

Abstract
Perimenopause marks initiation of female reproductive senescence with age-of-onset 47% heritable suggesting that factors other than inheritance regulate this endocrine aging transition. To elucidate these factors, we characterized transcriptional and epigenomic changes across endocrine aging transitions using a rat model recapitulating characteristics of human perimenopause. RNA-seq analysis revealed that hypothalamic aging precedes perimenopause. In the hypothalamus, global DNA methylation declined with both age and reproductive senescence. Treatment with the DNA-methyltransferase-1 inhibitor accelerated transition to reproductive senescence, menopause, whereas supplementation with the S-adenosyl-methionine precursor methionine delayed onset of perimenopause and endocrine aging. Genome-wide epigenetic analysis revealed DNA methylation of genes required for hormone signaling, glutamate signaling, and melatonin and circadian pathways. Specific epigenetic changes in these signaling pathways provide insight into origin of perimenopause-associated neurological symptoms such as insomnia. Collectively, these data provide evidence that female neuroendocrine aging precedes ovarian failure and that DNA methylation regulates the onset and duration of perimenopause.
Keywords: Menopause, Perimenopause, Aging, Epigenetics, DNA methylation, Hypothalamus
1. Introduction
Perimenopause marks the initiation of the transition into female reproductive senescence and is characterized by the exhaustion of oocytes, amenorrhoea, and the loss of cyclic estrogen production (Brinton, Yao et al. 2015). In women, the perimenopausal transition can span several years and timing of onset, duration, and completion has been linked to differential neurocognitive health outcomes (Geerlings 2001; Henderson 2007; Rocca 2011; Brinton, Yao et al. 2015). Age of onset is only 47% heriTable (Byars, Ewbank et al. 2010) and variability is present in monozygotic twins (Snieder, MacGregor et al. 1998) and inbred rat strains (Finch 2014), suggesting that epigenetics and environmental factors play an important role in reproductive aging. Indeed, recent studies in both human and rat have demonstrated an association between reproductive senescence and changes in the epigenome (Levine, Lu et al. 2016; Ionov 2017). In humans, post-menopausal women exhibit accelerated patterns of epigenetic aging compared to pre-menopausal women of the same biological age (Levine, Lu et al. 2016). However, the cause-effect relationship between epigenetics and reproductive senescence remains unclear. Untangling this relationship will prove challenging as differences in epigenetic patterns seen across the perimenopause transition most likely consist of environmental and age-related changes that initiate onset of reproductive senescence, as well as changes that occur as a direct result of fluctuating sex hormones.
Establishment, maintenance, and reorganization of the epigenome rely on the availability of methyl-donor molecules that are produced from one-carbon metabolism. The one-carbon cycle utilizes co-factors such as folate, choline, and various other B vitamins (B6, B12, riboflavin), to recycle homocysteine to produce S-adenosylmethionine (SAM), the universal methyl-donor that provides methyl-groups used for DNA, histone, and other protein methylation. Breakdown of the one-carbon cycle results in decreased production of SAM and an accumulation of the intermediate molecule, homocysteine (Herrmann and Obeid 2011, Tomizawa, Matsuzawa et al. 2015). Failure to produce sufficient levels of SAM can lead to global decline of DNA and histone methylation, resulting in dysregulation of the epigenome. One-carbon metabolism varies among individuals and has been shown to fluctuate with age as well as menopausal status (Zeisel 2009). Furthermore, elevated plasma homocysteine has been observed in both post-menopausal women (Hak, Polderman et al. 2000) and Alzheimer’s disease patients (Zeisel 2009 (Nazef, Khelil et al. 2014, Shen and Ji 2015), linking reproductive senescence to increased risk for neurocognitive disease later in life, possibly through epigenetic mechanisms.
The sequence of events leading up to reproductive senescence is complicated. Reproductive senescence is not solely initiated by ovarian depletion of oocytes but rather, is a function of both ovarian failure and hypothalamic aging. The hypothalamic-pituitary-gondal (HPG) axis, which is activated during puberty, is a negative feedback system in which pulsatile gonadotropin-releasing hormone (GnRH) produced in the hypothalamus stimulates luteinizing hormone (LH) and follicle stimulating hormone (FSH) production and secretion by the pituitary. LH and FSH then stimulate estrogen and production in the ovaries. Systemic estrogen then feeds back onto the pituitary and hypothalamus to modulate GnRH, LH and FSH production and secretion (Davis, Lambrinoudaki et al. 2015). Pituitary response to GnRH, and ovarian response to LH and FSH simultaneously decline with age resulting in the diminished sex steroid production characteristic of reproductive senescence, and a loss of negative feedback resulting in increased GnRH, LH and FSH production (Bottner, Leonhardt et al. 2007, Davis, Lambrinoudaki et al. 2015).
Preliminary evidence suggests that impaired one-carbon metabolism precedes decline in ovarian function may be partially responsible for initiating reproductive senescence. Dietary supplements of folate (vitamin B9) have been shown to increase luteal progesterone levels in pre-menopausal women and decrease risk for sporadic anovulatory cycles (Gaskins, Mumford et al. 2012). We hypothesize that one-carbon metabolism has the ability regulate the estrus cycle modulate initiation of reproductive senescence through loss of methyl-donor production needed to properly maintain the epigenome.
2. Methods
2.1. Animals
Animal studies were performed following National Institutes of Health guidelines on use of laboratory animals; protocols were approved by the University of Southern California Institutional Animal Care and Use Committee. A total of 201 young or middle-aged female Sprague-Dawley rats were obtained from Envigo Laboratories. Daily assessment of endocrine cycling of female rats was conducted from 5-12 months of age using rats that had given birth to at least one litter. Ovarian-function / cycle status was evaluated daily by the cytology of uterine cells obtained from lavage at 11am. The smear was morphologically characterized based on the four stages of the cycle: Estrus (E), Metestrus (M), Diestrus (D) and Proestrus (P). The regular 45 day estrus cycle is defined as the period between successive estrus smears (E, M, D, P, E, M, D, P, E). In addition to regular cycling animals, selected groups included middle-aged rodents at defined stages of “perimenopause” (Fig. 1B). The irregular group was defined as 2 contiguous cycles of >5 days characterized by prolonged diestrus stages. The acyclic (constant estrus) group was defined as persistent vaginal cornification lasting > 8 days. Rats at designated age (6m or 9-10m) and cycling status were euthanized at estrus or constant estrus. Five sets of animals were used in this study. The first set (85 rats in total) included all 4 experimental groups (Reg-6m, Reg-9-10m, Irreg-9–10m, and Acyc-9–10m). Of this set, N = 5–6 per group were used for DNA methylation analysis. A second set of rats, containing all four groups, (40 rats in total) was used for RNA-seq analysis. A third set of rats (30 rats in total) was used to assess timing of perimenopause onset, duration, and completion. Cycling status of these animals were monitored from 9 months regular cycling until they reached constant estrus. The fourth set of rats, containing only the Reg-6m group, (20 rats in total) was used for 5-aza-2’-deoxycitidine treatment. A fifth set of rats, containing only the Reg-6m group, (26 rats in total) was used for methionine treatment. Rats that did not meet the endocrine criteria for each group were excluded from analyses for this study.
Figure 1. Phenotypic characterization of endocrine aging of female rats during endocrine aging.

A) Transition of cycling stages with age: percentage of aging rats by cycling status from a cohort of 85 rats. B) Body and uterine weight of animals with different age and cycling status. Data were presented as average ± SEM, *p ≤ 0.0001, n = 9-20.
2.2. 5-aza-2’-deoxycytidine Treatment
0.1mg/ml 5-aza-2’-deoxycitadine-saline solution was prepared fresh for each use and stored on ice (Sigma Aldrich). Animals (N=6 per group) were subcutaneously injected with 0.25mg/kg drug, or an equivalent volume of saline, three times weekly (M,W,F) beginning at 6 months of age and until 9 months when cycle status was assessed.
2.3. Methionione Treatment
50 mg/ml Methionine-H2O stock solution will be prepared and stored (Sigma Aldrich). Regularly cycling animals were subcutaneously injected with 50mg/kg, 100mg/kg, 200mg/kg methionine, or an equivalent volume of H2O, three times weekly (M,W,F) beginning at 6 months of age and until 10 months when cycle status was assessed. All three methionine treatment groups were combined for analysis (n = 7) against the vehicle group (n = 4).
2.4. Tissue Collection
For the first two sets of animals, rats were euthanized and the brains rapidly dissected on ice. Cerebellum, brainstem, and hypothalamus were removed from each brain and the two hemispheres were separated. The cortical hemisphere was fully peeled laterally and hippocampus was then separated. Cerebellum, midbrain, brainstem, hypothalamus, and both cortexes and hippocampi were harvested and frozen at −80°C for. Ovaries and uterus were harvested and frozen at −80°C.
2.5. Nucleic Acid Extraction
Total RNA was extracted from tissue homogenized in trizol and purified using the PureLink® RNA Mini Kit (Thermo Fisher Scientific). RNA was DNase treated on column during purification (Thermo Fisher Scientific). DNA was extracted from tissue homogenized in lysis buffer [10mM Tris-HCl(pH 8.0), 1mM EDTA, 0.1% SDS], RNase treated (Zymo Research Corp., Irvine, CA), purified using phenol/chloroform/isoamyl alcohol, and then precipitated in isopropanol.
2.6. RNA-seq
Total RNA-seq libraries were constructed from RNA extracted from the hypothalamus of female rats. Samples were run on the Illumina HiSeq 2500 using 50bp PE to obtain a total read depth of roughly 50 million read pairs per sample. Raw data files in the FASTQ format underwent QA/QC and trimming procedure in the cloud-based Partek Flow environment (http://www.partek.com/). The paired end reads for each sample were then aligned using TopHat to the rat reference genome rn6 (Ensembl 80). Transcript assembly and quantification of aligned reads were carried out using Cufflinks. The Cufflinks output consisted of a list of differentially expressed genes (DEG) for each comparison.
