

Abstract
To achieve proper diagnosis of dogs based on acute clinical symptoms and poorly preserved field samples taken from animals that died due to canine distemper (CD), a new differential diagnostic test has been developed based on polymerase chain reaction (PCR). In this study, more than 150 samples collected from dogs showing respiratory, gastrointestinal and neurological signs suggesting canine distemper virus (CDV) infection were examined. The samples consisted of urine, blood and nasal swabs collected from clinically ill patients, sent to our laboratory by clinicians from various veterinary clinics throughout Hungary. Various organs collected during the necropsy of dogs with pathological changes that suggested CDV infection were also included. Three distinct PCRs were designed. For diagnostic purposes, a primer pair specific to a 409 bases-long segment within the conservative part of the large polymerase region (L) of the CDV genome was designed. Using this test, out of the 150 analyzed samples, 46 (30.66%) proved to be positive for CDV, indicating that CDV still represents a high risk to the canine population in Hungary. For the phylogenetical analysis, a primer pair that completely encompasses the hemagglutinin (H) gene of the CDV genome was designed. The amplicons of this region were sequenced in both directions using the appropriate primers. Our results indicate that several different CDV genotypes are currently present in Hungary. Nine of the analyzed Hungarian strains turned out to belong to the so-called Arctic group of CDVs, and were most closely related to non-European strains from North America, China and Greenland, as well as to the phocine distemper virus 2 (PDV-2) isolated from Baikal seals (Phoca sibirica). One of the Hungarian strains showed high similarity to other European isolates from Denmark, Germany, Italy and Turkey, as well as to other isolates from geographically more distant regions, such as the USA. Three Hungarian strains seem to join a new cluster that is formed by only a couple of strains, one isolated from a mink in Denmark, and another from a dog in North America. Using a third set of primers, a restriction fragment length polymorphism (RFLP) assay has also been designed for the fast and reliable differentiation of the wild-type CDVs from the vaccine strains.
Keywords: Canine distemper, Phylogenetic analysis, RFLP, Vaccine strain discrimination
1. Introduction
Despite the vaccination procedures applied in most European countries, canine distemper (CD) is still one of the most serious threats to the susceptible carnivore population in Central and Eastern Europe. CD is a highly contagious viral infection of different carnivores that belong to numerous animal families, such as Canidae, Mustelidae, Procyonidae, Felidae and many others (Kovács et al., 1983, Appel, 1987, Blixenkrone-Møller et al., 1993, Gemma et al., 1996, Barrett, 1999, Kim et al., 2001, Shin et al., 2004, Kabakci et al., 2004, Lan et al., 2006). CD is caused by the canine distemper virus (CDV) which belongs to the Morbillivirus genus of the Paramyxoviridae virus family. The clinical symptoms of the disease include severe respiratory, digestive and neurological signs with frequently fatal outcome (Appel and Summers, 1995; Stettler et al., 1997; Frisk et al., 1999; Ozkul et al., 2004). The clinical evolution of the infection is characterized by a specific biphasic fever curve, and other clinical signs that do not respond to symptomatic antimicrobial therapy. Domestic canine populations seem to act as reservoirs for other receptive carnivores (Carpenter et al., 1998, Lednicky et al., 2004).
In this study, the authors analyzed nucleic acid sequences of virus strains from different parts of Hungary along with other CDV sequences deposited in the GenBank by researchers from all over the world. Since the hemagglutinin (H) gene shows the greatest extent of genetic variation that allows distinction of various lineages (Haas et al., 1997, Harder and Osterhaus, 1997, Mochizuki et al., 1999, Iwatsuki et al., 2000; Uema et al., 2005; Martella et al., 2006), the authors designed a primer pair that allowed the complete amplification of the H gene. Due to the fact that several lineages can be present during the same time within one geographic region (Haas et al., 1999, Lan et al., 2005), and the fact that the possibility of point mutations at the attachment site of the primers is relatively high for the H gene, a more conserved region of the viral genome, the large polymerase (L) gene was targeted for diagnostic purposes.
The main objective of the work described in the present study was to design a reliable test that can be used on a routine basis for the fast and proper diagnosis of CDV infections. Another objective of the investigation was to clarify the phylogenetic relationships among the Hungarian-, and other CDV strains from surrounding countries and around the world. Due to the fact that cases of infection with CDV of vaccinated animals have been reported recently, when the infection was demonstrated by PCR using the viral RNA isolated from cell cultures infected with tissue homogenates (Lan et al., 2006), the authors also designed a restriction fragment length polymorphism assay that allows the fast and reliable differentiation of the wild-type viruses from the vaccine strains. The availability of a reliable test for the fast discrimination of wild-type viruses and vaccine strains is very important, since in some situations the vaccination itself could be blamed by the owner of the animal for certain clinical signs or manifestations in cases where recently vaccinated animals get infected before developing proper immunity. Without a specific test that can differentiate between the virus strains used in the vaccine and the one that actually infected the animal, the owner could interpret the incident as a failure of the vaccination, or even worse, he or she might think that the vaccine itself was the cause of the infection and eventual death of the animal. The differentiation is also useful in cases of abortion of pregnant females that were vaccinated against CD during gestation. Vaccine manufacturers rule out the possibility of this event, but in most situations the owners and the clinicians demand an irrefutable proof whether the abortion was linked or not to the virus strain present in the vaccine. Due to these and several other reasons, there is definite need for the reliable differentiation of the wild-type viruses and the virus strains used in vaccines.
