

Abstract
The distribution of FCoV Types I and II in a Portuguese cat population was studied by a RT-PCR assay targeting the 3′-end of the viral RNA. For a period of 3 years, 120 samples were collected and 57 were found positive for FCoV RNA. Within the positive samples the presence of FCoV Type I was found in 79%. Type II was only detected in 3.5% in animals with Feline Infectious Peritonitis. The remaining 17.5% could not be differentiated. These viral sequences, comprising a region within gene S were further subjected to a heteroduplex mobility assay (HMA) detecting the presence of viral quasispecies in 17% of the samples. Phylogenetic analysis for FCoV Type I revealed high genetic diversity between the Portuguese sequences and other previously characterized strains, while Type II tree showed a higher genetic homogeneity. This study confirmed the presence of FCoV Types I and II circulating in Portugal and detected high genetic diversity between circulating strains suggesting that the virus persists within the host as mixed viral populations.
Keywords: FCoV, Genetic diversity, Heteroduplex mobility assay
1. Introduction
Feline Coronaviruses (FCoVs) are members of genus Coronavirus, Family Coronaviridae. Two variants of FCoV have been described, Feline Enteric Coronavirus responsible for an enteric disease and Feline Infectious Peritonitis Virus (FIPV) the etiological agent of FIP, a fatal disease of domestic and wild cats. FIP development in a small percentage of animals infected with FCoV is related to the occurrence of genomic mutations supported by the close genetic relationship between the higher virulent virus and the parental FCoV (Herrewegh et al., 1997, Vennema et al., 1998, Vennema, 1999). Additionally, FCoVs were classified as Types I and II according to their serological and genomic features. Type I is strictly feline. Type II FCoV, although sharing with Type I a high degree of similarity in the 3′-end of the genome, is antigenic and genetically more related to Canine Coronavirus (CCoV) (Hohdatsu et al., 1991), suggesting that Type II resulted from a homologous and independent recombination event within the S and the M genes of Type I FCoV and CCoV (Herrewegh et al., 1998). The current information on the geographical distribution of FCoV indicates a high incidence of Type I in Europe, Japan and USA (Addie et al., 2003, Hohdatsu et al., 1992). However in Japan and Taiwan detection of FIP was associated with Type II FCoV (Hohdatsu et al., 1992, Lin et al., 2009). Presently there is no available information about the molecular epidemiology of FCoV in Portugal. Due to the pathogenic impact of this virus in the feline health it was our purpose to investigate the presence of FCoV Types in the Portuguese cat population using a RT-PCR–nested PCR assay previously reported (Addie et al., 2003) and to assess its genetic diversity by heteroduplex mobility assay (HMA) and phylogenetic analysis.
2. Materials and methods
2.1. Sampling collection
The biological materials included rectal swabs, stool, plasma, thoracic and ascite fluids collected from animals attending the Veterinary Hospital of the Faculty of Veterinary Medicine, Lisbon.
2.2. Detection of FCoV RNA
FCoV RNA was extracted using the QIAmp Viral RNA or Qiagen RNeasy mini kits (Qiagen), following the manufacturer's instructions. A fragment of 177 bp located in the 3′-UTR was amplified by RT-PCR followed by a nested PCR using two primer pairs described by Herrewegh et al. (1995). The 25 μL reactions were amplified with one step RT-PCR Fidelity Taq 2x Master Mix (GE Healthsciences), 50 pmol of each primer and 10 μL of RNA sample. The nPCR was performed with PCR Fidelity Taq 2x Master Mix (GE Healhsciences), 50 pmol of each primer and 5 μL of the previous amplification in a 25 μL reaction
2.3. Differentiation between Types I and II FCoV
Positive FCoV RNA samples were subjected to a RT-PCR assay to amplify a 376–283 bp fragment located in the 3′-region of the spike gene (S), followed by a nPCR to amplify a 360–218 bp internal fragment, for differentiation of Type I from Type II FCoV according to the fragment length (Addie et al., 2003). The amplification was performed with one step RT-PCR Fidelity Taq 2x Master Mix (GE Healthsciences), 5 μL of RNA sample and primers Iffs, Icfs, Iubs in a 25 μL reaction, followed by a nested PCR with PCR Fidelity Taq 2x Master Mix (GE Healthsciences), primers nIffles, nIcfs, nIubs (Addie et al., 2003) and 2.5 μL of the previous reaction in 25 μL final volume. The amplicons were analyzed in a 2.5% agarose gel stained with 0.5 μg/mL of ethidium bromide in 1× TBE.