2.7. RNA-seq Bioinformatic Analysis by Ingenuity Pathway Analysis (IPA)
Expression data for genes with the p-value < 0.05 were analyzed by IPA core analysis composed of a network analysis and an upstream regulator analysis. We used these relaxed criteria to maximize the coverage of the gene array results in the bioinformatic analyses. The network analysis identified biological connectivity among molecules in the dataset that were up- or down-regulated in a comparison (focus molecules that serve as “seeds” for generating networks) and their interactions with other molecules present in the Ingenuity Knowledge Base. Focus molecules were combined into networks that maximized their specific connectivity. Additional molecules from the Ingenuity Knowledge Base (interacting molecules) were used to specifically connect two or more smaller networks to merge them into a larger one. A network was composed of direct and indirect interactions among focus molecules and interacting molecules, with a maximum of 70 molecules per network. Generated networks were ranked by the network score according to their degree of relevance to the network eligible molecules from the dataset. The network score was calculated with Fisher’s exact test, taking into account the number of network eligible molecules in the network and the size of the network, as well as the total number of network eligible molecules analyzed and the total number of molecules in the Ingenuity Knowledge Base that were included in the network. Higher network scores are associated with lower probability of finding the observed number of network eligible molecules in a given network by chance.
IPA Upstream Regulator Analysis is a tool that predicts upstream regulators of observed gene expression changes based on the published literature and compiled in the Ingenuity Knowledge Base. The analysis determines known targets of each transcription regulator are present in the dataset, and also compares direction of change to expected based on published literature to predict likely relevant transcriptional regulators. If the observed direction of change is consistent with a particular activation state of the transcriptional regulator (“activated” or “inhibited”), then a prediction is made for that activation state (Z-score). For each potential regulator two statistical measures, an overlap p-value and an activation z-score are computed. The overlap p-value identifies likely upstream regulators based on significant overlap between dataset genes and known targets regulated by a regulator. The activation z-score is used to infer likely activation states of upstream regulators based on comparison with a model that assigns random regulation direction. The upstream regulator list is generated using computational predictions and is not based on experimental evidence in the rat model used in this study. Although in practice, z-scores greater than 2 or smaller than −2 can be considered significant, our particular interest was in identifying upstream regulators potentially controlling transcription across the endocrine transition states. Thus, all molecules with significant p-values regardless of z-score were further evaluated.
2.8. Global DNA methylation Analysis
100ng of genomic DNA from hypothalamus or blood was used to determine total 5-methylcytosine (5-mC) using the 5-mC DNA ELISA kit (Zymo Research Corp., Irvine, CA, USA) as per the manufacturer’s instruction.
2.9. Genome-Wide DNA Methylation Profiling
A modified reduced representative bisulfite sequencing (RRBS) protocol (Methyl-MiniSeq™) was used to prepare libraries from 200-500 ng of genomic DNA digested with 60 units of TaqαI and 30 units of MspI (NEB) sequentially and then extracted with Zymo Research (ZR) DNA Clean & ConcentratorTM-5 kit (Cat#: D4003). Fragments were ligated to pre-annealed adapters containing 5’-methyl-cytosine instead of cytosine according to Illumina’s specified guidelines (www.illumina.com). Adaptor-ligated fragments of 150–250 bp and 250–350 bp in size were recovered from a 2.5% NuSieve 1: 1 agarose gel (ZymocleanTM Gel DNA Recovery Kit, ZR Cat#: D4001). The fragments were then bisulfite-treated using the EZ DNA Methylation-LightningTM Kit (ZR, Cat#: D5020). Preparative-scale PCR was performed and the resulting products were purified (DNA Clean & ConcentratorTM - ZR, Cat#D4005) for sequencing on an Illumina HiSeq.
2.10. Reduced Representation Bisulfite Sequencing (RRBS) Sequence Alignments and Data Analysis
Sequence reads from bisulfite-treated MiniSeq™ libraries were identified using standard Illumina base-calling software and then analyzed using a Zymo Research proprietary analysis pipeline, which is written in Python and used Bismark (http://www.bioinformatics.babraham.ac.uk/projects/bismark/) to perform the alignment to the rn6 genome. Index files were constructed using the bismark_genome_preparation command and the entire reference genome. The --non_directional parameter was applied while running Bismark. All other parameters were set to default. Filled-in nucleotides were trimmed off when doing methylation calling. The methylation level of each sampled cytosine was estimated as the number of reads reporting a C, divided by the total number of reads reporting a C or T. Fisher’s exact test or t-test was performed for each CpG site which has at least five reads coverage, and promoter, gene body and CpG island annotations were added for each CpG included in the comparison.
3. Results
3.1. The Perimenopause Animal Model (PAM)
Cycle duration of five month-old female Sprague-Dawley rats was assessed longitudinally for 5 months. Based on cycle data, rats were stratified into groups according to the stage of ovarian senescence following the classification of the human perimenopause-menopause transition as per Stages of Reproductive Aging Workshop (STRAW) (Harlow, Gass et al. 2012, Finch 2014). The endocrine aging groups included regular cyclers (4–5 day cycles), irregular cyclers (5–8 day cycles), and acyclic (no cycling within 9 days) at 9–10 months.
Cycling patterns were assessed daily and stratification of endocrine status was analyzed at 6 months, 8 months, and 9 months of age. At 6 months, 82% of females were regular cyclers (reg 6m), 18% were irregular cyclers at 8 months, 62% were regular cyclers and the percentage of irregular cyclers increased to 43%, with the first appearance of acyclicity in 5%; at 9 months, the percent of regular cyclers declined to 37% (reg 9m), the percent of irregular cyclers decreased to 30% (irreg 9m), and the remaining 33% were acyclic (acyc 9m) (Fig. 1A). An increase in body weight was observed between the 6 month- and the 9 month-old regular cyclers (p < 0.0001) and was maintained across subsequent stages (Fig. 1D). There were no differences in body weight among 9 month-old animals at different endocrine stages (p = 0.0515). Uterine weight did not differ across the 5 endocrine phenotypes (p = 0.30) and was consistent with all animals being euthanized at estrus or constant estrus (Fig. 1B).
3.2. RNA-seq
A total of 20877 transcripts in the hypothalamus were identified, sequenced, and analyzed (Fig. 2). Between 6-9 months, 2094 (10%) genes were significantly different (p< 0.05). Between 9 month regular and irregular, and irregular and acyclic groups, 374 (2%) and 442 (2%) genes, respectively, were significantly different (p< 0.05) (Fig 2). Ingenuity Pathway Analysis (IPA) was used to identify canonical signaling pathways associated with changes in gene expression between endocrine groups. 446 significant signaling pathways were identified within reg 6m – reg 9m, 256 within reg 9m – irreg 9m, and 195 within irreg 9m – acyc 9m group comparisons. Selected top canonical pathways, and the genes involved, are summarized in the supplemental information (supplemental Table 1).
Figure 2. Hypothalamic gene expression across endocrine aging cycling phenotypes.

A total of 20877 transcripts in the hypothalamus were identified and sequenced using Illumina HiSeq 2500 using 50bp PE to obtain a total read depth of roughly 50 million read pairs per sample. Of the total number of transcripts sequenced, 2094 (10%), 374 (2%), 442 (2%) genes were significantly different between the 6 vs 9 month old regular cyclers, 9 month old regular vs irregular cyclers, and 9 month irregular vs acyclic groups, respectively (p< 0.05).
The top upstream transcriptional regulator across all group comparisons was beta-estradiol (E2) (Table 2). E2 signaling was predicted to be involved in the down-regulation of genes between 6-9 months, prior to perimenopause onset of irregular cycling. GnRH and KISS1 were also identified as regulators across all group comparisons. SAM and homocysteine were predicted to down-regulate related genes between the reg 6m and reg 9m groups, suggesting that impaired one carbon metabolism may be responsible for transcriptional changes seen during this time frame, possibly through epigenetic mechanisms. S-adenoslyhomocysteine, which is formed by the demethylation of SAM during methyl-group donation, was identified as a regulator during reg 9m – irreg 9m and irreg 9m – acyc 9m transitions.
Table 2: Correlation of Cytosine Methylation (r-value) -.
CpG, CHG, and CHH correlations between endocrine groups. For replicates, one would expect r = 1, demonstrating perfect 1:1 correlation. Considering regulatory role of CpGs and the inbred genetic background of the, it is expected that CpG methylation between two groups be close to r= 1. CHG and CHH correlations have a lower r-value between groups.
| Cytosine context |
Reg 6m – Reg 9m | Reg 9m – Irreg 9m | Irreg 9m – Acyc 9m |
|---|---|---|---|
| r-value | |||
| CpG | 0.9531 | 0.9521 | 0.9558 |
| CHG | 0.4441 | 0.4552 | 0.4978 |
| CHH | 0.4711 | 0.4760 | 0.5240 |
3.3. Epigenetic changes across perimenopause
Hypothalamic samples underwent RRBS analysis (n = 5 – 6 / group) with an average of 36 million (M) read pairs obtained for each sample (range = 31 – 46M read pairs). The percentage of the raw reads which were mapped to the rn6 genome ranged from 44 – 58%. An average of 4.7 million unique CpG sites were sequenced for each sample (range = 3.8 ‒ 5.4M) at an average depth of 13X (range = 10 – 16X).
