

Abstract
Transmissible gastroenteritis virus (TGEV) has been reported to induce apoptosis in swine testis (ST) cells. However, the mechanisms underlying TGEV-induced apoptosis are still unclear. In this study we observed that TGEV infection induced apoptosis in porcine kidney (PK-15) cells in a time- and dose-dependent manner. TGEV infection up-regulated FasL, activated FasL-mediated apoptotic pathway, leading to activation of caspase-8 and cleavage of Bid. In addition, TGEV infection down-regulated Bcl-2, up-regulated Bax expression, promoted translocation of Bax to mitochondria, activated mitochondria-mediated apoptotic pathway, which in turn caused the release of cytochrome c and the activation of caspase-9. Both extrinsic and intrinsic pathways activated downstream effector caspase-3, followed by the cleavage of PARP, resulting in cell apoptosis. Moreover, TGEV infection did not induce significant DNA fragmentation in ammonium chloride (NH4Cl) pretreated PK-15 cells or cells infected with UV-inactivated TGEV. In turn, block of caspases activation also did not affect TGEV replication. Taken together, this study demonstrates that TGEV-induced apoptosis is dependent on viral replication in PK-15 cells and occurs through activation of FasL- and mitochondria-mediated apoptotic pathways.
Keywords: TGEV, Apoptosis, FasL, Mitochondria, Viral replication
1. Introduction
Viruses have evolved various sophisticated strategies to manipulate the host's cells, limit anti-virus response of the host, which may be advantageous for virus replication and persistent infection (Everett and McFadden, 1999). Apoptosis, a tightly controlled physiological process, plays crucial roles not only in the normal development and homeostasis of multicellular organisms (Arch and Thompson, 1999, Shih et al., 2004), but also in the pathogenesis of several viral infections (Everett and McFadden, 1999, Lee and Kleiboeker, 2007). A number of viruses have been reported to induce apoptosis in immune cells, targeted cells or infected cells (Eleouet et al., 1998, Ruggieri et al., 2007, Umeshappa et al., 2010).
Transmissible gastroenteritis virus (TGEV) is animal virus belonging to family coronaviridae (Annamaria, 2011). According to present virus taxonomy of ICTV (international committee of taxonomy viruses), the coronaviridae family comprises the subfamily coronavirinae and torovirinae. The subfamily coronavirinae is classified into genera alpha-, beta- and gammacoronavirus, instead of phylogroups 1 through 3 (Annamaria, 2011, Carstens, 2010). TGEV, together with human coronavirus 229E (HCoV-229E), human coronavirus NL63 (HCoV-NL63), porcine epidemic diarrhea virus (PEDV), canine coronaviruses (CCoVs), feline coronavirus (FCoV), belongs to alphacoronavirus (Annamaria, 2011, Carstens, 2010). In phylogenetic analyses of the genomic region downstream of the S gene, TGEV consistently clusters with extant CCoV-II field strains, suggesting that TGEV is likely to be of canine coronavirus origin (Lorusso et al., 2008, Decaro et al., 2007). Like other coronaviruses, TGEV is an enveloped virus that contains a large, positive-sense, single-stranded RNA genome (Eleouet et al., 1998). TGEV replicates in enterocytes and provokes villous atrophy, resulting in lethal watery diarrhea and dehydration in piglets, which is considered to be a central event in the pathogenesis of TGEV infection (Weingartl and Derbyshire, 1993). In consistent with in vivo pathologic changes, TGEV also induces cytopathic effects (CPE) when propagated in vitro cultured cells (Eleouet et al., 1998, Solorzano et al., 1978). Further studies have demonstrated that TGEV infection caused CPE in swine testis (ST) cells by induction of apoptosis (Eleouet et al., 1998, Kim et al., 2000, Sirinarumitr et al., 1998). However, the exact molecular pathways of TGEV-induced apoptosis are not clear.
In the present study, we investigated the effects of TGEV infection in porcine kidney (PK-15) cells and associated molecular mechanisms. Our results demonstrated that TGEV infection could induce apoptosis through the activation of Fas/FasL- and mitochondria-mediated pathways, and that this apoptotic occurrence requires viral replication in PK-15 cells.
2. Materials and methods
2.1. Antibodies, cells and virus
Monoclonal antibodies against caspase-8 (sc-81662), caspase-9 (sc-56077), caspase-3 (sc-70497), Fas (sc-51885), FasL (sc-73974), Bid (sc-56025), Bcl-2 (sc-7382), Bax (sc-23959), cytochrome c (sc-13560), Cox4 (sc-58348), poly (ADP-ribose) polymerase (PARP) (sc-8007), Smac (sc-73039), β-actin (sc-69879) were purchased from Santa Cruz Biotechnology (Santa Cruz, Inc., CA, US). Horseradish peroxidase (HRP)-conjugated secondary antibody was purchased from Pierce (Pierce, Rockford, IL, US). PK-15 cells (ATCC, CCL-33) were grown in Dulbecco Minimal Essential Medium (D-MEM) (Gibco BRL, Gaithersburg, MD, US) supplemented with 10% heat-inactivated new born bovine serum (Gibco BRL, Gaithersburg, MD, US), 100 IU of penicillin and 100 μg of streptomycin per ml, at 37 °C in a 5% CO2 atmosphere incubator. The TGEV Shaanxi strain was isolated from intestinal tract contents of TGEV-infected piglets in Shaanxi Province of China, and identified by physicochemical test, neutralization test, RT-PCR and sequence analysis (Ding et al., 2011). Virus titers were determined by 50% tissue culture infective doses (TCID50) as described previously (Reed and Muench, 1938).
2.2. Cell viability assessment
Cell viability was determined by MTT assay as described previously (De Martino et al., 2010). Briefly, PK-15 cells were seeded in 96-well plates and treated with caspase inhibitors for 2 h. Then, cells were infected with TGEV at 10 multiplicity of infection (MOI) for 24 h and 48 h. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg/ml) was added to each well and incubated at 37 °C for 4 h. After dimethyl sulfoxide (DMSO) was added, the absorbance was measured by microplate spectrophotometer (BioTek Instruments, Inc., Winooski, US) at 570 nm.
