

Highlights
-
•
The KM22 genome sequence is similar to other B. bronchiseptica strains isolated from animals.
-
•
bopN, bvgA, fimB, and fimC were the most highly conserved BvgAS-regulated genes present in all strains analyzed.
-
•
BvgAS-regulated genes with the highest sequence divergence werefimN, fim2, fhaL, and fhaS.
-
•
A total of eight major fimbrial subunit genes were identified in KM22.
-
•
qPCR data demonstrated that seven of the eight fimbrial subunit genes are expressed and regulated by BvgAS.
Keywords: Bordetella, Genome sequence, Swine
Abstract
The well-characterized Bordetella bronchiseptica strain KM22, originally isolated from a pig with atrophic rhinitis, has been used to develop a reproducible swine respiratory disease model. The goal of this study was to identify genetic features unique to KM22 by comparing the genome sequence of KM22 to the laboratory reference strain RB50. To gain a broader perspective of the genetic relationship of KM22 among other B. bronchiseptica strains, selected genes of KM22 were then compared to five other B. bronchiseptica strains isolated from different hosts. Overall, the KM22 genome sequence is more similar to the genome sequences of the strains isolated from animals than the strains isolated from humans. The majority of virulence gene expression in Bordetella is positively regulated by the two-component sensory transduction system BvgAS. bopN, bvgA, fimB, and fimC were the most highly conserved BvgAS-regulated genes present in all seven strains analyzed. In contrast, the BvgAS-regulated genes present in all seven strains with the highest sequence divergence werefimN, fim2, fhaL, andfhaS. A total of eight major fimbrial subunit genes were identified in KM22. Quantitative real-time PCR data demonstrated that seven of the eight fimbrial subunit genes identified in KM22 are expressed and regulated by BvgAS. The annotation of the KM22 genome sequence, coupled with the comparative genomic analyses reported in this study, can be used to facilitate the development of vaccines with improved efficacy towards B. bronchiseptica in swine to decrease the prevalence and disease burden caused by this pathogen.
1. Introduction
The U.S. swine industry is the third largest producer of pork in the world, and respiratory disease in pigs is the most important health concern for swine producers today (U. S. Department of Agriculture, 2008). According to the recent survey conducted by the National Animal Health Monitoring System (NAHMS), respiratory disease is on the rise and is the greatest cause of mortality in nursery and grower/finisher swine, making the development of efficacious vaccines and therapeutic interventions that can protect against these infections a top research priority (U. S. Department of Agriculture, 2008). Bordetella bronchiseptica colonization is pervasive in swine herds and is an important contributor to respiratory disease in pigs. It is a primary cause of bronchopneumonia in young pigs, while it additionally contributes to secondary pneumonia in older pigs. It is also the primary etiologic agent of nonprogressive atrophic rhinitis, a mild to moderately severe reversible condition that promotes colonization by toxigenic strains of Pasteurella multocida and can lead to severe progressive atrophic rhinitis (Brockmeier et al., 2012). In swine exhibiting pneumonia, B. bronchiseptica is often isolated in combination with other pathogens (Brockmeier et al., 2012). Previous studies have demonstrated that co-infection with B. bronchiseptica increases colonization and exacerbates the severity of disease caused by both viral and bacterial pathogens, including swine influenza virus (SIV), porcine reproductive and respiratory syndrome virus (PRRSV), porcine respiratory coronavirus (PRCV), Haemophilus parasuis, P. multocida and Streptococcus suis(Brockmeier et al., 2012).
Various typing methods have been applied to B. bronchiseptica in an effort to elucidate the population structure of the bacterium. One of the approaches found to be highly discriminatory is multilocus sequence typing (MLST) based on comparison of DNA sequences from seven housekeeping genes (Diavatopoulos et al., 2005). The virulent B. bronchiseptica strain KM22 was originally isolated in Hungary in 1993 from a swine herd with atrophic rhinitis. Similar to the majority of B. bronchiseptica strains isolated from pigs exhibiting disease, KM22 is sequence type (ST) 7 in Clonal Complex 1 of an MLST-based Bordetella phylogeny (Diavatopoulos et al., 2005), and it harbors a PvuII ribotype (Register and Magyar, 1999). Additionally, KM22 contains a pertactin repeat region variant (Register, 2001) that is shared with the majority of strains isolated from swine. KM22 has been successfully used by our laboratory to develop a reproducible swine respiratory disease model reflective of clinical B. bronchiseptica infections within swine herds and host-to-host transmission (Brockmeier and Register, 2007, Nicholson et al., 2009, Nicholson et al., 2014a, Nicholson et al., 2012).
The complete genome sequence of the laboratory reference B. bronchiseptica strain RB50 was first made available in 2003 by Parkhill et al. (Parkhill et al., 2003). Recently Park et al. published the genome sequence of five B. bronchiseptica strains isolated from both human and non-human hosts (strains 253, 1289, MO149, Bbr77, and D445) (Park et al., 2012) and our laboratory reported the genome sequence of KM22 (Nicholson et al., 2014b). The goal of this study was to compare the genome sequence of KM22 to the complete genome sequence of the laboratory reference strain RB50 to identify genetic features unique to KM22. To gain a broader perspective of the genetic relationship of KM22 among other B. bronchiseptica strains, genetic sequences from KM22 were subsequently compared to five other B. bronchiseptica strains.
2. Materials and methods
2.1. Genome sequencing and annotation
A detailed list of the genome sequence information for the B. bronchiseptica isolates used in this study is provided in Table 1 . This table includes the NCBI genome accession number and the reference for the original genome publication.
Table 1.