Considering the regulatory role of CpG sites within the genome and the inbred genetic background of the animals used in this study, it was expected that the correlation (r) of CpG methylation patterns between two groups would approach 1 for the same tissue type. However, thousands of statistically significant differentially methylated CpG sites were evident indicative of epigenetic regulation of the transcriptional states in the different endocrine statuses. The regulatory role of non-CpG methylation is less clearly established and CHG and CHH sites (“H” refers to different cytosine methylation contexts, namely CpG, CHG, and CHH, where H means “not G” (A,T, or C) are more prone to mutation. This was reflected in our data by a lower r-value for CHG and CHH comparisons between groups (Table 3), demonstrating that CHG and CHH patterns are less similar between groups. However, heatmaps displaying hierarchical clustering of the top 100 significant CHG and CHH sites showed very little differences between groups, suggesting that the genome-wide differences observed are largely on an individual basis and that very few sites correlate with endocrine status (Supplemental figures 1-3).
Table 3: Correlation Between Changes in DNA Methylation and RNA Expression in Genes Related to the HPG-Axis and Hormone Signaling.
Fold changes and their respective p-values (RNA column) are listed for each endocrine group comparison. DM column at what endocrine status differential cytosine methylation was observed, within what region (promoter, exon, or intron), and whether the site(s) were hypo- or hyper-methylated. Hyper- or hypo-methylated is defined as 0-33% more, or less respectively, methylated than reference (p-value < 0.05).
| Reg 6m – Reg 9m |
Reg 9m – Irreg 9m |
Irreg 9m – Acyc 9m |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Symbol | RNA | DM | RNA | DM | RNA | DM | ||||||
|
| ||||||||||||
| GNRH1 | −1.7 | (0.0368) | - | 1.9 | (0.0114) | - | −1.6 | (0.0469) | - | |||
| FSHB | - | - | 1.8 | (0.0124) | - | −1.8 | (0.0157) | - | ||||
| PRL | 73.5 | (0.0106) | - | −3.3 | (0.00085) | - | - | - | ||||
| PRLHR | −1.6 | (0.00655) | - | - | - | - | - | |||||
| PRLR | −1.4 | (0.0223) | - | - | (1) Intron | HYPER | - | - | ||||
| TRH | −1.4 | (0.00005) | - | 1.3 | (0.00005) | - | −1.2 | (0.0007) | - | |||
| TSHR | 1.6 | (0.00005) | - | - | (1) Intron | HYPER | - | - | ||||
| DRD5 | −1.4 | (0.0464) | (1) Exon | HYPO | - | - | - | - | ||||
| DRD2 | −1.3 | (0.0011) | - | - | - | - | - | |||||
| ESR2 | −1.5 | (0.0138) | - | - | - | - | - | |||||
| ESRRG | −1.3 | (0.00035) | (15) Intron | (8) HYPO (7) HYPER |
- | (6) Intron | (5) HYPO (1) HYPER |
- | (4) Intron | (1) HYPO (3) HYPER |
||
| OXT | −3.5 | (0.00005) | - | 3.5 | (0.00005) | - | - | - | ||||
| CRH | −1.9 | (0.00005) | (1) Promoter | HYPO | 1.4 | (0.00845) | - | - | - | |||
| CRHBP | −1.3 | (0.0216) | (1) Intron | HYPER | - | - | - | - | ||||
| KISS1 | 1.4 | (0.00805) | - | - | - | - | - | |||||
3.4. GnRH signaling & the HPG Axis
In the hypothalamus, GnRH transcripts showed a dynamic pattern of expression. GnRH mRNA decreased by 1.7-fold between 6 to 9 months, increased by 1.9-fold at the onset of irregular cycling (irreg 9), and finally decreased once again by 1.5 fold in the acyclic animals (acyc 9) (Table 6). Hypothalamic expression of the follicle stimulating hormone subunit B (FSHB) was unchanged between 6 to 9 months but subsequently increased at the onset of irregular cycling followed by a decline in the acyclic group (Table 6). No significant methylation differences in the GnRH1 or FSHB genes were observed (Table 3).
Table 6: Correlation Between Changes in DNA Methylation and RNA Expression in Genes involved in Melatonin Signaling and Circadian Rhythm.
Fold changes and their respective p-values (RNA column) are listed for each endocrine group comparison. DM column denotes at what endocrine status differential cytosine methylation was observed, how many sites within what regions (promoter, exon, or intron), and whether the site(s) were hypo-, strongly(S) hypo, hyper-, or strongly hyper-methylated. Hyper- or hypo-methylated is defined as 0-33% more, or less respectively, methylated than reference (p-value < 0.05). Strongly hyper- or strongly hypo-methylated is defined as 33-100% more, or less respectively, methylated than reference (p-value < 0.05).
| Reg 6m – Reg 9m |
Reg 9m – Irreg 9m |
Irreg 9m – Acyc 9m |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Symbol | RNA | DM | RNA | DM | RNA | DM | ||||||
|
| ||||||||||||
| ABAT | −1.1 | (0.0358) | (1) Intron | - | - | - | - | |||||
| ADCY1 | −1.7 | (0.00005) | (3) Intron | (1)HYPO (2) HYPER |
- | - | - | (1) Exon | HYPER | |||
| ADCY3 | −1.1 | (0.0276) | (1) Intron | HYPO | - | - | - | (1) Intron | HYPER | |||
| ATF2 | −1.2 | (0.00675) | - | - | - | - | (1) Intron | HYPER | ||||
| AVP | −1.4 | (0.00005) | (1) Promoter | S HYPER | 1.7 | (0.00005) | - | - | - | |||
| BHLHE40 | 1.1 | (0.0142) | - | −1.2 | (0.0183) | - | - | - | ||||
| CAMK2D | −1.1 | (0.028) | (1) Intron | S HYPER | - | (1) Intron | S HYPER | - | (2) Intron | S HYPO | ||
| CAMK4 | −1.3 | (0.0216) | (3) Intron | (2) HYPO (1) HYPER |
- | - | - | (1) Intron | HYPO | |||
| CACNA1C | −1.1 | (0.0234) | (2) Exon (2) Intron |
(3)HYPO (1)HYPER |
- | (4) Exon (3) Intron |
(3) HYPO (4) HYPER |
- | (2) Exon (2) Promoter |
(1) HYPO (3) HYPER |
||
| CDH1 | 2.1 | (0.00005) | (1) Exon | HYPER | −1.7 | (0.00005) | (1) Intron | HYPO | - | - | ||
| CREB1 | −1.3 | (0.0491) | - | - | - | - | - | |||||
| CREB5 | - | - | 1.2 | (0.0365) | (5) Intron | (2) HYPO (3) HYPER |
- | (2) Intron | HYPER | |||
| ELMO1 | −1.2 | (0.0142) | - | - | (2) Intron | (1) HYPO (1) HYPER |
- | (2) Intron | (1) HYPO (1) HYPER |
|||
| EZR | 1.3 | (0.00005) | (2) Intron | HYPO | - | (1) Exon (4) Intron |
(3) HYPO (2) HYPER |
- | (2) Exon (3) Intron |
(4) HYPO (1) HYPER |
||
| GNAI1 | −1.1 | (0.0189) | - | - | - | - | - | |||||
| GNAQ | −1.2 | (0.00425) | (1) Intron | HYPO | - | - | - | (1) Intron | HYPO | |||
| GRIA1 | −1.2 | (0.0035) | (1) Intron | HYPO | - | (2) Intron | (1) S HYPER (1) HYPER |
- | - | |||
| GRIA3 | −1.1 | (0.043) | - | - | - | - | - | |||||
| GRIN2A | −1.7 | (0.00005) | - | - | (1) Intron | HYPO | −1.3 | (0.0361) | (2) Intron | (1) HYPO (1) HYPER |
||
| GRIN2B | −1.2 | (0.0035) | (6) Intron | (1) S HYPO (4) HYPO (1) HYPER |
- | - | - | - | ||||
| GRIN2D | −1.2 | (0.00595) | - | 1.2 | (0.0192) | - | −1.2 | (0.0222) | - | |||
| GRIN3A | −1.3 | (0.00065) | - | - | - | - | - | |||||
| IGF1R | −1.2 | (0.00685) | - | - | (2) Intron | (1) HYPO (1) HYPER |
- | (1) Exon (8) Intron |
(4) HYPO (5) HYPER |
|||
| ITPR3 | 1.3 | (0.00655) | (2) Intron | (1) HYPO (1) HYPER |
- | (4) Intron | (3) HYPO (1) HYPER |
- | (1) Intron | HYPO | ||
| MAPK3 | 1.1 | (0.0417) | - | - | - | - | - | |||||
| NOS1 | −1.2 | (0.0172) | (2) Intron | HYPO | - | (2) Intron | (1) HYPO (1) HYPER |
- | (1) Intron | HYPER | ||
| PCLO | −1.2 | (0.0002) | - | 1.1 | (0.0137) | (1) Intron | HYPER | - | (1) Exon | HYPO | ||
| PER3 | 1.1 | (0.014) | (1) Exon (1) Intron |
(1) HYPO (1) HYPER |
- | (1) Exon (1) Intron |
HYPO | - | - | |||
| PLCB1 | −1.2 | (0.00215) | (2) Intron | (1) HYPO (1) HYPER |
- | (2) Intron | HYPO | - | (2) Intron | (1) HYPO (1) HYPER |
||
| PLCB3 | 1.2 | (0.004) | - | - | - | - | - | |||||
| PLCB4 | −1.1 | (0.0376) | (3) Intron | (2) HYPO (1) HYPER |
- | (3) Intron | (1) HYPO (2) HYPER |
- | (2) Intron | HYPO | ||
| PLCD1 | 1.2 | (0.0253) | - | - | - | - | - | |||||
| PLCL2 | −1.1 | (0.0282) | - | - | - | - | (1) Intron | HYPO | ||||
| PPP2R2C | −1.2 | (0.0004) | (1) Intron | HYPO | - | - | - | - | ||||
| PRKACB | −1.3 | (0.00005) | (1) Promoter (1) Intron |
(1) HYPO (1) HYPER |
- | - | - | - | ||||
| PRKAR1B | −1.2 | (0.00485) | - | - | - | - | (2) Intron | HYPER | ||||
| PRKAR2A | −1.2 | (0.00245) | - | - | - | - | - | |||||
| PRKCB | −1.3 | (0.00005) | (4) Intron | (1) HYPO (3) HYPER |
- | - | - | (1) Intron | HYPER | |||
| PRKCD | −1.6 | (0.00005) | - | - | - | - | - | |||||
| PRKCE | −1.2 | (0.0104) | (6) Intron | (5) HYPO (1) HYPER |
(8) Intron | (4) HYPO (4) HYPER |
- | (5) Intron | (2) HYPO (1) S HYPER (2) HYPER |
|||
| PRKCH | −1.3 | (0.04) | (5) Intron | (1) HYPO (4) HYPER |
- | (3) Intron | (1) HYPO (2) HYPER |
- | (4) Intron | HYPO | ||
| PRKG2 | −1.2 | (0.025) | - | - | - | - | - | |||||
| SHC3 | −1.3 | (0.0019) | (2) Intron | HYPER | - | - | - | (1) Intron | HYPO | |||
| VIP | −1.4 | (0.00005) | - | - | - | 1.4 | (0.0024) | - | ||||
3.5. Increase in Prolactin Production
The hormone prolactin plays a key role in fertility by negatively regulating FSH and GnRH. Although primarily produced in the pituitary, the hypothalamus can produce significant amounts of prolactin (DeVito 1988). In the pituitary, activation of dopamine receptor D2 (DRD2) suppresses prolactin gene expression as well as secretion (Fitzgerald and Dinan 2008). Furthermore, thyropropin-releasing hormone (TRH) produced in the hypothalamus acts on the pituitary to increase prolactin expression and secretion (Fitzgerald and Dinan 2008).
