

Abstract
A single tube fluorogenic RT–PCR-based ‘TaqMan’ assay was developed for detection and classification of bovine viral diarrhea virus (BVDV). TaqMan–PCR was optimized to quantify BVD virus using the ABI PRISM 7700 sequence detection system and dual-labeled fluorogenic probes. Two different gene specific labeled fluorogenic probes for the 5′ untranslated region (5′ UTR) were used to differentiate between BVD types I and II. Sensitivity of the single tube TaqMan assay was compared with two-tube TaqMan assay and standard RT–PCR using 10-fold dilutions of RNA. Single tube TaqMan assay was 10–100-fold more sensitive than the two-tube TaqMan assay and the standardized single tube RT–PCR. Specificity of the assay was evaluated by testing different BVD virus strains and other bovine viruses. A total of 106 BVD positive and negative pooled or single serum samples, field isolates and reference strains were tested. Quantitation of cRNA from types I and II BVD virus was accomplished by a standard curve plotting cycle threshold values (CT) versus copy number. Single tube TaqMan–PCR assay was sensitive, specific and rapid for detection, quantitation and classification of BVD virus.
Keywords: BVD virus, TaqMan–PCR, Typing, Quantitation, Herd screening
1. Introduction
Bovine viral diarrhea virus (BVDV) is a common pathogen of cattle that causes major economic losses to dairy industry. The virus is a member of the Pestivirus genus in the family Flaviviridae (Wengler et al., 1995). BVDV isolates were segregated into two genotypes based on comparison of sequences from the 5′ untranslated region (5′ UTR) of the viral genome (Ridpath et al., 1994, Ridpath and Bolin, 1998). Infection with BVD virus causes gastrointestinal disease, reproductive failure, respiratory disorders, fetal malformation and persistent infections (PI). The PI animals act as reservoirs of virus and shed the virus for life exposing other animals in the herd. In addition to a good vaccination program, identification and removal of PI animals from the herd is an essential component of control measures for eradication of BVD virus (Fulton and Burge, 2000, Cortese and Victor, 1999).
RT–PCR proved to be sensitive and specific for detection of BVD virus (Laamanen et al., 1997, Drew et al., 1999, Renshaw et al., 2000). Nested PCR was used to simultaneously detect and type BVD virus from clinical specimens (El-Kholy et al., 1998, Gilbert et al., 1999, Ridpath and Bolin, 1998). In most of the studies, two-step RT–PCR reaction was followed by agarose gel analysis for the detection of amplicons. Other studies performed probe hybridization (Radwan et al., 1995, Letellier et al., 1999) and restriction enzyme analysis (Canal et al., 1996) to evaluate the specificity of the amplified fragments. The application of RT–PCR and its subsequent analysis in a diagnostic setting requires simple, rapid but reproducible procedures with few steps. The development of fluorogenic PCR utilizing 5′–3′ nuclease activity of Taq DNA polymerase made it possible to eliminate post PCR processing (Holland et al., 1991).
The ABI Prism 7700 sequence detection system (TaqMan) was shown to be a sensitive and rapid method for detection and quantitation of human factor VIII (Heid et al., 1996). The system uses oligonucleotide probes labeled with fluorescent dyes, a reporter at the 5′ end and a quencher at the 3′ end (Livak et al., 1995). The quencher suppresses the reporter dye emission when both are attached to the probe. During each PCR cycle, the Taq DNA polymerase cleaves the reporter dye from the annealed probe by its 5′–3′ nucleolytic activity (Gibson et al., 1996). The free reporter dye emits its characteristic signal that can be detected by the ABI Prism 7700 sequence detection system. The increase in fluorescence emission is proportional to the accumulated PCR products. In this study a TaqMan–PCR assay was developed for detection, classification and quantitation of BVD virus in single tube. Specific probes were designed to classify BVD virus as genotype I or II. Quantitation of BVD virus was performed using an RNA standard.
