

Abstract
High-resolution metabolomics (HRM) profiling of metabolic fingerprints can improve understanding of how poly and perfluoroalkyl substances (PFASs) induce metabolic alterations of in utero environment and impact fetal health. HRM profiling and quantification of PFASs were performed for 397 maternal perinatal serum samples collected from 1959–1967 in the Child Health and Development Studies (CHDS). We used Metabolome-Wide Association Studies (MWAS) and pathway enrichment analysis for metabolic associations with PFOS, its precursor EtFOSAA, and EtFOSAA-to-PFOS ratio. Distinct metabolic profiles were found with EtFOSAA and PFOS. Urea cycle metabolites such as arginine, lysine and creatine had opposite associations with EtFOSAA (negative) and PFOS (positive); whereas, carnitine shuttle metabolites were found to be exclusively and positively associated with PFOS indicating perturbation in fatty acid metabolism. These differential metabolic associations for precursor and end-product represent an important first step in identifying how PFASs alter the in utero environment and potentially leads to disease risk.
Keywords: PFOS, EtFOSAA, metabolomics, in utero, CHDS, breast cancer
Introduction
Perfluoroalkyl and polyfluoroalkyl substances (generally denoted as PFASs) have been widely used in numerous industrial and commercial applications since 1950 [1]. Their exceptional chemical and thermal stability, as well as hydrophobic and lipophobic nature, have led to extensive utilization in surfactants for resisting stain and grease, including nonstick cookware, packaging products, carpets, leather products, and textiles that enhance water, grease and soil repellency [1, 2]. Despite the phasing out of the production and use of several PFASs in the early 2000s, they are still present in all environmental media including air, water, soil and food, and can almost always be detected in human serum due to their persistence [3, 4]. The major exposure pathways are ingestion of contaminated food and water, while children are more susceptible than adults due to hand-to-mouth transfer of chemicals from carpets and ingestion of dust [5]. The most abundant PFASs compound detected in humans is perfluorooctane sulfonic acid (PFOS) followed by perofluorooctanoic acid (PFOA) [2], which may come from direct sources of manufacturing emissions and products themselves, and from indirect sources via precursor degradation [6]. With the decline of PFOS manufacturing over the last decade, investigating the indirect sources has become increasingly important [7].
Health concerns of PFASs compounds have attracted growing attention in recent years. Human epidemiology studies have suggested associations between PFASs exposure and multiple adverse health outcomes [2]. In particular, the developmental toxicity of PFASs have been well studied and recognized in humans including pregnancy-induced hypertension, pre-eclampsia, decreased fertility, decreased birth weight and thyroid disease [8–10]. Consistently in rodent experiments, exposure to PFOA and PFOS during pregnancy was shown to induce both early and later life adverse health outcomes such as delay in mammary gland development, neurodevelopmental and immune toxicity in offspring of exposed animals [11]. More recently, two human studies have suggested the associations between PFASs and breast cancer risks [12, 13]. These associations may extend to the connection between maternal exposure and offspring health. In the Child Health and Development Studies (CHDS) cohort, we measured the level of 2-(N-Ethyl-perfluorooctane sulfonamido) acetic acid (EtFOSAA), a precursor which is applied to a variety of consumer products and degraded to PFOS [6]. We found that high EtFOSAA exposure combined with high maternal cholesterol predicted a 3.6-fold increased risk of breast cancer in daughters. Conversely, maternal PFOS was associated with decreased breast cancer risk in daughters [14]. Although the underlying mechanisms are not well understood, the link between maternal endocrine disruptor exposure and offspring breast cancer risks is mostly established in mammary gland development of the offspring [10, 11, 15, 16]. This evidence speaks to the possibility of epigenetic programming as one of the key mechanisms linking early developmental exposures to PFAS, but additional research is needed [10].
Metabolic profiling of maternal samples is a newly developed tool to understand the alterations in fetal environment and make further links to the health outcomes of the offspring [17–19]. A limited number of human studies have reported that PFOS and other PFASs were associated with alteration in human serum metabolome involving redox signaling and multiple fatty acid metabolism pathways [20, 21], which are in line with findings from animal and cell culture models [22, 23]. However, no information is available on the metabolic effects of maternal PFASs exposure, in other words, changes in the in utero environment.
In this study, we performed a metabolome-wide association study (MWAS) using untargeted high-resolution mass spectrometry (HRM) to identify metabolic changes associated with serum PFOS concentrations in women during pregnancy or early postpartum. We also tested an a priori hypothesis that the PFOS precursor, EtFOSAA [1] would have different effects on the metabolome of the in utero environment and potentially on the risk of reproductive diseases than its their end-product, PFOS, because of structural differences and possibly different biological activity [24]. This possibility is corroborated by the findings of different effects of o,p’-DDT, p,p’-DDT and p,p’-DDE on multiple reproductive health outcomes [25–29]. Moreover, PFOS and EtFOSAA were differentially associated with daughter’s breast cancer risk in the CHDS [14]. To test this hypothesis, we assessed the metabolite profiles associated with serum EtFOSAA levels, PFOS levels and the ratio of EtFOSAA to PFOS (as an indicator of active exposure) in the CHDS F0 generation, and compared the similarity and difference of metabolic pathways.
Methods
Study population and exposures
In this study, we analyzed sera from 397 subjects as part of the Child Health and Development Studies (CHDS) cohort. The CHDS was designed to examine the association between prenatal exposures and health and development over the life course for parents and children. The CHDS recruited women residing in the Oakland, California area who were members of the Kaiser Foundation Health Plan and received obstetric care for pregnancies between 1959 and 1967 [30]. Previously we found a strong positive association between perinatal EtFOSAA exposure and offspring breast cancer risk among a subset of CHDS daughters (102 breast cancer cases and 310 controls of offspring). Age-matched, conditional logistic regression to estimate maternal EtFOSAA association with daughters’ breast cancer showed a 3.6-fold increased risk when accompanied by high levels of maternal cholesterol, while maternal PFOS was associated with decreased risk. Therefore, here we sought to follow up and test the hypothesis that EtFOSAA and PFOS induce different metabolic alterations in maternal serum that potentially may contribute to the health outcomes of the offspring. This paper includes 50 randomly selected cases (breast cancer in daughters) and 347 controls, roughly approximating the disease prevalence of 1/8 of the US population. The cases and controls were matched on birth year and trimester of maternal blood draw. All blood specimens were collected throughout all trimesters and early postpartum (1–3 days after delivery). The present study was reviewed and approved by The Institutional Review Board of the Public Health Institute and it has complied with all federal guidelines governing use of human subjects.
