

Abstract
Aging biology is intimately associated with dysregulated metabolism, which is one of the hallmarks of aging. Aging‐related pathways such as mTOR and AMPK, which are major targets of anti‐aging interventions including rapamcyin, metformin, and exercise, either directly regulate or intersect with metabolic pathways. In this review, numerous candidate bio‐markers of aging that have emerged using metabolomics are outlined. Metabolomics studies also reveal that not all metabolites are created equally. A set of core “hub” metabolites are emerging as central mediators of aging. The hub metabolites reviewed here are nicotinamide adenine dinucleotide, reduced nicotinamide dinucleotide phosphate, α‐ketoglutarate, and β‐hydroxybutyrate. These “hub” metabolites have signaling and epigenetic roles along with their canonical roles as co‐factors or intermediates of carbon metabolism. Together these hub metabolites suggest a central role of the TCA cycle in signaling and metabolic dysregulation associated with aging.
Keywords: aging, hub metabolites, metabolomics
1. Introduction
Aging is the organism‐wide loss in homeostasis and regenerative capacity, and this phenomenon is displayed across unicellular, invertebrate, and vertebrate model systems.[ 1 , 2 , 3 ] It is now well accepted that aging is the single greatest risk factor associated with an array of diseases including cancer, metabolic, and cardiac diseases.[ 4 , 5 ] A number of physiological processes and signaling pathways have been associated with aging, and of these, metabolic dysregulation (including mitochondrial dysfunction) is a key hallmark of aging.[ 6 , 7 , 8 ] Metabolism lies at the intersection of aging‐related pathways such as mTOR,[ 9 , 10 ] AMPK,[ 11 ] and anti‐aging interventions such as caloric restriction[ 11 ] and metformin treatment.[ 12 ] Therefore, in order to understand the process of aging, the comprehensive and systematic measurement of metabolic pathways is essential. This systems approach to metabolism is expected to illuminate the underlying mechanisms that mediate aging and aging‐associated diseases. Although metabolites can be measured comprehensively across the metabolic network using metabolomics technologies,[ 13 , 14 ] specific subsets of metabolites within the network have emerged as particularly significant mediators and targets of anti‐aging interventions (nicotinamide adenine dinucleotide [NAD+], reduced nicotinamide dinucleotide phosphate [NADPH], α‐ketoglutarate [αKG], and β‐hydroxybutyrate [βHB]), these will be further elucidated in this review. Here we discuss MS‐based metabolomics enabled aging biomarkers, followed by analysis of significant nodes in the metabolome that are emerging in the context of aging biology. Finally, we point to mitochondrial TCA as an integrator of hub metabolites and their signaling.
2. Tools and Technologies for Metabolomics/Biomarker discovery
Metabolomics involves the identification of small molecule metabolites (MW < 1500 Da), generated within a complex biological system, as a biochemical end‐point of genomics, transcriptomics, and proteomics.[ 14 , 15 ] The traditional goal of metabolomics has been the identification of biomarkers and underlying metabolic mechanisms that can be used to define the physiological or pathological state of biological systems.[ 16 , 17 ] Commonly tracked metabolites in biomarker research are lipids, amino acids, peptides, organic acids, vitamins, and carbohydrates, and the commonly used biological matrices include plasma, tissues, urine, cerebrospinal fluid (CSF), and fecal samples. The key analytical instruments used in sample analysis include mass spectrometers coupled to chromatographic separation techniques, and NMR. NMR is a non‐invasive technique and has an advantage over the mass spectrometer, in terms of accurate structure elucidation of the metabolite.[ 18 ] However, it requires at least an order of magnitude higher amount of sample as compared to MS, for example in the range from 5 to 25 mg for 1H NMR. Figure 1 briefly summarizes metabolomics workflows for analysis of biofluids and tissues using mass spectrometric approaches. These workflows have already been reviewed elsewhere[ 13 , 14 , 15 , 16 , 17 , 18 ] and are beyond the scope of the current review.
Figure 1.
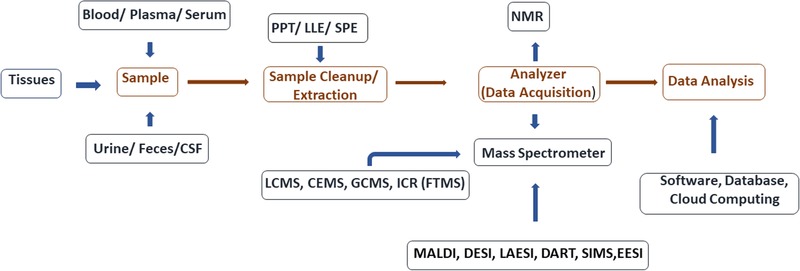
Outline of metabolomic workflow for biomarker discovery in aging biology. Analytical workflow for biomarker analysis involves a number of possible platforms for performing sample preparation (from tissues and bio fluids like urine, feces, and cerebrospinal fluid (CSF)), sample extraction (protein precipitation (PPT), solid phase extraction (SPE), and liquid–liquid extraction (LLE)). Data acquisition (NMR) followed by MS (using matrix associated laser desorption ionization [MALDI], desorption electrospray ionization [DESI] laser ablation electrospray ionization [LAESI], direct ionization in real‐time [DART], secondary ion MS [SIMS] or electrospray ionization [ESI]), and data analysis using local software resources or via cloud computing.
3. Application of Metabolomics for Biomarker Discovery in Aging
The discovery of biomarkers in a biological specimen relies on the identification and characterization of unique metabolites, which are statistically distinct among different groups. Table S1, Supporting Information summarizes recent biomarkers of aging discovered using metabolic profiling of biofluids and tissues derived from human and vertebrate models (not focusing on seminal studies performed using invertebrate models) along with the corresponding listing of metabolomics platforms used in their respective studies. For human studies on aging, longitudinal studies have been a transformative resource, 51 of which are in the National Institutes of Aging database.[ 19 ] Two of these longitudinal studies have been instrumental in metabolomics based biomarker discoveries, number of which are listed in Table S1, Supporting Information, the Baltimore longitudinal study of aging (https://www.blsa.nih.gov/)[ 20 ] and the Leiden longevity study.[ 21 ] One of the exciting insights that has emerged from these longevity studies is the association between the APOE4 genotype and aging.[ 22 , 23 ] Subjects with APOE4 genotype have substantially decreased extreme longevity unlike subjects with E2E3 genotype.[ 24 ] Though increased risk of AD pathology conferred by the APOE4 genotype has been studied, the basal metabolomic differences on “normal” aging remain to be elucidated and represent a significant opportunity.