2. Methods and materials
2.1. Viruses and clinical specimens
BVD viral reference strains NADL, New York-1, Singer (type Ia), Oregon-C24V (type Ia), Draper (type Ib), type II-125 and type II 890 were obtained from National Animal Disease Center (NADC, Ames, IA). The reference strains were propagated in MDBK cells. Reference strains of other bovine viruses were obtained from NADC: bovine respiratory syncycial virus (BRSV), parainfluenza type 3 (PI3), infectious bovine rhinotracheitus virus (IBR), bovine rotavirus, bovine coronavirus, bluetongue virus (BT) and bovine cytomegalovirus (BCMV).
Clinical specimens included individual and pooled serum samples. Samples were collected from dairy herds located throughout the Commonwealth of Pennsylvania, USA. Blood was allowed to clot and serum was removed and stored at −70°C. Viral RNA was extracted using QIAamp viral RNA purification kits (Qiagen, #52904). Extracted RNA was stored at −70°C until used.
2.2. Primers and probes
Primers and probes were designed based on sequence data of genotype I NADL strain (Collet et al., 1988) and genotype II (Ridpath and Bolin, 1995) BVD virus using Primer express-1.0 PE Biosystems (Foster City, CA). The primer set used for RT–PCR was from a previously reported study (Weinstock et al., 2000). Primers for TaqMan were common for BVD virus genotypes I and II. The reverse primer has 100% homology for type I BVD with one mismatch for type II. The probes for TaqMan were labeled with a fluorescent reporter dye at 5′ end and a quencher dye at 3′ end. The type I probe contained a fluorescent reporter (6-carboxyfluorescein) (FAM) and a fluorescent quencher (6-carboxytetramethylrhodamine) (TAMRA). The type II probe contained a fluorescent reporter (VIC) and TAMRA. Sequences of primers and probes were given Table 1 .
Table 1.
Sequences of the probes and primers and their genome position (5′–3′)
| Probes for TaqMan–PCR |
| 145–171 (FAM-AACAGTGGTGAGTTCGTTGGATGGCTT-TAMRA) (type I) |
| 147–172 (VIC-TAGCAGTGAGTCCATTGGATGGCCGA-TAMRA) (type II) |
| Common primer set for TaqMan–PCR |
| 103–123 (TAGCCATGCCCTTAGTAGGAC) |
| 176–196 (GACGACTACCCTGTACTCAGG) |
| Primers used for quantitation |
| 57–77 (TTAGGCCTAGGGAACAAA) (type I) |
| 421–441 (CCGACGGGTTTTTGTTTGTA) (type I) |
| 87–97 (AAGGCCGAAAAGAGGCTAGC) (type II) |
| 372–393 (ACTCCATGTGCCATGTACAGC) (type II) |
| T7–103, first 28 bp on 5′ end are T-polymerase site followed by forward primer sequence (GGATCCTAATACGACTCACTATAGGGAGTAGCCATGCCCTTAGTAGGAC) |
2.3. RT–PCR and TaqMan/RT–PCR methods
The RT–PCR was performed in single tube using a single enzyme, recombinant Thermus thermophilus (rTth) DNA polymerase according to Weinstock et al., 2000. TaqMan/RT–PCR assays were carried out in a 96-well flat-bottomed microtiter plate (Perkin-Elmer). The reaction mixture for each one-tube-TaqMan reaction mix consisted of 5 μl of TaqMan universal 10× buffer, 8 μl of 25 mM MgCl2, 1 μl of 10 μM forward primer, 1 μl of 10 μM reverse primer, 5 μl of 1 μM fluorogenic FAM labeled BVDV type I probe, 5 μl of 1 μM fluorogenic VIC labeled BVDV type II probe, 1.5 μl of each dNTP (A,C,G,U), 13 μl of DEPC water, 0.25 μl of AmpliTaq Gold DNA polymerase (5 U/μl), 0.25 μl MultiScribe reverse transcriptase (100 U/μl), 1.0 μl of RNAse inhibitor (20 U/μl), and 2.5 μl of BVD viral RNA sample to a total volume of 50 μl. Thermocycling conditions were as follows: 30 mm at 48°C for reverse transcription; 10 min at 95°C to activate DNA polymerase and to deactivate RT; 45 cycles of 15 s at 95°C to denature and 1 min at 60°C for annealing and extension.
