

The spatiotemporal dynamics of excitatory neurotransmission must be tightly regulated to achieve efficient synaptic communication. By limiting spillover, glutamate transporters are believed to prevent excessive activation of extrasynaptically located receptors that can impair synaptic plasticity.
Keywords: iGluSnFR, calcium imaging, glutamate transporter, long-term potentiation, synaptic plasticity, VGCCs
Abstract
The spatiotemporal dynamics of excitatory neurotransmission must be tightly regulated to achieve efficient synaptic communication. By limiting spillover, glutamate transporters are believed to prevent excessive activation of extrasynaptically located receptors that can impair synaptic plasticity. While glutamate transporter expression is reduced in numerous neurodegenerative diseases, the contributions of transporter dysfunction to disease pathophysiology remain ambiguous as the fundamental relationship between glutamate dynamics and plasticity, and the mechanisms linking these two phenomena, remain poorly understood. Here, we combined electrophysiology and real-time high-speed imaging of extracellular glutamate transients during LTP induction and characterized the sensitivity of the relationship between glutamate dynamics during theta burst stimulation (TBS) and the resulting magnitude of LTP consolidation, both in control conditions and following selective and nonselective glutamate transporter blockade. Glutamate clearance times were negatively correlated with LTP magnitude following nonselective glutamate transporter inhibition but not following selective blockade of a majority of GLT-1, the brain's most abundant glutamate transporter. Although glutamate transporter inhibition reduced the postsynaptic population response to TBS, calcium responses to TBS were greatly exaggerated. The source of excess calcium was dependent on NMDARs, L-type VGCCs, GluA2-lacking AMPARs, and internal calcium stores. Surprisingly, inhibition of L-type VGCCs, but not GluA2-lacking AMPARs or ryanodine receptors, was required to restore robust LTP. In all, these data provide a detailed understanding of the relationship between glutamate dynamics and plasticity and uncover important mechanisms by which poor glutamate uptake can negatively impact LTP consolidation.
SIGNIFICANCE STATEMENT Specific patterns of neural activity can promote long-term changes in the strength of synaptic connections through a phenomenon known as synaptic plasticity. Synaptic plasticity is well accepted to represent the cellular mechanisms underlying learning and memory, and many forms of plasticity are initiated by the excitatory neurotransmitter glutamate. While essential for rapid cellular communication in the brain, excessive levels of extracellular glutamate can negatively impact brain function. In this study, we demonstrate that pharmacological manipulations that increase the availability of extracellular glutamate during neural activity can have profoundly negative consequences on synaptic plasticity. We identify mechanisms through which excess glutamate can negatively influence synaptic plasticity, and we discuss the relevance of these findings to neurodegenerative diseases and in the aging brain.
Introduction
NMDARs are essential initiators of activity-dependent synaptic plasticity. Recently, emerging evidence suggests a dichotomy in NMDAR function that is at least partially explained by the receptor's subcellular localization; synaptic NMDAR (synNMDAR) activation promotes downstream signaling associated with synapse strengthening and cell survival, whereas extrasynaptic NMDAR (exNMDAR) activation promotes synapse weakening and cell death (Lu et al., 2001; Massey et al., 2004; Hardingham and Bading, 2010; Papouin et al., 2012; Parsons and Raymond, 2014). Intracellular signaling pathways associated with synapse strengthening, in the form of LTP, and synapse weakening, in the form of LTD, represent competing mechanisms that ultimately determine the strength of synaptic connectivity at a given synapse at a given time (Peineau et al., 2007; Shipton and Paulsen, 2014; Volianskis et al., 2015). Therefore, the precise extracellular glutamate dynamics during plasticity-inducing stimuli should govern the subsequent magnitude of synaptic plasticity. Glutamate transporters shape both the spatial and temporal dynamics of synaptically released glutamate (Rothstein et al., 1994; Danbolt, 2001). At the conventional tripartite synapse (Perea et al., 2009), glutamate transporters on astrocytes limit glutamate spillover, presumably facilitating LTP by maximizing the ratio of synNMDAR to exNMDAR activity (Katagiri et al., 2001; Koeglsperger et al., 2013). In addition, neuronal transporters help to reduce the activation of extrasynaptically located receptors by buffering glutamate released during synaptic activity (Scimemi et al., 2009).