2.3. Observation under optical, fluorescence and electron microscopy
Cells were seeded into 24-well culture plates and infected with TGEV at 10 MOI. Then the cells were washed with D-MEM and stained with 200 μl of phosphate-buffered saline (PBS) with acridine orange (AO, 100 μg/ml) and ethidium bromide (EB, 100 μg/ml) at indicated time. After incubated at room temperature for 5 min in the dark, the stained cells were observed under a fluorescence microscope (Nikon, Inc., Tokyo, Japan). The normal cells and early apoptotic cells can be stained by AO to appear bright green fluorescence, while the late apoptotic cells can be stained by EB to appear orange fluorescence.
To observe the ultrastructural changes of TGEV-infected cells, the mock- or TGEV-infected cells attached on cover slips were washed with PBS, and fixed in 2.5% glutaraldehyde in PBS for 30 min. Then, cells were washed with PBS for 3 times, and dehydrated in a series of ethanol solutions of decreasing dilution. After critical point dryer and ion sputter were performed, cells were observed under a scanning electron microscope (SEM) (JEOL, Tokyo, Japan).
2.4. DNA fragmentation assay
Mock-infected cells or TGEV-infected cells were harvested, washed and incubated with lysis buffer (20 mM EDTA, 100 mM Tris, pH 8.0, 0.8% SDS) at room temperature for 1 h. After centrifugation for 10 min at 12 000 × g, the supernatants were collected and treated with RNase A (500 μg/ml) for 1 h at 37 °C, followed by digestion with proteinase K (500 μg/ml) for 2 h at 55 °C. The DNA was extracted using the phenol/chloroform/isoamylol (25:24:1), precipitated with ethanol, dissolved in TE buffer (10 mM Tris, pH 8.0, 1 mM EDTA), and subjected to 2.0% agarose gel electrophoresis for DNA fragmentation analysis.
2.5. Caspase activity assay
Caspases activities were measured by colorimetric assay kits (BioVision, Inc., Mountain View, California, US) according to the manufacture's recommendations. Briefly, cell lysates were prepared and protein concentrations were measured using BCA Protein Assay Reagent (Pierce, Rockford, IL, US). Then 200 μg of protein in each sample was incubated with each caspase substrate (200 μM final concentration) at 37 °C in a microplate for 4 h. Samples were read at 405 nm in microplate spectrophotometer (BioTek Instruments, Inc., Winooski, US).
2.6. Western blot analysis
Cells were harvested and washed with ice-cold PBS, then treated with ice-cold RIPA lysis buffer with 1 mM phenylmethyl sulfonylfluoride (PMSF). Isolation of mitochondrial and cytosolic proteins was performed using the Mitochondria/cytosol Fractionation Kit (Pierce, Rockford, IL, US). Protein concentrations were measured using BCA Protein Assay Reagent (Pierce, Rockford, IL, US). Equivalent amounts of proteins were loaded and electrophoresed on 8–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Subsequently, proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore Corp, Atlanta, GA, US). The membranes were blocked with 5% nonfat dry milk at room temperature for 1 h, and then incubated with indicated primary antibodies over night at 4 °C, followed by HRP-conjugated secondary antibodies at room temperature for 1 h. The signal was detected using ECL reagent (Pierce, Rockford, IL, US).
2.7. Real-time quantitative PCR
Total RNA was extracted using TRIzol agent (Invitrogen, Carlsbad, CA, US), and 2 μg each RNA sample was reverse-transcribed using First-strand cDNA synthesis kit (Invitrogen, Carlsbad, CA, US). The expression of apoptosis regulating genes was quantified using Bio-Rad iQ5 Real Time PCR System by means of a real-time quantitative PCR assay (qRT-PCR) as described previously (Ravindra et al., 2009). The primers for qRT-PCR in this study were shown in Table 1 . Reactions were carried out in 25 μl volume containing 1× SYBR Premix Ex Taq™ II (Takara, Dalian, China), sense and anti-sense primers (0.4 μM) and target cDNA (4 ng). The cycling conditions were 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s, 60 °C for 30 s. A negative control was included in each run and the specificity of amplification reaction was checked by melting curve (T m value) analysis. The relative quantification of gene expression was analyzed by the two −ddCt method (Livak and Schmittgen, 2001).
Table 1.
Sequences of primer pairs used for qRT-PCR.
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) | Product (bp) | Accession no. |
|---|---|---|---|---|
| Bax | ATGATCGCAGCCGTGGA | GGGCCTTGAGCACCAGTTT | 139 | AJ606301 |
| Bcl-2 | TTGTGGCCTTCTTTGAGTTCG | CTACCCAGCCTCCGTTATCC | 150 | XM_003121700.1 |
| Fas | CCAGAATGAGAATGAAAGCTTGACC | AAGCGATGTTTACAGCCAGCA | 124 | NM_213839 |
| FasL | ACCAACACTCCTGCCATCAA | CAGCCCCAATCCAACCA | 136 | NM_213806 |
| β-Actin | GGACTTCGAGCAGGAGATGG | AGGAAGGAGGGCTGGAAGAG | 138 | XM_003124280.1 |
| TGEV | ACAAACACACCTGGAAGAGAACT | GAATGCTAGACACAGATGGAACAC | 165 | HQ462571 |
2.8. Absolute quantification of TGEV by real-time PCR
Viral RNA extraction and reverse transcription were performed as described above. The sequences of the primers for a 165 bp fragment of TGEV N gene were shown in Table 1. The purified PCR product of TGEV was cloned into pGEM-T easy vector (Promega, Madison, US) and the concentration of Plasmid DNA was measured using spectrophotometer (NanoDrop 2000, Wilmington, US). A standard curve obtained using serially diluted plasmid DNA standards of 101–109 copies/μl was used to quantify the TGEV viral genomic copy numbers. The amplification was carried out as described above.
2.9. Statistical analysis
Data are shown as mean ± SEM of three independent experiments done in triplicate. Results were analyzed by one-way analysis of variance (ANOVA). A value of P < 0.05 was considered significant.
