

Abstract
Cholesterol is involved in the life cycle of many viruses. Here, we examined the role of cholesterol for both viral envelope and target cell membrane for bovine herpesvirus type 1 (BoHV-1) infection. Cholesterol depletion by pretreatment of Madin–Darby bovine kidney (MDBK) cells with a cholesterol-sequestering drug methyl-β-cyclodextrin (MβCD), inhibited the production of BoHV-1 in a dose-dependent manner. This inhibitory effect was partially reversed by cholesterol replenishment, indicating that the reduction was caused by cholesterol depletion. Cholesterol depletion at the post-entry stage only had a mild effect on the virus production. However, cell membrane cholesterol depletion did not reduce the virus attachment. In addition, treatment of BoHV-1 particles with MβCD also reduced the virus infectivity significantly and the effect was partially reversed by addition of exogenous cholesterol. Taken together, these data implicated that cell membrane cholesterol mainly contributed to BoHV-1 entry into MDBK cells and the viral envelope cholesterol was also essential for the virus infectivity.
Keywords: BoHV-1, Lipid raft, Cholesterol, MβCD
1. Introduction
BoHV-1 is an alphaherpesvirus causing abortion, respiratory, and genital infections in cattle (Tikoo et al., 1995, Straub, 2001). For the latent infection mainly established in ganglion cells and tonsils, it becomes a reservoir of BoHV-1 allowing the virus spreading to susceptible animals (Winkler et al., 2000). Now it is worldwide distributed and tends to be endemic in most populations, though national and regional variations occur (Kampa et al., 2009).
Lipid rafts refer to putative microdomains distributed in the membrane, which are similar to the rafts floating in the sea. It is thought that lipid rafts are enriched in cholesterol, glycosphingolipids, sphingomyelin, phospholipids with long, unsaturated acyl chains, glycosylphosphatidylinositol (GPI)-linked proteins and at least some membrane-spanning proteins (Simons and van Meer, 1988, Simons and Ikonen, 1997, Simons and Toomre, 2000). Approximately 15–20% of the plasma membrane surface area is believed to consist of lipid rafts (Parolini et al., 1999, Schutz et al., 2000). The tight packaging domains are associated with numerous important biological processes, such as transmembrane signal transduction, apoptosis, cell adhesion, migration, synaptic transmission, organization of the cytoskeleton, and protein sorting during endocytosis and exocytosis (Brown and London, 1998, Simons and Toomre, 2000, Harris and Siu, 2002, Tsui-Pierchala et al., 2002). However, the extraction of cholesterol by various chemicals destroys the raft organization and consequently blocks biological processes that depend on lipid rafts. So the function of lipid rafts in some biological process can be evaluated by studying the role of cholesterol.
Accumulating evidence suggests that many pathogens, especially viruses require cholesterol at multiple stages of their lifecycles. Human immunodeficiency virus type-1 infection requires cholesterol, both in the target cell membrane and the viral envelope (Guyader et al., 2002, Liao et al., 2001, Liao et al., 2003). Many viruses require cholesterol in the target cell membrane, such as Semliki Forest virus (Phalen and Kielian, 1991, Ahn et al., 2002), severe acute respiratory syndrome-coronavirus (Li et al., 2007) and SV40 virus (Anderson et al., 1996). For influenza virus and duck hepatitis B virus, the presence of cholesterol in its viral envelope is critical, but it is not essential in the target cell (Sun and Whittaker, 2003, Funk et al., 2008). Whereas numerous strains of flavivirus dengue virus and yellow fever virus 17D enter and infect cells independent of cholesterol (Umashankar et al., 2008).
In this study, we sought to clarify the importance of cholesterol at cell membrane and viral envelope for BoHV-1 infection of MDBK cells. We found that intact cell membrane cholesterol was indispensable for the virus entry and the viral envelope cholesterol was also imperative for the virus infectivity.
2. Materials and methods
2.1. Cells and virus
MDBK cells were maintained in DMEM (Gibco BRL) supplemented with 10% horse serum, and passaged whenever they became confluent. The Colorado 1 strain of BoHV-1 was used for this study (Wang et al., 2003). Viruses were propagated in MDBK cells as previously described (Lawrence et al., 1986), except that the horse serum was used in place of calf serum. The collected culture medium from infected MDBK cells was titrated and stored at −80 °C.
