

Graphical abstract
Keywords: Predominant clonal evolution, Near-clade, Discrete typing unit, Recombinational load, Drug resistance
Highlights
-
•
We expose our view on Trypanosoma cruzi population genetics.
-
•
We show it is enlightened by our model of pathogen predominant clonal evolution.
-
•
We draw comparisons with other pathogens, in particular Leishmania.
-
•
We underline the implications of the model for applied research.
Abstract
Comparing the population structure of Trypanosoma cruzi with that of other pathogens, including parasitic protozoa, fungi, bacteria and viruses, shows that the agent of Chagas disease shares typical traits with many other species, related to a predominant clonal evolution (PCE) pattern: statistically significant linkage disequilibrium, overrepresented multilocus genotypes, near-clades (genetic subdivisions somewhat blurred by occasional genetic exchange/hybridization) and “Russian doll” patterns (PCE is observed, not only at the level of the whole species, but also, within the near-clades). Moreover, T. cruzi population structure exhibits linkage with the diversity of several strongly selected genes, with gene expression profiles, and with some major phenotypic traits. We discuss the evolutionary significance of these results, and their implications in terms of applied research (molecular epidemiology/strain typing, analysis of genes of interest, vaccine and drug design, immunological diagnosis) and of experimental evolution. Lastly, we revisit the long-term debate of describing new species within the T. cruzi taxon.
Although much progress has been made in vector control, Chagas disease remains a major health problem in Latin America, and is now threatening industrial countries, in particular Spain and the USA.
With the hope of developing radical control means for Chagas disease, extensive studies have been conducted on Trypanosoma cruzi population genetics, and have made this parasite probably one of the micropathogens whose intraspecific diversity is best known.
A comparative population genetic analysis of T. cruzi and of many other pathogens, including parasitic protozoa, fungi, bacteria and viruses, has broaden our view of the agent of Chagas disease’ population structure and has made it possible to reveal the evolutionary strategies used by this pathogen. Moreover, such a comparative population genetics has brought relevant considerations about the possibility to describe new species within the T. cruzi taxon.
1. The predominant clonal evolution (PCE) model of pathogens
The model will be recalled only briefly here, since it has been exposed at length in several recents articles (Tibayrenc and Ayala, 2012, Tibayrenc and Ayala, 2013, Tibayrenc and Ayala, 2014a, Tibayrenc and Ayala, 2014b).
PCE is not defined by any precise mating system or cytological mechanism, but only by strongly restrained genetic recombination* on an evolutionary scale. It therefore deals with population structure of the whole species in the long run only. This definition is accepted by most authors working on pathogen population genetics, including parasitic protozoa, fungi and bacteria (Tibayrenc and Ayala, 2012) and is well accepted in the T. cruzi literature (Barnabé et al., 2013, Flores-López and Machado, 2011, Minning et al., 2011). According to our definition, PCE includes, not only mitotic reproduction, but also selfing*/strong homogamy*, several cases of parthenogenesis* (Tibayrenc and Ayala, 1991, Tibayrenc and Ayala, 2002), and all cases of “unisex” (Feretzaki and Heitman, 2013). PCE would group all means used by micropathogens to escape the “recombinational load” (disrupting favorable multilocus genotypes by genetic recombination) (Agrawal, 2006).
Equating selfing/strong homogamy and many cases of parthenogenesis to clonality is not confined to microbiologists and extends to evolutionists working on higher organisms too (Avise, 2008). Some authors, working on Leishmania (Rougeron et al., 2009) or on T. cruzi (Llewellyn et al., 2009a) recommend to distinguish selfing/homogamy from “strict” clonality (=mitotic propagation). This is a matter of definition. The problem deals with the means used for distinguishing selfing/homogamy from “strict” clonality.
The main consequences of PCE are: (i) statistically significant linkage disequilibrium, or nonrandom association of genotypes occuring at diffferent loci (LD); (ii) overrepresented multilocus genotypes (MLGs), which can be lacking if the mutation rate of the marker is high (all strains have different genotypes); (iii) the presence of “near-clades” (genetic subdivisions somewhat blurred by occasional genetic exchange/hybridization); “Russian doll” patterns (RDPs-PCE is observed, not only at the level of the whole species, but also, within the near-clades) (Tibayrenc and Ayala, 2013).
We have coined the term “near-clade” (Tibayrenc and Ayala, 2012) because it can be suspected that virtually all micropathogen species undergo some bouts of recombination, including among different species. This makes the demands of a strict cladistic* analysis impossible. We have recommended to evidence the presence of near-clades with the tools of population genetics analysis (LD), then by a flexible phylogenetic approach relying on the “congruence principle” (Avise, 2004). The use of more reliable information supports more and more the working hypothesis (here: the presence of near-clades). For example, in the case of MLST*, the phylogenies of individual genes may exhibit some discrepancies. However, the use of the whole set of genes evidences a strong phylogenetic signal (Lauthier et al., 2012). Or different clustering/phylogenetic approaches, relying on different working hypotheses, give convergent results, which confirm the robustness of the branchings (Fig. 1 ). Or the phylogenies designed from different molecular markers corroborate each other (Brisse et al., 2000, Tibayrenc et al., 1993). Identification of PCE relies on the concept of a “clonality threshold”, beyond which the impact of clonal evolution overcomes that of recombination and leads to the inescapable evolutionary divergence of the near-clades. This concept of a “PCE border” makes it possible to replace vague and subjectives assertions (“substantial” recombination; de Paula Baptista et al., 2014; “Gross incongruences”, “multiple introgressions”: Messenger et al., 2012) with a clear-cut criterion. The alternative hypothesis to PCE is that recombination is abundant enough to erase the effects of clonal evolution in the long run. This corresponds to the “epidemic clonality” model (Maynard Smith et al., 1993) or “semiclonal model” (Maiden, 2006).
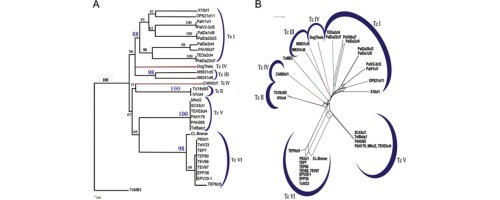
Fig. 1.
The six Trypanosoma cruzi near-clades evidenced by MLST analysis (after Lauthier et al., 2012). Left: neighbor-joining tree; right: split decomposition analysis. The two modes of analysis, which rely on different computing methods and different working hypotheses, show the same near-clading pattern. This is an indication that the clustering of the near-clades is robust, according to the congruence principle (Avise, 2004).
2. Some important points of the model
-
-
As recalled many times, the PCE model does not state that recombination is absent, but rather, that it is not frequent enough to break the prevalent PCE pattern. The model considers that recombination/hybridization could play a major role in pathogens’ evolution, but only on an evolutionary scale. It does not state by any means that recombination is “inconsequential” (Ramírez and Llewellyn, 2014) or “of little consequence” (Miles et al., 2009).
-
-
In a first step, PCE should be explored at the level of the entire species. The species concept, as we will see further, is a matter of debate in the case of micropathogens. However, the unit of analysis when PCE is explored should be the presently described species. It has been argued that “it makes little sense to address each parasite species as a whole”, because these “genetic subdivisions (i.e.: the near-clades) act as reproductive barriers” (Ramírez and Llewellyn, 2014). However, only the PCE approach is able to evidence the existence of reproductive barriers among the near-clades. Claiming that such reproductive barriers are self-evident (Ramírez and Llewellyn, 2014) is quite untrue. Evidencing them is the very goal of the PCE approach.
-
-
To explore PCE, sampling should be twofold: (i) in a classical population genetics strategy, it should be conducted in close sympatry* on very limited spans of time, in order to ascertain that the organism under study has actual opportunity for mating (de Meeûs et al., 2007). However, it is extremely difficult to ascertain a strict sympatry when micropathogens are concerned. Even sampling at the level of individual hosts is not a guarantee for it. If different genotypes of the species concerned do not have the same tropism for different tissues of the host, they could have little opportunity for mating. (ii) Since the PCE pattern is definitely a “bird's eye view” of the overall genetic variability of the whole species in the long term, we recommend a global sampling of the entire species on its complete ecogeographical range, including all known hosts, and relying on retrospective studies to evaluate the stability of the PCE pattern. Retrospective studies can concern the analysis of ancient culture collections, or of ancient publications, or both. Such a sampling strategy makes it possible to ascertain that ubiquitous MLGs, near-clades and RDPs are stable in space and time, are specific properties of the species under study, and cannot be explained by a Wahlund effect* (Rougeron et al., 2015). If inhibition of recombination were due to trivial physical obstacles (Wahlund), overrepresented MLGs, near-clades and RDPs would be strongly linked to space and/or time.
-
-
The PCE model predicts that different clonal genotypes, due to inhibition of recombination among them, will tend to accumulate divergent mutations, which could concern genes governing relevant phenotypes (virulence, resistance to drugs). This should be especially true when the evolutionary divergence among clones is high. However, the model does not predict that all phenotypes should be strictly linked to the clonal population structure. This should be only a general tendency, and could be not verified at microevolutionary levels, and when strongly selected phenotypes (drug resistance) are concerned. Such selected phenotypes are bound to emerge independently in distinct lineages. This is not an evidence for recombination in itself (Rougeron et al., 2015), although it can be consistent with recombination events. Chloroquine resistance is observed in different Plasmodium species. This is not the result of genetic exchange between these different species, but only of a common selective pressure.
-
-
The clonet concept: Identical MLG is a relative notion, and is highly dependent on the resolution power/mutation rate/molecular clock* of the marker employed. A set of strains that appears as identical for a given marker could reveal genetic heterogeneity when a more discriminative marker is used. If PCE obtains, this set should be considered as a family of closely related clones, rather than a true clone. The term “clonet” has been coined by us to refer to those sets of strains that appear to be identical with a given set of genetic markers in a species that undergoes PCE (Tibayrenc et al., 1991a). A typical example of clonet is the zymodeme* MON1 of Leishmania infantum. This MLEE* MLG is widespread in the ancient and new world. The use of a more discriminating marker (microsatellites) shows that MON1 is genetically heterogeneous (Amro et al., 2009, Chargui et al., 2009). One important point with the clonet concept is that the population genetic tests and phylogenetic analyses made on the basis of a given marker remain entirely valid when more discriminating markers evidence additional genetic variability by comparison with this marker. A given marker addresses a given level of resolution and evolutionary divergence and is better adapted for given studies.