Prior to perimenopause, between 6 to 9 months, there was a substantial 73.5-fold increase in hypothalamic prolactin expression. Accompanying this increase, factors that suppress prolactin secretion exhibited a modest −1.6-fold decrease in transcripts coding for the prolactin releasing hormone receptor (PRLHR) and a −1.4 fold prolactin receptor (PRLR), both. TRH declined −1.4 fold, and dopamine receptors D2 and D5 declined −1.3 and −1.4 fold, respectively. During the transition from regular to irregular cycling, prolactin decreased, but remained elevated compared to 6 months, and remained unchanged through the transition to acycling. However, prolactin remained elevated compared to pre-perimenopause levels. PRLHR, PRLR, DRD2, and DRD5 did not show any further changes in expression. TRH increased 1.2 fold from regular to irregular cycling and showed no significant changes during the irregular to acycling transition.
There were no observed DNA methylation alterations in PRL or its’ receptors PRLHR and PRLR that correlated to changes in expression levels. However, hypomethylation of DRD5 was observed during 6-9 months when DRD5 expression declined (Table 3).
3.6. Other hormones and receptors
Estrogen receptor β (Esr2) gene expression decreased −1.5 fold from 6-9 months and remained unchanged through perimenopause. Oxytocin (OXT) mRNA expression temporarily dropped during 6-9 months before recovering between RC9-IR9. Corticotorpin releasing hormone (CRH) and corticotorpin releasing hormone binding protein (CRHBP) expression decreased −1.9 and −1.3 fold respectively. A single site within the CRH promoter region was hypomethylated, and a single site within a CRHBP intron was hypermethylated between 6-9 months. No further changes in DNA methylation occurred in the two genes. CRH expression increased slightly (1.4 fold) between RC9-IR9. Estrogen Related Receptor Gamma (ESRRG) transcriptionally activates DNA cytosine-5-methyltransferases 1 (Dnmt1) via binding to estrogen response elements in the DNMT1 promoters. Between 6-9 months, ESRRG transcription drops −1.3 fold and remains unchanged through the perimenopause transition. This decreased expression was accompanied by changes in 15 DNA methylation sites throughout the gene. Although ESRRG RNA levels remained unchanged through the transition, DNA methylation continued to change throughout perimenopause (Table 3).
3.7. Changes in Glutamate Signaling
From the RNA expression data, we inferred a substantial decrease in glutamate receptor signaling between 6-9 months which did not recover during perimenopause. Fifteen glutamate receptors and 4 glutamate transporter genes were significantly down-regulated in the regular cycling 9 month group compared to 6 month. Of these, 3 receptors and 2 solute carriers also underwent changes in DNA methylation between 6-9 months. Only 1/15 receptor RNA levels (GRIND2) increase at the onset of irregularity before decreasing again at the ayclic stage. In addition, the glutamate decarboxylase, GAD2, increases in irregular cycling animals. Including GRIND2, two additional glutamate receptors continue to drop at acyclicity. An additional glutamate receptor (GRIK3) had unchanged expression until the onset of acyclicity, at which it declined. Several genes saw changes in DNA methylation across perimenopause that was not associated with changes in gene transcription (Table 4).
Table 4: Correlation Between Changes in DNA Methylation and RNA Expression in Genes Related to Glutamate Signaling.
Fold changes and their respective p-values (RNA column) are listed for each endocrine group comparison. DM column at what endocrine status differential cytosine methylation was observed, within what region (promoter, exon, or intron), and whether the site(s) were hypo-, strongly(S) hypo, hyper-, or strongly hyper-methylated. Hyper- or hypo-methylated is defined as 0-33% more, or less respectively, methylated than reference (p-value < 0.05). Strongly hyper- or strongly hypo-methylated is defined as 33-100% more, or less respectively, methylated than reference (p-value < 0.05).
| Reg 6m – Reg 9m | Reg 9m – Irreg 9m | Irreg 9m – Acyc 9m | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Symbol | RNA | DM | RNA | DM | RNA | DM | ||||||
| GRIA1 | −1.2 | (0.0035) | - | - | (2) Intron | S HYPER | - | - | ||||
| GRIA3 | −1.1 | (0.0430) | - | - | - | - | - | |||||
| GRIA4 | −1.3 | (0.0001) | - | - | - | - | (1) Promoter | HYPER | ||||
| Grid1 | −1.1 | (0.0331) | - | - | (6) Intron | (5) HYPO (1) HYPER |
- | (6) Intron | (3) HYPO (3) HYPER |
|||
| GRID2 | −1.2 | (0.0323) | (2) Intron | HYPER | - | (4) Intron | (3) HYPO (1) HYPER |
- | (2) Intron | (1) HYPO (1) HYPER |
||
| GRID2IP | −1.6 | (0.0002) | - | - | (1) Exon | HYPO | - | |||||
| GRIK1 | −1.2 | (0.0071) | (1) Intron (1) Exon |
HYPER | - | (1) Intron | HYPER | - | (1) Intron | HYPO | ||
| GRIK3 | - | (4) Intron | (2) HYPO (2) HYPER |
- | (4) Intron | HYPER | −1.2 | (0.0432) | (2) Intron | HYPO | ||
| GRIN2A | −1.7 | (0.0001) | - | - | (1) Intron | HYPO | −1.3 | (0.0361) | (2) Intron | (1) HYPO (1) HYPER |
||
| GRIN2B | −1.2 | (0.0035) | (6) Intron | (4) HYPO (1) HYPER (1) S HYPER |
- | - | - | - | ||||
| GRIN2D | −1.2 | (0.0060) | - | 1.1 | (0.0192) | - | −1.2 | (0.0222) | - | |||
| GRIN3A | −1.3 | (0.0007) | - | - | - | - | - | |||||
| GRM1 | −1.2 | (0.0003) | - | - | - | - | (1) Intron | HYPER | ||||
| GRM3 | −1.2 | (0.0058) | - | - | - | - | (1) Intron | HYPO | ||||
| GRM5 | −1.2 | (0.0086) | - | - | - | - | (1) Promoter | HYPER | ||||
| GRM7 | −1.2 | (0.0082) | - | - | (3) Intron | (1) S HYPO (2) HYPO |
- | (1) Intron | HYPO | |||
| VGLUT1 | −3 | (0.0095) | - | - | - | - | - | |||||
| VGLUT3 | −1.4 | (0.0236) | (1) Promoter | HYPO | - | - | - | - | ||||
| GLT1 | −1.2 | (0.0006) | (2) Intron | (1) HYPO (1) HYPER |
- | - | - | - | ||||
| NAT2 | −1.2 | (0.0011) | - | - | - | - | (1) Intron | HYPER | ||||
3.8. Changes in GABA Signaling
The RNA levels of 4 GABA receptors (GABRA1, GABRB2, GABRG2, and GABRG3) and 1 transporter gene (SLC32A1) were significantly down regulated between 6-9 months. SLC32A1 expression increased at the irregular cycling stage and decreased at the onset of acyclicity. In contrast, 3 GABA receptors (DBI, GABRA5, and GABRD) were up regulated between 6-9 months. GABRA5 expression decreased at irregular cycling and did not change further. A single site was hypermethylated and associated with decreased expression at 6-9 months in GABRG3. The remaining receptors and transporters exhibited no changes in DNA methylation across the perimenopause transition (Table 5).
Table 5: Correlation Between Changes in DNA Methylation and RNA Expression in Genes Related to GABA Signaling.
