

Abstract
Herein, we present a study on the oxidation of aldehydes to carboxylic acids using three recombinant aldehyde dehydrogenases (ALDHs). The ALDHs were used in purified form with a nicotinamide oxidase (NOx), which recycles the catalytic NAD+ at the expense of dioxygen (air at atmospheric pressure). The reaction was studied also with lyophilised whole cell as well as resting cell biocatalysts for more convenient practical application. The optimised biocatalytic oxidation runs in phosphate buffer at pH 8.5 and at 40 °C. From a set of sixty-one aliphatic, aryl-aliphatic, benzylic, hetero-aromatic and bicyclic aldehydes, fifty were converted with elevated yield (up to >99%). The exceptions were a few ortho-substituted benzaldehydes, bicyclic heteroaromatic aldehydes and 2-phenylpropanal. In all cases, the expected carboxylic acid was shown to be the only product (>99% chemoselectivity). Other oxidisable functionalities within the same molecule (e.g. hydroxyl, alkene, and heteroaromatic nitrogen or sulphur atoms) remained untouched. The reaction was scaled for the oxidation of 5-(hydroxymethyl)furfural (2 g), a bio-based starting material, to afford 5-(hydroxymethyl)furoic acid in 61% isolated yield. The new biocatalytic method avoids the use of toxic or unsafe oxidants, strong acids or bases, or undesired solvents. It shows applicability across a wide range of substrates, and retains perfect chemoselectivity. Alternative oxidisable groups were not converted, and other classical side-reactions (e.g. halogenation of unsaturated functionalities, Dakin-type oxidation) did not occur. In comparison to other established enzymatic methods such as the use of oxidases (where the concomitant oxidation of alcohols and aldehydes is common), ALDHs offer greatly improved selectivity.
Introduction
Traditional methods for the oxidation of aldehydes to carboxylic acids are no longer sustainable. In fact, they require stoichiometric amounts (or more) of chemical reagents such as toxic transition-metal oxidants (e.g. Cr or Ru oxides), or different types of salts (e.g. permanganate, bromine, hypochlorite, chlorite, perborate), or even Ag2O in combination with sodium cyanide.1–13 Furthermore, the use of strong oxidants in excess often produces side-products when multiple functional groups are available for oxidation. A classic example is the concomitant oxidation of an aldehyde group and the halogenation of aliphatic or aromatic alkene groups within the same molecule.9–11
With the goal of improving the environmental footprint for the selective oxidation of aldehydes, several alternative methods have been investigated using various metal salts or metal-complexes in catalytic amounts (e.g. CuCl, AgNO3, Bi2O3, CoCl2, pyridinium chlorochromate, VO(acac)2, Ni(dmp)2, CH3ReO3, Ag2O-IPr, Cu(acac)SIMes), and in the presence of stoichiometric amounts of an oxidant (e.g. tert-BuOOH, H2O2, H5IO6) or dioxygen.14–23 This second generation of methods provided some improvements in terms of chemoselectivity, relatively simple reaction procedures and complementary substrate scope (e.g. depending on the method of choice and related mechanism, substituted aromatic aldehydes bearing either electron donating or electro withdrawing substituents can be converted with high yields). The disadvantages in many cases are that a considerable amount of metal-catalyst is still required along with “ungreen” organic solvents as reaction medium.
Heterogeneous metal catalysts have also been developed for the catalytic oxidation of aldehydes to carboxylic acids at the expense of air, pure dioxygen or tert-BuOOH as oxidising agent. Notable heterogeneous systems include: Au–CeO2 and Au–C catalysts; polyoxometalates of Ni, Co, Mn and Cu; Co-acetylacetonates on organically modified 3-aminopropyl silica; Co(ii)-substituted heteropolyacid supported on silica; Fe(iii)-hybrid polyoxometalate.24–27 These methods are currently limited by either the relatively high loading of the heterogeneous metal-catalyst (typically 5 mol%) or problems related to long-term activity and recyclability or moderate selectivity and substrate scope.
Despite the large variety of available metal-based catalytic protocols, it was already shown more than seventy years ago that H2O2 can oxidise aldehydes to carboxylic acids without the need for a metal catalyst.28 However, the low reaction rate makes the oxidation with H2O2 alone synthetically non-applicable. Reaction rates were significantly enhanced by combining H2O2 with additional organic reagents such as stoichio-metric trifluoroacetic anhydride (i.e. generating 2-hydroperoxy-hexafluoro-2-propanol in situ).29 Unfortunately, even these types of method have disadvantages from a “green” chemistry perspective because of the requirement for halogenated solvents, the huge excess of carbonate salt and, additionally, a complicated reaction procedure. Similar drawbacks apply with other related methods in which H2O2 is used in combination with excess of either urea and formic acid,30 or formic acid alone,31 or monoperoxyphthalate and urea.32 Variations using H2O2 with a catalytic amount of an organic molecule are also known. Benzeneseleninic acid33 or N-methylpyrrolidin-2-one hydrotribromide (10 mol%),34 or quaternary acidic salt (in aqueous/organic biphasic conditions)35 can be used as catalysts with H2O2 for aldehyde oxidation. However, none of the systems based on H2O2 and organic catalyst is of general applicability. For instance, the H2O2/quaternary acid salt method developed by Noyori and coworkers is efficient with straight-chain aliphatic aldehydes and benzaldehydes, but chemo-selectivity is mediocre with α-branched aldehydes.35 Further research to develop a more general metal-free method for the chemoselective oxidation of aldehydes to carboxylic acids led others to consider the use of alternative peroxides such as peroxymonosulphate (Oxone)36 or even N-heterocyclic carbenes.37 On one hand, the oxidation using Oxone requires toxic and high boiling DMF as solvent,38 complicating the isolation of low-boiling aliphatic products. Furthermore, the Oxone oxidation method failed with para-methoxy and hydroxy substituted aromatic aldehydes because the related Dakin-type oxidation products (i.e. phenol esters) were obtained. On the other hand, the N-heterocyclic carbene-catalysed oxidation represents a valuable option for the synthesis of high value α-chiral, α-halo carboxylic acids, but the high loading of the sophisticated catalysts (10–20 mol%) makes it economically unprofitable and unsustainable for a more general oxidation of achiral aldehydes.
Biocatalytic methods for the oxidation of functional groups are attracting interest because of the elevated (and often perfect) chemo- and regio-selectivity, the mild reaction conditions (ambient temperature and atmospheric pressure) in aqueous environment and the possibility to use efficiently molecular oxygen as benign oxidant.39 Interestingly, biocatalytic oxidation of aldehydes to carboxylic acids in organic synthesis has mainly been conducted using flavin or copper-dependent oxidases (EC 1.2.3), and with an emphasis for converting bio-derived feedstock.40–44 Although aldehyde oxidases require only dioxygen to accomplish the oxidation, the reaction is not fully selective as also alcohol moieties are generally oxidised. Thus, aldehyde oxidases (and even some alcohol oxidases) have been applied for the two-step oxidation of alcohols to carboxylic acids.41,42,44 In contrast, synthetic applications of dehydrogenases during the past two decades mainly involved the reversible oxidation/reduction of alcohols to carbonyl compounds.45–48 Aldehyde dehydrogenases (EC 1.2.1), which catalyse the oxidation of aldehydes via the nicotinamide coenzyme (NAD), have been applied only very recently in biocatalysis,49–51 although fundamental biochemical studies date back since early 1980s.52–54
Although marginally investigated in biocatalysis so far, aldehyde dehydrogenases might become the first choice for oxidation of aldehydes to carboxylic acids, if coupled with an efficient system for the recycling of NAD+, because of their exquisite chemoselectivity. Herein, we present a study on the optimisation of the chemoselective biocatalytic oxidation of aldehydes to carboxylic acids catalysed by three recombinant aldehyde dehydrogenases (ALDHs) originated from bovine lens (ALDH-Bov),49,52 Escherichia coli (ALDH-Ec),49,53 and Pseudomonas putida (PP-ALDH).50 For regeneration of the catalytic NAD+ coenzyme, the H2O forming NAD-oxidase from Streptococcus mutans (NOx)55 was applied. The substrate scope of the three ALDHs was elucidated with a wide range of structurally diverse substrates (Scheme 1).