The two-tube TaqMan reaction involved RT and PCR steps in separate tubes with incorporation of fluorogenic probes in the PCR process. The RT mix consisted of 0.5 μl RNAse Inhibitor (40 U/μl), 2.0 μl of 10× TaqMan Universal master mix buffer, 3.6 μl MgCl2 (25 mM), 1.0 μl of reverse primer (10 μM), 1.0 μl each dNTPS A,C,G,U (10 mM), 5.0 μl DEPC water 0.44 μl of moloney murine leukemia virus (MMLV) reverse transcriptase (50 U/μl). The BVD viral RNA sample was heated at 65°C for 5 min and 2.5 μl (10–100 ng/μl) was added to mix. Thermocycler conditions for RT were 1 h at 42°C for RT and 2 min at 72°C for denaturing unused RNA sample. The PCR mix was made up to a volume of 50 μl, containing 5.0 μl of 10× TaqMan Universal master mix buffer, 80 μl MgCl2 (25 mM), 1.0 μl of forward primer (10 μM), 1.0 μl of reverse primer (10 μM), 5 μl of 1 μM fluorogenic FAM labeled BVDV type I probe, 5 μl of 1 μM fluorogenic VIC labeled BVDV type II probe, 1.0 μl of each dNTP, 0.25 μl of AmpliTaq Gold DNA polymerase (5 U/μl), 12.75 μl of DEPC water and 8 μl of cDNA. After 2 min incubation at 50°C, 10 min incubation at 95°C, the cDNA was amplified by 40 two-step cycles: 15 s at 95°C and 1 min at 60°C.
2.4. Post-PCR analysis
Amplification products from RT–PCR were visualized on 2.5% agarose gels stained with ethidium bromide. In TaqMan–PCR, ABI Prism 7700 sequence detector measured fluorescent signal generated by the sequence-specific probes. The plate was scanned at 518 nm (FAM), 552 nm (VIC) and 582 nm (TAMRA). ΔR n is the fluorescence signal increase due to template amplification. The amplification plots were generated with ΔR n mean value on the y-axis and cycle number on the x-axis (Gibson et al., 1996). The threshold cycle (C T) is the cycle number at which the reporter fluorescence generated by cleavage of the probe passed a fixed threshold above baseline. Standard deviation 10 above base line was used to determine the fixed threshold. For analysis of C T values, less than 15 cycles threshold need to be adjusted manually.
2.5. Sensitivity
Different RT–PCR methods were tested for detecting BVDV and sensitivities were compared. RT–PCR assay, one-tube TaqMan and two-tube TaqMan/RT–PCR assays were evaluated using tissue culture supernatants of BVDV type I (NADL) and type II (125) strains. Ten-fold serial dilutions of virus strains were prepared in DEPC water and RNA extracted from each dilution. In addition, serum of known virus titer from a cow persistently infected with genotype I virus was serially diluted in pooled BVD negative sera. RNA was extracted from each serial dilution and stored −70°C until evaluation.
