

Highlights
-
•
Sequence comparison of FIPV/FECV.
-
•
ORF3c genes of FIP animals heavily affected by nucleotide deletions/point mutations.
-
•
Genes encoding 3a, 3b, 7a and 7b displayed no mutations linked to the FCoV biotype.
-
•
Mutated 3c genes were found in ascites and faeces samples of FIP animals.
-
•
Slightly affected 3c genes were accompanied by mutations in S gene.
Keywords: Feline coronavirus, FIPV, FECV, Spike protein gene, Accessory genes
Abstract
The genes encoding accessory proteins 3a, 3b, 3c, 7a and 7b, the S2 domain of the spike (S) protein gene and the membrane (M) protein gene of feline infectious peritonitis virus (FIPV) and feline enteric coronavirus (FECV) samples were amplified, cloned and sequenced. For this faeces and/or ascites samples from 19 cats suffering from feline infectious peritonitis (FIP) as well as from 20 FECV-infected healthy cats were used. Sequence comparisons revealed that 3c genes of animals with FIP were heavily affected by nucleotide deletions and point mutations compared to animals infected with FECV; these alterations resulted either in early termination or destruction of the translation initiation codon. Two ascites-derived samples of cats with FIP which displayed no alterations of ORF3c harboured mutations in the S2 domain of the S protein gene which resulted in amino acid exchanges or deletions. Moreover, changes in 3c were often accompanied by mutations in S2. In contrast, in samples obtained from faeces of healthy cats, the ORF3c was never affected by such mutations. Similarly ORF3c from faecal samples of the cats with FIP was mostly intact and showed only in a few cases the same mutations found in the respective ascites samples. The genes encoding 3a, 3b, 7a and 7b displayed no mutations linked to the feline coronavirus (FCoV) biotype. The M protein gene was found to be conserved between FECV and FIPV samples. Our findings suggest that mutations of 3c and spike protein genes correlate with the occurrence of FIP.
1. Introduction
Feline coronavirus (FCoV) belongs to the genus Alphacoronavirus of the subfamily Coronavirinae, family Coronaviridae within the order Nidovirales (de Groot et al., 2011). The coronaviral genome consists of a single-stranded positive-sense RNA with a size of about 30 kb, representing the largest viral RNA genome (Gorbalenya et al., 2006). Two thirds of the genome (two large open reading frames (ORF1a and 1b)) encode the replication machinery. ORFs 2, 4, 5 and 6 encode the structural proteins S (spike protein), E (envelope protein), M (membrane protein) and N (nucleoprotein), respectively (Masters, 2006). The FCoV ORFs 3 and 7 encode the accessory proteins 3a, 3b, 3c, 7a and 7b with unknown functions (Haijema et al., 2004, Dye and Siddell, 2005).
FCoV is worldwide distributed and can infect all members of the Felidae (Heeney et al., 1990, Leutenegger et al., 1999, Kennedy et al., 2003). About 80–90% of cats in catteries and 10–50% of cats in single cat households are seropositive. Based on serology, two serotypes of FCoV (types I and II) can be distinguished. While type I FCoV accounts for the vast majority of infections in the field (Rottier, 1999), type II FCoV is less common. About 5–10% of FCoV infected cats sporadically develop FIP, mostly within the first two years of their life (Pedersen, 2009). It has been demonstrated that FECV and FIPV are virulence variants (biotypes or pathotypes) of the same virus.
FECV replicates mainly in intestinal epithelial cells, while FIPV replicates efficiently in macrophages and is disseminated throughout the host (Dewerchin et al., 2005, Rottier et al., 2005). Infection of cats with the enteric virus generally leads to inapparent infection or mild enteric symptoms. FIP, however, is characterized by fibrinous serositis with protein-rich effusions in body cavities (“wet form”) and/or granulomatous-necrotizing inflammatory lesions in several organs (“dry form”) (Pedersen, 1987, Pedersen et al., 2009). FIP represents the leading viral cause of death in domestic cats.
Comparative sequence analyses suggest that the highly virulent FIPV arises by mutation from the low virulent FECV (Vennema et al., 1998). Point mutations and/or deletions have been identified in the accessory genes, the spike protein gene and the membrane protein gene of FIPV (Herrewegh et al., 1995a, Vennema et al., 1998, Kennedy et al., 2001, Rottier et al., 2005, Brown et al., 2009, Pedersen et al., 2009, Pedersen et al., 2012, Chang et al., 2010, Chang et al., 2011, Chang et al., 2012, Brown, 2011, Takano et al., 2011, Licitra et al., 2013). Special attention has been paid to the ORF3c gene which is heavily affected in the majority of FIPVs but intact in FECVs (Pedersen et al., 2009, Pedersen et al., 2012, Chang et al., 2010).
The so-called “internal mutation theory” has been questioned by an alternative “circulating virulent–avirulent FCoV” hypothesis. According to the latter harmless as well as virulent strains of FCoV circulate in a population, and only individuals infected by virulent strains will develop the disease (Brown et al., 2009). This conclusion was based on sequencing ORF7 and the M gene. The results which led to this hypothesis were questioned by others (Chang et al., 2011).
To detect possible marker mutations that correlate with FIPV we conducted a comprehensive analysis of the accessory genes from several FCoV samples. Furthermore, the S2 domain of the S gene as well as the M gene was examined.
2. Materials and methods
2.1. Sample collection
For comparison of nucleotide (nt) and amino acid (aa) sequences of FECV and FIPV samples, faeces and/or ascites samples from clinically healthy cats as well as from cats suffering from FIP were collected (Table 1 ). Faecal samples from healthy FCoV-infected cats (n = 20, FECV-1 to -20) originated mostly from two different catteries (cattery 1: FECV-1 to -7; cattery 2: FECV-12 to -18). Samples from FIPV infected cats (n = 19) were provided from the Small Animal Clinic of the JLU Giessen (faeces and ascites) or were obtained from the diagnostic laboratory of our institute. For 10 FIP cats we could obtain both faecal and ascites samples, both being subjected to analysis.
Table 1.
Samples, regions sequenced and accession numbers. Isolates from healthy cats are designated as FECV. Isolates from cats with FIP are designated as FIPV (F = faeces derived, A = ascites derived). In the case of the S gene the S2 domain comprising HR1 and 2 or only HR1 and/or HR2 plus adjacent regions and/or a short fragment including nucleotide 23531 were sequenced as indicated in brackets. n.s.: not sequenced.
