

Abstract
Asthma remains the most prevalent chronic respiratory disorder, affecting people of all ages. The relationship between respiratory virus infection and asthma has long been recognized, though remains incompletely understood. In this article, we will address key issues around this relationship. These will include the crucial role virus infection plays in early life, as a potential risk factor for the development of asthma and lung disease. We will assess the impact that virus infection has on those with established asthma as a trigger for acute disease and how this may influence asthma throughout life. Finally, we will explore the complex interaction that occurs between the airway and the immune responses that make those with asthma so susceptible to the effects of virus infection.
Keywords: asthma, viruses, bronchiolitis, respiratory syncytial virus, rhinovirus, airway microbiota, genetic susceptibility
Early-Life Exposure to Viruses and the Development of Asthma
Viral Bronchiolitis and the Development of Asthma
Acute lower respiratory tract infections resulting in bronchiolitis remain among the most common cause of hospital admission in the developed world for children younger than the age of 2 years. In those younger than 1 year, bronchiolitis caused by respiratory syncytial virus (RSV) is the primary cause, 1 while for those older than 1 year, infection with rhinovirus (RV) is more prominent. 2
RSV bronchiolitis has been associated with an increase in subsequent wheezing illness, and it has long been considered that this association may play a causative role in the development of asthma. Research has now shown that severe early-life infection with RSV is associated with an increased risk of infrequent wheeze (odds ratio: 3·2 [95% confidence interval [CI]: 2·0–5·0], p < 0·001), and frequent wheeze (4·3 [2·2–8·7], p ≤ 0·001) by the age of 6 years, but was no longer associated with episodes of wheeze by the age of 13 years. 3 There was also no association between RSV lower respiratory tract illnesses and development of allergic disease, though RSV lower respiratory tract illnesses were associated with significantly lower lung function at the age of 13 years. 3 In support of these observations, Lu et al also went on to show that the risk of an asthma diagnosis at the age of 6 years was determined by the severity of the acute RSV episode along with a family history of asthma highlighting interaction with atopy. 4 Similarly, severe RSV bronchiolitis resulting in hospitalization was associated with a six- to eightfold increase in admission for wheeze in the first 2 months after RSV hospitalization, but this was no longer increased a year later. 5 However, in those with a diagnosis of asthma, the risk for hospitalization with RSV infection was increased threefold, and this risk was not time dependent, suggesting host factors associated with a diagnosis of asthma at this age increase susceptibility to RSV, but the effect RSV has alone on airway symptoms such as wheeze is transitory. 5
RV infection is the second most common virus associated with bronchiolitis, and the most common virus detected in association with wheezing illnesses in children by the age of 1 to 2 years. 6 7 8 Three species of RV (human RV [HRV]-A, HRV-B, and HRV-C) are now recognized to include 150+ antigenically distinct virus subtypes. Most of these are likely to also represent serotypes explaining life-long susceptibility to RV infections. 9 Differences in association of RV species with disease have been observed, with RV-A and the recently discovered RV-C responsible for more severe lower respiratory tract illnesses. 10 11 12 HRV-A and HRV-B can be grown in standard laboratory cell lines enabling isolation from clinical samples and classification according to neutralizing antibody responses (serotypes). This occurred in the 1960s and 1970s and led to the identification of approximately 100 strains. HRV-C was only discovered in 2006, 13 as they could not be cultured using standard techniques and hence has gone unappreciated as a pathogen. HRV-C has now been shown to infect both the youngest children and to cause the most severe acute RV-induced lower respiratory tract disease. 12 Once researchers were able to culture HRV-C using respiratory mucosal explants or air–liquid interface differentiated bronchial epithelium, they determined that it used the human transmembrane protein, cadherin-related family member 3 (CDHR3) for virus binding and replication. 14 Serendipitously, it had recently been shown that there were four distinct alleles associated with CDHR3 expression, with one of these representing a single nucleotide polymorphism (G→A) that converts a residue cysteine to tyrosine at position 529 (Cys 529 →Tyr, rs6967330). This leads to a rare, but phenotypically dominant, asthma-related A-encoded Tyr529 variant of the CDHR3 gene that was linked to greater airway epithelial cell (AEC)-surface expression of CDHR3, and an increased risk of wheezing illnesses and hospitalizations for children 2 to 5 years. 15 16 Bochkov et al then went on to show that compared with wild-type CDHR3, cells transfected with the asthma susceptible CDHR3-Y529 variant had a 10-fold increase in HRV-C binding, increasing infectious virus yields. 14 These findings are the first to link genetic susceptibility to respiratory tract infection with the development of childhood asthma.
Allergy Virus Infection and the Risk of Asthma
Allergic sensitization is also intertwined in the risk of developing asthma and virus-induced wheezing. Children who develop sensitization to multiple aeroallergens at a younger age are more prone to develop severe acute asthma needing hospitalization. 17 The relationship between virus infection and allergic sensitization was elegantly demonstrated by the investigators of the Childhood Origins of Asthma study (COAST) study who showed that children sensitized to aeroallergens and who wheezed with RV during the first 3 years of life had the greatest risk for developing asthma. 18 The investigators went on to demonstrate that there was a sequential relationship with allergic sensitization leading to viral wheezing, especially wheezing was associated with RV, but there was no evidence that viral wheezing itself led to allergic sensitization. 19 In fact, the impact of RV infection on early life has been the subject of a meta-analysis of 15 original studies, 10 reported on the results of 4 longitudinal cohort studies with different follow-up periods. RV wheezing illness in the first 3 years of life was associated with an increased risk of wheezing/asthma in later life (relative risk [RR] = 2.00, 95% CI: 1.62–2.49, p < 0.001). In subgroup analysis done by age at follow-up, the association was significant in those <10 years (RR = 2.02, 95% CI: 1.70–2.39, p < 0.001) and ≥10 years (RR = 1.92, 95% CI: 1.36–2.72, p < 0.001). 20 Therefore, as has been seen with RSV, it is a host susceptibility linked to atopy, as demonstrated by a family history of allergic asthma that confers a significant risk of RV-induced wheeze. The impact of viral infection in early life and the role this then plays in the inception of asthma was best summarized by Gern and Busse, when they stated “severe infections at this stage can result in 2 non-exclusive outcomes, the first being that they may impair normal lung development and the second being that they may trigger wheezing illnesses in susceptible individuals.” 21
Preventing Virus-Induced Wheeze
Given the close association with viral bronchiolitis and childhood asthma, interventions to prevent or at least reduce the severity of the disease, potentially could have important and long-lasting effects. Although this is not possible for RV infection, the development of a humanized monoclonal antibody that targets the RSV F protein, palivizumab, allows passive immunization that reduces the susceptibility and severity of RSV bronchiolitis in preterm, high-risk infants. 22 For this reason, investigators randomized 429 otherwise healthy preterm infants to treatment with palivizumab or placebo. 23 The treated infants had a 61% (95% CI: 56–65) relative reduction in total number of wheezing days in the first year of life, and the proportion of infants with recurrent wheeze was 10% points lower in those treated with palivizumab (11 vs. 21%, p = 0.01). 23 In a similarly sized study that looked at outcomes up to 6 years of age, following palivizumab treatment in the first year of life, there were fewer recorded episodes of wheeze, but no difference was seen in doctor diagnosis of asthma. 24 It will be interesting to determine whether this intervention will impact on either lung function or asthma at later ages.
