

Abstract
Sapoviruses (SaVs) within the Caliciviridae family are an important cause of gastroenteritis in both humans and animals. Although the widespread occurrence of divergent human SaV strains has been reported, there have only been a few studies of porcine SaVs examining their genetic diversity. The aim of this study was to assess the genetic diversity of porcine SaVs in piglets with diarrhea in South Korea. Two hundred and thirty-seven fecal specimens from piglets with diarrhea were examined from 78 farms over a 2-year period from six provinces in South Korea. Overall, 69 (29.1%) of the samples from five provinces tested positive for porcine SaVs by either RT-PCR or nested PCR with the primer pairs specific to porcine SaVs. An analysis of the partial capsid gene (757 bp) of 12 porcine SaVs detected from fecal samples showed genetic divergence, not only among the Korean porcine SaVs (67.7–99.1%), but also between Korean and American porcine SaVs (69.1–90.2%). Interestingly, one strain (Po/SaV/JN-MA113/05/Korea), formed a second porcine SaV/GIII genotype, and is tentatively called GIII/2. This strain had a 0.236–0.405 inter-cluster distance with the other strains in the same genogroup, which is comparable to the distances between the established GI and GII SaVs. Furthermore, two potential recombinant porcine SaVs were identified. In conclusion, porcine SaV infections are common in diarrheic piglets in South Korea. The infecting strains are genetically diverse, and include a newly recognized genotype and recombinant viruses within genogroup III.
Keywords: Sapovirus, Porcine, Genetic diversity, New genotype, Recombination
1. Introduction
Caliciviruses are small, non-enveloped viruses 27–38 nm in diameter and possess a single-stranded, 7.3–8.3 kb plus-sense RNA genome and a single 56–71 kDa capsid protein (Green et al., 2000). The family Caliciviridae is divided into at least four genera: Vesivirus, Lagovirus, Norovirus (NoV), and Sapovirus (SaV) with a new proposed genus, NB-like (Smiley et al., 2002), based on genomic organization and genetic analysis (Green et al., 2000). Caliciviruses are associated with a wide range of economically important diseases in animals, including humans (Geissler et al., 1997, Green et al., 2001, Ohlinger et al., 1990).
Noroviruses and sapoviruses have emerged as the leading cause of food- and waterborne, acute, non-bacterial gastroenteritis in humans worldwide (Green et al., 2001). Animal SaVs and NoVs also cause gastroenteritis in swine, calves and mink (Bridger, 1990, Guo et al., 2001a, Guo et al., 2001b, Saif et al., 1980). Several of porcine SaVs and NoVs are closely related to human SaVs and NoVs genetically, which has raised public health concerns of potential cross-species transmission and animal reservoirs for SaVs and NoVs related to human SaVs and NoVs (Guo et al., 1999, Wang et al., 2005a, Wang et al., 2005b). However, the occurrence of porcine enteric caliciviruses has been reported in only a few countries including the US (both SaVs and NoVs), Japan (only NoVs), Venezuela (both SaVs and NoVs), Netherlands (both SaVs and NoVs), and the United Kingdom (Bridger, 1990, Martínez et al., 2006, Sugieda et al., 1998; van Der Poel et al., 2000, Wang et al., 2005a, Wang et al., 2005b).
Human enteric caliciviruses cannot be cultured in vitro. Therefore, molecular studies have relied on the availability of stool samples collected during outbreaks or from human volunteer studies. Molecular analysis of human SaVs has demonstrated significant genetic diversity among the viruses within this genus: these have been genetically divided into four genogroups (GI, −II, −IV, and −V) and at least eight genetic clusters or genotypes (GI/1 to 3, GII/1 to −3, GIV/1, and GV/1) (Farkas et al., 2004, Schuffenecker et al., 2001). Porcine SaVs circulating in the US have been genetically analyzed using the major strains classified as SaV/GIII (Guo et al., 1999, Wang et al., 2005a), though one strain represents a potential new genogroup of porcine SaVs (Wang et al., 2005a). Since the discovery of porcine SaVs in the 1980s (Saif et al., 1980), only a few additional studies of their genetic diversity have been reported (Farkas et al., 2004, Guo et al., 1999, Schuffenecker et al., 2001, Wang et al., 2005a). In South Korea, we demonstrated that porcine SaVs cause diarrhea outbreaks (Kim et al., 2006). However, little is known about the genetic diversity of the porcine SaVs circulating in South Korea. Therefore, the aim of this study was to obtain information on the genetic heterogeneity of porcine SaVs circulating in South Korea.
2. Materials and methods
2.1. Specimens
From 2004 to 2005, a total of 237 fecal specimens from diarrheic pigs obtained from 78 herds were collected from six provinces in South Korea during the spring (105 samples/33 farms), summer (35 samples/14 farms), autumn (65 samples/16 farms) and winter (32 samples/15 farms). The ages of the pigs tested from all provinces ranged from 3 to 70 days. The fecal samples were examined for common bacterial enteric pathogens including Salmonella spp. using specific agar media. Brachyspira hyodysenteriae detection was performed by PCR using the specific primers (La et al., 2003). For the extraction of viral RNA, fecal suspensions of each sample were prepared immediately by diluting the feces 1:10 in 0.01 M phosphate-buffered saline (PBS), pH 7.2. The suspensions were then vortexed for 30 sec, centrifuged (1200 × g for 20 min), and the supernatants were collected and stored at −80 °C for further testing.
2.2. RNA extraction
The RNA was extracted from a 200 μl starting volume of centrifuged 10% fecal suspensions using the Trizol-LS (Gibco-BRL, Life Tech, Grand Island, NY) procedure. The total RNA recovered was suspended in 50 μl of RNase free water and stored at −80 °C until used.
2.3. RT-PCR and nested PCR
RT-PCR assays with different primer sets (Table 1 ) for the detection of group A, B and C porcine rotaviruses (PRV A–C), porcine SaV, porcine NoV, transmissible gastroenteritis coronavirus (TGEV) and porcine epidemic diarrhea coronavirus (PEDV) were performed using a standard one-step RT-PCR as previously described (Cho et al., 2001, Park et al., 2006). In order to increase the sensitivity and specificity of RT-PCR, nested PCR assays with the primer pairs specific to each capsid and RNA dependent RNA polymerase (RdRp) of porcine SaV, PRV A and PRV C (Table 1) were performed as previously described (Cho et al., 2001, Park et al., 2006). The amplification products were analyzed by 1.5 or 2% agarose gel electrophoresis and visualized by UV after ethidium bromide staining.