| FECV sample | S gene | ORF3 | M gene | ORF7 | FIPV sample | S gene | ORF3 | M gene | ORF7 |
|---|---|---|---|---|---|---|---|---|---|
| FECV-1 | n.s. | KJ665813 | n.s. | KJ665779 | FIPV-1F | KJ665867 (S2) | KJ665834 | n.s. | KJ665796 |
| FECV-2 | (23531) | KJ665814 | n.s. | n.s. | FIPV-1A | KJ665868 (S2) | KJ665833 | n.s. | KJ665795 |
| FECV-3 | n.s. | KJ665815 | n.s. | n.s. | FIPV-2A | KJ665869 (S2) | KJ665839 | KJ665811 | KJ665797 |
| FECV-4 | (23531) | KJ665816 | n.s. | n.s. | FIPV-3F | KJ665870 (S2) | KJ665836 | KJ665812 | KJ665799 |
| FECV-5 | KJ665862 (S2) | KJ665817 | KJ665806 | KJ665785 | FIPV-3A | KJ665871 (S2) | KJ665835 | n.s. | KJ665798 |
| FECV-6 | n.s. | KJ665818 | n.s. | KJ665786 | FIPV-4A | KJ665872 (HR2) | KJ665840 | n.s. | KJ665800 |
| FECV-7 | n.s. | KJ665819 | n.s. | n.s. | FIPV-5A | n.s. | KJ665841 | n.s. | n.s. |
| FECV-8 | n.s. | KJ665820 | n.s. | n.s. | FIPV-6A | n.s. | KJ665842 | n.s. | n.s. |
| FECV-9 | n.s. | KJ665821 | n.s. | n.s. | FIPV-7F | KJ665873 (S2) | KJ665837 | n.s. | KJ665801 |
| FECV-10 | n.s. | KJ665822 | n.s. | n.s. | FIPV-8F | n.s. | KJ665844 | n.s. | n.s. |
| FECV-11 | n.s. | KJ665823 | n.s. | n.s | FIPV-8A | n.s. | KJ665843 | n.s. | n.s. |
| FECV-12 | KJ665863 (HR1/2) | KJ665824 | KJ665802 | KJ665780 | FIPV-9F | n.s. | KJ665846 | n.s. | n.s. |
| FECV-13 | (23531) | KJ665825 | n.s. | n.s. | FIPV-9A | n.s. | KJ665845 | n.s. | n.s. |
| FECV-14 | KJ665864 (HR1/2) | KJ665826 | KJ665803 | KJ665781 | FIPV-10F | n.s. | KJ665848 | n.s. | n.s. |
| FECV-15 | n.s. | KJ665827 | n.s. | n.s. | FIPV-10A | n.s. | KJ665847 | n.s. | n.s. |
| FECV-16 | KJ665865 (HR1) | KJ665828 | n.s. | KJ665782 | FIPV-11A | n.s. | KJ665849 | n.s. | n.s. |
| FECV-17 | (23531) | KJ665829 | n.s. | n.s. | FIPV-12F | KJ665874 (HR2) | KJ665838 | KJ665808 | KJ665788 |
| FECV-18 | (23531) | KJ665830 | n.s. | n.s. | FIPV-12A | KJ665875 (HR2) | KJ665850 | KJ665807 | KJ665787 |
| FECV-19 | KJ665866 (S2) | KJ665831 | KJ665804 | KJ665783 | FIPV-13F | n.s. | KJ665851 | n.s. | n.s. |
| FECV-20 | n.s. | KJ665832 | KJ665805 | KJ665784 | FIPV-14F | KJ665876 (S2) | KJ665852 | n.s. | KJ665789 |
| FIPV-15F | KJ665881 (S2) | KJ665853 | n.s. | KJ665791 | |||||
| FIPV-15A | KJ665877 (S2) | KJ665854 | KJ665809 | KJ665790 | |||||
| FIPV-16F | KJ665878 (S2) | KJ665855 | n.s. | KJ665792 | |||||
| FIPV-17F | (23531) | KJ665857 | n.s. | n.s. | |||||
| FIPV-17A | KJ665879 (S2) | KJ665856 | n.s. | KJ665793 | |||||
| FIPV-18F | n.s. | KJ665859 | n.s. | n.s. | |||||
| FIPV-18A | KJ665880 (S2) | KJ665858 | KJ665810 | KJ665794 | |||||
| FIPV-19F | n.s. | KJ665861 | n.s. | n.s. | |||||
| FIPV-19A | n.s. | KJ665860 | n.s. | n.s. |
2.2. Isolation of RNA
Faecal samples were diluted 1:10 in phosphate buffered saline and vortexed. After centrifugation for 10 min at 4000 U/4 °C RNA was extracted from 140 μl of the supernatants using the Viral RNA Mini Kit (Qiagen) according to the manufacturer's instructions. Ascites samples were directly applied to RNA extraction. RNA was stored at −20 °C until use.
2.3. Amplification and cloning of ORF3, ORF7, M and S2 domain of spike
ORF3, ORF7, M and S gene sequences were amplified from RNA samples using nested RT-PCR protocols. Due to a limited amount of material in some samples analyses of all available genomes was not possible. We first concentrated on the 3c gene and subsequently further analyzed genomes from FIP samples with deletions in 3c as well as with a complete 3c gene. For FECV, samples were chosen randomly. Primers were designed using the PrimerExpress software (Applied Biosystems (Life Technologies)). Primer sequences are listed in Table 2 .
Table 2.
Primers used in this study. Numbers refer to position in FCoV laboratory strain Black genome (accession number EU186072).
| Primer | Sequence (5′–3′) | Position Black genome (EU186072) | Reference |
|---|---|---|---|
| SM-r1 | TTTGCATAAAGGTCTTATAGGCATC | 26012–26036 | This study |
| S-TMD-2 | TGGCTCAACAGGATTGAAACTTATG | 24583–24607 | This study |
| SM-r2 | ACCCAAATTGCAACATACCATGC | 25953–25975 | This study |
| S-TMD-1 | TGGCCTTGGTATGTGTGGCTAC | 24613–24634 | This study |
| N1-sense | GCCAACAAACACACCTGGAAG | 27569–27590 | This study |
| N2-sense | AACTTTGGTGATAGTGATCTCG | 27648–27669 | This study |
| 3a-8rev | GCCWATRGACTTGACAGTGTCCAT | 24809–24833 | This study |
| S702sense | TGCACTGATTATAACATATATGG | 22507–22529 | This study |
| S1464rev | GTGAATGTGAACCTTTTCAATAGG | 24772–24795 | This study |
| S726sense | GGCCTTTACTACACATCTATTAGTG | 22579–22603 | This study |
| SC-3a-sense | GGTCTTGGCACTGTGGATGATGATTA | 23374–23399 | This study |
| SC-3a-rev | ATCACTCATCTGAGGTTTTCTAGGTTG | 24259–24285 | This study |
| SC-3b-sense | TGTGCCCAATATTACAATGGCATAATGG | 23440–23467 | This study |
| SC-3b-rev | TTACGAGGAGTCACCATATACGT | 24229–24251 | This study |
| SC-4a-sense | GTGACGGCATGGTCAGGAATATGTGTT | 24142–24168 | This study |
| SC-4b-sense | TATGCATATGTGTTGAAAGAYTT | 24178–24200 | This study |
| FCoV-M-03F | ATGATGCCTATAAGACCTTTATGC | 26009–26032 | This study |
| FCoV-M-04R | ACGTCTTTTGGAAGGTTCATCTCC | 26919–26942 | This study |
| FCoV-M-06R | TGACGCGTTGTCCCTGTG | 26896–26913 | This study |
| p204 | GCTCTTCCATTGTTGGCTCGTC | 29138–29159 | Herrewegh et al. (1995a) |
| p211 | CACTAGATCCAGACGTTAGCTC | 29156–29177 | Herrewegh et al. (1995a) |
| Chang-sense | CAATATTACAATGGCATAATGG | 23446–23467 | Chang et al. (2012) |
| Chang-antisense | CCCTCGAGTCCCGCAGAAACCATACCTA | 24033–24060 | Chang et al. (2012) |
| Chang-nested-sense | GGCATAATGGTTTTACCTGGTG | 23458–23479 | Chang et al. (2012) |
| Chang-nested-antisense | TAATTAAGCCTCGCCTGCACTT | 23579–23600 | Chang et al. (2012) |
RT-PCR was performed following an in-house protocol essentially as described earlier (Becher et al., 1997). Reverse transcription of 2.5 μl RNA was performed after heat-denaturation (3 min/94 °C, 2 min/4 °C) in the presence of 0.5 μl reverse primer (50 mM) by adding RT mix (final concentration 50 mM Tris (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM DTT, 2 mM dNTPs), 10 U RNaseOUT™ recombinant Ribonuclease Inhibitor (Invitrogen) and 50 U reverse transcriptase (SuperScriptII, Invitrogen) for 30 min at 42 °C. Reverse transcription was terminated by heating to 80 °C for 2 min. Subsequently 0.5 μl forward primer (50 mM) and 1 U Taq Polymerase (Biotherm) were added together with PCR mix (final concentration 25 mM Tris (pH 8.3), 50 mM KCl, 2.5 mM MgCl2, 1.8 mM dNTPs, 0.1% Triton X 100, 0.02% BSA). Amplification conditions were 30 cycles of denaturation at 94 °C for 30 s, annealing at 50–55 °C for 30 s and extension at 72 °C for 30–90 s.