3. PCE and Trypanosoma cruzi
T. cruzi is a paradigmatic case of PCE. It is actually the parasitic model used by us for long to design the PCE approach (Tibayrenc et al., 1981, Tibayrenc et al., 1986), and to extend it later to various other pathogens (Tibayrenc et al., 1990, Tibayrenc et al., 1991a, Tibayrenc et al., 1991b, Tibayrenc and Ayala, 2012).
Apart from our own studies, many data are available in the literature, back to the 70 s (the pioneering studies by Miles et al., 1977, Miles et al., 1978, Miles et al., 1981), relying on various markers, which makes comparative and retrospective studies easy (Zingales et al., 2012). Retrospective studies (ancient strain collections and publications) are highly relevant to evaluate the persistence of T. cruzi PCE features (widespread MLGs, near-clades, RDPs). Many of the ancient studies dealing with the population genetics and strain typing of T. cruzi and various other micropathogens rely on MLEE. It is remarkable that MLEE results as a rule have been fully confirmed by modern DNA markers. So ancient MLEE data, when properly interpreted, give a reliable basis for retrospective studies. Lastly, the analysis of paleo DNA (Araujo et al., 2008) constitutes a promising approach to evaluate the antiquity of T. cruzi population structure.
The PCE indices rely on many raw data sets that can be easily verified by anybody. They are as follows:
-
-
LD: We do not consider LD and PCE as “synonymous” (Rougeron et al., 2015). However, we and many authors working on micropathogen population genetics (Tibayrenc and Ayala, 2012) consider LD as a valid circumstantial evidence for clonal evolution, as long as the bias due to Wahlund effect is excluded. LD in T. cruzi is observed: (i) among MLEE loci (Tibayrenc et al., 1981, Tibayrenc et al., 1986); (ii) among different markers (“test g” of LD; Tibayrenc et al., 1990): between MLEE and RAPD* (Tibayrenc et al., 1993, Brisse et al., 2000); between microsatellites and ribosomal DNA restriction fragment length polymorphism (Oliveira et al., 1998); between MLST*, MLEE and RAPD (Lauthier et al., 2012); between microsatellites and the sequence of the Gpi gene (Lewis et al., 2011). LD in T. cruzi is not limited to classical markers, and is observed between genetic markers and copy number variants (Minning et al., 2011); microsatellites and DNA content (Lewis et al., 2009); between MLEE and RAPD on one hand, 12 antigen genes (which are under strong selection) on the other hand (Rozas et al., 2007); between classical markers ad the cruzipain antigen gene (which is under strong selection too) (Lima et al., 2012). Lastly, a statistical association is observed between the variability revealed by molecular markers and the polymorphism of expressed genes (Telleria et al., 2004, Telleria et al., 2010, Telleria et al., 2013).
-
-
Overrepresented, widespread genotypes: we have called them: “major clones” (Tibayrenc and Brenière, 1988). This is the case for example of the MLEE MLGs n° 19, 20, 32 and 39 in Tibayrenc et al. (1986), which have been observed in diverse countries, years apart. For example; MLG 19 has been sampled in Brazil, Venezuela, Colombia, Bolivia, in Chagasic patients, as well as in various mammalian hosts and triatomine bug species, from 1977 to 1983. With more discriminating MLEE typing, ubiquitous MLGs still are observed. One MLG repeated 31 times has been observed in four different countries (Barnabé et al., 2000). The remarks made about the clonet concept (see above) should be recalled here. MLEE reveals ubiquitous MLGs. However, when more discriminating markers (microsatellites) are used, each strain may have a different genotype, due to the fast mutation rate of this marker (Oliveira et al., 1998).
-
-
Near-clades. The near-clade concept (Tibayrenc and Ayala, 2012) gives a clear evolutionary definition to the descriptive term DTU* (discrete typing unit) coined by us (Tibayrenc, 1998). On the basis of both MLEE and RAPD typing, it has been proposed that T. cruzi is subdivided into 6 discrete genetic lineages, or DTUs (Barnabé et al., 2000, Brisse et al., 2000). T. cruzi DTUs perfectly fit the definition of near-clades: well-individualized genetic lineages that are somewhat blurred up by occasional genetic exchange and/or hybridization. Hybridization events have been described since long in T. cruzi (Machado and Ayala, 2001, Brisse et al., 2003), although their precise evolutionary history still is a matter of debate (de Freitas et al., 2006, Ferreira and Briones, 2012, Subileau et al., 2009, Westenberger et al., 2005, Westenberger et al., 2006). The classification into 6 DTU/near-clades has been recently validated by a panel of international experts (Zingales et al., 2012). To a certain extent, T. cruzi near-clades correspond to the “principal zymodemes” early described by Miles et al., 1977, Miles et al., 1978. This illustrates their permanency in time. T. cruzi near-clades have been corroborated by: (i) MLEE, RAPD (Brisse et al., 2000); (ii) MLST (Lauthier et al., 2012; see Fig. 1); (iii) fluorescent fragment length barcoding (Hamilton et al., 2011); (iv) microsatellites, 3 mitochondrial genes and the 24Sα rRNA gene (de Freitas et al., 2006); (v) the SSU rDNA, Cytochrome B, Histone 2B genes, ITS1rDNA genotyping, RAPD (5 primers) and PFGE* (Marcili et al., 2009); (vi) the sequences of 32 genes (Flores-López and Machado, 2011). All T. cruzi near-clades are stable in time and widespread, although their geographical distribution is not the same (Barnabé et al., 2000, Zingales et al., 2012). Although the precise history of hybridization still is questioned (see above), it is considered that near-clades V and VI have a hybrid origin.
-
-
An additional near-clade, named “TC-Bat” (for it has been isolated from bats only until now) has been recently described. It has been recorded in Brazil and Panama years apart, in different species of bats (Marcili et al., 2009, Pinto et al., 2012). This is a striking illustration of the stability in space and time of the near-clades.
-
-
Russian doll patterns (RDPs). It is frequently inferred that genetic exchange is inhibited among the genetic clusters (=near-clades) that subdivide a species, but not within each of them (Campbell et al., 2005). When exploring this hypothesis, it is important to be conscious that a lower evolutionary scale is under study. The molecular markers used at the level of the whole species may lack resolution at such lesser levels. This, together with the fact that the sample size is generally much lowered when considering the near-clades separately, increases by far the risk of statistical type II error (impossibility to reject the null hypothesis of random genetic exchange, not because it is verified, but because the test lacks power). This bias being discarded, contrary to what has been recently stated (Ramírez and Llewellyn, 2014), RDP evidence is abundant in T. cruzi, as well as in other parasites (Leishmania, Giardia). The near-clade “TC I” (Zingales et al., 2012) has never been considered “homogeneous” (Guhl and Ramírez, 2011). On the contrary, MLEE as well as RAPD studies have always shown that it exhibits a high variability (Tibayrenc et al., 1993, Barnabé et al., 2000, Brisse et al., 2000). However, until recently, it had been not possible to evidence LD and a clear structuring within it (RDP). This is now done, thanks to the availability of more discriminating markers and ampler samplings. Geographical distance has an obvious role in the distribution of TCI genetic structuring (Llewellyn et al., 2009b). However, it cannot account for the data cited thereafter. Within TCI selvatic strains, strong LD evidences “widespread clonality, infrequent recombination” (Llewellyn et al., 2011). Contrary to what Ramírez et al. (2013b) state, within Colombian TCI strains, there is LD too. As a matter of fact, the p values for the LD test and the Ia index of association (another LD test) are 4 × 10 −4 and 0.037, respectively (Ramírez et al., 2013a, Ramírez et al., 2013b). As noted by Tomasini et al. (2014), this is evidence for LD, and not for the opposite, as wrongly concluded by Ramírez et al., 2013a, Ramírez et al., 2013b. Moreover, absence of LD would be inconsistent with the presence of 2 genetic subdivisions, clearly corroborated by bootstrap analysis, within this sample. This last result is evidence for RDP. As a matter of fact, not only TCI does exhibit LD, but it is also subdivided into lesser near-clades that are widespread throughout different countries and exhibit host specificities (Fig. 2 ; Guhl and Ramírez, 2011, Herrera et al., 2007). TCI lesser near-clades have been corroborated by various markers, including the Cytochrome b gene sequence (a mitochondrial gene) and the mini-exon gene (Ramírez et al., 2011), PCR-RFLP of 5 coding regions (Ramírez et al., 2012), mini-exon and MLST (Tomasini et al., 2011, Tomasini et al., 2014).
-
-
Population genetics within other T. cruzi near-clades has been less explored. However, in the near-clade TCIII, a strongly significant (p < 0.001) LD (measured using the Index of Association (IA)) was detected in all populations except one, where only marginal significance was observed (p = 0.032) (Llewellyn et al., 2009a). This supports the hypothesis that TCIII exhibits also a RDP.
-
-
Indices of frequent recombination within near-clades? A lack of congruence between mitochondrial and nuclear phylogenies (Ramírez et al., 2012) indicates at best limited introgression, as it can be observed even between different biological species. The null hypothesis of the RDP model is not occasional bouts of genetic exchange, which is consistent with PCE. It is random or quasi-random recombination, as it can be observed in human populations for example. In humans, the only obstacles to genetic exchange are geographical or cultural ones (quasi-random recombination). TCI populations isolated from selvatic triatomine bugs in Bolivia show data that are compatible with such frequent genetic exchange. However, according to the authors themselves, these data are prone to type II error and should be corroborated by stronger sampling (Barnabé et al., 2013). A survey in Ecuador (Ocaña-Mayorga et al., 2010) is consistent with recombination in a small TCI subpopulation. However, as we have shown (Tibayrenc and Ayala, 2013), (i) the “domestic” subpopulation is poorly defined (33% of selvatic strains are included in it, while the “selvatic” clade comprises 12% of domestic strains); (ii) the risk of type II error is high, due to very limited sampling (only 12 truly “domestic” strains). Additionally, the “selvatic” subpopulation exhibits strong LD and clonality, which fits a RDP. de Paula Baptista et al. (2014) have proposed that Brazilian strains of the TCII near-clade undergo substantial recombination. However, this study is based also on limited sampling and is subject to type II error. Moreover, there is a strong contradiction between the hypothesis of substantial recombination (with lack of LD) and the evidence for clear structuration of these populations, even in sympatry, as shown by STRUCTURE analysis. In conclusion: it is quite possible that some populations within T. cruzi near-clades undergo frequent recombination. However, the limited evidence presently available obviously has to be completed by far more extensive studies. On the contrary, the evidence above exposed for RDPs is robust.