3. Results
3.1. TGEV infection induces apoptosis in PK-15 cells
TGEV infection in PK-15 cells generally induces CPE and eventually causes cell death (Solorzano et al., 1978). To determine whether apoptosis occurs in TGEV infection-induced CPE and cell death, we first observed the morphological changes of TGEV-infected PK-15 cells. After infection with TGEV at 10 MOI, CPE appeared in PK-15 cells at 12 h post infection (p.i.), and became more evident at 24 and 48 h when compared to mock infection (Fig. 1A, upper panel). Consistent with this, the TGEV-infected cells showed typical apoptotic features including chromatin condensation and nuclear fragmentation after AO/EB staining (Fig. 1A, middle panel). Scanning electron microscope observation showed cell shrinkage, volume reduction, and microvilli disappearance at 12 h p.i., and cell blebbing and membrane embedded apoptotic bodies at 48 h p.i. (Fig. 1A, lower panel).
Fig. 1.
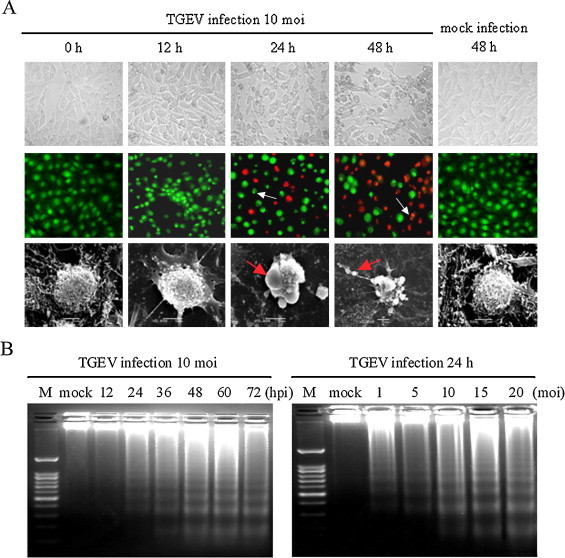
Apoptosis induced by TGEV infection in PK-15 cells. (A) Morphological changes in TGEV-infected cells. Cells were seeded into 24-well culture plates and infected with TGEV (MOI 10) for different times. Photomicrographs at inverted microscope of TGEV-induced CPE (upper panel). Morphological changes under fluorescence microscopy followed by AO and EB staining (middle panel). Normal and early apoptotic cells were stained by AO and showed green fluorescence, while late apoptotic cells were stained by EB and showed orange fluorescence. Arrows indicate condensed chromatin and nuclear fragmentation; scanning electron micrographs of TGEV-infected PK-15 cells (lower panel). Arrows indicate plasma membrane blebbing in the apoptotic cells and the formation of apoptotic body. (B) DNA fragmentation in TGEV-infected cells. DNA isolated from TGEV-infected PK-15 cells was subjected to 2% agarose gel electrophoresis, followed by visualization of bands and photography. Lane M, 100 bp DNA molecular weight marker. Lane mock (left panel), sham-infected for 72 h; lane mock (right panel), sham-infected for 24 h.
Next, we analyzed the chromosomal DNA fragmentation of TGEV-infected cells by agarose gel electrophoresis. PK-15 cells were infected with TGEV at 10 MOI for different times (12, 24, 36, 48, 60 and 72 h) or at various titers (1, 5, 10, 15 and 20 MOI) for 24 h. As shown in Fig. 1B, DNA ladders could be detected as early as 24 h p.i., when PK-15 cells were infected with TGEV at 10 MOI, and the intensity of the ladder bands increased with time and dose of infection. These results demonstrate that TGEV infection induces apoptosis in PK-15 cells in a time- and dose-dependent manner.
3.2. TGEV infection activates caspase-8, -9, -3, and cleaves PARP
To gain insight into the mechanism underlying TGEV-induced apoptosis, we investigated the contribution of caspases to TGEV-induced apoptosis in PK-15 cells. The protein levels of caspase-8, -9 and -3 were measured by Western blot. Upon TGEV infection, full-length procaspase-8, procaspase-9 and procaspase-3 decreased with the increased infection times and doses, while their activated form showed a time- and dose-dependent increase (Fig. 2A). PARP, a representative substrate for effector caspases, can be cleaved by caspase-3 (Thornberry and Lazebnik, 1998). Western blot analysis showed that the levels of full-length PARP decreased at 12 h p.i., while the levels of cleaved PARP were significantly increased at 12 h p.i., whereas no cleaved PARP were detected in mock-infected cells. The cleaved fragment of PARP was also increased with the infection dose in TGEV-infected cells at 24 h p.i. (Fig. 2B, right panel).
Fig. 2.
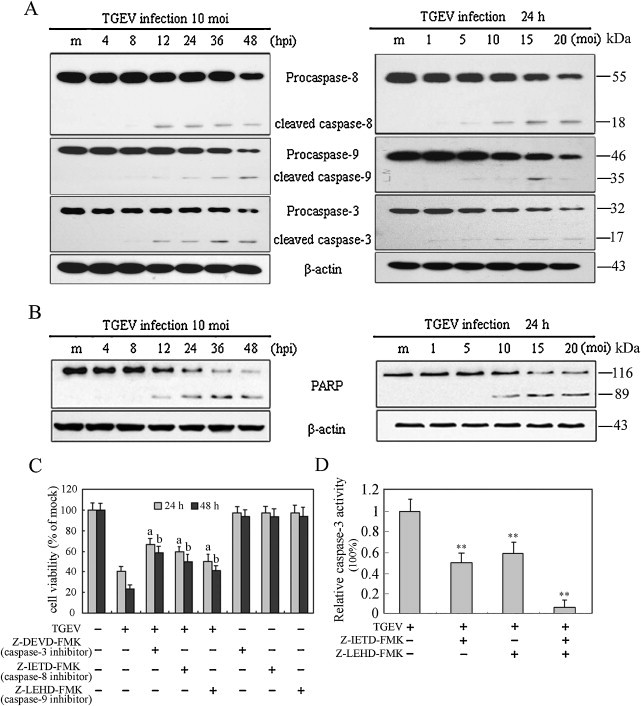
Effects of TGEV infection on caspases activation and PARP cleavage in PK-15 cells. (A) Western blot analysis for caspases activation in TGEV-infected cells (10 MOI) for different times (left panel) or at different MOIs for 24 h p.i. (right panel). The molecular weight (kDa) of protein size standards is shown on the right hand side. Lane m (left panel), sham-infected for 48 h; lane m (right panel), sham-infected for 24 h; β-actin was used as an internal loading control. (B) The cleavage of PARP was analyzed by Western blot. (C) Effect of caspase inhibitors on cell viability. The cells were incubated with 20 μM of each caspase inhibitor for 2 h prior to infection with TGEV (10 MOI) for 24 h and 48 h. Cell viability was evaluated by MTT assay. Values are shown as the mean ± SEM. a, P < 0.05 versus TGEV infection alone without inhibitors for 24 h; b, P < 0.05 versus TGEV infection alone without inhibitors for 48 h. (D) The Inhibitory efficacy of caspase-8 and -9 inhibitors on the activity of caspase-3. The cells were incubated with 20 μM of each caspase inhibitor (z-IETD-fmk and z-LEHD-fmk) for 2 h prior to infection with TGEV (10 MOI) for 24 h. The activity of caspase-3 was measured by colorimetric assay kit. The relative caspase-3 activity (caspase-3 activity with inhibitor/caspase-3 activity without inhibitor) was calculated, after the activity of caspase-3 in cells treated with TGEV or/and inhibitors subtracted the background value (caspase-3 activity in cells without virus and inhibitors). Values are mean ± SEM. **P < 0.01 versus TGEV infection alone without inhibitors.