Fold changes and their respective p-values (RNA column) are listed for each endocrine group comparison. DM column at what endocrine status differential cytosine methylation was observed, within what region (promoter, exon, or intron), and whether the site(s) were hypo- or hyper-methylated. Hyper- or hypo-methylated is defined as 0-33% more, or less respectively, methylated than reference (p-value < 0.05).
| Reg 6m – Reg 9m |
Reg 9m – Irreg 9m |
Irreg 9m – Acyc 9m |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Symbol | RNA | DM | RNA | DM | RNA | DM | ||||||
|
| ||||||||||||
| DBI | 1.2 | (0.00005) | - | - | - | - | ||||||
| GABRA1 | −1.3 | (0.00005) | - | - | - | - | - | |||||
| GABRA5 | 1.2 | (0.00005) | - | −1.1 | 0.0443 | - | - | - | ||||
| GABRB2 | −1.2 | (0.0041) | - | - | - | - | - | |||||
| GABRD | 1.1 | (0.008) | - | - | - | - | - | |||||
| GABRG2 | −1.3 | (0.00005) | - | - | - | - | - | |||||
| GABRG3 | −1.8 | (0.00015) | (1) Intron | HYPER | - | - | - | (1) Intron | HYPO | |||
| SLC32A1 | −1.4 | (0.00515) | - | 1.2 | 0.00035 | - | −1.2 | 0.00145 | - | |||
3.9. Melatonin and Circadian Rhythm signaling
Post-menopausal women exhibit a loss of circadian rhythm control (Gomez-Santos, Saura et al. 2016) and are more likely to exhibit sleep disturbances, such as insomnia or poor sleep quality, compared to pre-menopausal women (Jehan, Masters-Isarilov et al. 2015). Both RNA and DNA methylation pathway analyses identified melatonin signaling and circadian rhythm as systems undergoing change during the perimenopause transition. Alterations in these systems first appeared during early hypothalamic aging, between 6-9 months. A majority (36/44) of the genes showed decreased RNA levels while DNA methylation changes consisted of both hypo- and hyper- methylation in primarily intron regions. While transcriptional changes predominantly occurred prior to onset of irregular cycling, changes in the DNA methylation continued to accumulate throughout the transition (Table 6).
3.10. Epigenome Maintenance and One Carbon Metabolism
Multiple genes involved in epigenetic maintenance of DNA methylation and histone modifications changed in expression across the perimenopause transition (Table 7). In total, 15 genes were identified, including two DNA methyltransferases (DNMT), 2 ten-eleven translocation methylcytosine dioxygenases (Tet), 7 histone methyltransferases, 2 histone deacetylases, and 1 histone demethylase. The majority of the changes occurred between 6-9 months with 11 genes exhibiting change in genes involved in DNA methylation (Table 7). One gene, a histone methyltransferase, changed at the onset of irregular cycling whereas five genes were down regulated in the acyclic group. The temporal pattern for change in epigenetic gene expression suggest that epigenetic reorganization begins between 6 to 9 months, prior to the onset of perimenopause.
Table 7: Correlation Between Changes in DNA Methylation and RNA Expression in Genes in Epigenome Regulation.
Fold changes and their respective p-values (RNA column) are listed for each endocrine group comparison. DM column at what endocrine status differential cytosine methylation was observed, within what region (promoter, exon, or intron), and whether the site(s) were hypo- or hyper-methylated. Hyper- or hypo-methylated is defined as 0-33% more, or less respectively, methylated than reference (p-value < 0.05).
| Reg 6m – Reg 9m |
Reg 9m – Irreg 9m |
Irreg 9m – Acyc 9m |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Symbol | RNA | DM | RNA | DM | RNA | DM | ||||||
|
| ||||||||||||
| DNMT3A | - | - | - | - | −1.1 | (0.0392) | - | |||||
| DNMT3B | - | - | - | - | −1.5 | (0.0212) | - | |||||
| TET1 | −1.4 | (0.00565) | - | −1.1 | (0.0443) | - | - | - | ||||
| TET3 | - | - | - | - | −1.2 | (0.0232) | - | |||||
| MECP2 | −1.2 | (0.0106) | - | - | - | - | - | |||||
| EHMT1 | −1.2 | (0.0262) | - | - | - | - | ||||||
| PRMT8 | −1.2 | (0.041) | (1) Intron | HYPER | - | (1) Intron | HYPO | - | (1) Intron | HYPO | ||
| METTL7A | 1.2 | (0.00185) | - | 1.2 | (0.00035) | - | - | |||||
| METTL8 | −1.3 | (0.0311) | - | - | - | - | ||||||
| HDAC1 | 1.2 | (0.0089) | - | - | - | - | ||||||
| HDAC5 | −1.2 | (0.00025) | (1) Intron | HYPO | - | (1) Intron | HYPER | - | ||||
| KDM6B | 1.1 | (0.0137) | - | - | - | - | ||||||
| KMT2A | −1.1 | (0.0126) | - | 1.15 | (0.00615) | - | −1.16 | (0.0079) | ||||
| KMT2B | - | - | - | - | −1.12 | (0.038) | ||||||
Nine genes involved in one-carbon metabolism and SAM production were significantly changed across the groups (Table 8). Eight of these genes changed between 6-9 months. None of the identified genes were found to change during the transition to irregular cycling, and only one gene was found to change at the onset of acyclicity. Again, the data indicate that systems involved in epigenetic maintenance and reorganization are altered prior to manifestation of irregular cyclicity that characterizes the perimenopause.
Table 8: Correlation Between Changes in DNA Methylation and RNA Expression in Genes Related to One Carbon Metabolism.
Fold changes and their respective p-values (RNA column) are listed for each endocrine group comparison. DM column at what endocrine status differential cytosine methylation was observed, within what region (promoter, exon, or intron), and whether the site(s) were hypo- or hyper-methylated. Hyper- or hypo-methylated is defined as 0-33% more, or less respectively, methylated than reference (p-value < 0.05).
| Reg 6m – Reg 9m |
Reg 9m – Irreg 9m |
Irreg 9m – Acyc 9m |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Symbol | RNA | DM | RNA | DM | RNA | DM | ||||||
|
| ||||||||||||
| MTHFR | - | - | - | - | −1.2 | (0.0481) | - | |||||
| SLC44A2 | 1.1 | (0.0314) | - | - | (1) Intron | HYPO | - | (1) Intron | HYPER | |||
| FOLR2 | 2.5 | (0.003) | - | - | (1) Intron | HYPO | - | (1) Intron | HYPER | |||
| TYMS | −1.4 | (0.0067) | - | - | - | - | - | |||||
| MUT | −1.1 | (0.0462) | - | - | - | - | - | |||||
| PLD1 | 1.2 | (0.0144) | - | - | (2) Intron | HYPO | - | (2) Intron | HYPER | |||
| PLD2 | 1.2 | (0.00605) | - | - | - | - | (1) Exon (1) Intron |
(1) HYPO (1) HYPER |
||||
| PLD4 | 1.2 | (0.0273) | - | - | - | - | - | |||||
| NAPEPLD | −1.3 | (0.0252) | - | - | - | - | - | |||||
3.11. Changes in Hypothalamic Global DNA Methylation
In the hypothalamus, global DNA methylation significantly declined (p=0.0035) at the onset of irregular cycling which marks the onset of perimenopause (Fig. 3A). Decreased DNA methylation levels were sustained throughout the transition (p=0.0051) (Fig. 5A). Further analysis of the pattern of DNA methylation revealed that while animals were still regular cycling at 6-9 months, two distinct populations of global DNA methylation was apparent and were statistically different (p=0.0026) (Fig. 5B). One third (33%) of the 6-9 month old animals exhibited global DNA methylation levels comparable to 9 month regular cycling animals. In contrast, two thirds (66%) of the 6-9 month old animals exhibited global DNA methylation levels comparable to irregular (p=0.0028) (Fig. 5B). These findings suggest that individual differences in epigenetic profile may contribute to individual differences in endocrine aging.
Figure 3. Magnitude of global DNA methylation across endocrine aging cycling phenotypes.

A) Hypothalamic DNA methylation declined at the onset of perimenopause (p= 0.0035) and remains low through completion of the transition (p=0.0051). B) Two statistically different populations exist within the 6 month regular group (p=0.0026). When compared separately, the 6 month “low” population has statistically decreased levels of global DNA methylation (p=0.0028).
3.12. DNA Methylation and Menopause Timing
3.12.1. 5-aza-2’-deoxycitidine Treatment
To determine if DNA methylation directly influenced onset and progression of perimenopause, 9-month old animals with 5-aza-2’-deoxycitidine (5-aza), a de-methylating agent followed by assessment of cyclicity and perimenopause timing. 5-aza hypomethylates DNA by effectively depleting DNMT’s, preventing methylation of cytosines. If perimenopause is epigenetically regulated then treatment with 5-aza should disrupt timing of onset and/or completion of the perimenopause transition. Because the RNA-seq and global DNA methylation analyses suggested that hypothalamic aging occured between 6-9 months, before the onset of irregular cycling, treatment was initiated at 6 months. Regular cycling animals were injected with 5-aza or vehicle three times a week for three months. Because hypothalamic DNA methylation dramatically declined at the onset of perimenopause (Fig. 3), we hypothesized that 5-aza-induced hypomethylation would induce premature of onset and/or completion of the perimenopause transition.
To monitor the effectiveness of the 5-aza treatments, we assessed DNA methylation levels in peripheral blood to predict hypomethylation in the brain. To verify that animals were indeed responding to 5-aza treatments, we collected serial blood collections and measured DNA methylation levels for 3 weeks after initiation of treatment. During the 3 weeks, we observed a continuous decline in global DNA methylation that was not observed in saline-treated animals. In 5-aza treated animals, DNA methylation levels were significantly lower (p=0.0120) at week 3 compared to week 1 (Fig. 4). While 5-aza treatment was use to experimentally hypomethylate DNA, methionine treatment was intended to stabilize DNA methylation and slow epigenetic aging. Because we expected methionine treated animals to look nearly identical to their controls, we did not assess “effectiveness” of methionine during the course of the treatment.