The Airway Microbiome and Asthma in Early Life
The interaction between the developing immune system and viral infections in early life also occurs against the backdrop of exposure to bacteria or the airway microbiota. Until recently, the healthy lower airway was not considered to harbor bacteria. Our understanding of respiratory microbiota has changed with the advent of culture-independent techniques involving high-throughput sequencing of the 16S rRNA gene, a highly conserved locus of the bacterial genome. This has revealed a complex microbial community that varies between states of health and respiratory disease. 25 A relationship between bacterial colonization and early-life asthma was first demonstrated in the Copenhagen birth cohort that showed a heightened risk of wheeze and asthma in children whose nasopharynx was colonized with Streptococcus pneumoniae , Moraxella catarrhalis , and Haemophilus influenzae at 1 month of age, These same bacteria were also present during acute episodes of wheeze associated with viral infections and conferred a similar degree of risk for acute wheeze. 26
Teo et al collected nasopharyngeal aspirates from children at 2, 6, and 12 months of age and within 48 hours of a clinical respiratory tract infection and applied modern methods of 16S rRNA gene sequencing. 27 They then went on to assess changes in the upper airway microbiota and chronic wheeze at the age of 5 years. Staphylococcus and Corynebacterium , organisms known to be part of the skin microbiome, were the dominant species in the first 2 months of life before changing to Alloiococcus or Moraxella at 6 to 12 months, concomitant with a stabilization of the microbial population. They speculated that infants are likely to be colonized initially with skin bacteria from their parents and these populations are replaced over time by Moraxella or Alloiococcus . This upper respiratory microbiota then remains stable over time in healthy individuals. However, in infants who had virus respiratory infections, there was a greater abundance of Streptococcus , Moraxella , and Haemophilus. While early Moraxella colonization was associated with a younger age for the first viral upper respiratory infection, Streptococcus colonization was associated with earlier initial lower respiratory infection. The level of subsequent asthma risk then was found to inversely relate to the age at which initial Streptococcus colonization occurred. The investigators speculated that lower airway infection with RSV or RV contributes to an altered microbe featuring Streptococcus and the resulting effect on the airway and/or developing immune system predisposes to the development of asthma. 27 This intriguing area obviously requires further work, but it is fascinating to see how exposure in early life to pathogens, either viral or bacterial can alter both the airways and immune response so profoundly.
The Hygiene Hypothesis and Protection against Asthma
The hygiene hypothesis was developed to explain the relationship between microbial exposure in early life and protection against allergic and autoimmune diseases that continue to increase in prevalence particularly in westernized societies. It proposed that improvements in housing and changes to an urban lifestyle may be responsible for a lack of exposure to bacterial pathogens in early life resulting in an immune response that becomes skewed to the development of hypersensitivity and allergy. 28 29 Apart from infection, exposure to multiple bacterial antigens can occur from the indoor and outdoor environment. Bacterial products, such lipopolysaccharide (LPS) or endotoxin, are recognized as foreign by the innate immune system and their exposure, especially in early life activates a developing immune system. In support of this concept, high-level chronic exposure to LPS has been shown experimentally to induce immune tolerance. 30 This hypothesis was supported by German investigators who examined samples of dust from the mattresses of children and then related the levels of endotoxin to the prevalence of asthma and allergies and to serum levels of specific immunoglobulin E (IgE). They found that children exposed to “farm dust,” containing high levels of LPS were associated with a significant decrease in the risk of hay fever, atopic sensitization, atopic asthma, and atopic wheeze in childhood. 31 This work has been supported by strong epidemiological studies confirming early-life exposure to farming and bacterial LPS significantly reduces the risk of asthma. 31 32 Recently, the mechanisms behind this regulation have started to be understood, with AECs and the regulator protein, A20, playing a key role. 33 A20 is a deubiquitinase that is an essential negative regulator of nuclear factor-κB (NF-κB) activation and the subsequent induction of inflammatory responses. 34 A20 forms a unique ubiquitin-editing complex with other proteins that is required for the activation of NF-κB. 35 A20 knockout mice are highly susceptible to exposure to inflammatory mediators and LPS. 36 Recently, an important role has emerged for A20 in the regulation of early asthma. Mice when exposed to low levels of LPS in early life do not develop features of asthma, despite sensitization to house dust mite (HDM). Using knockout mice with selective A20 deficiency in the AECs, the protective effect of LPS was diminished, with increased epithelial expression of inflammatory mediators, chemokine (C–C motif) ligand (CCL)-20 and granulocyte macrophage colony-stimulating factor (GM-CSF) following HDM. 36 In human AECs, LPS treatment also blunted the inflammatory response (CCL-20, GM-CSF, and interleukin [IL]-1α) following HDM exposure. 37 Bringing these findings together, Schuijs et al demonstrated that LPS-exposed epithelial cell cytokines are responsible for activating dendritic cells (DCs), and suppressing type 2 immunity to HDM, while loss of the ubiquitin-modifying enzyme A20 in lung epithelium abolished this protective effect. They also demonstrated that a single-nucleotide polymorphism in the gene encoding A20 was associated with allergy and asthma risk in children growing up on farms. 33
While preventing exposure to RSV may be a risk for early-life asthma, exposure to bacterial polyproteins may conversely be protective. Some preliminary evidence hints at this. Recent studies have been performed using an extract from bacterial products OM-85 BV that can safely be given as an oral preparation. Preliminary studies suggest that children treated with OM-85 BV demonstrated an immune response that was skewed away from type 2 immunity, with increased interferon (IFN)- γ/IL-4 ratios. 38 A small clinical trial has also shown that its use decreased asthma exacerbations; in 1 to 6 years old, attacks were reduced 37.9% and 2 days shorter for OM-85 BV versus placebo ( p < 0.001). 39 In fact, OM-85 BV reduced attacks to a greater degree than inhaled corticosteroids.
Respiratory Viruses as Triggers of Asthma Exacerbations
Epidemiology of Acute Asthma
While viral infection is highly linked with wheeze in early life, and the subsequent development of asthma, the association between viral infections and exacerbation of asthma in adolescents and adults with an established diagnosis of asthma is particularly strong. Currently, the Global Initiative for Asthma (GINA) describes an exacerbation as an acute change in symptoms or lung function, typically requiring a change to management, though consensus on what defines an exacerbation is poor. 40 Acute exacerbations are the leading cause of asthma deaths and hospitalization. It is estimated that 80% of the costs associated with the treatment of asthma are due to exacerbations, despite them happening in a minority of patients. 41 Given the gravity associated with asthma exacerbations, there has been much work on the etiology of these acute events. It has been identified that viral infections are responsible for the majority of asthma exacerbations (80% in children and 76% in adults). 42 43 Thus, we will discuss the role of viruses in the epidemiology, risk factors to exacerbation, and influence on asthma control.
In general, infection with respiratory viruses remains the most common cause of upper respiratory tract infection (URTI). Of these infections, RV is the most common, representing up to 50% of documented cases. 44 Following this are infections with coronavirus, parainfluenza, influenza, RSV, enterovirus, and adenovirus. While the majority of these viruses tends to involve AEC infection of the upper respiratory tract and subsequent inflammation, influenza and RSV are distinguished by a propensity to infect the lower respiratory tract and cause particularly severe damage to the airway epithelial layer and, as such, lead to increased disease severity. 44
RV, while being the most common cause of URTI (and the most common viral infection of man), is also most common virus associated with exacerbations of asthma. Indeed, while upwards of 76 to 80% of acute exacerbations of asthma have been linked with virus infection, 60% of these are due to RV. 42 43 45 This represents a higher proportion of patients affected by RV than that found in the general population associated with URTI, which has led to insights that patients with asthma may actually be more susceptible to viral infection. 46 47 It has been observed that patients with asthma are more likely to develop more severe lower respiratory tract symptoms in the setting of viral infection than healthy individuals. 48 This is not uniformly associated with all RVs as there is heterogeneity associated with the responses to RV infection 49 among more than 150 strains identified. 45 In particular, it has been noted that lower respiratory tract infection with RV occurs a few days after initial symptoms begin, which correlates well with the timing of asthma exacerbations. 45 RV-induced asthma exacerbations tend to occur in autumn and spring, mirroring increasing incidence of infection in the general population. 47
RSV and influenza make up most of the remaining viral-induced asthma exacerbations. 50 As previously noted, RSV is a particularly important pathogen in children younger than 2 years of age in winter, 51 but in the past has thought to be less common in adults with acute exacerbations of asthma. 48 However, with more widespread testing of adults in hospital settings, it is becoming increasingly recognized as a serious and common cause of lower respiratory tract infection, especially in the elderly. 52 Influenza most commonly causes asthma exacerbations during the winter months, as this is also when it is most prevalent. 53 Asthma in the setting of influenza infection is associated with worse disease severity, and higher risk of hospitalization, need for intensive care, and mortality. 53 Although it is implicated in a very small proportion of asthma exacerbations (0.7–2.5%), adenovirus has similarly been linked with more severe disease. 48
Those at Risk of Acute Exacerbations of Asthma
Asthma is a heterogeneous disease of airway inflammation and airway hyperresponsiveness (AHR). Its multidimensional nature means we should look at the disease from many angles to assess how individuals will respond to a particular insult. This is especially true in the difference in response to viral infection between patients with asthma and healthy patients. In general, compared with healthy individuals without asthma, Corne et al showed in a community cohort study that while patients with asthma were no more susceptible to getting RV infection than healthy individuals, they were much more prone to develop lower respiratory tract involvement and more severe symptoms. 54 Differences in the immune response in asthma may account for this and there may be a complex interaction between the inflammation seen in asthma and the ability respond to virus infection in the airways.