Table 1.
RT-PCR and nested PCR primers used for the detection of the group A, B and C porcine rotaviruses (PRV A–C), porcine sapovirus, porcine enteric calicivirus (designed to detect sapovirus and norovirus), transmissible gastroenteritis coronavirus (TGEV) and porcine epidemic diarrhea coronavirus (PEDV) in the fecal samples from pigs with diarrhea
| Target viruses | Target genesa | Primer names | Primer sequence, 5′–3′b | Region (nt) | Size (bp) | Source or reference |
|---|---|---|---|---|---|---|
| PSaV and PNoV | RdRp | P289c | F: GATTACTCCAAGTGGGACTCCAC | 4568–4590 | 319 | Jiang et al. (1999a) |
| P290 | R: TGA CAATGTAAT ATCACCATA | 4865–4886 | ||||
| PSaV | Cap | CapsidF | F: GTGATCAACCCTTTTGAAAC | 5698–5717 | 757 | Kim et al. (2006) |
| CapsidR | R: AAAGCATGATGTTGTTAGGC | 6435–6454 | ||||
| PECVCnF | nF: CTCGTCATAGTAGGTGTGGC | 5890–5909 | 565 | |||
| PECVCnR | nR: AAAGCATGATGTTGTTAGGC | 6435–6454 | ||||
| RdRp | PEC45 | F: GTGCTCTATTGCCTGGACTA | 4312–4331 | 572 | ||
| PEC46 | R: TCTGTGGTGCGGTTAGCCTT | 4864–4883 | ||||
| PECnF | nF: CTCGTATGCTGAGGACACAC | 4392–4411 | 380 | |||
| PECnR | nR: GAGTGTCTGTTGGCTCAATG | 4752–4771 | ||||
| PRV A | VP6 | F: AAAGATGCTAGGGACAAAATTG | 58–78 | 308 | Elschner et al. (2002) | |
| R: TTCAGATTGTGGAGCTATTCCA | 344–365 | |||||
| nF: GACAAAATTGTCGAAGGCACATTATA | 69–94 | 121 | ||||
| nR: TCGGTAGATTACCAATTCCTCCAG | 166–189 | |||||
| PRV B | NSP2 | F: CTATTCAGTGTGTCGTGAGAGG | 18–40 | 434 | Gouvea et al. (1991) | |
| R: GCAGACAAGCTAGCCCGCTTCG | 429–451 | |||||
| PRV C | VP6 | F: CTCGATGCTACTACAGAATCAG | 994–1018 | 366 | Gouvea et al. (1991) | |
| R: AGCCACATAGTTCACATTTCATCC | 1339–1359 | |||||
| nF: CTCGATGCTACTACAGAATCAG | 994–1018 | 328 | ||||
| nR: GGGATCATCCACGTCATGCGT | 1300–1321 | |||||
| TGEV | ORF1b | F: GGGTAAGTTGCTCATTAGAAATAATGG | 7968–7994 | 1006 | Kim et al. (2000) | |
| Spike | R: CTTCTTCAAAGCTAGGGACTG | 920–940 | ||||
| PED | N | F: AGGAACGTGACCTCAAAGACATCCC | 812–836 | 540 | Kubota et al. (1999) | |
| R: CCAGGATAAGCCGGTCTAACATTG | 1328–1351 | |||||
RdRp: RNA dependent RNA polymerase; Cap: capsid; VP6: viral protein 6; NSP2: non-structural protein 2; ORF1b: open reading frame 1b; N: nucleocapsid.
F: upstream primer for RT-PCR; R: downstream primer for RT-PCR; nF: upstream primer for nested PCR; nR: downstream primer for nested PCR.
A primer pair p289/290 designed to detect both noroviruses and sapoviruses by RT-PCR.
2.4. DNA sequencing
The RT-PCR products for a portion of the porcine SaV capsid gene (757 bp), and the nested PCR products amplified by the PECnF and PECnR primers for a portion of the RdRp gene (380 bp) (Table 1) were selected from different test reactions and sequenced to verify reaction the specificity and to obtain genomic data for phylogenetic analysis. The RT-PCR products were purified using a GenClean II kit (BIO 101, Inc., LaJolla, CA) according to the manufacture's instructions. DNA sequencing was carried out using an automated DNA sequencer (ABI system 3700, Applied Biosystem Inc., Foster City, CA).
2.5. Molecular analysis
Using the DNA Basic module (DNAsis MAX, Alameda, CA), the nt and deduced amino acid (aa) sequences of the partial capsid gene (717 bp; did not contain primer pair sequences) and the p289/290 products were compared with those selected from other known caliciviruses (Table 2 ).
Table 2.