2. Materials and methods
2.1. Samples
A total number of 150 samples consisting of urine, blood and nasal swabs taken from clinically ill patients were investigated. Beside these, samples from various organs were collected and analyzed from animals that succumbed following clinical symptoms that suggested CDV infection, and when CD was suspected after the pathological and histopathological examination of carcasses.
The dogs from which the samples were taken had various clinical backgrounds and mostly unknown immunization histories. As part of a monitoring survey, samples were collected from animals living at the Dog Shelter of the City Council of Budapest. The samples were taken during the spring of 2005 and during the spring and summer of 2006. The interval of time between the collection of the first and the last samples lasted more than a month in both cases. In the phylogenetical study, a number of six positive samples from the first, and three from the second period were used. These samples consisted of blood, urine and nasal swabs. During the investigation of blood samples, both serum and separated leukocytes were used. The authors decided to collect blood samples, because a positive result obtained in case of a blood sample was considered to be the most reliable proof for the viremic status of the animal. For this purpose, blood was collected in two separate tubes from each animal: one contained an anticoagulant (EDTA), while the other did not. The serum was obtained from the clotted blood following centrifugation (20 min at 1500 × g). In case of the anticoagulant-treated samples, the leukocytes were separated from the rest of the blood components using the hypoosmotic shock technique.
Other samples used in the present study were taken from carcasses of dogs that died following clinical signs suggesting CD, or that were sent to the Department of Pathology and Forensic Veterinary Medicine. The rest of samples were collected at various small animal practices in Budapest and other parts of Hungary, and in the Department of Internal Medicine of our Faculty. Table 1 contains the description of the animals, their provenience, their clinical and immunological status, and the types of samples that were used in the present study.
Table 1.
Description of animals and samples
| Group | Sample code | Anamnesis | Analyzed sample |
|---|---|---|---|
| I | H04Bp1F | Five months old, female vizsla; normal temperature, only neurological signs; euthanized; histology: lymphoplasmocytic perivascular encephalitis; previously vaccinated against CD | Urine and urinary bladder |
| IIa | H05Bp2S | One year old, mixed breed male; respiratory signs; UVHa | Blood |
| H05Bp3S | Ten months old mixed breed female; respiratory signs; UVH | Blood | |
| H05Bp4S | Three years old, mixed breed male; respiratory signs; UVH | Blood | |
| H05Bp5F | Two years old, mixed breed male; respiratory signs; UVH | Urine | |
| H05Bp6F | Four months old, female labrador retriever; respiratory, digestive and neurological signs; UVH | Urine | |
| H05Bp7F | One year old, mixed breed female; respiratory signs; UVH | Urine | |
| IIb | H06Bp8F | 4.5 years old, male rottweiler; respiratory signs; UVH | Urine |
| H06Bp9S | One year old, mixed breed male; severe respiratory and digestive clinical signs; euthanized; pathological changes, characteristic to CD; UVH | Urine and urinary bladder | |
| H06Bp10S | 1.5 years old mixed breed female; respiratory signs; UVH | Nasal swab | |
| III | H06Ny11 | Two years old, mixed breed male; severe respiratory and digestive signs; not vaccinated | Urine |
| H06Ny12 | One year old mixed breed male; severe respiratory and digestive signs; UVH | Urine | |
| H06Ny13 | 2.5 years old mixed breed female; severe respiratory and digestive signs; UVH | Urine | |
Group I: sample obtained from Budapest (winter of 2004). Group IIa samples obtained from animals at, or retrieved from the Dog Shelter of the City Council of Budapest, during the first period (spring of 2005). Group IIb: samples obtained from animals at, or retrieved from the Dog Shelter of the City Council of Budapest, during the second period (spring and summer of 2006). Group III: samples obtained from Eastern Hungary (winter of 2005).
UVH: unknown vaccination history.
2.2. Purification of the nucleic acid
Tissue samples were homogenized in 10 ml phosphate buffered saline (PBS) and centrifuged at 1500 × g for 10 min. The nasal swabs were immersed in PBS solution with 10 ml/L antibiotic and antimycotic component (Sigma A5955 Antibiotic-Antimycotic suspension, stabilized solution) and then centrifuged, while the liquid samples (i.e. urine) were only centrifuged.
The viral RNA was isolated from the supernatants using the QIAamp viral RNA Mini Kit (Qiagen, Germany), according to the manufacturer's instructions.
2.3. Primers
In order to demonstrate the presence of the viral genome in samples, different pairs of primers were designed. For diagnostic purposes, primer pair “A” specific to a 409 bases long segment of the conservative region of the large polymerase gene (L) of the CDV genome was designed (Table 2 ). Samples that tested positive with these primers underwent another RT-PCR assay using the “B” set of primers that partially encompassed the hemagglutinin (H) gene of the CDV (Table 2). The phylogenetic analysis of the new Hungarian samples was based on the full nucleotide sequence of the H gene, determined by using another set of primers (“C”). The sensitivity of the PCRs was determined using serial dilutions of a vaccine virus strain with a previously determined number of CDV RNA copies (105).
Table 2.