Summary of sequenced B. bronchiseptica strain information.
| Strain | KM22 | RB50 | 1289 | 253 | MO149 | Bbr77 | D445 |
|---|---|---|---|---|---|---|---|
| Sequence type (ST) |
7 | 12 | 32 | 27 | 15 | 18 | 17 |
| Host | Pig | Rabbit | Monkey | Dog | Human | Human | Human |
| Size (bp) | 5,199,729 | 5,339,179 | 5,208,522 | 5,264,383 | 5,091,817 | 5,115,717 | 5,243,194 |
| G + C content (%) | 68.2% | 68.5% | 68.7% | 68.6% | 68.9% | 68.5% | 68.2% |
| Predicted CDSs | 4,843 | 5,009 | 4,785 | 4,845 | 4,669 | 4,667 | 4,775 |
| rRNA operons | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| tRNA | 54 | 55 | 54 | 54 | 54 | 54 | 54 |
| Accession number |
JNHR00000000 | NC_002927 | HE983626 | NC_019382 | HE965807 | HE983628 | HE983627 |
| Reference | This study | Parkhill et al. (2003) | Park et al. (2012) | Park et al. (2012) | Park et al. (2012) | Park et al. (2012) | Park et al. (2012) |
Assembly and annotation of the KM22 genome sequence data identified a total of 4853 predicted protein coding sequences (CDSs) (Nicholson et al., 2014b). Upon further investigation, seven CDSs were identified as likely representing a CDS split across two contiguous sequences (contigs) such that the 5′ end of one gene is located at the end of one contig and the 3′ end of the same gene is located at the start of the following contig, with some portion of the middle gene sequence missing, preventing joining of the contigs. PCR primers were designed for each end of the fourteen contigs, and amplification confirmed that each predicated pair was indeed one contiguous region. Additionally, 3CDSs were identified that have sequence homology at the nucleotide and translated protein level with a single large CDS present in RB50 (BB1186) that is homologous to KM22_01212. The three CDSs in KM22 (KM22_04905, KM22_04906 and KM22_04907) each comprise small contigs and align to highly repetitive regions of the RB50 sequence that are missing in the homologous KM22CDS (KM22_01212). Therefore, we believe these three annotated CDSs in KM22 actually comprise various portions of a single gene (KM22_01212) that was not correctly assembled due to the highly repetitive nature of the DNA sequence. As a result, our revised assembly and annotation of the KM22 genome sequence resulted in 4843 unique protein coding sequences in 46 contigs ranging from 679,114 bp to 686 bp, with a total length of 5119,729 bp.
2.2. Comparative genomic analysis
The KM22 genome sequence was analyzed using the RAST server, which mapped annotated protein coding sequences (CDS) to functional subsystems and performed a one-to-one BLASTP comparison to the genome sequence of RB50 (Parkhill et al., 2003) (Accession # NC_002927.3) to identify conserved protein coding sequences. Genome organization was examined by comparison to strain RB50 using the Artemis Comparison Tool (Carver et al., 2005) and progressiveMauve (Darling et al., 2010).
2.3. Comparison of individual genes
Genome sequences from six B. bronchiseptica isolates were available at the outset of our study, as listed in Table 1. The nucleotide sequences of selected genes were compared with BLASTN. Percent identity relative to RB50 was determined and used to generate a comparative heatmap using MeV software. Alignments of individual gene and protein sequences, as well as calculation of percent identities, were performed using the Geneious Alignment tool in Geneious Pro 5.0.4.
2.4. Phylogenetic analyses
A concatenated alignment composed of SNP sites in 31 BvgAS-associated genes, as well as a concatenated alignment of SNP sites from the 7 house-keeping genes (MLST), was generated for each isolate. Maximum-likelihood phylogenetic trees were constructed from the SNP alignment using RAxML v7.0.4 (Stamatakis, 2006) using a general time reversible substitution model with gamma correction (GTRGAMMA) for among-site rate variation. Support for nodes on the tree was assessed using 100 bootstrap replicates.
2.5. qPCR analysis of fimbrial subunit gene expression
Four independent cultures (biological replicates) of B. bronchiseptica strain KM22 and its phase-locked derivatives TN30 (Bvg−) (Nicholson et al., 2012) and TN31 (Bvgi) (Nicholson et al., 2012) were grown in SS broth at 37 °C with shaking until mid-log phase (OD600 0.7–0.8). Bacteria were harvested by centrifugation and total RNA was extracted using the Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, CA), treated with DNaseI (Life Technologies, Carlsbad, CA) and purified using RNA Clean & Concentrator-5 columns (Zymo Research, Irvine, CA). DNase-treated RNA from each biological replicate (500 ng) was reverse transcribed using SuperScript III reverse transcriptase (Life Technologies, Carlsbad, CA) and 100 ng of random primer oligonucleotides according to the manufacturer’s protocol. The cDNA was diluted 1:250 in water and 5 μl of the dilution was used for qPCR in reactions containing 500 nM each primer and 250 nM probe using the TaqMan Gene Expression Master Mix (Life Technologies, Carlsbad, CA). qPCR was performed on an Applied Biosystems 7300 Real Time PCR System (Applied Biosystems, Foster City, CA). Reactions lacking reverse transcriptase enzyme were performed to test for genomic DNA contamination. Primers were designed using the PrimerQuest web tool from IDT (Coralville, IA). Probes were designed as double-quenched, labelled with 5′- FAM (6-carboxyfluorescein) and the internal IDT ZEN and 3′ Iowa Black (IBFQ) Quenchers. All primers and probes used in this study are listed in Supplemental Table 1. Relative expression of the fimbrial gene mRNAs was determined using the 2−ΔΔCt method using the 16S rRNA gene as the endogenous control (Livak and Schmittgen, 2001).
3. Results and discussion
B. bronchiseptica isolate KM22 was originally isolated from a pig, and this isolate, along with KM22-derived mutants, have been used in many infection studies in its natural swine host to characterize the associated disease phenotypes. The phenotypes of some KM22-derived mutants used in these studies did not always concur with reciprocal studies using the well characterized rabbit RB50 isolate, along with RB50-derived mutants, in rodent infection models (Nicholson et al., 2009, Nicholson et al., 2012). To determine genomic differences that could account for such virulence-associated phenotypic differences, we compared the genomes from KM22 and RB50, as well genomes from five additional B. bronchiseptica strains (1289 isolated from a monkey, 253 from a dog, and MO149, Bbr77, and D445 from humans).
3.1. Phylogenetic analysis based on MLST target sequences
The phylogenetic relationship among the B. bronchiseptica strains used in this study was estimated based on a Maximum-likelihood tree derived from concatenated MLST target sequences for each strain (Supplemental Fig. 1). There are two distinct sub lineages, one including strains isolated from human hosts (MO149, D445, and Bbr77) and the other including isolates from non-human animal hosts (KM22, RB50, 1289, and 253). Strain 253, isolated from a dog, is highly polymorphict compared to the other non-human animal hosts.
