

Abstract
A reverse transcriptase-polymerase chain reaction (RT-PCR) assay for the detection of the feline coronavirus (FCoV) genome and a co-cultivation method for the isolation of field strains of FCoV are described. Using the RT-PCR assay to assess blood samples from cats with feline infectious peritonitis (FIP) (n=47) and healthy cats from households with endemic FCoV (n=69) it was shown that approximately 80% of the cats were viraemic, irrespective of their health status. It was also shown that, over a 12-month period, a similar percentage of healthy cats remained viraemic, and that the presence of viraemia did not appear to predispose the cats to the development of FIP. The co-cultivation system proved to be a suitable method for the culture of field strains of FCoV from blood samples, so long as the cultures were maintained for at least 4 weeks. Using this system, followed by the RT-PCR, viraemia was detected as frequently as by RT-PCR on RNA extracted directly from peripheral blood mononuclear cells.
Keywords: Feline coronavirus, Cat, Polymerase chain reaction, Cell culture, Co-cultivation
1. Introduction
FIP is a usually fatal, immune-mediated, disease triggered by infection with FCoV, which can affect both domestic and wild felines (Pedersen, 1987; Barlough and Scott, 1988). The elucidation of FCoV epidemiology is essential to our understanding of FIP, but to study the FCoV infection it is necessary to be able to detect the virus. Unfortunately, most attempts to culture FCoV from clinical cases of FIP or from cats believed to have been exposed to FCoV have failed (Pedersen, 1987; Pedersen et al., 1984; Hohdatsu et al., 1992). While, in a number of studies, methods of demonstrating the virus in cats with clinical FIP have been found (Pedersen, 1987; Hok, 1989a, Hok, 1989b; Cammarata Parodi et al., 1993; Martinez and Weiss, 1993; Tammer et al., 1995), they are generally of little use as reliable diagnostic tools, and at the start of the study, there were no commercially available tests for the detection of the virus within chronically-infected, but clinically healthy cats. Since persistently infected cats may harbour FCoV in their intestines, blood or possibly tonsils, and shed the virus in faeces or saliva, these cats may act as a continued source of infection (Pedersen, 1987; Addie et al., 1996; Foley et al., 1997). Being able to detect these cats is essential, not only for our understanding of their role in the dissemination of the FCoV infection, but also in determining what role chronic viraemia may play in the induction of clinical FIP.
While it was initially hypothesised that FIP-causing coronaviruses (FIPVs) differed from non-pathogenic enteric coronaviruses (FECVs) by their ability to replicate within macrophages, leave the gut and enter the bloodstream (Pedersen et al., 1981), it is now known that FCoV can be detected, using RT-PCR techniques, in the blood of up to 95% of healthy cats from households with endemic FCoV (Egberink et al., 1995; Herrewegh et al., 1995, Herrewegh et al., 1997; Richter et al., 1996). This current study involved developing a successful method for the culture of FCoV, and an RT-PCR assay for the detection of the FCoV genome. It was then possible to determine (i) whether cats remained viraemic over time, (ii) whether or not the presence of viraemia led ultimately to the development of FIP, and (iii) whether this type of methodology could be used in the diagnosis of FIP.
2. Materials and methods
2.1. Cat details
Four groups of cats were used in this study:
Group 1 (n=20) contained feline patients presented to The Feline Centre, University of Bristol Veterinary School, in which FIP was subsequently confirmed by histopathological examination of post mortem material. They represented eight different breeds, and were from 12 weeks to 8 years of age (average age of 8 months). They showed no sex predisposition. All were seropositive for FCoV.
Group 2 (n=14) contained clinically healthy, 1 year-old specific- pathogen free (SPF) cats, which came from a commercial supplier. The cats were naturally infected with an undefined, generally non-pathogenic FCoV. Seven of the cats were assessed on three separate occasions over a 6 month period; one cat was assessed only twice. All 14 cats were seropositive for FCoV.
Group 3 (n=27) contained 10 juvenile, FCoV antibody-positive, domestic short haired (DSH) cats from mixed backgrounds. The cats were group-housed with 17 FCoV antibody-negative, juvenile SPF cats. All 27 cats succumbed to FIP. The 23 cats still alive 10 weeks after the first sampling were re-sampled prior to euthanasia.