GenBank accession numbers of the reference sapovirus strains used in phylogenetic analysis
| Strain | Genus/genogroupe-genotype | Abbreviation | GenBank accession no. |
|---|---|---|---|
| Hu/Sapporo/82/JP | SaV/GI-1 | Sapporo | U65427 |
| Hu/Manchester/93/UK | SaV/GI-1 | Manchester | X86560 |
| Hu/Plymouth/92/UK | SaV/GI-1 | Plymouth | X86559 |
| Hu/Lyon30388/98/UK | SaV/GI-1 | Lyon30388 | AJ251991 |
| Hu/Houston/86/US | SaV/GI-1 | Houston86 | U95643 |
| Hu/Parkville/94/US | SaV/GI-2 | Parkville | U73124 |
| Hu/Houston/90/US | SaV/GI-2 | Houston90 | U95644 |
| Hu/Stockholm/97/SE | SaV/GI-3 | Stockholm | AF194182 |
| Hu/London/92/UK | SaV/GII-1 | London92 | U95645 |
| Hu/Lyon598/97/FR | SaV/GII-1 | Lyon598 | AJ271056 |
| Hu/Bristol/98/UK | SaV/GII-1 | Bristol | AJ249939 |
| Hu/Mex340/90/MX | SaV/GII-2 | Mex340 | AF435812 |
| Hu/Cruise ship/00/US | SaV/GII-3 | Cruise ship | AY289804 |
| Hu/Mc10/00/TH | SaV/GII-4 | Mc10 | AY237420 |
| Hu/C12/00/JP | SaV/GII-5 | C12 | AY603425 |
| Po/SaV/Cowden/80/US | SaV/GIII | Cowden | AF182760 |
| Po/SaV/LL14/02/US | SaV/GIII | LL14 | AY425671 |
| Po/SaV/OH-JJ259/00/US | SaV/GIII | JJ259 | AY826423 |
| Po/SaV/OH-MM280/03/US | SaV/GIII | MM280 | AY823308 |
| Po/SaV/OH-QW270/03/US | SaV/GIII | QW270 | AY826426 |
| Po/SaV/OH-JJ681/00/US | SaV/GVI | JJ681 | AY974192 |
| Hu/Hou7-1181/90/US | SaV/GIV | Hou7 | AF435814 |
| Hu/Argentina39/Arg | SaV/GV | Arg39 | AY289803 |
| Ra/RHDV/GH/1988/GE | Lagovirus | RHDV | M67473 |
| Fe/FCV/F9/1958/US | Vesivirus | FCV | M86379 |
| Hu/Norwalk/69/US | Norovirus | Norwalk | M87661 |
Phylogenetic analysis based on the nt and aa alignments were constructed using the neighbor-joining method and the UPGMA method of molecular evolutionary gentetics analysis (MEGA version 3.1) with a pairwise distance (Kumar et al., 2004). A sequence similarity search was performed for the porcine calicivirus capsid and the RDRP protein using the LALIGN Query program of the GENESTREAM network server at Institut de Génétque Humaine, Montpellier, France (http://www.eng.uiowa.edu/∼tscheetz/sequence-analysis/examples/LALIGN/lalign-guess.html). The recombinant viruses were identified using the recombinant identification program (RIP, http://hivweb.lanl.gov/RIP/RIPsubmit.html) (Siepel et al., 1995).
3. Results
3.1. Prevalence of porcine SaVs in pigs with diarrhea in Korea
In order to determine the prevalence of porcine SaVs in Korean piglets with diarrhea, a total of 237 fecal samples from diarrheic pigs in 78 farms were screened by RT-PCR and nested PCR using five sets of primer pairs (Table 1). The RT-PCR and nested PCR revealed different sensitivities for porcine SaVs according to each primer pair (Table 3 ). Between the RT-PCR and nested PCR analyses for the porcine SaVs, the PECnF/nR primer pair (targeting a 380 bp fragment of the RdRp region) used for nested PCR was the most sensitive (Table 3). The fecal samples were determined to be positive if at least one sample tested positive to each primer pair. Sixty-nine (29.1%) out of 237 diarrheic samples (22 [28.2%] of 78 herds) tested positive for porcine SaVs (Table 3).
Table 3.
Summary of the RT-PCR and nested PCR assay results for the individual swine fecal samples
| Period | Porcine fecal samples positive by using different PCR assaysa (no. of positive/total no. of samples [%]) |
No. of total positive samplesb | ||||
|---|---|---|---|---|---|---|
| RT-PCR P289/290 | RT-PCR PEC45/46 | Nested PCRc PECnF/nR | RT-PCR CapF/R | Nested PCRc CapnF/nR | ||
| 2004 | 4/80 (5%) | 3/80 (3.8%) | 23/80 (28.8%) | 8/80 (10.0%) | 11/80 (13.8%) | 26/80 (32.5%) |
| 2005 | 11/157 (7.1%) | 2/157 (1.3%) | 27/157 (17.2%) | 14/157 (8.9%) | 28/157 (17.8%) | 43/157 (27.3%) |
| Total | 15/237 (6.3%) | 5/237 (2.1%) | 50/237 (21.1%) | 22/237 (9.3%) | 39/237 (16.5%) | 69/237 (29.1%) |
Virus and target proteins for each primer pair are listed in Table 1.
Fecal samples were considered positive if at least one positive fecal sample was detected in the same sample by each primer pair.
The number of nested PCR positive samples also contained the number of RT-PCR positive samples.
A primer pair p289/290 was originally designed to detect both NoVs and SaVs by RT-PCR (Jiang et al., 1999b). Moreover, RT-PCR using this primer under low stringency was reported to be able to detect rotaviruses according to their cross-reactivity (Ludert et al., 2004). In order to determine if fragments amplified by this primer pair were from NoVs or rotaviruses, eight DNA fragments that had been amplified by RT-PCR with the same primer pair were sequenced (Table 3). Their nt sequences were similar to the porcine SaVs but not to noroviruses or rotaviruses (data not shown).
3.2. Other enteric pathogens
Of the 69 porcine SaV-positive fecal specimens from 22 porcine herds with diarrhea, 29 fecal samples (12.2%) from four herds (5.1%) tested positive for the porcine SaV alone, while the other 40 fecal samples (16.9%) from the 18 herds (23.1%) also tested positive for other enteric pathogens (Table 4 ). In addition, 105 fecal specimens from the 45 herds that tested negative for porcine SaV also tested positive for other enteric pathogens (Table 4). No enteric pathogens were detected in 63 fecal samples from 11 herds.
Table 4.
Summary of the enteric pathogens present in the fecal samples obtained from pigs with diarrhea
| Enteric pathogens presenta | No. of farms (%)b | No. of samples (%)c |
|---|---|---|
| PSaV alone | 4 (5.1) | 29 (12.2) |
| PSaV plus PRV A | 3 (3.8) | 19 (8.0) |
| PSaV plus PRV B | 1 (1.3) | 1 (0.4) |
| PSaV plus PRV C | 1 (1.3) | 2 (0.8) |
| PSaV, PRV A plus PRV B | 1 (1.3) | 1 (0.4) |
| PSaV, PRV A plus PRV C | 2 (2.6) | 6 (2.5) |
| PSaV, PRV A, PRV B plus PRV C | 1 (1.3) | 1 (0.4) |
| PSaV plus salmonellosis | 1 (1.3) | 1 (0.4) |
| PSaV, PRV A plus salmonellosis | 1 (1.3) | 2 (0.8) |
| PSaV, PRV C plus salmonellosis | 1 (1.3) | 1 (0.4) |
| PSaV, PRV A, PRV B plus salmonellosis | 1 (1.3) | 1 (0.4) |
| PSaV plus PEDV | 1 (1.3) | 1 (0.4) |
| PSaV, PRV A plus PEDV | 1 (1.3) | 1 (0.4) |
| PSaV, PRV A, PRV C plus PEDV | 1 (1.3) | 1 (0.4) |
| PSaV, PRV A, salmonellosis plus swine dysentery | 1 (1.3) | 1 (0.4) |
| PSaV, PRV C, salmonellosis plus swine dysentery | 1 (1.3) | 1 (0.4) |
| Other enteric pathogens detected | 45 (57.7) | 105 (44.3) |
| No enteric pathogens detected | 11 (14.1) | 63 (26.6) |
| Total | 78 (100) | 237 (100) |
PSaV: porcine sapovirus; PRV A–C: porcine groups A, B, C rotaviruses; PEDV: porcine epidemic diarrhea virus.