Scheme 1.

Oxidation of aldehydes catalysed by aldehyde dehydrogen-ases (ALDHs) using nicotinamide oxidase (NOx) for coenzyme recycling. The biocatalytic oxidation requires dioxygen from air as ultimate oxidant. No side-product or waste is produced.
Results and discussion
Optimisation of the reaction conditions
We started our study on the three ALDHs in highly purified form (Bov and Ec 5 µM; PP 20 µM) by determining the optimum temperature for the biocatalytic oxidation of two model substrates (20 mM): a linear aldehyde (1a, 2-methyl pentanal) and an aromatic aldehyde (2a, benzaldehyde) in Tris/ HCl buffer (50 mM, pH 9). The type of buffer and pH initially applied was known to be suitable for the biocatalytic reaction from our previous study.49 Nonetheless, the influence of buffer has been assessed thoroughly in this work. For regeneration of the NAD+ coenzyme (0.5 mM), the H2O forming NAD-oxidase from Streptococcus mutans (NOx) was applied (5 µM).55 DTT (100 µg mL–1) was also added in the reaction mixture in order to prevent any possible oxidation of the catalytic cysteine residue of the ALDHs. Indeed, addition of DTT was mandatory for obtaining reproducible results using different batches of the same enzyme (for details, see ESI section 4†). Fig. 1 summarises the results for the temperature study (for details see ESI, Tables S3 and S4†).
Fig. 1.

Temperature profile for the conversion of 1a (panel A to C) and 2a (panel D to F) by ALDH-Bov, ALDH-Ec and PP-ALDH. Experimental conditions: 1 mL final volume in Eppendorf tubes, buffer = 50 mM Tris/HCl pH 9, 180 rpm on orbital shaker, [1a] or [2a] = 20 mM, [NAD+] = 0.5 mM, [ALDH-Bov] = 5 µM for 1a and 10 µM for 2a, [ALDH-Ec] = 5 µM, [PP-ALDH] = 20 µM, [NOx] = 5 µM, [DTT] = 100 µg mL–1. Extraction with EtOAc under acidic conditions using toluene as IS and derivatisation of acids to the methyl esters with (trimethylsilyl)diazomethane. Conversions were measured by GC-FID (DB-1701).
ALDH-Bov converted >90% of 1a to 1b after 2 h reaction time at all three temperatures tested (Fig. 1A). The initial activity, however, was higher at 40 °C. The higher reaction rate at temperatures moderately above room temperature was also observed for the oxidation of 2a. In fact, the analytical yield of 2a into 2b already exceeded 90% after 90 min at 40 °C and 50 °C. In contrast, only 33% analytical yield into 2b was obtained after 90 min at 30 °C, and 4 h were required to reach a maximum of 88% (Fig. 1D). ALDH-Ec also operated well at 30 °C and 40 °C, having a slight better catalytic performance at 40 °C (Fig. 1B and E). In this case, 1b and 2b were obtained with >90% analytical yield in less than 90 min. Increasing the temperature to 50 °C, ALDH-Ec produced more than 90% of 1b only just after 90 min, whereas no more than 68% analytical yield of 2b could be obtained under the same reaction conditions. Observing in figure E the progress of the analytical yield of 2a over time revealed that 64% of 2b was obtained already within 15 min, but the reactions did not proceed significantly further after that time. Thus, it appeared that the choice of the temperature for performing the biocatalytic oxidation should be a compromise between activity and stability of the tested ALDHs. This observation is in agreement with our thermostability data (see next paragraph) and the data reported for ALDH-Ec in literature.53 PP-ALDH showed similar activity for the oxidation of 1a at 40 and 50 °C, with a slight preference for 50 °C (Fig. 1C, >90% analytical yield after 180 min). At 30 °C, only 48% analytical yield into 1b was obtained. An interesting first indication of the different substrate acceptance of the ALDHs was revealed already at this stage, since PP-ALDH prefers to oxidise aliphatic aldehydes over aromatic ones. Indeed, only a maximum of 10–13% of 2b was formed during the bio-oxidation, independent of the temperature applied (Fig. 1F).
In a second step, we investigated the influence of the pH and type of buffer used for the oxidation of 1a and 2a. Analytical yield values at 40 °C were compared for the reactions using Tris/HCl buffer at pH 7, 8 and 9, KPi buffer pH 8 and Gly/NaOH buffer at pH 9 (all buffers at 50 mM concentration) and the results are summarized in Fig. 2 and the ESI (Tables S5 and S6†). In general, all three enzymes are poorly active at pH 7, leading to maximum analytical yields of 14–40% for 1a and 15–30% for 2a after 4 h. The origin for the low activity at neutral pH might have been related to the protonation state of the catalytic essential cysteine residue in the active site of the ALDHs (pK a 8.6),52 as well as the required nucleophilicity of the attacking water molecule in the last step of the mechanism.54 In fact, the thiol group of the cysteine residue must be deprotonated in order to perform the first nucleophilic attack on the aldehyde substrate.
Fig. 2.

pH and buffer profile for the conversion of 1a (panel A to C) and 2a (panel D to F) by ALDH-Bov, ALDH-Ec and PP-ALDH. Experimental conditions: 1 mL final volume in Eppendorf tubes, buffer strength = 50 mM (Gly/NaOH buffer = 50 mM glycine titrated with NaOH to pH 9), T = 40 °C, 180 rpm on orbital shaker, [1a] or [2a] = 20 mM, [NAD+] = 0.5 mM, [ALDH-Bov] = 5 µM for 1a and 10 µM for 2a, [ALDH-Ec] = 5 µM, [PP-ALDH] = 20 µM, [NOx] = 5 µM, [DTT] = 100 µg mL–1. Extraction with EtOAc under acidic conditions using toluene as IS and derivatization of acids to the methylester with (trimethylsilyl)diazomethane. Conversions were measured by GC-FID (DB-1701).
Then, after the hydride shift from the first thiohemiacetal intermediate to the NAD+, a polarized water molecule is necessary to hydrolyse the second thioester intermediate. As both steps are enabled by Brønsted base catalysis, a basic pH buffer should accelerate the reaction.
Previous biochemical studies indicated that ALDH-Bov is inhibited in Tris/HCl buffer.52 However, the use of Tris/HCl buffer did not influence the outcome of our biocatalytic reactions since the same increase of the analytical yield over time was observed using both Tris/HCl and KPi buffer at pH 8 (>90% analytical yield after 60 min for 1a and 2a; Fig. 2A and D). Moreover, even at pH 9, the enzyme was found to perform better in Tris/HCl (>90% after 60 and 90 min for 1a and 2a) than in Gly/NaOH (70% and 60% for 1a and 2a after 60 and 90 min, respectively).
ALDH-Ec was described as a sensitive enzyme towards pH changes, reaching a maximum of activity at pH 8. Increasing or decreasing the pH by one unit led to 70% loss of activity.53 Indeed, using Gly/NaOH buffer at pH 9 (as reported in literature),53 the analytical yields did not exceed 40–45% for both substrates tested. However, when we applied Tris/HCl buffer at the same pH, the analytical yield increased over 90% after 60 and 30 min for the oxidation of 1a and 2a, respectively. The results for the oxidations of 1a and 2a in Tris/HCl were comparable with the results in KPi buffer at pH 8 (Fig. 2B and E).