2.2. Antibodies and reagents
Bovine sera against BoHV-1 was raised by the infection of animals, experimentally (kindly provided by Dr. Fei Xue, Harbin Veterinary Research Institute, China). Goat anti-bovine immunoglobulin secondary antibody coupled to fluorescein isothiocyanate (FITC) was purchased from Beijing Biosynthesis Biotechnology Co., Ltd. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), methyl-β-cyclodextrin (MβCD), cholesterol and fluorescent cholera toxin B subunit (CTB-FITC) were bought from Sigma. The reagents are water soluble.
2.3. Plaque formation assay
The process of plaque formation assay was followed as previously described with some modification (Dasika and Letchworth, 1999). In brief, the monolayer cells in 24-well plates were infected with 200 μl diluted virus and incubated for indicated times at 37 °C, the inoculum was removed and overlaid with DMEM supplemented with 1% horse serum and 0.5% agarose. The cytopathic effect of BoHV-1 is very evident such as causing the infected cells enlarged or ballooned and then become rounded, take on a refractile appearance. So the plaques were directly counted under microscope after 48 h incubation at 37 °C, when the CPE was clearly evident.
2.4. Assays for cell viability and cholesterol level
Cell viability was assessed by the MTT assay as previously described with some modification. (Guo et al., 2007). Briefly, MDBK cells were seeded in 96-well microplates at 1 × 104 cells/well. Eight replicates were mock treated or treated with MβCD at various concentrations of 2.5, 5, 10, 15 and 20 mM for 30 min at 37 °C in a humidified atmosphere of 5% CO2. After further incubation in a final volume of 200 μl DMEM for 48 h, 30 μl MTT solution (2 mg/ml in PBS) was added to each well. The cells were then incubated at 37 °C for 4 h, the supernatant was removed and 150 μl of DMSO was added to each well to solubilize the formazan. Then, the microplate was shaken on a rotary platform for 10 min. Finally, the absorbance value was measured at wave length of 550 nm using a Wellscan (Labsystems, Santa Fe, NM, USA). The mean optical density of the cell control wells was assigned a value of 100%.
Efficiency of cholesterol removal by MβCD was assessed with flow cytometry assay. MDBK cells seeded in six-well plates for three replicates were mock treated or treated with 2.5, 5, 10 and 20 mM MβCD for 30 min at 37 °C. After washing three times with ice-cold PBS, the detached cells were incubated with CTB-FITC at 10 μg/ml in serum-free DMEM for 30 min on ice. Flow cytometry was carried out using a Becton–Dickinson FACSCalibur flow cytometer with 10,000 events collected and data were analysed using FlowJo software, version 7.1.3.
2.5. Effect of the cell membrane cholesterol depletion on the virus entry
To determine if the cholesterol depletion at the virus entry stage affects the virus replication, the monolayer cells seeded in 24-well plates were mock treated or treated with MβCD at the concentrations of 2.5, 5, 10, 20 mM for 30 min at 37 °C before incubation for 1 h with BoHV-1 of 50TCID50. After treatment with citrate buffer (40 mM citric acid, 10 mM KCl, 135 mM NaCl, pH 3.0) for exactly 1 min to inactivate cell membrane bound but unpenetrated virions as described elsewhere (Chung et al., 2005, Highlander et al., 1987), the cells were further incubated to determine the virus yield with plaque formation assay.
For cholesterol replenishment, monolayer of MDBK cells in 24-well plates mock pretreated or pretreated with 10 mM MβCD for 30 min at 37 °C, were supplemented or mock supplemented with 400 μg/ml cholesterol in DMEM and incubated for 1 h at 37 °C (Li et al., 2007). After washing, the cells were subjected to BoHV-1 infection, followed by the treatment with citric buffer, the virus yield was determined with plaque formation assay.
2.6. Effect of cell membrane cholesterol depletion at the post-entry stage on the virus replication
To analyze whether the depletion of cholesterol after virus entry affects virus replication, MDBK cells seeded in 24-well plates were pre-incubated for 1 h with BoHV-1 of 50TCID50. Following the treatment with citrate buffer they were treated with 10 mM MβCD for 30 min at 37 °C (Treat: +1). In the controls, the cells were mock treated or treated with 10 mM MβCD before the virus infection (Mock and Treat: −0.5). The virus yield was determined with plaque formation assay.
2.7. Effect of MβCD treatment on the virus binding
To analyze whether cell membrane cholesterol reduction affects BoHV-1 binding to the target cells, MDBK cells in 6-well plates were either mock treated or were treated with 5 and 10 mM MβCD respectively for 30 min at 37 °C, washed twice with ice-cold medium. Thereafter, cells were incubated with BoHV-1 of 1000 TCID50 in ice-cold DMEM for 1 h at 4 °C allowing virus attaches to the cells. After extensive washing and freezing–thawing, the cell-associated virus was collected and titrated in 96-well microplates with MDBK cells. And the result was expressed as TCID50 calculated using Reed–Muench formula (Reed and Muench, 1932). Alternatively, the infected cells were fixed with 4% paraformaldehyde for 15 min at room temperature, and stained with bovine anti-BoHV-1 serum followed by FITC-conjugated anti-bovine immunoglobulin. After the incubation the cells were washed twice with ice-cold PBS, and the bound virion particles were analyzed on FACS.