2.6. Synthesis and quantitation of RNA standard
Quantitation of BVD viral RNA in samples containing unknown quantities depends on the accuracy with which the BVD viral RNA standard was measured. The RNA standard representing the partial 5′ UTR of BVD virus was synthesized in vitro. Viral RNA isolated from NADL strain of BVD virus was reverse transcribed and amplified as described in Section 2.3. The following primer set was used for the above reaction; forward 57 and reverse 421 designed from 5′ UTR of NADL strain (Collet et al., 1988). The cDNA obtained was amplified in a second round of PCR with the primer set T7-103 and 421. The forward primer T7-103 consists of a 27 bp T-7 promoter sequence (Vanden Heuvel et al., 1993) with 21 bp BVD specific sequence of primer 103. Sequences of the above primer sets were given in Table 1. Amplicons were separated on 2% agarose gel and purified using gel extraction kit (Qiagen, #28704). Pure cDNA was transcribed to cRNA with T-7 RNA polymerase (Ambion, #1356). Ethanol precipitation and DNase treatment I purified the transcript to remove unused cDNA. Similar experiment was repeated with Type II-125 strain of BVD virus genotype II. Primers 87 and 372 were designed from type II sequence (Ridpath and Bolin, 1995) for first round amplification followed by second round PCR with T-7-103 and primer 372. The cDNA obtained was transcribed to cRNA as explained above. The RNA transcripts were dissolved in DEPC treated water and quantified by spectrophotometer. The cRNA was stored at −70°C. TaqMan–PCR was performed with 10-fold serial dilutions (from 108 copies to 1 copy of RNA per reaction) as described in the above section.
3. Results
3.1. Specificity
BVD virus reference strains, field isolates and clinical diagnostic samples were tested using TaqMan–PCR assay. The tissue culture propagated reference strains including NADL, NewYork-1 Singer (type Ia), Oregon C24V (type Ib), Draper (type Ib), type-II 125, type II 890 and type II PA-1, tested positive by TaqMan–PCR. A total of 23 BVD positive field isolates obtained from New York and Wyoming were correctly identified by TaqMan–PCR. In previous the study, sera from 80 cattle herds were screened for BVD virus by RT–PCR and virus isolation methods. Twenty herds contained sera that were positive for BVD virus. RNA was extracted from pools of 30–100 sera. All pooled sera positive by RT–PCR were also positive by TaqMan–PCR. The optimized technique was applied on routine diagnostic serum samples from thirty five herds that were pooled in thirties. Pooled sera from four herds shown to be positive by TaqMan–PCR were tested with individual virus isolation to find positive animals in the pool. Results from TaqMan–PCR were correlated with individual virus isolation. TaqMan–PCR with BVD specific primers and probes was negative for bovine viruses BRSV, PI3, IBR, bovine rotavirus, bovine coronavirus, and BT-17.
BVD virus positive samples shown to be positive by TaqMan–PCR were simultaneously classified into genotypes I and II. Eighteen of the 20 herds were positive for type I and the other two herds were positive for type II BVD virus. All reference strains were correctly identified into genotypes. Some tissue culture propagated BVD field isolates, 5 samples of 23 obtained from Cornell University, New York, were positive for both types I and II. These five samples were submitted to NADC (Ames, IA) and shown to be genotype II BVD by nested PCR according to Ridpath and Bolin (1998).
3.2. Sensitivity
The sensitivity of detection of BVD virus strains by TaqMan–PCR assay and RT–PCR was compared. Viral RNA extracted from 10-fold dilutions of BVD virus strains was tested by one-tube and two-tube TaqMan–PCR and RT–PCR. Results were tabulated (Table 2 ). The sensitivity of detection was equal with RT–PCR and two-tube TaqMan–PCR but one-tube TaqMan–PCR was 10-fold more sensitive. Similar findings were observed with 10-fold serial dilutions of BVD virus positive serum from a persistently infected cow diluted in sera negative for BVD virus. One-tube TaqMan–PCR method was shown to be 10–100-fold more sensitive than the other two detection methods.
Table 2.