The preferred buffer conditions for the aldehyde oxidation catalysed by PP-ALDH were Tris/HCl and KPi at pH 8 (>90% after 120 min for 1a and 2a, Fig. 1C and F). Whilst similar analytical yields were obtained for 1a at pH 9 (>90% after 180 min), only 10% analytical yield was measured for 2a. In the case of PP-ALDH, we investigated also the oxidation at pH 8.5 in Tris/HCl as well as KPi buffer, which demonstrated the preference for KPi buffer for the oxidation of substrate 2a.
For the continuation of our study, we finally decided to use KPi buffer at pH 8.5. In fact, Tris(hydroxymethyl)aminomethane (i.e. Tris buffer) contains a primary amine moiety that is amenable for condensation with some aldehydes in aqueous solution as also observed by other authors.56,57 Consequently, the possible interaction between tris and some aldehyde substrates might influence negatively the efficiency of the biocatalytic oxidation. On the other hand, the buffer capacity of KPi buffer is maximum in the range of pH between 6.5 to 8. Thus, KPi buffer is not commonly applied above pH 8. However, in our specific cases, KPi at pH 8.5 is applicable, because the oxidation of the aldehyde generates only the carboxylic acid product. Thus, only buffering towards the production of Brønsted acid species is required. In fact, under our optimised reaction conditions, no significant variation of pH was observed between the start and the end of the reaction. The temperature was instead set to 40 °C for the continuation of the study, to avoid any possible partial evaporation of volatile aldehydes and to retain enzyme stability.
An interesting aspect is the possible enantiodiscrimination of the three ALDHs towards racemic substrate 1a, which contains a stereogenic centre in α-position to the aldehyde moiety. Our data clearly shows that all three ALDHs can efficiently oxidise both (S)-1a and (R)-1a, although the oxidation of (R)-1a is slightly kinetically favoured. However, when the oxidation reaches completion (for details, see ESI section 9†), product 1b is obtained in racemic form. Thus, significant spontaneous chemical racemisation of substrate 1a does not occur within the reaction time (1 h).
Finally, the irreversibility of the biocatalytic oxidation catalysed by the ALDHs under our reaction conditions was demonstrated by running the reverse reaction using carboxylic acids 1b and 2b (20 mM) as substrates (see ESI section 13†). The reduced nicotinamide coenzyme (1 mM) was recycled via the established formate dehydrogenase (FDH)/formate recycling system. Formation of aldehydes 1a and 2a was not detected at all.
Thermostability study on the ALDHs and NOx
The thermostability (melting temperature T m) of the three ALDHs as well as NOx was determined by differential scanning fluorometry. T m values were determined in Tris/HCl buffers (50 mM, pH 8, 8.5 and 9), KPi buffers (50 mM, pH 8 and 8.5) and Gly/NaOH buffer (50 mM, pH 9). Additionally, we measured the T m values of the three ALDHs in presence as well as absence of NAD+ and DTT, since stabilising effect of coenzyme is expected. The T m values of ALDH-Bov, ALDH-EC and PP-ALDH in KPi (50 mM, pH 8.5) were 47 °C, 44 °C and 42 °C (for details, see ESI section 14†). The T m value of NOx was 51 °C. These data are in agreement with the temperature profile of the biocatalytic reactions reported in Fig. 1 and 2.
Elucidation of the substrate scope
A panel of sixty-one structurally diverse substrates was tested with the three ALDHs under the optimised reaction conditions. The substrates investigated can be divided into four main structural groups: (1) aliphatic (linear and cyclic) aldehydes, (2) aryl-alkyl aldehydes (e.g. cinnamic/hydrocinnamic motif), (3) aromatic aldehydes (e.g. substituted benzaldehydes) and (4) heteroaromatic aldehydes (i.e. mono- and bicyclic). Moreover, we assessed the potential chemospecificity of the ALDHs for the oxidation of the aldehyde moiety to the carboxylic moiety by testing additional substrates that contain further oxidisable groups such as alcohols. Racemic phenyl-2-propanal was also tested. The progress of the reaction was monitored after 4 and 24 h. In general, we did not observe any significant differences in the analytical yield after 4 and 24 h reaction time. Either the reaction proceeded to high analytical yield within 4 h or did not proceed to a significant extent at all. The results of the substrate screening are summarized in Tables 1–4 (for the full dataset including the blank measurements see ESI, Tables S9 and S10†). Comparing the results of the biocatalytic reactions (i.e. in presence of ALDH) with the blank experiments (i.e. same conditions but without addition of enzyme), we could detect a minor spontaneous aerobic oxidation of some aldehydes to the related carboxylic acids under aerobic mixing in aqueous buffer at pH 8.5. This observation is in agreement with a recent publication by Zhang et al., in which the spontaneous oxidation of some aldehydes at 5–10 mM concentration was reported in aqueous environment at 1 bar of pure dioxygen pressure and 37 °C without any additional catalyst.58
Table 1. Biocatalytic aerobic oxidation of aliphatic/cyclic aldehydes to carboxylic acids catalysed by ALDHs.
| Substrate | ALDH | Yield [%] | Substrate | ALDH | Yield [%] | Substrate | ALDH | Yield [%] |
|---|---|---|---|---|---|---|---|---|
 3a
3a
|
Bov | 67a |
 6a
6a
|
Bov | 69a |
 10a
[b]
10a
[b]
|
Bov | 70a |
| Ec | 76a | Ec | 92 | Ec | 62a | |||
| PP | 72a | PP | 64a | PP | 61a | |||
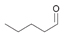 4a
4a
|
Bov | >99 |
 7a
[b]
7a
[b]
|
Bov | 76a |
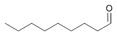 11a
11a
|
Bov | 75a |
| Ec | >99 | Ec | 76a | Ec | 69a | |||
| PP | >99 | PP | 70a | PP | 67a | |||
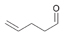 5a
5a
|
Bov | >99 |
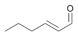 8a
8a
|
Bov | 10a |
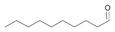 12a
12a
|
Bov | 77 |
| Ec | >99 | Ec | 92a | Ec | 78 | |||
| PP | >99 | PP | 94a | PP | 80 | |||
 1a
1a
|
Bov | >99a |
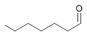 9a
[b]
9a
[b]
|
Bov | 60a |
 13a
[b]
13a
[b]
|
Bov | >99 |
| Ec | >99a | Ec | 54a | Ec | >99 | |||
| PP | >99a | PP | 55a | PP | >99 |
If not specified, reaction time was 4 h.
Reaction time was 24 h.
High analytical yield was observed without the addition of enzyme. Conversion of blank reactions (24 h, average of two samples): 7a, 20%; 9a, 15%; 13a, 10%. For full data set, see ESI section 8. Experimental conditions: 1 mL Eppendorf tubes in KPi buffer (50 mM, pH 8.5), T = 40 °C, 180 rpm on orbital shaker, [substrate] = 20 mM, [ALDH] = 10 µM, [NOx] = 5 µM, [DTT] = 100 µg mL–1, [NAD+] = 0.5 mM. Extraction with EtOAc under acidic conditions using toluene as IS and derivatization of acids to the methylester with (trimethylsilyl)diazomethane. Analytical yields were measured by GC-FID (DB-1701).