2.8. Virus envelope cholesterol sequestration, replenishment and infectivity
To determine if the virus cholesterol was essential for the virus infectivity, BoHV-1 of 1000 TCID50 was incubated with MβCD at the concentration of 5 and 10 mM respectively for 30 min at 37 °C. To remove MβCD, the virus was pelleted by ultracentrifugation at 20,000 rpm (Beckman SW28 rotor) for 1 h, 4 °C, as described elsewhere (Karger et al., 1998). The controls were treated with the same manner but without treatment with MβCD. Thereafter the virus was resuspended in DMEM and its infectivity was determined in 24-well plates with plaque formation assay.
In viral cholesterol replenishment, the virus was mock treated or treated with 10 mM MβCD for 30 min at 37 °C, and the supernatant was removed by ultracentrifugation as described above. The virus was suspended with DMEM or DMEM containing exogenous cholesterol of 400 μg/ml and incubated for 1 h at 37 °C. Then the virus was titrated by plaque formation assay.
3. Results
3.1. Depletion of cholesterol from the MDBK cell membrane by 10 mM MβCD had little effect on the cell viability
MβCD, a derivative of a cyclic oligomer of glucose with a lipophilic property (Pitha et al., 1988), is a cholesterol-sequestering agent. This drug is not incorporated into the membrane but selectively extracts membrane cholesterol by binding it in a central non-polar cavity (Ilangumaran and Hoessli, 1998). In this study, we used MβCD to investigate the role of cholesterol in BoHV-1 life cycle.
The cell toxicity of MβCD on MDBK cells was measured with MTT method. MDBK cells were mock or treated with various concentrations of MβCD in serum-free DMEM. As showed in Fig. 1a MβCD reduced the cell viability in a dose-dependent manner compared to the mock treated control cells. MβCD at the concentration of less than 15 mM, have little toxicity on MDBK cells with the cell viability of more than 90%.
Fig. 1.
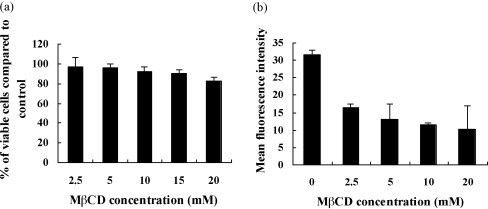
Depletion of cholesterol in MDBK cells by MβCD and the effect on cell proliferation. (a) Cell viability after 30 min treatment with MβCD at various concentrations was determined using the MTT method, with the number of viable cells being expressed as a % of mock treated cells. Experiments were repeated three times, and the error bars indicate the standard deviations of three independent experiments. (b) Depletion of cholesterol from the membrane of MDBK cells was measured by detecting CTB-FITC binding to GM1 of the cells. Cells were mock treated or treated with MβCD at various concentrations. CTB binding occurred for 30 min on ice and was measured by FACS where cellular autofluorescence (without CTB-FITC) was used as the control. Experiments were repeated twice, and the error bars indicate the standard deviations of two independent experiments.
The ganglioside GM1 is a commonly used raft marker and its association with membrane lipid microdomains has been proved by microscopy and biochemical experiments (Montixi et al., 1998, Janes et al., 1999). Here, depletion of MDBK cell membrane cholesterol by MβCD was measured by detecting fluorescent CTB-FITC binding to GM1 with FACS. As shown in Fig. 1b, CTB-FITC binding to drug-treated cells on ice was reduced in a dose-dependent manner compared to mock treated cells, confirming that MβCD effectively sequestered the membrane cholesterol. The treatment with 10 mM MβCD decreased the cholesterol level with an average range of 63% compared to mock treated control cells. So the drug concentration of 10 mM was appropriate for this study with negligible cell toxicity and efficiently cholesterol depletion.