Downregulation of glutamate transporters, particularly glutamate transporter-1 (GLT-1) (Danbolt, 2001), is observed in a variety of neurodegenerative diseases and other CNS disorders, including Alzheimer disease, Huntington disease, epilepsy, stroke, and traumatic brain injury (Yi and Hazell, 2006; Faideau et al., 2010; Parsons and Raymond, 2014; Soni et al., 2014; Petr et al., 2015). It has been proposed that the LTP impairment induced by amyloid β oligomers (Townsend et al., 2006; Welsby et al., 2007; Jo et al., 2011; S. Li et al., 2011; Varga et al., 2015; Lei et al., 2016) is mediated by GLT-1 downregulation and subsequent glutamate spillover, causing pathological overactivation of GluN2B-containing exNMDARs (S. Li et al., 2011; Varga et al., 2015) and downstream recruitment of intracellular kinases, such as GSK3 that promote LTD (Peineau et al., 2007; S. Li et al., 2009; Jo et al., 2011). In such a model, glutamate spillover, presumably during LTP induction (Townsend et al., 2006), recruits exNMDARs to promote LTD pathways that oppose LTP (S. Li et al., 2009, 2011; Varga et al., 2015). Considering the number of CNS conditions associated with reduced glutamate transporter expression (Yi and Hazell, 2006; Faideau et al., 2010; Parsons and Raymond, 2014; Soni et al., 2014; Petr et al., 2015) and impaired synaptic plasticity (Townsend et al., 2006; Welsby et al., 2007; Jo et al., 2011; S. Li et al., 2011; Paoletti et al., 2013; Parsons and Raymond, 2014; Varga et al., 2015; Lei et al., 2016), understanding how the spatiotemporal dynamics of endogenous glutamate impacts activity-dependent synaptic plasticity may reveal common mechanisms of disease pathophysiology. Despite the association of synNMDAR and exNMDAR activation with synapse strengthening and weakening, respectively, fundamental knowledge of the precise relationship between glutamate dynamics and plasticity is lacking.
Here, we monitored hippocampal extracellular glutamate dynamics and synaptic strength in real time by combining high-speed imaging of the optogenetic sensor iGluSnFR (intensity-based glutamate-sensing fluorescent reporter) (Marvin et al., 2013) with conventional electrophysiology at the CA3-CA1 synapse. We demonstrate the impact that pharmacological manipulations of glutamate uptake has on glutamate clearance rates, LTP, and associated downstream signaling pathways. Our data demonstrate that LTP is more sensitive to the slowing of glutamate clearance induced by nonselective glutamate transporter blockade compared with that induced by selective inhibition of GLT-1. Moreover, we found that pharmacologically induced glutamate spillover impairs LTP by a mechanism associated with a pathological increase in cytosolic calcium levels through L-type VGCCs (L-VGCCs). In all, our data characterize the relationship between glutamate dynamics and LTP, and suggest a novel mechanism underlying LTP impairment in cases where glutamate uptake is compromised.
Materials and Methods
Animals.
One to three-month old male C57BL/6NCrl mice (Charles River, strain code 027) were used in the present experiments. Mice had ad libitum access to food and water and were housed on a 12 h:12 h light:dark cycle. All procedures followed the guidelines of the Canadian Council on Animal Care and were approved by Memorial University's Institutional Animal Care Committee.
Stereotaxic surgery.
For the duration of the surgical procedure, mice were anesthetized with isoflurane. Viral injections of 1 μl AAV2/1.hSyn.iGluSnFr.WPRE.SV40 (a gift from Loren Looger; Addgene viral prep #98929-AAV1) for glutamate imaging or 1 μl AAV1.Syn.GCaMP6f.WPRE.SV40 (a gift from The Genetically Encoded Neuronal Indicator and Effector Project and Douglas Kim; Addgene viral prep #100837-AAV1) for calcium imaging were administered directly into the hippocampus. Adeno-associated virus was injected using a Neuros 7002 Hamilton syringe coupled to an infusion pump (Pump 11 Elite Nanomite; Harvard Apparatus), which allowed a constant infusion rate of 2 nl/s. The syringe was left in place for an additional 5 min. The following coordinates from bregma were used for hippocampal infusions: 2.6 mm posterior, 2.4 mm lateral (right), and 1.4 to 1.2 mm ventral to the brain surface.
Slice preparation.
Two to 5 weeks after adeno-associated virus injection, mice were anesthetized by isoflurane inhalation, and brains were quickly removed and placed in oxygenated ice-cold slicing solution containing the following (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 MgCl2, 0.5 CaCl2, and 10 glucose. Transverse hippocampal slices (350 μm) were obtained using a Lecia VT1000 vibratome. Slices were transferred to a holding chamber containing oxygenated ACSF, the same as slicing solution, except containing 1 mm MgCl2 and 2 mm CaCl2, for recovery (>60 min) before experimentation.