Table 4. Biocatalytic aerobic oxidation of mono- and bicyclic heteroaromatic aldehydes, bicyclic aromatic aldehydes and profen aldehydes to carboxylic acids catalysed by ALDHs.
| Substrate | ALDH | Yield [%] | Substrate | ALDH | Yield [%] | Substrate | ALDH | Yield [%] |
|---|---|---|---|---|---|---|---|---|
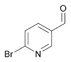 48a
48a
|
Bov | 40a |
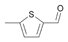 53a
53a
|
Bov | 99 |
 58a
58a
|
Bov | >99a |
| Ec | >99 | Ec | >99 | Ec | 17a | |||
| PP | 13a | PP | 4 | PP | n.m. | |||
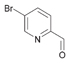 49a
49a
|
Bov | >99 |
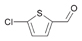 54a
54a
|
Bov | >99 |
 59a
59a
|
Bov | 3a |
| Ec | >99 | Ec | >99 | Ec | n.m.a | |||
| PP | 4 | PP | 3 | PP | n.m.a | |||
 50a
50a
|
Bov | >99 |
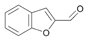 55a
55a
|
Bov | 11 |
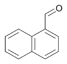 60a
60a
|
Bov | 94a |
| Ec | >99 | Ec | 8a | Ec | 3 | |||
| PP | >99 | PP | 1a | PP | 2a, b | |||
 51a
51a
|
Bov | 90 |
 56a
56a
|
Bov | 51a |
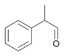 61a
61a
|
Bov | 25c |
| Ec | 91a | Ec | 8a | Ec | 27c | |||
| PP | 25 | PP | n.m.a | PP | 25c | |||
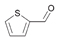 52a
52a
|
Bov | >99 |
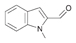 57a
57a
|
Bov | 67 | |||
| Ec | >99 | Ec | <1 | |||||
| PP | 6 | PP | n.m. | |||||
If not specified, reaction time was 4 h.
Reaction time was 24 h.
The blank measurement without the addition of substrate gives the same analytical yield.
For ALDH-Bov and ALDH-Ec, racemic product was obtained. PP-ALDH afforded the product in slightly enantioenriched form (ee 16%, (R)). Experimental conditions: 1 mL Eppendorf tubes in KPi buffer (50 mM, pH 8.5), T = 40 °C, 180 rpm on orbital shaker, [substrate] = 20 mM, [ALDH] = 10 µM, [NOx] = 5 µM, [DTT] = 100 µg mL–1, [NAD+] = 0.5 mM. Extraction with EtOAc under acidic conditions using toluene as IS and derivatization of acids to the methylester with (trimethylsilyl)diazomethane. Analytical yields were measured by GC-FID (DB-1701).
Among the set of 61 substrates investigated (20 mM), spontaneous oxidation afforded analytical yields in the range of 1–5% in 21 cases, and in the range of 6–10% in 7 cases. Spontaneous oxidation slightly above 10% was detected for substrates 2a, 7a, 9a and 21a. Aliphatic and aryl aliphatic aldehydes (see ESI Table S9† for data and Tables 1 and 2 for structures) were the substrates to be affected generally by spontaneous oxidation, with the exception of 16a. Instead, the occurrence of spontaneous oxidation with benzaldehyde derivatives depended on the type of substituent on the phenyl ring (see ESI Tables S9 and S10† for data and Table 3 for structures). In contrast, other homoaromatic (bicyclic) and heteroaromatic aldehydes did not show any spontaneous oxidation (see ESI Table S10† for data and Table 4 for structures).
Table 2. Biocatalytic aerobic oxidation of aromatic aldehydes (cinnamic/hydrocinnamic motif) to carboxylic acids catalysed by ALDHs. The reaction time was 24 h.
| Substrate | ALDH | Yield [%] | Substrate | ALDH | Yield [%] | Substrate | ALDH | Yield [%] |
|---|---|---|---|---|---|---|---|---|
 14a
14a
|
Bov | 83 |
15a |
Bov | 6 |
 16a
16a
|
Bov | 4 |
| Ec | 85 | Ec | 25 | Ec | 5 | |||
| PP | 79 | PP | 5a | PP | <1 |
The blank measurement without the addition of enzyme gives the same analytical yield. Experimental conditions: 1 mL Eppendorf tubes in KPi buffer (50 mM, pH 8.5), T = 40 °C, 180 rpm on orbital shaker, [substrate] = 20 mM, [ALDH] = 10 µM, [NOx] = 5 µM, [DTT] = 100 µg mL–1, [NAD+] = 0.5 mM. Extraction with EtOAc under acidic conditions using toluene as IS and derivatization of acids to the methylester with (trimethylsilyl) diazomethane. Analytical yields were measured by GC-FID (DB-1701).
Table 3. Biocatalytic aerobic oxidation of aromatic aldehydes (substituted benzaldehydes) to carboxylic acids catalysed by ALDHs.
| Substrate | ALDH | Yield [%] | Substrate | ALDH | Yield [%] | Substrate | ALDH | Yield [%] |
|---|---|---|---|---|---|---|---|---|
 2a
[c]
2a
[c]
|
Bov | >99 |
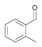
 27a
27a
|
Bov | >99 |
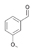 38a
38a
|
Bov | >99 |
| Ec | >99 | Ec | >99 | Ec | 49a | |||
| PP | >99a | PP | 7a | PP | n.d. | |||
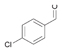
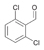
 17a
17a
|
Bov | 51 |
 28a
28a
|
Bov | n.d. |
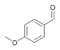 39a
39a
|
Bov | >99 |
| Ec | 28a | Ec | n.d. | Ec | 12a | |||
| PP | 1a,b | PP | n.d. | PP | <1a,b | |||
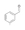 18a
18a
|
Bov | >99 |
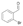 29a
29a
|
Bov | 15a |
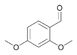 40a
40a
|
Bov | 2a |
| Ec | 25 | Ec | 8 | Ec | n.d. | |||
| PP | 2a,b | PP | 7b | PP | n.d. | |||
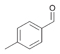 19a
19a
|
Bov | >99 |
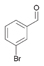 30a
30a
|
Bov | 97a |
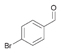
 41a
41a
|
Bov | 95a |
| Ec | >99 | Ec | >99 | Ec | 1a | |||
| PP | 3a,b | PP | 14 | PP | n.d. | |||
 20a
20a
|
Bov | 2b |
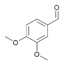
 31a
31a
|
Bov | >99 |
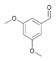 42a
42a
|
Bov | 91 |
| Ec | 2b | Ec | >99a | Ec | 8a | |||
| PP | 1b | PP | 8 | PP | 4a,b | |||
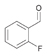 21a
[c]
21a
[c]
|
Bov | >99 |
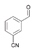 32a
32a
|
Bov | 94 |
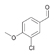 43a
43a
|
Bov | >99a |
| Ec | >99 | Ec | 93 | Ec | 16a | |||
| PP | 16 | PP | 92 | PP | 3a | |||
 22a
22a
|
Bov | >99 |
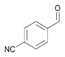 33a
33a
|
Bov | 98 |
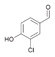 44a
44a
|
Bov | >99a |
| Ec | >99 | Ec | 96 | Ec | 3a | |||
| PP | 13 | PP | 96a | PP | 2a,b | |||
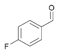 23a
23a
|
Bov | >99 |
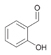 34a
34a
|
Bov | >99 |
 45a
45a
|
Bov | 57a |
| Ec | >99 | Ec | 2a | Ec | <1a | |||
| PP | 17 | PP | <1b | PP | n.d. | |||
 24a
24a
|
Bov | >99 |
 35a
35a
|
Bov | >99 |
 46a
46a
|
Bov | 2a,b |
| Ec | >99 | Ec | 20 | Ec | 2a,b | |||
| PP | 3a,b | PP | 1a,b | PP | 2a,b | |||
 25a
25a
|
Bov | 11a |
 36a
36a
|
Bov | 96a |
 47a
47a
|
Bov | >99a |
| Ec | 11a | Ec | 6a | Ec | >99 | |||
| PP | 5a,b | PP | 2a | PP | 18 | |||
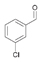 26a
26a
|
Bov | >99a |
 37a
37a
|
Bov | 10 | Experimental: 1 mL in KPi buffer (50 mM, pH 8.5), T = 40 °C, 180 rpm, [substrate] = 20 mM, [ALDH] = 10 µM, [NOx] = 5 µM, [DTT] = 100 µg mL–1, [NAD+] = 0.5 mM. Analytical yields were measured by GC-FID as methyl ester. | ||
| Ec | >99 | Ec | 2a | |||||
| PP | 20 | PP | 1a,b | |||||
If not specified, reaction time was 4 h.