Comparative detection of BVDV infected samples by different tests
| BVDV strain | Sample dilutions |
|||||||||||||||
| Neat | 10−1 | 10−2 | 10−3 | 10−4 | 10−5 | 10−6 | 10−7 | |||||||||
| NADL | ||||||||||||||||
| RT–PCR | + | + | + | + | + | − | − | − | ||||||||
| Two-tube TaqMan/RT−PCR | + | + | + | + | + | − | − | − | ||||||||
| One-tube TaqMan/RT–PCR | + | + | + | + | + | Wa+ | − | − | ||||||||
| Type II-125 | ||||||||||||||||
| RT–PCR | + | + | + | + | + | − | − | − | ||||||||
| Two-tube TaqMan/RT–PCR | + | + | + | + | + | − | − | − | ||||||||
| One-tube TaqMan/RT–PCR | + | + | + | + | + | + | − | − | ||||||||
| pi BVDV + serum | ||||||||||||||||
| RT–PCR | + | − | − | − | − | NAb | − | − | ||||||||
| Two-tube TaqMan/RT–PCR | + | − | − | − | − | NA | − | − | ||||||||
| One-tube TaqMan/RT–PCR | + | + | + | − | − | NA | − | − | ||||||||
W: weak.
NA: not available.
A total of 20 BVD virus positive serum samples were identified by both microplate virus isolation and RT–PCR and stored at −70°C for more than 2 years. After several freeze-thaw cycles, evaluation by TaqMan–PCR, virus isolation and RT–PCR were performed. Seventeen of 20 samples were positive by RT–PCR and virus isolation, but all samples were positive by TaqMan–PCR.
3.3. Quantitation of BVD virus
TaqMan–PCR amplifications were performed with serial dilutions of cRNA transcript prepared from NADL strain and Type II-125 strain (108 copies to 1 copy of RNA per reaction mixture). Fig. 1 shows the amplification plot with number of cycles versus ΔR n values for NADL strain. A detectable fluorescence signal above the threshold occurred at 12 cycles for 108 copies cRNA transcript. The ABI Prism 7700 detection system software generated a standard curve by plotting the C T values against each standard dilution of known concentration. A linear standard curve was obtained from 101 to 108 copies per reaction mixture, resulted in C T values ranging from 12 to 37 cycles (Fig. 2 ). The detection limit of the TaqMan/RT–PCR for cRNA transcripts was 10 copies per reaction mixture for NADL and 100 copies per reaction mixture for type II-125 at ΔR n=0.6. Samples with ΔR n values of 0.5 or less were interpreted as negative.
Fig. 1.
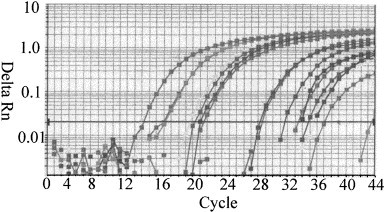
RT–PCR amplification plots of synthetic cRNA from NADL strain BVD virus. Amplification of known amount of cRNA starting from 108 copies to 1 copy per reaction. Cycle number was plotted vs. change in amplification fluorescence signal AR.
Fig. 2.
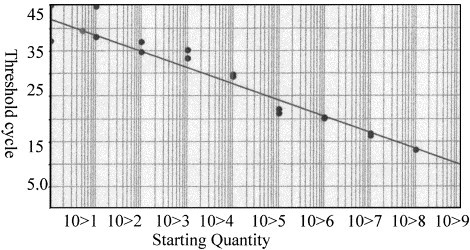
RT–PCR standard curve generated from cRNA amplification plots. Standard curve was plotted between starting copy number vs. threshold cycle (CT) (slope: 3.576, correlation coefficient: 0.946).
4. Discussion
RT–PCR has been reported to be a sensitive and specific technique for rapid detection of BVD virus (Hyndman et al., 1998, Laamanen et al., 1997, Renshaw et al., 2000). Quantitation of BVD virus by TaqMan–PCR described in this study has several advantages over conventional PCR. TaqMan–PCR is more sensitive and yields more information. Detection, classification and quantitation take place within one tube. The fluorogenic probes simultaneously classify genotypes I and II BVD virus time consuming post-PCR analysis such as gel electrophoresis or hybridization procedures were omitted since the amplification of a specific PCR product was measured during PCR cycling.