3.1. Hub Metabolites Emerging from Aging: Metabolomics at the Interface Cellular Signaling, Epigenetics, and Metabolism
The important insight that has emerged from metabolomics based studies of aging is the central impact of specific metabolic intermediates on aging. These “hub” metabolites represent nodes in the biochemical network that play a critical role in integrating the information flow in cells between metabolism and signaling pathways, to control aging (Figure 2 ). There are a number of candidate hub metabolites that might fulfill these criteria, but following is a summary of four conserved emerging hubs in the aging metabolome.
Figure 2.
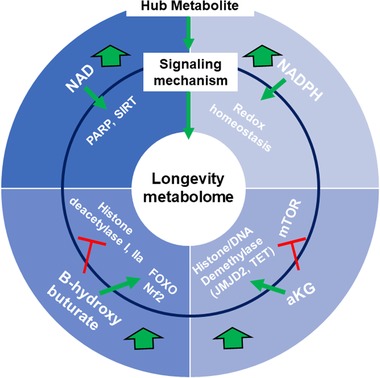
Hub metabolites and signaling mechanisms that promote longevity. The aging metabolome can be viewed as two layers comprised of i) hub metabolites (NAD+, NADPH, αKG, and ketone bodies such as βHB) and ii) signaling mechanism involving regulation of aging‐related signaling, epigenetic, and stress responsive pathways. Large green arrows indicate the pro‐longevity effects of their supplementation, red T's indicate inhibitory effects on enzymes/pathways and green arrows indicate activation of enzymes/pathways.
3.1.1. NAD+ Metabolome, at the Hub of Central Carbon Metabolism, DNA Damage, and Protein Deacetylation, and Cellular Senescence
Nicotinamide adenine dinucleotide (NAD+) is an essential electron carrier in cellular biochemistry, and the ratio of the oxidized and reduced forms is a readout of the redox status of cells. It is primarily generated via glycolytic and TCA cycle metabolism in cells.[ 25 ] Beyond its canonical role as a coenzyme for redox reactions, it serves as a cofactor for three enzymes that are of central interest in aging biology, i) Sirtuins (SIRT), ii) poly(ADP‐ribose) polymerases (PARP), and iii) cyclic ADP‐ribose synthases (e.g., CD38). NAD+ is continually consumed by these three enzymes and organisms to produce nicotinamide. Nicotinamide salvage pathways and biosynthetic pathways maintain stable levels of NAD+ in cells. The salvage pathways is mediated by the enzyme nicotinamide mononucleotide phosphoribosyl transferase (NAMPT) that recycles nicotinamide into nicotinamide mononucleotide (NMN) that is converted to NAD+ by nicotinamide mononucleotide adenylyl‐transferase (NMNAT) family of enzymes. In parallel, NAD+ can be biosynthesized from dietary sources such as nicotinic acid (a form of vitamin B3) via NMNAT enzymes in the Preiss‐Handler pathway.[ 25 ] NAD+ can also be synthesized from dietary tryptophan via the kynurenine pathway.[ 8 , 26 ] The three classes of NAD+ consuming enzymes have emerged as key players in aging. The family of sirtuin enzymes were identified as NAD+ dependent protein deacylases (modifying acetyl, succinyl, malonyl palmitoyl, and glutaryl groups[ 27 , 28 , 29 , 30 , 31 , 32 ]) producing nicotinamide and O‐acetyl‐ADP‐ribose as products of the deacylation reaction. Sirtuins serve as NAD+ sensors, and it has been shown that caloric restriction dependent increase in lifespan in SIRT1 dependent fashion.[ 33 ] There are seven mammalian sirtuins in the cytoplasm and the mitochondria.[ 34 , 35 ] The substrates of sirtuins range from acetylated histones to transcription factors (e.g., PGC‐1a, FOXO3a, and NF‐kb).[ 8 ] The other NAD+ consuming enzyme PARP is important for the cellular DNA damage sensing and the repair‐response. PARP1 and 2 transfer ADP‐ribose from NAD+ proteins to accepting proteins/nucleic acids generating long poly (ADP‐ribose) chains (PAR). PARP1 in response to DNA damage causes PAR chains at site of damage and leads to a significant decrease in intracellular NAD+ levels.[ 8 , 21 , 36 ]
The emerging evidence suggests that levels of NAD+ decrease with aging in both vertebrate and invertebrate systems, and the decline in these levels is associated with aging related pathologies.[ 37 , 38 ] A recent metabolomics study has comprehensively quantified the NAD+ metabolome in the plasma of human subjects ranging from 20 to 87 years of age.[ 39 ] Measurements using LC–MS/MS showed that levels of NAD+ and nicotinic acid adenine dinucleotide (NAAD) decreased significantly with age. This was not the case with the entire NAD+ metabolome as there was no change in nicotinic acid (NA), nicotinamide mononucleotide (NMN), and nicotinic acid mononucleotide (NAMN). An aging‐related increase in the reduced form on NAD, nicotinamide (NAM), and adenosine diphosphate ribose (ADPR) was seen. This suggests an aging‐related alteration in the plasma NAD+ metabolome, but the reasons for this, and specifically for the decrease in NAD+ during aging remains an area of intense study. It is clear that levels of NAD+ will be regulated by the complex interplay of pathways that produce and consume this metabolite. In order to address this, a landmark study has been reported by the Liu et al.,[ 40 ] where metabolomics was applied in conjunction with isotope tracer studies to elucidate the fluxes of NAD+ synthesis and breakdown across various tissues in mice. The mass spectrometric analysis was done using HILIC separation and a Q EXACTIVE PLUS mass spectrometer. Isotope abundances were quantified using the freely available AccuCor software (https://github.com/XiaoyangSu/Isotope-Natural-Abundance-Correction).[ 41 ] NAD+ precursors that were used was decided by plasma concentrations of at least 0.1 × 10−6 m. The authors selected U‐13C Trp, [2,4,5,6‐2H]NAM and U‐13C NA for in vivo labeling of NAD+. Labeled precursors were infused into C57BL/6 mice in order to quantify biosynthesis of NAD from Trp and NA, flux of the salvage pathway from NAM to NAD+, flux through NAD kinase, and exchange of serum and tissue NAM. An important insight that emerged from this study is that liver is the primary site of de novo synthesis of NAM from Trp, with insignificant fluxes in other tissues. On the other hand, NA seems to be an insignificant contributor to NAM in tissues as judged by infusion of 13C‐labeled NA. Next, the authors measured the incorporation of labeled NAM into NAD in various tissues, this was judged by formation of (M+3) NAM from infused (M+4) NAM. (M+3) NAM is formed by loss of the reducing hydrogen into NAD from the (M+4) NAM and subsequent cleavage to (M+3) NAM. The analysis revealed that liver is the major producer of NAM, which is consumed by peripheral tissues to generated NAD in a tissue‐dependent fashion with the small intestine and spleen having the highest NAD turnover and skeletal muscle with the least. Though these studies were carried out in young mice, nevertheless it emphasizes the need to study fluxes of the NAD metabolome in a tissue specific and temporal fashion to determine the source of the aging‐related dysfunction in the NAD metabolome.
