and 2HMD, cartoon representation generated in PyMol). (k) Indicated plasmids were replicated in Xenopus egg extract, repair intermediates were digested with NotI, 3’ labelled, and resolved by denaturing PAGE. Increase in intensity of the 44 nt band in time indicates ongoing replication and repair. A higher mobility band, probably generated from end-joining activity in some extracts is indicated (*). Independent experimental duplicate of Main Fig. 1e (l) Quantification of repair based on the intensity of 44 nt product on gel in (k), as described in methods (Supplementary Information Methods). Independent experimental duplicate of Main Fig 1f Additional replicates of this experiments are presented in Main Fig. 2a, Extended Data Figs. 2k-m, and Extended Data Fig. 4b.
Extended Data Figure 2. Acetaldehyde ICLs are repaired by Fanconi-dependent and independent mechanisms.

(a) Model for ICL repair by the FA pathway. Upon convergence of two replication forks at the crosslink, the CMG helicase is unloaded from the DNA to allow approach of one replication fork to the -1 position. Ubiquitylation of FANCD2 promotes the recruitment of the XPF-ERCC1-SLX4 (XES) complex to the ICL that enables nucleolytic incisions that unhook the crosslink. This step could be preceded by fork reversal of one of the stalled replication forks22. Incisions generate a broken strand and a strand with an adduct, the latter is bypassed by TLS while the broken strand is repaired by homologous recombination. In mammalian cells, it has been shown that a single fork can pass over the ICL without unhooking23, this ‘traverse’ gives rise to a structure that resembles the one generated after fork convergence and CMG unloading and could follow the same steps subsequently. (b) Indicated plasmids were replicated in Xenopus egg extract, reaction samples were analysed by western blot with FANCD2 antibody. Two independent experiments. (c) FANCD2 Western blot showing a titration of Xenopus egg extract compared to mock and FANCD2 depleted extract. Two independent experiments. (d) Indicated plasmids were replicated in Mock or FANCD2 depleted (ΔFANCD2) extract in the presence of α-32P-dCTP. Repair products were digested by AflIII, separated on a sequencing gel alongside a ladder derived from extension primer S, and visualized by autoradiography. White arrow denotes –1 product that is 2 nt larger in pICL-Pt due to the position of the ICL. Three independent experiments. (e) Indicated plasmids were replicated in Mock or FANCD2 depleted (ΔFANCD2) extract, repair intermediates were digested with NotI, 3’ labelled, and resolved by denaturing PAGE. Quantification of repair based on the intensity of 44 nt product is shown in main Fig. 1g. (f) Independent experimental duplicate of Main Fig. 1g. (g) Independent experimental triplicate of Main Fig. 1g, but only using pICL-AANAT. (h) Plasmid pICL-AANAT was replicated in FANCD2 depleted extract, or FANCD2 depleted extract supplemented with recombinant FANCI-FANCD2 complex (ID). Reaction samples were resolved by native agarose gel, visualised by autoradiography. Replication/repair intermediates (RRI), open circle (OC), and supercoiled (SC) products are indicated. Stalled repair product (grey arrow) is indicated. Two independent experiments. (i) Indicated plasmids were replicated in Xenopus egg extract in presence or absence of p97i and the intermediates were resolved by native agarose gel electrophoresis. Stalled repair products (grey arrow) are indicated. Seven independent experiments. (j) The indicated plasmids were replicated in Xenopus egg extracts in presence or absence of p97i and repair intermediates were digested with NotI, 3’ labelled, and resolved by denaturing PAGE. Increase in intensity of the 44 nt band (white arrow) in time indicates ongoing replication and repair. A higher mobility band, probably generated from end-joining activity in some extracts is indicated (*). (k) Quantification of repair based on the intensity of 44 nt product on gel in (j) as indicated in methods (Supplemetary Information Methods). (l) Quantified independent experimental duplicate of (j). (m) Quantified independent experimental triplicate of (j).
Extended Data Figure 3. Reduced acetaldehyde ICLs are repaired by the Fanconi Pathway.

(a) MALDI data confirms identity and stability of the reduced ICL. Three independent experiments. (b) The indicated plasmids were replicated in Xenopus egg extracts in presence or absence of p97i and the repair intermediates were analysed using the NotI digestion assay. Increase in intensity of the 44 nt band indicates ongoing replication and repair. A band with higher mobility, probably generated from end joining activity in some extracts is indicated (*). Quantification of repair based on this gel is shown in Main Figure 1j. Three independent experiments. (c) Independent experimental duplicate of main figure 1j. (d) Independent experimental triplicate of main figure 1j.
Extended Data Figure 4. AA-ICL repair requires DNA replication and replication fork convergence.

(a) Indicated plasmids were replicated in Xenopus egg extracts in presence or absence of Geminin. Repair intermediates were digested with NotI, 3’ labelled, and resolved by denaturing PAGE.