2.4. Heteroduplex mobility assay (HMA) and cloning
The amplicons were subjected to a HMA before cloning (Delwart et al., 1993). Briefly, 10 μL of the second round S amplicons were combined with 1 μL of 10× annealing buffer (1 M NaCl, 100 mM Tris pH 7.8, 20 mM EDTA), heated to 94 °C/5 min, cooled on ice, added of 1 μL of loading buffer (50% glycerol, 0.01% of xyleno cyanol) and loaded in a 5% polyacrylamide gel (29.2:0.8 acrylamide:bisacrylamide) poured into plates of 15 cm × 15 cm. The electrophoresis was carried at a constant 150 V/3 h in a vertical gel apparatus (Biorad) in 1× TBE. After electrophoresis the gel was stained with 0.5 μg/mL of ethidium bromide in 1× TBE and observed in a UV transiluminator. The visualized homoduplexes bands were cuted, eluted at 56 °C/5 min in 200 μL H2O of which 1 μL was re-amplified using the nPCR primers set described above. The resulting amplicons were cloned in pGEM®-T Easy Vector System (Promega) according to the manufacture's conditions and sequenced by STAB Vida Lda, Portugal. The nucleotide sequences were submitted to Genbank and were assigned the accession numbers EU327694–EU327752. Sample identification was included in Table 1 . FCoV. Types I and II assignment based on the amplicons length were in accordance with the nucleotide sequence of each sample.
Table 1.
Identification and origin of the Portuguese FeCoV sequences, amplified from healthy and FIP (S) animals. The genetic diversity of the amplicons was evaluated by heteroduplex mobility assay and each positive sample was re-amplified. The number of corresponding sequences is indicated (a–c).
| Identification | Heteroduplexes | Origin | |
|---|---|---|---|
| 2004 | |||
| 13LVI_2004 | Positive (a;b) | Lisbon, Portugal | |
| 19LVI_2004 | Positive (a;b) | Lisbon, Portugal | |
| 20LVI_2004 | Negative | Lisbon, Portugal | |
| 22LVI_2004 | Positive | Palmela, Portugal | Sharing household |
| 23LVI_2004 | Negative | Palmela, Portugal | |
| 25LVI_2004 | Positive (a;b) | Palmela, Portugal | |
| 2005 | |||
| 3LVI_2005 | Negative | Lisbon, Portugal | |
| 10LVI_2005 | Positive (a;b) | Lisbon, Portugal | Sharing household |
| 11LVI_2005 | Positive (a;b) | Lisbon, Portugal | |
| 14LVI_2005 | Negative | Lisbon, Portugal | |
| 27LVI_2005 | Negative | Lisbon, Portugal | |
| 33LVI_2005 | Positive (a;b) | Rio Mouro, Portugal | Multi-cat environment |
| 37LVI_2005 | Negative | Lisbon, Portugal | |
| 38LVI_2005 | Negative | Lisbon, Portugal | |
| 48LVI_2005 | Negative | Lisbon, Portugal | |
| 50LVI_2005 | Negative | Lisbon, Portugal | |
| 58LVI_2005 | Negative | Lisbon, Portugal | |
| 59LVI_2005 | Negative | Lisbon, Portugal | |
| 71LVI_2005 | Negative | Lisbon, Portugal | |
| 80LVI_2005 | Negative | Vila Nova de Gaia, Portugal | |
| 65LVI_2005 (S) | Negative | Lisbon, Portugal | Sharing household |
| 74LVI_2005 | Negative | Lisbon, Portugal | |
| 81LVI_2005 | Negative | Lisbon, Portugal | |
| 86LVI_2005 | Negative | Lisbon, Portugal | |
| 87LVI_2005 | Negative | Lisbon, Portugal | |
| 88LVI_2005 | Negative | Lisbon, Portugal | |
| 90LVI_2005 | Negative | Lisbon, Portugal | |
| 93LVI_2005 | Negative | Lisbon, Portugal | |
| 94LVI_2005 | Negative | Lisbon, Portugal | |
| 96LVI_2005 | Negative | Lisbon, Portugal | |
| 99LVI_2005 | Positive (a;b) | Lisbon, Portugal | |
| 102LVI_2005 | Negative | Lisbon, Portugal | |
| 2006 | |||
| 12LVI_2006 | Negative | Lisbon, Portugal | |