To further confirm the contribution of caspase-8, -9, -3 in apoptosis, we examined the cell viability of TGEV-infected cells pre-treated with or without caspase-8, -9 and -3 specific inhibitors z-IETD-FMK, z-LEHD-FMK and Z-DEVD-FMK, respectively. Results showed that caspases-8, -9 and -3 specific inhibitors significantly prevented cell death as expected (Fig. 2C). To further determine the effect of initiator caspase-8 or -9 on the activation of caspase-3, we analyzed the inhibitory efficacy of caspase-8 or caspase-9 inhibitor in caspase-3 activity in TGEV-infected cells. The activity of caspase-3 was significantly inhibited in cells in the presence of caspase-8 or caspase-9 inhibitor, and was more significantly inhibited by combining the two inhibitors (Fig. 2D). Taken together, these results suggest that both extrinsic and intrinsic pathways might contribute to caspase-3 activation in TGEV-induced apoptosis.
3.3. TGEV infection induces apoptosis through Fas/FasL dependent pathway and Bid cleavage
Caspase-8 plays a pivotal role in Fas/FasL mediated-apoptosis. The involvement of caspase-8 hints us to further test whether Fas and FasL are involved in TGEV-induced apoptosis. Thus, we investigated the expression of Fas and FasL in TGEV-infected PK-15 cells. As shown in Fig. 3A, the protein levels of FasL were notably increased in a time- and dose-dependent manner, whereas Fas did not show any changes after TGEV infection. To further determine the mechanism whereby FasL expression was increased, we examined FasL mRNA levels in TGEV-infected PK-15 cells by qRT-PCR. The results showed that the mRNA levels of FasL significantly increased as early as 4 h p.i., and continued to increase with infection time (Fig. 3B), suggesting that FasL expression is up-regulated in TGEV-infected cells. However, the mRNA levels of Fas maintained relatively stable levels in TGEV-infected cells compared to that of control (Fig. 3C), suggesting that Fas is expressed constitutively in PK-15 cells.
Fig. 3.
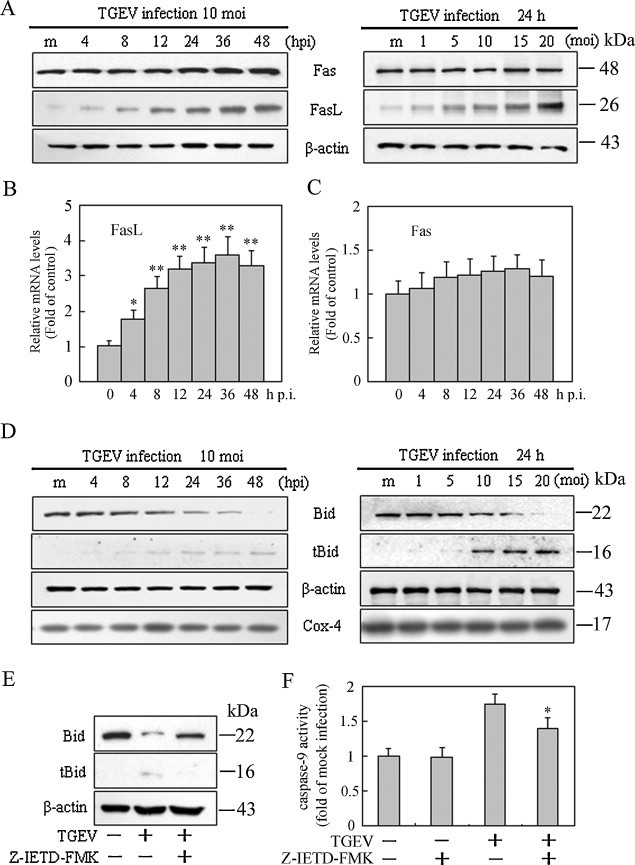
Effects of TGEV infection on FasL, Fas expression and Bid cleavage in PK-15 cells. (A) Cells were mock-infected or TGEV-infected at 10 MOI for different times (left panel) or at different MOIs for 24 h (right panel). Cell lysates were analyzed by Western blot. The molecular weight (kDa) of protein size standards is shown on the right hand side. (B, C) qRT-PCR assay for mRNA levels of FasL and Fas after cells were infected with TGEV at 10 MOI for different times. Values are shown as the mean ± SEM. *P < 0.05, **P < 0.01 versus control. (D) Bid cleavage and tBid translocation to mitochondria. Mitochondrial protein (tBid) and cytosolic protein (Bid) were isolated, and subjected to Western blot analysis. Cox4 and β-actin were used as internal controls for the mitochondrial fractions and the cytosolic fractions, respectively. The molecular weight (kDa) of protein size standards is shown on the right hand side. (E) Cells were pre-treated with the caspase-8 inhibitor for 2 h and infected with TGEV for 24 h. tBid expression in mitochondria was analyzed by Western blot. (Note: X-ray film exposure times were different in D left panel, D right panel and E). (F) Effect of caspase-8 inhibitor on the activity of caspase-9. Cells were pretreated with Z-IETD-FMK for 2 h, and then infected with TGEV for 24 h. Caspase-9 activity was measured by colorimetric assay kit. Values represent the mean ± SEM of three different experiments performed in triplicate. *P < 0.05 versus TGEV infection alone.