Reaction time was 24 h.
The blank measurement without the addition of enzyme gives the same analytical yield.
High analytical yield observed without the addition of enzyme. Conversion of blank reactions (24 h, average of two samples): 21a, 12%; 2a, 13%. For full data set, see ESI section 8.
Table 1 summarizes the results for the oxidation of aliphatic/cyclic aldehydes. The shortest chain aldehyde tested was butanal (3a), whose biocatalytic oxidation resulted in the formation of 67–76% analytical yield of acid 3b after 24 h. However, the actual productivity of the system might be improved as a fraction of volatile aldehyde 3a may have evaporated during the course of the reaction with the current experimental set-up. Indeed, homologous aldehydes but less volatile such as 4a, 5a and 1a (5 and 6 carbon-atom chains) gave quantitative analytical yield with all three ALDHs tested. The same applied for cyclohexancarbaldehyde 13a. The capability of converting short chain aliphatic aldehydes by ALDHs is of interest. In fact, a number of catalytic organic and metal–organic reagents are not applicable for the oxidation of these substrates because of the need for medium-high boiling solvents for the reaction, which complicates product isolation, or lack of activity.12,19,36 In the case of medium and long chain aldehydes from six to ten carbon atoms (6–12a), the ALDHs could oxidise the substrates with analytical yields ranging from 54% to 94%. A general trend was not revealed as the best performing ALDH depended on the particular substrate. The interesting exceptions were the bio-oxidation of 6a and 8a, which both contain a carbon–carbon double bond conjugated to the aldehyde moiety. 6a is preferentially converted by ALDH-Ec (92% analytical yield), whereas ALDH-Bov and PP-ALDH afforded 69% and 64% analytical yield, respectively. 8a was also well accepted by ALDH-Ec and PP-ALDH (92 and 94% analytical yield, respectively), but not by ALDH-Bov (10% analytical yield). This feature was also observed in our previous work, in which we exploited the higher activity of ALDH-Bov towards the saturated aldehyde rather than the unsaturated one in a dual-enzyme cascade process.49 The same effect is also evident in the case of other structurally related substrates (Table 2): the α,β-unsaturated aldehyde cinnamaldehyde (15a) and its saturated counterpart hydrocinnamaldehyde (14a) (Table 2). In fact, the analytical yield for the saturated substrate 14a (83, 85, 79% for Bov, Ec, and PP) was always higher than the analytical yield of the unsaturated one 15a (6, 25, 5% for Bov, Ec, and PP) with all three ALDHs.59 This discrimination between saturated and unsaturated aldehydes did not occur when testing substrates possessing the unsaturation moiety in further ω-positions. In this case, respectively saturated and unsaturated substrates 4a and 5a were converted equally (>99%). The substituted cinnamaldehyde 16a, which bears an additional methoxy group in meta and a hydroxyl group in para position was converted to low extend (4–5% max with ALDH-Bov and Ec). It might be that, in this case, steric hindrances become already a limiting factor.
Furthermore, a wide panel of 32 benzaldehyde derivatives was tested (Table 3). Chemoselective oxidations to carboxylic acids starting from benzaldehyde derivatives bearing electrodonating substituents such as hydroxy and methoxy, are problematic by chemocatalytic methods because of the frequent occurrence of competitive Dakin-type oxidation to phenols (and their oligomerisation).12,21,36 On the contrary, we found that all the biocatalytic oxidations of benzaldehyde derivatives catalysed by ALDHs were perfectly chemoselective as the expected carboxylic acids were the only products. As suggested above from the pH and temperature studies, PP-ALDH preferentially oxidises aliphatic aldehydes rather than aromatic aldehydes. Among the benzaldehyde derivatives tested, quantitative analytical yield was only observed for the oxidation of benzaldehyde 2a. For all other benzaldehyde derivatives bearing a substituent in meta or para position, there was at least one ALDH enzyme that was capable of catalysing the reaction above 90% analytical yield (18–19a, 22–23a, 26–27a, 30–31a, 32–33a, 35–36a, 38–39a, 47a). Moreover, the oxidation of meta and para di-substituted aromatic aldehydes worked well with analytical yields from 91–99% for 41–44a and 57% for 45a. In contrast, the oxidation of aldehydes possessing substituents in ortho position were generally more challenging. It seems that electron donating substituents in the ortho position still favour the reaction (51% for 17a and >99% 34a with ALDH-Bov), whereas analytical yields were moderate or poor with electron withdrawing groups in the ortho position (11% for 25a, 15% for 29a). The exceptions are the fluoro-substituted aromatic benzaldehyde derivatives 21a and 24a, which were fully converted by ALDH-Bov and ALDH-Ec. It might also be that steric effects can play a role in the bio-oxidation of ortho-substituted benzaldehyde derivatives, as the bulkier methoxy (37a) and acetyl (46a) groups in the ortho position gave only 10% and 2% analytical yield for ALDH-Bov, respectively. Di- or tri-substituted derivatives with at least one substituent in the ortho position, like 20a, 28a and 40a, were not accepted by any of the ALDHs used in this study. Table 4 demonstrates the broad applicability of the ALDHs since all the monocyclic six-membered and five-membered ring heteroaromatic compounds tested were converted quantitatively by at least one of the enzymes (48–54a). Interestingly, even some bicyclic heteroaromatic compounds, such as 56a and 57a, were converted (51–67%), whereas 55a and 59a were converted to a lesser extent (11 and 3%). It is important to note that the oxidation of these substrates proceeded with perfect chemoselectivity because sulphur heteroatoms (52–54a, 56a), nitrogen heteroatoms (48–49a, 57a, 59a), or additional hydroxyl groups (51a) were not oxidised at all by the ALDHs. However, it is known that all these functional groups can become oxidised when chemical catalysts and/or reagents are used for oxidation of aldehydes.2,3 Furthermore, the perfect chemoselectivity for the biocatalytic oxidation of furan-2-carbaldehydes (50–51a) and benzofuran-2-carbaldehyde (55a) is also noteworthy as formation of Dakin-type by-products is known using, among others, peroxides as chemical oxidising agent.36 In addition, ALDH-Bov converted bulkier bicyclic homoaromatic aldehydes such as 58a and 60a with elevated analytical yields (>99% and >90% respectively) and again perfect chemoselectivity. In contrast, strong chemical oxidising agents such as pyridinium chlorochromate/periodate fail in the oxidation of naphtaldehydes such as 58a and 60a.
Finally, racemic 2-phenylpropanal (61a) was tested for the oxidation with the three ALDHs, affording the carboxylic acid product 61b in 25–27% analytical yield and in racemic (ALDH-Bov, ALDH-Ec) or slightly enantioenriched form (PP-ALDH, ee 16% R).