PFASs levels were measured in non-fasting maternal perinatal serum samples including mostly samples (73.5%) collected right after delivery (within 2 days) and a smaller subset from the third trimester (17.9%). The number of samples from the first and second trimester were relatively small (3 and 15 samples, respectively) (Supplemental Table S1). Because PFASs cross the placenta and accumulate in the fetus, we used samples throughout pregnancy and postpartum yet with a preference on early postpartum samples which may best capture more prolonged in utero exposure. A family of PFAS compounds were measured including perfluorodecanoic acid (PFDeA), perfluorododecanoic acid (PFDoA), perfluoroheptanoic acid (PFHpA), perfluorohexane sulfonic acid (PFHxS), perfluorononanoic acid (PFNA), perfluorooctanoic acid (PFOA), perfluorooctanesulfonic acid (PFOS), perfluorooctanesulfonamide (PFOSA), perfluoroundecanoic acid (PFUdA), 2-(N-Methyl-perfluorooctane sulfonamido) acetic acid (MeFOSAA) and 2-(N-Ethyl-perfluorooctane sulfonamido) acetic acid (EtFOSAA). Briefly, 100 μL of serum were mixed with 0.1 M formic acid, and internal standards were added (MPFAC-MXA, Wellington Laboratories), then injected by an online SPE-HPLC system (Symbiosis Pharma system with Mistral CS Cool, Spark Holland Inc.) to a C18 cartridge (HySphere C18HD, 7 μm, 10 × 2 mm). After washing, the target analytes were eluted to a C8 HPLC column (BETASIL C8 column, Thermo Fisher Scientific) for separation. The eluate was then introduced to the MS/MS (ABSciex API 4000 QTrap) for multiple-reaction- monitoring (MRM) analysis. p,p’-DDE was measured in the laboratory of the California Department of Toxic Substances Control using methods developed previously [31]. Briefly, serum samples spiked with surrogate standards (tetrachloro-m-xylene, PCB-65, PCB-166) were extracted using Oasis HLB SPE cartridges (Waters Corp.; Milford, MA), and subsequently cleaned up with 33% sulfuric acid silica. DDE was analyzed on a DB-5ms column (30 m × 0.25 mm I.D., 0.25 μm film thickness; Agilent Technologies; Sunnyvale, CA) using gas chromatograph-tandem mass spectrometer (Agilent, Santa Clara, CA).
Total lipids were measured by total cholesterol and triglycerides assayed enzymatically as previously described [25] at the Clinical & Epidemiologic Research Laboratory (CERLab) at Boston Children’s Hospital (certified by the Centers for Disease Control and Prevention/National Heart, Lung, and Blood Institute Lipid Standardization).
High-resolution LC-MS metabolomics
Metabolite extracts of sera were analyzed as described previously to acquire untargeted metabolic profiling [32–34]. Briefly, 65 μL aliquots were taken from each thawed serum sample and maintained on ice. Ice-cold acetonitrile was added to the aliquots at a 2:1 ratio to precipitate the proteins. A mixture of 14 stable isotope internal standards was included for quality control prior to protein precipitation. Following 30 minutes incubation on ice, samples were centrifuged for 10 minutes at 16,100×g at 4°C. Supernatants were then randomized, and injected in triplicates to C18 chromatography (Higgins Analytical, 100×2.1mm, 5uL) coupled with Q-Exactive HF mass spectrometer (Thermo Fisher). Mass spectral data were collected with positive electrospray ionization from m/z (mass-to-charge ratio) 85 to 1275.
Bioinformatics and Biostatistics
Raw mass spectrometry data was extracted using apLCMS [35] and xMSanalyzer [36] for m/z, retention time, and integrated peak intensity. Data were pre-filtered from 12,855 to 3,121 metabolic features to retain only features with non-zero values in > 80% in all samples and >50% in each group. Principle component analysis and hierarchical clustering analysis were performed to decide whether the factors related to pregnancy time, delivery type, parity or breastfeeding affects the metabolism. Metabolome-wide-association study (MWAS) was conducted using linear regression models, which associate the serum PFOS and EtFOSAA concentration with metabolite intensities, adjusted for covariates including total cholesterol (continuous variable), age (continuous variable) and p,p’-DDE levels (continuous variable). EtFOSAA and PFOS were entered into models simultaneously and were alternatively entered as a ratio of precursor, EtFOSAA to PFOS to model individual relative exposures. PFOS and EtFOSAA concentrations as well as the ratio were log 2 transformed for normalizing for regression models.
All metabolic features were annotated with xMSannotator [37] using HMDB (Human Metabolome Database, [38]) at 5 ppm tolerance; confidence scores for annotation by xMSannotator are derived from a multistage clustering algorithm. Identities of selected metabolites were confirmed by accurate m/z match and co-elution with authentic standards (level 1 identification by criteria of Schymanski et al [39]).
Each PFASs variable (EtFOSAA, PFOS and ratio of EtFOSAA to PFOS) was categorized in quartiles of distribution based on the cohort population. Student’s t test (two-tailed with Welch’s correction for unequal variance) were used to test statistical significance among quartiles. Pathway enrichment analysis was performed in mummichog (version 2.0, features with P < 0.05 were treated as the significant input list), which uses a probabilistic algorithm different from feature level statistics and produces pathway p-values based on permutation [40, 41]. The version 2 of mummichog enforces retention time in grouping isotopes or adducts, thus producing more stringent results compared to version 1. All other bioinformatics analyses were performed in RStudio version 1.1.447 (RStudio, Inc.). The significance level was P < 0.05 for all tests; Benjamin and Hochberg false discovery rate was used for multiple comparisons [42].
Results
Distributions of study variables
Demographic information for this study is summarized in Supplemental Table 1. The present study sample has an average age of 25 years old, mostly non-obese and non-African-American. Other than age, we also adjusted for total serum cholesterol and p,p’-DDE levels which may exhibit similar endocrine disrupting effects. Information on parity, breast feeding, delivery type as well as time of blood collection during or after pregnancy is listed in Supplemental Table 1. The distributions of the aforementioned factors were primarily concentrated within one group: multiparous (69.5%) ; blood drawn within 2 days post-delivery (73.5%); vaginal delivery (94.5%); never breastfed (74.6%), with the latter two distributions reflecting characteristics of the general population around 1960s. These distributions suggested that the effects of these factors on metabolic profiles of this cohort were insignificant, which was confirmed by the lack of group separation by any of these factors (Supplemental Figure S1-S4).
Out of the 11 PFASs compounds we monitored, we found that PFOS was the most abundant chemical (median level 33.9 ng/mL), followed by perfluorohexane sulfonic acid (PFHxS) (median 2.29 ng/mL), PFOA (median 0.40 ng/mL) and EtFOSAA (median 0.28 ng/mL) in abundance (Supporting Table S1). Levels of other PFASs compounds, including PFOSA (the other PFOS precursor), were below detection limit for more than 50% of samples. Thus we restricted our attention to PFOS, and EtFOSAA as its precursor. Both PFOS and EtFOSAA showed positive skewed distribution, yet the skewness was lower for PFOS (1.9) than for EtFOSAA (4.2) (Figure 1A and B). This suggested difference in sources of exposure to EtFOSAA and PFOS, or differences in elimination rates of the two chemicals among individuals. Distribution of the ratio (EtFOSAA : PFOS) showed a number of outliers that have high EtFOSAA to PFOS ratio (Figure 1C), which may indicate an active exposure to the precursor or slow metabolism rate. Median levels of PFOS (33.9 ng/mL) and EtFOSAA (0.28 ng/mL) detected in this cohort were comparable to the 1999–2000 NHANES survey (30.4 ng/mL and 0.6 ng/mL, respectively) [4].
Figure 1.

Distribution of serum EtFOSAA (A), PFOS (B) concentrations and the ratio of EtFOSAA to PFOS (C) in 397 subjects from the Child Health and Development Studies (CHDS) cohort, visualized by histogram and density curves.