Deficient Innate Immune Response in Asthma
While not a risk factor that usually distinguishes patients with asthma from each other, asthmatics have been demonstrated to have deficient immune response to viral infection. 55 In vitro experiments with RV have shown that patients with asthma have higher levels of viral replication following infection than control subjects. Indeed, experimental studies imply there are deficiencies in IFN-mediated antiviral responses in patients with asthma that are contributory to this process, 47 56 though these findings have not always been observed, particularly in those with mild disease. 57 58 By their nature, all these studies have had limited numbers and findings in clinical asthma strongly suggest that the risk of acute asthma varies and could be restricted to a certain “at risk of acute asthma” phenotype. 59 This concept is supported by the finding that deficient type I IFN responses are found in those with severe asthma. 60 Recently, the hypothesis that correcting deficient airway antiviral responses would improve outcomes in acute asthma was tested by randomizing asthmatics to receive nebulized IFN-β after developing cold symptoms. 61 Subjects had moderate-to-severe asthma, and a history of cold induced acute asthma. While 91% of subjects developed a cold, the majority did not develop acute asthma and treatment with nebulized IFN-β overall did not result in a significant improvement. However, in those with poor control despite treatment, “cold” symptoms led to a greater worsening in asthma and nebulized IFN-β led to a significant attenuation. This may be partly explained by alterations in the immune system due to active inflammation since atopic children have been shown to be particularly at risk of more severe viral exacerbations, and this is mitigated by anti-IgE therapy. 62 Other risk factors that require further exploration include low vitamin D levels and higher levels of stress. It is suspected that these both play their part in reducing immunity against viral infection, contributing to risk of exacerbation. 27 Resultant higher levels of viral replication and insufficient IFN production and the associated airway inflammation that occurs concurrently could certainly explain this propensity for frequent exacerbations.
The importance of acute asthma is increasingly acknowledged in international guidelines that seek to minimize its risk. 63 The clinical factors that have been associated with an increased risk of acute asthma are (1) poor asthma symptom control, (2) more severe asthma (as defined by GINA and based on a lower forced expiratory volume in 1 second [FEV 1 ]), and (3) smoking. 64 Exacerbation frequency also has been linked to poor asthma control 59 and there appears to be a complex relationship between poor asthma control and susceptibility to virus infection. The majority of research into asthma exacerbations has included well-controlled individuals. Jackson et al demonstrated that uncontrolled asthmatics experience more frequent lower respiratory tract symptoms, a worse fall in FEV 1 and slower recovery compared with well-controlled individuals during exacerbation, independent of asthma severity. 65 Bateman et al have shown a clear link between asthma control and predicting future risk of exacerbation but have so far not been able to demonstrate improvement in exacerbation rate with better control. 66 Kupczyk et al have shown that in a cohort of patients with severe asthma, frequent exacerbations were correlated with markers of ongoing inflammation such as exhaled nitric oxide and higher sputum eosinophils. 59 We also have shown that asthmatics with moderate-to-severe disease and persistent eosinophilic inflammation showed reduced toll-like receptor (TLR)7 expression, along with impaired IFN expression, in endobronchial biopsies. 67 In support of the concept that poor asthma control along with persistent asthmatic inflammation is responsible for the increased susceptibility to virus infection, investigators demonstrated in a cohort of children at high risk for acute exacerbations of asthma that the addition of omalizumab (a monoclonal antibody to IgE), when added to a regimen of guidelines-based therapy further improved asthma control, nearly eliminated seasonal peaks in exacerbations previously shown to be associated with virus infection. 68 They also went on to show in a subgroup that treatment with omalizumab improved in vitro IFN responses to RV, and in those who demonstrated the greatest improvement, there was the greatest reduction in exacerbation frequency. 69
Smoking and Pollution
A history of smoking has been identified as a risk factor for frequent exacerbation. 59 Indeed, in vitro work has confirmed that cigarette smoke exposure is associated with impaired IFN production, suggesting a plausible method for higher exacerbation frequency due to impaired antiviral responses, thus predicting exacerbations. 70 Passive exposure to cigarette smoke has also been linked with risk of exacerbations in children. This extends to pollution in the environment in general, as exposure to pollutants, such as sulfur and nitrogen dioxide, is also a risk factor for viral exacerbation of asthma. 27
The Impact of Virus Infection on Asthma Control
Well-controlled asthma is defined by the current GINA guidelines as that which causes infrequent symptoms, needs for reliever therapy, and does not limit exercise tolerance, while optimizing adherence, psychosocial factors, exposures, comorbidities, and markers of inflammation. 40 A subset of patients who frequently exacerbate has been identified, and it is postulated that viral exacerbation may be one of the inciting factors in this recurrent loss of control and cycle of exacerbation. 71
In addition, there is evidence that in children who presented to hospital emergency rooms, persistence of RV was noted at 6 weeks in 44% of patients. This was associated with worse peak flow at presentation and likely contributes to severity of disease and thus, impacts on subsequent control. 72 In contrast, Chang et al have looked at the effects of documented viral infection on the course of asthma exacerbation and subsequent recovery. While viral infection did predict an increased severity of asthma exacerbations for clinically defined viral illness, it did not correlate with features of subsequent outcome. In particular, pediatric asthma quality of life, asthma, and cough diaries were not worsened by the presence of viral infection in the setting of acute asthma exacerbation after 6 weeks. 73 As a result, at present, while there are some suggestions of an impact of viral exacerbation on subsequent asthma control, this is not yet entirely defined and further research is needed in the area to be able to make any further conclusions on the effects on asthma control of viral exacerbation.
Bidirectional Interaction of Asthma and Virus Infection—The Mechanisms as to How One Predisposes to the Other
The airway epithelium acts as a physical and active immune barrier between the internal environment of the lung and the external environment that contains potentially noxious gases, agents, and pathogens including viruses. The pathogen-associated molecular patterns (PAMPs) such as viral or bacterial nucleic acids, fungal products, and bacterial endotoxins such as LPSs interact with the airway epithelium. These PAMPs are efficiently recognized by diverse range of innate germline encoded receptors in the cell surface or in the cytoplasm of the AECs and able to elicit a strong immune response. These receptors are known as pattern recognition receptors (PRRs). 74 75
Respiratory viruses such as HRVs, RSV, coronaviruses, and influenza virus are the most common infectious illnesses in the airways and usually self-limiting and confined to the respiratory tract. 44 The presence of these RNA virus infections are sensed via PRRs such as the TLRs, retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), and nucleotide-binding oligomerization domain-like receptors. Among TLRs, TLR3 expressed in the intracellular endosomes of bronchial epithelium cells and responds to the presence of double-stranded RNA, which is produced during replication of these viruses. 76 TLR3 triggers signaling via TIR-domain-containing adaptor-inducing IFN-β (TRIF) and then associates with tumor necrosis factor (TNF) receptor-associated factor 3 (TRAF3) and TRAF6. This leads to the activation of IFN regulatory factor 3, which translocates into the nucleus resulting in production of type I IFNs. TRIF also interact with receptor interacting protein I which activates NF-κB and production of inflammatory cytokines. 77 TLR 7/8 are also found within endosomes, activated by single-stranded RNA and leads NF-κB activation via MyD88-dependent pathway. 78 TLR4 is mainly responsible for the detection of bacterial endotoxins; however, it has been reported that some viral proteins (F protein of RSV) are detected by TLR4 and induce type I IFNs and proinflammatory cytokines via activating IRF-3, NF-κB, and activating protein I. 78
RLR signaling plays a major role in detecting several picornaviruses, influenza virus, and RSV ( Fig. 1 ). RIG I recognizes 5′-triphosphate (5′PPP) RNA and dsRNA during virus infection and melanoma differentiation-associated protein 5 detects long dsRNA of > 2 kb. 79 The components of viral RNA are recognized by these RLR receptors and initiate type I IFN production. 80 81
Fig. 1.