It is known that activated caspase-8 can cleave Bid to truncated Bid (tBid), which is important in the crosstalk between death receptor pathway and intrinsic pathway in some cell types (Yin, 2000). tBid translocation to mitochondria participates in the destruction of mitochondria integrity and cytochrome c release to cytosol, which further facilitates caspase-9 activation (Yin, 2000). To test whether Bid is involved in TGEV-induced apoptosis, we measured the changes of Bid in cells upon TGEV infection. Western blot analysis revealed that tBid appeared in mitochondrial fractions at 8 h p.i., and maintained a high level in subsequent times in TGEV-infected cells but not in mock-infected cells (Fig. 3D). However, following incubation with z-IETD-FMK, a specific inhibitor of caspase-8, tBid could not be detected in TGEV-infected cells (Fig. 3E). Moreover, the activation of caspase-9 was partly blocked in TGEV-infected cells in the presence of caspase-8 inhibitor compared to that in cells without the inhibitor (Fig. 3F), suggesting that block of caspase-8 activity could affect Bid cleavage and caspase-9 activation. Take together, these results suggest that apoptosis signals from the FasL are transmitted to activate caspase-8, which in turn cleaves Bid, activates mitochondria-mediated apoptotic pathway, followed by caspase-9 activation, and that other apoptosis signals might contribute to the activation of caspase-9.
3.4. TGEV infection regulates the expression of Bcl-2 family proteins and promotes the release of cytochrome c from mitochondria
Once activation of mitochondria-mediated apoptotic pathway, several toxic proteins such as cytochrome c and Smac can release from mitochondria to cytoplasm to induce the activation of downstream caspase cascade. This process is tightly controlled by members of Bcl-2 family. Pro-apoptotic Bcl-2 proteins such as Bax or Bak were activated upon upstream apoptosis signals, resulting in outer mitochondrial membrane permeabilization and toxic proteins release. In contrast, anti-apoptotic Bcl-2 proteins such as Bcl-2 or Bcl-XL can prevent this occurrence (Shimizu et al., 1999). In the present study, the expression of Bcl-2 and Bax were detected by Western blot assay and qRT-PCR. In TGEV-infected cells, protein levels of Bax exhibited an apparent increase in a time- and dose-dependent manner, whereas the protein levels of Bcl-2 were down-regulated in a time- and dose-dependent manner (Fig. 4A). Consistent with the changes of protein levels, the mRNA levels of Bax were up-regulated in TGEV-infected cells as early as 8 h p.i. and continuously increased until 48 h p.i. (Fig. 4B). Conversely, the mRNA levels of Bcl-2 were significantly decreased after infection with 10 MOI of TGEV for 8 h, and progressively declined at subsequent time points (Fig. 4B).
Fig. 4.
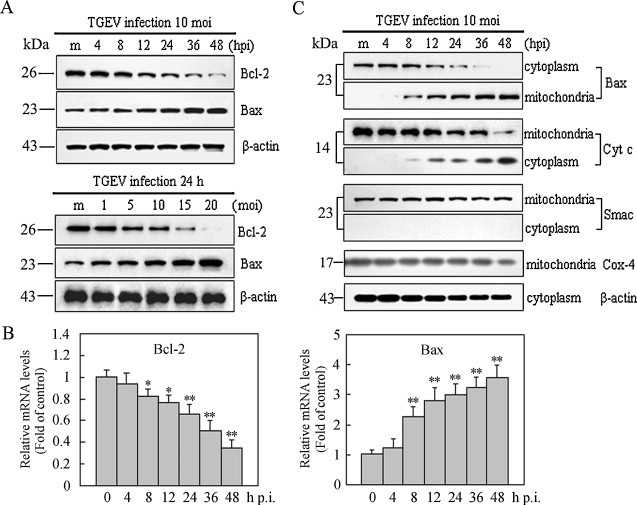
Effects of TGEV infection on the expression of Bcl-2 family proteins and release of cytochrome c. (A) The expression of Bcl-2 and Bax in TGEV-infected PK-15 cells. Cells were mock-infected or TGEV-infected (10 MOI) for different times (upper panel) or TGEV-infected at different MOIs for 24 h (lower panel) and cell lysates were analyzed by Western blot. The molecular weight (kDa) of protein size standards is shown on the left hand side. (B) qRT-PCR assay for mRNA levels of Bcl-2 (left panel), Bax (right panel). Values are mean ± SEM. *P < 0.05, **P < 0.01 versus control. (C) Bax translocation and cytochrome c release. The cells were mock-infected or TGEV-infected for different times at 10 MOI, and cell lysates were analyzed by Western blot. The molecular weight (kDa) of protein size standards is shown on the left hand side. Data are representative of three separate experiments.
Next, we observed that Bax translocated from cytosol to mitochondria at 8 h p.i., and steadily increased in mitochondria with the infection time (Fig. 4C). In coincidence with Bax translocation to mitochondria, cytochrome c but not Smac released from mitochondria to cytosol in TGEV-infected cells (Fig. 4C). In mock-infected cells, however, Bax translocation and cytochrome c release could not be detected. Taken together, these results suggest that Bax translocation and Bcl-2 down-regulation might play important roles in promoting the release of cytochrome c from mitochondria in TGEV-infected cells.
3.5. Virus replication is required for apoptosis induction
To test the effects of caspases activation on the replication of TGEV, virus progeny release from cells in the presence or absence of the caspases inhibitors was determined by measuring TCID50. Results showed that the titer of TGEV did not appear significant changes at 24 h p.i., but progressive increase in subsequent times and apparently increased at 48 h p.i. in inhibitors-treated cells compared with that in cells without inhibitors, especially in the presence of caspase-3 inhibitor (Fig. 5A). These changes might be attributed to cell viability increase in the presence of caspases inhibitors.
Fig. 5.