3.2. Pretreatment of MDBK cells with MβCD inhibited BoHV-1 production and the effect was partially reversed by cholesterol replenishment
To determine whether cholesterol on the target-cell surface was important for BoHV-1 entry, MDBK cells were treated with various concentrations of MβCD. The pretreated or mock pretreated cells were infected with BoHV-1, followed by treatment with citric buffer to remove the cell bound but unpenetrated virions. They were cultured for the examination of virus yield with plaque formation assay. As shown in Fig. 2a, a significant impairment of the virus production by MβCD treatment in a dose-dependent manner was observed, suggesting that cell membrane cholesterol is necessary for the replication of BoHV-1 in MDBK cells, especially at the virus entry stage.
Fig. 2.
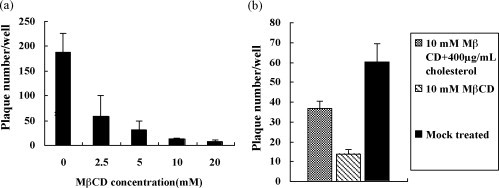
Cholesterol depletion reduced BoHV-1 entry (a) and the inhibitory effect was partially reversed by exogenous cholesterol (b). (a) Productive BoHV-1 entry into MDBK cells treated with MβCD at various concentrations was measured by plaque formation assay. Mock treated cells were used as a control. (b) MβCD treated cells were treated with 400 μg/ml cholesterol for 1 h before BoHV-1 infection. After treatment with citric buffer, the productive BoHV-1 entry was measured by plaque formation assay. Mock treated cells and the cells treated with MβCD but not with cholesterol were used as controls. Experiments were repeated three times, and the error bars indicate the standard deviations of three independent experiments.
To confirm that the inhibitory effects were due to cholesterol depletion, cell membrane cholesterol was replenished with exogenous cholesterol. After treatment with 10 mM MβCD for 30 min, cholesterol at the final concentration of 400 μg/ml was used for the replenishment and the virus yield was investigated with plaque formation assay. As shown in Fig. 2b, this inhibitory effect was partially reversed, and virus production was restored by the cholesterol replenishment. The production of BoHV-1 was restored to an average of 50% by the replenishment with 400 μg/ml exogenous cholesterol, compared to the mock treated cells. These results confirmed that the reduction of virus production was specifically due to the cholesterol depletion, and this inhibitive effect was partially reversible.
3.3. Cholesterol depletion at the post-entry stage had a mild effect on BoHV-1 production
We attempted to investigate whether cell membrane cholesterol depletion at the post-entry stage affected virus production. MDBK cells were infected with BoHV-1, after incubation for 1 h the cell-bound virus was inactivated, and the cells were treated with 10 mM MβCD for 30 min at 37 °C (Treat: +1). Then the virus production was determined with plaques formation assay. As shown in Fig. 3 , compared to the treatment before virus entry (Treat: −0.5), the treatment after virus entry (Treat: +1) had only a mild inhibitory effect, suggesting that cholesterol was mainly required during the virus entry stage.
Fig. 3.
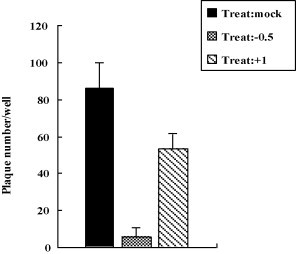
Cholesterol depletion at the post-entry stage mildly affected the virus yield. MDBK cells were first infected with BoHV-1, 1 h post-inoculation followed by treatment with citric buffer they were then treated with 10 mM MβCD for 30 min (Treat: +1). In the controls cells were mock treated (Treat: mock) or pretreated with 10 mM MβCD for 30 min (Treat: −0.5), and then infected with BoHV-1, followed by treatment with citric buffer. The virus production was measured by plaque formation assay. Experiments were repeated three times, and the error bars indicate the standard deviations of three independent experiments.
3.4. Cholesterol depletion did not impair the virus binding
To further pinpoint the stage at which cell membrane cholesterol was required for the virus infection, we carried out virus binding assay. Equal amounts of BoHV-1 were added to the mock treated or MβCD-treated MDBK cells for 1 h at 4 °C. After extensive washing the amount of cell-bound virus was titrated and expressed as TCID50. As shown in Fig. 4a, no apparent decrease in virus adsorption to MβCD-treated MDBK cells compared to the mock treated cells was observed. In addition, another method FACS was used to determine the amount of cell bound virions (Fig. 4b). It confirmed that it was essentially the same in MβCD-treated cells and mock treated cells. Therefore, cholesterol depletion did not affect virus binding.
Fig. 4.