Group 4 (n=55) contained clinically healthy pet cats from nine separate pedigree breeding catteries. They represented nine different breeds and were from 8 weeks to 14 years of age (average age 3.5 years); 23 were male, 32 were female. Fifty-three of the 55 cats were seropositive for FCoV (the range of titres by indirect immunofluorescence was 80 to ≥5120, with a median of 1280). Within the 6 months preceding the start of the study, at least one case of FIP had been confirmed within each of the households. Thirty-five of the cats were re-assessed up to 16 months after sampling. Thirty-five of the cats were followed over time; three for 4 months, 13 for 9 months, 14 for 12 months and five for 16 months. Four of the cats in the latter group were assessed 4 months after the initial sampling, as well as after 16 months. The cultures of the samples collected in the first 4 months of the study were assessed within 2 months of being frozen at −70°C in RNA extraction solution (TRI REAGENT). Unfortunately, the later samples were held in storage for more protracted periods, resulting in degradation of the RNA in many of the samples. Despite numerous attempts at salvage, most of the samples were not recoverable.
For each sample, 2–5 ml of blood was collected from each cat by cephalic or jugular venepuncture into ethylenediaminetetraacetic acid (EDTA) tubes.
2.2. Cultivation of FCoV
Blood samples were first centrifuged at 2000×g for 10 min, and the plasma was stored prior to assessment of anti-FCoV antibody by indirect immunofluorescence (Stoddart et al., 1988). The buffy coat cells were then diluted to a final volume of 10 ml with RPMI (Dutch modification) cell culture medium, supplemented with 5% (v/v) foetal bovine serum (Sigma), 0.1 μg ml−1 l-glutamine (Sigma), 50 μg ml−1 gentamycin, and Concanavalin A (Con A)(Sigma) and lipopolysaccharide (LPS) from E. coli serotype 0127:B8 (Sigma), both at 10 μg ml−1 of medium. The cells were cultured overnight at 37°C, 5% CO2 in 25 cm2 flasks (Corning, UK). Non-adherent cells were rinsed off with multiple washes of warm sterile phosphate buffered saline (sPBS), and the adherent cell sheet was seeded with 5 ml of whole feline embryo (WFE) cells, re-suspended at ∼5×104 cells ml−1 in Eagle's Minimum Essential Medium (MEM) (Sigma), supplemented with 10% (v/v) foetal bovine serum, 0.23% (w/v) sodium bicarbonate, 0.1 μg ml−1 L-glutamine, 1% (v/v) non-essential amino acids (GibcoBRL), 0.05% (v/v) lactalbumin hydrolysate (GibcoBRL) and 50 μg ml−1 gentamycin. Cultures were maintained for 4 weeks, being transferred to 75 cm2 flasks (Corning, UK) after 4 days and the cells being subcultured approximately one-in-three every 7 days. At each subculture two-thirds of the cells were pelleted by centrifugation (2000×g for 5 minutes), rinsed with sPBS, lysed in 1 ml of RNA extraction solution (TRI REAGENT [Sigma]) and frozen at −70°C pending further processing.
A number of cell culture-adapted serotype 1 FCoV strains were a kind gift from Dr. N.C. Pedersen, including FIPV TN406, FIPV UCD-1, FIPV UCD-3 and FIPV UCD-4, while serotype 2 FCoVs consisted of FIPV 79-1146, FECV 79-1683, FIPV Wellcome and FIPV Primucell™. For each of the viruses, a culture of confluent WFE cells was inoculated with virus at ∼0.5 multiplicity of infection (MOI) and incubated for ∼48 h. The cells were then frozen at −70°C, pending further processing. The cell culture-adapted virus strains generated identifiable cytopathic effect (CPE) in WFE cell cultures, but significant changes were rarely seen with the field strains during the 4 week culture period.
2.3. RNA extraction and first strand synthesis of cDNA
RNA was extracted using 1 ml of TRI REAGENT (Sigma) per ∼5×106 culture cells or ∼200 μl of whole anticoagulated blood, buffy coat, plasma or serum. Cells were lysed by repeated pipetting until the lysate changed from a viscous to a watery consistency, then each lysate was vortexed for 30 s prior to adding 200 μl of HPLC grade chloroform (Sigma). Each sample was vortexed for a further 30 s, then centrifuged for 15 min at 13000×g, at 4°C (Hettich Zentrifugen EBA12R). The upper aqueous layer was transferred to a fresh 1.5 ml microcentrifuge tube and an equivalent volume of cold isopropanol (Sigma) was added. The sample was vortexed, then chilled at −20°C for at least 2 hours. After cold chilling, the sample was centrifuged for 15 min at 13000×g, 4°C. The supernatant was discarded and the RNA pellet was washed by adding 1 ml of 75% ethanol (molecular grade Hayman), vortexed, then centrifuged for 10 minutes at 8000×g, 4°C. The supernatant was again discarded and the RNA was dried briefly in air, prior to being re-suspended in diethylpyrocarbonate (DEPC)-treated water (∼20 μl per 5×106 cell equivalents).
The amount of total RNA extracted varied from 5–30 μg per 5×106 cells for co-cultured WFE cells (usually 5–15 μg per 5×106 cells), and 1–50 μg per 5×106 cells when the flasks were infected with cell culture-adapted FCoV.