| 13LVI_2006 | Negative | Lisbon, Portugal | |
| 15LVI_2006 | Negative | Lisbon, Portugal | |
| 19LVI_2006 | Negative | Lisbon, Portugal | |
| 20LVI_2006L | Negative | Lisbon, Portugal | |
| 20LVI_2006PBMC | Negative | Lisbon, Portugal | |
| 20LVI_2006Z | Negative | Lisbon, Portugal | |
| 24LVI_2006 | Negative | Lisbon, Portugal | |
| 24LVI_2006D1 | Negative | Lisbon, Portugal | |
| 1FG_2006 (S) | Positive (a;b) | Lisbon, Portugal | |
| 1LG_2006 | Negative | Lisbon, Portugal | |
| 28LVI_2006 | Negative | Lisbon, Portugal | |
| 29LVI_2006 | Negative | Lisbon, Portugal | |
| 32LVI_2006 (S) | Positive (a;b,c) | Lisbon, Portugal | |
| 38LVI_2006 | Negative | Lisbon, Portugal | |
2.5. Phylogenetic analysis
The nucleotide sequences were aligned (PILEUP from GCG Wisconsin Package v. 10.0), including sequences of FCoV Type I (KU_2 AN-D32044; UCD1 AN-AB088222; Black AN-AB088223; C2_0301UK AN-AY159737; J8_0196UK AN-AY159758; FQ1_1099UK AN-AY159754; H9_1100UK AN-AY159770; FUI_1191UK AN-AY159735) and FCoV Type II (79_1683 AN-X80799; 79_1146 AN-X06170; G6_1095 AN-AY159784). Due to its recombinant status FCoV Type II sequences were aligned and submitted to phylogenetic analysis separately from Type I sequences.
The phylogenetic relationship between sequences was inferred by genetic distances between pairs of sequences using Kimura's two parameter method (DNADIST, Phylip Package). The values of transition/transversion ratio and nucleotide frequencies were calculated from the data sets (TREE-PUZZLE) (Strimmer and von Haeseler, 1997). The resulting trees were constructed by Neighbor Joining (NEIGHBOR, Phylip Package) and the branching order reliability was evaluated by bootstrap analysis of 1000 replicates (SEQBOOT, Phylip Package). To maximize the phylogenetic information of the nucleotide alignment closely related sequences were excluded but were indicated between brackets following the sequence chosen to represent each set of viruses as summarized; 65LVI_05 [74LVI_05, 81LVI_05, 86LVI_05, 87LVI_05]; 14LVI_05 [10LVI_05a]; 19LVI_04a [19LVI_04b]; 58LVI_05 [59LVI_05a/b]; 1Lg_06 [1Fg_06a].
3. Results
3.1. Detection of Types I and II FCoV
For 3 years 120 samples were collected from domestic cats to detect FCoV RNA due to clinical suspicion of FIP and to confirm viral excretion from healthy animals. Fifty-seven samples were found FCoV positive (47.5%) and afterwards processed for Types I and II differentiation. Out of those 57 positive samples, 45 (79%) were classified as Type I FCoV and were obtained from healthy animals or with FIP. Only two samples (3.5%) from animals with FIP, were classified as Type II. No amplification was obtained from ten samples (17.5%).
3.2. Detection of viral quasispecies
The presence of viral quasispecies within the animal, was evaluated by HMA for each sample (Fig. 1 ). This assay detects the presence of fully (homoduplexes) and partially (heteroduplexes) complementary DNA chains formed between highly related sequences during the process of denaturing and annealing. Homoduplexes molecules tend to migrate faster, while heteroduplexes show a reduced mobility in a non-denaturing PAGE. The observation of two or more homoduplexes bands, plus the corresponding heteroduplexes bands indicates the presence of different DNA populations within the sample, named viral quasispecies and mirror the in vivo genetic diversity of the virus (Delwart et al., 1993).
Fig. 1.