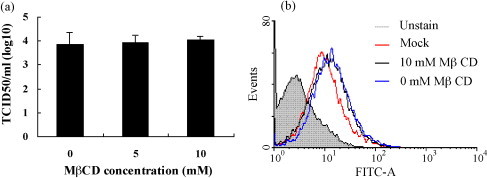
Cholesterol depletion did not reduce attachment of BoHV-1. (a) Binding of BoHV-1 to the target cells was determined with virus titration. MDBK cells were mock treated or were treated with 5 and 10 mM MβCD for 30 min, and then binding of BoHV-1 to the cells was processed at 4 °C for 1h. The amount of cell-associated virus was titrated in MDBK cells. (b) Binding of BoHV-1 to the target cells was determined with FACS. Cells were mock treated or treated with 10 mM MβCD and then infected or mock infected with BoHV-1 at 4 °C. At 1h later, the cells were fixed with 4% paraformaldehyde and stained with bovine anti-BoHV-1 serum followed by FITC-conjugated anti-bovine immunoglobulin. Experiments were repeated three times, and the error bars indicate the standard deviations of three independent experiments. Image is one of three independent experiments.
3.5. Viral cholesterol was required for BoHV-1 infectivity
To analysis whether the viral envelope cholesterol was required for BoHV-1 infectivity, the virus were mock treated or treated with MβCD of 5 and 10 mM for 30 min respectively, and the reagent MβCD was removed by ultracentrifugation. Then the virus was titrated by plaque formation assay. As shown in Fig. 5a, the exposure of BoHV-1 to MβCD resulted in a dose-dependent manner of inhibitory effect on the virus infectivity. Also the inhibitory effect of MβCD could be partially reversed by the replenishment of viral envelope cholesterol. Cholesterol replenishment restored virus infectivity to an average of 23% compared to the mock treated control (Fig. 5b). These results suggested that the envelope cholesterol of BoHV-1 was also required for the virus infectivity.
Fig. 5.
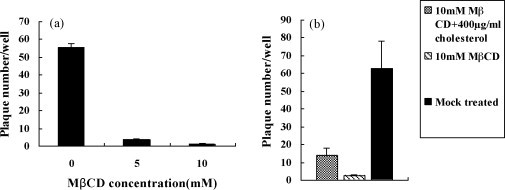
Viral envelope cholesterol was required for the virus infectivity. (a) Infectivity of BoHV-1 which was mock treated or treated with various concentrations of MβCD, was measured by plaque formation assay. (b) Infectivity of BoHV-1 which was replenished with exogenous cholesterol was measured by plaque formation assay. Cell was first mock pretreated or pretreated with 10 mM MβCD and then mock replenished or replenished with 400 μg/ml cholesterol for 1 h before BoHV-1 infection. Experiments were repeated three times, and the error bars indicate the standard deviations of three independent experiments.
4. Discussion
The importance of cholesterol, an essential constituent of lipid rafts, in the target cell membrane or in the viral envelope during the virus entry has been demonstrated for several viruses (Choi et al., 2005, Huang et al., 2006, Hambleton et al., 2007, Li et al., 2007, Funk et al., 2008). Depletion of cellular or viral cholesterol could destruct the lipid rafts structure and as a result reduced viral entry into cells. For some virus in alphaherpesvirus subfamily of the herpesviruses, such as herpes simplex virus 1 (HSV-1), varicella-zoster virus (VZV), porcine pseudorabies virus (PRV) cell membrane cholesterol is required during the entry (Bender et al., 2003, Hambleton et al., 2007, Desplanques et al., 2008). Cholesterol in both membranes was shown here for the first time to be required for BoHV-1 infection of MDBK cells. And subsequent experiments revealed that the inhibitory events occurred at the early replication process.
In this study a cholesterol-sequestering reagent MβCD was used to deplete cholesterol in both viral envelope and target cell membrane, respectively. BoHV-1 is an enveloped virus which is rich in lipid rafts. So the role of cholesterol for the virus infection in both viral envelope and target cell membrane should be investigated, respectively. Firstly our observation showed that productive virus entry was significantly reduced when the MDBK cells membrane cholesterol was reduced by the treatment of MβCD. During the investigation the drug-treated cells were washed extensively before the addition of virus, allowing cholesterol from the target cells to be reduced exclusively, whilst the virus particle cholesterol remained intact. Similarly, when investigated the role of viral envelope cholesterol for the virus infectivity, the drug treated virions were separated from the reagent by ultracentrifugation. So that the disruption of cell membrane lipid rafts by MβCD was excluded. Depletion of cholesterol from viral membrane and cell membrane by MβCD pretreatment significantly inhibited the virus production, and the inhibitory effect could be partially reversed by exogenous cholesterol replenishment indicating that the decreased infectivity was at least in part due to the depletion of cholesterol from the membranes.