Number of positive farms.
Number of positive fecal samples.
3.3. Seasonal and geographic distribution of BNoVs in calves with diarrhea in Korea
Seasonally, porcine SaV infections are more prevalent in fecal samples of pigs in winter than in the other seasons: 26 (24.8%) out of 105 fecal samples (8/33 farms) were positive in spring; 10 (28.6%) out of 35 fecal samples (4/14 farms) were positive in summer; 19 (29.2%) out of 65 fecal samples (5/16 farms) were positive in autumn; and 14 (43.8%) out of 32 fecal samples (5/15 farms) were positive in winter. The geographic distribution of the porcine SaVs in each province revealed Jeonnam to be the most affected province, where 38 (34.2%) of 111 samples tested positive for porcine SaVs (Table 5 ). In contrast, there were no positive fecal samples obtained from Chungnam province. However, this might have been due to the low number of fecal samples analyzed.
Table 5.
Geographic distribution of Korean porcine sapoviruses and genotype classification of the strains according to phylogenetic analysis
| Provinces | No. of samples | No. of farms | Strains | Characterization ofa |
Seasonsb | Genotypesc | Accession no. | |
|---|---|---|---|---|---|---|---|---|
| Capsid | RdRp | |||||||
| Jeonnam | 38/111 (34.2%) | 12/37 (32.4%) | Po/SaV/JN-SC58/04/Korea | x | x | Autumn | GIII/1 | DQ389599/DQ389612 |
| Po/SaV/JN-YG69/04/Korea | x | x | Winter | GIII/1 | DQ389600/DQ389613 | |||
| Po/SaV/JN-YG71/04/Korea | x | Winter | DQ389614 | |||||
| Po/SaV/JN-GS110/05/Korea | x | Spring | DQ389620 | |||||
| Po/SaV/JN-MA113/05/Korea | x | Spring | GIII/2 | DQ389602 | ||||
| Po/SaV/JN-MA128/05/Korea | x | x | Spring | GIII/1 | DQ389605/DQ389622 | |||
| Po/SaV/JN-MA130/05/Korea | x | Spring | GIII/1 | DQ389606 | ||||
| Po/SaV/JN-YG137/05/Korea | x | Spring | GIII/1 | DQ389608 | ||||
| Po/SaV/JN-GH143/05/Korea | x | Spring | DQ389624 | |||||
| Po/SaV/JN-GH144/05/Korea | x | x | Spring | GIII/1 | DQ389609/DQ389625 | |||
| Jeonbuk | 27/89 (30.3%) | 7/26 (26.9%) | Po/SaV/JB-SC53/04/Korea | x | Autumn | DQ389611 | ||
| Po/SaV/JB-JE91/05/Korea | x | x | Winter | GIII/1 | DQ389601/DQ389615 | |||
| Po/SaV/JB-JE92/05/Korea | x | Winter | DQ389616 | |||||
| Po/SaV/JB-JE100/05/Korea | x | Spring | DQ389617 | |||||
| Po/SaV/JB-JE101/05/Korea | x | Spring | DQ389618 | |||||
| Po/SaV/JB-JE102/05/Korea | x | Spring | DQ389619 | |||||
| Po/SaV/JB-IS122/05/Korea | x | x | Spring | GIII/1 | DQ389603/DQ389631 | |||
| Po/SaV/JB-IS123/05/Korea | x | Spring | GIII/1 | DQ389604/DQ389621 | ||||
| Po/SaV/JB-JE133/05/Korea | x | x | Spring | GIII/1 | DQ389607/DQ389623 | |||
| Po/SaV/JB-GC155/05/Korea | x | Spring | GIII/1 | DQ389610 | ||||
| Chungnam | 0/13 (0.0%) | 0/2 (0.0%) | ||||||
| Chungbuk | 1/9 (11.1%) | 1/6 (16.7%) | ||||||
| Gyeonggi | 2/10 (20.0%) | 1/5 (20.0%) | ||||||
| Jeju | 1/5 (20.0%) | 1/2 (50.0%) | ||||||
| Total | 69/237 (29.1%) | 22/78(28.2%) | ||||||
Strains characterized by sequence comparison and phylogenetic analysis of capsid gene and/or RdRP region.
Strains detected in each season.
Genogroup classification of porcine sapoviruses by the intra- and inter-cluster distances of capsid gene sequences.
3.4. Genetic diversity of porcine SaVs
The genetic diversity of the porcine SaVs and their genetic relatedness to human strains was investigated by sequencing 757 nt of the nt 5698 to nt 6454 region of the capsid gene from 12 porcine SaVs which were amplified strongly by RT-PCR. By drawing comparisons of the deduced aa sequences among the caliciviruses within Caliciviridae, the porcine SaVs showed low aa identity with the human SaV (17.7–35.1%), the human NoV (11.1–13.3%), rabbit hemorrhagic disease virus (RHDV) (13.9–18.2%), and feline calicivirus (FCV) (10.6–14.3%) strains. Among the porcine SaVs examined, the paired comparisons showed the two strains with the most similar sequence identity to be the Po/SaV/JB-IS122/05/Korea (GIII-1) and Po/SaV/JN-MA128/05/Korea (GIII-1) strains (99.1%), whereas the Po/SaV/JN-MA113/05/Korea (GIII-2) and Po/SaV/JN-MA130/05/Korea (GIII-1) strains (67.7%) showed the lowest shared sequence identities. In addition, the most distant strains between the Korean and American porcine SaVs were those of the Po/SaV/JN-SC58/04/Korea (GIII-1) and Po/SaV/OH-MM280/03/US (GIII-1) strains, sharing 69.1% aa identity. On the other hand, the closest strains between the Korean and American porcine SaVs were the Po/SaV/JB-JE133/Korea (GIII-1) and Po/SaV/OH-JJ259/00/US (GIII-1) strains, sharing 90.2% aa identity.