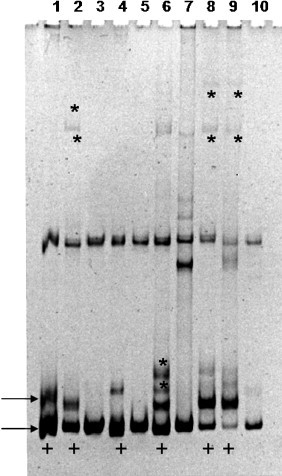
Heteroduplex mobility assay of S amplicons from samples 10LVI_05 (1); 11LVI_05 (2); 3LVI_05 (3); 25LVI_04 (4); 23LVI_04 (5); 22LVI_04 (6); 20LVI_04 (7); 10LVI_04 (8); 13LVI_04 (9); 12LVI_04 (10), included in Type I FCoV. Lanes 1, 2, 4, 6, 8 and 9 (+) show the presence of two homoduplexes bands (→), corresponding to fully complementary DNA chains. The heteroduplexes bands (*) consistent to partially complementary DNA chains with reduced mobility are clearly visible in lanes 2, 6, 8 and 9. In lanes 3, 5 and 7 no homo/heteroduplexes are present. The photograph of the ethidium bromide-stained gels is displayed as reversed image.
Within the 47 S gene amplicons 37 samples (78.7%) showed no genetic diversity whereas in 10 samples (21.3%) two homoduplexes bands were observed and individually amplified. Comparison of the nucleotide sequences of the heteroduplexes positive samples revealed similarities of 90–99% along the 320nt in Type I and 170nt in Type II FCoV. Although very similar, six sequences (13LVI_04b; 19LVI_04b; 10LVI_05a; 11LV_05a; 33LVI_05b; 99LVI_05b) showed a 5′-end variation length of 28–56 nucleotides justifying the homo/heteroduplexes formation. Sequences 33LVI_05a/b, 25LVI_04a/b, 10LVI_05a/b and 11LVI_05a/b were amplified from animals with multi-cat environment (Table 2 ).
Table 2.
Genetic diversity between amplicons subjected to HMA. The length and the percentages of genetic similarity between homoduplexes are shown.
| Identification | Sequence | Similarity (%) | FCoV |
|---|---|---|---|
| 13LVI_04a | 320 bp | 317/320 (99%) | Type I |
| 13LVI_04b | 348 bp | Extra 5′ 28nt | |
| 19LVI_04a | 320 bp | 320/320 (100%) | |
| 19LVI_04b | 348 bp | Extra 5′ 28nt | |
| 22LVI_04a | 347 bp | 344/347 (99%) | |
| 22LVI_04b | 347 bp | ||
| 25LVI_04a | 320 bp | 290/320 (90%) | |
| 25LVI_04b | 320 bp | ||
| 10LVI_05a | 348 bp | 316/320 (98%) | |
| 10LVI_05b | 320 bp | Extra 5′ 28nt | |
| 11LVI_05a | 348 bp | 303/320 (94%) | |
| 11LVI_05b | 320 bp | Extra 5′ 28nt | |
| 33LVI_05a | 320 bp | 319/320 (99%) | |
| 33LVI_05b | 348 bp | Extra 5′ 28nt | |
| 99LVI_05a | 320 bp | 319/320 (99%) | |
| 99LVI_05b | 376 bp | Extra 5′ 56nt | |
| 1FLVI_06a | 320 bp | 299/320 (93%) | |
| 1FLVI_06b | 320 bp | ||
| 32LVI_06a | 170 bp | 168/170 (98%) | Type II |
| 32LVI_06b | 170 bp |
3.3. Phylogenetic analysis
The genetic relationships between the Portuguese sequences of FCoV and previously assigned sequences from isolates of Type revealed a heterogeneous clustering of sequences with several tree branches supported by high bootstrap values (Fig. 2 ). Sequences from distinct viral quasispecies obtained from the same animal as well as sequences from cats sharing the same household were included in branch A indicating the presence of different viral strains within each confined cat population and within the same individual host. Sequences 83 LVI_05, 88LVI_05, 90LVI_05, 94LVI_05 and 96LVI_05 came from cats sharing the same household than the source animal of sequence 65LVI_05 [74LVI_05, 81LVI_05, 86LVI_05, 87LVI_05]. However sequence 65LVI_05 was positioned outside group A, in a separate branch with 80% of bootstrap value. The above-mentioned sequences were obtained from a multi-cat household where a clinical FIP episode (65LVI_05) had been reported. Although all the other animals were FCoV positive, none developed FIP. Sequence 11LVI_05b was also included in branch A but sequence 11LVI_05a, obtained from the same cat was positioned in branch B. Sequence 25LVI_04a was grouped in branch A though it was obtained from the same animal as sequence 25LVI_04b placed in a different branch. These two viral quasispecies (25LVI_04a/b) came from the same household of the source cats of sequences 22LVI_04a/b and 23LVI_04, which in this analysis were found distributed in outlier branches supported by 100% of bootstrap value. Sequences 20LVI_06Z (stool), 20LVI_06PBMC (blood) and 20LVI_06L (ascites) amplified from the same animal, were placed in two separate branches, one with 100% bootstrap value (20LVI_06PBMC and 20LVI_06L).