Fig. 2.
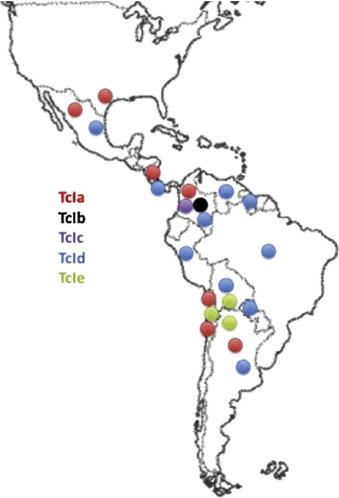
Geographic distribution of the five lesser near-clades evidenced within the Trypanosoma cruzi near-clade TCI (see Fig. 1) (after Guhl and Ramírez, 2011). The lesser near-clades are widespread and occur sympatrically, which is an evidence of a “Russian doll Pattern” (Tibayrenc and Ayala, 2013).
From the results above exposed, it is hardly questionable that T. cruzi at the level of the whole species undergoes PCE. Moreover, when enough data are available, it is apparent that PCE operates also within the near-clades (RDP). Genetic recombination/hybridization in the agent of Chagas disease has probably an important impact at an evolutionary scale. However, it is unable to counter the effects of clonal evolution in the long run (“clonality threshold”) and to prevent the evolutionary divergence of the near-clades. When looking for recombination in a given species amounts to “searching for a needle in a haystack” (Ramírez and Llewellyn, 2014), it does show that recombination is strongly restrained, in other words, that the PCE model is corroborated. When it is not, like in the highly recombining bacterium Helicobacter pylori, one finds easily clear indications for recombination without searching a haystack.
In the framework of PCE, it is quite possible that local populations of genetically related strains undergo more genetic exchange than clonal propagation (Barnabé et al., 2013, Ocaña-Mayorga et al., 2010, de Paula Baptista et al., 2014). However, this has to be better explored on more extensive samples. It is possible that in T. cruzi, recombination is less restrained among identical genotypes (=selfing) or similar genotypes (tending to selfing) than among more distantly related genotypes. This happens in bacteria, where recombination is more frequent among “sister cells” than among cells that have radically different genotypes (Caugant and Maiden, 2009). This leads to “invisible sex” (Balloux, 2010) and is explained by the fact that the higher the genetic divergence, the more the restriction systems are different (Caugant and Maiden, 2009). Moreover, local introgressions of mitochondrial genomes are quite possible in T. cruzi (Messenger et al., 2012, Ramírez et al., 2012). All this does not challenge the PCE model. As exposed earlier, the alternative hypothesis to PCE is that recombination is abundant enough to overcome the effects of clonal evolution in the long run.
Lastly, restrained recombination in T. cruzi cannot be attributed to a lack of opportunity for mating. As a matter of fact, multiclonal infections in the same host, either vectors (Tibayrenc et al., 1985) or chagasic patients (Brenière et al., 1985) are very frequent.
4. Aneuploidy and the selfing/homogamy debate
We recall here that selfing/homogamy is included in our PCE concept, a view shared by many, if not most, specialists of pathogen population genetics (Tibayrenc and Ayala, 2012) as well as by evolutionist working on metazoa undergoing uniparental reproduction (Avise, 2008). However, other scientists recommend distinguishing selfing/homogamy from “strict” (=mitotic) clonality (Rougeron et al., 2009, Rougeron et al., 2015). When the macroevolution scale recommended by the PCE approach is considered, the distinction in terms of population structure has limited relevance, since both “strict” clonality and selfing/homogamy lead to restrained recombination and LD. Now selfing as a mode of restrained recombination has the evolutionary advantage of permitting DNA repair, which is not the case of a strictly mitotic mode of propagation. If one wants to distinguish selfing from “strict” clonality, the problem lies on the way to do it. When bacteria are considered, since they are haploid, selfing, which is certainly very frequent in these pathogens (Maiden, 2008, Caugant and Maiden, 2009), is impossible to distinguish from mitotic propagation (“invisible sex”; Balloux, 2010). When protozoa are concerned, the evidence proposed for selfing relies on heterozygote deficit (Rougeron et al., 2009). However, apart from selfing, many phenomena could cause heterozygote deficit, such as null alleles, allelic drop-out, homoplasy (a strong concern in the case of microsatellites) (Alam et al., 2009, Lehmann et al., 2004, Pearson et al., 2009), and genome-wide mitotic gene conversion (Llewellyn et al., 2009a, Llewellyn et al., 2011). Experiments in the strictly asexual species Daphnia pulex have shown that a loss of heterozygosity by mitotic recombination is 1000 times more frequent than an accumulation of divergent mutations (Omilian et al., 2006). The probability of the possible causes of heterozygote deficit should not be evaluated separately (Rougeron et al., 2009), since they are not mutually exclusive. Moreover, heterozygote deficit inferred from microsatellite analysis clashes with SNP* data for Leishmania mexicana and L. braziliensis (Rogers et al., 2011) and T. cruzi (Yeo et al., 2011), where an excess of heterozygotes is recorded. However, selfing should cause a heterozygote deficit for all loci, including the ones involved in SNPs. The main problem with the way to explore heterozygote deficit is that all tests proposed for it (de Meeûs et al., 2007) are based on the hypothesis that the organism under survey is diploid*. Now strong convergent results suggest that Leishmania undergoes widespread aneuploidy (Downing et al., 2011, Lachaud et al., 2014, Mannaert et al., 2012, Rogers et al., 2011, Sterkers et al., 2011, Sterkers et al., 2012, Sterkers et al., 2014), which renders the tests invalid, and at the same time, is an explanation for apparent heterozygote deficit. As a matter of fact, widespread aneuploidy should cause a rapid elimination of heterozygosity by frequent passage through a haploid state (Sterkers et al., 2012). This should happen even if aneuploidy is transitory in Leishmania, as proposed by Rougeron et al. (2015). The evidence for aneuploidy in T. cruzi still is more limited than for Leishmania, although progress is rapid (Minning et al., 2011, Souza et al., 2011). When population genetic data are concerned, contrary to what has been claimed (Ramírez et al., 2013a), in a survey dealing with more than 200 cloned stocks from the same host, in mammals from Venezuela and Bolivia, “multiple aneuploid clones were isolated across those infrapopulations studied” (Llewellyn et al., 2011). It has been proposed that microsatellite data do not suggest aneuploidy in Leishmania (Rougeron et al., 2015). However, Lachaud et al. (2014) disagree with this statement. The conclusion of all this is that selfing in Leishmania and T. cruzi is a reasonable hypothesis. However, the tests proposed to explore it may not be reliable. As stated by Ramírez et al. (2012): “In general, we urge caution in the interpretation of heterozygosity statistics at STR loci in the context of parasite sexuality, especially given that strong evidence for linkage disequilibrium often accompanies both negative and positive values for FIS”. Such difficulties in evidencing selfing in parasitic protozoa give additional ground to our proposal of including it in the PCE model.
It should be noted that LD tests do not suffer from this problem. As a matter of fact, they can be practiced whatever be the ploidy of the organism under study, and even, when this ploidy level is not known (Tibayrenc et al., 1990).
5. Evolutionary features that could mimic recombination
Recombination can be confounded with several evolutionary features. They include: (i) homoplasy; (ii) lack of phylogenetic signal; (iii) natural selection; (iv) different molecular clocks; (v) long branch attraction; (vi) different modes of inheritance.
Homoplasy is a concern for many, if not most, molecular markers. Microsatellites are considered as especially prone to it (Alam et al., 2009, Lehmann et al., 2004, Pearson et al., 2009). This could lead to overestimate both recombination and heterozygote deficit.
Lack of phylogenetic signal: if the population is close to genetic monomorphism, or if the molecular marker has too low a resolution power, it is impossible to evidence LD and the presence of near-clades. This should not be taken as evidence for recombination. A lack of phylogenetic signal could be due also to a saturation of the molecular marker. As an example, microsatellites are not adapted to survey higher levels of evolutionary divergence, since their molecular clock is too fast. MLSTs and MLEE could reveal a strong phylogenetic signal where microsatellites cannot.
Natural selection is difficult to distinguish from recombination (Didelot and Maiden, 2010). Strongly selected characters, such as drug resistance, should be especially prone to this bias. Resistance to the same antibiotics can be present in different bacterial lines, and even in different species. This can be the result of genetic exchange, but it is also easily explained by an identical selective pressure. Such convergent selected characters should be therefore considered as evidence for recombination only carefully (Rougeron et al., 2015).
Differences in molecular clocks could lead to discrepancies between trees designed from different genes. In Giardia duodenalis, Wielinga and Thompson (2007) have attributed the discrepancies observed among trees built from different genes to both homoplasy and molecular clock differences, not to recombination. Similarly, in the case of the SARS coronavirus, phylogenetic incongruence among trees has been attributed to different molecular clocks (Moya et al., 2004).
Long branch attraction: analyzing the phylogeny of the SARS coronavirus, Yip et al. (2009) have recommended to not confound recombination with both homoplasy and long branch attraction.