Figure 4. Impact of 5-aza-2’-deoxycitidine (5-aza) on global DNA methylation in blood following systemic treatment.

DNA methylation in peripheral blood was significantly hypo-methylated by week 3 of 5-aza treatment (p=0.0120; N=6)
At 9 months of age a significantly higher proportion of 5-aza-treated animals had transitioned into constant estrus and were no longer cycling (acyc), as assessed by Chi-square analysis (p = 0.033). In contrast, all of the vehicle treated animals continued to cycle (regularly or irregularly) (Fig. 5). Survival curve analyses, using log-rank (Mantel-Cox) test followed by Gehan-Breslow-Wilcoxon test, indicated a non-significant trend towards accelerated onset of perimenopause in the 5-aza treated animals (Fig. 6A). More striking acceleration was evident in the earlier age to exit perimenopause and enter into menopause of the 5-aza treated animals (Mantel-Cox p= 0.0043 ; Gehan-Brewslow-Wilcoxon p= 0.006) (Fig. 6B). These results indicate that perimenopause duration and menopause timing is regulated by epigenetic mechanisms including DNA methylation.
Figure 5. Inhibition of DNA methylation and impact on endocrine aging phenotype.

Using Reduced Representation Bisulfite Sequencing (RRBS), DNA methylation at cytosine residues were determined. At 9 months of age, the 5-aza treated group had a significantly higher proportion of acyclic animals, whereas the vehicle treated group had a significantly higher proportion of irregular cycling animals (p= 0.033; Chi-square test).
Figure 6. Impact of 5-aza-2’-deoxycitidine (5-aza) on initiation and completion of perimenopause.
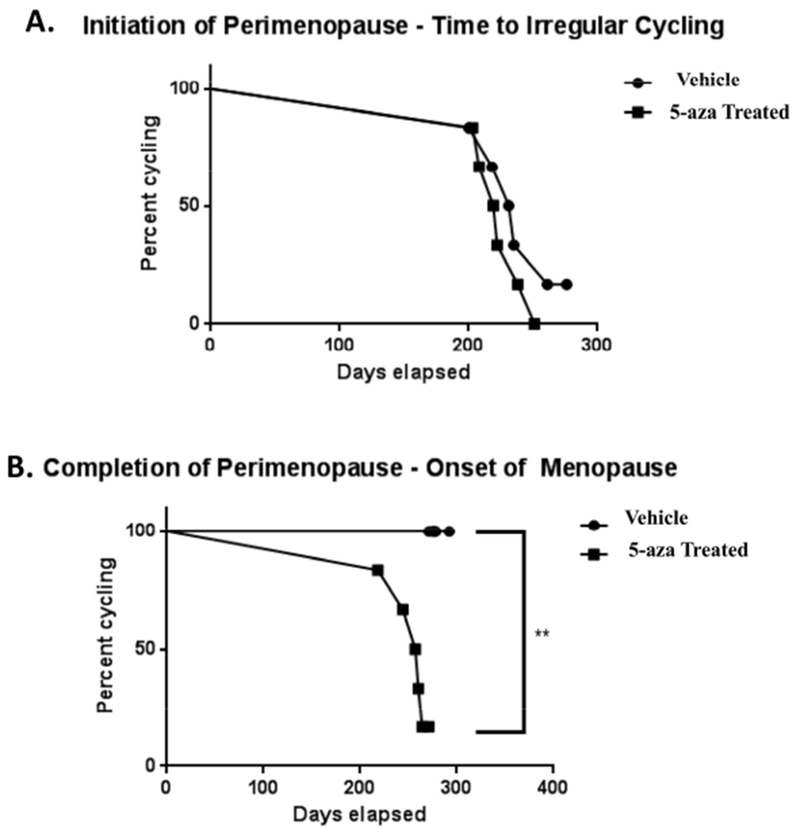
A) Survival curve analysis indicated a non-significant acceleration to initiation of perimenopause in 5-aza-treated animals. B) 5-aza significantly accelerated progression through the perimenopausal transition to menopausal acyclicity (Mantel-Cox p= 0.0043 ; Gehan-Brewslow-Wilcoxon p= 0.006).
3.12.2. Methionine Treatment
RNA-seq and genome-wide DNA methylation analyses suggested that impaired one-carbon metabolism could be involved in the onset of perimenopause. Impaired one-carbon metabolism, resulting in a decrease of SAM production may be responsible for the loss of DNA methylation observed in perimenopausal animals. To test whether perimenopause onset and/or completion could be delayed, animals were treated with methionine (a precursor of SAM), in an attempt to supplement the aging epigenome and prevent DNA hypomethylation. Treatment was initiated at 6 months and continued until 10 months at which time cyclicity was assessed. At 10 months a significantly larger proportion of methionine treated animals (86%) were still regularly cycling compared to the control group (25%) as assessed by Chi-square test (p = 0.044). There were no irregularly cyclers and all remaining animals were acyclic (Fig. 6).
4. Discussion
4.1. Hypothalamic Endocrine Aging Begins Prior to Perimenopause:
Reproductive senescence is a function of both hypothalamic and ovarian aging, and is influenced by environmental, genetic, and lifestyle factors, as well as systemic diseases. The majority of changes in hypothalamic gene expression occurred during the 6- to 9-month period when animals were still cycling regularly, indicating that hypothalamic aging begins prior to the phenotypic manifestation of perimenopause. Furthermore regulators involved in HPG-axis signaling, GnRH, E2, and KISS1, were identified as upstream regulators during this time suggesting that a in HPG signaling precedes ovarian aging.
Prolactin expression is remarkably upregulated prior to irregular cycling, making it a promising candidate as a perimenopause initiator. Hyper-prolactinemia is a known reproductive inhibitor in both males and females and is responsible for loss of libido as well as infertility in humans (Anderson, Kieser et al. 2008). Although prolactin signaling is not well described within the hypothalamus, we propose that hypothalamic hyper-prolactinemia plays a similar role in reproductive senescence - by negatively regulating GnRH and FSH. We saw no changes in the DNA methylation patterns in the genes coding for prolactin, or prolactin’s receptors indicating that the observed changes were regulated by other factors, such as histone modifications or transcription factor binding. In the pituitary, prolactin secretion and gene expression are regulated by multiple factors including dopamine and TRH and their receptors (Fitzgerald and Dinan 2008, Yu, Murao et al. 2010). Consistent with what is known for pituitary prolactin, data reported herein indicate that both TRH and dopamine receptor D5 (DRD5) expression change across the transition.
However, multiple genes coding for histone modifying proteins during this period were modified. Candidate epigenetic regulator genes that could play role in prolactin’s increased expression included one demethylase (KDM6B), five methyltransferases (EHMT1, PRMT8, METTL7A, METTL8, and KMT2A), two deacetylases (HDAC1 and HDAC5), the methyl-binding domain protein (MECP2).
4.2. Hypothalamic Neurological Aging Begins Prior to Irregular Cycling
Prior to irregular cycling, multiple hypothalamic genes related to one-carbon metabolism and epigenetic regulation exhibited altered expression. Both glutamatergic and GABAergic signaling pathways fluctuate substantially across the perimenopause. Prior to perimenopause, numerous glutamate transporters and receptors were down regulated while GABA receptors exhibited changes in both increased and decreased expression. Glutamate transporters are responsible for removing glutamate from the extracellular/synaptic space to prevent tonic activation of post-synaptic glutamate receptors and excitotoxicity. In particular, the transporter GLT1 is crucial to glutamate re-uptake has been reported to constitute 1% of total brain protein (Sheldon and Robinson 2007). We observed a significant decrease in four glutamate transporter genes, including GLT1 between 6-9 months (Table 4). Expression changes were accompanied by differential DNA methylation in two of the transporters, VGLUT3 and GLT1. It’s possible that the changes in glutamate and GABA signaling prior to perimenopause sensitize the brain to environmental or hormonal insult, modifying the risk and/or rate of neurological decline. Glutamate-mediated excitotoxicity has been linked to several neurodegenerative disorders such as Alzheimer’s, amyotrophic lateral sclerosis, multiple sclerosis, as well as Parkinson’s (Hynd, Scott et al. 2004, Lau and Tymianski 2010). Impaired GABAergic signaling is reported to be a key feature of all neurodegenerative etiologies (Blaszczyk 2016).
4.3. Accelerated Epigenomic Aging in Early Transitioners
Global DNA methylation is reported to decline with age (Wilson and Jones 1983, Pogribny, Muskhelishvili et al. 2011). In the hypothalamus, we do not observe a significant change in DNA methylation between 6-9 months (Fig 3A). However, within the regular 6 month animals, we see two distinct populations – with “high” and “low” global DNA methylation levels that are significantly different from each other. Individual animals appear to be aging at various rates and a subset of the 6 month animals’ epigenomes appear to be biologically and endocrinologically “older”. Indeed, at 6 months two-thirds of the animals showed low DNA methylation levels equivalent to 9 month irregular and acyclic groups and the remaining one-third had DNA methylation levels similar to regular cycling 9 month animals (Fig 2B). Furthermore, this ratio of 1:3/2:3 matches the percentage of 9 month animals regular cycling (37) versus irregular (30%) or acyclic (33%) (Fig. 1A), suggesting that if left undisturbed, animals with lower global DNA methylation levels at 6 month would become irregular or acyclic and animals with higher levels would continue to cycle regularly at 9 months of age. This data suggest that individual epigenetic differences that are present prior to perimenopause predispose an individual towards a particular perimenopause outcome (late vs. early).