Description of the primer pairs
| Primer pair | Sequence 5′–3′ | Target | Sense | Position | Purpose | Amplicon size (bp) |
|---|---|---|---|---|---|---|
| A | ATCCGCTCATCGATCAAGAC | L | + | 12400–12420 | Diagnostic | 409 |
| CAAGCCTCTTGCCAAGATTC | − | 12788–12808 | ||||
| B | AGGCCGTACATCACCAAGTC | H | + | 7323–7343 | RFLP | 1110 |
| TGGTAAGCCATCCGGAGTTC | − | 8412–8432 | ||||
| C | AACTTAGGGCTCAGGTAGTC | H | + | 6994–7014 | Phylogenetic | 2023 |
| AGATGGACCTCAGGGTATAG | − | 8996–9016 | ||||
2.4. RT-PCR assays
Reverse transcription and amplifications were performed in a continuous RT-PCR method by using the QIAGEN OneStep RT-PCR Kit (Qiagen, Germany). The 25 μl reaction mixtures contained 5 μl of 5× buffer (final MgCl2 concentration 1.5 mM), 0.4 mM of each deoxynucleozide triphosphate (dNTP), 10 U rRNasin™ RNase Inhibitor (Promega, USA), 0.8 μM of the appropriate forward and reverse primers, 1 μl of enzyme mix and 2.5 μl of template RNA. Reverse transcription was carried out at 50 °C for 30 min. Following an initial denaturation at 95 °C for 15 min, the reaction mixture was subjected to 40 cycles of heat denaturation at 94 °C for 45 s, primer annealing at 52 °C for 45 s and DNA extension at 72 °C for 1 min, followed by a final extension of 10 min at 72 °C. The reactions were performed in a PCR Sprint Thermal Cycler SPRT001 (Hybaid Limited, UK).
Following RT-PCR, 7.5 μl of the amplicons were electrophoresed in a 1.2% Tris acetate–EDTA–agarose gel (EM Science, Merck KGaA, Germany) at 80 V for 80 min. The gel was stained with ethidium bromide and the bands were visualized at 312 nm using a TFX 35M UV transilluminator (Life Technologies, UK), and photographed with a Kodak DS Electrophoresis Documentation and Analysis System using the Kodak Digital Science 1D software program. Product sizes were determined with reference to a 100 bp and a 1 kb molecular weight ladder (Gene Ruler 100 bp DNA Ladder, Fermentas Life Sciences, Lithuania).
2.5. RFLP analysis
The RFLP was performed on products amplified by the primer set “B” as follows: 5 μl of the PCR amplicons was mixed with 3.5 μl ddH2O, 1 μl of PsiI enzyme (SibEnzyme, Russia) and 1 μl of B9006S buffer solution provided by the manufacturer. The mixture was then heated to 36 °C for 60 min and mixed at 300 rpm, at intervals of 30 s in a Thermomixer Comfort device (Eppendorf, Germany). The amplicons were then detected by electrophoresis in a 2% Tris acetate–EDTA–agarose gel at 80 V for 80 min. The bands were visualized and photographed as described above for PCR products, and fragment sizes were determined using the same procedure.
2.6. Sequence analysis and phylogeny
Thirteen samples that proved to be positive while using primer pair “A” were selected for sequence analysis (Table 1). The samples used in the phylogenetic analysis were selected based on their geographical provenience. Only a limited number of samples obtained from the Dog Shelter of the City Council of Budapest have been selected for the phylogenetic study. We selected six samples collected during the spring of 2005, and three samples collected during the spring and summer of 2006. We also determined the partial H gene sequence of several other samples obtained from the same shelter (data not shown). Previous investigations (Demeter et al., 2006) demonstrated that there is an endemic infection at the establishment, since the sequences from the two different examination periods differ only in the same four or five nucleotides. Due to the confined space and the level of identity observed among the virus strains obtained from the same establishment, we did not feel it was necessary to include even more samples in the present study, since those would have only underlined the endemic infection. All other samples obtained from other establishments from different parts of Hungary were included in the study.
Following electrophoresis in a 2% Low Melting Agarose Gel (Bio-Rad Laboratories, USA), the amplicons produced by the “C” set of primers were cut out from the gel, and DNA was extracted with the QiaQuick Gel Extraction Kit (Qiagen, Germany). Fluorescence-based direct sequencing was performed in both directions on the amplicons at BIOMI Kft. (Gödöllő, Hungary), using an ABI 3100 genetic analyzer (AppliedBiosystems, Foster City, CA, USA). The nucleotide sequences were identified using the Basic Local Alignment Search Tool (BLAST, http://www.ncbi.nlm.nih.gov/BLAST/). The two direction sequences were compiled and aligned to the complete genome sequence of the Onderstepoort reference strain (AF378705) using the Align Plus 4 software (Scientific & Educational Software, Cary, NC, USA). Phylogenetic trees of the nucleic acid and putative amino acid (aa) sequences were established by using sequence data from 13 positive samples.
Phylogenetic analyses were performed with the help of the ClustalX (multiple sequence alignment) program, with the Onderstepoort strain (AF378705) used as reference. The phylogenetic tree based on the full sequence of the H gene was constructed by neighbor-joining with two parameters distance matrix (Kimura, 1980), using the PHYLIP program. The robustness of the groupings in the neighbor-joining analysis was assessed with 1000 bootstrap resampling, and computed with PAUP. Besides our own, a total of 42 sequences retrieved from the GenBank database were used in this study, including those of the currently used vaccine strains (Onderstepoort, Snyder Hill, Convac). Since the nucleotide sequence of the Lederle strain was not present in the GenBank, we sequenced the full length of the H gene of the virus present in a vaccine (Canigen DH(A2)PPi/L, Ceva Sante Animale, France), that according to the manufacturer contained the above mentioned strain (accession number: DQ903854).
3. Results
3.1. PCR
The PCRs performed according to conditions described above resulted in amplicons of the expected size of 409 bp when using the “A” set of primers. When using the “B” and “C” primers, the amplicons were of 1110 and 2023 bp, respectively (Fig. 1 ).
Fig. 1.