3.2. Annotation of the KM22 genome sequence
Initially, assembly and annotation of the KM22 genome sequence data identified a total of 4853 predicted protein coding sequences (CDSs). Seven CDSs were identified as likely representing a CDS split across two contiguous sequences (contigs) and PCR amplification was used to subsequently confirm that each of CDS was indeed one contiguous region. Additionally, three annotated CDSs in KM22 actually comprise various portions of a single gene (KM22_01212) that was not correctly assembled due to the highly repetitive nature of the DNA sequence. Our revised assembly and annotation of the KM22 genome sequence resulted in 4843 unique protein coding sequences and shares many similarities with other B. bronchiseptica strains evaluated in this study, including size (bp) and predicted CDSs (Table 1).
3.3. Comparison of the genome sequences of B. bronchiseptica strains RB50 and KM22
We compared the KM22 genome sequence to the genome sequence of the laboratory reference strain RB50, given that it was the first complete B. bronchiseptica genome sequence available and the most widely studied B. bronchiseptica strain. A one-to-one BLASTP comparison of the protein coding sequences in KM22 and RB50 was performed to identify shared CDS regions. Of the 4843 predicted protein coding sequences identified in KM22, 4674 were identified as shared with RB50. The remaining 169 predicted protein coding sequences from KM22, which lacked a reciprocal match in RB50, were designated KM22-specific coding sequences (Table 2 in the Supplement). KM22-specific coding sequences include tracheal colonization factor (tcfA), serum resistance protein (brkA), several autotransporters including bapC, adhesion (KM22_00109), pilus assembly protein (KM22_04913), fimbrial subunit gene (KM22_03370), genes encoding the alternative or O2-antigen, and seven transcriptional regulators. This list includes genes that are predicted to be expressed as full-length functional proteins in KM22 and therefore contains coding sequences annotated as a pseudogene in RB50, such as tcfA and brkA. Of the 4994 total protein coding sequences in RB50, 320 were identified as RB50-specific, many of which are phage-related and located in discrete blocks. RB50 was previously shown to have a number of phage-related genes that are not conserved among other Bordetella strains (Parkhill et al., 2003).
Comparison of the linear organization of the genomes of B. bronchiseptica strains RB50 and KM22 revealed a high degree of synteny within contiguously assembled regions of the genome (Fig. 1 ). Four potential regions of rearrangement were identified. In three of these regions, the rearrangement is supported by genome sequence assembly data from both strains, such that the position at which the organization changes in KM22 relative to RB50 occurs within a single contig on at least one edge of the region (Fig. 1). The fourth rearrangement identified occurs at contig boundaries such that it is not possible to determine if a true genome rearrangement has occurred at that site or if the contig ordering is incorrect (Fig. 1). Genome rearrangements can have profound phenotypic effects on bacterial niche adaptation by impacting gene expression or loss of gene function resulting from genes located near rearrangement breakpoints being placed under control of new transcriptional regulatory elements (Suyama and Bork, 2001). Genome rearrangements have been previously identified in other Bordetella isolates, including B. bronchiseptica strains (Brinig et al., 2006, Park et al., 2012, Parkhill et al., 2003). A comparison of the linear organization of the genomes of all seven B. bronchiseptica strains evaluated in this study is provided in Supplemental Fig. 2.
Fig. 1.
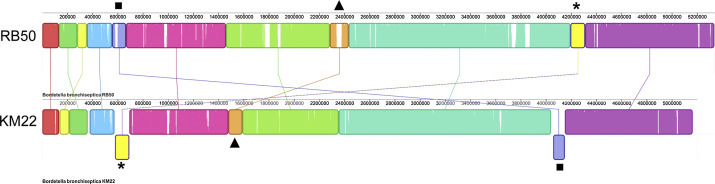
Comparison of the linear organization of the genomes of B. bronchiseptica strains RB50 and KM22. progressiveMauve was used to compare KM22 to RB50 (NC_002927.3). Locally collinear blocks (LCBs) representing regions of sequence that align in each genome are illustrated as colored rectangles connected by lines. For the KM22 sequence, blocks above the center line are in the same orientation as RB50, blocks below the center line align in the inverse orientation. White areas reflect regions that are not conserved. Symbols are used to mark LCBs whose rearrangement in KM22 relative to RB50 occurs within a single contig, likely reflecting a true rearrangement.
3.4. Comparison of bvgAS amongst sequenced B. bronchiseptica strains
The bvgAS locus encodes a two-component sensory transduction system that controls the expression of the majority of genes encoding virulence factors and is a universal regulatory mechanism shared among Bordetella strains (Cotter and Jones, 2003). This locus comprises a histidine kinase sensor protein, BvgS, and a DNA-binding response-regulator protein, BvgA.
The nucleotide sequences of bvgS from strains KM22, 1289 and 253 are 100% identical. Nucleotide substitutions were observed in bvgS from strain RB50 compared to bvgS from strains KM22, 1289 and 253 and are predicted to result in only 4 amino acid replacements in the 1238 amino acid protein. These amino acid changes in RB50 relative to KM22, 1289 and 253 are: T50A in the periplasmic region, between the signal peptide and Periplasmic Substrate Binding domain 1 or VFT1; L395M in the central region of PBPb 2; A651S in the central region of the PAS intracellular domain; and V1144L in the histidine phosphotransferase domain (HPT). It is unclear whether these amino acid differences could affect the activity of BvgS given that none are located within regions previously studied by mutation (Dupre et al., 2013, Herrou et al., 2010). Similar to the results reported by Herrou et al., numerous nucleotide substitutions were observed in bvgS from strains isolated from human hosts (MO149, D445, and Bbr77) compared to bvgS from strain RB50 further supporting the conclusion that BvgS has evolved into two different types: a “pertussis-type” found in Bordetella pertussis strains and in a lineage of B. bronchiseptica strains isolated from humans, and a “bronchiseptica-type” associated with the majority of B. bronchiseptica strains (Herrou et al., 2009).