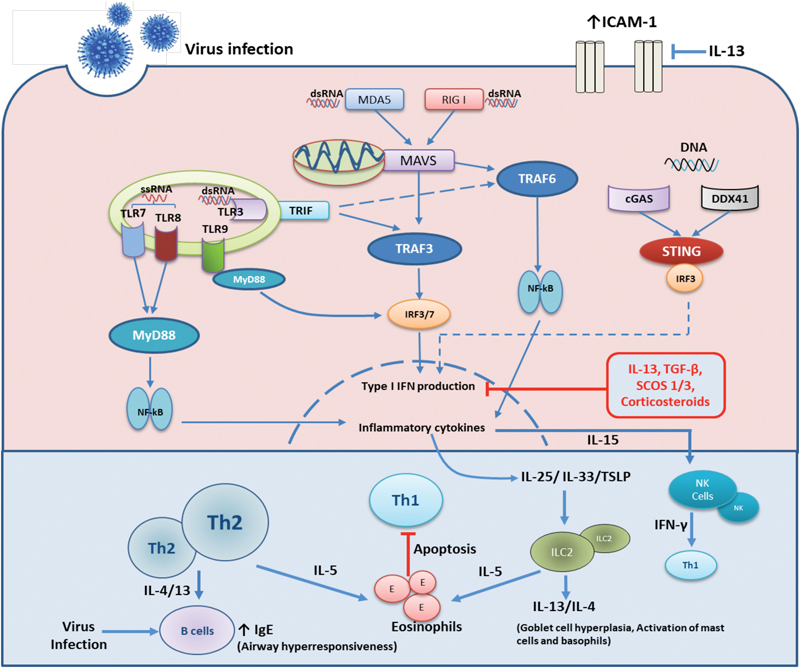
Innate and adaptive immune responses to respiratory virus infections in the airways. Under normal circumstances the innate immune response to virus infection is triggered by infection of airway epithelial cells and recognition of viral pathogen associated molecular patterns (PAMPs) by both the infected epithelium and resident immune cells. These cells contain various pathogen recognition receptors (PRRs) that recognise a diverse range of PAMPs and elicit innate then adaptive immune responses to eliminate virus infections. Viral dsRNA are recognised by RIG I, MDA5 or TLR3 receptors whiles sRNA are mainly sensed by TLR7/8. Various DNA viruses are recognised by cytoplasmic DNA sensors; cGAS, DDX41 or by endosomal TLR9. Upon recognition downstream signaling cascades are initiated to produce type I interferons via IRF3/7 activation that induces antiviral proteins that inhibit viral replication within infected cells and spread to neighboring cells. Inflammatory cytokines production via NF-kB activation, results in release of IL-6, CXCL8 and the recruitment and activation of neutrophils and macrophages. Infected cells release IL-15 that activates NK cells that produce IFN-β and that target infected cells. This supports a robust Th1 environment for recruitment of TH-1 and type 1 innate lymphoid cells (ILC-1), resulting in viral clearance. In asthma there is a pre-existent state of active airway inflammation. In the case of active type 2 inflammation, the presence of increased type 2 lymphocytes, both TH-2 cells and Ilc-2 cells promote this abnormal state. Following infection of the asthmatic epithelium there is heightened release of IL-25/IL-33 and TSLP that further activate ILC2 cells. Activated ILC2s and Th2 cells largely produce more IL-4/13 and IL-5 all of which activate other inflammatory cells such as eosinophils and this results in worsened inflammation of the airways. IL-4/13 also enhance the IgE production by B lymphocytes which may further impair activation of the innate immune cells such as dendritic cells to virus infection. Increased expression of type 2 cytokines, TGF-β and SOCS1/3 negatively regulates type I interferon production while type 2 cytokines also enhance ICAM-1 expression all of which results increased virus replication.
Type I and type III IFNs have a crucial role in controlling virus infection in the airway epithelium. Type I IFNs and IFN-α/β act on the type I IFN receptor and via JAK-STAT1/2 pathway activates a series of IFN-stimulatory genes which produce potent antiviral proteins and eliminate virus infections. Type III IFNs (λ IFNs) such as IFN λ1, 2, and 3 (also known as IL-29, IL-28A, and IL-28B) also elicit an antiviral response; however, their cellular receptor distribution on different tissues is limited. 82 83
Besides IFNs, there are several cytokines and chemokines produced by airway epithelium during virus infections. These cytokines/chemokines possess different roles in innate and adaptive immunity. Cytokines such as IL-6, TNF-α, granulocyte colony-stimulating factor (G-CSF), and granulocyte macrophage-CSF (GM-CSF) are secreted upon various respiratory virus infections. IL-6 is a proinflammatory cytokine that modulate lung neutrophils, monocytes, and T cells activity. 84 Increased secretion of IL-6 can result in excessive inflammation and damage. In contrast, the absence of IL-6 in the setting of influenza infection results in increased viral replication and reduced antiviral T cell activity. 85 Therefore, proper balance of IL-6 in the lung is necessary for the balance of innate and adaptive immunity. G-CSF modulates production and recruitment of neutrophils. GM-CSF involves in expansion and activation of pulmonary DCs and macrophages. Also, GM-CSF has shown a protective role against influenza virus infection. 86 TNF-α, while a potent proinflammatory cytokine, also has demonstrated an antiviral activity against influenza and RSV infections. 87 88
Chemokines such as CXCL-8/IL-8, CXCL-10, and RANTES/CCL-5 are also induced by respiratory virus infections. CXCL-8/IL-8 mainly recruits neutrophils into lungs and modulates their activity. In addition, it has various functions on other types of immune cells as well. 89 IL-8 is induced by various virus infections such as RSV, influenza A, and RV in airway epithelium. 90 91 92 CXCL-10/inducible protein-10 is produced by several cells such as monocytes, neutrophils, eosinophils, epithelial, endothelial, and fibroblast with response to IFN-γ. It can be induced by RV, RSV, influenza virus, etc., and have shown a protective role against pathogen infections. 93 94 95 96 CCL-5/RANTES is a chemokine induced by viral infections. 97 It recruits monocytes, T cells, and eosinophils into the lungs 98 and induces viral clearance.