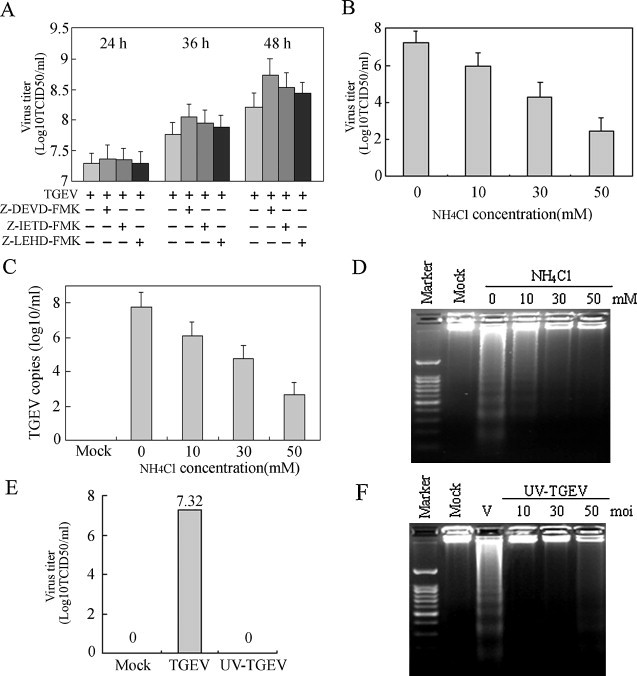
Inhibitory experiments of TGEV-induced apoptosis. (A) PK-15 cells were pre-treated with 20 μM of each caspase inhibitor for 2 h, and then infected with TGEV (10 MOI) for 24 h, 36 h and 48 h, respectively. After incubation, virus was collected and virus titers were shown as log10TCID50/ml. (B) PK-15 cells were pretreated with NH4Cl at different concentrations for 2 h before infection and then infected with TGEV (10 MOI) for 24 h. Cells infected in the same conditions without NH4Cl were used as positive controls. Virus was collected and virus titers were shown as log10TCID50/ml. (C) PK-15 cells were treated as in (B), and the copies of TGEV were assessed by qRT-PCR. PCR data was presented as follows: mean log10 genomic copies/ml ± SEM. (D) DNA fragmentation in TGEV-infected cells in the presence of NH4Cl was examined by agarose gel electrophoresis. (E, F) TGEV was inactivated by exposing to a 30 W UV germicidal light at a distance of 30 cm for 30 min at 4 °C. PK-15 cells were infected with UV-inactivated virus at different MOIs for 24 h at 37 °C, and then viral titers (E) and DNA fragmentation (F) were examined.
To determine whether the entry of virion into the cells was necessary for the induction of apoptosis, we used NH4Cl to inhibit the endosomal acidification to impede the release of virion to the cytoplasm. We observed that NH4Cl treatment not only reduced viral progeny production in a dose-dependent manner (Fig. 5B and C), but also attenuated TGEV-induced apoptosis (Fig. 5D), suggesting that TGEV induction of apoptosis requires the viral particles to undergo uncoating.
To further examine whether apoptosis induction by TGEV required virus replication, we used UV-treatment to abrogate replication of virus to investigate the capacity of UV-treated TGEV to induce apoptosis. After TGEV was subjected to UV treatment, no viral progeny was detected (Fig. 5E). Consistently, apoptosis induction disappeared in cells infected with UV-inactivated TGEV (Fig. 5F). Taken together, our results suggest that viral replication is needed for apoptosis induction in TGEV-infected cells.
4. Discussion
Apoptosis is a predominant component for contributing to the pathogenesis process of animal virus infectious diseases (Chen et al., 2008, Everett and McFadden, 1999, Shih et al., 2004). Previous studies have reported that apoptosis occurs in response to TGEV infection in ST cells and human rectal tumor adenocarcinoma cell line HRT18 modified to express the porcine aminopeptidase N (pAPN, a cellular receptor for porcine coronaviruses) in vitro (Eleouet et al., 1998, Eleouet et al., 2000, Kim et al., 2000, Sirinarumitr et al., 1998). In the present study, we detected the effects of TGEV infection in PK-15 cells and analyzed possible mechanisms involved in apoptosis of TGEV-infected cells. The results showed that TGEV-infected PK-15 cells exhibited evident apoptotic features including chromatin condensation, DNA fragmentation. Further results demonstrated that caspases activation, Fas/FasL and mitochondrial pathways were involved in the apoptotic process in TGEV-infected PK-15 cells.
Caspases, a family of cysteine-dependent aspartate-directed proteases, play pivotal roles in initiation and execution of apoptosis by cleaving a large number of proteins (Thornberry and Lazebnik, 1998). The activation of the caspase cascades is a trigger for apoptosis in some viruses-infected cells (Chen and Makino, 2002, De Martino et al., 2010, Lee and Kleiboeker, 2007). In TGEV-infected PK-15 cells, the activation of caspase-8, -9, and -3 and cleavage of PARP were observed, while inhibition of these caspases activities significantly attenuated the apoptotic effects of TGEV infection. These results suggest that the activation of caspase cascades is involved in TGEV-induced apoptosis in PK-15 cells. Similar results have been found in other coronaviruses, including canine coronavirus and infectious bronchitis virus (De Martino et al., 2010, Liu et al., 2001).
There are two main signaling pathways contributing to caspases activation, death receptor and mitochondrial pathways. Death receptor pathway is initiated by death receptor ligation followed by recruitment of adaptor molecules and activation of upstream caspases (Ashkenazi and Dixit, 1998). Mitochondrial pathway is dependent on the process of mitochondrial outer membrane permeabilization, leading to the release of toxic proteins from mitochondria, which is subject to the regulation by Bcl-2 family proteins (Shimizu et al., 1999). Fas/FasL-mediated signaling has been reported in apoptosis in response to infection of several viruses such as Bovine ephemeral fever virus (BEFV) (Lin et al., 2009) and murine coronavirus (Liu and Zhang, 2007). Our current study revealed that the induction of apoptosis by TGEV infection was preceded by increased expression of FasL, which was followed by subsequent activation of caspase-8 and -3. This finding demonstrates that caspase-8 activation in TGEV-infected cells can be mediated by ligation of Fas and FasL. Mitochondrial apoptotic signals are controlled prominently by the Bcl-2 family proteins, which include a number of pro-apoptotic and anti-apoptotic proteins to regulate apoptosis progress by changing relative levels (Shimizu et al., 1999). Some viruses can encode proteins homologous to Bcl-2 to inhibit apoptosis, while some viruses can induce apoptosis by modulating the expression of Bcl-2 family proteins (Cheng et al., 1997, De Martino et al., 2010, Umeshappa et al., 2010). Our results showed that TGEV infection down-regulated Bcl-2, up-regulated Bax expression, promoted Bax translocation from cytosol to mitochondria, and induced the release of cytochrome c, resulting in PK-15 cell apoptosis. These results are consistent with the observation in murine and canine coronaviruses (Chen and Makino, 2002, De Martino et al., 2010), suggesting that modulation of Bcl-2 members might be a key step for coronaviruses induction of CPE and eventually cell death.