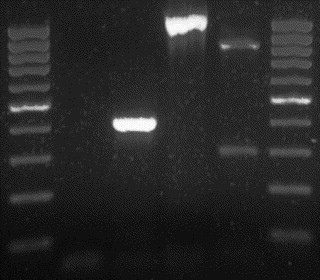
Results of the PCRs with two of the primer pairs used in the present study, and result of the RFLP analysis based on PsiI restriction enzyme recognition site of the CDV H gene amplicons produced by primer pair “B”. Lanes 1 and 6: 100 bp molecular weight marker (Fermentas Life Sciences, Lithuania); lane 2: negative control; lane 3: positive control for primer pair “A”; lane 4: positive control for primer pair “B”; lane 5: vaccine strain (Canigen, Ceva Sante Animale, France) digested by the PsiI enzyme.
When testing the newly designed primers (“A”, “B” and “C”) by using serial dilutions (105, 104, 103, 102, 101 and 100) of a vaccine sample with a previously determined content of viral particles (105), we obtained visible bands even in samples with a concentration of up to 102 copies of the CDV nucleic acid.
3.2. RFLP analysis
Following the previously described protocol, the authors obtained two, clearly differentiable bands for the currently used vaccine strains at the predicted sizes of 294 and 816 bp, while the amplicons obtained from all tested wild-type viruses remained undigested. Therefore, in these cases only one band was visible at the UV examination of the agarose gels in which the electrophoresis was performed (Fig. 1).
The alignment of the sequences with the cleavage site of the PsiI enzyme is shown in Fig. 2 . It demonstrates that the vaccine strains (Onderstepoort, Snyder Hill, Convac, Lederle) contain at the position of interest a thymine (T), while the wild-type strains contain a cytosine (C). This nucleotide change is present in all of the examined nucleotide sequences of wild-type strains deposited in the GenBank.
Fig. 2.
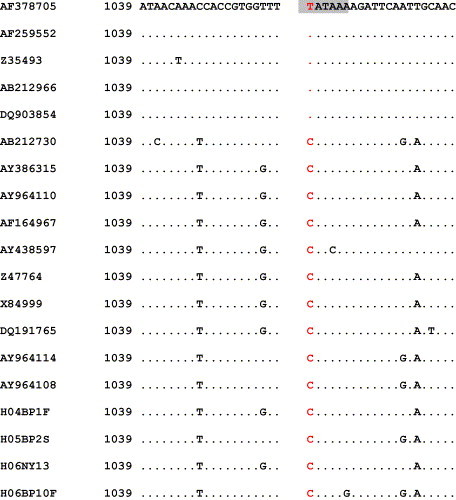
The PsiI enzyme cleavage site (shaded) and the exact location of the genetic marker (red) that allows the differentiation of wild-type viruses from the vaccine strains. The nucleotide position number corresponds to the position in the H gene sequence of the Onderstepoort reference strain (AF378705, Onderstepoort strain; AF259552, Snyder Hill strain; Z35493, Convac strain; AB212966, Vaccine strain, Japan; DQ903854, Lederle strain). One wild-type genotype from each branch of the phylogenetic tree (Fig. 3) is represented.
3.3. Molecular characterization, sequence analysis and phylogeny of the Hungarian CDV strains
The study demonstrates that the Hungarian CDV strains are similar to other typical CDVs reported in different parts of the world (Haas et al., 1997). The nucleotide sequences of the amplicons were determined and deposited into the GenBank under the accession numbers: DQ889177–DQ889189. Following BLAST searches, the sequences obtained showed the highest identity with the nucleotide sequence of the H gene region of the CDV strains presented on the phylogenetic tree (Fig. 3 ). After the alignment of the sequences to the Onderstepoort strain (accession number AF378705), the nucleotide identity levels varied between 90.86 and 91.91%.
Fig. 3.
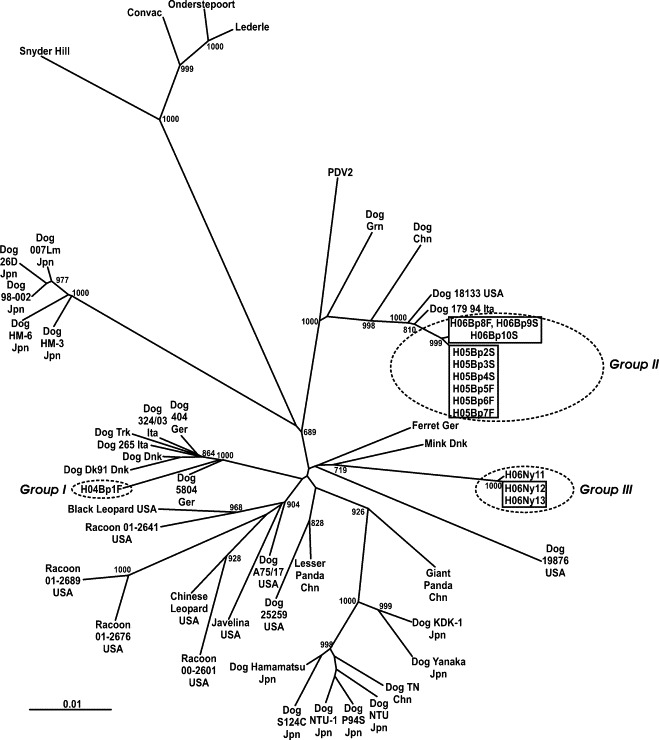
Phylogenetic tree constructed upon the complete nucleotide sequences of the H genes of representative CDVs and the selected Hungarian strains. The code, accession number and provenience for the sequences used in the present study are as follow: Dog 98-002 Jpn (AB025270, Japan), Dog KDK1 Jpn (AB025271, Japan), Dog 26D Jpn (AB040766, Japan), Dog Hm3 Jpn (AB040767, Japan), Dog Hm6 Jpn (AB040768, Japan), Dog 00Lm Jpn (AB212730, Japan), Dog P945 Jpn (AB212964, Japan), Dog S124C Jpn (AB212965, Japan), Dog A75/17 USA (AF164967, USA), Dog Chn (AF172411, China), Giant Panda Chn (AF178038, China), Lesser Panda Chn (AF178039, China), Snyder Hill (AF259552, Germany), Onderstepoort (AF378705, South Africa), Dog DK91 Dnk (AF478544, Denmark), Dog Trk (AY093674, Turkey), Dog 5804 Ger (AY386315, Germany), Dog TN Chn (AY390347, China), Racoon 00-2601 USA (AY438597, USA), Racoon 01-2676 USA (AY498692, USA), Racoon 01-2641 USA (AY526496, USA), Racoon 01-2689 USA (AY649446, USA), Dog 18133 USA (AY964108, USA), Dog 19876 USA (AY964110, USA), Dog 25259 USA (AY964114, USA), Dog NTU 1 Jpn (DQ191175, Japan), Dog NTU Jpn (DQ191765, Japan), Dog 324-03 Ita (DQ494317, Italy), Dog 265 Ita (DQ494318, Italy), Ferret Ger (X84999, Germany), Convac (Z35493, Denmark), Mink Dnk (Z47759, Denmark), Dog Dnk (Z47761, Denmark), Black Leopard USA (Z47763, USA), Javelina USA (Z47764, USA), Chinese Leopard USA (Z54156, USA), Dog 404 Ger (Z77671, Germany), Dog Grn (Z47760, Greenland), Seal PDV2 Rus (X84998, Russia), Dog 179-94 Ita (DQ226087, Italy), Dog Yanaka Jpn (D85755, Japan), Dog Hamamatsu Jpn (D85754, Japan).
The sequenced genome fragments were translated, resulting in 607 aa long polypeptides, representing the complete sequences of the H protein. The amino acid sequence alignments indicated 89.71 and 91.14% aa identity between the investigated Hungarian viruses and the Onderstepoort strain.
4. Discussion