Metabolome wide association of PFOS and EtFOSAA
In order to investigate the global metabolic alterations associated with varying EtFOSAA and PFOS concentrations, we performed a metabolome wide association study (MWAS) of the untargeted HRM data from the serum samples collected during pregnancy or early postpartum. Using a linear regression model, 483 out of 3121 metabolic features were found significantly associated with EtFOSAA concentrations (P < 0.05, Figure 2A). Similarly, 301 features were associated with PFOS concentrations (Figure 2B), while the ratio of the two showed the significant association with 360 features (Figure 2C). With FDR under 0.05, 34 features remained significant in association with PFOS, and 49 with EtFOSAA. Among the significant features (P < 0.05), EtFOSAA and PFOS overlapped on 81 features; EtFOSAA and the ratio overlapped on 161 features; and PFOS and the ratio overlapped on 143 features. Altogether, 63 features were commonly associated with EtFOSAA, PFOS and the ratio. Interestingly, correlations of m/z features (Figure 2, red or blue indicating positive or negative) showed different patterns between EtFOSAA and PFOS, indicating distinct metabolic effects (Figure 2).
Figure 2.
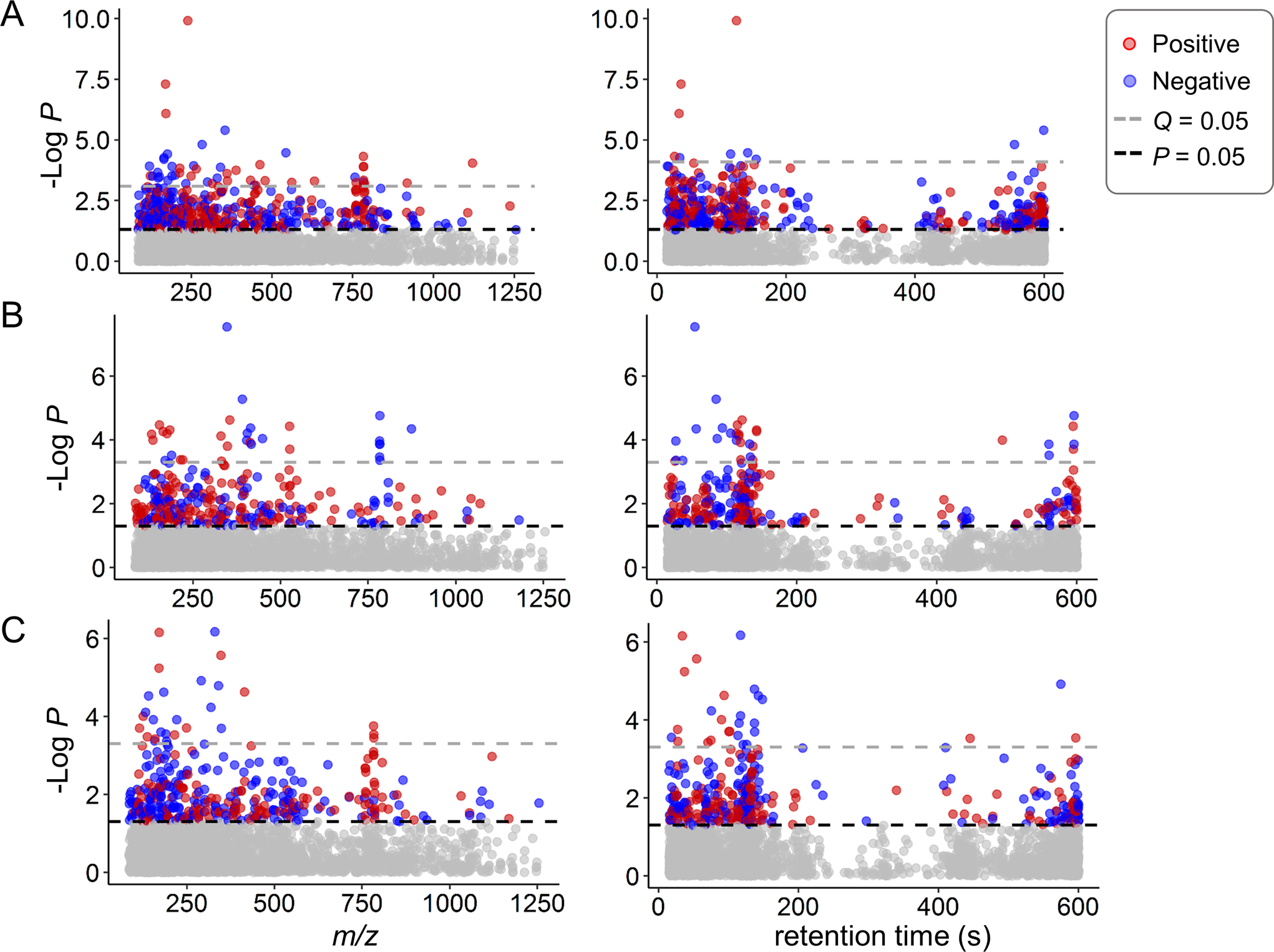
Metabolome-wide association study (MWAS) of serum EtFOSAA (A), PFOS (B) and the EtFOSAA/PFOS ratio (C) in 397 subjects from the CHDS cohort. Type 1 Manhattan plot for –log P vs m/z features and type 2 Manhattan plot for –log P vs retention time with C18 chromatography separation are presented left and right, respectively. Red indicates positive correlation between significant features (P < 0.05) and exposure; blue indicates negative correlation.
The results from the regression analysis were used to perform pathway enrichment analysis with mummichog, to understand the functional alterations of the metabolome. Results showed that EtFOSSA and PFOS shared two major metabolic pathways that were significantly enriched (P < 0.05) and contained ≥6 detected metabolites within the biological pathway. These two common pathways were glycine, threonine, alanine and serine metabolism, and urea cycle/amino group metabolism (Figure 3). On the other hand, carnitine shuttle was most strongly enriched (P = 0.00064) and also exclusively associated with PFOS exposure as the top metabolic pathway. Consistently other unique pathways from PFOS exposure were physiologically linked to carnitine shuttle, including lysine metabolism which generates carnitine, and branched chain amino acid (BCAA) metabolism (i.e. valine, leucine and isoleucine degradation) since both BCAA and carnitine facilitate generation of energy precursors in mitochondria [43, 44]. This cluster of pathways suggested a mechanistic link of PFOS to energy production that is not associated with EtFOSAA.
Figure 3.