Markers that have a different mode of inheritance will exhibit different evolutionary patterns. For example, in humans, the population structures inferred from autosomal genes, mitochondrial genes and Y chromosome genes, respectively, are quite different (Zoossmann-Diskin, 2010). In T. cruzi, experimental mating has shown that nuclear genes are inherited from both parental cells, while the kinetoplast is inherited from only one parental cell (Gaunt et al., 2003). In the framework of the PCE model, in case of occasional bouts of genetic exchange, this phenomenon is bound to maximize the discrepancies between mitochondrial and nuclear phylogenies.
Lastly, these different phenomena are not exclusive of each other and could combine their effects. In the case of T. cruzi, microsatellites are subject to homoplasy. They have a fast molecular clock, they are explored at the level of the nucleus, whose inheritance is biparental, and they are considered as selectively neutral. Mitochondrial maxicircle genes correspond to coding sequences, which are subject to natural selection. They have a slower molecular clock than microsatellites, and their mode of inheritance is uniparental. So, although the discrepancies between microsatellite and mitochondrial gene phylogenies are consistent with recombination (Ramírez et al., 2012), alternative explanations are possible.
6. A brief comparison with other micropathogens
We have performed a compared population genetic approach dealing with viruses (22 species and categories), bacteria (43 species), parasitic protozoa (30 species) and fungi (13 species) (Tibayrenc and Ayala, 2012 and in preparation). This shows that T. cruzi population structure is not unique and, on the contrary, is very similar to that of many major pathogen species. Each species exhibits specific traits. However, the common denominator among many of them is strong, and suggests the existence of common evolutionary strategies, which could be either ancestral or the result of convergence.
Well-established T. cruzi “siblings” in terms of population structure are the species for which the PCE features (LD, widespread MLGs, near-clades and RDPs) have been recorded. It is assumed that these species have crossed the “clonality threshold” beyond which the impact of clonal evolution will overcome the effects of recombination. Such species include: (i) parasitic protozoa: the Leishmania infantum complex (Tibayrenc and Ayala, 2013), Giardia duodenalis, (Tibayrenc and Ayala, 2014b), Toxoplasma gondii (Tibayrenc and Ayala, 2014a); (ii) fungi: Candida albicans, the Cryptococcus neoformans complex; (iii) bacteria: Escherichia coli, Mycobacterium tuberculosis, Neisseria meningitidis, Staphylococcus aureus. Many other species exhibit clonality traits, but the PCE pattern is not complete, either because these species undergo more recombination, or because the studies are less advanced than for the species above listed, or both. When parasitic protozoa are concerned, such examples include: Leishmania braziliensis/peruviana, the Trypanosoma brucei complex, T. congolense, Cryptosporidium spp.
Plasmodium falciparum and P. vivax constitute distinctive cases. Since P. falciparum undergoes meiosis in the vector, it has been long assumed that the agent of the most malignant form of malaria was a panmictic* organism. Our cautious proposal that “uniparental and biparental lineages could coexist within this species”, based on the observation of departures from panmixia in some populations (Tibayrenc et al., 1990), had been rejected but it was subsequently confirmed (Anderson et al., 2000). Many P. falciparum and P. vivax populations exhibit clonality traits. Selfing is probably at the origin of them. However, the hypothesis that selfing is strictly correlated to the level of transmission is at odds with many data (Tibayrenc and Ayala, 2014a). This is epidemiologically relevant, since LD and the degree of clonality in P. falciparum cannot be taken as an indication for the transmission rate (Tibayrenc and Ayala, 2014a), as it had been proposed (Volkman et al., 2012). Now P. falciparum and P. vivax definitely do not fit the PCE model. Clonality traits (LD, widespread MLGs and near-clades) that are observed in these species are labile and change over time, due to frequent genetic recombination. Still the fact remains that clonal propagation introduces a major stratification effect in P. falciparum and P. vivax natural populations, that should be taken into account in studies dealing with malaria epidemiology, drug resistance and the analysis of genes of interest.
When considering the various species that fit the PCE model, two facts should be underlined: (i) for several of them, PCE was not expected. Neisseria meningitidis was considered a “highly recombining” bacterium (Maiden, 2008). However, widespread MLGs and near-clades in this species are extremely stable, as verified by retrospective studies. Moreover, whole genome sequencing has revealed the existence of deep phylogenies (Budroni et al., 2011), which is incompatible with the “epidemic clonality model” (occasional bouts of clonal propagation in an otherwise recombining species; Maynard Smith et al., 1993). Toxoplasma gondii constitutes another unexpected case of PCE, since this parasite undergoes meiosis in the mammalian host. We have proposed that T. gondii undergoes frequent, and possibly predominant clonal evolution (Tibayrenc et al., 1991a, Tibayrenc et al., 1991b). This has been fully confirmed by further studies (Sibley and Boothroyd, 1992). High resolution typing with many different markers (Su et al., 2012) has shown the existence of deep phylogenies, with 6 major “clades” (=near-clades), which definitely includes T. gondii in the PCE model (Tibayrenc and Ayala, 2014a). (ii) The species listed pertain to phylogenetically highly diverse categories (bacteria, fungi, and parasitic protozoa), and exhibit various host ranges, modes of transmission (air-, vector-, food- and water-borne) and tissue tropisms. This strongly suggests that PCE represents a common evolutionary strategy of many pathogens, probably linked to parasitism.
7. Experimental mating, meiosis genes and PCE
The seminal work by Gaunt et al. (2003) has demonstrated that the potential for recombination is present in T. cruzi. However, these experiments dealt only with related strains pertaining to the near-clade TCI. It remains to be established whether mating can also be obtained between strains pertaining to different near-clades. More important: these experiments provide no indication about the frequency of genetic exchange in natural populations, which matters when population structure is concerned. Successful mating experiments cannot therefore be used to challenge the PCE model.
Genes homologous to meiosis genes are observed in many micropathogen species, including T. cruzi (El-Sayed et al., 2005). However, this does not imply that T. cruzi undergoes meiosis (Ramírez and Llewellyn, 2014), and if it did, the presence of these genes does not give indications about the frequency of it. Moreover, genes analogous to meiosis genes may have acquired totally different functions through evolution. “Evolution is constantly re-using old genes for new purposes” (Birky, 2009). We have suggested that, in micropathogens undergoing PCE, the meiosis kit could actually correspond to a “sexuality/clonality machinery that could allow micropathogens to use either one or the other mode of propagation when facing different evolutionary challenges (Tibayrenc and Ayala, 2012). In conclusion: like for mating experiments, the presence of genes homologous to meiosis genes is therefore not evidence against the PE model.
8. The PCE model and the problem of pathogen species definition
Defining species in micropathogens has been always a challenge, since the mixiological species concept (Dobzhansky, 1937), which addresses only organisms with obligatory mating, cannot be applied to them. Moreover, the phylogenetic species concept (Cracraft, 1983) is difficult to apply, since it is most probable that some genetic exchange always occurs in micropathogens natural populations, even between different species. The near-clade concept, which relaxes the cladistic demands, makes it possible to revisit the phylogenetic species concept for micropathogens (Tibayrenc and Ayala, 2014b). Two situations occur. The first case is when presently described species can be equated to near-clades in an evolutionary point of view. Many, if not most, Leishmania species are in this case. As a matter of fact, in general, they are clearly discriminated by molecular markers. However, hybridizations are frequent among them. Actually, many Leishmania species are perfectly equivalent to T. cruzi near-clades from an evolutionary point of view. The second case is when presently observed near-clades have not been described as Linnean species yet. This is the case for the Giardia duodenalis “assemblages” (=near-clades) (Tibayrenc and Ayala, 2014b). T. cruzi near-clades could constitute the basis for splitting the agent of Chagas disease into several species. We do not recommend it for several reasons. First, the inventory of T. cruzi genetic diversity could still evidence new features and unknown near-clades. Second, the advent of whole genome sequencing could lead to reconsider the presently accepted near-clade classification (Zingales et al., 2012). Third, the epidemiological and pathological specificity of T. cruzi near-clades is not clear-cut. The Chagas disease scientific community will decide whether the description of these new species is desirable. The near-clade concept may constitute a necessary, but not sufficient, criterion for it. Since near-clading is widespread among micropathogens, it would be misleading to equate all of them to Linnean species. This should be done only when a given near-clade exhibits strong phenotypic specificities dealing with relevant characters (host range, drug resistance, pathogenicity for example).
9. The PCE model and applied research
The PCE model provides applied research with relevant units of analysis that are based on simple concepts, and are sharply defined. Their stability in space and time has been fully ascertained by retrospective studies, as well as by population genetics and phylogenetic analysis. Clonal MLGs (clonets) can be used for epidemiological tracking. To characterize them, markers with different mutation rates and level of resolution can be selected according to the study concerned: microsatellites for microepidemiology, MLST and whole genome sequencing plus SNPs (when these two last tools become more accessible) for epidemiological surveys at broader scales. Near-clades and their subdivisions (RDPs) can, and should be used as units of analysis for epidemiological tracking, drug design (Zingales et al., 2014), clinical studies and experimental evolution. Taking the near-clades as units of analysis, we have conducted long-term experimental evolution studies, which have shown that many phenotypic characters (including drug sensitivity, virulence in mice and cell infectivity), as well as gene expression patterns, were linked to the near-clades (Revollo et al., 1998, Telleria et al., 2004, Telleria et al., 2010, Telleria et al., 2013).
A major application of the PCE model for strain typing/molecular epidemiology is indirect typing, which is a remarkable property of LD. Since all genes are linked together (“block of genes”), the near-clades can be identified by only one or two markers (“tags”; Tibayrenc, 1998). Several RAPD and isoenzyme tags have been identified for the 6 near-clades, including near-clade TCI, (Brisse et al., 2000). By the amplification of a unique gene (TcSC5D), Cosentino and Agüero (2012) were able to identify the T. cruzi near-clades I, II, II, IV, V + VI, TC-Bat, and the subspecies T. cruzi marinkellei. We recommend this “phylogenetic character mapping” (PCM) approach (Avise, 2004). A convenient population genetics framework is first designed through the PCE approach. Once the relevant units of analysis (near-clades) are clearly identified, phenotypes of interest (drug resistance, virulence) are mapped onto this framework. This makes it possible to see the links between the overall evolution of the organism under study and its phenotypes, and at the same time, to show the specific phenotypic properties of the near-clades.