DNAse treatment was performed by heating each aliquot of RNA solution to 94°C for 5 minutes, then chilling it quickly in ice. The reaction mixture was then adjusted to contain 0.1 μg μl−1 total RNA, 20 mM Tris–HCl (pH 8.3), 50 mM KCl, 2.5 mM MgCl2, 0.1 U μl−1 DNAse I (molecular biology grade, Gibco BRL) and DEPC-treated water to a final volume of 50 μl. The mixture was incubated for 15 min at room temperature, then the reaction was terminated by adding 2.5 mM EDTA and incubating at 65°C for 15 min.
First strand synthesis of cDNA was performed using a commercially available kit (SuperScript Preamplification System for First strand cDNA synthesis [Gibco BRL]), according to the manufacturer's recommendations, using oligodeoxythymidine triphosphate (dT)12-18 primers, the only adaptation being raising the incubation temperature from 42 to 45°C.
2.4. Reverse transcriptase-polymerase chain reaction (RT-PCR)
The primers and probe were designed to the highly conserved C-terminus of the FCoV-S gene (De Groot et al., 1987). Potential sites for PCR primers were identified and consensus sequences aligned using computer assistance (MacVector™ 5.0, Kodak). The oligonucleotides were made by Genosys. Their sequences are shown in Table 1 .
Table 1.
Details of FCoV primers and probe
| Oligos | Size (nt) | TTm (°C) | G+C% | Priming site | Prod. size (nt) | Sequence 5′→3′ |
| FCoV | ||||||
| No.66 (F) | 18 | 50 | 39 | 2720→2737 | CTG CAT GTC AAA CTA TTG | |
| No.67 (R) | 17 | 52 | 53 | 3521←3504 | CTT GTG CAT CAG CAC TC | |
| No.30 (R) | 16 | 48 | 50 | 2923←2981 | TGC TAT TAT GGG ACG G | |
| No.66 (F) + No.67 (R) | 800 | |||||
| No.66 (F) + No.30 (R) | 217 | |||||
| Probe | ||||||
| FCoV-S | 18 | 66 | 50 | 2816→2834 | ATT GGC ATC TGT TGA GGC | |
nt = nucleotides.
TTm = theoretical annealing temperature based on 100% identity.
F = forward primer; R = reverse primer.
The components for the first round of the PCR were combined together into a master mix adjusted to give reaction conditions: 20 mM Tris–HCl (pH 8.4), 50 mM KCl, 2.0 mM MgCl2, 200 μM dNTP mix, 0.1 U μl−1 Taq DNA polymerase (AmpliTaq Gold™ Perkin Elmer), 2 μl (5% (v/v)) of the cDNA solution and 0.3 μM FCoV primers (Nos. 66 and 67). DEPC-treated water was added to make a final volume of 50 μl and the PCR was performed in a temperature cycler with a heated lid (Touchdown–Hybaid). The PCR protocol consisted of an initial incubation of 94°C (10 min), followed by 35 cycles of melting 94°C (1 min), annealing 50°C (1 min) and extension 72°C (2 min), then a final extension of 72°C (10 min). The second round of PCR was performed, where necessary, as per the first round, using the semi-nested primers Nos.66 and 30 and 2 μl of the first round product.
Controls were incorporated with each set of PCRs. Positive controls included amplifying cDNA generated from kit-supplied control RNA using kit-supplied PCR primers at 0.1 μM, and using experimental cDNA samples known to be strong positives for FCoV. Negative controls included using cDNA prepared from known negative cell cultures or cats at the same time as the experimental material was processed, plus no RT, no primer and no cDNA controls.
2.5. Detection of RT-PCR products
PCR products were detected on agarose gels by ethidium bromide staining, then Southern blotted to nylon membrane (Hybond™-N, Amersham Life Science), and hybridised probe was detected using the Enhanced Chemiluminescence detection system (ECL™, Amersham Life Science) as per the manufacturer's recommendations.
3. Results
3.1. Optimisation of the PCR
The ability to detect FCoV was assessed with a number of cell culture-adapted strains of FCoV. Uninfected cell cultures were included as negative controls. Serotype 2 FCoVs (FIPV 79-1146, FIPV-Wellcome, FIPV Primucell and FECV 79-1683) were detected after 35 cycles of PCR. However, the serotype 1 FCoVs; FIPV UCD-1, FIPV UCD-3, FIPV UCD-4 or FIPV TN406 were not detected after 35 PCR cycles, but FIPV UCD-4 and FIPV TN406 were detected after a further 35 cycles (results not shown). No product was detected in uninfected cultures even after two rounds of 35 cycles.