Fig. 2.
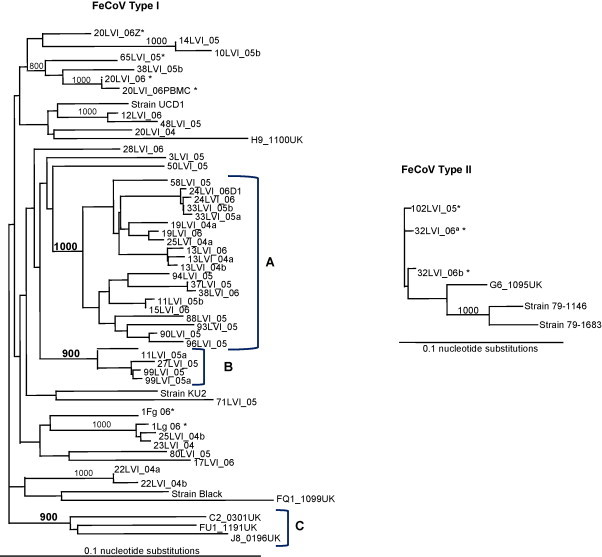
Phylogenetic analysis of FCoV Types I and II sequences based on the 3′-end of the S gene. The trees were constructed by Neighbor Joining based on the genetic distances between the nucleotide sequences, calculated by Kimura two-parameter using a transition/transversion rate of 3.23 for Type I sequences and 9.99 for Type II sequences. A boostrap analysis of 1000 replicates was performed and values above 80% are indicated on the branches. Branches A–C include FCoV sequences with the lowest value of nucleotide substitutions within each branch. Sequences marked with an asterisk (*) were amplified from animals with FIP.
Type II FCoV phylogenetic tree included previously assigned FCoV Type II sequences and two Portuguese sequences obtained during this study, one positive to viral quasispecies (32LVI_06; 32LVI_06a/b). All the sequences clustered in one branch, although FIPV 79-1146 and 79-1683 grouped together with 100% of bootstrap value. Due to the low number of nucleotides considered in the alignment (170b), a likelihood mapping analysis was preformed (TREE-PUZZLE) to assess its phylogenetic content, revealing 62.86% of fully resolved, 0% of partially resolved and 37.14% of unresolved quartets demonstrating therefore its reliable use to estimate phylogenetic relationships.
4. Discussion
Until now, no previous study on the presence and prevalence of FCoV Types I and II in domestic cats had been done in Portugal. In our analysis 47.5% of the samples were positive in the RT-PCR assay (57/120) and were obtained either from healthy animals or from cats with FIP. Moreover, the vast majority of the amplicons was classified as Type I. Type II was found only in two samples both from cats with FIP. The values found for FCoV Types I and II were in accordance with the distribution reported in Europe and USA (Addie et al., 2003, Benetka et al., 2004) although in Japan and Taiwan a higher prevalence of FCoV Type II was observed in animals with FIP (Hohdatsu et al., 1992, Lin et al., 2009). In our study even though the two FCoV Type II sequences were obtained from sick cats, the low incidence of this viral biotype does not allow any relationship between FCoV II and symptomatic disease.
The evaluation of FCoV genetic diversity revealed 21% of the amplicons positive for viral quasispecies. These viral variants were found within the same animal but also in samples collected from animals sharing a multi-cat environment. This finding supports the viral internal mutation theory (Dye and Siddell, 2007) and confirms the animal exposure to divergent viral strains. The significance of FCoV quasispecies had already been studied (Battilani et al., 2003) suggesting a correlation between high viral genetic diversity within the host and development of disease. However from our data it was not possible to correlate viral diversity with virus subtype or disease therefore a more extensive study should be performed to assess this connection.
The phylogenetic analysis of FCoV Types I and II sequences revealed an assorted distribution of sequences in Type I tree, showing high genetic diversity of the FCoV Portuguese sequences, its divergence toward European and Japanese sequences (Fq1_1099UK, H9_1100UK, and strains KU2, Black and UCD1) confirming the previous findings.
Interestingly, FCoV Type II phylogenetic tree although with a limited number of Portuguese and previously characterized Type II sequences, showed a homogeneous distribution of sequences as also reported by Lin et al. (2009) in a similar study, indicative of high similarity between distantly related sequences (UK; Netherlands; Portugal). The genetic diversity of the Portuguese FCoV sequences confirmed and strengthens the evidence that this virus persists within the host as mingled viral populations whose interaction with the animal may or not result in severe disease.