Overall, these results highlight the perfect chemoselectivity of the oxidation catalysed by aldehyde dehydrogenases.18 This level of chemospecificity in the oxidation of the aldehyde moiety is unrivalled by any chemical or other enzymatic method (i.e. oxidase enzymes).
Biocatalytic oxidation using E. coli/ALDH whole cells
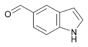
For practical applications, the use of whole cells rather than purified enzymes is the preferred solution because time consuming purification steps are omitted. At this stage, we decided to focus on the oxidation of two heterocyclic compounds with lyophilised whole cells containing overexpressed ALDH (E. coli cells devoid of expressed ALDH were also tested as negative control reactions). We chose 5-hydroxymethyl-2-furancarbaldehyde (51a) as substrate because it is a starting material derivable from renewable fructose. Furthermore, the related oxidation product 5-hydroxymethyl-2-furancarboxylic acid (HMFCA, 51b) is a versatile building block for the synthesis of polyesters as well as an interleukin inhibitor.60,61 In addition, it was reported that HMFCA possesses antitumor activity.40 As second test substrate thiophene-2-carbaldehyde (54a) was chosen as its chemoselective oxidation is challenging, due to the fact that both, sulphur heteroatom and carbon–carbon double bonds are prone to oxidation by conventional chemical oxidants.62,63 Additionally, 54b was quantitatively recovered after bio-oxidation catalysed by ALDH-Bov and ALDH-Ec on analytical scales and further extraction of the reaction mixture. Before scaling-up the biocatalytic oxidation, we carried out a preliminary set of experiments at analytical scale. The bio-oxidation (substrate 20 mM, reaction volume 1 mL) was performed using lyophilised E. coli overexpressing the ALDHs (10 mg mL–1) in presence and absence of NOx (0–5 µM) and NAD+ (0–0.5 mM) in order to assess the influence of the exogenous recycling system and NAD coenzyme in a whole cell environment (ESI, Tables S12 and S13†).
According to the results for the biocatalytic oxidation of 51a in analytical scale using purified ALDHs (Table 4), the substrate must be converted to the product 51b with a maximum of 91% analytical yield. In that case, no starting material was detected after the reaction with purified ALDHs. Using lyophilised E. coli cells overexpressing the ALDHs, the aldehyde substrate 51a was converted again to a similar level (ca. 80% analytical yield, see ESI Table S12†). Surprisingly, the same conversion of 80% from 51a to 51b was also obtained using lyophilised E. coli cells/ALDH but without the addition of NAD+ and NOx. For substrate 51a, also a considerable level of oxidation to the carboxylic acid (36%) was observed when E. coli whole cells devoid of any overexpressed ALDHs were tested.
Conversely, the bio-oxidation of 54a with lyophilized cells overexpressing ALDH-Ec required the mandatory addition of either NAD+ or NOx in order to reach elevated analytical yields. In fact, a drop in the analytical yield of 54a was observed when both NOx and NAD+ were excluded from the reaction (26–38%). In any case, the analytical yield was slightly lower using ALDH-Ec as lyophilised whole cells biocatalyst as the maximum analytical yield with addition of either NAD+ or NOx was 89% (see ESI, Table S13†) In comparison, the same oxidation using the purified ALDH-EC or ALDH-Bov gave >99% analytical yield (Table 4). The reason for that might be the probable interaction between the reactive aldehyde moiety of the substrate with abundant nucleophilic groups of the whole cell environment, hence reducing free substrate availability in solution. Interestingly, the bio-oxidation of 54a using ALDH-Bov required mandatory addition of both NOX and NAD+ to achieve elevated analytical yield (87%). Without NAD+ or NOx the analytical yield using ALDH-Bov fell to 42–46%. Absence of both, coenzyme and recycling enzyme led to only 15% analytical yield. Thus, it seems that the need for addition of coenzyme to lyophilized whole cells systems depends from the particular overexpressed ALDH and the substrate converted.
In the following step, lyophilized whole cells (ALDH-Bov and Ec) were tested for the oxidation at increased substrate concentrations of 51a (20–100 mM) either with (Table S14†) or without (Table S15†) addition of NAD+ and NOx. Already at 40 mM substrate concentration, the analytical yield dropped down from >80% to 35% (addition of NOx and NAD+) or 5–18% (without coenzyme and recycling enzyme). Thus, 20 mM substrate concentration was kept for the preparative scale oxidation.
In the final set of experiments, we tested both substrates 51a and 54a for the biocatalytic oxidation using resting E. coli cells (i.e. wet cells) overexpressing either ALDH-Bov or ALDH-Ec. The study was conducted following the same systematic methodology as for the biocatalytic oxidation with lyophilised cells. In general, we observed a correlation between the yields for the oxidation of substrate 51a using either resting cells (Table S16†) or lyophilised cells (Table S14†). In contrast, few minor discrepancies were observed for the oxidation of substrate 54a (Tables S17 and S15,† respectively). The major discrepancy was revealed for the oxidation of 54a catalysed by ALDH-Ec in absence of NOX and NAD+. Resting cells afforded higher yields (89–90%) than lyophilised cells (26–38%). This difference may be attributed to the fact that a significant level of endogenous NAD+ is available to sustain the reaction in resting cells, whereas it is mainly degraded during lyophilisation procedures.
Preparative scale biocatalytic oxidation
A preparative scale bio-oxidation of 51a (>500 mg) using lyophilised whole cells was conducted without the addition of NAD+ and recycling enzyme NOx, resulting in 74% and 75% analytical yield for ALDH-Bov and ALDH-Ec, respectively. After extraction and solvent evaporation, the isolated yields of 51b were 63% and 56% (i.e. 356 and 315 mg). We further increased the scale of the reaction up to 2 g of substrate using ALDH-Ec lyophilised whole cells; that resulted in 71% analytical yield and 61% isolated yield of 51b (1.37 g). Thus, applicability of the ALDHs in chemical synthesis and potentially in large scale can be demonstrated.
Conclusions
The oxidation of aldehydes to carboxylic acids is a relatively simple organic chemical transformation, but a “green” and general method for performing this reaction with perfect chemoselectivity (>99%) and on a wide range of structurally diverse substrates is needed. Some chemical methods offer elevated efficiencies in terms of yields, selectivities and substrate acceptance but these are accompanied by one or more of the following drawbacks: (i) a requirement for increasingly unsustainable transition metal-catalysts; (ii) the need for organic oxidants in stoichiometric amounts (or even large excess); (iii) the need for strong acidic or basic conditions; (iv) the requirement for elevated temperatures or pressures.
Thus, there is still a need for new methods of aldehyde oxidation which are both chemically efficient, chemoselective and green.
In this work, we have performed the oxidation of aldehydes using three recombinant aldehyde dehydrogenases that originated from bacteria and bovine lens. The enzymes were applied in highly purified form in combination with a nicotinamide oxidase for the efficient recycling of NAD+ at the expense of molecular oxygen from air. The biocatalytic oxidation was optimised in terms of temperature, pH and type of buffer. The final bio-oxidation runs in aqueous phosphate buffer, under mild reaction conditions (40 °C and atmospheric pressure) and consumes only dioxygen from air as benign oxidant. Sixty-one structurally diverse aldehydes (aliphatic, aryl aliphatic, benzylic-, hetero-aromatic and bicyclic aldehydes) were tested. With the exception of only a few ortho-substituted benzaldehydes and two bicyclic heteroaromatic aldehydes, all the other substrates were converted with elevated yield (up to >99%). In all cases, the chemoselectivity was perfect: no other product was detected except the expected carboxylic acid. Oxidisable functionalities such as the hydroxyl moiety, alkene groups, aryl groups, and sulphur as well as nitrogen heteroatoms remained untouched. The bio-oxidation was also investigated with lyophilised whole cell as well as resting E. coli cell biocatalysts for a more convenient practical application. Interestingly, supplementation of NAD+ and NOx recycling enzyme can be omitted in some cases as the microbial host produces sufficient amount of coenzyme, which can be recycled by endogenous enzymes. The reaction was upscaled to the oxidation of bio-based starting material 5-(hydroxymethyl)furfural (2 g) to afford 5-(hydroxymethyl)furoic acid in 61% isolated yield.