Tissue culture propagated viruses and validated field samples were used to prove the specificity of the TaqMan–PCR assay. The specific probes designed in this study correctly identified all reference samples and classified them into genotypes. Once the technique was optimized with validated reference strains and field isolates, the assay was applied to pooled positive serum samples and routine diagnostic samples. Sera collected from 80 herds across the Commonwealth of Pennsylvania were screened for BVD virus by RT–PCR and virus isolation. Pooled sera from 20 herds identified as positive for BVD virus by these two methods were also positive by TaqMan–PCR. Pooled sera from an additional 35 herds were screened for BVD virus by TaqMan–PCR followed by individual virus isolation only from positive pools.
Some of the tissue culture propagated field isolates tested in this study were identified as both types I and II virus. Sequence analysis of the amplified 5′ UTR segment identified nucleotide variation that was intermediate between published types I and II sequences, suggesting there was hybridization of both types I and II probes. Further evaluation of these samples (NADC, IA) using nested PCR to a different segment of the 5′ UTR (Ridpath and Bolin, 1998), were shown to be BVD genotype II. Result of both types I and II with TaqMan–PCR warrants further evaluation of the sample.
The comparison of TaqMan–PCR with conventional RT–PCR proved to be useful in assessing the sensitivity of the newly developed method. Ten-fold dilutions of tissue culture supernatants in DEPC water and BVD positive serum from a persistently infected cow diluted in negative serum were used to define the sensitivity of the assays. One-tube TaqMan–PCR proved to be 10–100-fold more sensitive than two-tube TaqMan–PCR and RT–PCR. This was further proved using BVD positive diagnostic samples. After several freeze-thaw cycles, 3 of 20 BVD virus positive serum samples tested negative by virus isolation and RT–PCR, but were positive by TaqMan–PCR. False negatives were attributed to the degradation of the samples over time and the lower detection limits of RT–PCR and virus isolation.
Quantitation of BVD virus reported in this study was sensitive and reproducible. Since 5′ UTR is the highly conserved part of the genome, the assay allows absolute quantitation of BVD virus in clinical samples. Quantitation of standards demonstrates sensitivity and precision of the assay for distinguishing between positive and negative clinical samples. While quantitation is not required for a diagnostic test, but is useful for several applications.
The prevalence of BVD infected cows within herds was reported to be 1.2–2% (Houe, 1995). Screening of each individual cow by virus isolation or RT–PCR is expensive. Testing of pooled sera was proposed as a cost efficient approach for low prevalence (Munoz-Zanzi et al., 2000). Single tube single enzyme RT–PCR evaluation of up to 100 pooled sera was validated as a useful method (Weinstock et al., 2000). TaqMan–PCR is a more sensitive method to detect BVD virus in pooled serum samples. The positive pooled sera can be further tested in smaller pools followed by testing of individual serum by virus isolation.
TaqMan–PCR offers several advantages over the other two PCR approaches: quantitative competitive RT–PCR where an exogenous competitor for each target is used as a control (Gilliland et al., 1990), or semi-quantitative RT PCR where an endogenous housekeeping gene (for example, β-actin) is used as a control concurrently with the target gene (Kaufman et al., 1995). TaqMan–PCR method is performed in a closed tube and requires no post-PCR analyses. Therefore, contamination with amplicons can be avoided. But contamination may occur during RNA extractions and pre-PCR preparations. Several quality assurance guidelines were instituted to avoid false positives. These include separate locations for sample preparation, RNA extraction and amplification procedures. In addition, each PCR run included positive and negative controls.
In conclusion, the TaqMan–PCR described here for detection, classification and quantitation of BVD virus has been shown to be sensitive and specific. These features make it an excellent large-scale screening tool for individual or pooled serum samples. Rapid turnaround time, reproducibility and ease of use make this technique a valuable diagnostic tool for detection of BVD virus.