Despite the vaccination procedures applied in Hungary, CDV is still a serious threat to the susceptible animal population (Chappuis, 1995). In order to determine the frequency of this infection, we analyzed a total number of 150 samples taken from dogs that presented clinical symptoms suggesting CD, and from carcasses of dogs that succumbed following CD clinical signs, or that presented pathological changes that could have been attributed to CDV infection. For fast and reliable diagnosis we designed a pair of “diagnostic” primers that targeted the L conserved domain of the viral genome. Due to the conservative aspect of the amplified segment, this primer pair could probably be used to detect all CDV strains currently present in Hungary. Out of the 150 analyzed samples, 46 (30.66%) proved to be positive for CDV, indicating that CDV still represents a high risk to the canine population in Hungary. Despite widespread vaccination, CDV infects a high number of animals, causing severe illness, frequently resulting in fatal outcome. The high number of CDV negative samples obtained from animals showing respiratory signs can be explained by the fact that, in many cases, animals are misdiagnosed with CD when showing respiratory and other related clinical signs. In many situations, the cause of the clinical signs is not the CDV infection, but infections caused by other viral (canine parainfluenza virus, canine adenovirus 2, canine respiratory coronavirus, etc.) and/or bacterial (Bordetella sp., Pasteurella sp., etc.) agents. Because the initial clinical signs following infection can be indicative not only of CD, the signs can be misleading and the regular use of the specific diagnostic tests, such as those based on PCR, are indicated for the proper diagnosis of CDV. Based on the nucleotide sequences deposited in the GenBank, and the conserved character of the L gene, primer pair “A” theoretically attaches to the genome of all currently circulating wild-type viruses. Since the sensitivity test demonstrated that it can detect CDV RNA concentrations of up to 102, primer pair “A” can be used as a specific diagnostic test for CDV infections.
Like in other cases when modified live vaccines (MLV) are used for the prevention of certain infectious diseases, the immunization of dogs with MLV against CDV infection sometimes gives misleading results in the PCR-based test due to the presence of the vaccine strain in the clinical samples. Since the introduction of vaccinations for the prevention of CD, several virus strains (such as the Onderstepoort, Rockborn, Snyder Hill, Lederle or the Convac strains) have been used (Chappuis, 1995). Due to the fact that primer pair “A” also attaches to the genome of vaccine strains, and that recent immunization of animals with an attenuated live vaccine might cause difficulties in the evaluation of the PCR test, we developed an RFLP test to accurately differentiate the vaccine strains from the wild-type strains. This test is based on the presence of a constant enzyme cleavage site on the region amplified by primer pair “B”. Since the nucleotide sequence of the Lederle strain is not present in the GenBank, to make sure that the result of our RFLP test is reliable, prior to designing the test, we sequenced the amplicons obtained using primer pair “C” on one of the vaccines (Canigen DH(A2)PPi/L, Ceva Sante Animale, France) that according to the manufacturer contains the above mentioned strain. Based on the obtained nucleotide sequence (accession number: DQ903854), and on the sequences of other vaccine strains already available in the GenBank, the newly designed RFLP test works on all vaccine strains currently applied to prevent CD in Hungary. According to these sequences, the cleavage site on which the newly designed RFLP test is based is present on all investigated vaccine strains, and it is absent from all the analyzed wild-type strains retrieved from the GenBank. More precisely, it is based on the change of the nucleotide at the position 8139 of the viral genome from thymine (T) in the vaccine strains to cytosine (C) in the wild-type strains. This point mutation can be found in the nucleotide sequences of all wild-type CDVs deposited in the GenBank. The presence of the mutation does not cause any change in the amino acid sequence of the wild-type strains at the given position. The nucleotide sequence that suffered the described mutation is also the recognition sequence for the PsiI endonuclease. Due to this fact, the PsiI enzyme will cut only the amplicons of the vaccine strains and will leave the amplicons of all wild-type strains undigested, therefore it can be used for the fast and reliable differentiation of the wild-type CDV strains from the currently used vaccine strains. The positive result of the enzymatic digestion was clearly visible in case of the virus strains from vaccines as a double band (294 and 816 bp) by transillumination of the gels in UV light. Meanwhile, the amplicons of the field samples remained uncut and formed a single characteristic band (1110 bp).