Real-time imaging of glutamate or calcium biosensors.
Following recovery, slices were transferred to a recording chamber and visualized with an Olympus BX51 microscope. Oxygenated ACSF was continuously perfused at a flow rate of 1.5–2 ml/min and maintained at 25°C (except where noted) with a TC-344C temperature controller (Warner Instruments). Glass pipettes were pulled using a Narishige PB-7 pipette puller to a resistance of 1–3 mΩ and filled with ACSF. The Schaffer collateral pathway was stimulated by controlling an Iso-flex stimulator (A.M.P.I.) with the digital outputs of a Digidata 1550 digitizer (Molecular Devices). iGluSnFR or gCAMP6f responses were evoked by stimulating the Schaffer collateral pathway with either single pulses or theta burst stimulation (TBS). TBS consisted of 10 bursts of 4 pulses at 100 Hz with a 200 ms interburst interval (pulse width = 0.1 ms; stimulation intensity = 75 μA). Electrodes were placed at least 50 μm below the surface of the slice. iGluSnFR or gCAMP6f fluorescence was excited by a LED (Prior Scientific, Lumen 300) passed through a 460–490 nm bandpass filter. Fluorescence emission was captured with an Olympus 4×/0.28 NA objective, filtered through a 510 nm high-pass emission filter, and visualized using an EM-CCD camera (Andor, iXon Ultra 897). Exposure time was 0.0047 s, and 4 × 4 binning was used to achieve a framerate of 205 Hz. Clampex software was used to synchronize the LED, stimulus isolator, and image acquisition. Images were acquired using Andor Solis software.
Image analysis.
Evoked iGluSnFR or GCaMP6f responses were analyzed in ImageJ. For single pulses, a total of 5 stimulus trials (20 s intervals) were averaged together. Between each stimulation trial, a blank trial consisting of LED exposure but no stimulation was acquired. The average of 5 blank trials was subtracted from the average of 5 stimulus trials to account for the mild photobleaching effect observed. Trial averaging and subtraction were performed using the “intrinsic signal processor VSD” plugin for ImageJ. Changes in fluorescence intensity were converted to %ΔF/F. For TBS, a single stimulation and single blank trial were recorded. A 10 × 10 pixel (160 × 160 μm) ROI in the stratum radiatum of CA1 was used to measure the %ΔF/F response. All values were calculated as the average %ΔF/F within the ROI for each frame. Using GraphPad Prism, decay tau was analyzed by fitting a nonlinear regression single-exponential curve to the iGluSnFR or GCaMP6f response transients, beginning at the response's peak.
Electrophysiology.
For electrophysiological recordings of LTP, acute slices were placed in the recording chamber and were left for a minimum of 10 min before electrode placement. fEPSPs were evoked by 0.1 ms single pulses with a glass electrode, generated at a frequency of 0.33 Hz, and the stimulus intensity was adjusted to elicit fEPSPs that were 30%–40% of the maximal response. Field potentials were recorded with a glass electrode in CA1 stratum radiatum, ∼400 μm from the site of stimulation, and signals were amplified and low-pass filtered at 10 kHz with a Multiclamp 700B amplifier (Molecular Devices). LTP was induced with TBS as described above. Recordings continued for 30 or 60 min after LTP induction, as indicated, and LTP magnitude was quantified by averaging the fEPSP slope throughout the last 5 min of recording. Percent potentiation was expressed as the percentage increase in the average fEPSP slope compared with baseline. All data were collected and analyzed using pClamp10 software (Molecular Devices).
Immunofluorescence.