3.5. Potential new genotypes of porcine SaVs
The intra- and inter-cluster distances among the 17 porcine SaV capsid sequences were compared in order to determine if genogroup III porcine SaVs belongs to a new genotypes. The UPGMA phylogenetic analysis based on the 717 nt sequence and its deduced 239-aa capsid sequence showed that all the Korean SaVs were clustered within genogroup III (Fig. 1 ). Genogroup III of the porcine SaVs could be further divided into at least two genotypes. The inter-cluster distance between the Po/SaV/JN-MA113/05/Korea (potential GIII-2 genotype) and GIII-1 strains was 0.236–0.405. When the inter-cluster distance between the Po/SaV/JN-MA113/05/Korea and GIII-1 strains was compared, the inter-cluster distance became 0.302–0.405 with the exception of the Po/SaV/JB-JE91/05/Korea strain. The inter-cluster distances were also comparable to the distances between the established GI and GII because the distance between GII-1 and GII-4, and GII-2 and GII-3 was 0.254–0.273 and 0.273–0.283, respectively. This data supports the assignment of two new genetic clusters within genogroup III of the porcine SaVs.
Fig. 1.
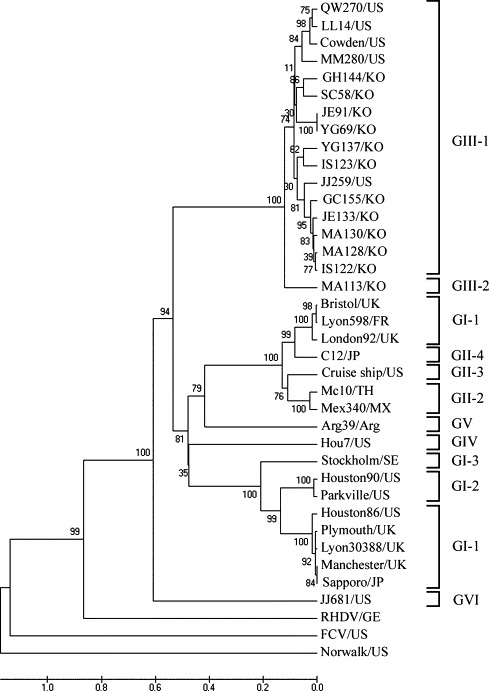
UPGMA phylogenetic tree based on the partial capsid region of the human and animal caliciviruses. The Po/SaV/JN-MA113/05/Korea newly identified porcine sapovirus genotype strain is underlined.
3.6. Potential recombinant porcine SaVs
Seven strains, which were amplified by both primer pairs for the partial RdRp region (380 bp) and the capsid gene (757 bp), respectively, were selected to determine if the potential recombination events occurred between the porcine SaVs (Table 5). Although the Po/SaV/JB-IS123/05/Korea strain shared the highest nt identity (93.1%) with the Po/SaV/Cowden/80/US strain in the RdRp region, it shared only 84.6% nt identity with the Po/SaV/Cowden/80/US strain in the capsid region. However, the Po/SaV/JB-IS123/05/Korea strain shared the highest nt identity (92.9%) with the Po/SaV/JN-YG69/05/Korea capsid region. In contrast, the Po/SaV/JN-YG69/05/Korea strain shared the highest nt identity (92.7%) with the Po/SaV/JB-JE91/05/Korea strain in the RdRp region but shared only 83.8% nt identity with the Po/SaV/JB-JE91/05/Korea strain in the capsid region. Nevertheless, the Po/SaV/JN-YG69/05/Korea strain shared the highest nt identity (92.9%) with the Po/SaV/JB-IS123/05/Korea capsid region. This suggests that Po/SaV/JB-IS123/05/Korea is a potential recombinant between the Po/SaV/Cowden/80/US-like (RdRp region) and Po/SaV/JN-YG69/05/Korea-like (capsid region) SaVs. Moreover, a recombination event of Po/SaV/JN-YG69/05/Korea probably occurred for the Po/SaV/JB-JE91/05/Korea-like (RdRp region) and Po/SaV/JB-IS123/05/Korea-like (capsid region) strains.
In order to determine if recombination occurred between the porcine SaVs, RIP analysis was performed by placing the partial RdRp (380 bp) and partial capsid (757 bp) sequence of Po/SaV/JB-IS123/05/Korea, Po/SaV/JN-YG69/05/Korea, Po/SaV/JB-JE91/05/Korea and Po/SaV/Cowden/80/US strains (Fig. 2A and B). The Po/SaV/JB-IS123/05/Korea strain showed high homology with the Po/SaV/Cowden/80/US strain in the RdRp region but it changed to a shared high homology with the Po/SaV/JN-YG69/05/Korea strain in the capsid region (Fig. 2A). The Po/SaV/JN-YG69/05/Korea strain changed abruptly to a high identity with the Po/SaV/JB-JE91/05/Korea strain in the capsid region (Fig. 2B).
Fig. 2.
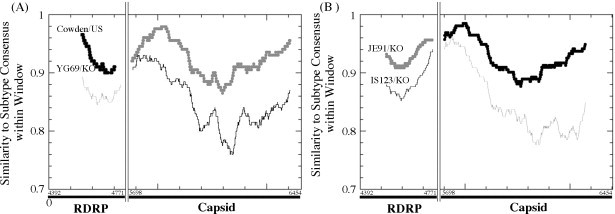
Indication of the Po/SaV/JN-YG69/04/Korea and Po/SaV/JB-IS123/05/Korea strains as potential recombinant viruses. (A) The Po/SaV/JB-IS123/05/Korea was a query sequence compared with the background Po/SaV/Cowden/80/US and Po/SaV/JN-YG69/04/Korea. RIP analysis was performed based on the partial RdRp and partial capsid nucleotide sequences. (B) The Po/SaV/JN-YG69/04/Korea was a query sequence compared with the background Po/SaV/JB-JE91/05/Korea and Po/SaV/JB-IS123/05/Korea. The homologous region are indicated as thick dots on the plot when the query sequence is similar to the background sequence. The nucleotide position of the partial RdRp and partial capsid is indicated. Window size of 70 and statistical of 90%.