Nested PCR was done using 2.5 μl of the first round product, 0.5 μl of both forward and reverse primer (50 μM), 2 mM dNTPs and 1 U Taq Polymerase (Biotherm) in PCR buffer (Biotherm). Amplification conditions were 30 cycles of denaturation at 94 °C for 30 s, annealing at 50–55 °C for 30 s and extension at 72 °C for 30–90 s.
For ORF3, a cDNA was generated using reverse primer SM-r1 at 42 °C for 30 min. Subsequently, a PCR was performed (94 °C, 30 s/55 °C, 30 s/72 °C, 90 s; 30 cycles) by addition of forward primer S-TMD-2. In a second step, 2.5 μl of the reaction was applied to a nested PCR using reverse primer SM-r2 and forward primer S-TMD-1 amplifying a 1.3 kb fragment. For amplification of ORF 7, reaction conditions were as described above. cDNA was obtained using reverse primer 211 (Herrewegh et al., 1995a); for PCR forward primer N1-sense was added. Nested PCR was performed using reverse primer 204 (Herrewegh et al., 1995a) and forward primer N2-sense which led to a 1.4 kb fragment. The 3′-terminal part of the spike gene was amplified as described above with primers 3a-8rev and S702sense in the first round at an annealing temperature of 50 °C (product of 2.3 kb), and primers S1464rev and S726sense in the nested PCR step with annealing at 53 °C (product of 2.2 kb). In some cases amplification of the complete S2 domain failed; therefore only putative heptad repeat regions HR1 and/or HR2 were amplified. For amplification of HR1, primers SC-3a-sense and SC-3a-rev, and in a nested step SC-3b-sense and SC-3b-rev were used. For amplification of HR2, primers SC-4a-sense and 3a-8rev, and for nested PCR SC-4b-sense and S1464rev were utilized.
For amplification of the genomic region comprising position 23531 (nt 23535 in reference strain Black (accession number EU186072; Tekes et al., 2008)), primers Chang-sense and Chang-antisense were used amplifying a 615 bp fragment (reference: Black (EU186072)) followed by a nested PCR using primers Chang-nested-sense and Chang-nested-antisense (product of 143 bp) (Chang et al., 2012). In both RT-PCR and nested PCR an annealing temperature of 50 °C was used according to the protocol by Chang et al. (2012).
The matrix protein gene was amplified using primers FCoV-M-03F and FCoV-M-04R amplifying a 934 bp fragment. A semi-nested PCR was conducted using primers FCoV-M-03F and FCoV-M-06R (905 bp).
PCR and nested PCR products were analyzed on 1–1.5% agarose gels stained with ethidium bromide following standard protocols. Nested RT-PCR products were further processed by cloning into vector pGEM-T easy using the pGEM-T easy Cloning Kit (Promega) following the manufacturer's instructions. Plasmid DNA was obtained using the QIAprep Spin Miniprep Kit (Qiagen) according to the manufacturer's instructions.
2.4. Sequencing of ORF3, ORF7, M and spike cDNA
Sequencing was performed using the Thermo Sequenase Kit (GE Healthcare) with IRD-800 labelled standard primers M13 and M13reverse (94 °C, 2 min, 1 cycle; 94 °C, 15 s/56 °C, 15 s/70 °C, 30 s; 30 cycles) for at least three independent cDNA clones each in a DNA sequencing device Li-COR 4000L (MWG Biotech). Sequences were analyzed using BaseImagIR V4.2 ImageAnalysis (LICOR) and afterwards analyzed using the HUSAR package (DKFZ, Heidelberg, Germany). Later samples were applied to external sequencing (Qiagen). Consensus sequences of three independent clones for each reaction were created and compared to each other. Sequences have been submitted to GenBank; the referring accession numbers are listed in Table 1.
3. Results
Faecal samples from 20 healthy FECV-infected cats were applied to sequence analyses of the accessory protein genes 3a, 3b, 3c, 7a and 7b, the S2 region of the spike (S) protein gene and the membrane (M) protein gene. Faecal (F) samples and ascites (A) from 19 cats with FIP were also tested including 10 animals, where paired samples comprised ascites as well as faeces from the same animal (n = 20) and nine animals, where only either ascites (n = 5) or faeces (n = 4) was available. Sequences were submitted to GenBank; samples, regions sequenced and the respective accession numbers are listed in Table 1. In order to obtain reliable data each sequence represents a consensus sequence of three independent clones.
3.1. Analysis of accessory genes
Comparison of the ORF3a of FECV and FIPV field viruses revealed nucleotide exchanges. In five out of 29 FIPV samples (FIPV-6A, -10F/-10A, -11A, -15A), the putative 3a translation product was truncated due to point mutations resulting in stop codons (Table 3 ).
Table 3.