Quantification of repair based on the intensity of 44 nt product is shown in main Figure 2a. (b) Independent duplicate experiment of main Figure 2a. (c) As in (b). Independent triplicate experiment of main Fig. 2a but only representing pICL-AANAT (d) pICL-Pt-LacO was replicated in Xenopus egg extract containing α-32P-dCTP, in presence or absence of LacR. The repair intermediates were digested by AflIII and EcoRI, separated on a sequencing gel and visualized by autoradiography. Two independent experiments. (e) The indicated plasmids were replicated in extract in presence or absence of LacR. Repair products were digested with NotI, 3’ labelled, and resolved by denaturing PAGE. (f) Quantification of repair based on the intensity of 44 nt product on gel in (e), as described in methods. (g) Independent experimental duplicate of (f). (h) Independent experimental triplicate (f).
Extended Data Figure 5. The alternate route of AA-ICL repair does not involve DNA excision.

(a) NEIL3 Western blot showing a titration of Xenopus egg extract compared to NEIL3 depleted extract and NEIL3 depleted extract supplemented with recombinant wildtype NEIL3 (WT) or catalytically inactive NEIL3 (MUT). Three independent experiments. (b) Indicated plasmids were replicated in NEIL3-depleted extract (ANEIL3) containing α-32P-dCTP, supplemented with wild-type (WT) or catalytically inactive (MUT) NEIL3. Replication intermediates were resolved by native agarose gel electrophoresis and visualised by autoradiography. Three independent experiments. (c) Clonogenic survival of WT, NEIL3, FANCL or NEIL3/FANCL deficient human HAP1 cells after 2 h exposure to acetaldehyde. Three independent experiments. Measure of centre represents the average values, error bars were calculated as S.E.M. (d) The LD50 for the survival of WT and deficient HAP1 cells was calculated by regression analysis of the curves presented in (c). Measure of centre represents the LD50, error bars were calculated as standard error. Three independent experiment. (e) Quantification of the arm fragments resulting from APE1 treatment (AP sites) from the gel in Main Figure 2e. (f) Quantification of the APE1 arms as in (e), from an independent duplicate experiment without p97i addition. (g) Quantification of the APE1 arms as in (e), from an independent triplicate experiment without p97i addition. As a positive control we used a plasmid containing an abasic site-induced interstrand crosslink (pICL-AP) that is also repaired via the glycosylase NEIL3. (h) Quantification of the HincII arm fragments from the gel in Main Figure 2f. (i) Quantification of the HincII arms as in (h), from an independent duplicate experiment. (j) Quantification of the HincII arms as in (h), from an independent triplicate experiment. (k) Schematic representation of the formation of DNA adducts by unhooking incisions during ICL repair (left). These adducts are not removed during ICL repair in Xenopus egg extracts14 and can therefore be visualized. Plasmids were replicated in Xenopus egg extracts in the presence or absence of p97i. Late reaction samples were digested with AflIII and AseI and separated on a sequencing gel. Adducts on either the top or bottom strand (white arrow heads) were detected by strand-specific Southern blotting. Three independent experiments.
Extended Data Figure 6. Both routes of AA-ICL repair are mediated by REV1 and REV7.

(a) Indicated plasmids were replicated in Mock or REV1 depleted (ΔREV1) extracts containing α-32P-dCTP, in the presence or absence of p97i. Reaction samples were digested by either AflIII or AflIII and BamHI, separated on a sequencing gel alongside a ladder derived from extension of primer S, and visualized by autoradiography. White arrows denote 0 products, dark grey arrows indicate -1 products, and light grey arrows indicate 0/-1 products (not separated). Two independent experiments. (b) Western blot detection of REV7 in REV1 (ΔREV1) or Mock depleted Xenopus egg extract. Two independent experiments. (c) Western blot detection of REV7 and REV1 in REV7 (ΔREV7) or Mock depleted Xenopus egg extract. Two independent experiments. (d) Indicated plasmids were replicated in mock or REV7 depleted (ΔREV7) extracts containing α-32P-dCTP. Reaction samples were digested by either AflIII or AflIII and BamHI, separated on a sequencing gel and visualized by autoradiography. Two independent experiments. (e) Indicated plasmids were replicated in mock or REV1 depleted (ΔREV1) extracts containing α-32P-dCTP. Reaction samples were digested by AflIII and BamHI, separated on a denaturing PAGE gel alongside a ladder derived from extension primer S, and visualized by autoradiography. (*) Indicate 121 nt background fragment caused by a second BamHI restriction site in the leftward fork. Two independent experiments.
Extended Data Figure 7. Mutagenic outcome of acetaldehyde crosslink repair.