The molecular analysis of the amplicons obtained by the use of primer pair “C” helped to clarify the relationship of the Hungarian strains with other CDV strains reported in other parts of the world. Following the criteria mentioned earlier, the authors have selected 13 Hungarian wild-type virus strains, and performed a phylogenetic analysis on the full segment of nucleotide sequence of the H gene. The analysis has shown that the diversity of the Hungarian strains is very high, and that they are placed on different branches of the phylogenetic tree. Molecular analyses performed in other countries demonstrate similar findings: in a given geographical region more than one genotype of CDV can be present at the same time (Haas et al., 1999, Lednicky et al., 2004, Martella et al., 2006). The studies indicate that genotypes from far away countries appear sooner or later in the dog population due to the intense, uncontrolled trade and travel of dogs. This observation is also validated by the fact that Hungarian strains are closely related to other CDVs currently present in other parts of the world, including Europe and other distant regions, such as China and North America. The similarity of the Hungarian strains to the Onderstepoort reference strain varied between 90.86 and 91.91%.
The Hungarian strain H04Bp1F from Group I (Table 1) obtained from Budapest in the winter of 2004 is closely related to virus strains reported in other European countries, such as Denmark, Italy, Germany and Turkey (Group I).
Although the diversity of the CDV in the total canine population is obvious, one large, relatively homogenous cluster (Group II) is formed based on the sequences obtained from one population, that of the Dog Shelter of the City Council of Budapest. The investigations demonstrated that there is a high incidence of CD at the above mentioned establishment: from the 99 clinically ill dogs examined during the 16 months period, 25 turned out to be positive for CD. The authors have analyzed several samples from different periods of time. We determined the complete sequence of the H gene of six samples from the spring of 2005 and three samples from the spring and summer of 2006, as well as the partial H gene sequence of many other samples collected from the same shelter. The relatively long period of time between the collection of the first and last samples, and the level of similarity among the obtained nucleotide sequences demonstrate that there is an endemic infection at the above mentioned establishment (Demeter et al., 2006). Based on this conclusion, the authors considered that further investigation of other samples obtained at the same establishment was not necessary, since they would have only confirmed the endemic infection. In the present study we have included only the sequences that had the full sequence of the H gene determined. The analyses also demonstrate that the investigated virus strains from the second period of examination had suffered a homologous amino acid change when compared to the strains from the first period of the investigation, specifically, the change of lysine to arginine at position 282 of the H protein.
The investigation revealed that in the case of large, open population, such as in dog shelters, nasal swab samples are not always reliable in the diagnosis of CD. Out of the 99 clinically ill dogs that were examined in the shelter during a 1-year period, there were 16 cases when both blood samples and nasal swab samples were analyzed. For six dogs, both the blood (serum and separated leukocytes) and nasal swab samples proved to be positive, and hence the results were regarded as proof of viremia caused by the CDV infection. Animals that had the PCR test using their serum positive, also tested positive on the separated leukocytes test. We could not identify animals with only one of their blood tests (serum or leukocytes) positive. On the other hand, for 10 dogs, only the nasal swab samples turned out to be positive, while the repeated examination of both blood samples gave only negative results. This means that the virus remained at the entrance site, on the nasal mucosa and did not actually cause viremia, or it could mean that viremia had been overcome. Therefore, in 10 out of 16 of these situations (62.5%), the positive results obtained using only the nasal swab samples gave misleading, diagnostically false positive results. These results, as well as the decreasing number of clinical cases at the shelter also demonstrate the efficiency of the CD preventive program started during the fall of 2005 (Demeter et al., 2006).
Interestingly, the Hungarian CDV strains obtained from the above mentioned establishment belong to the so-called Arctic group (Martella et al., 2006), closely related to virus strains from countries geographically distant from Hungary, such as the United States of America (98.95–99.01%), Italy (99.23–99.28%), China (97.86–97.91%) and Greenland (97.31–97.36%), as well as to the PDV-2 (X84998), isolated from seals from Lake Baikal (96.71–96.76%). These findings seem to emphasize the unfortunate consequences of uncontrolled animal movement and trade.
The rest of the Hungarian strains are located in other parts of the phylogenetic tree. Three viruses (Group III) from samples collected in Eastern Hungary (H06Ny11, H06Ny12 and H06Ny13) seem to join a different cluster which presently includes only three other strains isolated from a Danish mink (96.82–96.92%), from a ferret in Germany (96.27–96.38%) and from a dog in North America (94.62–94.73%). These findings are in accordance with the conclusions of other authors who concluded that there are strong interconnections among the susceptible wild and domestic animals, and that they can act as reservoirs one for the other (Kovács et al., 1983, Haas et al., 1997, Frölich et al., 2000; Martella et al., 2002; Rzezutka and Mizak, 2002, Lednicky et al., 2004). The H proteins of these three strains have eight potential glycosylation sites, same as all virus strains belonging to the Arctic group.