10. Conclusion
Due to the dynamism of many research groups throughout the world, with a major contribution from Latin American teams, the intraspecific genetic variability of T. cruzi is probably one of the best known among micropathogens. The agent of Chagas disease could be considered a star model for evolution, like Drosophila melanogaster, Mus musculus, Caenorabditis elegans and Escherichia coli.
The PCE model, which has been elucidated and enriched by wide surveys dealing with many pathogens (Tibayrenc and Ayala, 2012), sets in perspective the particular case of T. cruzi population genetics. It shows that T. cruzi shares many features with other major pathogens, including parasitic protozoa, fungi and bacteria.
Promising avenues of evolutionary research are represented by experimental mating (less advanced for T. cruzi than for T. brucei), within near-clade population genetics, experimental evolution, and the development of more powerful tools of analysis (whole genome sequencing, SNP analysis). These tools constitute a challenge for analysis, since the computing power required for treating these data is huge. However, the use of these tools becomes routine for viruses and bacteria, and should be more and more accessible when parasitic protozoa are concerned, making it possible to develop a population genomics of these pathogens.
Glossary
- Allopatry
a situation where different populations occur at different locations; cf sympatry*
- Aneuploidy
a situation in which the chromosome number of an individual is not an exact multiple of the haploid set of the species. Copy numbers may vary among chromosomes.
- Clade
evolutionary line defined by cladistic analysis. A clade is monophyletic* and is genetically differentiated (i.e., evolves independently) from other clades.
- Cladistic analysis
phylogenetic analysis based on the distribution of characters that are shared between ancestral (plesiomorphic) and derived (apomorphic) characteristics. Only apomorphic characters shared by all members of a given clade* (synapomorphic character) convey relevant phylogenetic information.
- Diploid
possessing 2 copies of each chromosome.
- DTU = Discrete Typing Unit
sets of strains that are genetically closer to each other than to any other stock and are identifiable by common genetic, molecular or immunological markers called tags (Tibayrenc, 1998).
- Haploid
possessing only one copy of each chromosome.
- Homogamy
preferential mating between individuals that are genetically identical or extremely similar to each other.
- Homoplasy
distinct evolutionary units possess similar characters that do not originate from a common ancestor.
- MLEE = Multilocus Enzyme Electrophoresis
Analysis by electrophoresis of protein extracts from biological samples. MLEE banding reflects the aminoacid sequence of the proteins, coded by the DNA sequences. It is an efficient genetic typing method. However, identical MLEE bands may correspond to different DNA sequences (homoplasy*).
- MLST = multilocus sequence typing
a highly standardized typing technique based on the sequencing of 450-bp segments of a set of housekeeping genes (usually seven). It has been widely used for a high number of bacterial species and some eukaryotic pathogens as well.
- Molecular clock
in its strict, original sense (more correctly named: “DNA clock hypothesis”), the concept that the rate of nucleotide substitutions in DNA remains constant. In a broader sense, simply the rate of evolution of the genomic part that codes for the variability of a given marker. This fastness is driven by the rate of substitution/mutation. It may be regular or irregular.
- Monophyletic
the case where a given phylogenetic lineage has only one ancestor (see clade*).
- Panmixia, panmictic
a situation where genetic exchange occurs at random.
- Parthenogenesis
Reproduction by the development of a single gamete without fertilization by a gamete of the opposite sex.
- PFGE = Pulse Field Gel Electrophoresis
separation of large DNA fragments by a particular electrophoresis technique using alternately pulsed, perpendicularly oriented electrical fields. Strains that share the same PFGE are referred to as pulse types. In the case of bacteria, the large DNA fragments result from the action of a “low-frequency cutter” (a bacterial endonuclease which restriction action has a low frequency) on the bacterial chromosome. In the case of parasitic protozoa (Trypanosoma and Leishmania) and of yeasts, the large DNA fragments correspond to entire chromosomes (“molecular karyotype”).
- RAPD = Random Amplified Polymorphic DNA
In the classical PCR method, the primers used are known DNA sequences, whereas the RAPD technique relies on primers whose sequence is arbitrarily determined.
- Recombination, recombining
Reassortment of genotypes occurring at different loci.
- Selfing
self-fertilization of an organism. If the organism is haploid (bacteria), or if all loci are homozygous, selfing is undistiguishable from mitotic clonality. In diploid organisms, selfing leads to a deficit of heterozygotes, and to a lack of genetic recombination.
- SNP = Single Nucleotide Polymorphism
a DNA sequence variation occurring commonly within a population, in which a single nucleotide – A, T, C or G differs between individuals.
- Sympatry
a situation where different populations are present in the same place; cf allopatry*.
- Wahlund effect
when the sampled population is composed of several subpopulations (isolated in time or space or both) that have different genotype compositions, population genetic tests performed at the level of the whole population show departures from panmictic expectations, even if each subpopulation is panmictic*.
- Zymodeme
MLEE* MLG.
Footnotes
See glossary of specialized terms.
References
- Agrawal A.F. Evolution of sex: why do organisms shuffle their genotypes? Curr. Biol. 2006;16:R696–R704. doi: 10.1016/j.cub.2006.07.063. [DOI] [PubMed] [Google Scholar]
- Alam M.Z., Kuhls K., Schweynoch C., Shyam Sundar S., Rijal S., Shamsuzzaman A.K.M., Raju B.V.S., Salotra P., Dujardin J.C., Schönian G. Multilocus microsatellite typing (MLMT) reveals genetic homogeneity of Leishmania donovani strains in the Indian subcontinent. Infect. Genet. Evol. 2009;9:24–31. doi: 10.1016/j.meegid.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Amro A., Schönian G., Barakat Al-Sharabati M.B., Azmi K., Nasereddin A., Abdeen Z., Schnur L.F., Baneth G., Jaffe C.L., Kuhls K. Population genetics of Leishmania infantum in Israel and the Palestinian Authority through microsatellite analysis. Microbes Infect. 2009;11:484–492. doi: 10.1016/j.micinf.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Anderson T.J., Haubold B., Williams J.T., Estrada-Franco J.G., Richardson L., Mollinedo R., Bockarie M., Mokili J., Mharakurwa S., French N., Whitworth J., Velez I.D., Brockman A.H., Nosten F., Ferreira M.U., Day K. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol. Biol. Evol. 2000;17(10):1467–1482. doi: 10.1093/oxfordjournals.molbev.a026247. [DOI] [PubMed] [Google Scholar]
- Araujo A., Reinhard K., Ferreira L.F. Parasite findings in archeological remains: diagnosis and interpretation. Quat. Int. 2008;180:17–21. [Google Scholar]
- Avise J.C. Natural History and Evolution. 2nd ed. Chapman & Hall; New York, London: 2004. Molecular markers. [Google Scholar]
- Avise J. Oxford University Press; 2008. Clonality. The Genetics, Ecology and Evolution of Sexual Abstinence in Vertebrate Animals. [Google Scholar]
- Balloux F. Demographic influences on bacterial population structure. In: Robinson D.A., Falush D., Feil E.J., editors. Bacterial Population Genetics in Infectious Disease. Wiley-Blackwell; Hoboken: 2010. pp. 103–120. [Google Scholar]
- Barnabé C., Brisse S., Tibayrenc M. Population structure and genetic typing of Trypanosoma cruzi, the agent of Chagas’ disease: a multilocus enzyme electrophoresis approach. Parasitology. 2000;150:513–526. doi: 10.1017/s0031182099005661. [DOI] [PubMed] [Google Scholar]
- Barnabé C., Buitrago R., Brémond P., Aliaga C., Salas R., Vidaurre P., Herrera C., Cerqueira F., Bosseno M.F., Etienne Waleckx E., Brenière S.F. Putative panmixia in restricted populations of Trypanosoma cruzi isolated from wild Triatoma infestans in Bolivia. PLOS ONE. 2013;8(11):e82269. doi: 10.1371/journal.pone.0082269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birky C.W. Giardia sex? Yes, but how and how much? Trends Parasitol. 2009;26:70–74. doi: 10.1016/j.pt.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Brenière S.F., Tibayrenc M., Antezana G., Pabon J., Carrasco R., Selaès H., Desjeux P. Résultats préliminaires en faveur d’une relation faible ou inexistante entre les formes cliniques de la maladie de Chagas et les souches isoenzymatiques de Trypanosoma cruzi. C. R. Acad. Sci. Paris. 1985;300(15):555–558. [PubMed] [Google Scholar]
- Brisse S., Barnabé C., Tibayrenc M. Identification of six Trypanosoma cruzi phylogenetic lineages by random amplified polymorphic DNA and multilocus enzyme electrophoresis. Int. J. Parasitol. 2000;30:35–44. doi: 10.1016/s0020-7519(99)00168-x. [DOI] [PubMed] [Google Scholar]