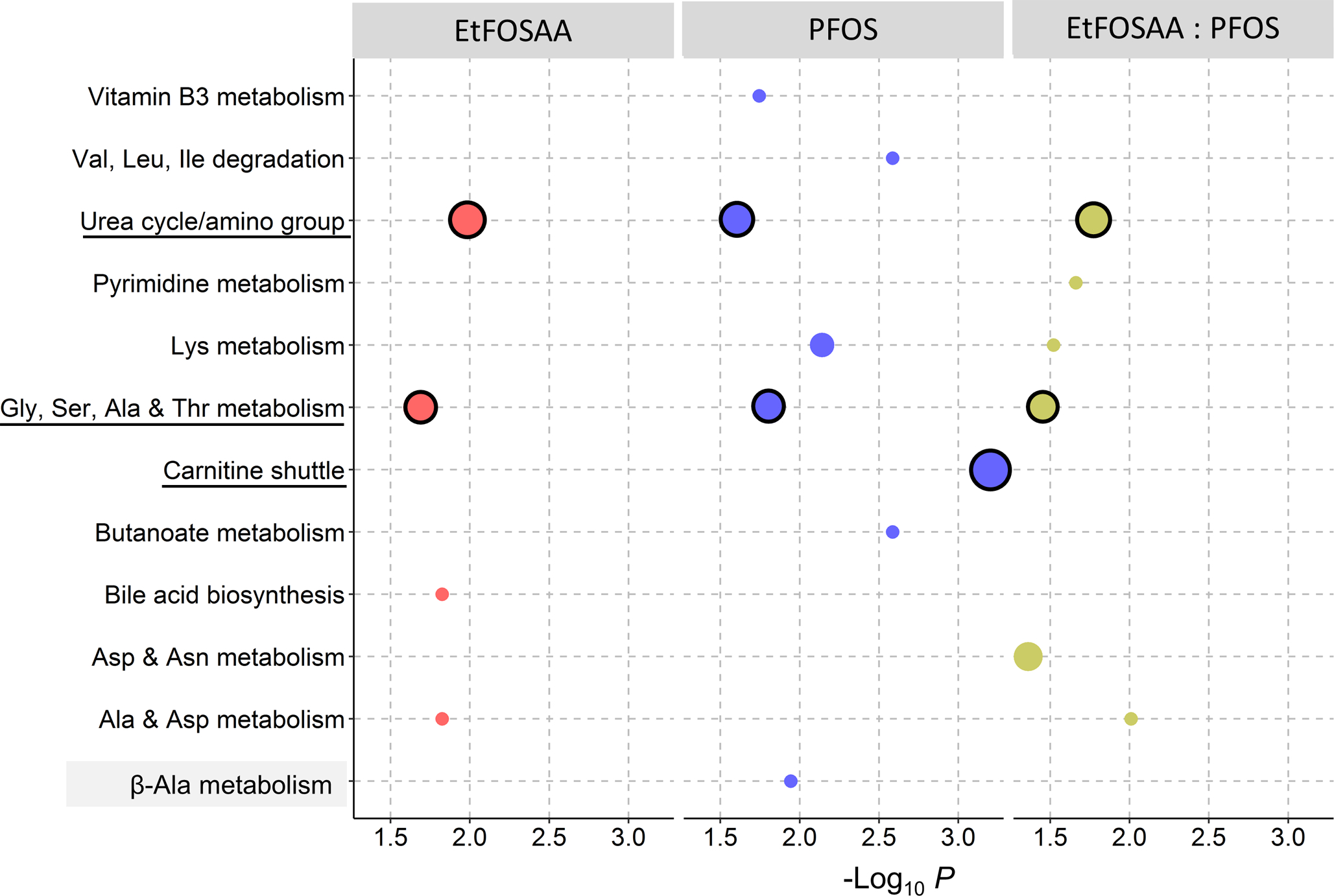
Metabolic pathways enriched by significant features associated with EtFOSAA, PFOS concentrations and the EtFOSAA/PFOS ratio. Size of circles is proportional to the overlap size (i.e. number of significant metabolites in each pathway). Major pathways (≥ 6 metabolites) are circled in black outline.
Metabolic effects of recent active exposure were examined by MWAS of EtFOSAA to PFOS ratio (Figure 2C and Figure 3). Two major pathways were shared by EtFOSAA and EtFOSAA to PFOS metabolic associations, on metabolism of glycine, serine, alanine, threonine, and urea cycle/amino groups, which reflected the metabolic alterations caused by EtFOSAA.
Key Metabolites in network associated with EtFOSAA and PFOS exposure
We further investigated the key metabolites that were in the common pathways shared by EtFOSAA, PFOS and the ratio of the two. Figure 4A shows the quartile average abundance of all significant 360 metabolites in association with EtFOSAA to PFOS ratio. We annotated a total of nine metabolites that contributed to enrichment of the two common pathways (Figure 3) and found that they clustered according to their biological pathways. For example, metabolites in urea cycle and amino group metabolism such as β-alanine, creatinine, pipecolate, lysine, arginine, creatine and adrenochrome were all decreased with increasing ratio of EtFOSAA to PFOS (Figure 4A). These metabolites showed similar negative association with EtFOSAA levels, whereas positive associations were found for all of them with PFOS levels (indicated by t statistics in Figure 4B). On the contrary, the two metabolites (homocysteine and betaine) representing glycine, serine and threonine metabolism were negatively correlated with PFOS yet positively correlated with EtFOSAA and the ratio of the two. This discrepancy in directionality of correlation was also obvious with other metabolites such as phosphoserine and dimethyl-lysine (details of all metabolites listed in Supplemental Table S2). Thus, it is evident that even within the commonly affected pathways, PFOS and its precursor exhibited differential effects.
Figure 4.
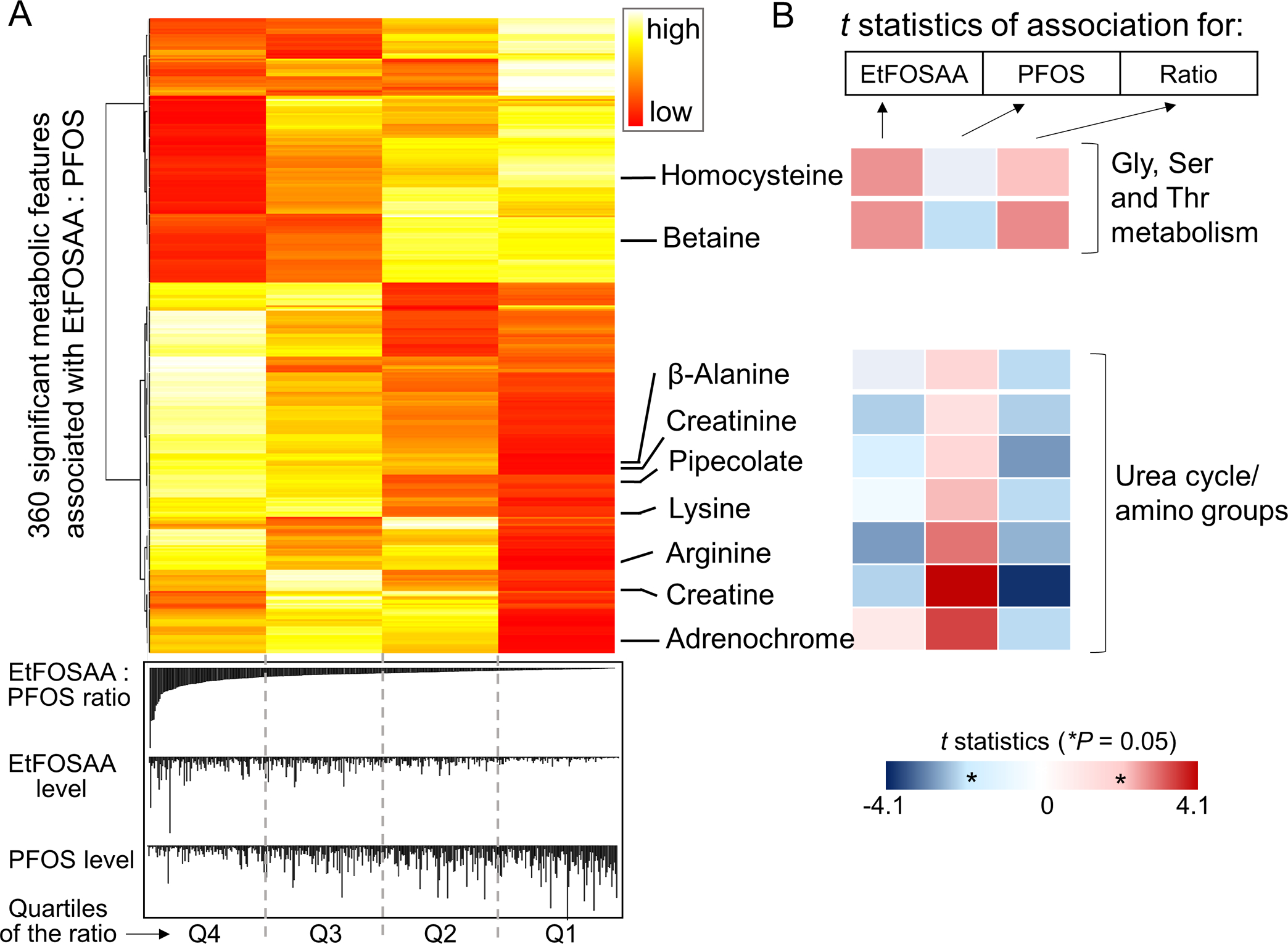
Key metabolites in two major common pathways associated with EtFOSAA and PFOS exposure. A: Heat map of quartile average abundance of metabolites significantly associated with the EtFOSAA/PFOS ratio. Metabolites in glycine, serine and threonine metabolism pathway and urea cycle pathway are annotated. The metabolites here are annotated by accurate mass (5 ppm, details in Supplemental Table S2), and selected metabolites were further confirmed by authentic chemical standards (arginine, lysine, creatine, creatinine, β-alanine). B: Colors are coded by significance of the association (t statistics) showing differences in metabolic responses associated with the exposure (left to right: EtFOSAA, PFOS and the EtFOSAA/PFOS ratio).
Because PFOS exposure showed a unique effect in carnitine shuttle compared to EtFOSAA, we examined the metabolites in the carnitine pathway affected by PFOS and presented the corresponding levels of representative metabolites for the women in each quartile of PFOS exposure in Figure 5. We found that women with higher PFOS levels (quartile 4) had lower intensities of N6,N6-dimethyl-L-lysine (a biosynthesis precursor of carnitine [45], Figure 5A), and higher intensities of carnitine and acyl-carnitines (Figure 5B,C and D). The alterations of carnitine-related metabolites indicated a possible interference of PFOS with fatty acid metabolism and confirmed the findings of PFOS toxicity in lipid regulation [2].
Figure 5.

Levels of selected carnitine-related metabolite in CHDS subjects categorized by quartile 1, 2, 3 and 4 of PFOS serum levels. A: Protein N6,N6-dimethyl-lysine; B: carnitine; C: clupanodonyl carnitine; D: tetracosapentaenyl carnitine. The metabolites here are annotated by accurate mass (5 ppm, details in Supplemental Table S2), and carnitine was further confirmed by authentic chemical standards. *P < 0.05, #P =0.07, by student’s t test compared to quartile 1 (Q1).