For an enveloped virus, the virus entry begins with attachment to the target cell and ends with fusion between the viral envelope and the cellular cytoplasmic or endosomal membrane. In this study, the role of cholesterol played in the three important stages including, binding, entry and post-entry was separately investigated. As a result, the two independent methods, virus titration and FACS indicated that MβCD treatment did not inhibit the virus binding to the MDBK cells. To investigate whether lipid rafts play a role in the post-entry step(s), we treated the cells with MβCD after the virus entry. The results on virus production revealed that Treat: +1 also inhibited virus replication, but compared to Treat: −0.5 the effect of the later was much more significant. These results suggested that cholesterol was mainly required at the early replication process, especially for the entry events, as reported for other viruses (Hambleton et al., 2007, Li et al., 2007).
The sensitivity of the virus to MβCD treatment may be explained by the association of virus receptors or co-receptors with lipid rafts similar to the findings obtained about HIV-1 virus (Popik et al., 2002, Kozak et al., 2002). The virus receptor(s) for BoHV-1 has not yet been well defined. Two proteins as the candidate receptors with molecular weight of 56 and 60 kDa had been reported (Thaker et al., 1994, Varthakavi and Minocha, 1996). But the amino acid sequence for the two proteins is unknown. Connolly et al. (2001) reported that BoHV-1 gD could bind to human nectin-1 with low affinity. However, the affinity of BoHV-1 gD for bovine nectin-1 is unknown. And there is no evidence reported by the others, indicating that nectin-1 on the native bovine cells surface could bind to BoHV-1 gD. And based on their data Connolly et al. (2001) predicted that BoHV-1 infection of its natural host may be mediated by a receptor other than a bovine nectin-1 homolog. Their study also indicated that BoHV-1 likely used multiple receptors for entry into bovine cells. So if the bovine nectin-1 homolog serves as receptor for BoHV-1 remains to be determined. Under this condition, whether MβCD treatment affected the receptor(s) and coreceptor(s) expression or the interaction between BoHV-1 and receptor(s) or coreceptor(s) could not be determined until the virus receptor(s) and co-receptors were discovered in the future. In addition, we proposed that some signal molecules located in lipid raft and MβCD treatment destructed corresponding signal transduction process, which was crucial in the virus life cycle. Based on this presume further study is being carried out.
The entry of virus into target cells is a complicated process and series of components were involved. Here for the first time we reported that cell membrane cholesterol was required for BoHV-1 entry into MDBK cells and the viral envelope cholesterol was essential to the virus infectivity.
Acknowledgements
The authors are grateful to Dr. Leonard J. Bello, University of Pennsylvania, for providing the MDBK cells, Colorado 1 strain of BoHV-1. This work was supported by the Science and Technology Department of Jiangsu province (grant no. BE2008363), the Chinese National Science Foundation Grant (No. 30771603), Jiangsu High Education Key Basic Science Foundation (08KJA230002) and Ministry of Agriculture of the People's Republic of China (grant no. 200803020).
References
- Ahn A., Gibbons D.L., Kielian M. The fusion peptide of Semliki Forest virus associates with sterol-rich membrane domains. J. Virol. 2002;76:3267–3275. doi: 10.1128/JVI.76.7.3267-3275.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson H.A., Chen Y., Norkin L.C. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell. 1996;7:1825–1834. doi: 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender F.C., Whitbeck J.C., Ponce de Leon M., Lou H., Eisenberg R.J., Cohen G.H. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 2003;77:9542–9552. doi: 10.1128/JVI.77.17.9542-9552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.A., London E. Structure and origin of ordered lipid domains in biological membranes. J. Membr. Biol. 1998;164:103–114. doi: 10.1007/s002329900397. [DOI] [PubMed] [Google Scholar]