There is an intense interest in the role of senescent cells in aging and the emerging possibility of targeting these cells as an anti‐aging therapy.[ 42 , 43 ] Senescence is a non‐proliferative state of cells that accumulate with aging and at sites of aging‐related pathologies including arthritis, atherosclerosis, obesity, etc.[ 43 ] Apart from replicative senescence, this state can also be caused by DNA damage and oncogenes.[ 42 , 43 ] A number of hallmarks of senescent cells have been recognized (including upregulated expression of p16Ink4A and/or p21)[ 44 ] of which the senescence‐associated secretory phenotype (SASP) is a well‐characterized one.[ 45 ] It is important to note that depending on the tissues of origin and inducers of senescence, cells display a range of non‐overlapping hallmarks including secreted SASP factors (e.g., TGF‐β, IL‐6, and IL‐8). An important feature of senescent cells is also their dysregulated metabolome. A subtype of senescence is driven by mitochondrial dysfunction that is termed mitochondrial dysfunction‐induced senescence (MIDAS).[ 46 , 47 ] Studies on how the metabolome of senescent cells contributes to this cell‐state has led to insights on the role of NAD+ as a central metabolic node in senescence. Nacarelli et.al. have recently shown that SASP is controlled by NAD+ metabolism.[ 48 ] Specifically, the authors showed that the salvage arm of NAD metabolism, which is controlled by the enzyme nicotinamide phosphoribosyl transferase (NAMPT) controls the proinflammatory SASP. Using an oncogene induced senescence model and LC–MS/MS assays, the authors showed that the senescent cells have significantly increased levels of NAD+, and NAMPT mediated synthesis of NAD+ (using cells with NAMPT inhibition), and was required for the secretion of SASP factors (IL‐10 and TNF‐α). Feeding senescent cells 13C‐labeled glucose followed by metabolic profiling showed that senescent cells had enhanced glycolysis via pyruvate and lactate, and the TCA cycle as a pathway for maintaining NAD+ levels. It was proposed that high NAD+ levels support senescence‐related metabolic changes, and also promote AMPK and NF‐κB signaling pathways that activate the expression of inflammatory cytokines. In contrast to the high NAD+ levels observed in this study, the MIDAS cells were driven by lower NAD+ levels. Wiley et al.[ 47 ] showed that mitochondrial dysfunction (by ethidium bromide induced mitochondrial depletion) had low NAD/NADH ratios as expected, and displayed the induction of senescence. This study also showed the importance of NAD sensing mitochondrial sirtuins (SIRT3) in MIDAS, as overexpression of these proteins significantly delayed the induction of senescence. The authors showed that induction of AMPK and its phosphorylation of p53 is the mechanism by which decreased NAD/NADH levels induce MIDAS. An important issue that is underlined by this study is the importance of investigating the cellular compartments which have perturbed NAD/NADH ratios. Unexpectedly the compartment with the altered ratio is the cytoplasm, and not the mitochondria suggesting the important role of NAD+ transport into the cytosol that might be perturbed by mitochondrial dysfunction. Finally, the in vivo significance of this metabolic study was shown using a POLGD257A mouse that has a mutation in the proof reading domain of the mitochondrial DNA polymerase, causing accumulation of mutations in the mitochondrial DNA, leading to mitochondrial dysfunction. The skin and fat tissue of these mice showed a significant accumulation of senescent cells, suggesting that MIDAS could occur in vivo.
3.1.2. NADP Metabolome at the Hub of the Pentose Phosphate Shunt and Redox Homeostasis
Compared to NAD+, its phosphorylated cousin, NADP, has received lesser attention until recently in aging biology. It is well known that the reduced form NADPH is the donor of reductive equivalents to glutathione reductase and thioredoxin reductase, in the synthesis of compounds in the oxidative stress response (reduced glutathione and reduced thioredoxin, respectively).[ 49 ] NADPH is produced by the oxidative arm of the pentose phosphate shunt, by the enzyme glucose 6‐phosphate dehydrogenase and 6‐phosphogluconate reductase. It can also be produced by the mitochondrial enzymes isocitrate dehydrogenase 1 and 2, NADP dependent malic enzyme, and one carbon cycle enzyme.[ 49 , 50 ]. Liu et al.[ 40 ] have shown that the NAD kinase (a source of NADP) only acts on about 10% of the total NAD+ pool. A comprehensive fluxomics of the NADP metabolome remains to be accomplished and is now necessary to elucidate how pools of this metabolite are maintained in various tissues.