Discussion
Metabolic phenotyping with high-resolution metabolomics provides a new approach to understand the effects of mixed exposure and underlying toxic mechanisms. Our study showed differential metabolic effects associated with serum EtFOSAA and PFOS levels in women during pregnancy and early postpartum. MWAS of PFOS and EtFOSAA exposure in women showed different directionality of impacts on critical metabolites in urea cycle and mostly non-essential amino acid metabolism pathway (glycine, serine and threonine). MWAS of PFOS also showed a metabolic signature involving carnitine metabolism that was distinct from the EtFOSAA signatures, revealing a unique PFOS association with fatty acid metabolism [34, 46–48]. This finding supported the hypothesis that PFOS precursors would have different metabolic effects and therefore may contribute to different health risk than their end-product, PFOS [24]. Interestingly, MWAS of the ratio of EtFOSAA to PFOS showed similar metabolic alterations of pathways and metabolites compared to that of EtFOSAA, suggesting this pattern can be a reproducible and relatively unique marker discriminating effects of recent active exposure from the PFOS metabolite.
Urea cycle is metabolically indispensable in the elimination of unneeded nitrogen, and produces endogenous arginine and citrulline as the sole source outside liver. Recent studies have revealed essential roles for urea cycle intermediates and related non-essential amino acids in cancer cells which reprogram metabolism to maximize the use of nitrogen and carbon [49]. In many types of tumor cells including invasive breast carcinoma, synthesis of arginine from citrulline via arginosuccinase synthase and arginosuccinase lyase is silenced or deficient [49–51]. Thus, it is possible that metabolic alterations with these amino acids can be indicative of a cell’s transformation to a malignant state or responses to activation of specific oncogenes. In particular, arginine depletion by myeloid-derived suppressor cells in breast cancer patients has been shown to play a critical role promoting tumor growth and metastasis via suppressing activated T cells [52]. These suppressor cells produce arginase 1 and NO synthase 2 metabolizing arginine to citrulline and nitric oxide, which also plays an important role in cytotoxicity and vasodilatation in cancer [53, 54]. Supplementation of arginine was able to enhance immune response and inhibit tumor growth in mice [55]. Therefore, the depletion of arginine (Figure 4B) and increased level of citrulline (Supplemental Table S2) with high EtFOSAA exposure was consistent with our previous finding highlighting a clear relationship between perinatal exposure to EtFOSAA and breast cancer risk in the CHDS cohort [14]. Further study that includes assessment of offspring health outcomes is needed to validate the role of this metabolic alteration in linking perinatal EtFOSAA exposure to breast cancer
Perhaps the more direct impact of maternal metabolic alteration is on fetal development via disruption of intrauterine environmental nutrition [56]. Although our study did not assess metabolic profiles or health status in the offspring, our findings provide information on the changes of intrauterine environment that further affects the fetal development. For example, arginine is nutritionally essential for embryonic survival, fetal and neonatal growth [57]. Due to increasing demand by the growing fetus, pregnancy is relatively susceptible to arginine deficiency [57]. Maternal deficiency of arginine causes intrauterine growth restriction and supplementation of arginine has shown benefits in pregnancy outcomes, premature neonate survival and enhanced immune function [57–59]. As growing evidence supports the recent theory of “fetal origins of adult disease” and the link of fetal growth restriction to abnormal mammary gland development is now established [11, 60, 61], our finding calls for more studies to identify the molecular mechanism of nutritional programming of fetal development and its long-term health consequences.