We found the response regulator BvgA to be more highly conserved among the strains analyzed, which is also similar to the findings of Herrou et al. (Herrou et al., 2009). The nucleotide sequences of bvgA from strains KM22, 1289 and 253 are 100% identical. A single synonymous substitution was observed in bvgA from strain RB50 compared to bvgA from strains KM22, 1289 and 253. A varying small number of single nucleotide substitutions (SNPs) were observed in bvgA from strains Bbr77, D445, and MO149 compared to bvgA from strain RB50. Specifically, six SNPs were identified in bvgA from strain Bbr77, five from strain D445, and seven from strain D445. These SNPs are predicted to result in only 2 amino acid replacements in the BvgA protein and are shared by strains Bbr77, D445, and MO149. One amino acid change is M1T and the second is L12S. It is worth noting that the M1T amino acid change is due to the presence of an ACG (Thr) at the 5′ prime end of the bvgA nucleotide sequence from strains Bbr77, D445, and MO149, instead of the ATG (Met) present at the same location in RB50, KM22, 1289, and 253. All seven strains analyzed have a second ATG located downstream at position 55 that is in frame (Met19) and could be used as the start site of translation.
3.5. Comparison of Bvg-regulated genes amongst sequenced B. bronchiseptica strains
The BvgAS regulon defines regions of the genome of importance to host-pathogen interactions (Cotter and Jones, 2003); therefore to examine genomic differences that may influence how B. bronchiseptica strains interact with their host and environment, we compared the nucleotide sequences of 31 genes encoding BvgAS-regulated factors, such as adhesins, toxins, and other known virulence factors, as well as the flagellin structural gene flaA. The percent identity for each of the BvgAS-regulated genes was determined for each isolate relative to RB50 (Fig. 2 ). Compared to RB50, the average percent identity of all the BvgAS-regulated genes examined was higher in the strains isolated from animal hosts (1289, KM22, and 253) than from strains isolated from human hosts (MO149, D445, and Bbr77), with strain D445 containing the lowest average percent identity of 94.9% consistent with its distance from RB50 in the phylogentic reconstruction from the MLST data (Supplemental Fig. 1). Focusing on specific genes present in all strains, the genes with the highest average percent identity across all strains are bopN (99.5% ± 0.39), (99.4% ± 0.30), bvgA (99.4% ± 0.41), and fimC (99.1% ± 0.40). In contrast, genes present in all strains with the highest sequence divergence are fimN (89.8% ± 1.1), fim2 (93.1% ± 6.6), fhaL(94.6% ± 4.0), and fhaS (94.9% ± 3.3). flaA is present in all strains and contains an average percent identity of 96.4%±4.8, with flaA from strains KM22 and 1289 both having a lower percent identity of 89.1%.
Fig. 2.
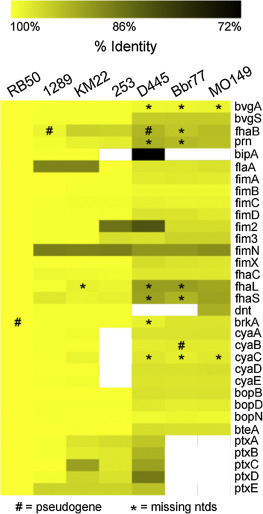
Sequence conservation of Bvg-regulated genes. The nucleotide sequences of selected genes from B. bronchiseptica strains 1289, KM22, 253, Bbr77, D445 and MO149 were compared to genes from RB50 using BLASTN. The percent identity of the genes (rows) from each strain (columns) is represented using the color scale at top, while genes not present within a strain are indicated by white.
Among the BvgAS-regulated genes, including genes not present in all strains, bipA from strain D445 is the most divergent with an identity of 71.4% and is not present in strains 253, Bbr77, and MO149. Seven SNPs were observed in bipA from strain KM22 compared to RB50 and are predicted to result in 4 amino acid replacements: S322G, V813A, E912G, and S1131P. These findings are similar to the results reported in a recent study evaluating virulence gene polymorphisms among B. bronchiseptica seal isolates, in which bipA was found to have the greatest sequence divergence of the genes examined (Register et al., 2015).
dnt, encoding the dermonecrotic toxin (DNT), is not present in strains D445 and Bbr77 and contains a lower percent identity in strain MO149 (93.0%) compared to strains isolated from animal hosts (1289, KM22, and 253) (99.6–99.8%).
The ptx locus is present in KM22 with an average identity of 97.3% ± 0.01 compared to RB50. Consistent with that previously reported (Park et al., 2012), the ptx locus was absent in strains Bbr77 and MO149 isolated from human hosts. Additionally, strain D445 contains a high sequence divergence in the ptx locus compared to RB50, with ptxD containing the most sequence divergence with an identity of 88.2% compared to ptxD from RB50.
As previously reported, the cya operon encodes genes responsible for the production and secretion of adenylate cyclase toxin (ACT) and is not present in strain 253 (Fig. 2) (Buboltz et al., 2008, Park et al., 2012). The start codon for the cyaB gene in the KM22 annotation is ATG, which is identical to the start codon for the cyaB gene in the B. pertussis Tohama I annotation (Parkhill et al., 2003). In contrast, the predicted start codon for the cyaB gene in the RB50 annotation is GTG and is located slightly upstream of the predicted ATG start codon for KM22. This ATG codon is present in all the B. bronchiseptica strains, but is not annotated as the start codon. Instead, similar to the RB50 annotation, GTG is located slightly upstream of this ATG and is predicted to be the start codon for the other B. bronchiseptica strains. When this GTG is used as the start codon, it does not encode a full length protein sequence due to a resulting frameshift mutation. This GTG codon only encodes a full length protein sequence in RB50. However, if the conserved ATG is used as the start codon, then a full length protein sequence is capable of being produced by all the B. bronchiseptica strains. Therefore it is possible that the predicted ATG start codon for KM22 is the start codon for the cyaB gene in the other B. bronchiseptica strains and this may also explain the results reported by Park et al. (Park et al., 2012) that strains 1289, D445, Bbr77, and MO149 are beta-hemolytic despite containing a frameshift mutation in the cyaB gene.
There is a large disparity in average sequence identity between genes predicted or known to encode for proteins that function in attachment or adhesion. Specifically, fimB (99.4%) is one of the most conserved genes, while fimN (89.8%) is the most divergent of the genes predicted to encode for proteins that function in attachment. The fhaB gene, encoding the well-characterized FhaB adhesin, was comparatively more divergent (93.7% in strain D445 and 94.3% in strain MO149) and is predicted to be a pseudogene in strain D445 due to a frame shift mutation.