As already discussed, deficient innate IFN responses in asthmatic epithelial cell release of IFN-β/λ have been described 47 56 though not in all cases. 58 99 100 Some studies suggest this impairment correlates with the disease severity and more prominent in severe, less controlled asthma. 61 However, the molecular mechanisms of impaired innate immune responses in asthma still remain obscure. It has been proposed that the Th2/type 2 airway inflammatory environment present in the asthmatic airway may have a role in impaired innate antiviral immunity. Treatment with recombinant IL-13 in epithelial cells has been shown to enhance RV replication and suppress types I and III IFN production. 101 102 In addition, IL-13 has been shown to increase Intracellular adhesion molecule (ICAM)-1 expression on epithelial cells resulting increased RV infection. 103 Transforming growth factor-β (TGF-β) is a potent mediator of airway remodeling in asthma and is highly expressed in the asthmatic epithelium. 104 Expression of TGF-β can suppress both IFN-β/λ in airways 105 and its overexpression enhances RV infection. 106 Suppressor of cytokine signaling (SOCS) proteins are negative regulators of cytokine signaling. Intriguingly, SOCS1 and SOCS3 appear to be increased in asthma. 107 108 Moreover, RV infection and Th2 cytokines further increased the SOCS1 and SOCS3 in asthmatic individuals resulting dampened IFN production. 109 Continuous corticosteroid treatments have also demonstrated a negative effect on innate antiviral effect. Peripheral blood mononuclear cells pretreated with budesonide promoted RV infection by reducing type I IFN production. 110 In addition, influenza A and RV demonstrated enhanced viral replication in AECs treated with glucocorticosteroids, while exogenous IFN adjuvant markedly reduced the glucocorticosteroid amplified virus infections. 111
Respiratory Virus Infection and Activation of Innate Lymphoid Cells and Natural Killer Cells in Asthma
Following viral infection, AECs release numerous immune activating mediators ( Fig. 1 ). It is reasonable to suggest that the composition of the epithelial immune response will have a profound impact on the downstream immune pathways that regulate inflammation and ultimately disease severity. Relevant to infections in asthma are reports showing that epithelium expresses mediators that can drive both innate type 2 and antiviral immunity. For example, IL-25 and IL-33 activate group 2 innate lymphoid cells (ILC2), 112 113 while release of IFN-α/β and IL-15 activates natural killer (NK) cells. 114
ILC2 is recently discovered innate immune cells of lymphoid origin that lack lineage-specific markers including T and B cell receptors. 115 116 They are an important source of type 2 cytokines. ILC2 expresses an array of cytokine receptors and upon activation with airway epithelial derived cytokines such as IL-33, IL-25, and thymic stromal lymphopoietin, ILC2s produce large amounts of type 2 cytokines such as IL-4, IL-5, IL-9, and IL-13. 117 Type 2 cytokines play an important role in the activation of mast cells, eosinophils, and basophils; induce goblet cell hyperplasia and mucus production; and thus directly stimulate the characteristic pathological responses in asthma. 118
RV infection of the AECs stimulates the release of IL-25 and IL-33 both of which activate NF-kB and STAT5 pathways that drive the expression of IL-5 and IL-13 from ILC2. 119 Influenza virus can also induce AHR by activating ILC2 via IL-33 secretion from alveolar macrophages 120 and NKT cells both of which stimulate ILC2-mediated production of IL-5 and IL-13 and accumulation of eosinophils in the lungs. 121
NK cells are another class of innate immune cell that arises from a common lymphoid progenitor. These play an important role in defense against infectious agents including viruses. 122 Following viral infection, NK cells produce large amounts of IFN-γ that supports a Th1/type 1 immune environment in the lung that promotes viral clearance, and plays a critical role in the subsequent development of an effector CD4 Th1 response. This may occur indirectly through NK cell priming of DCs; with bidirectional cross-talk, IFN-γ released by NK cells activates DCs to produce IL-12, which in turn feeds back on the NK cell to further amplify IFN-γ secretion. 123 However, when NK cell responses are impaired to viral infection, reduced levels of IFN-γ may promote type 2 immune environment in the lungs with increased inflammation, viral replication, and clinical features of asthma. 124 125 Interestingly, impaired NK cell responses appear to be a strong feature of acute severe RSV bronchiolitis, 126 and as we have previously discussed, this is an independent predictor for recurring wheeze up to the age of 6 years.
During RV infection, IL-15 is expressed by macrophages, DCs, and epithelial cells. 127 IL-15 mediates the activation, expansion, and recruitment of NK cells to the lungs to eliminate the virally infected cells by producing granzyme and IFN-γ. 128 Impairment in IL-15 signaling can affect the NK cell protective feature against the virus, hence promoting asthma. Influenza virus can worsen airway inflammation in a murine asthma model of H1N1 strain. NK cells similarly play an important role in the response to influenza, with increased NK cells infiltrating within the first few days of infection with influenza virus. 129 NKp46 is an important NK cell receptor that binds directly with influenza virus HA protein resulting in cell activation and virus clearance. 130
Conclusion
Respiratory virus infections, especially RSV and RV, are a major cause of early childhood wheezing illnesses. As acute events, these infections are enough to trigger recurring episodes of wheeze (particularly with recurrent RV infections), are likely to influence permanently lung development, and in those susceptible, predispose to the development of asthma in later life. In established asthma, viruses remain important triggers of acute exacerbations, with those most susceptible tending to have either pre-existent poor asthma control or more severe underlying asthma. The reason for this susceptibility to respiratory virus infection remains unclear, but is very clearly closely related to the imbalanced inflammatory process active in asthma that can involve impaired type I/III IFN production coupled with excessive expression of cytokines that activate neutrophilic and type-2/eosinophilic inflammatory pathways ( Fig. 1 ). Originally thought to be primarily involved in control of viral replication, there is growing evidence that type I IFN is also an important negative regulator of inflammation. The role of the airway epithelium as the site of viral replication and initiator of the inflammatory pathways that cause exacerbations is now well established. Given the diversity of viruses that can precipitate an asthma exacerbation, antivirals and vaccines are unlikely to be feasible treatment options. As a result, research is now focused on redressing the asthmatic airway epithelial innate immune response to viral infection.
References
- 1.Hall C B, Weinberg G A, Iwane M K et al. The burden of respiratory syncytial virus infection in young children. N Engl J Med. 2009;360(06):588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meissner H C. Viral bronchiolitis in children. N Engl J Med. 2016;374(18):1793–1794. doi: 10.1056/NEJMc1601509. [DOI] [PubMed] [Google Scholar]
- 3.Carroll K N, Wu P, Gebretsadik T et al. Season of infant bronchiolitis and estimates of subsequent risk and burden of early childhood asthma. J Allergy Clin Immunol. 2009;123(04):964–966. doi: 10.1016/j.jaci.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu S, Hartert T V, Everard M L et al. Predictors of asthma following severe respiratory syncytial virus (RSV) bronchiolitis in early childhood. Pediatr Pulmonol. 2016;51(12):1382–1392. doi: 10.1002/ppul.23461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stensballe L G, Simonsen J B, Thomsen S F et al. The causal direction in the association between respiratory syncytial virus hospitalization and asthma. J Allergy Clin Immunol. 2009;123(01):131–1370. doi: 10.1016/j.jaci.2008.10.042. [DOI] [PubMed] [Google Scholar]
- 6.Lemanske R F, Jr, Jackson D J, Gangnon R E et al. Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J Allergy Clin Immunol. 2005;116(03):571–577. doi: 10.1016/j.jaci.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 7.Conickx G, Mestdagh P, Avila Cobos F et al. MicroRNA profiling reveals a role for microRNA-218-5p in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;195(01):43–56. doi: 10.1164/rccm.201506-1182OC. [DOI] [PubMed] [Google Scholar]