In humans, menopause has been shown to accelerate epigenetic patterns of aging in blood (Levine, Lu et al. 2016). Women with an earlier age of menopause onset have been found to be “epigenetically older” than women with a later onset (Levine, Lu et al. 2016). Here we show that, in rats, global DNA methylation declines at the onset of perimenopause supporting the hypothesis that menopause and epigenetic age are inversely correlated. However, the cause-effect relationship of epigenetic and menopausal age remains ambiguous. To better understand this relationship, we sought to perturb the epigenome in our animals and assess if perimenopause timing was altered. We used 5-aza, which has been shown to shorten the lifespan of cells via hypomehtylation of DNA (Fairweather, Fox et al. 1987), to induce an accelerated aging phenotype. In doing so, we were able to accelerate the perimenopause transition, bringing on an early “menopause” status in our animals (Fig. 5B).
Because an “older” epigenome is correlated with earlier menopause and associated with impaired one carbon metabolism and loss of SAM, we also supplemented animals with methionine in an attempt to slow epigenetic aging and prolong reproductive competency. Methionine, a precursor to SAM, was chosen rather than SAM itself because it is much less volatile, has a greater half-life, and a lower dose is required to obtain systemic effects (Young and Shalchi 2005). Regular cycling 6 month animals that were regularly supplemented with methionine remained reproductively competent longer than their vehicle treated counterparts (Fig. 6). Together these data further clarify the cause-effect relationship of epigenetic and menopause age and provide evidence that epigenetic mechanisms regulate the perimenopause transition.
4.4. Hypothalamic Aging Precedes Hippocampal Aging
The greatest change in both gene expression and DNA methylation in the hypothalamus occurred prior to changes in the phenotypic feature of perimenopause, irregular cycling. Subsequent to gene and DNA methylation changes between 6-9 months of age in the rat hypothalamus, minor changes were evident indicating that mechanisms within the hypothalamus that control reproductive senescence occur prior to and are predictive of onset perimenopause.
These early initiating events in the hypothalamus precede gene expression changes in the hippocampus which are coincident with the onset of irregular cycling (Yin, Yao et al. 2015). In the hippocampus, we reported evidence of decline in bioenergetic systems and synaptic plasticity in the hippocampus during the transition from regular to irregular cycling (Yin, Yao et al. 2015). Additionally during the perimenopausal transition, we observed decline in brain glucose uptake and deficits in mitochondrial function. Further dynamic changes in genes and pathways related to glucose metabolism, fatty acid metabolism, inflammation, and mitochondrial function occurred across the perimenopausal to menopausal to post-menopausal transitions (Yin, Yao et al. 2015). Unlike the hypothalamus, the critical period of change in the hippocampus begins at the initiation of irregular cycling. It is likely that hippocampal aging is driven by the hormonal changes that occur as a result of reproductive senescence initiated by hypothalamic changes that occur prior to irregular cycling.
4.5. DNA Methylation Controls Specific Aspects of Endocrine Aging and Reproductive Senescence
Treatment with either 5-aza or methionine, known epigenetic modifiers, impacted the timing of reproductive senescence with inhibition of DNA methylation accelerating endocrine aging whereas promoting DNA methylation with methionine delayed endocrine aging. Numerous studies have shown that peripheral 5-aza treatment, which crosses the blood brain barrier, suppresses DNA methylation in all regions of the brain that have been studied (Lomniczi and Ojeda 2016, Zhang, Yang et al. 2016, Fonteneau, Filliol et al. 2017, Li, Ma et al. 2017).
The use of 5-aza to induce hypomethylation as a model of accelerated aging was a means to test our hypothesis that epigenetic regulates reproductive senescence. Because 5-aza globally hypomethylates DNA in a non-specific manner, genome-wide methylation studies are likely to be inconclusive. For this reason, we focused on investigating patterns of genome-wide DNA methylation in animals undergoing biological endocrine aging to identify specific pathways directly related to natural reproductive senescence. Conversely, we hypothesized that the DNA methylation and RNA expression patterns in the methionine treated animals to be comparable to a naturally endocrine aging animal, as methionine treatment delayed advance to the next stage in endocrine aging.. Studies have shown that methionine can effectively reverse and prevent disease-associated epigenetic patterns in numerous models (Fuso, Nicolia et al. 2012, Parrish, Buckingham et al. 2015, Wright, Hollis et al. 2015, Gregoire, Millecamps et al. 2017).
4.6. Conclusion
Collectively, the transcriptional and epigenetic reported herein indicate that the hypothalamus undergoes endocrine aging prior to onset of symptoms of reproductive senescence. Importantly this phenotypically silent period of genomic regulation represents a critical period in endocrine aging. Inhibition of DNA methylation accelerated transition through the perimenopause to complete reproductive senescence. In contrast, promoting DNA methylation through methionine supplementation delayed progression of endocrine aging and markedly sustained regular cycling, an endocrine phenotype of reproductive competence.
From a translational perspective, data presented herein indicate that hypothalamic aging begins prior to the onset of the perimenopausal phenotype of irregular cycling. Initiation of interventions to sustain epigenetic mechanisms, specifically DNA methylation, could be a strategy to sustain endocrine and neurological function in women (Fuso, Nicolia et al. 2008, Fuso, Nicolia et al. 2011, Fuso, Nicolia et al. 2012).
Supplementary Material
Figure 7. Methionine treatment delayed onset of perimenopause.

At 10 months 86% of methionine treated animals continued to cycle regularly compared to 25% of vehicle treated animals (p = 0.044). Remaining animals were all acyclic.
Table 1: Upstream regulators identified by Ingenuity Pathway Analysis (IPA).
Activation z-score infers the activation states of predicted transcriptional regulators. Dependent on the observed expression of a gene in the dataset, the activation state of a regulator is determined by the direction associated with the relationship to the regulator. Positive and negative z-scores indicate predicted activation or inhibition, respectively. Z-scores are not available for when no directional prediction can be made.
| Symbol | Gene Name | Reg 6m – Reg 9m | Reg 9m – Irreg 9m | Irreg 9m – Acyc 9m | |||
|---|---|---|---|---|---|---|---|
| z-score | p-value | z-score | p-value | z-score | p-value | ||
| GnRH | Gonadotropin releasing hormone 1 | −0.286 | 5.87E-05 | 1.647 | 4.36E-07 | −0.971 | 8.47E-05 |
| KISS1 | Kisspeptin | −1.969 | 0.153 | 1.969 | 0.000456 | −1.969 | 0.000779 |
| SAH | S-adenosylhomocysteine | 0.0334 | 0.0385 | ||||
| SAM | S-adenosylmethionine | −1.287 | 0.0336 | 0.0258 | |||
| Hcy | Homocysteine | −0.043 | 0.0206 | ||||
| PRL | Prolactin | 3.936 | 8.25E-15 | ||||
| E2 | 17β-estradiol | −2.08 | 8.65E-25 | 0.569 | 4.7E-18 | −0.042 | 1.05E-19 |
Highlights.
Epigenetic DNA methylation regulates endocrine aging in the female brain.
Perimenopause is accompanied by a decline in global DNA methylation in the hypothalamus.
Hypothalamic transcriptional changes underlying endocrine aging precede perimenopause.
DNA methylation changes in glutamate signaling genes are associated with perimenopause.
Treatment with a hypomethylating chemical agent accelerated transition through the perimenopause to menopause whereas increasing DNA methylation delayed onset of perimenopause / endocrine aging.
5. Acknowledgements:
This study has been supported by NIA P01AG026572 to RDB: Project 1 to RDB, Analytic Core C to FY. The authors declare no competing financial interests and nothing to disclose.