The observed genetic heterogeneity of the CDV strains is intriguing, since Hungary is a relatively small country. The presence of more than one virus genotype in such a small territory can be explained by the epizootological openness of the canine population, maintained by the lack of geographic barriers, as well as the high number of foreign citizens who own dogs and are living in Hungary or visit the country. The import of exotic canine breeds and other receptive species, as well as the uncontrolled movement of the receptive wild species also contribute to the heterogeneity of the Hungarian CDV strains.
The phylogenetic analyses of the deduced aa sequences show a similar clustering: the strains from the same establishment are positioned very close to each other (99.83% identity between the strains from the two examination periods), while the positions of the other aa sequences are quite similar to that of the nucleotide sequences. The identity between the aa sequence of the H protein of the Hungarian Arctic strains and PDV-2 varies from 95.71 to 96.04%. Bootstrap resampling analyses support most of the clustering, both on the nucleotide and on the aa based phylogenetic trees.
In conclusion, the results of our study demonstrate that based on the nucleotide sequences available in the GenBank, the RFLP analysis using the PsiI enzyme allows the reliable differentiation of vaccine, and wild-type virus strains. On the other hand, the study demonstrates that more than one genotype of CDV is present currently in Hungary. Furthermore, point mutations resulting in the emergence of new variants can be observed in populations where endemic infection is present.
Acknowledgements
The authors would like to express their gratitude to Dr. Tamás Bakonyi, Dr. Ákos Hornyák, Dr. Elemér Demeter, Melinda Demeter and Zsuzsanna Tapaszti for their help and support. The present work was supported by National Grants OTKA M027651 and M041852. Dr. Zoltán Demeter's PhD studies are supported by a fellowship of the Hungarian Ministry of Education, the Márton Áron Szakkollégium and the Agora Foundation.
References
- Appel M.J.G. Canine distemper virus. In: Horzinek M.C., editor. vol. 1. Elsevier Science Publishers; Amsterdam: 1987. pp. 133–159. (Virus Infections of Carnivores). [Google Scholar]
- Appel M.J.G., Summers B.A. Pathogenicity of morbilliviruses for terrestrial carnivores. Vet. Microbiol. 1995;44:187–191. doi: 10.1016/0378-1135(95)00011-x. [DOI] [PubMed] [Google Scholar]
- Barrett T. Morbillivirus infections, with special emphasis on morbilliviruses of carnivores. Vet. Microbiol. 1999;69:3–13. doi: 10.1016/s0378-1135(99)00080-2. [DOI] [PubMed] [Google Scholar]
- Blixenkrone-Møller M., Svansson V., Have P., Orvell C., Appel M., Pedersen I.R., Dietz H.H., Henriksen P. Studies on manifestations of canine distemper virus infection in an urban dog population. Vet. Microbiol. 1993;37:163–173. doi: 10.1016/0378-1135(93)90190-i. [DOI] [PubMed] [Google Scholar]
- Carpenter M.A., Appel M.J.G., Roelke-Parker M.E., Munson L., Hofer H., East M., O’Brien S.J. Genetic characterization of canine distemper virus in Serengeti carnivores. Vet. Immunol. Immunopathol. 1998;65:259–266. doi: 10.1016/s0165-2427(98)00159-7. [DOI] [PubMed] [Google Scholar]
- Chappuis G. Control of canine distemper. Vet. Microbiol. 1995;44:351–358. doi: 10.1016/0378-1135(95)00028-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeter Z., Palade E.A., Lakatos B., Kozma T., Rusvai M. Experiences on the control of endemic distemper at the flaying-house of Budapest. Magyar Állatorvosok Lapja. 2006;128:665–673. [Google Scholar]
- Frisk A.L., Konig M., Moritz A., Baumgartner W. Detection of canine distemper virus nucleoprotein RNA by reverse transcription-PCR using serum, whole blood, and cerebrospinal fluid from dogs with distemper. J. Clin. Microbiol. 1999;37:3634–3643. doi: 10.1128/jcm.37.11.3634-3643.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frölich K., Czupalla O., Haas L., Hentschke J., Dedek J., Fickel J. Epizootiological investigations of canine distemper virus in free-ranging carnivores from Germany. Vet. Microbiol. 2000;74:283–292. doi: 10.1016/s0378-1135(00)00192-9. [DOI] [PubMed] [Google Scholar]
- Gemma T., Watari T., Akiyama K., Miyashita N., Shin Y.S., Iwatsuki K., Kai C., Mikami T. Epidemiological observations on recent outbreaks of canine distemper in Tokyo area. J. Vet. Med. Sci. 1996;58:547–550. doi: 10.1292/jvms.58.547. [DOI] [PubMed] [Google Scholar]
- Haas L., Liermann H., Harder T.C., Barrett T., Löchelt M., von Messling V., Baumgärtner W., Greiser-Wilke I. Analysis of the H gene, the central untranslated region and the proximal coding part of the F gene of wild-type and vaccine canine distemper viruses. Vet. Microbiol. 1999;69:15–18. doi: 10.1016/s0378-1135(99)00081-4. [DOI] [PubMed] [Google Scholar]