4. Discussion
Like other RNA viruses, human SaVs show broad genomic sequence diversity between circulating strains, which is partly due to the poor template fidelity of their RNA polymerases (Green et al., 2001). The genetic divergence of porcine SaVs in different countries is unavailable because the sequence data of only a few porcine SaVs (mainly from the US) is available. In this study, genetically variable porcine SaVs from swine were detected in South Korea and their genetic relationship with human and other porcine SaVs as well as to the other three genera (Lagovirus, Vesivirus, and Norovirus) in the Caliciviridae were examined. Analyses of the partial capsid genes of the 12 porcine SaVs showed that they shared a lower aa and nt sequence identity with the human SaVs, human and animal NoVs, FCV and RHDV, which is consistent with a previous report (Wang et al., 2005a). The aa identity within the GIII porcine SaVs ranged from 67.7 to 99.1%, in which the genetic divergence was observed not only among Korean porcine SaVs but also between the Korean and American porcine SaVs. This suggests there is genetic variability in the porcine SaVs belonging to genogroup III. In addition, all the Korean and most US porcine SaVs sequenced belonged to the SaV/GIII genotype. Therefore, these results, along with others (Farkas et al., 2004, Guo et al., 1999, Schuffenecker et al., 2001, Wang et al., 2005a), indicated that SaV/GIII is the dominant genogroup of porcine SaVs.
Pairwise nt sequence and phylogenetic distance analysis of the partial capsid gene sequences provide useful information for identifying potential new genetic clusters for SaVs (Farkas et al., 2004, Schuffenecker et al., 2001). Although pairwise and phylogenetic analyses allowed the human SaVs within all genogroups to be divided into at least nine genotypes, the porcine SaVs could not be classified into genotypes because the sequences of only a few porcine SaVs have been reported. Interestingly, this study identified one strain (Po/SaV/JN-MA113/05/Korea) forming a second new porcine SaV/GIII genotype, which is tentatively called GIII/2. This strain had a 0.236–0.405 inter-cluster distance with the other strains in the same genogroup, which is comparable to the distances between the established GI and GII SaVs. Whether or not the two genotypes among the GIII porcine SaVs identified in this study represent sero- or antigen-types cannot be predicted based on their sequence alone. Therefore, more detailed antigenic analysis of porcine SaVs will be needed to understand their diversity and evolution as well as to develop classification schemes that can reduce the level of confusion and assist international molecular epidemiological studies.
All the recombination events occurred in the RdRp–capsid junction region, whose nucleotide sequence is generally well conserved among the NoV and SaV genotypes (Jiang et al., 1999a, Katayama et al., 2004, Wang et al., 2005a). This conservation is likely to facilitate a recombination event when the nucleic acid sequences of the parental strains come into physical contact with infected cells, e.g., during copy choice recombination (Worobey and Holmes, 1999). Therefore, genetic recombination may be a common mechanism for generating diversity among the genotypes or genogroups. Recombinant viruses within SaVs and NoVs have been reported in humans, cattle and pigs (Han et al., 2004, Jiang et al., 1999a, Katayama et al., 2004, Wang et al., 2005a). In this study, it was found that the Po/SaV/JN-YG69/04/Korea was a potential recombinant strain between the Po/SaV/JB-JE91/05/Korea-like strain (RdRp region) and the Po/SaV/JB-IS123/05/Korea-like strain (capsid, after RdRp) of the same genotype. In contrast, the Po/SaV/JB-IS123/05/Korea was a potential recombinant strain between the Po/SaV/Cowden/80/US-like strain (RdRp region) and the Po/SaV/JN-YG69/04/Korea-like strain (capsid, after RdRp) of the same genotype. In South Korea, post-weaning piglets introduced into each farm are frequently sourced from different farms in different provinces, which suggest that the Po/SaV/JN-YG69/04/Korea strain has different geographic origins. Because South Korea imports live pigs from the US, American porcine SaV strains may be the parent strains of the recombinant viruses such as the Po/SaV/JB-IS123/05/Korea strain. Therefore, the capsid gene diversity of the porcine SaVs detected in these farms is most likely to be a reflection of this diverse geographic sourcing.
Since a porcine SaV infection was first reported in the US in 1980s (Saif et al., 1980), it has only been reported in a few countries including the US, Netherlands, Venezuela and the United Kingdom (Bridger, 1990, Martínez et al., 2006, Saif et al., 1980). However, the precise epidemiology of porcine SaV infections in the other countries is not known. We previously reported the prevalence of porcine SaV infections in South Korea during the 2001–2002, in which 8.8% of diarrheic fecal samples tested positive by nested PCR (Kim et al., 2006). However, the more recent status of porcine SaV infections is unknown. This paper presents a large-scale epidemiological study of the prevalence of porcine SaV infections in diarrheic piglets in South Korea during 2004–2005. Although five sets of primer pairs were used to detect those porcine SaV strains, it is likely that some strains escaped detection. At this time, no primer pair or combination of primers appears suitable for the universal detection of porcine SaVs. However, among those primer pairs, the PECnF/nR primer pair used for nested PCR showed high sensitivity in detecting porcine SaVs. When fecal samples were determined to be positive based on at least one positive fecal sample detected by each primer pair, porcine SaVs were detected in 29.1% fecal samples obtained from six provinces. Overall, the high prevalence and widespread geographical distribution of porcine SaVs suggest that these viruses are widespread in piglets with diarrhea in South Korea. In addition, it is not known why there was a higher prevalence of porcine SaV infections during 2004–2005 compared with during 2001–2002. One possible explanation is that the SaVs infections might have been spread out to many more farms after 2001–2002.