Acute slices (350 μm) were obtained and maintained in ACSF as described above. Slices were transferred to the recording chamber and perfused (1–2 ml/min) with oxygenated ACSF either with or without 15 μm DL-threo-beta-benzyloxyaspartate (DL-TBOA). Once stimulating and recording electrodes were placed in the slice as above, a single pulse of stimulation was used to confirm the presence of a clean fEPSP, indicative of a healthy slice. Then slices were stimulated with TBS as above. Slices were transferred to 4% PFA 10, 20, or 30 min following TBS. Control slices were subject to electrode placements, and a clean fEPSP evoked by a single pulse was confirmed, but no TBS was administered. Slices remained in 4% PFA overnight at 4°C and then transferred to 30% sucrose in 0.01 m PBS at 4°C for at least 5 h. Slices were then flash-frozen using liquid nitrogen and sectioned on a cryostat (Leica Microsystems, CM3050 S) to 16 μm. Slide-mounted resectioned slices were washed three times using 0.01 m PBS for 5 min each and then incubated in blocking solution for 1 h at room temperature. Blocking solution contained 10% goat serum and 0.4% Triton X-100 in 0.01 m PBS. Slides were then incubated in rabbit anti-pERK primary antibody (1:500; Cell Signaling Technology #4370) or mouse anti-ERK (1:500; Cell Signaling Technology, catalog #4696S) overnight at 4°C. The primary antibody was diluted in 0.4% Triton X-100 in 0.01 m PBS. Slides were then washed three times in 0.01 m PBS and incubated in AlexaFluor-594 goat anti-rabbit secondary antibody (1:250; Thermo Fisher Scientific, A-11037) or AlexaFluor-488 goat anti-mouse (1:250, Thermo Fisher Scientific, A11029) for 2 h at room temperature. Slides were again washed three times with PBS and coverslipped with DAKO fluorescent mounting medium containing DAPI (Abcam, ab104139). All slides were then stored at 4°C until image analysis.
Immunofluorescence imaging and analysis.
Acute slices that were resectioned and processed for pERK immunofluorescence were imaged with an Axiovert inverted microscope (Carl Zeiss). To obtain clear images of the entire hippocampus while minimizing LED intensity, a 20×/0.8 NA objective lens was used and a 4 × 4 array of images were tiled and stitched using the Zen Pro software. Images were exported as TIFF files and analyzed in ImageJ. For each acute slice, all resectioned images were examined for evidence of clear placements of the stimulating and recording electrodes. pERK intensity was measured in a 100 μm × 100 μm ROI placed in CA1 stratum radiatum, between the recording and stimulating electrode placements. Mean pERK fluorescence intensity within this ROI was normalized to the background pERK intensity level in an ROI placed in an adjacent, nonstimulated area. Analysis for total ERK staining was conducted using the same approach. This approach was similar to that described previously (Zhu et al., 2015).
Drugs.
dl-TBOA, DHK, d-APV, AIDA, and MDL 28170 were purchased from Tocris Bioscience. Ifenprodil tartrate, SB 415286, dantrolene, nifedipine, and naspm were purchased from Sigma Millipore. (+)-MK 801 maleate was purchased from Thermo Fisher Scientific. For SB 415286 experiments, slices were incubated for 1–2 h before recording. For MK-801 experiments, slices were incubated for 3 h before recording. For AIDA experiments, slices were bath-applied with AIDA for a half hour before LTP induction. For MDL-28170 experiments, MDL 28170 was bath-applied starting 10 min after LTP induction. For all other experiments, drugs were bath-applied for at least 10 min before imaging or LTP induction.
Statistical analysis.
All data are represented as mean ± SEM. All statistical analyses were conducted using GraphPad Prism. Since all data were normally distributed, parametric statistics were used throughout. The statistical tested used include the following: t test, paired t test, one-way ANOVA, two-way ANOVA, repeated-measures (RM) two-way ANOVA, and linear regression. The specific statistical test used is listed within Results. To be considered significant, p values <0.05 were used. Reported n values indicate the number of acute slices used; and for every dataset, a minimum of 3 mice were used per group.
Results
Visualizing real-time extracellular glutamate dynamics in the hippocampus
To help understand the influence glutamate dynamics has on activity-dependent plasticity, we visualized extracellular glutamate in real time using the single-wavelength fluorescent reporter termed iGluSnFR (Marvin et al., 2013). iGluSnFR was virally expressed in the hippocampus under the synapsin promoter (Fig. 1A), allowing us to measure glutamate sensed at the neuronal surface. Endogenous synaptic release was evoked in acute hippocampal slices by electrically stimulating the Schaffer collateral pathway, and the resultant iGluSnFR transients were captured at 205 frames per second with an EM-CCD camera (Fig. 1B). Rapid iGluSnFR transients with a high signal-to-noise ratio were readily observed following brief electrical stimulation consisting of a single pulse (Fig. 1C,D). We then monitored extracellular glutamate dynamics in response to TBS, a commonly used and physiologically relevant (Otto et al., 1991) LTP induction paradigm. iGluSnFR imaging during TBS revealed 10 clear iGluSnFR peaks corresponding to each burst of the TBS. Glutamate was rapidly cleared from the extracellular space in between bursts, and we typically observed a noticeable reduction in the peak %ΔF/F of iGluSnFR responses throughout the TBS protocol (Fig. 1E,F).
Figure 1.