In most case of virus-induced apoptosis, apoptosis is a result of crosstalk between extrinsic and intrinsic pathways (De Martino et al., 2010, Lee and Kleiboeker, 2007). In this process, activated caspase-8 cleaves Bid to tBid, which translocates to mitochondria and initiates the release of cytochrome c into the cytosol, leading to caspase-9 activation (Yin, 2000). Our results also provided evidences for the potential crosstalk between extrinsic and intrinsic pathways in TGEV-induced apoptosis. Blocking caspase-8 activity using z-IETD-FMK, a specific inhibitor of caspase-8, did not completely abrogate caspase-9 activation, suggesting that caspase-9 is activated not only by caspase-8 activation but also by other upstream apoptotic signals. In our system, the higher expression of p53 in TGEV-induced apoptotic cell death (our unpublished data) might be involved in the regulation of this process.
Some viruses can trigger apoptosis by early steps of virus infectious cycle in the absence of viral replication, such as vesicular stomatitis virus and reovirus (Clarke et al., 2000, Gadaleta et al., 2002). However, some other viruses-induced apoptosis depends on viral replication, such as BEFV and human cytomegalovirus (Lin et al., 2009, Reboredo et al., 2004). Our results showed that inhibition of virus uncoating prevented the occurrence of apoptosis and that UV-inactivated TGEV lost the ability to induce apoptosis, suggesting that TGEV-induced cell apoptosis depends on viral replication. Then, we investigated whether caspases activation in cells is required for TGEV replication in cells. Although our results showed that inhibition of caspases activation could increase total viral titer, the increase of viral titer is proportionate with the effects of cell viability increase derived from the inhibition of caspases activation, suggesting that block of caspases activation does not affect TGEV replication in fact. All these findings suggest that viral replication is crucial for the induction of apoptosis in TGEV-infected PK-15 cells.
In conclusion, our results demonstrate that TGEV induces apoptosis through Fas/FasL-mediated caspase-8 activation and mitochondria-dependent caspase-9 activation. In addition, TGEV replication may be necessary for the induction of apoptosis. Based on these findings, a schematic model of possible apoptotic mechanisms of TGEV-infected cells is shown in Fig. 6 .
Fig. 6.
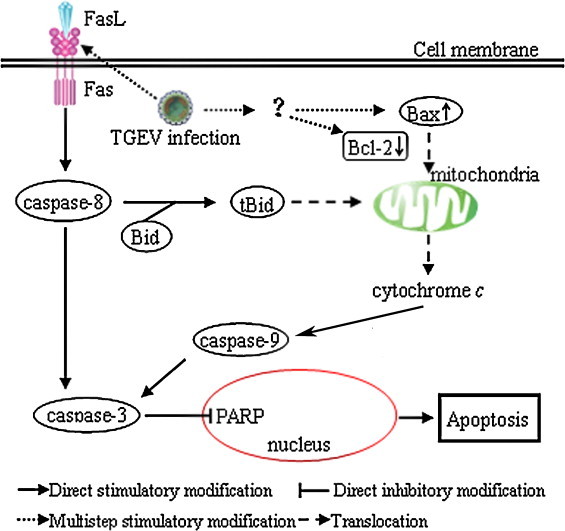
Schematic representation of apoptotic pathways in TGEV-infected PK-15 cells. TGEV infection promotes the activation of Fas/FasL signaling pathway, and then leads to activation of caspase-8. Activated caspase-8 could lead to the cleavage of Bid. tBid then translocates to mitochondria, resulting in the release of cytochrome c. Bcl-2 down-regulation and Bax up-regulation in TGEV-infected cells also induce cytochrome c release to cytoplasm, which in turn causes subsequently activation of caspase-9. The activation of caspase-8 and caspase-9 further activates caspase-3, leading to apoptosis.
Authors contribution
Li Ding, Dewen Tong and Xingang Xu designed the experiments; Li Ding and Yong Huang interpreted the data and wrote the article; Li Ding performed the experiments with assistance and advice from Zhaocai Li, Kuan Zhang, Guangda Chen, Gaoshui Yu, Zhisheng Wang, Wei Li. All authors have read the manuscript and approved for the submission.
Conflict of interest
There is no conflict of interest of any authors in relation to the submission.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 31072108/C1802), the Doctoral Program of Higher Education of China (Grant Nos. 20090204110016 and 20110204110014), and the Youth academic backbone support program of Northwest A&F University (No. E111020901).