3.6. Comparison of the fimNX locus a mongst sequenced B. bronchiseptica strains
B. bronchiseptica expresses fimbriae of at least four serotypes, Fim2, Fim3, FimX, and FimA, all of which belong to the chaperone-usher family of fimbriae (Boschwitz et al., 1997, Cuzzoni et al., 1990, Savelkoul et al., 1996). While these major fimbrial subunit genes are unlinked on the chromosome, assembly and secretion of their protein products are mediated by the products of three genes, fimBCD, encoding the usher, the chaperone, and an adhesin, respectively (Locht et al., 1992, Willems et al., 1994). The fimBCD locus is located between the fhaB and fhaC genes, which are required for production and secretion of filamentous hemagglutinin (FHA) (Locht et al., 1992, Willems et al., 1994). Similar to RB50, the fimBCD genes were identified in KM22 and located between the fhaB and fhaC genes. A total of eight major fimbrial subunit genes (fimA, fim2, fim3, fimN, fimX, fim2-like KM22_03370, KM22_03368, and KM22_03128) were identified in KM22 and located at unlinked positions along the chromosome. Of the chromosomal regions containing fimbrial subunit genes, we observed the greatest sequence divergence within the fimNX locus. In strain RB50, the fimNX locus contains three genes (fimbrial protein,fimN, fimX), while four genes were identified within this locus in KM22, annotated in order as KM22_03368 (fimbrial protein), KM22_03369 (fimN), KM22_03370 (fim2-like), KM22_03371 (fimX). KM22_03370 is a fimbrial subunit gene identified in KM22 and is not present in RB50. As shown in Supplemental Fig. 3, the gene organization present in the fimNX locus of KM22 is also present in strains 253, Bbr77 and D445. An additional fimbrial subunit gene is annotated at the beginning of the fimNX locus in 1289; however, it is predicted to be a pseudogene.
3.7. Quantitative real-time PCR analysis of fimbrial subunit genes from B. bronchiseptica KM22
Due to the variation in sequence divergence observed in fimbrial genes, we sought to determine if those fimbrial genes encoded in KM22 are expressed and whether or not they are regulated by BvgAS. Gene expression in two Bvg phase-locked derivatives of strain KM22, TN30 (Bvg¯) andTN31 (Bvgi), was analyzed by qPCR and compared to the wild-type KM22 strain. The TN30 (Bvg¯) mutant does not express the BvgAS-regulated virulence genes and is avirulent, while the TN31 (Bvgi) mutant has a reduced expression of these genes and has an avirulent phenotype in swine (Nicholson et al., 2012). The phase-locked mutants and the wild-type KM22 were grown under non-modulating conditions at 37 °C. With the exception of KM22_03128, the expression of all the fimbrial genes analyzed was decreased in the Bvg¯ phase-locked mutant, TN30, relative to wild-type KM22, while fimA, fim3, and KM22_03368 were increased in the Bvgi phase-locked mutant TN31 relative to the wild-type KM22 (Fig. 3 ). These data indicate that fimA, fim2, fim3, KM3368, fimN, KM22_3370, and fimX are all BvgAS-regulated genes.
Fig. 3.
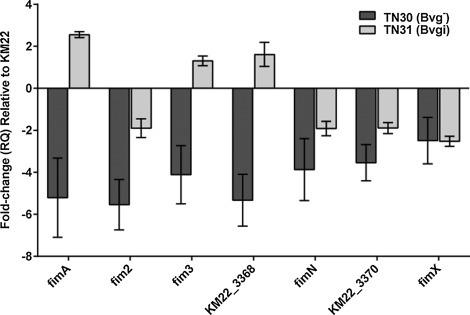
qPCR analysis of fimbrial subunit genes. Quantitative real-time PCR was used to determine mRNA expression of fimbrial subunit genes in wild-type KM22 and the phase-locked mutants TN30 (Bvg−) and TN31 (Bvgi). Gene expression relative to KM22 and standardized to 16S rRNA is plotted along the y-axis and fimbrial genes analyzed are shown along the x-axis. Data shown are averages obtained from triplicate cultures. The error bars represent ± the standard error.
KM22_03128 is located separately along the KM22 chromosome and is not in proximity to any other fimbrial genes. A homologous gene is present in the other B. bronchiseptica strains; however the homologous CDS in strain Bbr77 is truncated. Expression of KM22_03128 could not be detected in any of the strains, including wild-type KM22, indicating that this gene is either not expressed or expressed at a level too low to detect in these strains and/or under the growth conditions used in the study.
Poly-cytosine or poly(C) tracts are polymeric tracts of C residues located in the promoter region of genes that can result in the reversible switching ON or OFF of protein expression. This process is referred to as phase-variation, which is mediated by slipped-strand mispairing during DNA replication at these repetitive tracts (Stibitz et al., 1989, Willems et al., 1990). Promoter polyC tracts were identified in the KM22 sequence upstream of fimN, KM22_03370, fimX, fim2, and fim3. A stretch of 13C nucleotides with 3 G nucleotides interspersed was additionally identified upstream of KM22_03368. No polyC tracts were identified upstream of the annotated start codon for start of fimA and KM22_03128.
Overall, qPCR data demonstrated that all of the major fimbrial subunit genes identified in KM22, with the exception of KM22_03128, are expressed and regulated by BvgAS. The inability to detect expression of KM22_03128 by any of the strains analyzed suggests that this gene is either not expressed or expressed below detectable levels. Excluding fimA and KM22_03128, promoter polyC tracts were identified upstream of all of the major fimbrial subunit genes, suggesting that these genes have the ability to exhibit phase-variation mediated by slipped-strand mispairing.