- 8.Turunen R, Koistinen A, Vuorinen T et al. The first wheezing episode: respiratory virus etiology, atopic characteristics, and illness severity. Pediatr Allergy Immunol. 2014;25(08):796–803. doi: 10.1111/pai.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmenberg A C, Gern J E. Classification and evolution of human rhinoviruses. Methods Mol Biol. 2015;1221:1–10. doi: 10.1007/978-1-4939-1571-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee W M, Lemanske R F, Jr, Evans M D et al. Human rhinovirus species and season of infection determine illness severity. Am J Respir Crit Care Med. 2012;186(09):886–891. doi: 10.1164/rccm.201202-0330OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller E K, Williams J V, Gebretsadik T et al. Host and viral factors associated with severity of human rhinovirus-associated infant respiratory tract illness. J Allergy Clin Immunol. 2011;127(04):883–891. doi: 10.1016/j.jaci.2010.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bizzintino J, Lee W M, Laing I A et al. Association between human rhinovirus C and severity of acute asthma in children. Eur Respir J. 2011;37(05):1037–1042. doi: 10.1183/09031936.00092410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arden K E, McErlean P, Nissen M D, Sloots T P, Mackay I M. Frequent detection of human rhinoviruses, paramyxoviruses, coronaviruses, and bocavirus during acute respiratory tract infections. J Med Virol. 2006;78(09):1232–1240. doi: 10.1002/jmv.20689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bochkov Y A, Watters K, Ashraf S et al. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc Natl Acad Sci U S A. 2015;112(17):5485–5490. doi: 10.1073/pnas.1421178112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Husby A, Pasanen A, Waage J et al. CDHR3 gene variation and childhood bronchiolitis . J Allergy Clin Immunol. 2017;140(05):1469–1.471E10. doi: 10.1016/j.jaci.2017.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bønnelykke K, Sleiman P, Nielsen K et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. 2014;46(01):51–55. doi: 10.1038/ng.2830. [DOI] [PubMed] [Google Scholar]
- 17.Simpson A, Tan V Y, Winn J et al. Beyond atopy: multiple patterns of sensitization in relation to asthma in a birth cohort study. Am J Respir Crit Care Med. 2010;181(11):1200–1206. doi: 10.1164/rccm.200907-1101OC. [DOI] [PubMed] [Google Scholar]
- 18.Jackson D J, Gangnon R E, Evans M D et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. 2008;178(07):667–672. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson D J, Evans M D, Gangnon R E et al. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am J Respir Crit Care Med. 2012;185(03):281–285. doi: 10.1164/rccm.201104-0660OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu L, Pan Y, Zhu Y et al. Association between rhinovirus wheezing illness and the development of childhood asthma: a meta-analysis. BMJ Open. 2017;7(04):e013034. doi: 10.1136/bmjopen-2016-013034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gern J E, Busse W W. Relationship of viral infections to wheezing illnesses and asthma. Nat Rev Immunol. 2002;2(02):132–138. doi: 10.1038/nri725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Top F H, Jr, Connor E M, Carlin D A.Prophylaxis against respiratory syncytial virus in premature infants. IMpact-RSV Study Group Lancet 2000355(9208):1014. [DOI] [PubMed] [Google Scholar]
- 23.Blanken M O, Rovers M M, Molenaar J M et al. Respiratory syncytial virus and recurrent wheeze in healthy preterm infants. N Engl J Med. 2013;368(19):1791–1799. doi: 10.1056/NEJMoa1211917. [DOI] [PubMed] [Google Scholar]
- 24.Mochizuki H, Kusuda S, Okada K, Yoshihara S, Furuya H, Simões E AF; Scientific Committee for Elucidation of Infantile Asthma.Palivizumab prophylaxis in preterm infants and subsequent recurrent wheezing. Six-year follow-up study Am J Respir Crit Care Med 20171960129–38. [DOI] [PubMed] [Google Scholar]
- 25.Huang Y J, Charlson E S, Collman R G, Colombini-Hatch S, Martinez F D, Senior R M. The role of the lung microbiome in health and disease. A National Heart, Lung, and Blood Institute workshop report. Am J Respir Crit Care Med. 2013;187(12):1382–1387. doi: 10.1164/rccm.201303-0488WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bisgaard H, Hermansen M N, Bønnelykke K et al. Association of bacteria and viruses with wheezy episodes in young children: prospective birth cohort study. BMJ. 2010;341:c4978. doi: 10.1136/bmj.c4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teo S M, Mok D, Pham K et al. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe. 2015;17(05):704–715. doi: 10.1016/j.chom.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabesch M, Lauener R P. Why Old McDonald had a farm but no allergies: genes, environments, and the hygiene hypothesis. J Leukoc Biol. 2004;75(03):383–387. doi: 10.1189/jlb.1003468. [DOI] [PubMed] [Google Scholar]
- 29.Strachan D P.Hay fever, hygiene, and household size BMJ 1989299(6710):1259–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.West M A, Heagy W. Endotoxin tolerance: a review. Crit Care Med. 2002;30:S64–S73. [PubMed] [Google Scholar]
- 31.Braun-Fahrländer C, Riedler J, Herz U et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N Engl J Med. 2002;347(12):869–877. doi: 10.1056/NEJMoa020057. [DOI] [PubMed] [Google Scholar]
- 32.Liu A H. Revisiting the hygiene hypothesis for allergy and asthma. J Allergy Clin Immunol. 2015;136(04):860–865. doi: 10.1016/j.jaci.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 33.Schuijs M J, Willart M A, Vergote Ket al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells Science 2015349(6252):1106–1110. [DOI] [PubMed] [Google Scholar]
- 34.Shembade N, Parvatiyar K, Harhaj N S, Harhaj E W. The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-kappaB signalling. EMBO J. 2009;28(05):513–522. doi: 10.1038/emboj.2008.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parvatiyar K, Barber G N, Harhaj E W. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem. 2010;285(20):14999–15009. doi: 10.1074/jbc.M110.109819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang N I, Yoon H Y, Lee Y R et al. A20 attenuates allergic airway inflammation in mice. J Immunol. 2009;183(02):1488–1495. doi: 10.4049/jimmunol.0900163. [DOI] [PubMed] [Google Scholar]
- 37.Kelly C, Shields M D, Elborn J S, Schock B C. A20 regulation of nuclear factor-κB: perspectives for inflammatory lung disease. Am J Respir Cell Mol Biol. 2011;44(06):743–748. doi: 10.1165/rcmb.2010-0339TR. [DOI] [PubMed] [Google Scholar]
- 38.Lu Y, Li Y, Xu L, Xia M, Cao L.Bacterial lysate increases the percentage of natural killer T cells in peripheral blood and alleviates asthma in children Pharmacology 201595(3-4):139–144. [DOI] [PubMed] [Google Scholar]
- 39.Razi C H, Harmancı K, Abacı A et al. The immunostimulant OM-85 BV prevents wheezing attacks in preschool children. J Allergy Clin Immunol. 2010;126(04):763–769. doi: 10.1016/j.jaci.2010.07.038. [DOI] [PubMed] [Google Scholar]
- 40.Global Initiative for Asthma.Global Strategy for Asthma Management and Prevention2017. Available at:www.ginasthma.org. June 2017
- 41.Dougherty R H, Fahy J V. Acute exacerbations of asthma: epidemiology, biology and the exacerbation-prone phenotype. Clin Exp Allergy. 2009;39(02):193–202. doi: 10.1111/j.1365-2222.2008.03157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnston S L, Pattemore P K, Sanderson Get al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children BMJ 1995310(6989):1225–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gibson P G, Grootendor D C, Henry R L et al. Sputum induction in children. Eur Respir J Suppl. 2002;37:44s–46s. doi: 10.1183/09031936.02.00004402. [DOI] [PubMed] [Google Scholar]
- 44.See H, Wark P. Innate immune response to viral infection of the lungs. Paediatr Respir Rev. 2008;9(04):243–250. doi: 10.1016/j.prrv.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jamieson K C, Warner S M, Leigh R, Proud D. Rhinovirus in the pathogenesis and clinical course of asthma. Chest. 2015;148(06):1508–1516. doi: 10.1378/chest.15-1335. [DOI] [PubMed] [Google Scholar]
- 46.Matsumoto K, Inoue H. Viral infections in asthma and COPD. Respir Investig. 2014;52(02):92–100. doi: 10.1016/j.resinv.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 47.Wark P A, Johnston S L, Bucchieri F et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201(06):937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sandrock C E, Norris A. Infection in severe asthma exacerbations and critical asthma syndrome. Clin Rev Allergy Immunol. 2015;48(01):104–113. doi: 10.1007/s12016-014-8435-x. [DOI] [PubMed] [Google Scholar]
- 49.Wark P A, Grissell T, Davies B, See H, Gibson P G. Diversity in the bronchial epithelial cell response to infection with different rhinovirus strains. Respirology. 2009;14(02):180–186. doi: 10.1111/j.1440-1843.2009.01480.x. [DOI] [PubMed] [Google Scholar]