- Brisse S., Henriksson J., Barnabé C., Douzery E.J.P., Berkvens D., Serrano M., De Carvalho M.R.C., Buck G.A., Dujardin J.C., Tibayrenc M. Evidence for genetic exchange and hybridization in Trypanosoma cruzi based on nucleotide sequences and molecular karyotype. Infect. Genet. Evol. 2003;2(3):173–183. doi: 10.1016/s1567-1348(02)00097-7. [DOI] [PubMed] [Google Scholar]
- Budroni S., Siena E., Dunning Hotopp J.C., Seib K.L., Serruto D., Nofroni C., Comanducci M., Riley D.R., Daugherty S.C., Angiuoli S.V., Covacci A., Pizza M., Rappuoli R., Moxon E., Tettelin E.R., Medini H.D. Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proc. Natl. Acad. Sci. U. S. A. 2011;108:4494–4499. doi: 10.1073/pnas.1019751108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell L.T., Currie B.J., Krockenberger M., Malik R., Meyer W., Heitman J., Carter D. Clonality and recombination in genetically differentiated subgroups of Cryptococcus gattii. Eukaryot. Cell. 2005;4:1403–1409. doi: 10.1128/EC.4.8.1403-1409.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caugant D., Maiden M.C.J. Meningococcal carriage and disease—population biology and evolution. Vaccine. 2009;27S:B64–B70. doi: 10.1016/j.vaccine.2009.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chargui N., Amro A., Haouas N., Schönian G., Babba H., Schmidt S., Ravel C., Lefebvre M., Bastien P., Chaker E., Aoun K., Zribi M., Kuhls K. Population structure of Tunisian Leishmania infantum and evidence for the existence of hybrids and gene flow between genetically different populations. Int. J. Parasitol. 2009;39:801–811. doi: 10.1016/j.ijpara.2008.11.016. [DOI] [PubMed] [Google Scholar]
- Cosentino R.O., Agüero F. A simple strain typing assay for Trypanosoma cruzi: discrimination of major evolutionary lineages from a single amplification product. PLoS Negl. Trop. Dis. 2012;6:1–11. doi: 10.1371/journal.pntd.0001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cracraft J. Species concept and speciation analysis. In: Johnson R.F., editor. Current Ornithology. Plenum Press; New York: 1983. pp. 159–187. [Google Scholar]
- de Freitas J.M., Augusto-Pinto L., Pimenta J.R., Bastos-Rodrigues L., Gonçalves .V.F., Teixeira S.M.R., Chiari E., Junqueira A.C.V., Fernandes O., Macedo A.M., Machado C.R., Pena S.D.J. Ancestral genomes, sex, and the population structure of Trypanosoma cruzi. PLoS Pathog. 2006;2(3):e24. doi: 10.1371/journal.ppat.0020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Meeûs T., McCoy K., Prugnolle F., Chevillon C., Durand P., Hurtrez-Boussès S., Renaud F. Population genetics and molecular epidemiology or how to “débusquer la bête”. Infect. Genet. Evol. 2007;7:308–332. doi: 10.1016/j.meegid.2006.07.003. [DOI] [PubMed] [Google Scholar]
- de Paula Baptista R., Alchaar D’Ávila D., Segatto M., Faria do Valle I., Regina Franco G., Silva Valadares H.M., Dias Gontijo E., da Cunha Galvão L.M., Pena S.D.J., Chiari E., Machado C.R., Macedo A.M. Evidence of substantial recombination among Trypanosoma cruzi II strains from Minas Gerais. Infect. Genet. Evol. 2014;22:183–191. doi: 10.1016/j.meegid.2013.11.021. [DOI] [PubMed] [Google Scholar]
- Didelot X., Maiden M.C.J. Impact of recombination on bacterial evolution. Trends Microbiol. 2010;182:315–322. doi: 10.1016/j.tim.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T. Columbia University Press; New York: 1937. Genetics and the Origin of Species. [Google Scholar]
- Downing T., Imamura H., Decuypere S., Clark T.G., Coombs G.H., Cotton J.A., Hilley J.D., de Doncker S., Maes L., Mottram J.C., Quail M.A., Rijal S., Sanders M., Schönian G., Stark O., Sundar S., Vanaerschot M., Hertz-Fowler C., Dujardin J.C., Berriman M. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011;21:2143–2156. doi: 10.1101/gr.123430.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed N.M., Myler P.J., Bartholomeu D.C., Nilsson D., Aggarwal G., Tran A.N., Ghedin E., Worthey E.A., Delcher A.L., Blandin G., Westenberger S.J., Caler E., Cerqueira G.C., Branche C., Haas B., Anupama A., Arner E., Aslund L., Attipoe P., Bontempi E., Bringaud F., Burton P., Cadag E., Campbell D.A., Carrington M., Crabtree J., Darban H., da Silveira J.F., de Jong P., Edwards K., Englund P.T., Fazelina G., Feldblyum T., Ferella M., Frasch A.C., Gull K., Horn D., Hou L., Huang Y., Kindlund E., Klingbeil M., Kluge S., Koo H., Lacerda D., Levin M.J., Lorenzi H., Louie T., Machado C.R., McCulloch R., McKenna A., Mizuno Y., Mottram J.C., Nelson S., Ochaya S., Osoegawa K., Pai G., Parsons M., Pentony M., Pettersson U., Pop M., Ramirez J.L., Rinta J., Robertson L., Salzberg S.L., Sanchez D.O., Seyler A., Sharma R., Shetty J., Simpson A.J., Sisk E., Tammi M.T., Tarleton R., Teixeira S., Van Aken S., Vogt C., Ward P.N., Wickstead B., Wortman J., White O., Fraser C.M., Stuart K.D., Andersson B. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- Feretzaki F., Heitman J. Unisexual reproduction drives evolution of eukaryotic microbial pathogens. PLoS Pathog. 2013;9(10):e1003674. doi: 10.1371/journal.ppat1003674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R.C., Briones M.R.S. Phylogenetic evidence based on Trypanosoma cruzi nuclear gene sequences and information entropy suggest that inter-strain intragenic recombination is a basic mechanism underlying the allele diversity of hybrid strains. Infect. Genet. Evol. 2012;12:1064–1071. doi: 10.1016/j.meegid.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Flores-López C.A., Machado C.A. Analyses of 32 loci clarify phylogenetic relationships among Trypanosoma cruzi lineages and support a single hybridization prior to human contact. PLoS Negl. Trop. Dis. 2011;5(8):e1272. doi: 10.1371/journal.pntd0001272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaunt M.W., Yeo M., Frame I.A., Tothard J.R., Carrasco H.J., Taylor M.C., Mena S.S., Veazey P., Miles G.A., Acosta N., Rojas de Arias A., Miles M.A. Mechanism of genetic exchange in American trypanosomes. Nature. 2003;421:936–939. doi: 10.1038/nature01438. [DOI] [PubMed] [Google Scholar]
- Guhl F., Ramírez J.D. Trypanosoma cruzi I diversity: towards the need of genetic subdivision? Acta Trop. 2011;119:1–4. doi: 10.1016/j.actatropica.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Hamilton P.B., Lewis M.D., Cruickshank C., Gaunt M.W., Yeo M., Llewellyn M.S., Valente S.A., Maia da Silva F., Stevens J.R., Miles M.A., Teixeira M.M.G. Identification and lineage genotyping of South American trypanosomes using fluorescent fragment length barcoding. Infect. Genet. Evol. 2011;11:44–51. doi: 10.1016/j.meegid.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Herrera C., Bargues M.D., Fajardo A., Montilla M., Triana O., Vallejo G.A., Guhl F. Identifying four Trypanosoma cruzi I isolate haplotypes from different geographic regions in Colombia. Infect. Genet. Evol. 2007;7:535–539. doi: 10.1016/j.meegid.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Lachaud L., Bourgeois N., Kuk N., Morelle C., Crobu L., Merlin G., Bastien P., Pagès M., Sterkers Y. Constitutive mosaic aneuploidy is a unique genetic feature widespread in the Leishmania genus. Microbes Infect. 2014;16:61–66. doi: 10.1016/j.micinf.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Lauthier J.J., Tomasini N., Barnabé C., Monje Rumi M.M., Alberti D’Amato A.M., Ragone P.G., Yeo M., Lewis M.D., Llewellyn M.S., Basombrío M.A., Miles M.A., Tibayrenc M., Diosque P. Candidate targets for multilocus sequence typing of Trypanosoma cruzi: validation using parasite stocks from the Chaco Region and a set of reference strains. Infect. Genet. Evol. 2012;12:350–358. doi: 10.1016/j.meegid.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Lehmann T., Graham D.H., Dahl E.R., Bahia-Oliveira L.M.G., Gennari S.M., Dubey J.P. Variation in the structure of Toxoplasma gondii and the roles of selfing, drift, and epistatic selection in maintaining linkage disequilibria. Infect. Genet. Evol. 2004;4:107–114. doi: 10.1016/j.meegid.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Lewis M.D., Llewellyn M.S., Gaunt M.W., Yeo M., Carrasco H.J., Miles M.A. Flow cytometric analysis and microsatellite genotyping reveal extensive DNA content variation in Trypanosoma cruzi populations and expose contrasts between natural and experimental hybrids. Int. J. Parasitol. 2009;39:1305–1317. doi: 10.1016/j.ijpara.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis M.D., Llewellyn M.S., Yeo M., Acosta N., Gaunt M.W., Miles M.A. Recent independent and anthropogenic origins of Trypanosoma cruzi hybrids. PLoS Negl. Trop. Dis. 2011;5:1–13. doi: 10.1371/journal.pntd.0001363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima L., Ortiz P.A., da Silva F.M., Alves J.M.P., Serrano M.G., Cortez A.P., Alfieri S.C., Buck G.A., Teixeira M.M.G. Repertoire, genealogy and genomic organization of Cruzipain and homologous genes in Trypanosoma cruzi, T. cruzi-like and other trypanosome species. PLoS ONE. 2012;7(6):e38385. doi: 10.1371/journal.pone.0038385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn M.S., Lewis M.D., Acosta N., Yeo M., Carrasco H.J., Segovia M., Vargas J., Torrico F., Miles M.A., Gaunt M.W. Trypanosoma cruzi IIc: phylogenetic and phylogeographic insights from sequence and microsatellite analysis and potential impact on emergent Chagas disease. PLoS Negl. Trop. Dis. 2009;3(9):e510. doi: 10.1371/journal.pntd.0000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn M.S., Miles M.A., Carrasco H.J., Lewis M.D., Yeo M., Vargas J., Torrico F., Diosque P., Valente V., Valente S.A., Gaunt M.A. Genome-scale multilocus microsatellite typing of Trypanosoma cruzi discrete typing unit I reveals phylogeographic structure and specific genotypes linked to human infection. PLoS Pathog. 