In aging biology, NADPH has been recognized in the context of the free radical theory of aging proposed by Hannan in 1950. The theory suggested that free radicals create cumulative damage to macromolecules and nucleic acid causing aging. New biological insights regarding the positive role of free radicals in stem cell activation and are essential signaling factors[ 51 , 52 , 53 , 54 ] show that the theory needs refinement or revision. None the less molecules that play a critical role in redox homeostasis are expected to play a critical role in aging. It has been shown in D. melanogaster that overexpression of G6PD leads to higher levels of NADPH, higher reduced to oxidized glutathione as expected, and leads to increase in lifespan.[ 55 ] A recent study by Wang et al.[ 56 ] has used metabolomics in combination with proteomics to suggest a role for this pathway in a long lived mutant fly. In this study, the authors used metabolomics to compare the relative levels of stead state metabolites form the head of flies that were either wild‐type or mutants with one copy of the puc phosphatase resulting in the partial activation of the stress response JNK kinase. It has been previously shown that these puc mutants had a longer lifespan, but the underlying metabolic mechanisms were unclear. Steady state metabolomics revealed a significant increase in glucose 6‐phosphate (G6P) and ribose 5‐phosphate (R5P) in the young puc mutants are compared to wild‐type mutants, suggesting that a metabolic phenotype even in the young state might enable the mutants to live longer. The authors developed an approach to inject U‐13C glucose in the haemocytes of flies to monitor flux of glucose and TCA metabolism in the fly heads. This revealed that the puc mutants had an efficient flux of carbon into the TCA, higher levels of NADPH, and higher mRNA levels of G6PD. Finally, these mutants showed an improved protein homeostasis that was recapitulated by the over expression of G6PD. The two studies suggest that G6PD derived NADPH may be an important node in the metabolic network that can extent lifespan in flies. It is now clear that overexpression of G6PD and resultant increase NADPH can also display benefits in a transgenic mouse.[ 57 ] The transgenic mouse with a mild overexpression of G6PD via its endogenous promotor increased the median lifespan of female mice by 14%, including improved glucose tolerance and decreased weight gain. Knowing that NAD+ and NADP can be interconverted with one another, it is possible that there are overlapping benefits of these metabolites in aging, a quantitative fluxomics of both the NAD+ and NADP metabolome in the future will be critical to decouple each of their effects.
3.1.3. αKG Metabolome at the Hub of Mitochondrial Metabolism and Epigenetic Histone/DNA Methylation
Changes in nutritional and epigenetic state are integral to aging. This can be judged by the central role of the mTOR pathway[ 9 , 10 ] and histone/DNA methylation[ 58 , 59 ] in organismal aging. αKG has emerged as master regulatory metabolite in aging biology as it is a key TCA intermediate, a cofactor for numerous transamination reactions,[ 60 , 61 ] a cofactor for the prolyl hydroxylase dependent hydroxylation of the transcription factor hypoxia inducible factor‐1 (HIF‐1)[ 62 , 63 , 64 ] and a cofactor for epigenetic enzymes, histone, and DNA demethylases. Though this metabolite has been known to be at the intersection of important physiological pathways, its direct impact on aging became clear in the work of Chin et al.[ 65 ] who discovered that treatment with a cell permeable ester of αKG extended the lifespan of C. elegans by ≈50% in a concentration‐dependent fashion with a maximal extension at 8 × 10−3 m treatment. The authors identified mitochondrial ATP synthase as a direct target of αKG, and that the downstream inhibition of the mTOR kinase was mediated via a partial inhibition of the ATP synthase. The inhibition of mitochondrial ATPase and the resultant increase in AMP/ATP ratios activates the AMP‐activated protein kinase (AMPK) that directly inhibits the mTOR pathway. It is now well known that inhibition of mTOR is directly implicated in lifespan extension.[ 9 , 10 ] Further signifying the role of the mTOR pathway in the effects of αKG, the authors found that the transcription factor Fork head box Other (FOXO) (which has been shown to play a critical role in the mTOR inhibition mediated lifespan extension[ 10 , 66 ]), was required for the lifespan extending effect of αKG. The authors also found that αKG had no further effect on the lifespan of calorie‐restricted C. elegans. This suggests that αKG is also a key mediator of caloric restriction mediated lifespan extension.
A dramatic observation about αKG is that the metabolite is sufficient to maintain the pluripotency of ES cells by promoting a hypomethylated state via the regulation of the DNA and histone demethylases,[ 67 ] although the in vivo relevance of this effect remains unclear. Ability to maintain the undifferentiated state by intracellular αKG has also been shown in mesenchymal precursor cells.[ 68 ] The remarkable effects of this single metabolite suggest that αKG is at the interface of metabolism and epigenetics and may regulate maintenance of the undifferentiated state.
There is great interest in the role of epigenetics in aging, especially that mediated by DNA and histone methylation.[ 59 ] αKG could therefore be a central mediator since it is a cofactor for DNA and histone demethylating enzymes in both vertebrate and invertebrate systems. It is possible that the major mechanism by which αKG controls lifespan is by genome‐wide demethylation, but this possibility remains to be comprehensively examined. It is known that aging and cancer are accompanied by significant changes in genome‐wide methylation levels.[ 69 ] It is interesting that insignificant levels of DNA methylation are found in embryonic and induced pluripotent stem cells, and found at significant levels in developing and adult cells and tissues.[ 70 ] Using 8000 publicly available microarray datasets from multiple tissues from various age groups, a DNA methylation based estimator of chronological age (now known as the Horvath clock) has been put forth,[ 71 ] this clock identified 193 CpG that were positively correlated, and 160 that were negatively correlated with age. Epigenetic control of aging is an area of active investigation, and αKG may serve as a hub metabolite that connects aging‐metabolism with this clock, by controlling the activity of DNA demethylases. The mechanism underlying this hypothesis is based on the discovery that ten‐eleven translocation 1–3 (TET1‐3) proteins are αKG‐ and Fe (II)‐dependent dioxygenases that mediate the conversion of 5‐methylcytosine (5‐mC) to 5‐hydroxymethylcytosine (5‐hmC) (an intermediate in the demethylation process).[ 72 ] It has been presumed that increased methylation of gene‐promoters could prevent transcription factor binding to promoters, but it has now been recognized that actively transcribed genes have high levels of methylation in the gene body. It is also quite intriguing that high levels of Tet1 and 5hmC are associated with transcriptionally permissive histone bivalent domain (H3K27me3 marks).[ 73 ] Relationship between αKG levels and distribution of 5hmC and Tet enzymes in the aging genome remain to be elucidated. In conjunction with Tet enzymes, enzymes that mediated histone demethylation, JMJD2 (Jumanji domain‐containing protein 2), and JARID1 (Jumonji, AT rich interactive domain 1) families of the N ε‐trimethyl‐lysine demethylases are also Fe (II) and αKG dependent.[ 74 , 75 , 76 ] These demethylases target histone H3K9Me3 marks that are a characteristic of heterochromatin, and loss of these marks has been associated with Hutchinson–Gilford Progeria Syndrome, a disease of accelerated aging.[ 77 , 78 ] Therefore control of levels of histone methylation associated with the H3K9Me3 is expected to have profound impacts on aging. Uncovering the relationship between αKG metabolism and JMJD2 activity in aging is an unmet need and opportunity in aging biology.