We conclude that aldehyde dehydrogenases have the potential to become the first choice for chemoselective oxidation of aldehydes into carboxylic groups. This biocatalytic method is particularly attractive for the oxidation of aldehyde moieties within molecules possessing further oxidisable groups. Future research will focus on improving the enzymes tolerance to substrate concentration and long-term stability in order to enable even broader application of these enzymes.
Experimental
The expression and purification of the enzymes, influence of DTT, temperature and pH studies, calculation of response factors, the full dataset for the substrate scope using lyophilised cells and resting cells, thermostability studies as well as the analytical methods can be found in the ESI.†
General procedure for the biocatalytic oxidation of aldehydes in analytical scale
Unless otherwise stated, standard reactions (1.0 mL, in Eppendorf tubes) were performed in KPi buffer (50 mM KH2PO4/K2HPO4, pH 8.5), containing ALDH purified (10 µM), NOx purified (5 µM), NAD+ (0.5 mM) and DTT (100 µg mL–1). The reactions were started by the addition of substrate (1–61a, 20 mM final concentration), added pure or as DMSO stock solutions (2 M, 1 M or 0.5 M, depending on their solubility). The reactions were shaken at 40 °C at 170 rpm in an orbital shaker for 4 and 24 h. Reactions were stopped by the extraction with EtOAc (2 × 500 µL) under acidic conditions (addition of 100 µL HCl 3 N). The extracts were dried using anhydrous MgSO4 and derivatised to the methyl ester as described below. Analytical yields were measured by GC-FID.
Derivatisation of carboxylic acids to methyl esters
The extracts were diluted to obtain a final concentration of 5 mM in 1 mL reaction. To the organic phase (250 µL), EtOAc (540 µL), MeOH (200 µL) and (trimethylsilyl)diazomethane (10 µL) were added and the reaction was shaken at 30 °C, 160 rpm for 60 min. The excess of the derivatisation reagent was destroyed by the addition of acetic acid (2 µL) and incubation for further 30 min at 30 °C. Analysis was performed by GC-FID.
Preparative biocatalytic oxidation of 51a
A total volume of 200 mL consisted of KPi buffer (50 mM, pH 8.5), lyophilised whole cells of E. coli overexpressing ALDH-Bov or ALDH-Ec (2 g, 10 mg mL–1, resuspended in buffer), DTT (20 mg, final concentration 100 µg mL–1) and 51a (504 mg, 4 mmol for ALDH-Bov and 502 mg, 3.98 mmol for ALDH-Ec; final concentration 20 mM). Biotransformations were performed in 500 mL glass bottles, incubated at 40 °C in an orbital shaker. After 21 h, a sample was taken and, after derivatisation, it was analysed by GC. The reaction mixture was acidified to pH 2–4 via addition of HCl (3 M). NaCl was added (36 g) to enhance the extraction of the acid. The aqueous reaction mixture was then extracted with methyl tert-butyl ether (3 × 70 mL). Phase separation was facilitated by centrifugation. The organic fractions were combined and dried over MgSO4. After filtration and evaporation of the solvent, the product was obtained in a pure form. Column chromatography was not required. The authenticity of the product was confirmed by 1H-NMR (ESI, paragraph 11†). Upscaling of 2 g (15.86 mmol) 51a was performed similar to the reaction described above using lyophilized E. coli cells overexpressing AldDH-Ec. The reaction was performed in 2 L Erlenmeyer flasks, covered with aluminium foil.
Supplementary Material
Acknowledgements
This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 638271, BioSusAmin). Dutch funding from the NWO Sector Plan for Physics and Chemistry is also acknowledged.
Footnotes
Conflicts of interest
The authors declare to have no competing interests, or other interests that might be perceived to influence the results and/ or discussion reported in this article.
Notes and references
- 1.Tojo G, Fernández M. Oxidation of Primary Alcohols to Carboxylic Acids, chapter 7: Oxidation of Alcohols to Carboxylic Acids via Isolated Aldehydes. Springer; New York: 2007. [Google Scholar]
- 2.Hudlicky M. Oxidations in Organic Chemistry, American Chemical Sociecty. Washington DC: 1990. [Google Scholar]
- 3.Hainess AH. Methods for the Oxidation of Organic Compounds, Academic. New York: 1988. [Google Scholar]
- 4.Heilbron I, Jones ERH, Sondheimer F. J Chem Soc. 1949:604–607. [Google Scholar]
- 5.Zhao M, Li J, Song Z, Desmond R, Tschaen DM, Grabowski EJJ, Reider PJ. Tetrahedron Lett. 1998;39:5323–5326. [Google Scholar]
- 6.Webster FX, Rivas-Enterrios J, Silverstein RM. J Org Chem. 1987;52:689–691. [Google Scholar]
- 7.Sedelmeier J, Ley SV, Baxendale IR, Baumann M. Org Lett. 2010;12:3618–3621. doi: 10.1021/ol101345z. [DOI] [PubMed] [Google Scholar]
- 8.Abiko A, Roberts JC, Takemasa T, Masamune S. Tetrahedron Lett. 1986;27:4537–4540. [Google Scholar]
- 9.Barker IRL, Dahm RH. J Chem Soc B. 1970:650–653. [Google Scholar]
- 10.Nwaukwa SO, Keehn PM. Tetrahedron Lett. 1982;23:3131–3134. [Google Scholar]
- 11.Dalcanale E, Montanari F. J Org Chem. 1986;51:567–569. [Google Scholar]
- 12.McKillop A, Kemp D. Tetrahedron. 1989;45:3299–3306. [Google Scholar]
- 13.Corey EJ, Gilman NW, Ganem BE. J Am Chem Soc. 1968;90:5616–5617. [Google Scholar]
- 14.Mannam S, Sekar G. Tetrahedron Lett. 2008;49:1083–1086. [Google Scholar]
- 15.Chakraborty D, Gowda RR, Malik P. Tetrahedron Lett. 2009;50:6553–6556. [Google Scholar]
- 16.Malik P, Chakraborty D. Tetrahedron Lett. 2010;51:3521–3523. [Google Scholar]
- 17.Bhatia B, Punniyamurthy T, Iqbal J. J Org Chem. 1993;58:5518–5523. [Google Scholar]
- 18.Hunsen M. Synthesis. 2005:2487–2490. [Google Scholar]
- 19.Thakur A, Talukdar D, Sharma K, Bharadwaj S. Synlett. 2013;24:963–966. [Google Scholar]
- 20.Yamada T, Rhode O, Takai T, Mukaiyama T. Chem Lett. 1991;20:5–8. [Google Scholar]