2009;5(5):e1000410. doi: 10.1371/journal.ppat.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn M.S., Rivett-Carnac J.B., Fitzpatrick S., Lewis M.D., Yeo M., Gaunt M.W., Miles M.A. Extraordinary Trypanosoma cruzi diversity within single mammalian reservoir hosts implies a mechanism of diversifying selection. Int. J. Parasitol. 2011;41:609–614. doi: 10.1016/j.ijpara.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado C.A., Ayala F.J. Nucleotide sequences provide evidence of genetic exchange among distantly related lineages of Trypanosoma cruzi. Proc. Natl. Acad. Sci. U. S. A. 2001;98:7396–7401. doi: 10.1073/pnas.121187198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden M.C.J. Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 2006;60:561–588. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- Maiden M.C.J. Population genomics: diversity and virulence in the Neisseria. Curr. Opin. Microbiol. 2008;11:467–471. doi: 10.1016/j.mib.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaert A., Downing T., Imamura H., Dujardin J.C. Adaptive mechanisms in pathogens: universal aneuploidy in Leishmania. Trends Parasitol. 2012;28:370–376. doi: 10.1016/j.pt.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Marcili A., Lima L., Cavazzsana M., Junqueira A.C.V., Veludo H.H., Maia da Silva F., Campaner M., Paiva F., Nunes V.L.B., Teixeira M.M.G. A new genotype of Trypanosoma cruzi associated with bats evidenced by phylogenetic analyses using SSU rDNA, cytochrome b and Histone H2B genes and genotyping based on ITS1 rDNA. Parasitology. 2009;136:641–655. doi: 10.1017/S0031182009005861. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J., Smith N.H., O’Rourke M., Spratt B.G. How clonal are bacteria? Proc. Natl. Acad. Sci. U. S. A. 1993;90:4384–4388. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messenger L.A., Llewellyn M.S., Bhattacharyya T., Franzén O., Lewis M.D., Ramírez J.D., Carrasco H.J., Andersson B., Miles M.A. Multiple mitochondrial introgression events and heteroplasmy in Trypanosoma cruzi revealed by maxicircle MLST and next generation sequencing. PLoS Negl. Trop. Dis. 2012;6:e1584. doi: 10.1371/journal.pntd0001584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles M.A., Llewellyn M.S., Lewis M.D., Yeo M., Baleela R., Fitzpatrick S., Gaunt M.W., Mauricio I.L. The molecular epidemiology and phylogeography of Trypanosoma cruzi and parallel research on Leishmania: looking back and to the future. Parasitology. 2009;136:1509–1528. doi: 10.1017/S0031182009990977. [DOI] [PubMed] [Google Scholar]
- Miles M.A., Souza A., Povoa M., Shaw J.J., Lainson R., Toyé P.J. Isozymic heterogeneity of Trypanosoma cruzi in the first autochtonous patients with Chagas’ disease in Amazonian Brazil. Nature. 1978;272:819–821. doi: 10.1038/272819a0. [DOI] [PubMed] [Google Scholar]
- Miles M.A., Povoa M., Prata A., Cedillos R.A., De Souza A.A., Macedo V. Do radically dissimilar Trypanosoma cruzi strains (zymodemes) cause Venezuelan and Brazilian forms of Chagas’ disease? Lancet. 1981;8234:1336–1340. doi: 10.1016/s0140-6736(81)92518-6. [DOI] [PubMed] [Google Scholar]
- Miles M.A., Toyé P.J., Oswald S.C., Godfrey D.G. The identification by isoenzyme patterns of two distinct strain-groups of Trypanosoma cruzi, circulating independently in a rural area of Brazil. Trans. R. Soc. Trop. Med. Hyg. 1977;71(3):217–225. doi: 10.1016/0035-9203(77)90012-8. [DOI] [PubMed] [Google Scholar]
- Minning T.A., Weatherly D.B., Flibotte S., Tarleton R.L. Widespread, focal copy number variations (CNV) and whole chromosome aneuploidies in Trypanosoma cruzi strains revealed by array comparative genomic hybridization. BMC Genomics. 2011;12:139. doi: 10.1186/1471-2164-12-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya A., Holmes E.C., González-Candelas F. The population genetics and evolutionary epidemiology of RNA viruses. Nat. Rev. Microbiol. 2004;2:279–288. doi: 10.1038/nrmicro863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocaña-Mayorga S., Llewellyn M.S., Costales J.A., Miles M.A., Grijalva M.J. Sex subdivision, and domestic dispersal of Trypanosoma cruzi Lineage I in Southern Ecuador. PLoS Negl. Trop. Dis. 2010;4:1–8. doi: 10.1371/journal.pntd.0000915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira R.P., Broude N.E., Macedo A.M., Cantor C.R., Smith C.L., Pena S.D.J. Probing the genetic population structure of Trypanosoma cruzi with polymorphic microsatellites. Proc. Natl. Acad. Sci. U. S. A. 1998;95:3776–3780. doi: 10.1073/pnas.95.7.3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omilian A.R., Cristescu M.E.A., Dudycha J.L., Lynch M. Ameiotic recombination in asexual lineages of Daphnia. Proc. Natl. Acad. Sci. U. S. A. 2006;103:18638–18643. doi: 10.1073/pnas.0606435103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson T., Okinaka R.T., Foster J.T., Keim P. Phylogenetic understanding of clonal populations in an era of whole genome sequencing. Infect. Genet. Evol. 2009;9:1010–1019. doi: 10.1016/j.meegid.2009.05.014. [DOI] [PubMed] [Google Scholar]
- Pinto C.M., Kalko E.K.V., Cottontail I., Wellinghausen N., Cottontail V.M. TcBat a bat-exclusive lineage of Trypanosoma cruzi in the Panama Canal Zone, with comments on its classification and the use of the 18S rRNA gene for lineage identification. Infect. Genet. Evol. 2012;12:1328–1332. doi: 10.1016/j.meegid.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Ramírez J.D., Duque M.C., Guhl F. Phylogenetic reconstruction based on Cytochrome b (Cytb) gene sequences reveals distinct genotypes within Colombian Trypanosoma cruzi I populations. Acta Trop. 2011;119:61–65. doi: 10.1016/j.actatropica.2011.04.009. [DOI] [PubMed] [Google Scholar]
- Ramírez J.D., Guhl F., Messager L.A., Lewis M.D., Montilla M., Cucunubá Z., Miles M.A., Llewellyn M.S. Contemporary cryptic sexuality in Trypanosoma cruzi. Mol. Ecol. 2012;17:4216–4226. doi: 10.1111/j.1365-294X.2012.05699.x. [DOI] [PubMed] [Google Scholar]
- Ramírez J.D., Herrera C., Bogotá Y., Duque M.C., Suárez-Rivillas A., Guhl F. Validation of a Poisson-distributed limiting dilution assay (LDA) for a rapid and accurate resolution of multiclonal infections in natural Trypanosoma cruzi populations. J. Microbiol. Methods. 2013;92:220–225. doi: 10.1016/j.mimet.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Ramírez J.D., Tapia-Calle G., Guhl F. Genetic structure of Trypanosoma cruzi in Colombia revealed by a High-throughput Nuclear Multilocus Sequence Typing (nMLST) approach. BMC Genet. 2013;14:96. doi: 10.1186/1471-2156-14-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez J.D., Llewellyn M.S. Reproductive clonality in protozoan pathogens—truth or artefact? Mol. Ecol. 2014;23:4195–4202. doi: 10.1111/mec.12872. [DOI] [PubMed] [Google Scholar]
- Revollo S., Oury B., Laurent J.P., Barnabé C., Quesney V., Carrière V., Noël S., Tibayrenc M. Trypanosoma cruzi: impact of clonal evolution of the parasite on its biological and medical properties. Exp. Parasitol. 1998;89:30–39. doi: 10.1006/expr.1998.4216. [DOI] [PubMed] [Google Scholar]
- Rogers M.B., Hilley J.D., Dickens N.J., Wilkes J., Bates P.A., Depledge D.P., Harris D., Her Y., Herzyk P., Imamura H., Otto T.D., Sanders M., Seeger K., Dujardin J.C., Berriman B., Smith D.F., Hertz-Fowler C., Mottram J.C. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011;21:2129–2142. doi: 10.1101/gr.122945.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougeron V., De Meeûs T., Hide M., Waleckx E., Bermudez H., Arevalo J., Llanos-Cuentas A., Dujardin J.C., De Doncker S., Le Ray D., Ayala F.J., Bañuls A.L. Extreme inbreeding in Leishmania braziliensis. Proc. Natl. Acad. Sci. U. S. A. 2009;106:10224–10229. doi: 10.1073/pnas.0904420106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougeron V., De Meeûs T., Bañuls A.L. A primer for Leishmania population genetic studies. Trends Parasitol. 2015;31:52–59. doi: 10.1016/j.pt.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Rozas M., De Doncker S., Adaui V., Coronado X., Barnabé C., Tibayrenc M., Solari A., Dujardin J.C. Multilocus polymerase chain reaction restriction fragment–length polymorphism genotyping of Trypanosoma cruzi (Chagas disease): taxonomic and clinical applications. J. Infect. Dis. 2007;195:1381–1388. doi: 10.1086/513440. [DOI] [PubMed] [Google Scholar]
- Sibley L.D., Boothroyd J.C. Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature. 1992;359:82–85. doi: 10.1038/359082a0. [DOI] [PubMed] [Google Scholar]
- Souza R.T., Lima F.M., Moraes Barros R., Cortez D.R., Santos M.F., Cordero E.M., Conceiçao Ruiz J., Goldenberg S., Teixeira M.M.G., Franco da Silveira J. Genome size, karyotype polymorphism and chromosomal evolution in Trypanosoma cruzi. PLoS ONE. 2011;6(8):e23042. doi: 10.1371/journal.pone.0023042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterkers Y., Crobu L., Lachaud L., Pagès M., Bastien P. Parasexuality and mosaic aneuploidy in Leishmania: alternative genetics. Trends Parasitol. 2014;30:429–435. doi: 10.1016/j.pt.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Sterkers Y., Lachaud L., Bourgeois N., Crobu L., Bastien P., Pagès M. Novel insights into genome plasticity in Eukaryotes: mosaic aneuploidy in Leishmania. Mol. Microbiol. 2012;86:15–23. doi: 10.1111/j.1365-2958.2012.08185.x. [DOI] [PubMed] [Google Scholar]