βHB at the Hub of Caloric Restriction and Histone Deacylation
Caloric restriction has been shown to extend lifespan via the inhibition of mTOR and activation of FOXO transcription factors.[ 10 , 66 , 79 ] The ketone body βHB has emerged as an energetic and signaling metabolite that has been shown to mediate this effect in both invertebrate and vertebrate model systems.[ 80 ] Ketone bodies were first discovered in the urine of diabetic patients and later found to increase in subjects undergoing starvation.[ 80 ] Lehninger and Greville showed that the βHB formed during ketosis was the d‐isoform. Levels of this metabolite have been shown to increase to ≈2 × 10−3 m in the fasting state or 0.6 × 10−3 m during caloric restriction. The metabolite has a number of signaling roles in epigenetics and stress response signaling[ 81 ] that explain its pleiotropic effects on aging. A recent study has shown that ketogenic diet can improve the midlife mortality of mice and provide neurological benefits such as improved memory in aging mice.[ 82 ] The caloric restriction/anti‐aging mechanism of βHB has been ascribed to two overlapping mechanisms. i) in an energetic role, βHB provides an alternative (anaplerotic) source of carbon to the mitochondrial TCA cycle in the context of an aging‐associated mitochondrial dysfunction, ii) direct inhibition of HDAC I and IIa histone deacetylases (IC50 ≈1 × 10−3 m) causing the deacetylation of FOXO3a promoter leading to transcriptional activation.[ 81 ] It has been shown that treatment/supplementation with βHB esters can also promote the expression of ROS detoxification genes such as metallothionein and reduction in NADP/NADPH ratios.[ 83 ] The protective properties of this metabolite requires the nuclear respiratory factor 2 (Nrf2) and AMP kinase (AMPK) signaling pathways.[ 81 , 83 ] Ketone bodies are now being explored as therapeutics for a number of cardiac and neurological disorders,[ 84 ] therefore a metabolomic understanding of the ketone bodies and their fates in various tissues is an unmet need for effective application of ketone body esters to improve health‐span.
3.2. Emergence of Mitochondrial TCA Cycle as an Integrator of Hub Metabolites, and Anti‐Aging Interventions such as Exercise and Fasting
It is clear that the four hub‐metabolites presented in this review on the aging metabolome (NAD+, NADPH, αKG, and βHB) have significant metabolic and signaling roles in aging biology. These metabolites are in fact metabolically interconnected (Figure 3 ). An initial analysis of this interconnectedness reveals the central role of the mitochondrial TCA cycle as an integrator of these four signals, and an important target in aging biology. NAD+ in cells is maintained as separate nucleo‐cytoplasmic pools and mitochondrial pools. NAD+ in the cytoplasm is generated during glycolysis, and mitochondrial NADH produced by the TCA is utilized by the electron transport chain and converted back to NAD+. The two pools can interact via the malate aspartate shuttle, and possibly via the direct transporter of NAD into the mammalian mitochondria.[ 85 ] The age‐dependent dysregulation in NAD+ metabolism[ 37 , 38 ] and the well‐characterized mitochondrial loss associated with aging[ 86 , 87 ] are consistent with the loss of mitochondrial TCA function. Based on this observation there is significant interest in supplementation of NAD+ precursors (e.g., nicotinamide mononucleotide (NMN)) to compensate for aging‐related metabolic dysfunction in NAD metabolism.[ 88 , 89 ] The second hub metabolite, NADPH is generated primarily via the pentose phosphate shunt via the precursor NADP. NADP is not membrane permeable and similar to NAD+, both cytosolic and mitochondrial NADP pools are maintained separately. NADP is generated via the enzyme NAD kinase (NADK), with the cytosolic and mitochondrial localized NADK.[ 90 ] The physiological roles of these enzymes and how levels of the cytosolic and mitochondrial NADP pools are regulated is less understood, and it has been recently shown that knockout of the mitochondrial NADK caused stress induced hepatic steatosis.[ 91 ] Therefore, it is becoming clear that the mitochondrial NAD+ pools play a critical role in NADP homeostasis and physiology. The third hub metabolite αKG is itself a TCA intermediate and a substrate for numerous transmethylation reaction in cells, is primarily synthesized from pyruvate or glutamate. It is expected that aging‐related loss of mitochondrial function will have significant effects the TCA cycle and hence on αKG levels. Finally, βHB is a ketone body that is a primary source of acetyl‐CoA in the TCA cycle during starvation. Therefore, this hub metabolite is intimately linked with the TCA cycle and may help ameliorate aging related declines in mitochondrial function. It is not surprising that a number of anti‐aging interventions like exercise and starvation/caloric restriction act directly on the TCA cycle. It has been well known that exercise results in a significant increase in TCA cycle intermediates[ 92 , 93 ] generated from both glucose and fatty acid oxidation. It has recently become clear that branched chain amino acids (BCAA) are a major source of TCA cycle intermediates[ 94 , 95 ] during exercise, and that BCAA catabolism is required during exercise. Therefore, it is clear that multiple carbon substrates including BCAA are mobilized to maintain TCA intermediates that play important metabolic and signaling roles. The hub metabolites, βHB as reviewed in the previous section is not only a metabolic substrate for the TCA (giving rise to acetoacetate and acetyl‐CoA) but also a signaling molecule that helps cells adapt to starvation. In summary, the TCA cycle serves to integrate signals from hub metabolites to regulate aging‐associated metabolic dysfunction and is important for anti‐aging interventions.
Figure 3.