The association of PFOS exposure with and BCAA pathways and carnitine shuttle metabolites was indicative of effects on energy metabolism [44]. It has been shown that PFOA lowered carnitine concentration and increased acyl-carnitine levels while upregulating carnitine shuttle enzymes in human liver cell line [23]. In renal tubular epithelial cells, carnitine was able to alleviate PFOS–induced fibrosis [62]. The elevation of carnitine and the related metabolites could reflect either mitochondrial dysfunction [34, 46–48], or an adaptive change to disturbed lipid metabolism. PFOS is known to activate peroxisome proliferator activated receptor alpha (PPARα), an important transcription factor regulating lipid and carbohydrate metabolism, and cause metabolic alteration in fatty acid and cholesterol [2, 63–66]. Considering the importance of carnitine in fatty acid metabolism, more research is warranted to understand the contribution of carnitine interference with PFOS toxicity.
The opposite effects of PFOS and EtFOSAA on urea cycle and non-essential amino acids are interesting and suggest crosstalk between amino acid and lipid metabolism in response to environmental exposure. Concomitant to the impact of PFOS exposure on fatty acid and carbohydrate metabolism, enzymes in urea cycle including arginosuccinase synthase were also affected by PFOS in primary rat and human hepatocytes [67]. In a parallel analysis of the CHDS cohort, we have found perinatal exposure to EtFOSAA was associated with increase in breast cancer risk in contrast to the negative association with PFOS. We also found that maternal perinatal cholesterol level modified these associations [14]. This evidence supports the theory that lipid regulation may indicate and/or influence development of cancer cells [68]. It highlights that the interplay among different metabolic components is critical in developmental toxicity caused by environmental exposure. Another possible confounding factor is dietary effect on metabolomics as the subjects were not fasted for serum collection, which may decrease the sensitivity of statistical testing. Our past research showed comparable levels of nutritional metabolites in two different cohorts (fasting vs non-fasting) [69]. Others have shown similar reliability of measuring serum sugars and amino acids in fasting and non-fasting samples over time [70], thus we assumed that the effect of this potential confounder was minor.
In conclusion, this study of a pregnancy population shows that HRM is able to inform dysregulation of maternal endogenous metabolism and in utero environment associated with PFOS and EtFOSAA exposure. The results showed differential metabolic effects associated with PFOS and its precursor in urea cycle and amino acid metabolism, which may reflect an interference by lipid metabolism. Whereas our study was limited by the lack of assessment of offspring health outcomes, as the first proof-of-concept study, the metabolic associations represent an important first step in identifying how PFAS alters in utero environment and potentially leads to disease risk. MWAS of the EtFOSAA/PFOS ratio also suggests the metabolic alterations of EtFOSAA can be a reproducible and relatively unique marker discriminating recent active exposure. Future studies are needed on how these changes, in addition to transplacental and lactational PFAS exposure, can affect the metabolome of both the mother and the fetus and predispose both to future disease.
Supplementary Material
Acknowledgement:
This project has been funded in part by the California Breast Cancer Research Program 21UB-8002 (Cohn), and supported by the National Institutes of Health through the HERCULES exposome research center (P30ES019776), S10OD18006 (Jones), U2COD026485 (Jones) and U01OD026489 (Li).
Reference
- [1].Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, De Voogt P, Jensen AA, Kannan K, Mabury SA, van Leeuwen SP, Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins, Integrated environmental assessment and management 7(4) (2011) 513–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].ATSDR, Toxicological Profile for Perfluoroalkyls, (2009). [PubMed]
- [3].Ye X, Kato K, Wong LY, Jia T, Kalathil A, Latremouille J, Calafat AM, Per- and polyfluoroalkyl substances in sera from children 3 to 11 years of age participating in the National Health and Nutrition Examination Survey 2013–2014, International journal of hygiene and environmental health 221(1) (2018) 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Calafat AM, Wong LY, Kuklenyik Z, Reidy JA, Needham LL, Polyfluoroalkyl chemicals in the U.S. population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000, Environ Health Perspect 115(11) (2007) 1596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Trudel D, Horowitz L, Wormuth M, Scheringer M, Cousins IT, Hungerbuhler K, Estimating consumer exposure to PFOS and PFOA, Risk analysis : an official publication of the Society for Risk Analysis 28(2) (2008) 251–69. [DOI] [PubMed] [Google Scholar]
- [6].Paul AG, Jones KC, Sweetman AJ, A First Global Production, Emission, And Environmental Inventory For Perfluorooctane Sulfonate, Environmental Science & Technology 43(2) (2009) 386–392. [DOI] [PubMed] [Google Scholar]
- [7].Lindstrom AB, Strynar MJ, Libelo EL, Polyfluorinated Compounds: Past, Present, and Future, Environmental Science & Technology 45(19) (2011) 7954–7961. [DOI] [PubMed] [Google Scholar]
- [8].Rappazzo KM, Coffman E, Hines EP, Exposure to Perfluorinated Alkyl Substances and Health Outcomes in Children: A Systematic Review of the Epidemiologic Literature, International journal of environmental research and public health 14(7) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Whitworth KW, Haug LS, Baird DD, Becher G, Hoppin JA, Skjaerven R, Thomsen C, Eggesbo M, Travlos G, Wilson R, Longnecker MP, Perfluorinated compounds and subfecundity in pregnant women, Epidemiology (Cambridge, Mass.) 23(2) (2012) 257–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].White SS, Fenton SE, Hines EP, Endocrine disrupting properties of perfluorooctanoic acid, The Journal of steroid biochemistry and molecular biology 127(1–2) (2011) 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Osborne G, Rudel R, Schwarzman M, Evaluating chemical effects on mammary gland development: A critical need in disease prevention, Reproductive Toxicology 54 (2015) 148–155. [DOI] [PubMed] [Google Scholar]
- [12].Bonefeld-Jorgensen EC, Long M, Fredslund SO, Bossi R, Olsen J, Breast cancer risk after exposure to perfluorinated compounds in Danish women: a case-control study nested in the Danish National Birth Cohort, Cancer causes & control : CCC 25(11) (2014) 1439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bonefeld-Jorgensen EC, Long M, Bossi R, Ayotte P, Asmund G, Kruger T, Ghisari M, Mulvad G, Kern P, Nzulumiki P, Dewailly E, Perfluorinated compounds are related to breast cancer risk in Greenlandic Inuit: a case control study, Environmental health : a global access science source 10 (2011) 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Barbara Cohn MLM, Krigbaum, Wang, Park June-Soo, Petreas Myrto, Yeh Greg, Hovey Russell, Johnson Eileen, Lauren, In Utero Exposure to Poly and Perfluoroalkyl Substances (PFASs) and Subsequent Breast Cancer, 2018. [DOI] [PubMed]