- Sterkers Y., Lachaud L., Crobu L., Bastien P., Pagès M. FISH analysis reveals aneuploidy and continual generation of chromosomal mosaicism in Leishmania major. Cell. Microbiol. 2011;139:274–283. doi: 10.1111/j.1462-5822.2010.01534.x. [DOI] [PubMed] [Google Scholar]
- Su C., Khan A., Zhou P., Majumdara D., Ajzenberg D., Dardé M.L., Zhu X.Q., Ajioka J.W., Rosenthal B.M., Dubey J.P., Sibley D. Globally diverse Toxoplasma gondii isolates comprise six major clades originating from a small number of distinct ancestral lineages. Proc. Natl. Acad. Sci. U. S. A. 2012;109:5844–5849. doi: 10.1073/pnas.1203190109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subileau M., Barnabé C., Douzery E., Diosque P., Tibayrenc M. Trypanosoma cruzi: new insights on ecophylogeny and hybridization by Multigene sequencing of three nuclear and one maxicircle genes. Exp. Parasitol. 2009;122:328–337. doi: 10.1016/j.exppara.2009.04.008. [DOI] [PubMed] [Google Scholar]
- Telleria J., Barnabé C., Hide M., Bañuls A.L., Tibayrenc M. Predominant clonal evolution leads to a close parity between gene expression profiles and subspecific phylogeny in Trypanosoma cruzi. Mol. Biochem. Parasitol. 2004;137(1):133–141. doi: 10.1016/j.molbiopara.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Telleria J., Biron D.G., Brizard J.P., Demettre E., Seveno M., Barnabé C., Ayala F.J., Tibayrenc M. Phylogenetic Character Mapping of proteomic diversity shows high correlation with subspecific phylogenetic diversity in Trypanosoma cruzi, the agent of Chagas disease. Proc. Natl. Acad. Sci. U. S. A. 2010;107:20411–20416. doi: 10.1073/pnas.1015496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telleria J., Barnabé C., Ayala F.J., Tibayrenc M. Phylogenetic character mapping of RADES Probing, a new marker for exploring the clonal evolution of expressed coding sequences in Trypanosoma cruzi, the agent of Chagas disease. Infect. Genet. Evol. 2013;19:287–291. doi: 10.1016/j.meegid.2013.03.027. [DOI] [PubMed] [Google Scholar]
- Tibayrenc M. Genetic epidemiology of parasitic protozoa and other infectious agents: the need for an integrated approach. Int. J. Parasitol. 1998;28(1):85–104. doi: 10.1016/s0020-7519(97)00180-x. [DOI] [PubMed] [Google Scholar]
- Tibayrenc M., Ayala F.J. Towards a population genetics of microorganisms: the clonal theory of parasitic protozoa. Parasitol. Today. 1991;7(9):228–232. doi: 10.1016/0169-4758(91)90234-f. [DOI] [PubMed] [Google Scholar]
- Tibayrenc M., Ayala F.J. The clonal theory of parasitic protozoa: 12 years on. Trends Parasitol. 2002;18(9):405–410. doi: 10.1016/s1471-4922(02)02357-7. [DOI] [PubMed] [Google Scholar]
- Tibayrenc M., Ayala F.J. Reproductive clonality of pathogens: a perspective on pathogenic viruses, bacteria, fungi, and parasitic protozoa. Proc. Natl. Acad. Sci. U. S. A. 2012;109(48):E3305–E3313. doi: 10.1073/pnas.1212452109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M., Ayala F.J. How clonal are Trypanosoma and Leishmania? Trends Parasitol. 2013;29:264–269. doi: 10.1016/j.pt.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Tibayrenc M., Ayala F.J. New insights into Clonality and Panmixia in Plasmodium and Toxoplasma. Adv. Parasitol. 2014;84:253–268. doi: 10.1016/B978-0-12-800099-1.00005-3. [DOI] [PubMed] [Google Scholar]
- Tibayrenc M., Ayala F.J. Cryptosporidium, Giardia, Cryptococcus, Pneumocystis genetic variability: cryptic biological species or clonal near-clades? PLoS Pathog. 2014;10(4):e1003908. doi: 10.1371/journal.ppat.1003908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M., Brenière S.F. Trypanosoma cruzi: major clones rather than principal zymodemes. Mem. Inst. Oswaldo Cruz, Rio De Janeiro. 1988;83(Suppl. 1):249–255. doi: 10.1590/s0074-02761988000500005. [DOI] [PubMed] [Google Scholar]
- Tibayrenc M., Cariou M.L., Solignac M., Carlier Y. Arguments génétiques contre l’existence d’une sexualité actuelle chez Trypanosoma cruzi; implications taxinomiques. C. R. Acad. Sci. Paris. 1981;293:207–209. [Google Scholar]
- Tibayrenc M., Cariou M.L., Solignac M., Dédet J.P., Poch O., Desjeux P. New electrophoretic evidence of genetic variation and diploidy in Trypanosoma cruzi, the causative agent of Chagas’ disease. Genetica. 1985;67:223–230. [Google Scholar]
- Tibayrenc M., Kjellberg F., Ayala F.J. A clonal theory of parasitic protozoa: the population structure of Entamoeba, Giardia, Leishmania, Naegleria, Plasmodium, Trichomonas and Trypanosoma, and its medical and taxonomical consequences. Proc. Natl. Acad. Sci. U. S. A. 1990;87:2414–2418. doi: 10.1073/pnas.87.7.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M., Kjellberg F., Ayala F.J. The clonal theory of parasitic protozoa: a taxonomic proposal applicable to other clonal organisms. Bioscience. 1991;41(11):767–774. [Google Scholar]
- Tibayrenc M., Kjellberg F., Arnaud J., Oury B., Brenière S.F., Dardé M.L., Ayala F.J. Are eukaryotic microorganisms clonal or sexual? A population genetics vantage. Proc. Natl. Acad. Sci. U. S. A. 1991;88:5129–5133. doi: 10.1073/pnas.88.12.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M., Neubauer K., Barnabé C., Guerrini F., Skarecki D., Ayala F.J. Genetic characterization of six parasitic protozoa: parity of random-primer DNA typing and multilocus isoenzyme electrophoresis. Proc. Natl. Acad. Sci. U. S. A. 1993;90:1335–1339. doi: 10.1073/pnas.90.4.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibayrenc M., Ward P., Moya A., Ayala F.J. Natural populations of Trypanosoma cruzi, the agent of Chagas’ disease, have a complex multiclonal structure. Proc. Natl. Acad. Sci. U. S. A. 1986;83:115–119. doi: 10.1073/pnas.83.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasini N., Lauthier J.J., Monje Rumi M.M., Ragone P.G., Alberti D’Amato A.A., Pérez Brandan C., Cura C.I., Schijman A.G., Barnabé C., Tibayrenc M., Basombrıó M.A., Falla A., Herrera C., Guhl F., Diosque P. Interest and limitations of Spliced Leader Intergenic Region sequences for analyzing Trypanosoma cruzi I phylogenetic diversity in the Argentinean Chaco. Infect. Genet. Evol. 2011;11:300–307. doi: 10.1016/j.meegid.2010.10.020. [DOI] [PubMed] [Google Scholar]
- Tomasini N., Lauthier J.J., Monje Rumi M.M., Ragone P.G., Alberti D’Amato A.M., Pérez Brandán C., Basombrío M.A., Diosque P. Preponderant clonal evolution of Trypanosoma cruzi I from Argentinean Chaco revealed by Multilocus Sequence Typing (MLST) Infect. Genet. Evol. 2014;27:348–354. doi: 10.1016/j.meegid.2014.08.003. [DOI] [PubMed] [Google Scholar]
- Volkman S.K., Neafsey D.E., Schaffner S.F., Park D.J., Wirth D.F. Harnessing genomics and genome biology to understand malaria biology. Nat. Rev. Genet. 2012;13:315–328. doi: 10.1038/nrg3187. [DOI] [PubMed] [Google Scholar]
- Westenberger S.J., Barnabé C., Campbell D.A., Sturm N.R. Two hybridization events define the population structure of Trypanosoma cruzi. Genetics. 2005;171:527–543. doi: 10.1534/genetics.104.038745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenberger S.J., Sturm N.R., Campbell D.A. Trypanosoma cruzi 5S rRNA arrays define five groups and indicate the geographic origins of an ancestor of the heterozygous hybrids. Int. J. Parasitol. 2006;36:337–346. doi: 10.1016/j.ijpara.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Wielinga C.M., Thompson R.C.A. Comparative evaluation of Giardia duodenalis sequence data. Parasitology. 2007;134:1795–1821. doi: 10.1017/S0031182007003071. [DOI] [PubMed] [Google Scholar]
- Yeo M., Mauricio I.L., Messenger L.A., Lewis M.D., Llewellyn M.S., Acosta N., Bhattacharyya T., Diosque P., Carrasco H.J., Miles M.A. Multilocus sequence typing (MLST) for lineage assignment and high resolution diversity studies in Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2011;5(6):e1049. doi: 10.1371/journal.pntd.0001049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip C.W., Hon C.C., Shi M., Lam T.T., Chow K.Y., Zeng F., Leung F.C. Phylogenetic perspectives on the epidemiology and origins of SARS and SARS-like coronaviruses. Infect. Genet. Evol. 2009;9:1185–1196. doi: 10.1016/j.meegid.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingales B., Miles M.A., Campbell D., Tibayrenc M., Macedo A.M., Teixeira M.M., Schijman A., Llewellyn M.S., Lages-Silva E., Machado C.R., Andrade S.G., Sturm N.R. The revised Trypanosoma cruzi subspecific nomenclature: rationale, epidemiological relevance and research applications. Infect. Genet. Evol. 2012;12:240–253. doi: 10.1016/j.meegid.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Zingales B., Miles M.A., Moraes C.B., Luquetti A., Guhl F., Schijman A.G., Ribeiro I. Drug discovery for Chagas disease should consider Trypanosoma cruzi strain diversity. Mem. Inst. Oswaldo Cruz, Rio de Janeiro. 2014;109:828–833. doi: 10.1590/0074-0276140156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoossmann-Diskin A. The origin of Eastern European Jews revealed by autosomal, sex chromosomal and mtDNA polymorphisms. Biol. Direct. 2010;5:57. doi: 10.1186/1745-6150-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]