3.8. Comparison of FlaA protein sequences amongst sequenced bronchiseptica strains
One mechanism used by bacterial pathogens to survive and persist for long periods of time in their environment, both within and between hosts, is to exist as biofilms. It has recently been demonstrated that flagella are necessary and enhance the cell-surface interactions during the initial stages of biofilm development for B. bronchiseptica. The flagellin structural gene flaA encoding the flagellin monomer is present in all strains evaluated in this study and has a range of nucleotide identity relative to RB50 from 99.9% in 253 to 89.1% in strain 1289. An alignment of FlaA predicted protein sequences from all strains reveals that the protein sequence polymorphisms are confined to the middle section of the protein sequence, with the N-terminal and C-terminal sections highly conserved (identical in all 7 strains) (Supplemental Fig. 4). These conserved regions correspond to the N-terminal (pfam00669) and C-terminal region (pfam007000) of other bacterial flagellins, which form packed α-helix structures. This structural pattern is typical for bacterial flagellin proteins where the conserved termini regions flank a central region that is hypervariable both in residue composition and size (Ramos et al., 2004). This suggests flagella may play a strain-specific role within their respective environments and/or hosts.
3.9. Comparison of the Type 3 secretion system (T3SS) locus am ongst sequenced B. bronchiseptica strains
Genes encoding the components of the Bordetella T3SS, including the secretion apparatus, secreted effector proteins, and putative chaperones, are located within the bsc locus (Mattoo et al., 2004). Directly adjacent to this locus is the btr locus, which contains genes encoding the regulatory elements for T3SS (Mattoo et al., 2004). A broad comparison of the region encompassing both loci among strains RB50, KM22, 1289 and 253 showed that overall, the region is highly conserved and over 98% identical. One difference was identified among these strains; one CDS annotated in strains RB50 (BB1639) and KM22 (KM22_01795) as a hypothetical protein is annotated as a pseudogene in strains 1289 (BN113_1619) and 253 (BN112_1817) due to a frameshift. In strain 1289 the CDS is annotated as a hypothetical protein (pseudogene) over 2 ORFs and as an unnamed gene only (no CDS annotated) in strain 253.
3.10. Comparison of the Type 6 secretion system (T6SS) locus am ongst sequenced B. bronchiseptica strains
Comparison of the T6SS locus containing 26 genes among strains KM22, RB50 and 1289 revealed that the region is highly conserved in these 3 strains (∼99% identical). The only region that shows any sequence variation is one intergenic region, which differs in size in each strain. This intergenic region is located between CDSs BB0798 and BB0799 for strain RB50 and KM22_00821 and KM22_00822 for strain KM22. The overall conservation among the other strains that encode the T6SS compared to RB50 is 98% for strain 253 and 96% for strain MO149. Possible pseudogenes were identified in strains 1289 and KM22. In 1289, locus BN113_0788 is split into 2 coding regions in different reading frames, but the same CDS is full-length in strains RB50 and KM22. In KM22, two sets of possible pseudogenes were identified: M22_00819/KM22_00820 and KM22_00833/KM22_00834. These genes are full-length in strains RB50 and 1289, but the coding sequence switches reading frames in KM22. No possible pseudogenes were identified in strains RB50, MO149, and 253. Strains D445 and Bbr77, isolated from human hosts, are missing either most or the entire T6SS region.
3.11. The O-antigen locus of B. bronchiseptica strain KM22
Most B. bronchiseptica strains produce one of two immunologically distinct O-antigen serotypes encoded by either the classical O-antigen locus (O1) or the alternative O-antigen locus (O2), while strain MO149 produces a poorly immunogenic O-antigen referred to as O3 (Buboltz et al., 2009, Hester et al., 2013). The O-antigen locus from the B. bronchiseptica strains evaluated here typically contains 24 genes, except for the O3 locus in MO149, which contains 15 genes (Hester et al., 2013, Park et al., 2012). The O-antigen locus of KM22 is highly similar to that of strain 1289, with the annotated CDSs sharing between 99.7-100 percent identity and, therefore contains the genes encoding the alternative or O2-antigen.
3.12. Phylogenetic analysis based on BvgAS-regulated genes
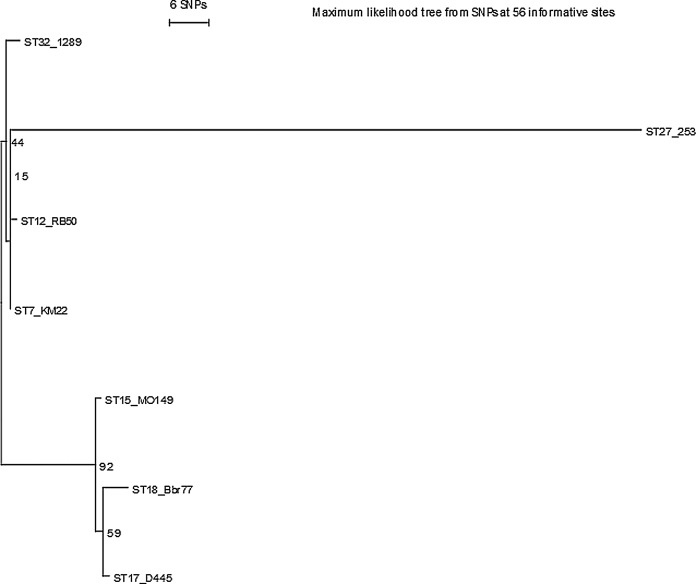
Given that the gene set analyzed in Fig. 2 encodes factors that influence how B. bronchiseptica strains interact with their host and/or environment, we sought to determine how a phylogenetic tree based on this gene set would compare to the MLST-based phylogenetic tree. Therefore the phylogenetic relationship among the B. bronchiseptica strains used in our analyses was reconstructed based on single nucleotide polymorphisms in the BvgAS-regulated genes using a Maximum Likelihood method (Fig. 4 ). We were intrigued by the similarity of a phylogeny based on BvgAS-regulated genes (Fig. 4) to one derived from MLST sequences (Supplemental Fig. 1). Both trees contain two distinct sublineages, one that includes all of the isolates from animal hosts (253, 1289, RB50, and KM22) and a second that includes the strains isolated from human hosts. Focusing on the phylogeny based on BvgAS-regulated genes (Fig. 4), the phylogenetic separation among the strains isolated from human hosts (MO149, D445, and Bbr77) was not well resolved based on the low support from the bootstrap values indicated at the nodes; this suggests that mechanisms of diversity, such as recombination, insertions, deletions, or horizontal gene transfer may have occurred within these genes. Although this is a small sample size it is not the first time this observation has been made. Based on SNP densities, Park et al. (Park et al., 2012) recently demonstrated that such mechanisms of diversity, specifically horizontal gene transfer, likely have occurred within the ptx and ptl loci. In addition, recombination could explain why strain 253 is so highly divergent from the other strains of animal origin in the MLST tree (Supplemental Fig. 1), but not in the trees based on the BvgAS-regulated genes (Fig. 4).