Acknowledgements
We thank Dr. Debby Grove, Nucleic Acid Facility at Penn State University, for performing the TaqMan–PCR reactions. We appreciate Dr. E.J. Dubovi, Cornell University, Dr. H. Van Campen, University of Wyoming for providing field isolates of BVDV. This study was supported by the Pennsylvania Department of Agriculture, PA, USA.
References
- Canal C.W, Hotzel I, de Almeida L.L, Michel Roehe P, Aoi M. Differentiation of classical swine fever virus from ruminant pestiviruses by reverse transcription and polymerase chain reaction. Vet. Microbiol. 1996;48:373–379. doi: 10.1016/0378-1135(95)00156-5. [DOI] [PubMed] [Google Scholar]
- Collet M.S, Larson R, Gold C, Strick D, Anderson D.K, Purchio A.F. Molecular cloning and nucleotide sequence of the pestivirus bovine viral diarrhea virus. Virology. 1988;165:191–199. doi: 10.1016/0042-6822(88)90672-1. [DOI] [PubMed] [Google Scholar]
- Cortese, V.S., 1999. Food animal practice. Curr. Vet. Ther. 286–290.
- Drew T.W, Fenella Y, Pato D.J. The detection of bovine viral diarrhea virus in bulk milk samples by the use of a single-tube RT–PCR. Vet. Microbiol. 1999;64:145–154. doi: 10.1016/s0378-1135(98)00266-1. [DOI] [PubMed] [Google Scholar]
- El-Kholy A.A, Bolin S.R, Ridpath J.F, Arab R.M.H, Abou-Zeid A.A, Hammam H.M, Platt K.B. Use of polymerase chain reaction to simultaneously detect and type bovine viral diarrhea viruses isolated from clinical specimens. Rev. Sci. Tech. Off. Int. Epiz. 1998;17(3):733–742. doi: 10.20506/rst.17.3.1137. [DOI] [PubMed] [Google Scholar]
- Fulton R.W, Burge L.J. Bovine viral diarrhea virus types 1 and 2 antibody response in calves receiving modified live virus or inactivated vaccines. Vaccine. 2000;19(2/3):264–274. doi: 10.1016/s0264-410x(00)00168-7. [DOI] [PubMed] [Google Scholar]
- Gibson U.E.M, Heid C.A, Williams P.M. A novel method for real time quantitative RT–PCR. Genome Res. 1996;6:995–1001. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- Gilbert S.A, Burton K.M, Prins S.E, Deregt D. Typing of bovine viral diarrhea viruses directly from blood of persistently infected cattle by multiplex PCR. J. Clin. Microbiol. 1999;37(6):2020–2023. doi: 10.1128/jcm.37.6.2020-2023.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland, G.S., Perrin, K., Bunn, H.F., 1990. Competitive PCR for quantitation of mRNA. In: Innis, et al. (Eds.), PCR Protocols: A Guide to Methods and Applications. Academic Press, San Diego, p. 60.
- Heid C.A, Stevens J, Livak K.J, Williams M.K. Real Time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Holland P.M, Abramson R.D, Watson R, Gelfand D.H. Detection of specific polymerase chain reaction product by utilizing the 5′–3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 1991;88:7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houe H. Veterinary clinic in North America. Food Anim. Pract. 1995;11:521–548. doi: 10.1016/s0749-0720(15)30465-5. [DOI] [PubMed] [Google Scholar]
- Hyndman L, Vilcek S, Conner J, Nettleton P.F. A novel nested reverse transcription PCR detects bovine viral diarrhea virus in fluids from aborted bovine fetuses. J. Virol. Methods. 1998;71:69–76. doi: 10.1016/s0166-0934(97)00206-1. [DOI] [PubMed] [Google Scholar]
- Kaufman, P.B., Wu, W., Kim, D., Cseke, L.J., 1995. Analysis of gene expression by semiquantitative PCR. In: Molecular and Cellular Methods in Biology and Medicine. CRC Press, Boca Raton, FL, p. 245.