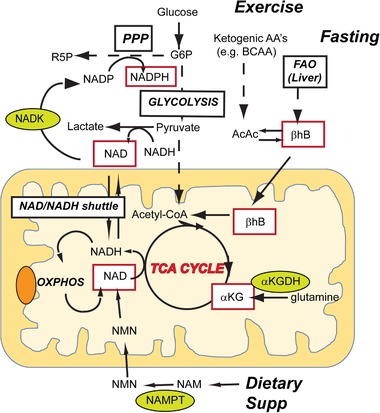
Mitochondrial TCA as the hub for integrating metabolism and signaling in the aging network. The four hub metabolites (red boxes) are interconnected via the mitochondrial TCA cycle. NAD+ is utilized during the TCA cycle to generate NADH and is regenerated during oxidative phosphorylation and during formation of lactate from pyruvate. NAD can also be produced by dietary supplementation of precursors of NAD such as nicotinamide (NAM) that is converted to nicotinamide mononucleotide (NMN) by the enzyme nicotinamide phosphoribosyl transferase (NAMPT). NADP is synthesized from NAD via the enzyme NAD kinase (NADK) that is localized to the cytoplasm and mitochondria. NADPH is generated during the pentose phosphate pathway (PPP) and used in biosynthetic and redox responsive pathways. αKG is a TCA intermediate, either generated from pyruvate or from glutamine by the enzyme αKG dehydrogenase (αKGDH). βHB, a ketone body, serves as an anaplerotic source for acetyl‐CoA in the TCA cycle. It is generated during starvation/fasting via the β‐oxidation of lipids in the liver and transported to tissues such as skeletal muscle and brain or can be generated by ketogenic amino acid (e.g., BCAA during exercise).
4. Conclusions
Metabolomics has revealed biomarkers and underlying hub metabolites that might mediate aging related loss of metabolic homeostasis. There is a need to translate the role of the four hub metabolites NAD+, NADPH, αKG, and βHB into human biology using multi‐tissue flux measurements of these metabolic hubs. An important insight gained is the role of the mitochondrial TCA cycle as an integrator of hub metabolite metabolism and signaling. Aging biology is rapidly moving from the understanding of basic mechanisms to translating these findings to improve health‐spans of human subjects. For example, the translation of the basic understanding of senescent cells toward identifying senolytics (compounds that selectively kill senescent cells) for targeting senescent cells in aging and aging‐associated diseases. The success of these interventions will depend on metabolomics based biochemical markers that can directly measure efficacy on aging related pathways like the TCA cycle. Metabolomics is therefore poised to play a transformative role in translating basic understanding of aging biology into the clinic.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
The study was supported by NIH grant R01AG057353.
Biographies
Rishi Sharma graduated with a Ph.D. in medicinal chemistry and drug development from Northeastern University, Boston in 2011. His graduate research was focused on the design, synthesis, and biological evaluation of novel cannabinoids with controlled metabolic detoxification. He secured a postdoctoral research associate position at same university, wherein he performed the metabolite identification of lead novel cannabinoids using LC–MS/MS platforms and NMR. He joined the Buck Institute in 2018 as a postdoctoral research fellow in Metabolomics. He utilizing HPLC–MS/MS based platforms for global measurement of metabolites and their fluxes in Drosophila flies, tissues, and neuronal cultures fed with 13C6‐glucose.

Arvind Ramanathan obtained his Ph.D. from NYU in Chemistry in 2002. In his post‐doctoral research at the Broad Institute, he pioneered the use of metabolomics for elucidating the regulation of metabolism in cancer cells in response to oncogenic and metabolic perturbations. He joined the Buck Institute as a faculty member in 2011. As the director of metabolomics at the Buck Institute his recent focus is on the metabolic basis of Aging especially on the role of lipid signaling in senescent cells and aging related biomarker discovery.

Sharma R., Ramanathan A., The Aging Metabolome—Biomarkers to Hub Metabolites. Proteomics 2020, 20, 1800407 10.1002/pmic.201800407
References
- 1. Cornelissen G., Otsuka K., Gerontology 2017, 63, 118. [DOI] [PubMed] [Google Scholar]
- 2. Vijg J., Kennedy B. K., Gerontology 2016, 62, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. López‐Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G., Cell 2013, 153, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Niccoli T., Partridge L., Curr. Biol. 2012, 22, R741. [DOI] [PubMed] [Google Scholar]
- 5. Fontana L., Bioch. Biophys. Acta 2009, 1790, 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Azzu V., Valencak T. G., Gerontology 2017, 63, 327. [DOI] [PubMed] [Google Scholar]
- 7. Feng Z., Hanson R. W., Berger N. A., Trubitsyn A., Oncotarget 2016, 7, 15410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Verdin E., Science 2015, 350, 1208. [DOI] [PubMed] [Google Scholar]
- 9. Weichhart T., Gerontology 2018, 64, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Antikainen H., Driscoll M., Haspel G., Dobrowolski R., Aging Cell 2017, 16, 1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burkewitz K., Zhang Y., Mair W. B., Cell Metab. 2014, 20, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barzilai N., Crandall J. P., Kritchevsky S. B., Espeland M. A., Cell Metab. 2016, 23, 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. J. M. Hoffman, Lyu Y., Pletcher S. D., Promislow D. E. L., Essays Biochem. 2017, 61, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu X., Locasale J. W., Trends Biochem. Sci. 2017, 42, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rinschen M. M., Ivanisevic J., Giera M., Siuzdak G., Nat. Rev. Mol. Cell Biol. 2019, 20, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yin P., Xu G., Methods Mol. Biol. 2017, 1619, 467. [DOI] [PubMed] [Google Scholar]