The PCR methodology was optimised to ensure that, as far as possible, the products generated produced single bands on 2% agarose gels, ran at the expected molecular weight and probed specifically for FCoV (Fig. 1 ). A second band was seen in a number of samples amplified. This product was of a higher molecular weight than expected when primers Nos.66 and 30 were used, and of a lower than expected molecular weight when primers Nos. 66 and 67 were used (Lanes 1, 2, 4 and 6). Occasionally, as shown in Lane 1, the extra band probed positive for FCoV. For some samples, (Lanes 23 and 28), the band running at the expected molecular weight failed to hybridise with the FCoV probe.
Fig. 1.
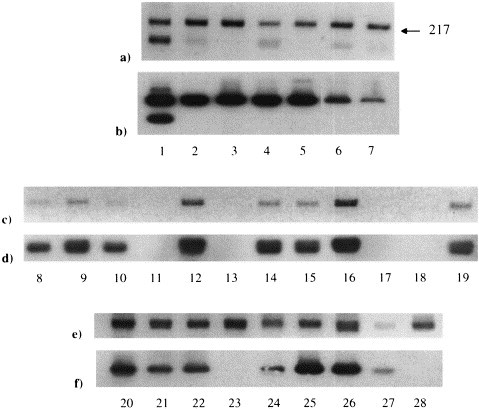
FCoV RT-PCR products run on ethidium bromide-stained agarose gels and autoradiographed. (a), (c) and (e) show the FCoV RT-PCR products run on ethidium bromide-stained agarose gels. Primers FCoV Nos. 66 and 67. (b), (d) and (f) show the autoradiographs of the gels seen in (a), (c) and (e), respectively, after Southern blotting to nylon membrane and hybridisation with the probe. Lanes 1–28 contain samples from 28 separate healthy cats (Group 4).
3.2. Detection of FCoV in blood of cats with FIP
Blood samples from 33 cats with clinical FIP were assessed, without prior co-cultivation, for the presence of FCoV by RT-PCR and virus-specific probe. Twenty-three of the cats were from Group 3, the other 10 were from Group 1. For the Group 3 cats, only whole EDTA anticoagulated blood was assessed. For the Group 1 cats, where possible, whole anticoagulated blood (EDTA or heparin), separated plasma, and serum, were each evaluated for the presence of FCoV.
Twenty of the 23 Group 3 cats were found to have FCoV in their blood. All 10 Group 1 cats were found to have FCoV in at least one of their blood fractions. Marginally better virus detection was achieved using EDTA anticoagulation than with heparin anticoagulation, and the intensity of the product was usually greater (Table 2 ).
Table 2.
Detection of FCoV, by RT-PCR and virus-specific probe, in blood samples from cats with clinical FIP
| Group 3 | Group 1 cats |
|||||
| Blood fraction | Whole blood (EDTA) | Whole blood (EDTA) | Whole blood (heparin) | Plasma (EDTA) | Plasma (heparin) | Serum |
| FCoV + | 20 (87%) | 4 (80%) | 3 (75%) | 5 (83%) | 4 (67%) | 6 (75%) |
| FCoV − | 3 | 1 | 1 | 1 | 2 | 2 |
3.3. Detection of FCoV following co-cultivation in vitro
3.3.1. Detection of FCoV in clinical FIP cases
Blood samples from 11 cats from Group 1 (only one of which was assessed without prior cultivation, see 3.2) were co-cultivated with WFE cells for 4 weeks prior to RT-PCR and hybridisation with virus-specific probe. Blood samples from 27 cats in Group 3 were co-cultivated with WFE cells for 2 weeks prior to RT-PCR. (Table 3 ). RT-PCR samples from two cats in Group 1 produced a band of the correct molecular weight that failed to hybridise with the virus-specific probe.
Table 3.
Blood samples from FIP cases tested for the presence of FCoV
| Duration of culture | Group 1 |
Group 3 | |
| 4 weeks | 2 weeks | ||
| By PCR | By Probe | By PCR | |
| FCoV + | 10 (91%) | 8 (73%) | 15 (56%) |
| FCoV − | 1 | 3 | 12 |
3.3.2. Detection of FCoV in clinically healthy, FCoV antibody-positive, SPF cats
Fourteen cats in Group 2 were sampled initially. After 2 weeks in culture, samples from six of the cats tested positive for FCoV, by PCR. After a further week in culture, only 3 of the 14 cultures tested positive for FCoV (Table 4 ). Interestingly, one of the cultures which tested positive at 3 weeks had been negative for FCoV by RT-PCR and probe a week previously; another had previously tested negative by RT-PCR, but positive by probe. Uninfected cell cultures remained negative throughout.
Table 4.