Abbreviations
- 5-aza
5-aza-2’-deoxycitidine
- 5-mC
5-methylcytosine
- Acyc
Acyclic
- AD
Alzheimer’s Disease
- D
Diestrus
- DNMT
DNA methyltransferase
- E
Estrus
- E2
beta-estradiol
- Esr2
Estrogen receptor β gene
- FSH
Follicle Stimulating Hormone
- GnRH
Gonadotropin-Releasing Hormone
- Hcy
Homocysteine HDAC1
- HPG
Hypothalamic-Pituitary-Gondal
- HYPER
Hypermethylated
- HYPO
Hypomethylated
- IPA
Ingenuity Pathway Analysis
- Irreg
Irregular Cycling
- KISS1
Kisspeptinl
- LH
Luteinizing Hormone
- M
Metestrus
- OXT
Oxytocin
- P
Proestrus
- PAM
Perimenopause Animal Model
- Reg
Regular cycling
- RRBS
Reduced Representative Bisulfite Sequencing
- S HYPER
Strongly Hypermethylated
- S HYPO
Strongly Hypomethylated
- SAM
S-adenosylmethionine
- Tet
Ten-eleven translocation methylcytosine dioxygenase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson GM, Kieser DC, Steyn FJ and Grattan DR (2008). “Hypothalamic prolactin receptor messenger ribonucleic acid levels, prolactin signaling, and hyperprolactinemic inhibition of pulsatile luteinizing hormone secretion are dependent on estradiol.” Endocrinology 149(4): 1562–1570. [DOI] [PubMed] [Google Scholar]
- Blaszczyk JW (2016). “Parkinson’s Disease and Neurodegeneration: GABA-Collapse Hypothesis.” Front Neurosci 10: 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottner M, Leonhardt S, Wuttke W and Jarry H (2007). “Changes of expression of genes related to the activity of the gonadotrophin-releasing hormone pulse generator in young versus middle-aged male rats.” J Neuroendocrinol 19(10): 779–787. [DOI] [PubMed] [Google Scholar]
- Brinton RD, Yao J, Yin F, Mack WJ and Cadenas E (2015). “Perimenopause as a neurological transition state.” Nat Rev Endocrinol 11(7): 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byars SG, Ewbank D, Govindaraju DR and Stearns SC (2010). “Colloquium papers: Natural selection in a contemporary human population.” Proc Natl Acad Sci U S A 107 Suppl 1: 1787–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SR, Lambrinoudaki I, Lumsden M, Mishra GD, Pal L, Rees M, Santoro N and Simoncini T (2015). “Menopause.” Nat Rev Dis Primers 1: 15004. [DOI] [PubMed] [Google Scholar]
- DeVito WJ (1988). “Distribution of immunoreactive prolactin in the male and female rat brain: effects of hypophysectomy and intraventricular administration of colchicine.” Neuroendocrinology 47(4): 284–289. [DOI] [PubMed] [Google Scholar]
- Fairweather DS, Fox M and Margison GP (1987). “The in vitro lifespan of MRC-5 cells is shortened by 5-azacytidine-induced demethylation.” Exp Cell Res 168(1): 153–159. [DOI] [PubMed] [Google Scholar]
- Finch CE (2014). “The menopause and aging, a comparative perspective.” J Steroid Biochem Mol Biol 142: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald P and Dinan TG (2008). “Prolactin and dopamine: what is the connection? A review article.” J Psychopharmacol 22(2 Suppl): 12–19. [DOI] [PubMed] [Google Scholar]
- Fonteneau M, Filliol D, Anglard P, Befort K, Romieu P and Zwiller J (2017). “Inhibition of DNA methyltransferases regulates cocaine self-administration by rats: a genome-wide DNA methylation study.” Genes Brain Behav 16(3): 313–327. [DOI] [PubMed] [Google Scholar]
- Fuso A, Nicolia V, Cavallaro RA, Ricceri L, D’Anselmi F, Coluccia P, Calamandrei G and Scarpa S (2008). “B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice.” Mol Cell Neurosci 37(4): 731–746. [DOI] [PubMed] [Google Scholar]
- Fuso A, Nicolia V, Pasqualato A, Fiorenza MT, Cavallaro RA and Scarpa S (2011). “Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency.” Neurobiol Aging 32(2): 187–199. [DOI] [PubMed] [Google Scholar]
- Fuso A, Nicolia V, Ricceri L, Cavallaro RA, Isopi E, Mangia F, Fiorenza MT and Scarpa S (2012). “S-adenosylmethionine reduces the progress of the Alzheimer-like features induced by B-vitamin deficiency in mice.” Neurobiol Aging 33(7): 1482 e1481–1416. [DOI] [PubMed] [Google Scholar]
- Gaskins AJ, Mumford SL, Chavarro JE, Zhang C, Pollack AZ, Wactawski-Wende J, Perkins NJ and Schisterman EF (2012). “The impact of dietary folate intake on reproductive function in premenopausal women: a prospective cohort study.” PLoS One 7(9): e46276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Santos C, Saura CB, Lucas JA, Castell P, Madrid JA and Garaulet M (2016). “Menopause status is associated with circadian- and sleep-related alterations.” Menopause 23(6): 682–690. [DOI] [PubMed] [Google Scholar]
- Gregoire S, Millecamps M, Naso L, Do Carmo S, Cuello AC, Szyf M and Stone LS (2017). “Therapeutic benefits of the methyl donor S-adenosylmethionine on nerve injury-induced mechanical hypersensitivity and cognitive impairment in mice.” Pain 158(5): 802–810. [DOI] [PubMed] [Google Scholar]
- Hak AE, Polderman KH, Westendorp IC, Jakobs C, Hofman A, Witteman JC and Stehouwer CD (2000). “Increased plasma homocysteine after menopause.” Atherosclerosis 149(1): 163–168. [DOI] [PubMed] [Google Scholar]
- Harlow SD, Gass M, Hall JE, Lobo R, Maki P, Rebar RW, Sherman S, Sluss PM, de Villiers TJ and Group SC (2012). “Executive summary of the Stages of Reproductive Aging Workshop + 10: addressing the unfinished agenda of staging reproductive aging.” Menopause 19(4): 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann W and Obeid R (2011). “Homocysteine: a biomarker in neurodegenerative diseases.” Clin Chem Lab Med 49(3): 435–441. [DOI] [PubMed] [Google Scholar]
- Hynd MR, Scott HL and Dodd PR (2004). “Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease.” Neurochem Int 45(5): 583–595. [DOI] [PubMed] [Google Scholar]
- Jehan S, Masters-Isarilov A, Salifu I, Zizi F, Jean-Louis G, Pandi-Perumal SR, Gupta R, Brzezinski A and McFarlane SI (2015). “Sleep Disorders in Postmenopausal Women.” J Sleep Disord Ther 4(5). [PMC free article] [PubMed] [Google Scholar]
- Lau A and Tymianski M (2010). “Glutamate receptors, neurotoxicity and neurodegeneration.” Pflugers Arch 460(2): 525–542. [DOI] [PubMed] [Google Scholar]
- Levine ME, Lu AT, Chen BH, Hernandez DG, Singleton AB, Ferrucci L, Bandinelli S, Salfati E, Manson JE, Quach A, Kusters CD, Kuh D, Wong A, Teschendorff AE, Widschwendter M, Ritz BR, Absher D, Assimes TL and Horvath S (2016). “Menopause accelerates biological aging.” Proc Natl Acad Sci U S A 113(33): 9327–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ma Q, Dasgupta C, Halavi S, Hartman RE, Xiao D and Zhang L (2017). “Inhibition of DNA Methylation in the Developing Rat Brain Disrupts Sexually Dimorphic Neurobehavioral Phenotypes in Adulthood.” Mol Neurobiol 54(6): 3988–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomniczi A and Ojeda SR (2016). “The Emerging Role of Epigenetics in the Regulation of Female Puberty.” Endocr Dev 29: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazef K, Khelil M, Chelouti H, Kacimi G, Bendini M, Tazir M, Belarbi S, El Hadi Cherifi M and Djerdjouri B (2014). “Hyperhomocysteinemia is a risk factor for Alzheimer’s disease in an Algerian population.” Arch Med Res 45(3): 247–250. [DOI] [PubMed] [Google Scholar]
- Parrish RR, Buckingham SC, Mascia KL, Johnson JJ, Matyjasik MM, Lockhart RM and Lubin FD (2015). “Methionine increases BDNF DNA methylation and improves memory in epilepsy.” Ann Clin Transl Neurol 2(4): 401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogribny IP, Muskhelishvili L, Tryndyak VP and Beland FA (2011). “The role of epigenetic events in genotoxic hepatocarcinogenesis induced by 2-acetylaminofluorene.” Mutat Res 722(2): 106–113. [DOI] [PubMed] [Google Scholar]
- Sheldon AL and Robinson MB (2007). “The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention.” Neurochem Int 51(6-7): 333–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L and Ji HF (2015). “Associations between Homocysteine, Folic Acid, Vitamin B12 and Alzheimer’s Disease: Insights from Meta-Analyses.” J Alzheimers Dis. [DOI] [PubMed] [Google Scholar]
- Snieder H, MacGregor AJ and Spector TD (1998). “Genes control the cessation of a woman’s reproductive life: a twin study of hysterectomy and age at menopause.” J Clin Endocrinol Metab 83(6): 1875–1880. [DOI] [PubMed] [Google Scholar]
- Tomizawa H, Matsuzawa D, Ishii D, Matsuda S, Kawai K, Mashimo Y, Sutoh C and Shimizu E (2015). “Methyl-donor deficiency in adolescence affects memory and epigenetic status in the mouse hippocampus.” Genes Brain Behav 14(3): 301–309. [DOI] [PubMed] [Google Scholar]
- Wilson VL and Jones PA (1983). “DNA methylation decreases in aging but not in immortal cells.” Science 220(4601): 1055–1057. [DOI] [PubMed] [Google Scholar]
- Wright KN, Hollis F, Duclot F, Dossat AM, Strong CE, Francis TC, Mercer R, Feng J, Dietz DM, Lobo MK, Nestler EJ and Kabbaj M (2015). “Methyl supplementation attenuates cocaine-seeking behaviors and cocaine-induced c-Fos activation in a DNA methylation-dependent manner.” J Neurosci 35(23): 8948–8958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Yao J, Sancheti H, Feng T, Melcangi RC, Morgan TE, Finch CE, Pike CJ, Mack WJ, Cadenas E and Brinton RD (2015). “The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity.” Neurobiol Aging 36(7): 2282–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SN and Shalchi M (2005). “The effect of methionine and S-adenosylmethionine on S-adenosylmethionine levels in the rat brain.” J Psychiatry Neurosci 30(1): 44–48. [PMC free article] [PubMed] [Google Scholar]
- Yu X, Murao K, Imachi H, Li J, Nishiuchi T, Dobashi H, Hosomi N, Masugata H, Zhang GX, Iwama H and Ishida T (2010). “The transcription factor prolactin regulatory element-binding protein mediates prolactin transcription induced by thyrotropin-releasing hormone in GH3 cells.” Endocrine 38(1): 53–59. [DOI] [PubMed] [Google Scholar]
- Zeisel SH (2009). “Importance of methyl donors during reproduction.” Am J Clin Nutr 89(2): 673S–677S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yang J, Liu X, Jia X, Xu S, Gong K, Yan S, Zhang C and Shao G (2016). “Effects of 5-Aza-2’-deoxycytidine on expression of PP1gamma in learning and memory.” Biomed Pharmacother 84: 277–283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.