- 17. Johnson C H., Ivanisevic J., Siuzdak G., Nat. Rev. Mol. Cell Biol. 2016, 17, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gowda G. A., Djukovic D., Methods Mol. Biol. 2014, 1198, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stanziano D. C., Whitehurst M., Graham P., Roos B. A., J. Am. Geriatr. Soc. 2010, 58, S292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferrucci L., J. Gerontol., Ser. A 2008, 63, 1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murabito J. M., Yuan R., Lunetta K. L., J. Gerontol., Ser. A 2012, 67A, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morris J. K., Uy R. A. Z., Vidoni E. D., Wilkins H. M., Archer A. E., Thyfault J. P., Miles J. M., Burns J. M., J. Alzheimer's Dis. 2017, 58, 1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thambisetty M., Metter E. J., Yang A., Dolan H., Marano C., Zonderman A. B., Troncoso J. C., Zhou Y., Wong D. F., Ferrucci L., Egan J., Resnick S. M., O'Brien R. J., JAMA Neurol. 2013, 70, 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sebastiani P., Gurinovich A., Nygaard M., Sasaki T., Sweigart B., Bae H., Andersen S. L., Villa F., Atzmon G., Christensen K., Arai Y., Barzilai N., Puca A., Christiansen L., Hirose N., Perls T. T., J. Gerontol., Ser. A 2019, 74, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yaku K., Okabe K., Nakagawa T., Ageing Res. Rev. 2018, 47, 1. [DOI] [PubMed] [Google Scholar]
- 26. Nikiforov A., Dölle C., Niere M., Ziegler M., J. Biol. Chem. 2011, 286, 21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wątroba M., Dudek I., Skoda M., Stangret A., Rzodkiewicz P., Szukiewicz D., Ageing Res. Rev. 2017, 40, 11. [DOI] [PubMed] [Google Scholar]
- 28. Satoh A., Imai S.‐I., Guarente L., Nat. Rev. Neurosci. 2017, 18, 362. [DOI] [PubMed] [Google Scholar]
- 29. Alleyn M., Breitzig M., Lockey R., Kolliputi N., Am. J. Physiol.: Cell Physiol. 2018, 314, C228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mathias R. A., Greco T. M., Oberstein A., Budayeva H. G., Chakrabarti R., Rowland E. A., Kang Y., Shenk T., Cristea I. M., Cell 2014, 159, 1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan M., Peng C., Anderson K. A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., Ro J., Wagner G. R., Green M. F., Madsen A. S., Schmiesing J., Peterson B. S., Xu G., Ilkayeva O. R., Muehlbauer M. J., Braulke T., Mühlhausen C., Backos D. S., Olsen C. A., McGuire P. J., Pletcher S. D., Lombard D. B., Hirschey M. D., Zhao Y., Cell Metab. 2014, 19, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hirschey M. D., Zhao Y., Mol. Cell. Proteomics 2015, 14, 2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cantó C., Auwerx J., Trends Endocrinol. Metab. 2009, 20, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Callaghan C., Vassilopoulos A., Aging Cell 2017, 16, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Singh C. K., Chhabra G., Ndiaye M. A., Garcia‐Peterson L. M., Mack N. J., Ahmad N., Antioxid Redox Signal. 2018, 28, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murata M., Kong X., Moncada E., Chen Y., Wang P., Berns M. W., Yokomori K., Digman M. A., Mol. Biol. Cell 2019, 30, 2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schultz M. B., Sinclair D. A., Cell Metab. 2016, 23, 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson S., Imai S.‐I., F1000Res. 2018, 7, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Clement J., Wong M., Poljak A., Sachdev P., Braidy N., Rejuvenation Res. 2019, 22, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu L., Su X., Quinn W. J., Hui S., Krukenberg K., Frederick D. W., Redpath P., Zhan L., Chellappa K., White E., Migaud M., Mitchison T. J., Baur J. A., Rabinowitz J. D., Cell Metab. 2018, 27, 1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Su X., Lu W., Rabinowitz J. D., Anal. Chem. 2017, 89, 5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Magalhāes J. P., Passos J. F., Mech. Ageing Dev. 2018, 170, 2. [DOI] [PubMed] [Google Scholar]
- 43. Kirkland J. L., Tchkonia T., EBioMedicine 2017, 21, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coppé J.‐P., Rodier F., Patil C K., Freund A., Desprez P.‐Y., Campisi J., J. Biol. Chem. 2011, 286, 36396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Basisty N., Kale A., Jeon O. H., Kuehnemann C., Payne T., Rao C., Holtz A., Shah S., Sharma V., Ferrucci L., Campisi J., Schilling B., PLoS Biol. 2020, 18, e3000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wiley C. D., Campisi J., Cell Metab. 2016, 23, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wiley C. D., Velarde M. C., Lecot P., Liu S., Sarnoski E. A., Freund A., Shirakawa K., Lim H. W., Davis S. S., Ramanathan A., Gerencser A. A., Verdin E., Campisi J., Cell Metab. 2016, 23, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nacarelli T., Lau L., Fukumoto T., Zundell J., Fatkhutdinov N., Wu S., Aird K. M., Iwasaki O., Kossenkov A. V., Schultz D., Noma K.‐I., Baur J. A., Schug Z., Tang H.‐Y., Speicher D. W., David G., Zhang R., Nat. Cell Biol. 2019, 21, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ying W., Antioxid. Redox Signal. 2008, 10, 179. [DOI] [PubMed] [Google Scholar]
- 50. Fernandez‐Marcos P. J., Nóbrega‐Pereira S., Oncotarget 2016, 7, 50814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou G., Meng S., Li Y., Ghebre Y. T., Cooke J. P., Cell Rep. 2016, 15, 919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hochmuth C. E., Biteau B., Bohmann D., Jasper H., Cell Stem Cell 2011, 8, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cieślar‐Pobuda A., Yue J., Lee H.‐C., Skonieczna M., Wei Y.‐H., Oxid. Med. Cell. Longevity 2017, 2017, 5047168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bigarella C. L., Liang R., Ghaffari S., Development 2014, 141, 4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Legan S. K., Rebrin I., Mockett R. J., Radyuk S. N., Klichko V. I., Sohal R. S., .Orr W C., J. Biol. Chem. 2008, 283, 32492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang L., Davis S. S., Borch Jensen M., Rodriguez‐Fernandez I. A., Apaydin C., Juhasz G., Gibson B. W., Schilling B., Ramanathan A., Ghaemmaghami S., Jasper H., Aging Cell 2019, 18, e12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nóbrega‐Pereira S., Fernandez‐Marcos P J., Brioche T., Gomez‐Cabrera M. C., Salvador‐Pascual A., Flores J. M., Viña J., Serrano M., Nat. Commun. 2016, 7, 10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Field A. E., Robertson N. A., Wang T., Havas A., Ideker T., Adams P. D., Mol. Cell 2018, 71, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McCauley B. S., Dang W., Bioch. Biophys. Acta 2014, 1839, 1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wu N., Yang M., Gaur U., Xu H., Yao Y., Li D., Biomol. Ther. 2016, 24, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. He L., Xu Z., Yao K., Wu G., Yin Y., Nyachoti C., Kim S., Curr. Protein Pept. Sci. 2015, 16, 576. [DOI] [PubMed] [Google Scholar]