References
- Annamaria P. The evolutionary processes of canine coronaviruses. Adv. Virol. 2011 doi: 10.1155/2011/562831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arch R.H., Thompson C.B. Lymphocyte survival—the struggle against death. Annu. Rev. Cell Dev. Biol. 1999;15:113–140. doi: 10.1146/annurev.cellbio.15.1.113. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A., Dixit V.M. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Carstens E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch. Virol. 2010;155:133–146. doi: 10.1007/s00705-009-0547-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.J., Makino S. Murine coronavirus-induced apoptosis in 17Cl-1 cells involves a mitochondria-mediated pathway and its downstream caspase-8 activation and bid cleavage. Virology. 2002;302:321–332. doi: 10.1006/viro.2002.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Cheng A., Wang M., Peng X. Detection of apoptosis induced by new type gosling viral enteritis virus in vitro through fluorescein annexin V-FITC/PI double labeling. World J. Gastroenterol. 2008;14:2174–2178. doi: 10.3748/wjg.14.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng E.H.Y., Nicholas J., Bellows D.S., Hayward G.S., Guo H.G., Reitz M.S., Hardwick J.M. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. U. S. A. 1997;94:690–694. doi: 10.1073/pnas.94.2.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke P., Meintzer S.M., Gibson S., Widmann C., Garrington T.P., Johnson G.L., Tyler K.L. Reovirus-induced apoptosis is mediated by TRAIL. J. Virol. 2000;74:8135–8139. doi: 10.1128/jvi.74.17.8135-8139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Martino L., Marfe G., Longo M., Fiorito F., Montagnaro S., Iovane V., Decaro N., Pagnini U. Bid cleavage, cytochrome c release and caspase activation in canine coronavirus-induced apoptosis. Vet. Microbiol. 2010;141:36–45. doi: 10.1016/j.vetmic.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N., Martella V., Elia G., Campolo M., Desario C., Cirone F., Tempesta M., Buonavoglia C. Molecular characterisation of the virulent canine coronavirus CB/05 strain. Virus Res. 2007;125:54–60. doi: 10.1016/j.virusres.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L., Chen G.D., Xu X.G., Tong D.W. Isolation and identification of porcine transmissible gastroenteritis virus Shaanxi strain and sequence analysis of its N gene. Chin. J. Vet. Med. 2011;47:9–12. [Google Scholar]
- Eleouet J.F., Chilmonczyk S., Besnardeau L., Laude H. Transmissible gastroenteritis coronavirus induces programmed cell death in infected cells through a caspase-dependent pathway. J. Virol. 1998;72:4918–4924. doi: 10.1128/jvi.72.6.4918-4924.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eleouet J.F., Slee E.A., Saurini F., Castagne N., Poncet D., Garrido C., Solary E., Martin S.J. The viral nucleocapsid protein of transmissible gastroenteritis coronavirus (TGEV) is cleaved by caspase-6 and-7 during TGEV-induced apoptosis. J. Virol. 2000;74:3975–3983. doi: 10.1128/jvi.74.9.3975-3983.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett H., McFadden G. Apoptosis: an innate immune response to virus infection. Trends Microbiol. 1999;7:160–165. doi: 10.1016/s0966-842x(99)01487-0. [DOI] [PubMed] [Google Scholar]
- Gadaleta P., Vacotto M., Coulomb F. Vesicular stomatitis virus induces apoptosis at early stages in the viral cycle and does not depend on virus replication. Virus Res. 2002;86:87–92. doi: 10.1016/s0168-1702(02)00049-7. [DOI] [PubMed] [Google Scholar]
- Kim B., Kim O., Tai J., Chae C. Transmissible gastroenteritis virus induces apoptosis in swine testicular cell lines but not in intestinal enterocytes. J. Comp. Pathol. 2000;123:64–66. doi: 10.1053/jcpa.2000.0386. [DOI] [PubMed] [Google Scholar]
- Lee S.M., Kleiboeker S.B. Porcine reproductive and respiratory syndrome virus induces apoptosis through a mitochondria-mediated pathway. Virology. 2007;365:419–434. doi: 10.1016/j.virol.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.H., Shih W.L., Lin F.L., Hsieh Y.C., Kuo Y.R., Liao M.H., Liu H.J. Bovine ephemeral fever virus-induced apoptosis requires virus gene expression and activation of Fas and mitochondrial signaling pathway. Apoptosis. 2009;14:864–877. doi: 10.1007/s10495-009-0371-5. [DOI] [PubMed] [Google Scholar]
- Liu C., Xu H.Y., Liu D.X. Induction of caspase-dependent apoptosis in cultured cells by the avian coronavirus infectious bronchitis virus. J. Virol. 2001;75:6402–6409. doi: 10.1128/JVI.75.14.6402-6409.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhang X. Murine coronavirus-induced oligodendrocyte apoptosis is mediated through the activation of the Fas signaling pathway. Virology. 2007;360:364–375. doi: 10.1016/j.virol.2006.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−[Delta][Delta] CT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lorusso A., Decaro N., Schellen P., Rottier P.J.M., Buonavoglia C., Haijema B.J., De Groot R.J. Gain, preservation, and loss of a group 1a coronavirus accessory glycoprotein. J. Virol. 2008;82:10312–10317. doi: 10.1128/JVI.01031-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindra P., Tiwari A.K., Ratta B., Chaturvedi U., Palia S.K., Chauhan R. Newcastle disease virus-induced cytopathic effect in infected cells is caused by apoptosis. Virus Res. 2009;141:13–20. doi: 10.1016/j.virusres.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Reboredo M., Greaves R.F., Hahn G. Human cytomegalovirus proteins encoded by UL37 exon 1 protect infected fibroblasts against virus-induced apoptosis and are required for efficient virus replication. J. Gen. Virol. 2004;85:3555–3567. doi: 10.1099/vir.0.80379-0. [DOI] [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938;27:493–497. [Google Scholar]
- Ruggieri A., Di Trani L., Gatto I., Franco M., Vignolo E., Bedini B., Elia G., Buonavoglia C. Canine coronavirus induces apoptosis in cultured cells. Vet. Microbiol. 2007;121:64–72. doi: 10.1016/j.vetmic.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih W.L., Hsu H.W., Liao M.H., Lee L.H., Liu H.J. Avian reovirus [sigma] C protein induces apoptosis in cultured cells. Virology. 2004;321:65–74. doi: 10.1016/j.virol.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Shimizu S., Narita M., Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Sirinarumitr T., Kluge J., Paul P. Transmissible gastroenteritis virus induced apoptosis in swine testes cell cultures. Arch. Virol. 1998;143:2471–2485. doi: 10.1007/s007050050477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solorzano R., Morin M., Morehouse L. The use of immunofluorescence techniques for the laboratory diagnosis of transmissible gastroenteritis of swine. Can. J. Comp. Med. 1978;42:385–391. [PMC free article] [PubMed] [Google Scholar]
- Thornberry N.A., Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Umeshappa C.S., Singh K.P., Nanjundappa R.H., Pandey A.B. Apoptosis and immuno-suppression in sheep infected with bluetongue virus serotype-23. Vet. Microbiol. 2010;144:310–318. doi: 10.1016/j.vetmic.2010.02.033. [DOI] [PubMed] [Google Scholar]
- Weingartl H., Derbyshire J. Binding of porcine transmissible gastroenteritis virus by enterocytes from newborn and weaned piglets. Vet. Microbiol. 1993;35:23–32. doi: 10.1016/0378-1135(93)90113-L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X.M. Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res. 2000;10:161–167. doi: 10.1038/sj.cr.7290045. [DOI] [PubMed] [Google Scholar]