Sequence analyses of 3abc genes FIPV samples. Deletions affecting two genes are listed in both columns. Numbering of the nucleotides refers to complete ORF3, beginning with the first nucleotide of gene 3a and ending with the last nucleotide of gene 3c. The impact of mutations on putative translation products is given as number of encoded amino acids. Alterations of 3a (70 aa), 3b (73 aa) or 3c (237 aa) are indicated by grey colour.
| Sample | Gene 3a |
Gene 3b |
Gene 3c |
|||
|---|---|---|---|---|---|---|
| Changes in nt sequence | Number of encoded aa | Changes in nt sequence | Number of encoded aa | Changes in nt sequence | Number of encoded aa | |
| FIPV-Black | – | 70 | Δ334–395 | 289 | Δ334–395 | See 3b |
| FIPV-1F | – | 70 | – | 73 | – | 237 |
| FIPV-1A | – | 70 | – | 73 | – | 237 |
| FIPV-2A | – | 70 | – | 73 | Δ833–834, Δ884–907 | 161 |
| FIPV-3F | – | 70 | – | 73 | – | 237 |
| FIPV-3A | – | 70 | – | 73 | – | 237 |
| FIPV-4A | – | 70 | – | 73 | Δ744–748 | 132 |
| FIPV-5A | – | 70 | Δ348–393 | 64 | Δ348–393 | – |
| FIPV-6A | 184–186 stop | 61 | Δ341–395 | 61 | Δ341–395, Δ835–836, Δ1001–1003 | – |
| FIPV-7F | – | 70 | – | 73 | – | 237 |
| FIPV-8F | – | 70 | – | 73 | 955–957 stop | 192 |
| FIPV-8A | – | 70 | – | 73 | 955–957 stop | 192 |
| FIPV-9F | – | 70 | – | 73 | Δ884–908 | 172 |
| FIPV-9A | – | 70 | – | 73 | Δ884–908 | 172 |
| FIPV-10F | 190–192 stop | 63 | – | 73 | – | 237 |
| FIPV-10A | 190–192 stop | 63 | – | 73 | 427–429 stop, Δ884–885 | 16 |
| FIPV-11A | 106–108 stop | 35 | – | 73 | 694–696stop | 105 |
| FIPV-12F | – | 70 | – | 73 | – | 237 |
| FIPV-12A | – | 70 | – | 73 | Δ586–587 | 74 |
| FIPV-13F | – | 70 | Δ337–339 | 81 | Start codon mutated, Δ886–912 | – |
| FIPV-14F | – | 70 | Δ337–339 | 72 | – | 237 |
| FIPV-15F | – | 70 | Δ337–339 | 72 | – | 237 |
| FIPV-15A | 142–144 stop | 47 | Δ337–339 | 72 | Δ739–745 | 127 |
| FIPV-16F | – | 70 | Δ337–339 | 72 | – | 237 |
| FIPV-17F | – | 70 | Δ337–339 | 72 | – | 237 |
| FIPV-17A | – | 70 | Δ337–339 | 72 | Δ834 | 155 |
| FIPV-18F | – | 70 | – | 73 | Start codon mutated, Δ749–751 | – |
| FIPV-18A | – | 70 | – | 73 | Start codon mutated, Δ749–751 | – |
| FIPV-19F | – | 70 | Δ337–339 | 72 | – | 237 |
| FIPV-19A | – | 70 | Δ337–339 | 72 | Δ705–730 | 108 |
For ORF3b, 10 FIPV samples showed nucleotide deletions at the 3′-terminus resulting in early termination of the translation product, while FECV samples did not exhibit deletions. For three FIPVs some of these deletions were also found in the respective faeces samples (deletion in 3b of FIPV-15F/A, -17F/A, -19F/A). In case of FIPV-13F the mutation affected the stop codon of 3b leading to elongation of this gene (Table 3).
According to earlier studies ORF3c of FIPV is often affected by various mutations while FECV exhibits an intact 3c gene (Chang et al., 2010, Pedersen et al., 2012). In the case of FIPV-5A and FIPV-6A, deletions comprised the termination signal of 3b as well as the start codon of 3c, which abolishes translation of 3c. Similarly, FIPV-13F and FIPV-18F/A displayed point mutations which destroyed the start codon of 3c. Deletions found in ORF3c in the other samples always led to a shift in the reading frame with subsequent early termination. In three cases (FIPV8F/A, -9F/A and -18F/A) these deletions were also present in the respective faecal samples. However, like all FECV samples, most viruses derived from faecal samples of cats with FIP (FIPV-1F, -3F, -7F, -10F, -12F, -14F, -15F, -16F, -17F and -19F) showed intact 3c genes. Two ascites derived viruses from cats with FIP (FIPV-1A, -3A) exhibited an intact ORF3c. A summary of the mutations detected within ORF3 of FIPV samples is listed in Table 3.
For selected samples ORF7 was analyzed. Eight FECV samples were chosen randomly (FECV-1, -5, -6, -12, -14, -16, -19 and -20), while FIPV sequences were determined from either genomes with deletions in 3c (FIPV-2A, -4A, -12A, -15A, -17A and -18A) as well as with a complete 3c gene (FIPV-1F, 1A, -3F, -3A, -7F, -12F, -14F, -15F and -16F). A comparison of FECV and FIPV samples revealed changes in the nucleic acid sequences which led to single aa exchanges. Within ORF7a, aa exchanges were found at positions 9, 12 and 47 (Fig. 1A). At position 9, the analyzed FECV samples and five FIPV samples possess a phenylalanine (F) residue, while the other FIPV samples have a leucine (L). Phenylalanine (F) instead of leucine (L) was also present for three FECV viruses (FECV-1, -5 and 6) at position 29. In some samples (FECV as well as FIPV) alanine (A) was present instead of valine (V) (position 12) and a change from serine (S) to asparagine (N) could be detected at position 47.
Fig. 1.

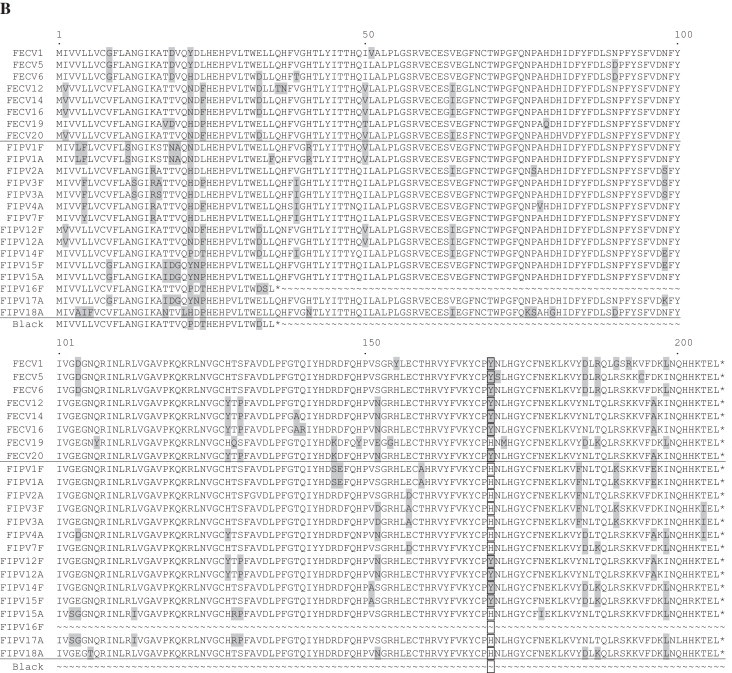
Sequence alignments of accessory proteins 7a (A) and 7b (B). Complete 7a comprises 101 aa, complete 7b comprises 206 aa residues. Residues diverging from the consensus are highlighted by grey boxes. For 7a, aa at positions 9, 12 and 47 and for 7b at position 170 are highlighted by translucent boxes (see text). 7b of FIPV-Black and FIPV-16F is truncated to 35 residues. Accession number of reference strain Black: EU186072.