- 62. Kuo C.‐Y., Cheng C.‐T., Hou P., Lin Y.‐P., Ma H., Chung Y., Chi K., Chen Y., Li W., Kung H.‐J., Ann D. K., Oncotarget 2016, 7, 34052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Semenza G. L., Science STKE 2007, 2007, cm8. [DOI] [PubMed] [Google Scholar]
- 64. Bruegge K., Jelkmann W., Metzen E., Curr. Med. Chem. 2007, 14, 1853. [DOI] [PubMed] [Google Scholar]
- 65. Chin R. M., Fu X., Pai M. Y., Vergnes L., Hwang H., Deng G., Diep S., Lomenick B., Meli V. S., Monsalve G. C., Hu E., Whelan S. A., Wang J. X., Jung G., Solis G. M., Fazlollahi F., Kaweeteerawat C., Quach A., Nili M., Krall A. S., Godwin H. A., Chang H. R., Faull K. F., Guo F., Jiang M., Trauger S. A., Saghatelian A., Braas D., Christofk H. R., Clarke C. F., Teitell M. A., Petrascheck M., Reue K., Jung M. E., Frand A. R., Huang J., Nature 2014, 510, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stefanetti R. J., Voisin S., Russell A., Lamon S., F1000Res. 2018, 7, 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Carey B. W., Finley L. W. S., Cross J. R., Allis C. D., Thompson C. B., Nature 2015, 518, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Singh K., Krug L., Basu A., Meyer P., Treiber N., Vander Beken S., Wlaschek M., Kochanek S., Bloch W., Geiger H., Maity P., Scharffetter‐Kochanek K., Stem Cells 2017, 35, 1704. [DOI] [PubMed] [Google Scholar]
- 69. Ahuja N., Li Q., Mohan A. L., Baylin S. B., Issa J. P., Cancer Res. 1998, 58, 5489. [PubMed] [Google Scholar]
- 70. Pawlak M., Jaenisch R., Genes Dev. 2011, 25, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Horvath S., Genome Biol. 2013, 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. An J., Rao A., Ko M., Exp. Mol. Med. 2017, 49, e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Verma N., Pan H., Doré L. C., Shukla A., Li Q. V., Pelham‐Webb B., Teijeiro V., González F., Krivtsov A., Chang C.‐J., Papapetrou E. P., He C., Elemento O., Huangfu D., Nat. Genet. 2018, 50, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zheng W., Huang Y., MedChemComm 2014, 5, 297. [Google Scholar]
- 75. Chen Z., Zang J., Whetstine J., Hong X., Davrazou F., Kutateladze T. G., Simpson M., Mao Q., Pan C.‐H., Dai S., Hagman J., Hansen K., Shi Y., Zhang G., Cell 2006, 125, 691. [DOI] [PubMed] [Google Scholar]
- 76. Tran K. A., Dillingham C. M., Sridharan R., J. Biol. Chem. 2019, 294, 5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shumaker D. K., Dechat T., Kohlmaier A., Adam S. A., Bozovsky M. R., Erdos M. R., Eriksson M., Goldman A. E., Khuon S., Collins F. S., Jenuwein T., Goldman R. D., Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Arancio W., Pizzolanti G., Genovese S. I., Pitrone M., Giordano C., Gerontology 2014, 60, 197. [DOI] [PubMed] [Google Scholar]
- 79. Madeo F., Pietrocola F., Eisenberg T., Kroemer G., Nat. Rev. Drug Discov. 2014, 13, 727. [DOI] [PubMed] [Google Scholar]
- 80. Veech R. L., Bradshaw P. C., Clarke K., Curtis W., Pawlosky R., King M. T., IUBMB Life 2017, 69, 305. [DOI] [PubMed] [Google Scholar]
- 81. Newman J. C., Verdin E., Trends Endocrinol. Metab. 2014, 25, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Newman J. C., Covarrubias A. J., Zhao M., Yu X., Gut P., Ng C.‐P., Huang Y., Haldar S., Verdin E., Cell Metab. 2017, 26, 547 e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shimazu T., Hirschey M. D., Newman J., He W., Shirakawa K., Le Moan N., Grueter C. A., Lim H., Saunders L. R., Stevens R. D., Newgard C. B., Farese R. V., de Cabo R., Ulrich S., Akassoglou K., Verdin E., Science 2013, 339, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Puchalska P., Crawford P. A., Cell Metab. 2017, 25, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Davila A., Liu L., Chellappa K., Redpath P., Nakamaru‐Ogiso E., Paolella L. M., Zhang Z., Migaud M. E., Rabinowitz J. D., Baur J. A., eLife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Loeb L. A., Wallace D. C., Martin G. M., Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 18769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wallace D. C., Annu. Rev. Genet. 2005, 39, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Khan N. A., Auranen M., Paetau I., Pirinen E., Euro L., Forsström S., Pasila L., Velagapudi V., Carroll C. J., Auwerx J., Suomalainen A., EMBO Mol. Med. 2014, 6, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yoshino J., Mills K. F., Yoon M. J., Imai S.‐I., Cell Metab. 2011, 14, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhang R., J. Cell Physiol. 2015, 230, 1697. [DOI] [PubMed] [Google Scholar]
- 91. Zhang K., Kim H., Fu Z., Qiu Y., Yang Z., Wang J., Zhang D., Tong X., Yin L., Li J., Wu J., Qi N. R., Houten S. M., Zhang R., Gastroenterology 2018, 154, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lundsgaard A.‐M., Fritzen A. M., Kiens B., Trends Endocrinol. Metab. 2018, 29, 18. [DOI] [PubMed] [Google Scholar]
- 93. Gaster M., Nehlin J. O., Minet A. D., Arch. Physiol. Biochem. 2012, 118, 156. [DOI] [PubMed] [Google Scholar]
- 94. She P., Zhou Y., Zhang Z., Griffin K., Gowda K., Lynch C. J., J. Appl. Physiol. 2010, 108, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Xu M., Kitaura Y., Ishikawa T., Kadota Y., Terai C., Shindo D., Morioka T., Ota M., Morishita Y., Ishihara K., Shimomura Y., PLoS One 2017, 12, e0180989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information