Fig. 4.
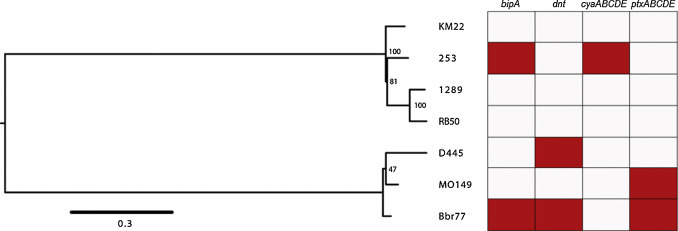
A Maximum Likelihood tree derived from BvgAS-regulated genes from each of the B. bronchiseptica strains used in this study. For each isolate, gene sequences from the BvgAS-regulated genes listed in Fig. 2 were concatenated and used to construct a Maximum Likelihood tree using 6870 SNPs from 34 genes with 100 bootstrap replicates. Grid indicates the genes or loci (rows) absent (red) from each strain (columns).
4. Conclusion
Despite widespread use of B. bronchiseptica vaccines by swine producers throughout the world, Bordetella-associated respiratory diseases remain a significant problem for the industry (U. S. Department of Agriculture, 2008, Brockmeier et al., 2012). Our hope is that the assembly and annotation of the KM22 genome sequence, coupled with the comparative genomic analyses reported in this study, will help identify genetic elements that could underlie and influence phenotypic differences among the isolates. Together, this information can aid in the development of vaccines with improved efficacy towards B. bronchiseptica in swine to decrease the prevalence and disease burden caused by this respiratory pathogen.
Acknowledgements
We thank Tibor Magyar (Institute for Veterinary Medical Research, Centre for Agricultural Research, Hungarian Academy of Sciences, Budapest, Hungary) for kindly providing B. bronchiseptica strain KM22. We additionally thank the support of the Wellcome Trust Sanger Institute core sequencing team for their assistance and David Alt (USDA/ARS/National Animal Disease Center) for his assistance. The work described here was funded in part by the Wellcome Trust through core funding grant 098051 for The Sanger Institute Pathogen Variation Group. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.vetmic.2015.10.026.
Appendix A. Supplementary data
The following are Supplementary data to this article:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Boschwitz J.S., van der Heide H.G., Mooi F.R., Relman D.A. Bordetella bronchiseptica expresses the fimbrial structural subunit gene fimA. J. Bacteriol. 1997;179:7882–7885. doi: 10.1128/jb.179.24.7882-7885.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinig M.M., Cummings C.A., Sanden G.N., Stefanelli P., Lawrence A., Relman D.A. Significant gene order and expression differences in Bordetella pertussis despite limited gene content variation. J. Bacteriol. 2006;188:2375–2382. doi: 10.1128/JB.188.7.2375-2382.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmeier S.L., Register K.B. Expression of the dermonecrotic toxin by Bordetella bronchiseptica is not necessary for predisposing to infection with toxigenic Pasteurella multocida. Vet. Microbiol. 2007;125:284–289. doi: 10.1016/j.vetmic.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Brockmeier S.L., Register K.B., Nicholson T.L., Loving C.L. Bordetellosis. In: Zimmerman J.J., Karriker L.A., Ramirez A., Schwartz K.J., Stevenson G.W., editors. Diseases of Swine. Wiley-Blackwell; 2012. pp. 670–708. [Google Scholar]
- Buboltz A.M., Nicholson T.L., Karanikas A.T., Preston A., Harvill E.T. Evidence for horizontal gene transfer of two antigenically distinct O antigens in Bordetella bronchiseptica. Infect. Immun. 2009;77:3249–3257. doi: 10.1128/IAI.01448-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buboltz A.M., Nicholson T.L., Parette M.R., Hester S.E., Parkhill J., Harvill E.T. Replacement of adenylate cyclase toxin in a lineage of Bordetella bronchiseptica. J. Bacteriol. 2008;190:5502–5511. doi: 10.1128/JB.00226-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T.J., Rutherford K.M., Berriman M., Rajandream M.A., Barrell B.G., Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- Cotter P.A., Jones A.M. Phosphorelay control of virulence gene expression in Bordetella. Trends Microbiol. 2003;11:367–373. doi: 10.1016/s0966-842x(03)00156-2. [DOI] [PubMed] [Google Scholar]
- Cuzzoni A., Pedroni P., Riboli B., Grandi G., de Ferra F. Nucleotide sequence of the fim3 gene from Bordetella pertussis and homology to fim2 and fimX gene products. Nucleic Acids Res. 1990;18:1640. doi: 10.1093/nar/18.6.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A.E., Mau B., Perna N.T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diavatopoulos D.A., Cummings C.A., Schouls L.M., Brinig M.M., Relman D.A., Mooi F.R. Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. PLoS Pathog. 2005;1:e45. doi: 10.1371/journal.ppat.0010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre E., Wohlkonig A., Herrou J., Locht C., Jacob-Dubuisson F., Antoine R. Characterization of the PAS domain in the sensor-kinase BvgS: mechanical role in signal transmission. BMC Microbiol. 2013;13:172. doi: 10.1186/1471-2180-13-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrou J., Bompard C., Wintjens R., Dupre E., Willery E., Villeret V., Locht C., Antoine R., Jacob-Dubuisson F. Periplasmic domain of the sensor-kinase BvgS reveals a new paradigm for the Venus flytrap mechanism. Proc. Natl. Acad. Sci. U.S.A. 2010;107:17351–17355. doi: 10.1073/pnas.1006267107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrou J., Debrie A.S., Willery E., Renauld-Mongenie G., Locht C., Mooi F., Jacob-Dubuisson F., Antoine R. Molecular evolution of the two-component system BvgAS involved in virulence regulation in Bordetella. PLoS One. 2009;4:e6996. doi: 10.1371/journal.pone.0006996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hester S.E., Park J., Goodfield L.L., Feaga H.A., Preston A., Harvill E.T. Horizontally acquired divergent O-antigen contributes to escape from cross-immunity in the classical bordetellae. BMC Evol. Biol. 2013;13:209. doi: 10.1186/1471-2148-13-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−△△CT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Locht C., Geoffroy M.C., Renauld G. Common accessory genes for the Bordetella pertussisfilamentous hemagglutinin and fimbriae share sequence similarities with the papC and papD gene families. EMBO J. 1992;11:3175–3183. doi: 10.1002/j.1460-2075.1992.tb05394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattoo S., Yuk M.H., Huang L.L., Miller J.F. Regulation of type III secretion in Bordetella. Mol. Microbiol. 2004;52:1201–1214. doi: 10.1111/j.1365-2958.2004.04053.x. [DOI] [PubMed] [Google Scholar]