- Choi K.S., Aizaki H., Lai M.M.C. Murine coronavirus requires lipid rafts for virus entry and cell–cell fusion but not for virus release. J. Virol. 2005;79:9862–9871. doi: 10.1128/JVI.79.15.9862-9871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C.S., Huang C.Y., Chang W. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J. Virol. 2005;79:1623–1634. doi: 10.1128/JVI.79.3.1623-1634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly S.A., Whitbeck J.J., Rux A.H., Krummenacher C., van Drunen Littel-van den Hurk S., Cohen G.H., Eisenberg R.J. Glycoprotein D homologs in herpes simplex virus type 1, pseudorabies virus, and bovine herpes virus type 1 bind directly to human HveC(nectin-1) with different affinities. Virology. 2001;280:7–18. doi: 10.1006/viro.2000.0747. [DOI] [PubMed] [Google Scholar]
- Dasika G.K., Letchworth G.J., 3rd Cellular expression of bovine herpesvirus 1 gD inhibits cell-to-cell spread of two closely related viruses without blocking their primary infection. Virology. 1999;254:24–36. doi: 10.1006/viro.1998.9553. [DOI] [PubMed] [Google Scholar]
- Desplanques A.S., Nauwynck H.J., Vercauteren D., Geens T., Favoreel H.W. Plasma membrane cholesterol is required for efficient pseudorabies virus entry. Virology. 2008;376:339–345. doi: 10.1016/j.virol.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk A., Mhamdi M., Hohenberg H., Heeren J., Reimer R., Lambert C., Prange R., Sirma H. Duck hepatitis B virus requires cholesterol for endosomal escape during virus entry. J. Virol. 2008;82:10532–10542. doi: 10.1128/JVI.00422-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Xiao B., Zhang S., Liu D., Liao Y., Sun Q. Growth inhibitory effects of gastric cancer cells with an increase in S phase and alkaline phosphatase activity repression by aloe-emodin. Cancer Biol. Ther. 2007;6:85–88. doi: 10.4161/cbt.6.1.3553. [DOI] [PubMed] [Google Scholar]
- Guyader M., Kiyokawa E., Abrami L., Turelli P., Trono D. Role for human immunodeficiency virus type 1 membrane cholesterol in viral internalization. J. Virol. 2002;76:10356–10364. doi: 10.1128/JVI.76.20.10356-10364.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambleton S., Steinberg S.P., Gershon M.D., Gershon A.A. Cholesterol dependence of varicella-zoster virion entry into target cells. J. Virol. 2007;81:7548–7558. doi: 10.1128/JVI.00486-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris T.J., Siu C.H. Reciprocal raft–receptor interactions and the assembly of adhesion complexes. Bioessays. 2002;24:996–1003. doi: 10.1002/bies.10172. [DOI] [PubMed] [Google Scholar]
- Huang H., Li Y., Sadaoka T., Tang H., Yamamoto T., Yamanishi K., Mori Y. Human herpesvirus 6 envelope cholesterol is required for virus entry. J. Gen. Virol. 2006;87:277–285. doi: 10.1099/vir.0.81551-0. [DOI] [PubMed] [Google Scholar]
- Highlander S.L., Sutherland S.L., Gage P.J., Johnson D.C., Levine M., Glorioso J.C. Neutralizing monoclonal antibodies specific for herpes simplex virus glycoprotein D inhibit virus penetration. J. Virol. 1987;61:3356–3364. doi: 10.1128/jvi.61.11.3356-3364.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilangumaran S., Hoessli D.C. Effects of cholesterol depletion by cyclodextrin on the sphingolipid microdomains of the plasmamembrane. Biochem. J. 1998;335:433–440. doi: 10.1042/bj3350433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes P.W., Ley S.C., Magee A.J. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell. Biol. 1999;147:447–461. doi: 10.1083/jcb.147.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa J., Alenius S., Emanuelson U., Chanlun A., Aiumlamai S. Bovine herpesvirus type 1 (BHV-1) and bovine viral diarrhoea virus (BVDV) infections in dairy herds: self clearance and the detection of seroconversions against a new atypical pestivirus. Vet J. 2009;182:223–230. doi: 10.1016/j.tvjl.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Karger A., Bettin B., Granzow H., Mettenleiter T.C. Simple and rapid purification of alphaherpesviruses by chromatography on a cation exchange membrane. J. Virol. Methods. 1998;70:219–224. doi: 10.1016/s0166-0934(97)00200-0. [DOI] [PubMed] [Google Scholar]
- Kozak S.L., Heard J.M., Kabat D. Segregation of CD4 and CXCR4 into distinct lipid microdomains in T lymphocytes suggests a mechanism for membrane destabilization by human immunodeficiency virus. J. Virol. 2002;76:1802–1815. doi: 10.1128/JVI.76.4.1802-1815.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence W.C., D’urso R.C., Kundel C.A., Whitbeck J.C., Bello L.J. Map location of the gene for a 130,000-dalton glycoprotein of bovine herpesvirus 1. J. Virol. 1986;60:405–414. doi: 10.1128/jvi.60.2.405-414.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.M., Li Y.G., Yamate M., Li S.M., Ikuta K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect. 2007;9:96–102. doi: 10.1016/j.micinf.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z., Graham D.R., Hildreth J.E. Lipid rafts and HIV pathogenesis: virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS. Res. Hum. Retroviruses. 2003;19:675–687. doi: 10.1089/088922203322280900. [DOI] [PubMed] [Google Scholar]