- Haas L., Martens W., Greiser-Wilke I., Mamaev L., Butina T., Maack D., Barrett T. Analysis of the haemagglutinin gene of current wild-type canine distemper virus isolates from Germany. Virus Res. 1997;48:165–171. doi: 10.1016/s0168-1702(97)01449-4. [DOI] [PubMed] [Google Scholar]
- Harder T.C., Osterhaus A.D.M.E. Canine distemper virus—A morbillivirus in search of new hosts? Trends Microbiol. 1997;5:120–124. doi: 10.1016/S0966-842X(97)01010-X. [DOI] [PubMed] [Google Scholar]
- Iwatsuki K., Tokiyoshi S., Hirayama N., Nakamura K., Ohashi K., Wakasa C., Mikami T., Kai C. Antigenic differences in the H proteins of canine distemper viruses. Vet. Microbiol. 2000;71:281–286. doi: 10.1016/s0378-1135(99)00172-8. [DOI] [PubMed] [Google Scholar]
- Kabakci N., Yarim M., Karahan S., Guvenc T., Yagci B.B., Gurcan I.S. Immunohistochemical investigation of cerebellum in dogs infected with canine distemper virus. Acta Vet. Hungarica. 2004;52:327–337. doi: 10.1556/AVet.52.2004.3.8. [DOI] [PubMed] [Google Scholar]
- Kim Y.H., Cho K.W., Youn H.Y., Yoo J.S., Han H.R. Detection of canine distemper virus (CDV) through one step RTPCR combined nested PCR. J. Vet. Sci. 2001;2:59–63. [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Kovács G., Mocsári E., Sztojkov V., Vetési F., Kiss J.H. Canine distemper epizooty on a large scale mink ranch. Epizootiological and diagnostic experiences. Magyar Állatorvosok Lapja. 1983;38:305–308. [Google Scholar]
- Lan N.T., Yamaguchi R., Furuya Y., Inomata A., Ngamkala S., Naganobu K., Kai K., Mochizuki M., Kobayashi Y., Uchida K., Tateyama S. Pathogenesis and phylogenetic analyses of canine distemper virus strain 007 Lm, a new isolate in dogs. Vet. Microbiol. 2005;110:197–207. doi: 10.1016/j.vetmic.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Lan N.T., Yamaguchi R., Inomata A., Furuya Y., Uchida K., Sugano S., Tateyama S. Comparative analyses of canine distemper viral isolates from clinical cases of canine distemper in vaccinated dogs. Vet. Microbiol. 2006;115:32–42. doi: 10.1016/j.vetmic.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Lednicky J.A., Dubach J., Kinsel M.J., Meehan T.P., Bocchetta M., Hungerford L.L., Sarich N.A., Witecki K.E., Braid M.D., Pedrak C., Houde C.M. Genetically distant American Canine distemper virus lineages have recently caused epizootics with somewhat different characteristics in raccoons living around a large suburban zoo in the USA. Virol. J. 2004;1:2. doi: 10.1186/1743-422X-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella V., Pratelli A., Cirone F., Zizzo N., Decaro N., Tinelli A., Foti M., Buonavoglia C. Detection and genetic characterization of canine distemper virus (CDV) from free-ranging red foxes in Italy. Mol. Cell. Probes. 2002;16:77–83. doi: 10.1006/mcpr.2001.0387. [DOI] [PubMed] [Google Scholar]
- Martella V., Cirone F., Elia G., Lorusso E., Decaro N., Campolo M., Desario C., Lucente M.S., Bellacicco A.L., Blixenkrone/Moller M., Carmichael L.E., Buonavoglia C. Heterogeneity within the hemagglutinin genes of canine distemper virus (CDV) strains detected in Italy. Vet. Microbiol. 2006;116:301–309. doi: 10.1016/j.vetmic.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Mochizuki M., Hashimoto M., Hagiwara S., Yoshida Y., Ishiguro S. Genotypes of canine distemper virus determined by analysis of the hemagglutinin genes of recent isolates from dogs in Japan. J. Clin. Microbiol. 1999;37:2936–2942. doi: 10.1128/jcm.37.9.2936-2942.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkul A., Sancak A.A., Gungor E., Burgu I. Determination and phylogenetic analysis of canine distemper virus in dogs with nervous symptoms in Turkey. Acta Vet. Hungarica. 2004;52:125–132. doi: 10.1556/AVet.52.2004.1.12. [DOI] [PubMed] [Google Scholar]
- Rzezutka A., Mizak B. Application of N-PCR for diagnosis of distemper in dogs and fur animals. Vet. Microbiol. 2002;88:95–103. doi: 10.1016/s0378-1135(02)00097-4. [DOI] [PubMed] [Google Scholar]
- Shin Y.J., Cho K.O., Cho H.S., Kang S.K., Kim H.J., Kim Y.H., Park H.S., Park N.Y. Comparison of one-step RT-PCR and a nested PCR for the detection of canine distemper virus in clinical samples. Aust. Vet. J. 2004;82:83–86. doi: 10.1111/j.1751-0813.2004.tb14651.x. [DOI] [PubMed] [Google Scholar]
- Stettler M., Beck K., Wagner A., Vandevelde M., Zurbriggen A. Determinants of persistence in canine distemper viruses. Vet. Microbiol. 1997;57:83–93. doi: 10.1016/s0378-1135(96)01281-3. [DOI] [PubMed] [Google Scholar]
- Uema M., Ohashi K., Wakasa C., Kai C. Phylogenetic and restriction fragment length polymorphism analyses of hemagglutinin (H) protein of canine distemper virus isolates from domestic dogs in Japan. Virus Res. 2005;109:59–63. doi: 10.1016/j.virusres.2004.10.008. [DOI] [PubMed] [Google Scholar]