- 50.Rowe R K, Gill M A. Asthma: the interplay between viral infections and allergic diseases. Immunol Allergy Clin North Am. 2015;35(01):115–127. doi: 10.1016/j.iac.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 51.Gavala M L, Bashir H, Gern J E. Virus/allergen interactions in asthma. Curr Allergy Asthma Rep. 2013;13(03):298–307. doi: 10.1007/s11882-013-0344-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pastula S T, Hackett J, Coalson J et al. Hospitalizations for respiratory syncytial virus among adults in the United States, 1997-2012. Open Forum Infect Dis. 2017;4(01):ofw270. doi: 10.1093/ofid/ofw270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ritchie A I, Farne H A, Singanayagam A, Jackson D J, Mallia P, Johnston S L. Pathogenesis of viral infection in exacerbations of airway disease. Ann Am Thorac Soc. 2015;12 02:S115–S132. doi: 10.1513/AnnalsATS.201503-151AW. [DOI] [PubMed] [Google Scholar]
- 54.Corne J M, Marshall C, Smith Set al. Frequency, severity, and duration of rhinovirus infections in asthmatic and non-asthmatic individuals: a longitudinal cohort study Lancet 2002359(9309):831–834. [DOI] [PubMed] [Google Scholar]
- 55.Hewitt R, Farne H, Ritchie A, Luke E, Johnston S L, Mallia P. The role of viral infections in exacerbations of chronic obstructive pulmonary disease and asthma. Ther Adv Respir Dis. 2016;10(02):158–174. doi: 10.1177/1753465815618113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Contoli M, Message S D, Laza-Stanca V et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12(09):1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 57.Lopez-Souza N, Favoreto S, Wong H et al. In vitro susceptibility to rhinovirus infection is greater for bronchial than for nasal airway epithelial cells in human subjects. J Allergy Clin Immunol. 2009;123(06):1384–9000. doi: 10.1016/j.jaci.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bochkov Y A, Hanson K M, Keles S, Brockman-Schneider R A, Jarjour N N, Gern J E. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010;3(01):69–80. doi: 10.1038/mi.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kupczyk M, ten Brinke A, Sterk P J et al. Frequent exacerbators–a distinct phenotype of severe asthma. Clin Exp Allergy. 2014;44(02):212–221. doi: 10.1111/cea.12179. [DOI] [PubMed] [Google Scholar]
- 60.Edwards M R, Regamey N, Vareille M et al. Impaired innate interferon induction in severe therapy resistant atopic asthmatic children. Mucosal Immunol. 2013;6(04):797–806. doi: 10.1038/mi.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Djukanović R, Harrison T, Johnston S L et al. The effect of inhaled IFN-β on worsening of asthma symptoms caused by viral infections. A randomized trial. Am J Respir Crit Care Med. 2014;190(02):145–154. doi: 10.1164/rccm.201312-2235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coleman L, Laing I A, Bosco A. Rhinovirus-induced asthma exacerbations and risk populations. Curr Opin Allergy Clin Immunol. 2016;16(02):179–185. doi: 10.1097/ACI.0000000000000245. [DOI] [PubMed] [Google Scholar]
- 63.Global Initiative for Asthma.Global Strategy for AsthmaManagement and Prevention. 2014Available at:www.ginasthma.org
- 64.Bateman E D, Buhl R, O'Byrne P M et al. Development and validation of a novel risk score for asthma exacerbations: the risk score for exacerbations. J Allergy Clin Immunol. 2015;135(06):1457–640000. doi: 10.1016/j.jaci.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 65.Jackson D J, Trujillo-Torralbo M B, del-Rosario J et al. The influence of asthma control on the severity of virus-induced asthma exacerbations. J Allergy Clin Immunol. 2015;136(02):497–500000. doi: 10.1016/j.jaci.2015.01.028. [DOI] [PubMed] [Google Scholar]
- 66.Bateman E D, Reddel H K, Eriksson Get al. Overall asthma control: the relationship between current control and future risk J Allergy Clin Immunol 201012503600–608., 608.e1–608.e6 [DOI] [PubMed] [Google Scholar]
- 67.Hatchwell L, Collison A, Girkin J et al. Toll-like receptor 7 governs interferon and inflammatory responses to rhinovirus and is suppressed by IL-5-induced lung eosinophilia. Thorax. 2015;70(09):854–861. doi: 10.1136/thoraxjnl-2014-205465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Busse W W, Morgan W J, Gergen P J et al. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N Engl J Med. 2011;364(11):1005–1015. doi: 10.1056/NEJMoa1009705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Teach S J, Gill M A, Togias A et al. Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. J Allergy Clin Immunol. 2015;136(06):1476–1485. doi: 10.1016/j.jaci.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sonnenfeld G, Hudgens R W. Effect of sidestream and mainstream smoke exposure on in vitro interferon-alpha/beta production by L-929 cells. Cancer Res. 1986;46(06):2779–2783. [PubMed] [Google Scholar]
- 71.Greenberg S. Asthma exacerbations: predisposing factors and prediction rules. Curr Opin Allergy Clin Immunol. 2013;13(03):225–236. doi: 10.1097/ACI.0b013e32836096de. [DOI] [PubMed] [Google Scholar]
- 72.Kling S, Donninger H, Williams Z et al. Persistence of rhinovirus RNA after asthma exacerbation in children. Clin Exp Allergy. 2005;35(05):672–678. doi: 10.1111/j.1365-2222.2005.02244.x. [DOI] [PubMed] [Google Scholar]
- 73.Chang A B, Clark R, Acworth J P, Petsky H L, Sloots T P. The impact of viral respiratory infection on the severity and recovery from an asthma exacerbation. Pediatr Infect Dis J. 2009;28(04):290–294. doi: 10.1097/INF.0b013e31819067b1. [DOI] [PubMed] [Google Scholar]
- 74.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30(01):16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 75.Kumagai Y, Takeuchi O, Akira S. Pathogen recognition by innate receptors. J Infect Chemother. 2008;14(02):86–92. doi: 10.1007/s10156-008-0596-1. [DOI] [PubMed] [Google Scholar]
- 76.Bowie A G, Haga I R. The role of toll-like receptors in the host response to viruses. Mol Immunol. 2005;42(08):859–867. doi: 10.1016/j.molimm.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 77.Häcker H, Redecke V, Blagoev Bet al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6 Nature 2006439(7073):204–207. [DOI] [PubMed] [Google Scholar]
- 78.Thompson A J, Locarnini S A. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol Cell Biol. 2007;85(06):435–445. doi: 10.1038/sj.icb.7100100. [DOI] [PubMed] [Google Scholar]
- 79.Kato H, Takeuchi O, Mikamo-Satoh E et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205(07):1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Seth R B, Sun L, Ea C K, Chen Z J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(05):669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 81.Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20(01):17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 82.Meager A, Visvalingam K, Dilger P, Bryan D, Wadhwa M. Biological activity of interleukins-28 and -29: comparison with type I interferons. Cytokine. 2005;31(02):109–118. doi: 10.1016/j.cyto.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 83.Zhou Z, Hamming O J, Ank N, Paludan S R, Nielsen A L, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the JAK-STAT pathway and the mitogen-activated protein kinases. J Virol. 2007;81(14):7749–7758. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jones S A. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175(06):3463–3468. doi: 10.4049/jimmunol.175.6.3463. [DOI] [PubMed] [Google Scholar]
- 85.Lauder S N, Jones E, Smart K et al. Interleukin-6 limits influenza-induced inflammation and protects against fatal lung pathology. Eur J Immunol. 2013;43(10):2613–2625. doi: 10.1002/eji.201243018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang F F, Barnes P F, Feng Y et al. GM-CSF in the lung protects against lethal influenza infection. Am J Respir Crit Care Med. 2011;184(02):259–268. doi: 10.1164/rccm.201012-2036OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neuzil K M, Tang Y W, Graham B S. Protective role of TNF-alpha in respiratory syncytial virus infection in vitro and in vivo. Am J Med Sci. 1996;311(05):201–204. doi: 10.1097/00000441-199605000-00001. [DOI] [PubMed] [Google Scholar]
- 88.Seo S H, Webster R G. Tumor necrosis factor alpha exerts powerful anti-influenza virus effects in lung epithelial cells. J Virol. 2002;76(03):1071–1076. doi: 10.1128/JVI.76.3.1071-1076.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mukaida N. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol. 2003;284(04):L566–L577. doi: 10.1152/ajplung.00233.2002. [DOI] [PubMed] [Google Scholar]