- Laamanen U.I, Neuvonen E.P, Yliviuhkola E.M, Veijalainen P.M.L. Comparison of RT–PCR assay and virus isolation in cell cultures for the detection of BVDV in field samples. Res. Vet. Sci. 1997;63:199–203. doi: 10.1016/s0034-5288(97)90020-5. [DOI] [PubMed] [Google Scholar]
- Letellier C, Kerkhofs P, Wellemans G, Vanopdenbosch E. Detection and genotyping of bovine viral diarrhea virus by reverse transcription–polymerase chain amplification of the 5′ untranslated region. Vet. Microbiol. 1999;64:155–167. doi: 10.1016/S0378-1135(98)00267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J, Flood S.J.A, Marmaro J, Giusti W, Deetz K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl. 1995;4:357–362. doi: 10.1101/gr.4.6.357. [DOI] [PubMed] [Google Scholar]
- Munoz-Zanzi C.A, Johnson W.O, Thurmond M.C, Hietala S.K. Pooled-sample testing as a herd-screening tool for detection of bovine viral diarrhea virus persistently infected cattle. J. Vet. Diagn. Invest. 2000;12:195–203. doi: 10.1177/104063870001200301. [DOI] [PubMed] [Google Scholar]
- Radwan G.S, Brock K.V, Hogan J.S, Smith K.L. Development of a PCR amplification assay as a screening test using bulk milk samples for identifying dairy herds infected with bovine viral diarrhea virus. Vet. Microbiol. 1995;44:77–92. doi: 10.1016/0378-1135(94)00121-c. [DOI] [PubMed] [Google Scholar]
- Renshaw R.W, Ray R, Dobovi E.J. Comparison of virus isolation and RT–PCR assay for detection of bovine viral diarrhea virus in bulk milk tank samples. J. Vet. Diagn. Invest. 2000;12:184–186. doi: 10.1177/104063870001200219. [DOI] [PubMed] [Google Scholar]
- Ridpath J.F, Bolin S.R. The genomic sequence of a virulent bovine viral diarrhea virus from the type II genotype: detection of a large genomic insertion in a noncytopathic BVDV. Virology. 1995;212:39–46. doi: 10.1006/viro.1995.1451. [DOI] [PubMed] [Google Scholar]
- Ridpath J.F, Bolin S.R. Differentiation of types Ia, Ib and II bovine viral diarrhea virus (BVDV) by PCR. Mol. Cell. Probes. 1998;12:101–106. doi: 10.1006/mcpr.1998.0158. [DOI] [PubMed] [Google Scholar]
- Ridpath J.F, Bolin S.R, Dubovi L.J. Segregation of bovine viral diarrhea virus into genotypes. J. Virol. 1994;205:66–74. doi: 10.1006/viro.1994.1620. [DOI] [PubMed] [Google Scholar]
- Vanden Heuvel J.P, Tyson F.L, Bell D.A. Constructions of recombinant RNA templates for use as internal standards in quantitative RT–PCR. Biotechniques. 1993;14:395–398. [PubMed] [Google Scholar]
- Weinstock D, Castro A.E, Bhudevi B. Single tube single enzyme RT–PCR assay for detection of bovine viral diarrhea virus in pooled bovine serum. J. Clin. Microbiol. 2000;39(1):343–346. doi: 10.1128/JCM.39.1.343-346.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengler, G., Bradley, D.W., Collett, M.S., Heinz, F.X., Schlesinger, R.W., Strauss, J.H., 1995. Family flaviviridae. In: Murphy, F.A., Fauquet, C.M., Bishop, C.M., et al. (Eds.), Viral Taxonomy. Springer, New York, pp. 27–45.