- [15].Rodgers KM, Udesky JO, Rudel RA, Brody JG, Environmental chemicals and breast cancer: An updated review of epidemiological literature informed by biological mechanisms, Environmental Research 160 (2018) 152–182. [DOI] [PubMed] [Google Scholar]
- [16].Soto AM, Sonnenschein C, DDT, endocrine disruption and breast cancer, Nature Reviews Endocrinology 11 (2015) 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Laine JE, Bailey KA, Olshan AF, Smeester L, Drobna Z, Styblo M, Douillet C, Garcia-Vargas G, Rubio-Andrade M, Pathmasiri W, McRitchie S, Sumner SJ, Fry RC, Neonatal Metabolomic Profiles Related to Prenatal Arsenic Exposure, Environ Sci Technol 51(1) (2017) 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu J, Liu G, Li Z, Importance of metabolomics analyses of maternal parameters and their influence on fetal growth, Experimental and Therapeutic Medicine 14(1) (2017) 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ciborowski M, Zbucka-Kretowska M, Bomba-Opon D, Wielgos M, Brawura-Biskupski-Samaha R, Pierzynski P, Szmitkowski M, Wolczynski S, Lipinska D, Citko A, Bauer W, Gorska M, Kretowski A, Potential first trimester metabolomic biomarkers of abnormal birth weight in healthy pregnancies, Prenatal diagnosis 34(9) (2014) 870–7. [DOI] [PubMed] [Google Scholar]
- [20].Wang X, Liu L, Zhang W, Zhang J, Du X, Huang Q, Tian M, Shen H, Serum metabolome biomarkers associate low-level environmental perfluorinated compound exposure with oxidative /nitrosative stress in humans, Environmental pollution (Barking, Essex : 1987) 229 (2017) 168–176. [DOI] [PubMed] [Google Scholar]
- [21].Salihovic S, Fall T, Ganna A, Broeckling CD, Prenni JE, Hyötyläinen T, Kärrman A, Lind PM, Ingelsson E, Lind L, Identification of metabolic profiles associated with human exposure to perfluoroalkyl substances, Journal of Exposure Science & Environmental Epidemiology (2018). [DOI] [PubMed] [Google Scholar]
- [22].Yu N, Wei S, Li M, Yang J, Li K, Jin L, Xie Y, Giesy JP, Zhang X, Yu H, Effects of Perfluorooctanoic Acid on Metabolic Profiles in Brain and Liver of Mouse Revealed by a High-throughput Targeted Metabolomics Approach, Scientific Reports 6 (2016) 23963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Peng S, Yan L, Zhang J, Wang Z, Tian M, Shen H, An integrated metabonomics and transcriptomics approach to understanding metabolic pathway disturbance induced by perfluorooctanoic acid, Journal of pharmaceutical and biomedical analysis 86 (2013) 56–64. [DOI] [PubMed] [Google Scholar]
- [24].O’brien TM, Wallace KB, Mitochondrial permeability transition as the critical target of N-acetyl perfluorooctane sulfonamide toxicity in vitro, Toxicological Sciences 82(1) (2004) 333–340. [DOI] [PubMed] [Google Scholar]
- [25].Cohn BA, La Merrill M, Krigbaum NY, Yeh G, Park J-S, Zimmermann L, Cirillo PM, DDT Exposure in Utero and Breast Cancer, The Journal of Clinical Endocrinology & Metabolism 100(8) (2015) 2865–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cohn BA, Wolff MS, Cirillo PM, Sholtz RI, DDT and breast cancer in young women: new data on the significance of age at exposure, Environ Health Perspect 115(10) (2007) 1406–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cohn BA, Cirillo PM, Christianson RE, Prenatal DDT exposure and testicular cancer: a nested case-control study, Archives of environmental & occupational health 65(3) (2010) 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cohn BA, Developmental and environmental origins of breast cancer: DDT as a case study, Reproductive Toxicology 31(3) (2011) 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kezios K, Liu X, Cirillio P, Yu H, Cohn B, Kalantzi O, Wang Y, Petreas M, Park J-S, Factor-Litvak P, DDT DDT Metabolites and Birth Weight, American journal of epidemiology, OXFORD UNIV PRESS INC JOURNALS DEPT, 2001 EVANS RD, CARY, NC 27513 USA, 2011, pp. S11–S11. [Google Scholar]
- [30].van den Berg BJ, Christianson RE, Oechsli FW, The California Child Health and Development Studies of the School of Public Health, University of California at Berkeley, Paediatr Perinat Epidemiol 2(3) (1988) 265–82. [DOI] [PubMed] [Google Scholar]
- [31].Whitehead TP, Crispo Smith S, Park JS, Petreas MX, Rappaport SM, Metayer C, Concentrations of persistent organic pollutants in California women’s serum and residential dust, Environ Res 136 (2015) 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chandler JD, Hu X, Ko E-J, Park S, Lee Y-T, Orr M, Fernandes J, Uppal K, Kang S-M, Jones DP, Go Y-M, Metabolic pathways of lung inflammation revealed by high-resolution metabolomics (HRM) of H1N1 influenza virus infection in mice, American Journal of Physiology - Regulatory, Integrative and Comparative Physiology 311(5) (2016) R906–R916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hu X, Chandler JD, Fernandes J, Orr ML, Hao L, Uppal K, Neujahr DC, Jones DP, Go Y-M, Selenium supplementation prevents metabolic and transcriptomic responses to cadmium in mouse lung, Biochimica et Biophysica Acta (BBA) - General Subjects (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hu X, Chandler JD, Orr ML, Hao L, Liu K, Uppal K, Go Y-M, Jones DP, Selenium Supplementation Alters Hepatic Energy and Fatty Acid Metabolism in Mice, The Journal of Nutrition 148(5) (2018) 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yu T, Park Y, Johnson JM, Jones DP, apLCMS—adaptive processing of high-resolution LC/MS data, Bioinformatics 25(15) (2009) 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Uppal K, Soltow QA, Strobel FH, Pittard WS, Gernert KM, Yu T, Jones DP, xMSanalyzer: automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data, BMC Bioinformatics 14(1) (2013) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Uppal K, Walker DI, Jones DP, xMSannotator: An R Package for Network-Based Annotation of High-Resolution Metabolomics Data, Analytical Chemistry 89(2) (2017) 1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, Fung C, Nikolai L, Lewis M, Coutouly MA, Forsythe I, Tang P, Shrivastava S, Jeroncic K, Stothard P, Amegbey G, Block D, Hau DD, Wagner J, Miniaci J, Clements M, Gebremedhin M, Guo N, Zhang Y, Duggan GE, Macinnis GD, Weljie AM, Dowlatabadi R, Bamforth F, Clive D, Greiner R, Li L, Marrie T, Sykes BD, Vogel HJ, Querengesser L, HMDB: the Human Metabolome Database, Nucleic Acids Res 35(Database issue) (2007) D521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, Hollender J, Identifying small molecules via high resolution mass spectrometry: communicating confidence, Environ Sci Technol 48(4) (2014) 2097–8. [DOI] [PubMed] [Google Scholar]
- [40].Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, Jones DP, Pulendran B, Predicting Network Activity from High Throughput Metabolomics, PLOS Computational Biology 9(7) (2013) e1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gardinassi LG, Xia J, Safo SE, Li S, Bioinformatics Tools for the Interpretation of Metabolomics Data, Current Pharmacology Reports 3(6) (2017) 374–383. [Google Scholar]