- 90.Garofalo R, Sabry M, Jamaluddin M et al. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J Virol. 1996;70(12):8773–8781. doi: 10.1128/jvi.70.12.8773-8781.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Choi A M, Jacoby D B. Influenza virus A infection induces interleukin-8 gene expression in human airway epithelial cells. FEBS Lett. 1992;309(03):327–329. doi: 10.1016/0014-5793(92)80799-m. [DOI] [PubMed] [Google Scholar]
- 92.Chun Y H, Park J Y, Lee H et al. Rhinovirus-infected epithelial cells produce more IL-8 and RANTES compared with other respiratory viruses. Allergy Asthma Immunol Res. 2013;5(04):216–223. doi: 10.4168/aair.2013.5.4.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lindell D M, Lane T E, Lukacs N W. CXCL10/CXCR3-mediated responses promote immunity to respiratory syncytial virus infection by augmenting dendritic cell and CD8(+) T cell efficacy. Eur J Immunol. 2008;38(08):2168–2179. doi: 10.1002/eji.200838155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zaheer R S, Proud D. Human rhinovirus-induced epithelial production of CXCL10 is dependent upon IFN regulatory factor-1. Am J Respir Cell Mol Biol. 2010;43(04):413–421. doi: 10.1165/rcmb.2009-0203OC. [DOI] [PubMed] [Google Scholar]
- 95.Spurrell J C, Wiehler S, Zaheer R S, Sanders S P, Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2005;289(01):L85–L95. doi: 10.1152/ajplung.00397.2004. [DOI] [PubMed] [Google Scholar]
- 96.Lam W Y, Yeung A C, Chu I M, Chan P K. Profiles of cytokine and chemokine gene expression in human pulmonary epithelial cells induced by human and avian influenza viruses. Virol J. 2010;7:344. doi: 10.1186/1743-422X-7-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Culley F J, Pennycook A M, Tregoning J S et al. Role of CCL5 (RANTES) in viral lung disease. J Virol. 2006;80(16):8151–8157. doi: 10.1128/JVI.00496-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schall T J, Bacon K, Toy K J, Goeddel D V.Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES Nature 1990347(6294):669–671. [DOI] [PubMed] [Google Scholar]
- 99.Sykes A, Macintyre J, Edwards M R et al. Rhinovirus-induced interferon production is not deficient in well controlled asthma. Thorax. 2014;69(03):240–246. doi: 10.1136/thoraxjnl-2012-202909. [DOI] [PubMed] [Google Scholar]
- 100.Sykes A, Edwards M R, Macintyre J et al. TLR3, TLR4 and TLRs7-9 induced interferons are not impaired in airway and blood cells in well controlled asthma. PLoS One. 2013;8(06):e65921. doi: 10.1371/journal.pone.0065921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moriwaki A, Matsumoto K, Matsunaga Y et al. IL-13 suppresses double-stranded RNA-induced IFN-λ production in lung cells. Biochem Biophys Res Commun. 2011;404(04):922–927. doi: 10.1016/j.bbrc.2010.12.082. [DOI] [PubMed] [Google Scholar]
- 102.Contoli M, Ito K, Padovani A et al. Th2 cytokines impair innate immune responses to rhinovirus in respiratory epithelial cells. Allergy. 2015;70(08):910–920. doi: 10.1111/all.12627. [DOI] [PubMed] [Google Scholar]
- 103.Bianco A, Sethi S K, Allen J T, Knight R A, Spiteri M A. Th2 cytokines exert a dominant influence on epithelial cell expression of the major group human rhinovirus receptor, ICAM-1. Eur Respir J. 1998;12(03):619–626. doi: 10.1183/09031936.98.12030619. [DOI] [PubMed] [Google Scholar]
- 104.Redington A E, Madden J, Frew A Jet al. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid Am J Respir Crit Care Med 1997156(2 Pt 1):642–647. [DOI] [PubMed] [Google Scholar]
- 105.Bedke N, Sammut D, Green B et al. Transforming growth factor-beta promotes rhinovirus replication in bronchial epithelial cells by suppressing the innate immune response. PLoS One. 2012;7(09):e44580. doi: 10.1371/journal.pone.0044580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Thomas B J, Lindsay M, Dagher H et al. Transforming growth factor-beta enhances rhinovirus infection by diminishing early innate responses. Am J Respir Cell Mol Biol. 2009;41(03):339–347. doi: 10.1165/rcmb.2008-0316OC. [DOI] [PubMed] [Google Scholar]
- 107.Harada M, Nakashima K, Hirota T et al. Functional polymorphism in the suppressor of cytokine signaling 1 gene associated with adult asthma. Am J Respir Cell Mol Biol. 2007;36(04):491–496. doi: 10.1165/rcmb.2006-0090OC. [DOI] [PubMed] [Google Scholar]
- 108.Seki Y, Inoue H, Nagata N et al. SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat Med. 2003;9(08):1047–1054. doi: 10.1038/nm896. [DOI] [PubMed] [Google Scholar]
- 109.Gielen V, Sykes A, Zhu J et al. Increased nuclear suppressor of cytokine signaling 1 in asthmatic bronchial epithelium suppresses rhinovirus induction of innate interferons. J Allergy Clin Immunol. 2015;136(01):177–1.88E13. doi: 10.1016/j.jaci.2014.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Davies J M, Carroll M L, Li H et al. Budesonide and formoterol reduce early innate anti-viral immune responses in vitro. PLoS One. 2011;6(11):e27898. doi: 10.1371/journal.pone.0027898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thomas B J, Porritt R A, Hertzog P J, Bardin P G, Tate M D. Glucocorticosteroids enhance replication of respiratory viruses: effect of adjuvant interferon. Sci Rep. 2014;4:7176. doi: 10.1038/srep07176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jackson D J, Makrinioti H, Rana B M et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. 2014;190(12):1373–1382. doi: 10.1164/rccm.201406-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Beale J, Jayaraman A, Jackson D J et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci Transl Med. 2014;6(256):256ra134. doi: 10.1126/scitranslmed.3009124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Colucci F, Caligiuri M A, Di Santo J P. What does it take to make a natural killer? Nat Rev Immunol. 2003;3(05):413–425. doi: 10.1038/nri1088. [DOI] [PubMed] [Google Scholar]
- 115.Artis D, Spits H.The biology of innate lymphoid cells Nature 2015517(7534):293–301. [DOI] [PubMed] [Google Scholar]
- 116.Eberl G, Colonna M, Di Santo J P, McKenzie A N.Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology Science 2015348(6237):aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Camelo A, Rosignoli G, Ohne Y et al. IL-33, IL-25, and TSLP induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Advances. 2017;1:577–589. doi: 10.1182/bloodadvances.2016002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wilhelm C, Stockinger B. Innate lymphoid cells and type 2 (th2) mediated immune responses - pathogenic or beneficial? Front Immunol. 2011;2:68. doi: 10.3389/fimmu.2011.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Guo L, Junttila I S, Paul W E. Cytokine-induced cytokine production by conventional and innate lymphoid cells. Trends Immunol. 2012;33(12):598–606. doi: 10.1016/j.it.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chang Y J, Kim H Y, Albacker L A et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(07):631–638. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gorski S A, Hahn Y S, Braciale T J. Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog. 2013;9(09):e1003615. doi: 10.1371/journal.ppat.1003615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Biron C A. Activation and function of natural killer cell responses during viral infections. Curr Opin Immunol. 1997;9(01):24–34. doi: 10.1016/s0952-7915(97)80155-0. [DOI] [PubMed] [Google Scholar]
- 123.Mailliard R B, Son Y I, Redlinger R et al. Dendritic cells mediate NK cell help for Th1 and CTL responses: two-signal requirement for the induction of NK cell helper function. J Immunol. 2003;171(05):2366–2373. doi: 10.4049/jimmunol.171.5.2366. [DOI] [PubMed] [Google Scholar]
- 124.Karimi K, Forsythe P. Natural killer cells in asthma. Front Immunol. 2013;4:159. doi: 10.3389/fimmu.2013.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kaiko G E, Phipps S, Angkasekwinai P, Dong C, Foster P S. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J Immunol. 2010;185(08):4681–4690. doi: 10.4049/jimmunol.1001758. [DOI] [PubMed] [Google Scholar]
- 126.Welliver T P, Garofalo R P, Hosakote Y et al. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J Infect Dis. 2007;195(08):1126–1136. doi: 10.1086/512615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stonier S W, Schluns K S. Trans-presentation: a novel mechanism regulating IL-15 delivery and responses. Immunol Lett. 2010;127(02):85–92. doi: 10.1016/j.imlet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Leavy O. Maturation and function of NK cells. Nat Rev Immunol. 2012;12(03):150. doi: 10.1038/nri3172. [DOI] [PubMed] [Google Scholar]
- 129.Ennis F A, Meager A, Beare A Set al. Interferon induction and increased natural killer-cell activity in influenza infections in man Lancet 19812(8252):891–893. [DOI] [PubMed] [Google Scholar]
- 130.Gazit R, Gruda R, Elboim M et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol. 2006;7(05):517–523. doi: 10.1038/ni1322. [DOI] [PubMed] [Google Scholar]