Porcine SaVs are believed to be the causative agent of piglet diarrhea because they induce enteric disease in gnotobiotic piglets and cause pathology in the small intestine (Flynn et al., 1988, Guo et al., 2001a, Guo et al., 2001b). However, it is possible that other porcine enteric pathogens play a role in the clinical and pathological presentation of this disease. This is because many other enteric viruses have been found to be associated with diarrhea in piglets and cause lesions of villous atrophy [transmissible gastroenteritis virus, porcine epidemic diarrhea virus (PEDV), groups A, B, C porcine rotaviruses (PRV A–C), etc.]. In this study, 12.2% diarrheic fecal samples tested positive for porcine SaVs alone, while 16.9% of diarrheic fecal samples testing positive for porcine SaVs and other pathogens including the PRV A–C, PEDV, etc. This suggests that a number of enteric pathogens, either singly or in combination, can influence the clinical course of porcine SaV infections.
Porcine NoVs have been detected in the fecal samples of adult pigs (Sugieda et al., 1998, van Der Poel et al., 2000, Wang et al., 2005b). In order to determine if porcine NoVs are involved in piglet diarrhea, a RT-PCR assay was performed with the primer pair p280/290, which was designed to detect NoVs, SaVs and rotaviruses (Jiang et al., 1999b, Ludert et al., 2004) and all the positive fragments were sequenced. Their nt sequences were similar to the porcine SaVs but not to the NoVs or to the rotaviruses, which suggests that NoVs are less likely to be involved in piglet diarrhea in the age groups tested. This might explain why porcine NoVs have only been detected in the normal feces of adult or finisher pigs but not in piglets (Sugieda et al., 1998, Wang et al., 2005b).
In summary, this study identified genetically diverse porcine SaVs consisting of at least two genotypes and two potential recombinant porcine SaVs (Po/SaV/JB-IS123/05/Korea and Po/SaV/JN-YG69/04/Korea strains), which were analogous to a previously reported human and porcine SaV recombinant. In addition, these results demonstrate that porcine SaV infections are widespread in piglets with diarrhea in South Korea.
Acknowledgments
This study was supported by the National Veterinary Research and Quarantine Service (NVRQS), Ministry of Agriculture and Forestry, Korea Health 21 R&D (01-PJ10-PG6-01GM02-002) by the Ministry of Health and Welfare, and the Regional Technology Innovation Program (RTI05-01-01) of the Ministry of Commerce, Industry and Energy (MOCIE), Republic of Korea. The authors acknowledge a graduate fellowship provided by the Korean Ministry of Education and Human Resources Development through the Brain Korea 21 project.
References
- Bridger J.C. Small viruses associated with gastroenteritis in animals. In: Saif L.J., Theil K.W., editors. Viral Diarrheas of Man and Animals. CRC Press; Boca Raton: 1990. pp. 161–182. [Google Scholar]
- Cho K.O., Hasoksuz M., Nielsen P.R., Chang K.O., Lathrop S., Saif L.J. Cross-protection studies between respiratory and calf diarrhea and winter dysentery coronavirus strains in calves and RT-PCR and nested PCR for their detection. Arch. Virol. 2001;146:2401–2419. doi: 10.1007/s007050170011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elschner M., Prudlo J., Hotzel H., Otto P., Sachse K. Nested reverse transcriptase-polymerase chain reaction for the detection of group A rotaviruses. J. Vet. Med. B. 2002;49(2):77–81. doi: 10.1046/j.1439-0450.2002.00510.x. [DOI] [PubMed] [Google Scholar]
- Farkas T., Zhong W.M., Jing Y., Huang P.W., Espinosa S.M., Martinez N., Morrow A.L., Ruiz-Palacios G.M., Pickering L.K., Jiang X. Genetic diversity among sapoviruses. Arch. Virol. 2004;149:1309–1323. doi: 10.1007/s00705-004-0296-9. [DOI] [PubMed] [Google Scholar]
- Flynn W.T., Saif L.J., Moorhead P.D. Pathogenesis of porcine enteric calicivirus-like virus in four-day-old gnotobiotic pigs. Am. J. Vet. Res. 1988;49:819–825. [PubMed] [Google Scholar]
- Geissler K., Schneider K., Platzer G., Truyen B., Kaaden O.R., Truyen U. Genetic and antigenic heterogeneity among feline calicivirus isolates from distinct disease manifestations. Virus Res. 1997;48:193–206. doi: 10.1016/s0168-1702(97)01440-8. [DOI] [PubMed] [Google Scholar]
- Gouvea V., Allen J.R., Glass R.I., Fang Z.Y., Bremont M., Cohen J., McCrae M.A., Saif L.J., Sinarachatanant P., Caul E.O. Detection of group B and C rotaviruses by polymerase chain reaction. J. Clin. Microbiol. 1991;29(3):519–523. doi: 10.1128/jcm.29.3.519-523.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green K.Y., Ando T., Balayan M.S., Berke T., Clarke I.N., Estes M.K., Matson D.O., Nakata S., Neill J.D., Studdert M.J., Thiel H.J. Taxonomy of the caliciviruses. J. Infect. Dis. 2000;18:S322–S330. doi: 10.1086/315591. [DOI] [PubMed] [Google Scholar]
- Green K.Y., Chanock R.M., Kapikian A.Z. Human caliciviruses. In: Knipe D.M., Howley P.M., editors. Field Virology. 4th ed. Lippinocott Williams & Wilkins; Philadelphia: 2001. pp. 841–874. [Google Scholar]