- [42].Benjamini Y, Hochberg Y, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing, Journal of the Royal Statistical Society. Series B (Methodological) 57(1) (1995) 289–300. [Google Scholar]
- [43].Manoli I, Venditti CP, Disorders of branched chain amino acid metabolism, Translational science of rare diseases 1(2) (2016) 91–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Paul HS, Adibi SA, Effect of carnitine on branched-chain amino acid oxidation by liver and skeletal muscle, The American journal of physiology 234(5) (1978) E494–9. [DOI] [PubMed] [Google Scholar]
- [45].Kanehisa M, Goto S, KEGG: kyoto encyclopedia of genes and genomes, Nucleic acids research 28(1) (2000) 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Go YM, Fernandes J, Hu X, Uppal K, Jones DP, Mitochondrial network responses in oxidative physiology and disease, Free radical biology & medicine 116 (2018) 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance, Cell metabolism 7(1) (2008) 45–56. [DOI] [PubMed] [Google Scholar]
- [48].Go Y-M, Sutliff RL, Chandler JD, Khalidur R, Kang B-Y, Anania FA, Orr M, Hao L, Fowler BA, Jones DP, Low-dose cadmium causes metabolic and genetic dysregulation associated with fatty liver disease in mice, Toxicological Sciences 147(2) (2015) 524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Keshet R, Szlosarek P, Carracedo A, Erez A, Rewiring urea cycle metabolism in cancer to support anabolism, Nature Reviews Cancer 18(10) (2018) 634–645. [DOI] [PubMed] [Google Scholar]
- [50].Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA, Incidence and distribution of argininosuccinate synthetase deficiency in human cancers, Cancer 100(4) (2004) 826–833. [DOI] [PubMed] [Google Scholar]
- [51].Lind DS, Arginine and Cancer, The Journal of Nutrition 134(10) (2004) 2837S–2841S. [DOI] [PubMed] [Google Scholar]
- [52].Rodríguez PC, Ochoa AC, Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives, Immunological reviews 222 (2008) 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Amber IJ, Hibbs JB Jr., Parker CJ, Johnson BB, Taintor RR, Vavrin Z, Activated macrophage conditioned medium: identification of the soluble factors inducing cytotoxicity and the L-arginine dependent effector mechanism, Journal of leukocyte biology 49(6) (1991) 610–20. [DOI] [PubMed] [Google Scholar]
- [54].Hibbs JB Jr., Taintor RR, Vavrin Z, Macrophage cytotoxicity: role for L-arginine deiminase and imino nitrogen oxidation to nitrite, Science 235(4787) (1987) 473–6. [DOI] [PubMed] [Google Scholar]
- [55].Cao Y, Feng Y, Zhang Y, Zhu X, Jin F, L-Arginine supplementation inhibits the growth of breast cancer by enhancing innate and adaptive immune responses mediated by suppression of MDSCs in vivo, BMC cancer 16 (2016) 343–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Manta-Vogli PD, Schulpis KH, Dotsikas Y, Loukas YL, The significant role of amino acids during pregnancy: nutritional support, The Journal of Maternal-Fetal & Neonatal Medicine (2018) 1–7. [DOI] [PubMed] [Google Scholar]
- [57].Wu G, Bazer FW, Davis TA, Kim SW, Li P, Marc Rhoads J, Carey Satterfield M, Smith SB, Spencer TE, Yin Y, Arginine metabolism and nutrition in growth, health and disease, Amino acids 37(1) (2009) 153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Badurdeen S, Mulongo M, Berkley JA, Arginine depletion increases susceptibility to serious infections in preterm newborns, Pediatric research 77(2) (2015) 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lee Y-C, Su Y-T, Liu T-Y, Tsai C-M, Chang C-H, Yu H-R, L-Arginine and L-Citrulline Supplementation Have Different Programming Effect on Regulatory T-Cells Function of Infantile Rats, Frontiers in Immunology 9(2911) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wu G, Bazer FW, Cudd TA, Meininger CJ, Spencer TE, Maternal Nutrition and Fetal Development, The Journal of Nutrition 134(9) (2004) 2169–2172. [DOI] [PubMed] [Google Scholar]
- [61].Beinder L, Faehrmann N, Wachtveitl R, Winterfeld I, Hartner A, Menendez-Castro C, Rauh M, Ruebner M, Huebner H, Noegel SC, Doerr HG, Rascher W, Fahlbusch FB, Detection of expressional changes induced by intrauterine growth restriction in the developing rat mammary gland via exploratory pathways analysis, PLoS One 9(6) (2014) e100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chou H-C, Wen L-L, Chang C-C, Lin C-Y, Jin L, Juan S-H, From the Cover: l-Carnitine via PPARγ- and Sirt1-Dependent Mechanisms Attenuates Epithelial-Mesenchymal Transition and Renal Fibrosis Caused by Perfluorooctanesulfonate, Toxicological Sciences 160(2) (2017) 217–229. [DOI] [PubMed] [Google Scholar]
- [63].Nelson JW, Hatch EE, Webster TF, Exposure to polyfluoroalkyl chemicals and cholesterol, body weight, and insulin resistance in the general U.S. population, Environ Health Perspect 118(2) (2010) 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Haughom B, Spydevold O, The mechanism underlying the hypolipemic effect of perfluorooctanoic acid (PFOA), perfluorooctane sulphonic acid (PFOSA) and clofibric acid, Biochimica et biophysica acta 1128(1) (1992) 65–72. [DOI] [PubMed] [Google Scholar]
- [65].Shabalina IG, Kramarova TV, Mattsson CL, Petrovic N, Rahman Qazi M, Csikasz RI, Chang S-C, Butenhoff J, DePierre JW, Cannon B, Nedergaard J, The Environmental Pollutants Perfluorooctane Sulfonate and Perfluorooctanoic Acid Upregulate Uncoupling Protein 1 (UCP1) in Brown-Fat Mitochondria Through a UCP1-Dependent Reduction in Food Intake, Toxicological Sciences 146(2) (2015) 334–343. [DOI] [PubMed] [Google Scholar]
- [66].Matilla-Santander N, Valvi D, Lopez-Espinosa M-J, Manzano-Salgado CB, Ballester F, Ibarluzea J, Santa-Marina L, Schettgen T, Guxens M, Sunyer J, Vrijheid M, Exposure to Perfluoroalkyl Substances and Metabolic Outcomes in Pregnant Women: Evidence from the Spanish INMA Birth Cohorts, Environmental Health Perspectives 125(11) (2017) 117004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bjork JA, Butenhoff JL, Wallace KB, Multiplicity of nuclear receptor activation by PFOA and PFOS in primary human and rodent hepatocytes, Toxicology 288(1) (2011) 8–17. [DOI] [PubMed] [Google Scholar]
- [68].McKeown-Eyssen G, Epidemiology of colorectal cancer revisited: are serum triglycerides and/or plasma glucose associated with risk?, Cancer Epidemiology Biomarkers & Prevention 3(8) (1994) 687–695. [PubMed] [Google Scholar]
- [69].Accardi CJ, Walker DI, Uppal K, Quyyumi AA, Rohrbeck P, Pennell KD, Mallon CTM, Jones DP, High-Resolution Metabolomics for Nutrition and Health Assessment of Armed Forces Personnel, Journal of occupational and environmental medicine 58(8) (2016) S80–S88. [DOI] [PubMed] [Google Scholar]
- [70].Carayol M, Licaj I, Achaintre D, Sacerdote C, Vineis P, Key TJ, Onland Moret NC, Scalbert A, Rinaldi S, Ferrari P, Reliability of Serum Metabolites over a Two-Year Period: A Targeted Metabolomic Approach in Fasting and Non-Fasting Samples from EPIC, PloS one 10(8) (2015) e0135437–e0135437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.