ORF7b displayed a higher number of aa differences when FECV and FIPV samples are analyzed (Fig. 1B). Interestingly, one particular change was detected at position 170. The tyrosine (Y) residue found in seven out of eight investigated FECV strains (except FECV-19) and three FIPV samples originating from faeces (FIPV-12F, -14F, -15F) was replaced by a histidine (H) in seven out of eight ascites-derived FIPV samples (except -12A). Almost all investigated genomes contained complete 7a and 7b genes resulting in full-length translation products except for FIPV-16F, which encodes a translational stop codon in 7b.
3.2. Analysis of S2-domain of spike protein
For analysis of the spike protein gene we focussed on 2.2 kb of the C-terminal half since this region has been suggested to be the determinant for efficient infection of macrophages by FCoV (Rottier et al., 2005). The analyses comprised FIPV strains with deletions in 3c (FIPV-2A, -4A (only HR2), -12A, -15A, -17A and -18A) as well as FIPV strains with intact 3c genes (FIPV-1F/A, -3F/A, -7F, -12F (only HR2), -14F, -15F and -16F). S2 sequences were also analyzed from FECV samples -5, -12, -14, 16 (only HR1) and -19.
The sequences comprised the functionally important heptad repeat regions called HR1 and HR2 (Fig. 2A and B). Several single nucleotide exchanges, some resulting in aa exchanges, were found, but did not correlate with either FECV or FIPV. Special attention was paid to aa position 1045 (nt 23535 of the complete genome of reference strain Black (EU186072)) within the putative fusion peptide which was found to be a hotspot for differentiation between FECV and FIPV biotype (Chang et al., 2012). 10 FECV (FECV-2, -4, -5, -12, -13, -14, -16, -17, -18 and -19) and 13 FIPV (FIPV-1F, -1A, -2A, -3F, -3A, -7F, -14F, -15F, -15A, -16F, -17A, -17F and -18A) samples were analyzed. All but one (FECV-16) FECV samples harboured an A residue leading to methionine (M), while all but one (FIPV-3A) ascites-derived FIPV samples displayed a T residue resulting in leucine (L). Three faeces-derived FIP viruses (FIPV-14F, -15F and -17F) encode methionine (M) at the respective position just like the FECV samples (Table 5).
Fig. 2.
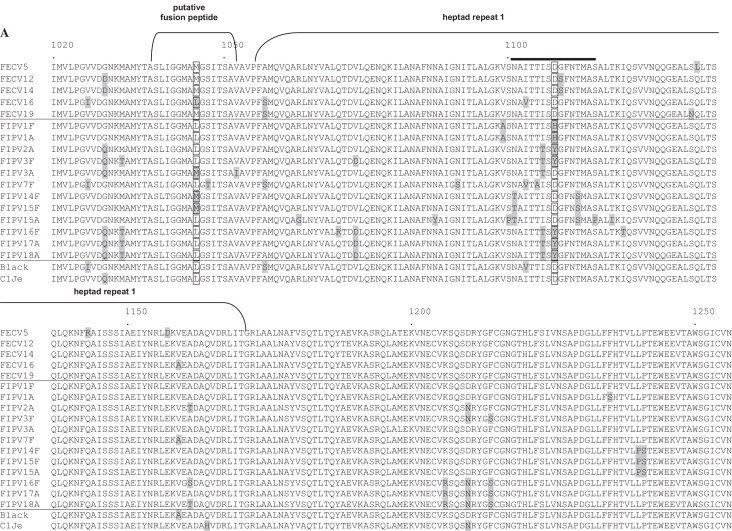
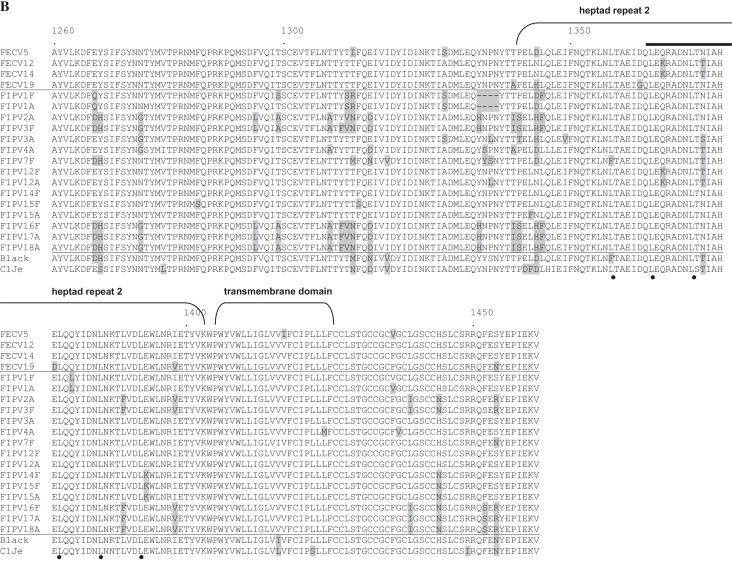
Sequence alignment of the putative fusion peptide and adjacent heptad repeat 1 (HR1) domain (A) and a part of the linker region and adjacent heptad repeat 2 (HR2) (B) of the spike protein. Residues diverging from the consensus are highlighted by grey boxes; aa at positions 1045 and 1108 are highlighted by translucent boxes. The putative fusion peptide and HR 1 and HR 2 domains are indicated by brackets. The leucine zipper domain in HR 2 is highlighted by black dots. A stretch of 14 aa comprising exactly two heptad repeats which is only found in alphacoronaviruses is indicated by a black bar. Numbers refer to amino acid position in spike protein of strain Black. Accession numbers of reference strains: Black: EU186072, C1Je: dq848678. Amino acids EY at position 1217/1218 of FECV were replaced with DH or QY in most FIPV spike proteins. Further noticeable changes were D1210N, E1267D/Q, Y1268H, N1275G, F1295L, T1299A/S, T1308A, YTT/I to FV/SN/R/M (1310–1312), E1315D/N, Y1334H, T/AP to I/TS/T (1341/1342), N/H/DL to HF/L (1345/1346), H1444N and S1454R/N (B).
Table 5.
Amino acid sequence alignment of mutation 23531 (corresponding to nt 23535) (aa 1045–1047 in spike protein) in reference strain Black (accession number EU186072).

Interestingly the aspartate (D) residue (aa position 1108 in spike protein of strain Black) found in all FECV sequences was substituted by an aromatic tyrosine (Y) residue in FIPV-2A, -3F, -16F, -17A, and -18A (Fig. 2A). It is located within a stretch of 14 aa unique to alphacoronaviruses which is present within HR1 (Fig. 2A).
Table 4 displays a summary of the different mutations found in ORF3, ORF7 and spike protein genes of FIPV samples including enteric and systemic viruses of the same animals.
Table 4.