Blood samples from healthy SPF cats (Group 2) tested for the presence of FCoV over a 6 month period
| Duration of culture | Initial blood sample |
+2 mo | 6 mo later |
|||||
| 2 weeks | 3 weeks | 3 weeks | None | 2 weeks | 3 weeks | 4 weeks | ||
| By PCR | By probe | By PCR | By PCR | By PCR | By PCR | By PCR | By PCR | |
| FCoV + | 6 (43%) | 11 (79%) | 3 (21%) | 4 (57%) | 5 (63%) | 3 (60%) | 2 (40%) | 4 (80%) |
| FCoV − | 8 | 3 | 11 | 3 | 3 | 2 | 3 | 1 |
mo=months.
Two months after the initial blood samples, further samples were collected from seven of the 14 cats. After 3 weeks of culture, virus was detected by RT-PCR in four of the seven (57%) cultures (Table 4). After a further 4 months, samples were collected from the seven cats previously re-assessed, plus a further cat. When assessed by RT-PCR prior to culture, FCoV was detected in five of the eight samples. The number of samples to test positive fell over the first 3 weeks of culture, then rose again by the time the cultures had been established 4 weeks (Table 4).
3.3.3. Detection of FCoV in healthy cats from households with endemic FCoV
Fifty-five cats were assessed initially, all of which were clinically healthy (Table 5 ). Of the seven cats re-sampled 4 months after the initial sample, all had initially been positive for FCoV by RT-PCR and probe. Five of the seven cats remained positive. Of the samples collected later in the study, RNA was successfully extracted from only six. All six of these cats were initially positive for FCoV by RT-PCR and probe. Five of the six were still positive after 12 months (Table 5). During the study no new cases of FIP developed within the cats of any of the test households.
Table 5.
Blood samples from healthy cats from households with endemic FCoV, tested for the presence of FCoV over a 12 month period
| Duration of culture | Initial blood sample 1–4 weeks |
+4 months 1 week | +12 months 4 weeks | |
| By PCR | By Probe | By PCR + probe | By PCR | |
| FCoV + | 49 (89%) | 43 (78%) | 5 (71%) | 5 (83%) |
| FCoV − | 6 | 12 | 2 | 1 |
4. Discussion
Using the RT-PCR assay, the easily grown serotype 2 FCoVs, FIPV 79–1146, FECV 79–1683, FIPV Wellcome and FIPV Primucell, as well as the serotype 1 FCoVs, FIPV TN406 and UCD4 were detected in cell culture. Since the primers were designed against a highly conserved region they should bind most FCoV variants. The inability to detect UCD1 and UCD3 may have resulted from failure of these strains to grow in WFE cells, since they are known to grow poorly in cell culture (Pedersen et al., 1984). The high detection rate in samples from field cases suggests that most, if not all, FCoV strains can be detected with the RT-PCR assay.
By performing hybridisation with a virus-specific probe, a greater level of specificity was achieved, plus a further degree of amplification. The significance of the amplification can best be seen in Table 4, where samples from healthy SPF cats were assessed for the presence of FCoV. From the initial blood sample only six of the 14 samples tested positive by RT-PCR alone, but a further five cats were found to be infected when the products were transferred to membranes and probed.
After amplification, not all of the bands seen on ethidium bromide-stained gels hybridised with the virus-specific probe. Where some of the samples demonstrated multiple bands on ethidium bromide staining, few of these extra bands probed positively. Interestingly, some of the bands seen running at the desired molecular weight were not detected by probing. There could be two reasons for this lack of hybridisation; (i) that the bands were due to mis-priming and were not FCoV-specific, or (ii) that while the bands were due to FCoV, mutations within the S gene prevented the probe from binding. In support for the latter theory is the finding that at least some of the extra bands did hybridise with the probe, and that coronaviruses are known to have a high level of mutation (Chen and Baric, 1995).
In a number of studies, RT-PCR techniques have been used to detect FCoV in samples from clinical FIP cases (Li and Scott, 1994; Egberink et al., 1995; Herrewegh et al., 1995, Herrewegh et al., 1997; Fehr et al., 1996; Richter et al., 1996). While some of these studies have involved fresh or formalin-fixed tissue (Li and Scott, 1994), in others plasma, serum, ascitic or pleural fluids have been examined (Egberink et al., 1995; Herrewegh et al., 1995 Herrewegh et al., 1997; Fehr et al., 1996; Richter et al., 1996). We found virus in approximately 80% of blood samples from confirmed cases of FIP. This value is in agreement with the previous authors, who found 78–92% of cases to be virus-positive. Herrewegh et al. (1995)found plasma to be more sensitive than serum when analysing EDTA anti-coagulated blood for the presence of FCoV. In our study a similar trend was noted.