- Guo M., Evermann J.E., Saif L.J. Detection and molecular characterization of cultivable caliciviruses from clinically normal mink and enteric caliciviruses associated with diarrhea in mink. Arch. Virol. 2001;146:479–493. doi: 10.1007/s007050170157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M., Hayes J., Cho K.O., Parwani A.V., Lucas L.M., Saif L.J. Comparative pathogenesis of tissue culture-adapted and wild-type Cowden porcine enteric calicivirus (PEC) in gnotobiotic pigs and induction of diarrhea by intravenous inoculation of wild-type PEC. J. Virol. 2001;75:9239–9251. doi: 10.1128/JVI.75.19.9239-9251.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M., Chang K.O., Hardy M.E., Zhang Q., Parwani A.V., Saif L.J. Molecular characterization of a porcine enteric calicivirus genetically related to Sapporo-like human caliciviruses. J. Virol. 1999;73:9625–9631. doi: 10.1128/jvi.73.11.9625-9631.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M.G., Smiley J.R., Thomas C., Saif L.J. Genetic recombination between two genotypes of genogroup III bovine noroviruses (BoNVs) and capsid sequence diversity among BoNVs and Nebraska-like bovine enteric caliciviruses. J. Clin. Microbiol. 2004;42:5214–5224. doi: 10.1128/JCM.42.11.5214-5224.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Espul C., Zhong W.M., Cuello H., Matson D.O. Characterization of a novel human calicivirus that may be a naturally occurring recombinant. Arch. Virol. 1999;144:2377–2387. doi: 10.1007/s007050050651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Huang P.W., Zhong W.M., Farkas T., Cubitt D.W., Matson D.O. Design and evaluation of a primer pair that detects both Norwalk- and Sapporo-like caliciviruses by RT-PCR. J. Virol. Meth. 1999;83:145–154. doi: 10.1016/s0166-0934(99)00114-7. [DOI] [PubMed] [Google Scholar]
- Katayama K., Miyoshi T., Uchino K., Oka T., Tanaka T., Takeda N., Hansman G.S. Novel recombinant sapovirus. Emerg. Infect. Dis. 2004;10:1874–1876. doi: 10.3201/eid1010.040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.J., Cho H.S., Cho K.O., Park N.Y. Detection and molecular characterization of porcine enteric calicivirus in Korea, genetically related to sapoviruses. J. Vet. Med. B: Infect. Dis. Vet. Public Health. 2006;53:155–159. doi: 10.1111/j.1439-0450.2006.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim L., Chang K.O., Sestak K., Parwani A., Saif L.J. Development of a reverse transcription-nested polymerase chain reaction assay for differential diagnosis of transmissible gastroenteritis virus and porcine respiratory coronavirus from feces and nasal swabs of infected pigs. J. Vet. Diagn. Invest. 2000;12(4):385–388. doi: 10.1177/104063870001200418. [DOI] [PubMed] [Google Scholar]
- Kumar S., Tamura K., Nei M. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Kubota S., Sasaki O., Amimoto K., Okada N., Kitazima T., Yasuhara H. Detection of porcine epidemic diarrhea virus using polymerase chain reaction and comparison of the nucleocapsid protein genes among strains of the virus. J. Vet. Med. Sci. 1999;61(7):827–830. doi: 10.1292/jvms.61.827. [DOI] [PubMed] [Google Scholar]
- La T., Phillips N.D., Hampson D.J. Development of a duplex PCR assay for detection of Brachyspira hyodysenteriae and Brachyspira pilosicoli in pig feces. J. Clin. Microbiol. 2003;41:3372–3375. doi: 10.1128/JCM.41.7.3372-3375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludert J.E., Alcala A.C., Liprandi F. Primer pair p289–p290, designed to detect both noroviruses and sapoviruses by reverse transcription-PCR, also detects rotaviruses by cross-reactivity. J. Clin. Microbiol. 2004;42:835–836. doi: 10.1128/JCM.42.2.835-836.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez, M.A., Alcalá, A.C., Carruyo, G., Botero, L., Liprandi, F., Ludert, J.E., 2006. Molecular detectionof porcine enteric caliciviruses in Venezuelan farms. Vet. Microbiol. (Epub ahead of print). [DOI] [PubMed]
- Ohlinger V.F., Haas B., Meyers G., Weiland F., Thiel H.J. Identification and characterization of the virus causing rabbit hemorrhagic disease. J. Virol. 1990;64:3331–3336. doi: 10.1128/jvi.64.7.3331-3336.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.J., Jeong C., Yoon S.S., Choy H.E., Saif L.J., Park S.H., Kim Y.J., Jeong J.H., Park S.I., Kim H.H., Lee B.J., Cho H.S., Kim S.K., Kang M.I., Cho K.O. Detection and characterization of bovine coronaviruses in fecal specimens of adult cattle with diarrhea during the warmer seasons. J. Clin. Microbiol. 2006;44:3178–3188. doi: 10.1128/JCM.02667-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saif L.J., Bohl E.H., Theil K.W., Cross R.F., House J.A. Rotavirus-like, calicivirus-like, and 23-nm virus-like particles associated with diarrhea in young pigs. J. Clin. Microbiol. 1980;12:105–111. doi: 10.1128/jcm.12.1.105-111.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuffenecker I., Ando T., Thouvenot D., Lina B., Aymard M. Genetic classification of “Sapporo-like viruses”. Arch. Virol. 2001;146:2115–2132. doi: 10.1007/s007050170024. [DOI] [PubMed] [Google Scholar]
- Siepel A.C., Halpern A.L., Macken C., Korber B.T. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res. Hum. Retroviruses. 1995;11:1413–1416. doi: 10.1089/aid.1995.11.1413. [DOI] [PubMed] [Google Scholar]
- Smiley J.R., Chang K.O., Hayes J., Vinje J., Saif L.J. Characterization of an enteropathogenic bovine calicivirus representing a potentially new calicivirus genus. J. Virol. 2002;76:10089–10098. doi: 10.1128/JVI.76.20.10089-10098.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugieda M., Nagaoka H., Kakishima Y., Ohshita T., Nakamura S., Nakajima S. Detection of Norwalk-like virus genes in the caecum contents of pigs. Arch. Virol. 1998;143:1215–1221. doi: 10.1007/s007050050369. [DOI] [PubMed] [Google Scholar]
- van Der Poel W.H., Vinje J., van Der Heide R., Herrera M.I., Vivo A., Koopmans M.P. Norwalk-like calicivirus genes in farm animals. Emerg. Infect. Dis. 2000;6:36–41. doi: 10.3201/eid0601.000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.H., Han M.G., Funk J.A., Bowman G., Janies D.A., Saif L.J. Genetic diversity and recombination of porcine sapoviruses. J. Clin. Microbiol. 2005;43:5672–5963. doi: 10.1128/JCM.43.12.5963-5972.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.H., Han M.G., Cheetham S., Souza M., Funk J.A., Saif L.J. Porcine noroviruses related to human noroviruses. Emerg. Infect. Dis. 2005;11:1874–1881. doi: 10.3201/eid1112.050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobey M., Holmes E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999;80:2535–2543. doi: 10.1099/0022-1317-80-10-2535. [DOI] [PubMed] [Google Scholar]