Synopsis of relevant aa changes in ORF3, ORF7 and spike protein and resulting translation products of FIPV samples. Numbers in brackets indicate number of truncated aa. Onlychanges affecting more than 50% of examined samples are listed. The change in the respective sample is underlined. Numbers refer to positions according to reference strain Black (EU186072). n.s.: not sequenced.
| Sample | Gene 3a | Gene 3b | Gene 3c | Gene 7a | Gene 7b | Gene S |
|---|---|---|---|---|---|---|
| FIPV-1F | – | – | – | F9L | Y170H | Deletions (4), M1045L/D1108H/Y, E1267D/Q, T1299A/S, T1311V/S, L1345F |
| FIPV-1A | – | – | – | F9L | Y170H | Deletions (4), M1045L/D1108H/Y, E1267D/Q,T1311V/S |
| FIPV-2A | – | – | Truncation (76) | F9L | Y170H | M1045L/D1108H/Y/D1210N, E1267D/Q, Y1268H, N1275G, T1299A/S, T1308A, T1311V/S, E1315D/N, P1341S/T, L1345F, H1444N, S1454R/N |
| FIPV-3F | – | – | – | F9L | Y170H | M1045L/D1108H/Y/D1210N, E1267D/Q, Y1268H, N1275G, T1299A/S, T1308A, T1311V/S, E1315D/N, P1341S/T, L1345F, H1444N, S1454R/N |
| FIPV-3A | – | – | – | F9L | Y170H | M1045L, N1275G, P1341S/T |
| FIPV-4A | – | – | Truncation (105) | F9L | Y170H | Only HR2: N1275G, T1308A, E1315D/N |
| FIPV-5A | – | Truncation (6) | No translation product | n.s. | n.s. | n.s. |
| FIPV-6A | Truncation (9) | Truncation (9) | No translation product | n.s. | n.s. | n.s. |
| FIPV-7F | – | – | – | F9L | Y170H | E1267D/Q, Y1268H, E1315D/N, S1454R/N |
| FIPV-8F | – | – | Truncation (45) | n.s. | n.s. | n.s. |
| FIPV-8A | – | – | Truncation (45) | n.s. | n.s. | n.s. |
| FIPV-9F | – | – | Truncation (65) | n.s. | n.s. | n.s. |
| FIPV-9A | – | – | Truncation (65) | n.s. | n.s. | n.s. |
| FIPV-10F | Truncation (7) | – | – | n.s. | n.s. | n.s. |
| FIPV-10A | Truncation (7) | – | Truncation (221) | n.s. | n.s. | n.s. |
| FIPV-11A | Truncation (35) | – | Truncation (132) | n.s. | n.s. | n.s. |
| FIPV-12F | – | – | – | – | – | Only HR2 |
| FIPV-12A | – | – | Truncation (163) | – | – | Only HR2 |
| FIPV-13F | – | Elongation (8) | No translation product | n.s. | n.s. | n.s. |
| FIPV-14F | – | Truncation (1) | – | F9L | – | M1045L, H1444N |
| FIPV-15F | – | Truncation (1) | – | – | – | M1045L, H1444N |
| FIPV-15A | Truncation (22) | Truncation (1) | Truncation (110) | – | Y170H | H1444N |
| FIPV-16F | – | Truncation (1) | – | F9L | Truncation (171) | D1108H/Y/D1210N, E1267D/Q, Y1268H, N1275G, T1299A/S, T1308A, T1311V/S, E1315D/N, P1341S/T, L1345F, H1444N, S1454R/N |
| FIPV-17F | – | Truncation (1) | – | n.s. | n.s. | Only 23531: M1045L |
| FIPV-17A | – | Truncation (1) | Truncation (82) | – | Y170H | D1108H/Y, D1210N, E1267D/Q, Y1268H, N1275G, T1299A/S, T1308A, T1311V/S, E1315D/N, P1341S/T, L1345F, H1444N, S1454R/N |
| FIPV-18F | – | – | No translation product | n.s. | n.s. | n.s. |
| FIPV-18A | – | – | No translation product | – | Y170H | D1108H/Y/D1210N, E1267D/Q, Y12668H, N1275G, T1299A/S, T1308A, T1311V/S, E1315D/N, P1341S/T, L1345F, H1444N, S1454R/N |
| FIPV-19F | – | Truncation (1) | – | n.s. | n.s. | n.s. |
| FIPV-19A | – | Truncation (1) | Truncation (129) | n.s. | n.s. | n.s. |
3.3. Analysis of M protein gene
Five aa positions within the M protein have been suggested to be hotspots for mutations (positions 108, 120, 138, 163 and 199) (Brown et al., 2009). We analyzed the M gene of the following samples: FECV-5, -12, -14, -19 and -20; FIPV-2A, -3F, -12F, -12A, -15A and -18A. After RT-PCR a 677 bp fragment of the M protein gene was obtained; the region examined encompassed 207 of 263 aa of the M protein (numbers for serotype I laboratory strain Black). Sequence comparison revealed few aa exchanges which did not discriminate between FECV and FIPV biotype (Fig. 3 ).
Fig. 3.

Sequence alignment of aa 57–263 of the membrane protein. Residues diverging from the consensus are highlighted by grey boxes. Numbers refer to amino acid position in spike protein of strain Black. Clear boxes indicate five aa positions formerly described as being marker mutations. Accession numbers of reference strains: p04135: TGEV, 79–1146 (serotype II): AY994055, Black (serotype I): EU186072.
4. Discussion
The study presented here was undertaken to identify mutations in feline coronavirus genomes which might trigger a switch from the harmless FECV biotype to the highly virulent FIPV biotype. Our work involved the sequence analysis of several genes (3a, 3b, 3c, 7a, 7b, S2 of S protein gene, M protein gene).
The changes of ORF3c in the FIPV samples investigated in this study were present as deletions and point mutations (early termination or deletions comprising the start codon of 3c) in accordance with the literature (Vennema et al., 1998, Chang et al., 2010, Pedersen et al., 2012). Such mutations could not be detected in ORF3c of FECV samples. It has been shown before that an intact 3c gene is considered to be essential for efficient FECV replication in the intestinal tract, but dispensable for systemic FIPV replication (Chang et al., 2010, Le Poder, 2011, Pedersen et al., 2012).
Previous studies described deletions in ORF7a in FIPV samples compared to FECV samples suggesting that mutations of 7a might correlate with the development of FIP (Kennedy et al., 2001). ORF7b has been speculated to play a determining role in virulence (Vennema et al., 1992, Herrewegh et al., 1995b). The most recent work on ORF7b revealed small in-frame deletions in the 3′-terminal region of 7b in some of the virulent and avirulent FCoV strains (Lin et al., 2008). Our analysis of ORF7 showed only minor changes in the nucleic acid sequence resulting in single aa exchanges (Fig. 1B and Table 4). However, all investigated genomes except for FIPV-16F, which encodes a stop codon in 7b at the same position as laboratory strain Black (position 36), contained complete 7a and 7b genes.