In this study, healthy cats living within households with endemic FCoV were also shown to be viraemic for FCoV. This finding is in agreement with those of Egberink et al. (1995), Herrewegh et al., 1995, Herrewegh et al., 1997and Richter et al. (1996). However, while Egberink et al. (1995)found only 2% of cats to be viraemic, Herrewegh et al., 1995, Herrewegh et al., 1997found between 29–86% of cats to be affected, and Richter et al. (1996)found 95% of cats to be positive. Our results, finding between 80–90% of the cats to be viraemic, are in best agreement with the latter studies, in which, as in our study, primers against the S region of the virus were used.
The presence of viraemia was successfully monitored for up to 12 months in 21 of the 63 healthy cats. We were able to show that viraemia persisted in five of seven cats for at least 4 months, in five of eight cats for at least 6 months and in five of six cats for at least 12 months. None of the persistently-infected cats developed signs of clinical disease within this period of observation. Given this, and the overall lack of new FIP cases within the catteries, it appears logical to surmise that in most cats the persistence of viraemia is not associated with the development of disease. Further study would be needed to confirm this.
Most of the cats in this study were both viraemic and seropositive for FCoV. While the presence of antibody generally correlated well with the presence of the virus, virus was not detected in all antibody-positive cats, and two of the viraemic cats lacked specific antibody. Seronegativity has been reported previously in cats with clinical FIP (Scott, 1986; Sparkes et al., 1991; Fehr et al., 1996) and healthy, but FCoV-infected individuals (Fehr et al., 1996; Richter et al., 1996; Herrewegh et al., 1997). Fehr et al. (1996), and Herrewegh et al. (1997)found a considerable proportion of healthy cats from mixed backgrounds to be seronegative for FCoV, despite being RT-PCR positive; eight of 18 cats (44%), and five of 16 cats (31%), respectively. Richter et al. (1996)found 10 out of 23 healthy but viraemic cats to be antibody-negative (44%), along with six of 28 cats with clinical FIP (21%). There are a number of possibilities for this apparent lack of serological response. In clinically ill cats, the lack of detectable antibody may be due to it being bound up in the form of immune complexes (Scott, 1986). However, since the seronegative cats in our study were not clinically ill, this appears to be an unlikely explanation. It is perhaps more likely that the lack of antibody relates to recent infection. This theory is supported by Richter et al. (1996), who found seven of the 10 viraemic, but seronegative cats, to be less than 12 weeks of age. At this age kittens are likely to have lost their maternally derived antibody, but may have only recently been exposed to the virus (Addie and Jarrett, 1992; Foley et al., 1997). The possibility of recent infection may also explain three of the five cases identified by Herrewegh et al. (1997), since all three were under 10 months of age. The remaining healthy, seronegative, but viraemic cats, in the studies by Richter et al. (1996)and Herrewegh et al. (1997), were of a wide range of ages. Interestingly, three of the cats had previously tested seropositive for FCoV. Of the two seronegative cats in our study, one was only 6 months of age, and not available for further study, while the other was 3 years of age, and remained seronegative during the following year of observation, but became seropositive 18 months later. From these findings, it would appear that while some viraemic cats may be intermittently seropositive, others may fail to produce detectable antibody. The significance of these findings to development of disease remains unclear.
From this study we can see that most cats living within large groups or pedigree breeding catteries are likely to be viraemic if they have been exposed to FCoV. Since there was no difference in the incidence of viraemia between healthy cats and those suffering from FIP, the RT-PCR assay cannot be considered a suitable test for the diagnosis of FIP. However, the test has much to recommend it for the detection of persistently-infected `carrier' cats, and in the screening of cats prior to their introduction to FCoV-free breeding colonies. It will also be an important asset in the ongoing study into FCoV epidemiology.
The co-cultivation system was shown to be a successful in vitro method for growing field strains of FCoV. A similar percentage of samples tested positive for FCoV by direct RT-PCR and after 4 weeks of co-cultivation. This can be seen in the clinical FIP samples, where approximately 80% were positive by direct RT-PCR and 91% were positive after co-cultivation and RT-PCR, (73% being positive after co-cultivation, RT-PCR and probe). Interestingly, when samples from individual cats were assessed, the culture system was less predictable. Presumably, the viruses had to adapt to the cell-line in which they had been placed, and different isolates had differing abilities to do this. It is possible that, during the initial period of adaptation, most of the input virus failed to replicate, resulting in a reduced number of positive samples. As the adapted virus then began to grow more strongly, it again became detectable. The adaptation period appeared to take at least 4 weeks. As of this, for most samples the `without culture' samples gave the best chance of virus detection, followed by those assessed after either 1 or 4 weeks of culture. As the virus was difficult to detect in samples that had been cultured for between 2–3 weeks, the prevalence of FCoV viraemia may have been underestimated in groups of cats that were assessed at these times. An example of this may be seen with the cats of Group 3 (Table 3). These cats were assessed after their samples had been cultured for only 2 weeks. Fifteen of 27 cats (56%) were found to be infected by RT-PCR, as compared to ten of the 11 cats (91%) in Group 1, which were assessed after 4 weeks of culture.