- 21.Bernini R, Coratti A, Provenzano G, Fabrizi G, Tofani D. Tetrahedron. 2005;61:1821–1825. [Google Scholar]
- 22.Liu M, Wang H, Zeng H, Li CJ. Sci Adv. 2015;1:e1500020. doi: 10.1126/sciadv.1500020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M, Li CJ. Angew Chem, Int. 2016;55:10806–10810. doi: 10.1002/anie.201604847. [DOI] [PubMed] [Google Scholar]
- 24.Sodhi RK, Paul S, Clark JH. Green Chem. 2012;14:1649. [Google Scholar]
- 25.Biella S, Prati L, Rossi M. J Mol Catal A: Chem. 2003;197:207–212. [Google Scholar]
- 26.Kharat AN, Pendleton P, Badalyan A, Abedini M, Amini MM. J Mol Catal A: Chem. 2001;175:277–283. [Google Scholar]
- 27.Yu H, Ru S, Dai G, Zhai Y, Lin H, Han S, Wei Y. Angew Chem, Int Ed. 2017;56:3867–3871. doi: 10.1002/anie.201612225. [DOI] [PubMed] [Google Scholar]
- 28.Späth E, Pailer M, Schmid M. Chem Ber. 1941;74:1552–1556. [Google Scholar]
- 29.Ganem B, Heggs RP, Biloski AJ, Schwartz DR. Tetrahedron Lett. 1980;21:685–688. [Google Scholar]
- 30.Balicki R. Synth Commun. 2001;31:2195–2198. [Google Scholar]
- 31.Dodd RH, Le M. Hyaric, Synthesis. 1993:295–297. [Google Scholar]
- 32.Heaney H, Newbold AJ. Tetrahedron Lett. 2001;42:6607–6609. [Google Scholar]
- 33.Choi J-K, Chang Y-K, Yeap Hong S. Tetrahedron Lett. 1988;29:1967–1970. [Google Scholar]
- 34.Joseph JK, Jain SL, Sain B. Catal Commun. 2007;8:83–87. [Google Scholar]
- 35.Sato K, Hyodo M, Takagi J, Aoki M, Noyori R. Tetrahedron Lett. 2000;41:1439–1442. [Google Scholar]
- 36.Travis BR, Sivakumar M, Hollist GO, Borhan B. Org Lett. 2003;5:1031–1034. doi: 10.1021/ol0340078. [DOI] [PubMed] [Google Scholar]
- 37.Vora HU, Rovis T. J Am Chem Soc. 2010;132:2860–2861. doi: 10.1021/ja910281s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byrne FP, Jin S, Paggiola G, Petchey THM, Clark JH, Farmer TJ, Hunt AJ, Robert McElroy C, Sherwood J. Sustainable Chem Processes. 2016;4:7. [Google Scholar]
- 39.Faber K, Fessner W-D, Turner NJ. Science of Synthesis, Biocatalysis in Organic Synthesis 3. Georg Thieme Verlag KG; Stuttgart (Germany): 2015. [Google Scholar]
- 40.Zhang X-Y, Zong M-H, Li N. Green Chem. 2017;19:4544–4551. [Google Scholar]
- 41.McKenna SM, Leimkuhler S, Herter S, Turner NJ, Carnell AJ. Green Chem. 2015;17:3271–3275. [Google Scholar]
- 42.Dijkman WP, Binda C, Fraaije MW, Mattevi A. ACS Catal. 2015;5:1833–1839. [Google Scholar]
- 43.Bechi B, Herter S, McKenna S, Riley C, Leimkühler S, Turner NJ, Carnell AJ. Green Chem. 2014;16:4524–4529. [Google Scholar]
- 44.Díaz-Rodríguez A, Lavandera I, Kanbak-Aksu S, Sheldon RA, Gotor V, Gotor-Fernández V. Adv Synth Catal. 2012;354:3405–3408. [Google Scholar]
- 45.Hollmann F. In: Science of Synthesis, Biocatalysis in Organic Synthesis 3, Chapter 3.3.1 Oxidation of Alcohols, Aldehydes, and Carboxylic Acids; Oxidation Using Dehydrogenases. Faber K, Fessner W-D, Turner NJ, editors. Georg Thieme Verlag KG; Stuttgart (Germany): 2015. pp. 115–138. [Google Scholar]
- 46.Moody TS, Mix S, Brown G, Beecher D. In: Science of Synthesis, Biocatalysis in Organic Synthesis 2, Chapter 2.5.1 Carbonyl Reduction; Ketone and Aldehyde Reduction. Faber K, Fessner W-D, Turner NJ, editors. Georg Thieme Verlag KG; Stuttgart (Germany): 2015. pp. 421–458. [Google Scholar]
- 47.Musa MM, Phillips RS. Catal Sci Technol. 2011;1:1311. [Google Scholar]
- 48.Romano D, Villa R, Molinari F. ChemCatChem. 2012;4:739–749. [Google Scholar]
- 49.Knaus T, Mutti FG, Humphreys LD, Turner NJ, Scrutton NS. Org Biomol Chem. 2015;13:223–233. doi: 10.1039/c4ob02282c. [DOI] [PubMed] [Google Scholar]
- 50.Winkler T, Gröger H, Hummel W. ChemCatChem. 2014;6:961–964. [Google Scholar]
- 51.Wu S, Zhou Y, Seet D, Li Z. Adv Synth Catal. 2017;359:2132–2141. [Google Scholar]
- 52.Ting HH, Crabbe MJ. Biochem J. 1983;215:361–368. doi: 10.1042/bj2150361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jo JE, Mohan Raj S, Rathnasingh C, Selvakumar E, Jung WC, Park S. Appl Microbiol Biotechnol. 2008;81:51–60. doi: 10.1007/s00253-008-1608-x. [DOI] [PubMed] [Google Scholar]
- 54.Coitinho JB, Pereira MS, Costa DMA, Guimarães SL, Araújo SS, Hengge AC, Brandão TAS, Nagem RAP. Biochemistry. 2016;55:5453–5463. doi: 10.1021/acs.biochem.6b00614. [DOI] [PubMed] [Google Scholar]
- 55.Matsumoto J, Higuchi M, Shimada M, Yamamoto Y, Kamio Y. Biosci, Biotechnol, Biochem. 1996;60:39–43. doi: 10.1271/bbb.60.39. [DOI] [PubMed] [Google Scholar]
- 56.Bubb WA, Berthon HA, Kuchel PW. Bioorg Chem. 1995;23:119–130. [Google Scholar]
- 57.Ogilvie JW, Whitaker SC. Biochim Biophys Acta. 1976;445:525–536. doi: 10.1016/0005-2744(76)90107-8. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y, Cheng Y, Cai H, He S, Shan Q, Zhao H, Chen Y, Wang B. Green Chem. 2017;19:5708–5713. [Google Scholar]
- 59.Alberty RA. Thermodynamics of biochemical reactions. Wiley-Interscience; 2003. The higher stability of the carbonyl moiety in α,β-unsaturated aldehydes cannot be a reason for the lower conversion. The overall reaction is driven from a thermo-dynamic point of view by the reoxidation of NADH to NAD+ catalysed by the NOx. In fact, the calculated ΔrG′° for the reaction: O2 + 2NADH = 2NAD+ + 2H2O, in aqueous buffer at pH 8.5 and buffer concentration 50 mM is –443.9 ± 6.5 KJ mol–1. http://equilibrator.weizmann.ac.il/ [Google Scholar]
- 60.Braisted AC, Oslob JD, Delano WL, Hyde J, McDowell RS, Waal N, Yu C, Arkin MR, Raimundo BC. J Am Chem Soc. 2003;125:3714–3715. doi: 10.1021/ja034247i. [DOI] [PubMed] [Google Scholar]
- 61.Munekata M, Tamura G. Agric Biol Chem. 1981;45:2149–2150. [Google Scholar]
- 62.Brown KN, Espenson JH. Inorg Chem. 1996;35:7211–7216. doi: 10.1021/ic960607+. [DOI] [PubMed] [Google Scholar]
- 63.Treiber A. J Org Chem. 2002;67:7261–7266. doi: 10.1021/jo0202177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.