Subsequently, we analyzed the S2 domain of the S glycoprotein from FECV as well as from several FIPV samples. Previously, Vennema et al. (1998) found deletions and point mutations of the S genes of FIPV compared to FECV. By comparison of the highly virulent strain 79–1146 (FIPV) and the enteric strain 79–1683 (FECV) 10 aa exchanges were detected in the S2 domain (Rottier et al., 2005). In our analyses, we could not identify mutations of these residues. Recent results obtained by full genome sequencing of 11 viruses of each pathotype revealed two aa differences in the putative fusion peptide of the S protein gene as promising hotspots for differentiation between FECV and FIPV, namely at positions 1045 and 1047 (M1058L and S1060A for strain FIPV strain C1Je (DQ848678)) (Chang et al., 2012). We could confirm that residue 1045 represents a hotspot since all but one FECV samples analyzed harboured an A residue leading to a methionine (M), while all but one ascites-derived FIPV samples display a T residue resulting in a leucine (L) (Table 5).
At aa position 1108 (Fig. 2A) within HR1 the aspartate (D) residue found in all FECV sequences was substituted by a tyrosine (Y) residue in five FIPV samples. Interestingly, some virus isolates from FIP cats that had no major mutations in 3c showed differences at this aa position (Table 4). Moreover, the linker region was subject to several aa substitutions when FECV and FIPV sequences were compared.
A recent paper hypothesized that changes of the furin recognition motif, located between S1 and S2 domains within the S gene, might represent a contributing factor for development of FIP (Licitra et al., 2013). All FECVs analyzed in their study contained a conserved furin cleavage motif, while the FIPVs showed substitutions in the motif. Therefore, the authors suggested that the presence/absence of this cleavage site correlates with the biotype. We analyzed the furin cleavage site of two FECV and 13 FIPV samples. While both FECV samples had an intact furin cleavage site, nine FIPVs showed alterations at positions P4 and/or P5. Positions P2 and P1 were conserved (RR) except for one FIPV sample (data not shown).
Brown et al. (2009) reported differences at five aa positions within the M protein which allow differentiation between FCoV biotypes. Like Chang et al. (2011), we were not able to confirm these aa signatures in our samples. The same group also promoted a so-called “circulating virulent–avirulent FCoV” hypothesis of viral pathogenesis suggesting that non-pathogenic and pathogenic strains circulate in a population based on phylogenetic analyses of the ORF7b and M genes (Brown et al., 2009). Therefore, we applied our sequences obtained for ORF7 and M to phylogenetic analyses using the HUSAR package provided by DKFZ (Heidelberg, Germany) (data not shown). For ORF7, we could not determine a clear segregation between FECV and FIPV isolates, however, this data was not supported by high bootstrap values in all branchings. Phylogenetic analyses of 645 b of the M gene of both FECV and FIPV samples did also not allow clustering into biotypes (data not shown). Our data support the clustering of FCoVs according to geographic origin irrespective of their pathotype as shown by others (Vennema et al., 1998, Chang et al., 2011).
It is still an open question whether FIPV can be transmitted horizontally. An intriguing finding within this study is that viruses with a mutated 3c gene could not only be found in ascites but also in the respective faecal samples of FIP animals (FIPV-8, -9 and -18). This is surprising in the light of a recent study where FIPVs with a mutated 3c were not shed in the faeces after either oronasal or intraperitoneal inoculation (Pedersen et al., 2012). Furthermore, we could detect viruses with an intact 3c gene but mutations within the S protein gene (M1045L) in both faeces and ascites (FIPV-1) or only in the faecal sample (FIPV-3).
For five animals the viruses in faeces and ascites were different (FIPV-10, -12, -15, -17 and -19): In the ascites a truncated 3c gene could be detected but in the faeces an intact 3c. This finding is in accordance with other studies (Chang et al., 2010). Whether the enteric virus has been acquired by FECV superinfection as suggested (Chang et al., 2010) or represents the original persisting FECV is not known. Accordingly horizontal transmission of FIP viruses cannot be excluded.
Considering all sequence data obtained in this study as well as the literature, the 3c gene is often affected by mutations. 13 out of 15 FIPV ascites samples displayed changes in 3c. The other two showed alterations in the S protein gene. Furthermore, only one out of eight faeces derived FIPV samples with an intact 3c and one ascites derived FIPV sample with a truncated 3c did not display mutations in the S protein gene. These findings might reflect an interplay between 3c and S proteins.
Our findings provide further support for the involvement of mutations in 3c and S genes in the biotype switch from FECV to FIPV. Furthermore, our data suggests that the involvement of the other investigated genes (3a, 3b, 7a, 7b and M) in the development of FIP is negligible. In conclusion, the data obtained in this study support the “internal mutational theory” rather than the “circulating virulent/avirulent FCoV” hypothesis. However, the significance of specific mutation events in the generation of FIPV needs to be further evaluated using reverse genetic systems for both serotype I and II feline coronaviruses followed by animal experiments (Tekes et al., 2008, Tekes et al., 2010, Tekes et al., 2012).
Acknowledgements
This study was supported by the “Bundesministerium für Bildung und Forschung” of the German Government (Zoonosis Network, Consortium on ecology and pathogenesis of SARS, project code 01KI1005A-F) as well as by the Collaborative Research Centre 1021: “RNA viruses: RNA metabolism, host response and pathogenesis”. We thank Dr. Matthias König from the diagnostic laboratory of our institute for providing samples.