Perhaps surprisingly, a number of samples tested negative initially, but became positive after 4 weeks of culture. Controls against cross-contamination were stringent, so the most likely scenario is that the level of virus was undetectable in the initial samples, but co-cultivation allowed for its amplification. It may have been the case that, in these samples, the number of FCoV-infected white blood cells or the amount of intracellular virus was very small (Weiss and Scott, 1981). In vitro cultivation of the cells may have stimulated virus expression and/or replication, and so aided detection (Weiss and Scott, 1981). Studies in mice have been performed that show that antigen- or mitogen-stimulated lymphocytes acquire the capacity to replicate exogenous or endogenous viruses (Hirsch, 1976). Also, in vitro maintenance of PBMCs from infected animals can activate the expression or replication of certain viruses producing persistent infections (Stock and Ferrer, 1972; Rowe et al., 1997). Similarly, PBMCs from FCoV-infected cats may have acquired the capacity to express or replicate virus in vitro, to co-infect other cells, or to release soluble factors (e.g. cytokines) into the culture medium to activate virus within other cells (Weiss and Scott, 1981). The co-cultivation system has been shown to be a suitable method for the growth of field strain FCoVs, and particularly when combined with the RT-PCR assay will allow for the continued study of these viruses.
Acknowledgements
The authors would particularly like to thank the cat owners and referring veterinary surgeons without whose help this study could not have been completed. D.G-M. was supported by a University of Bristol research Scholarship.
References
- Addie D.D., Jarrett J.O. A study of naturally occurring feline coronavirus infection in kittens. Vet. Rec. 1992;130:133–137. doi: 10.1136/vr.130.7.133. [DOI] [PubMed] [Google Scholar]
- Addie D.D., Toth S., Herrewegh A.A.P.M., Jarrett J.O. Feline coronavirus in the intestinal contents of cats with feline infectious peritonitis. Vet. Rec. 1996;139:522–523. doi: 10.1136/vr.139.21.522. [DOI] [PubMed] [Google Scholar]
- Barlough, J.E., Scott, F.W., 1988, Feline infectious peritonitis. In: Barlough, J.E. (Ed.), Manual of Small Animal Infectious Diseases, Churchill Livingston, New York, pp 63–78
- Cammarata Parodi M., Cammarata G., Paltrinieri S., Lavazza A., Ape F. Using direct immunofluorescence to detect coronaviruses in peritoneal and pleural effusions. J. Small Ani. Prac. 1993;34:609–613. [Google Scholar]
- Chen W., Baric R.F. Function of a 5′ end genomic mutation that evolves during persistent mouse hepatitis infection in vitro. J. Virol. 1995;69(12):7529–7540. doi: 10.1128/jvi.69.12.7529-7540.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot R.J., Maduro J., Lenstra J.A., Horzinek M.C., Van Der Zeijst B.A., Spaan W.J. cDNA cloning and sequence analysis of the gene encoding the peplomer protein of feline infectious peritonitis virus. Journal of General Virolology. 1987;68:2639–2646. doi: 10.1099/0022-1317-68-10-2639. [DOI] [PubMed] [Google Scholar]
- Egberink, H.F., Herrewegh, A.A.P.M., Schuurman, N.M.P., Van Der Linde-Sipman, Horzinek, M.C., De Groot, R.J., 1995. FIP, Easy to Diagnose? Vet. Quart. 17(1), S24–S25 [PubMed]
- Feh, D., Bolla, S., Herrewegh, A.A.P.M., Horzinek, M.C., Lutz, H., 1996. Nachweis feliner Coronaviren mittels RT-PCR: Grundlage zum Studium der Pathogenese der Felinen Infektiosen Peritonitis (FIP), Schweizer Archiv fur Tierheilkunde (Zurich), 138, 74–79 [PubMed]
- Foley J.E., Poland A., Carlson J., Pedersen N.C. Patterns of feline coronavirus infection and fecal shedding from cats in multiple-cat environments. J. Amer. Vet. Med. Associ. 1997;210:1307–1312. [PubMed] [Google Scholar]