In conclusion this study provided information on the FCoV molecular epidemiology in Southern Europe which was unavailable until now and revealed high genetic diversity between the Portuguese and previously characterized FCoV sequences.
Acknowledgments
This work was sponsored by the Center for Interdisciplinary Research in Animal Health (CIISA), FMV, UTL, Portugal. We are grateful to our colleagues from the Veterinary Hospital (FMV) for their collaboration in sampling collection.
References
- Addie D.D., Schaap I.A., Nicolson L., Jarrett O. Persistence and transmission of natural type I feline coronavirus infection. J. Gen. Virol. 2003;84(Pt 10):2735–2744. doi: 10.1099/vir.0.19129-0. [DOI] [PubMed] [Google Scholar]
- Battilani M., Coradin T., Scagliarini A., Ciulli S., Ostanello F., Prosperi S., Morganti L. Quasispecies composition and phylogenetic analysis of feline coronaviruses (FCoVs) in naturally infected cats. FEMS Immunol. Med. Microbiol. 2003;39:141–147. doi: 10.1016/S0928-8244(03)00237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benetka V., Kubber-Heiss A., Kolodziejek J., Nowotny N., Hofmann-Parisot M., Mostl K. Prevalence of feline coronavirus types I and II in cats with histopathologically verified feline infectious peritonitis. Vet. Microbiol. 2004;99:31–42. doi: 10.1016/j.vetmic.2003.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwart E.L., Shpaer E.G., Louwagie J., McCutchan F.E., Grez M., Rubsamen-Waigmann H., Mullins J.I. Genetic relationships determined by a DNA heteroduplex mobility assay: analysis of HIV-1 env genes. Science. 1993;262:1257–1261. doi: 10.1126/science.8235655. [DOI] [PubMed] [Google Scholar]
- Dye C., Siddell S.G. Genomic RNA sequence of feline coronavirus strain FCoV C1Je. J. Feline Med. Surg. 2007;9:202–213. doi: 10.1016/j.jfms.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A., de Groot R.J., Cepica A., Egberink H.F., Horzinek M.C., Rottier P.J. Detection of feline coronavirus RNA in feces, tissues, and body fluids of naturally infected cats by reverse transcriptase PCR. J. Clin. Microbiol. 1995;33:684–689. doi: 10.1128/jcm.33.3.684-689.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A., Mahler M., Hedrich H.J., Haagmans B.L., Egberink H.F., Horzinek M.C., Rottier P.J., de Groot R.J. Persistence and evolution of feline coronavirus in a closed cat-breeding colony. Virology. 1997;234:349–363. doi: 10.1006/viro.1997.8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A., Smeenk I., Horzinek M.C., Rottier P.J., de Groot R.J. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 1998;72:4508–4514. doi: 10.1128/jvi.72.5.4508-4514.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohdatsu T., Sasamoto T., Okada S., Koyama H. Antigenic analysis of feline coronaviruses with monoclonal antibodies (MAbs): preparation of MAbs which discriminate between FIPV strain 79-1146 and FECV strain 79-1683. Vet. Microbiol. 1991;28:13–24. doi: 10.1016/0378-1135(91)90096-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohdatsu T., Okada S., Ishizuka Y., Yamada H., Koyama H. The prevalence of types I and II feline coronavirus infections in cats. J. Vet. Med. Sci. 1992;54:557–562. doi: 10.1292/jvms.54.557. [DOI] [PubMed] [Google Scholar]
- Lin C.-N., Su B.-L., Wang C.-H., Hsieh M.-W., Chueh T.-J., Chueh L.-L. Genetic diversity and correlation with feline infectious peritonitis of feline coronavirus type I and II: a 5-year study in Taiwan. Vet. Microbiol. 2009;136:233–239. doi: 10.1016/j.vetmic.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K., von Haeseler A. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6815–6819. doi: 10.1073/pnas.94.13.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H. Genetic drift and genetic shift during feline coronavirus evolution. Vet. Microbiol. 1999;69:139–141. doi: 10.1016/S0378-1135(99)00102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Poland A., Foley J., Pedersen N.C. Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses. Virology. 1998;243:150–157. doi: 10.1006/viro.1998.9045. [DOI] [PMC free article] [PubMed] [Google Scholar]