- Liao Z., Cimakasky L.M., Hampton R., Nguyen D.H., Hildreth J.E. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS. Res. Hum. Retroviruses. 2001;17:1009–1019. doi: 10.1089/088922201300343690. [DOI] [PubMed] [Google Scholar]
- Montixi C., Langlet C., Bernard A.M., Thimonier J., Dubois C., Wurbel M.A., Chauvin J.P., Pierres M., He H.T. Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J. 1998;17:5334–5348. doi: 10.1093/emboj/17.18.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolini I., Topa S., Sorice M., Pace A., Ceddia P., Montesoro E., Pavan A., Lisanti M.P., Peschle C., Sargiacomo M. Phorbol ester-induced disruption of the CD4-Lck complex occurs within a detergent-resistant microdomain of the plasma membrane. Involvement of the translocation of activated protein kinase C isoforms. J. Biol. Chem. 1999;274:14176–14187. doi: 10.1074/jbc.274.20.14176. [DOI] [PubMed] [Google Scholar]
- Pitha J., Irie T., Sklar P.B., Nye J.S. Drug solubilizers to aid pharmacologists: amorphous cyclodextrin derivatives. Life Sci. 1988;43:493–502. doi: 10.1016/0024-3205(88)90150-6. [DOI] [PubMed] [Google Scholar]
- Phalen T., Kielian M. Cholesterol is required for infection by Semliki Forest virus. J. Cell. Biol. 1991;112:615–623. doi: 10.1083/jcb.112.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popik W., Alce T.M., Au W.C. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4+ T cells. J. Virol. 2002;76:4709–4722. doi: 10.1128/JVI.76.10.4709-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L.J., Muench H. A simple method for estimating 50% endpoints. Am. J. Hyg. 1932;27:493–497. [Google Scholar]
- Schutz G.J., Kada G., Pastushenko V.P., Schindler H. Properties of lipid microdomains in a muscle cell membrane visualized by single molecule microscopy. EMBO J. 2000:892–901. doi: 10.1093/emboj/19.5.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K., Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simons K., Toomre D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Simons K., van Meer G. Lipid sorting in epithelial cells. Biochemistry. 1988;27:6197–6202. doi: 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- Straub O.C. Advances in BOHV-1 (IBR) research. Dtsch. Tierarztl. Wochenschr. 2001;108:419–422. [PubMed] [Google Scholar]
- Sun X., Whittaker G.R. Role for influenza virus envelope cholesterol in virus entry and infection. J. Virol. 2003;77:12543–12551. doi: 10.1128/JVI.77.23.12543-12551.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker S.R., Stine D.L., Zamb T.J., Srikumaran S. Identification of a putative cellular receptor for bovine herpesvirus 1. J. Gen. Virol. 1994;75:2303–2309. doi: 10.1099/0022-1317-75-9-2303. [DOI] [PubMed] [Google Scholar]
- Tikoo S.K., Campos M., Babiuk L.A. Bovine herpesvirus 1 (BOHV-1): biology, pathogenesis and control. Adv. Virus. Res. 1995;45:191–223. doi: 10.1016/s0065-3527(08)60061-5. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala B.A., Encinas M., Milbrandt J., Johnson E.M., Jr. Lipid rafts in neuronal signaling and function. Trends Neurosci. 2002;25:412–417. doi: 10.1016/s0166-2236(02)02215-4. [DOI] [PubMed] [Google Scholar]
- Umashankar M., Sanchez-San Martin C., Liao M., Reilly B., Guo A., Taylor G., Kielian M. Differential cholesterol binding by class II fusion proteins determines membrane fusion properties. J. Virol. 2008;82:9245–9253. doi: 10.1128/JVI.00975-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varthakavi V., Minocha H.C. Identification of a 56 kDa putative bovine herpesvirus 1 cellular receptor by anti-idiotype antibodies. J. Gen. Virol. 1996;77:1875–1882. doi: 10.1099/0022-1317-77-8-1875. [DOI] [PubMed] [Google Scholar]
- Wang L., Menon S., Bolin S.R., Bello L.J. A hepadnavirus regulatory element enhances expression of a type 2 bovine viral diarrhea virus E2 protein from a bovine herpesvirus 1 vector. J. Virol. 2003;77:8775–8782. doi: 10.1128/JVI.77.16.8775-8782.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler M.T., Doster A., Jones C. Persistence and reactivation of bovine herpesvirus 1 in the tonsils of latently infected calves. J. Virol. 2000;74:5337–5346. doi: 10.1128/jvi.74.11.5337-5346.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]