- Herrewegh A.A.P.M., De Groot R.J., Cepica A., Egberink H.F., Horzinek M.C., Rottier P.J.M. Detection of feline coronavirus RNA in feces, tissues, and body fluids of naturally infected cats by reverse transcriptase PCR. J. Clin. Microbiol. 1995;33(3):684–689. doi: 10.1128/jcm.33.3.684-689.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A.P.M., Mahler M., Hedrich H.J., Haagmans B.L., Egberink H.F., Horzinek M.C., Rottier P.J.M., De Groot R.J. Persistence and evolution of feline coronavirus in a closed cat-breeding colony. Virology. 1997;234:349–363. doi: 10.1006/viro.1997.8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, M.S., 1976, Immune activation of endogenous viruses, In: Crowell, R.L., Friedman, H., Prier, J.E. (Eds.), Tumour Virus Infections and Immunity. Baltimore, University Park Press, pp 175–186
- Hohdatsu T., Okada S., Ishizuka Y., Yamada H., Koyama H. The prevalence of types I and II feline coronavirus infections in cats. J. Vet. Medi. Sci. 1992;54:557–562. doi: 10.1292/jvms.54.557. [DOI] [PubMed] [Google Scholar]
- Hok, K., 1989a, Demonstration of feline coronavirus (FCV) antigen in organs of cats suspected of feline infectious peritonitis (FIP) disease. APMIS 98, 659–664 [DOI] [PubMed]
- Hok, K., 1989b, Demonstration of feline infectious peritonitis virus in conjunctival epithelial cells from cats. APMIS 87, 820–824 [DOI] [PubMed]
- Li X., Scott F.W. Detection of feline coronaviruses in cell cultures and in fresh and fixed feline tissues using polymerase chain reaction. Vet. Microbiol. 1994;42:65–77. doi: 10.1016/0378-1135(94)90078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez M.L., Weiss R.C. Detection of feline infectious peritonitis virus in cell culture and peripheral blood mononuclear leukocytes of experimentally infected cats using a biotinylated cDNA probe. Vet. Microbiol. 1993;34:259–271. doi: 10.1016/0378-1135(93)90016-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen, N.C., 1987, Coronavirus diseases (coronavirus enteritis, feline infectious peritonitis), In: Holzworth, J. (Ed.), Diseases of the Cat. Medicine and Surgery Volume 1., Saunders, W.B., Philadelphia, pp 193–214
- Pedersen N.C., Boyle J.F., Floyd K., Fudge A., Barker J. An enteric coronavirus infection of cats and its relationship to feline infectious peritonitis. Ameri. J. Vet. Res. 1981;42:368–377. [PubMed] [Google Scholar]
- Pedersen N.C., Black J.W., Boyle J.F., Evermann J.F., McKeirnan A.J., Ott R.L. Pathogenic differences between various feline coronavirus isolates. Ad. in E. Med. Biol. 1984;173:365–380. doi: 10.1007/978-1-4615-9373-7_36. [DOI] [PubMed] [Google Scholar]
- Richter, M., Schinkinger, M.F., Mostl, K., 1996, Nachweis von Infektionen mit felinen Coronaviren Typ II im Blut von Katzen mittels Reverser Transcritase Polymerase-Kettenreaktion (RT-PCR), Wiener Tierarztliche Monatsschrift, 83, 263–268
- Rowe C.L., Baker S.C., Nathan M.J., Fleming J.O. Evolution of mouse hepatitis virus: Detection and characterisation of spike deletion variants during persistent infection. J. Virol. 1997;71(4):2959–2969. doi: 10.1128/jvi.71.4.2959-2969.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, F.W., 1986, Feline Infectious Peritonitis and other feline coronaviruses, In: Kirk, R.W., Saunders, W.B. (Eds), Current Veterinary Therapy IX., Philadelphia, pp 1059–1062
- Sparkes A.H., Gruffydd-Jones T.J., Harbour D.A. Feline infectious peritonitis: A review of clinico-pathological changes in 65 cases, and a critical assessment of their diagnostic value. Vet. Rec. 1991;129:209–212. doi: 10.1136/vr.129.10.209. [DOI] [PubMed] [Google Scholar]
- Stock N.D., Ferrer J.F. Replicating C type virus in phytohemagglutinin treated buffy coat cultures of bovine origin. Journal of the National Cancer Institute. 1972;48:985–996. [PubMed] [Google Scholar]
- Stoddart M.E., Gaskell R.M., Harbour D.A., Pearson G.R. The sites of early viral replication in feline infectious peritonitis. Vet. Microbiol. 1988;18:259–271. doi: 10.1016/0378-1135(88)90092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammer R., Evensen O., Lutz H., Reinacher M. Immunohistological demonstration of feline infectious peritonitis virus antigen in paraffin-embedded tissues using feline ascites or murine monoclonal antibodies. Vet. Immun. and Immunopathol. 1995;49:177–182. doi: 10.1016/0165-2427(95)05459-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R.C., Scott F.W. Pathogenesis of feline infectious peritonitis: nature and development of viraemia. American Journal of Veterinary Research. 1981;42:382–390. [PubMed] [Google Scholar]