(a) Frequency of nucleotide misincorporation in a 15 bp region flanking the lesions present in the indicated plasmids. Co-plotted are the mutation frequencies for the same plasmid that has not been replicated in Xenopus egg extract (NR) and for control vector, pCon. Strand specificity is lost because sample preparation involves PCR amplification, therefore, only the top sequence is indicated below the graphs. Related to Fig. 4. (b) Distribution and frequency of nucleotide misincorporations in a 15 bp region flanking the lesions present in the indicated plasmids. Independent duplicate sequencing experiment. The height of the bars represents the mutation frequency minus baseline mutations found in pCon. (c) Frequency of nucleotide misincorporations in a 15 bp region flanking the lesions present in the indicated plasmids (data from the same duplicate sequencing experiment as in b). Co-plotted is the mutation frequency for pCon.
Extended data table 1. Amplicon sequence for experiment 1 and 2. 622.
Extended data table 1 Sequence and read numbers for high-throughput sequencing experiments.
A, Amplicon sequence for sequencing experiments 1 and 2. b, Total and specific read numbers for sequencing experiment 1 (see Fig. 4). c, Total and specific read numbers for sequencing experiment 2 (see extended data Fig. 7).
| Amplified amplicon | |
|---|---|
| Experiment 1 | AGAACCAATGCATGCGGCCGCGAAGACAGCCCTCTTCCGCTCTT CTTTCGTGCGCGGCCGCGATCCGCTGCATTAATGAAT |
| Experiment 2 | CTCGAGCGGAAGTGCAGAACCAATGCATGCGGCCGCGAAGACA GCCCTCTTCCGCTCTTCTTTCGTGCGCG GCCGCGATCCGCTGCA TTAATGAATCGGCCAACGC GCGGGGAGAGGCGGTTTGCGTATT |
Extended data table 2. Total and specific read numbers for sequencing experiment 1 (to support Fig. 4).
| Sample | Total reads | Pair-matched reads | Reads with indels | Reads with substitutions | Perfect match reads |
|---|---|---|---|---|---|
| plCL-Pt | 8886804 | 7928127 | 2649263 | 249138 | 5029726 |
| pICL-AA | 14852974 | 13541980 | 2396503 | 1458825 | 9686652 |
| pPdG | 10122605 | 9331173 | 69747 | 565567 | 8695859 |
| pCon | 10449164 | 9644715 | 6466 | 240998 | 9397251 |
| pICL-AA+p97i | 32164058 | 29539112 | 491796 | 1600097 | 27447219 |
| plCL-Pt-NR | 2571879 | 2385136 | 102534 | 58870 | 2223732 |
| pICL-AA-NR | 7316392 | 6729595 | 170216 | 200250 | 6359129 |
| pPdG-NR | 8857493 | 8137089 | 52886 | 270870 | 7813333 |
Extended data table 3. Total and specific read numbers for sequencing experiment 2 (to support Fig. S4).
| Sample | Total reads | Pair-matched reads | Reads with indels | Reads with substitutions | Perfect match reads |
|---|---|---|---|---|---|
| pICL-AA | 13877258 | 4566626 | 388256 | 538491 | 3639879 |
| pPdG | 15334790 | 6649408 | 32855 | 585454 | 6031099 |
| pCon | 15403988 | 6714207 | 7772 | 171840 | 6534595 |
| pICL-AA + p97i | 15455175 | 5803423 | 75922 | 284148 | 5443353 |
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank G.P. Crossan, J.T.P. Yeeles and members of the Knipscheer and Patel laboratories for critically reading the manuscript. We thank Hubrecht animal caretakers for animal support, J.C. Walter and D.R. Semlow (Harvard Medical School) for providing us with recombinant xlNEIL3 proteins, the AP-ICL and LacO plasmids, and S.Y Peak-Chew, S. Maslen and M. Skehel (MRC-LMB) for mass spectrometry analysis.
Funding
This work was supported by a project grant from the Dutch Cancer Society (KWF HUBR 2015-7736 to P.K.), the Gravitation program CancerGenomiCs.nl from the Netherlands Organisation for Scientific Research (NWO), part of the Oncode Institute, which is partly financed by the Dutch Cancer Society. KS was supported by the Uehara Memorial Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, and the JSPS Postdoctoral Fellowship for Research Abroad. ANK-L was supported by the Wellcome Trust. JS was supported by CRUK. This work was supported by a ERC Starting grant (ERC-STG 678423-EpiID) to JK and KR and a Nederlandse organisatie voor Wetenschappelijk Onderwijs (NWO) veni grant (016.Veni.181.013) to KR.
All animal procedures and experiments were performed in accordance with national animal welfare laws and were reviewed by the Animal Ethics Committee of the Royal Netherlands Academy of Arts and Sciences (KNAW). All animal experiments were conducted under a project license granted by the Central Committee Animal Experimentation (CCD) of the Dutch government and approved by the Hubrecht Institute Animal Welfare Body (IvD), with project license number AVD80100201711044.