References
- Becher P., Orlich M., Shannon A.D., Horner G., Konig M., Thiel H.J. Phylogenetic analysis of pestiviruses from domestic and wild ruminants. J. Gen. Virol. 1997;78(Pt 6):1357–1366. doi: 10.1099/0022-1317-78-6-1357. [DOI] [PubMed] [Google Scholar]
- Brown M.A. Genetic determinants of pathogenesis by feline infectious peritonitis virus. Vet. Immunol. Immunopathol. 2011;143(3/4):265–268. doi: 10.1016/j.vetimm.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.A., Troyer J.L., Pecon-Slattery J., Roelke M.E., O’Brien S.J. Genetics and pathogenesis of feline infectious peritonitis virus. Emerg. Infect. Dis. 2009;15(9):1445–1452. doi: 10.3201/eid1509.081573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.W., de Groot R.J., Egberink H.F., Rottier P.J. Feline infectious peritonitis: insights into feline coronavirus pathobiogenesis and epidemiology based on genetic analysis of the viral 3c gene. J. Gen. Virol. 2010;91(Pt 2):415–420. doi: 10.1099/vir.0.016485-0. [DOI] [PubMed] [Google Scholar]
- Chang H.W., Egberink H.F., Halpin R., Spiro D.J., Rottier P.J. Spike protein fusion peptide and feline coronavirus virulence. Emerg. Infect. Dis. 2012;18(7):1089–1095. doi: 10.3201/eid1807.120143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.W., Egberink H.F., Rottier P.J. Sequence analysis of feline coronaviruses and the circulating virulent/avirulent theory. Emerg. Infect. Dis. 2011;17(4):744–746. doi: 10.3201/eid1704.102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot R., Baker S., Baric R., Enjuanes L., Gorbalenya A., Holmes K., Perlman S., Poon L., Rottier P., Talbot P., Woo P., Ziebuhr J. Family Coronaviridae. In: King M.Q., Lefkowitz E., Adams M.J., Carstens E.B., editors. Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier; Oxford: 2011. pp. 806–828. [Google Scholar]
- Dewerchin H.L., Cornelissen E., Nauwynck H.J. Replication of feline coronaviruses in peripheral blood monocytes. Arch. Virol. 2005;150(12):2483–2500. doi: 10.1007/s00705-005-0598-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye C., Siddell S.G. Genomic RNA sequence of Feline coronavirus strain FIPV WSU-79/1146. J. Gen. Virol. 2005;86(Pt 8):2249–2253. doi: 10.1099/vir.0.80985-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Enjuanes L., Ziebuhr J., Snijder E.J. Nidovirales: evolving the largest RNA virus genome. Virus Res. 2006;117(1):17–37. doi: 10.1016/j.virusres.2006.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haijema B.J., Volders H., Rottier P.J. Live, attenuated coronavirus vaccines through the directed deletion of group-specific genes provide protection against feline infectious peritonitis. J. Virol. 2004;78(8):3863–3871. doi: 10.1128/JVI.78.8.3863-3871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeney J.L., Evermann J.F., McKeirnan A.J., Marker-Kraus L., Roelke M.E., Bush M., Wildt D.E., Meltzer D.G., Colly L., Lukas J. Prevalence and implications of feline coronavirus infections of captive and free-ranging cheetahs (Acinonyx jubatus) J. Virol. 1990;64(5):1964–1972. doi: 10.1128/jvi.64.5.1964-1972.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A., de Groot R.J., Cepica A., Egberink H.F., Horzinek M.C., Rottier P.J. Detection of feline coronavirus RNA in feces, tissues, and body fluids of naturally infected cats by reverse transcriptase PCR. J. Clin. Microbiol. 1995;33(3):684–689. doi: 10.1128/jcm.33.3.684-689.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A., Vennema H., Horzinek M.C., Rottier P.J., de Groot R.J. The molecular genetics of feline coronaviruses: comparative sequence analysis of the ORF7a/7b transcription unit of different biotypes. Virology. 1995;212(2):622–631. doi: 10.1006/viro.1995.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M., Boedeker N., Gibbs P., Kania S. Deletions in the 7a ORF of feline coronavirus associated with an epidemic of feline infectious peritonitis. Vet. Microbiol. 2001;81(3):227–234. doi: 10.1016/S0378-1135(01)00354-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M., Kania S., Stylianides E., Bertschinger H., Keet D., van Vuuren M. Detection of feline coronavirus infection in southern African nondomestic felids. J. Wildl. Dis. 2003;39(3):529–535. doi: 10.7589/0090-3558-39.3.529. [DOI] [PubMed] [Google Scholar]
- Le Poder S. Feline and canine coronaviruses: common genetic and pathobiological features. Adv. Virol. 2011;2011:609465. doi: 10.1155/2011/609465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutenegger C.M., Hofmann-Lehmann R., Riols C., Liberek M., Worel G., Lups P., Fehr D., Hartmann M., Weilenmann P., Lutz H. Viral infections in free-living populations of the European wildcat. J. Wildl. Dis. 1999;35(4):678–686. doi: 10.7589/0090-3558-35.4.678. [DOI] [PubMed] [Google Scholar]
- Licitra B.N., Millet J.K., Regan A.D., Hamilton B.S., Rinaldi V.D., Duhamel G.E., Whittaker G.R. Mutation in spike protein cleavage site and pathogenesis of feline coronavirus. Emerg. Infect. Dis. 2013;19(7):1066–1073. doi: 10.3201/eid1907.121094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.N., Su B.L., Huang H.P., Lee J.J., Hsieh M.W., Chueh L.L. Field strain feline coronaviruses with small deletions in ORF7b associated with both enteric infection and feline infectious peritonitis. J. Feline Med. Surg. 2009;11(6):413–419. doi: 10.1016/j.jfms.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen N.C. Virologic and immunologic aspects of feline infectious peritonitis virus infection. Adv. Exp. Med. Biol. 1987;218:529–550. doi: 10.1007/978-1-4684-1280-2_69. [DOI] [PubMed] [Google Scholar]
- Pedersen N.C. A review of feline infectious peritonitis virus infection: 1963-2008. J. Feline. Med. Surg. 2009;11(4):225–258. doi: 10.1016/j.jfms.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen N.C., Liu H., Dodd K.A., Pesavento P.A. Significance of coronavirus mutants in feces and diseased tissues of cats suffering from feline infectious peritonitis. Viruses. 2009;1(2):166–184. doi: 10.3390/v1020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen N.C., Liu H., Scarlett J., Leutenegger C.M., Golovko L., Kennedy H., Kamal F.M. Feline infectious peritonitis: role of the feline coronavirus 3c gene in intestinal tropism and pathogenicity based upon isolates from resident and adopted shelter cats. Virus Res. 2012;165(1):17–28. doi: 10.1016/j.virusres.2011.12.020. Epub 2012 Jan 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier P.J. The molecular dynamics of feline coronaviruses. Vet. Microbiol. 1999;69(1/2):117–125. doi: 10.1016/S0378-1135(99)00099-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottier P.J., Nakamura K., Schellen P., Volders H., Haijema B.J. Acquisition of macrophage tropism during the pathogenesis of feline infectious peritonitis is determined by mutations in the feline coronavirus spike protein. J. Virol. 2005;79(22):14122–14130. doi: 10.1128/JVI.79.22.14122-14130.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T., Tomiyama Y., Katoh Y., Nakamura M., Satoh R., Hohdatsu T. Mutation of neutralizing/antibody-dependent enhancing epitope on spike protein and 7b gene of feline infectious peritonitis virus: influences of viral replication in monocytes/macrophages and virulence in cats. Virus Res. 2011;156(1/2):72–80. doi: 10.1016/j.virusres.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekes G., Hofmann-Lehmann R., Bank-Wolf B., Maier R., Thiel H.J., Thiel V. Chimeric feline coronaviruses that encode type II spike protein on type I genetic background display accelerated viral growth and altered receptor usage. J. Virol. 2010;84(3):1326–1333. doi: 10.1128/JVI.01568-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekes G., Hofmann-Lehmann R., Stallkamp I., Thiel V., Thiel H.J. Genome organization and reverse genetic analysis of a type I feline coronavirus. J. Virol. 2008;82(4):1851–1859. doi: 10.1128/JVI.02339-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekes G., Spies D., Bank-Wolf B., Thiel V., Thiel H.J. A reverse genetics approach to study feline infectious peritonitis. J. Virol. 2012;86(12):6994–6998. doi: 10.1128/JVI.00023-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Poland A., Foley J., Pedersen N.C. Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses. Virology. 1998;243(1):150–157. doi: 10.1006/viro.1998.9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennema H., Rossen J.W., Wesseling J., Horzinek M.C., Rottier P.J. Genomic organization and expression of the 3′ end of the canine and feline enteric coronaviruses. Virology. 1992;191(1):134–140. doi: 10.1016/0042-6822(92)90174-N. [DOI] [PMC free article] [PubMed] [Google Scholar]