- Nicholson T.L., Brockmeier S.L., Loving C.L. Contribution of Bordetella bronchiseptica filamentous hemagglutinin and pertactin to respiratory disease in swine. Infect. Immun. 2009;77:2136–2146. doi: 10.1128/IAI.01379-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson T.L., Brockmeier S.L., Loving C.L., Register K.B., Kehrli M.E.J., r, Shore S.M. The Bordetella bronchiseptica type III secretion system is required for persistence and disease severity but not transmission in swine. Infect. Immun. 2014;82:1092–1103. doi: 10.1128/IAI.01115-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson T.L., Shore S.M., Bayles D.O., Register K.B., Kingsley R.A. Draft Genome Sequence of the Bordetella bronchiseptica Swine Isolate KM22. Genome Announc. 2014;2 doi: 10.1128/genomeA.00670-14. e00670-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson T.L., Brockmeier S.L., Loving C.L., Register K.B., Kehrli M.E., Jr, Stibitz S.E., Shore S.M. Phenotypic modulation of the virulent Bvg phase is not required for pathogenesis and transmission of Bordetella bronchiseptica in swine. Infect. Immun. 2012;80:1025–1036. doi: 10.1128/IAI.06016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Zhang Y., Buboltz A.M., Zhang X., Schuster S.C., Ahuja U., Liu M., Miller J.F., Sebaihia M., Bentley S.D., Parkhill J., Harvill E.T. Comparative genomics of the classical Bordetella subspecies: the evolution and exchange of virulence-associated diversity amongst closely related pathogens. BMC Genomics. 2012;13:545. doi: 10.1186/1471-2164-13-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J., Sebaihia M., Preston A., Murphy L.D., Thomson N., Harris D.E., Holden M.T., Churcher C.M., Bentley S.D., Mungall K.L., Cerdeno-Tarraga A.M., Temple L., James K., Harris B., Quail M.A., Achtman M., Atkin R., Baker S., Basham D., Bason N., Cherevach I., Chillingworth T., Collins M., Cronin A., Davis P., Doggett J., Feltwell T., Goble A., Hamlin N., Hauser H., Holroyd S., Jagels K., Leather S., Moule S., Norberczak H., O'Neil S., Ormond D., Price C., Rabbinowitsch E., Rutter S., Sanders M., Saunders D., Seeger K., Sharp S., Simmonds M., Skelton J., Squares R., Squares S., Stevens K., Unwin L., Whitehead S., Barrell B.G., Maskell D.J. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 2003;35:32–40. doi: 10.1038/ng1227. [DOI] [PubMed] [Google Scholar]
- Ramos H.C., Rumbo M., Sirard J.C. Bacterial flagellins: mediators of pathogenicity and host immune responses in mucosa. Trends Microbiol. 2004;12:509–517. doi: 10.1016/j.tim.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Register K.B. Novel genetic and phenotypic heterogeneity in Bordetella bronchiseptica pertactin. Infect. Immun. 2001;69:1917–1921. doi: 10.1128/IAI.69.3.1917-1921.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Register K.B., Ivanov Y.V., Harvill E.T., Davison N., Foster G. Novel, host-restricted genotypes of Bordetella bronchiseptica associated with phocine respiratory tract isolates. Microbiol. 2015;161:580–592. doi: 10.1099/mic.0.000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Register K.B., Magyar T. Optimized ribotyping protocol applied to Hungarian Bordetella bronchiseptica isolates: identification of two novel ribotypes. Vet. Microbiol. 1999;69:277–285. doi: 10.1016/s0378-1135(99)00118-2. [DOI] [PubMed] [Google Scholar]
- Savelkoul P.H., de Kerf D.P., Willems R.J., Mooi F.R., van der Zeijst B.A., Gaastra W. Characterization of the fim2 and fim3 fimbrial subunit genes of Bordetella bronchiseptica: roles of Fim2 and Fim3 fimbriae and flagella in adhesion. Infect. Immun. 1996;64:5098–5105. doi: 10.1128/iai.64.12.5098-5105.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stibitz S., Aaronson W., Monack D., Falkow S. Phase variation in Bordetella pertussis by frameshift mutation in a gene for a novel two-component system. Nature. 1989;338:266–269. doi: 10.1038/338266a0. [DOI] [PubMed] [Google Scholar]
- Suyama M., Bork P. Evolution of prokaryotic gene order: genome rearrangements in closely related species. Trends Genet. 2001;17:10–13. doi: 10.1016/s0168-9525(00)02159-4. [DOI] [PubMed] [Google Scholar]
- U. S. Department of Agriculture . Part III: Reference of Swine Health and Health Management in the United States. USDA/APHIS/VS CEAH N478.0308. USDA; Fort Collins, CO: 2008. Swine 2006. [Google Scholar]
- Willems R., Paul A., van der Heide H.G., ter Avest A.R., Mooi F.R. Fimbrial phase variation in Bordetella pertussis: a novel mechanism for transcriptional regulation. EMBO J. 1990;9:2803–2809. doi: 10.1002/j.1460-2075.1990.tb07468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems R.J., Geuijen C., van der Heide H.G., Renauld G., Bertin P., van den Akker W.M., Locht C., Mooi F.R. Mutational analysis of the Bordetella pertussis fim/fha gene cluster: identification of a gene with sequence similarities to haemolysin accessory genes involved in export of FHA. Mol. Microbiol. 1994;11:337–347. doi: 10.1111/j.1365-2958.1994.tb00314.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.