Footnotes
Author Contributions
KJP and PK initiated and supervised the study, MRH co-ordinated key aspects of the project. MRH, MM, JS, MP, DMW and JC designed the strategy and synthesised the AA-ICL and PdG adducts. AKL performed cell toxicity data. AB, MW and PK designed biochemical assays in Xenopus egg extracts. AB, KS and MW conducted Xenopus extract assays. KR and JK performed bioinformatic analysis. KJP and PK wrote the manuscript and MH designed and created the figures.
Competing interests
Authors declare no competing interests.
Data availability
All relevant data are available from the authors or are included with this article. Source images are available in Supplementary Information 3.
Code availability
The custom code used for data analysis are available from the corresponding authors on reasonable request.
References
- 1.Brooks PJ, Enoch MA, Goldman D, Li TK, Yokoyama A. The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009;6:258–263. doi: 10.1371/journal.pmed.1000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai CL, et al. Dominance of the Inactive Asian Variant Over Activity and Protein Contents of Mitochondrial Aldehyde Dehydrogenase 2 in Human Liver. Alcoholism-Clinical and Experimental Research. 2014;38:44–50. doi: 10.1111/acer.12215. [DOI] [PubMed] [Google Scholar]
- 3.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–8. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 4.Garaycoechea JI, Patel KJ. Why does the bone marrow fail in Fanconi anemia? Blood. 2014;123:26–34. doi: 10.1182/blood-2013-09-427740. [DOI] [PubMed] [Google Scholar]
- 5.Hira A, et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood. 2013;122:3206–9. doi: 10.1182/blood-2013-06-507962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garaycoechea JI, et al. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489:571–5. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- 7.Garaycoechea JI, et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature. 2018;553:171–177. doi: 10.1038/nature25154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang M, et al. Identification of DNA adducts of acetaldehyde. Chemical Research in Toxicology. 2000;13:1149–1157. doi: 10.1021/tx000118t. [DOI] [PubMed] [Google Scholar]
- 9.Cho YJ, et al. Stereospecific formation of interstrand carbinolamine DNA cross-links by crotonaldehyde- and acetaldehyde-derived alpha-CH3-gamma-OH-1,N-2-propano-2'-deoxyguanosine adducts in the 5 '-CpG-3 ' sequence. Chemical Research in Toxicology. 2006;19:195–208. doi: 10.1021/tx050239z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loudon GM. Organic Chemistry. Oxford University Press; Oxford: 2002. pp. 874–878. [Google Scholar]
- 11.Duxin JP, Walter JC. What is the DNA repair defect underlying Fanconi anemia? Curr Opin Cell Biol. 2015;37:49–60. doi: 10.1016/j.ceb.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knipscheer P, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long DT, Raschle M, Joukov V, Walter JC. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333:84–7. doi: 10.1126/science.1204258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raschle M, et al. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Semlow DR, Zhang JQ, Budzowska M, Drohat AC, Walter JC. Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell. 2016;167:498–511. doi: 10.1016/j.cell.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duxin JP, Dewar JM, Yardimci H, Walter JC. Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell. 2014;159:346–57. doi: 10.1016/j.cell.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang JQ, et al. DNA interstrand cross-link repair requires replication-fork convergence. Nature Structural Molecular Biology. 2015;22:242–247. doi: 10.1038/nsmb.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Douwel DK, et al. XPF-ERCC1 Acts in Unhooking DNA Interstrand Crosslinks in Cooperation with FANCD2 and FANCP/SLX4. Molecular Cell. 2014;54:460–471. doi: 10.1016/j.molcel.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Budzowska M, Graham TGW, Sobeck A, Waga S, Walter JC. Regulation of the Rev1-pol zeta complex during bypass of a DNA interstrand cross-link. Embo Journal. 2015;34:1971–1985. doi: 10.15252/embj.201490878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. Eukaryotic polymerases i and z act sequentially to bypass DNA lesions. Nature. 2000;406:1015–1019. doi: 10.1038/35023030. [DOI] [PubMed] [Google Scholar]
- 21.Matsuda T, Kawanishi M, Yagi T, Matsui S, Takebe H. Specific tandem GG to TT base substitutions induced by acetaldehyde are due to intra-strand crosslinks between adjacent guanine bases. Nucleic Acids Res. 1998;26:1769–74. doi: 10.1093/nar/26.7.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amunugama R, et al. Replication Fork Reversal during DNA Interstrand Crosslink Repair Requires CMG Unloading. Cell Rep. 2018;23:3419–3428. doi: 10.1016/j.celrep.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang J, et al. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell. 2013;52:434–46. doi: 10.1016/j.molcel.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the authors or are included with this article. Source images are available in Supplementary Information 3.
Code availability
The custom code used for data analysis are available from the corresponding authors on reasonable request.