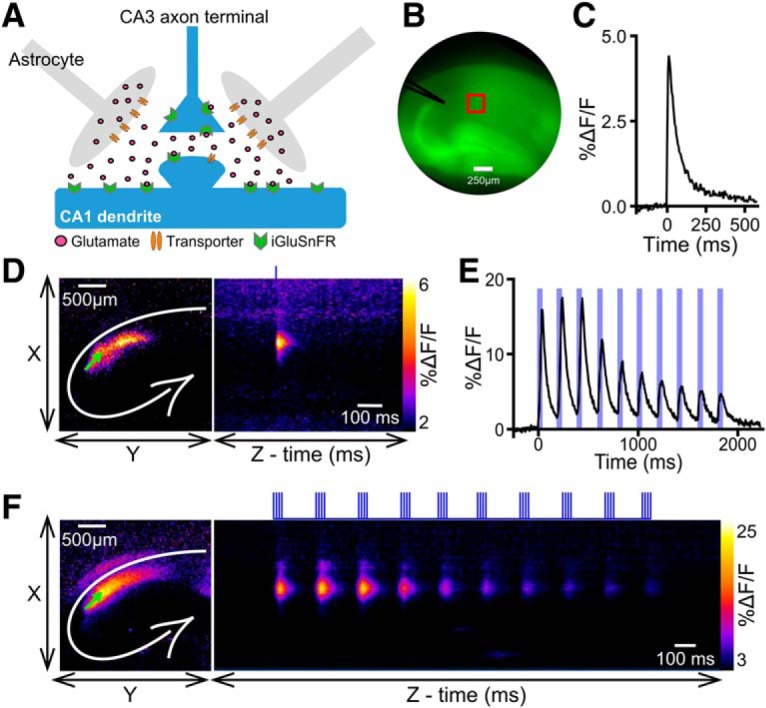
iGluSnFR expression and typical response profile in hippocampus. A, Schematic of a tripartite synapse with iGluSnFR expressed exclusively on neurons. B, Representative image of an acute hippocampal slice expressing iGluSnFR. The approximate locations of the stimulating electrode (black) and the region of interest (red) used to quantify the iGluSnFR response are shown. C, Representative iGluSnFR response to a single pulse of electrical stimulation. D, Heat map represents the maximal iGluSnFR response frame (left) and the iGluSnFR response profile over time (right) in response to a single pulse of stimulation. Left, White lines indicate the approximate locations of the hippocampal cell body layers. Green arrow indicates the stimulation electrode. Right, Vertical blue line indicates the timing of the pulse of electrical stimulation. E, Representative iGluSnFR response to TBS. Ten distinct iGluSnFR response peaks are synchronized with the 10 bursts of stimulation associated with the TBS protocol. Vertical blue bars represent electrical stimulation. F, Heat map showing maximal iGluSnFR frame (left) and response profile over time (right) during TBS. Vertical blue bars above image represent electrical stimulation.
Nonselective glutamate transporter blockade increases extracellular glutamate accumulation and impairs TBS-LTP
We first used TBOA to investigate the effect of nonselective glutamate transporter inhibition on glutamate dynamics during plasticity induction as well as the strength of TBS-LTP. TBOA bath application from 1 to 15 μm resulted in a concentration-dependent effect on both the peak and decay tau of iGluSnFR transients evoked by a single pulse (peak: Fig. 2A,B; n = 6, RM one-way ANOVA, p = 0.002; decay tau: Fig. 2A,C; n = 6, RM one-way ANOVA, p < 0.001). TBOA concentrations >15 μm (i.e., 50 and 100 μm) decreased iGluSnFR peak responses (Fig. 2A,B), consistent with the observation that TBOA concentrations of ≥50 μm can be toxic to hippocampal neurons (Bonde et al., 2003). In many cases, the iGluSnFR response in 50 or 100 μm TBOA degraded to the point where a decay tau could not be reliably measured; thus, decay kinetics were not quantified for these concentrations. The relative ambient (unstimulated) glutamate level, measured by changes in iGluSnFR basal fluorescence from baseline as described previously (Parsons et al., 2016; Pinky et al., 2018), increased steadily from 1 to 100 μm TBOA as expected (Pinky et al., 2018) (Fig. 2D; n = 6, RM one-way ANOVA, p < 0.001). As 15 μm TBOA achieved the maximal response on iGluSnFR decay tau values without degrading the response size, we decided to further explore the effect of 15 μm TBOA on both glutamate dynamics during TBS and the magnitude of TBS-LTP 1 h after induction. We also explored the effect of 5 μm TBOA on the same parameters, representing a moderate concentration of TBOA that still exerted an observable effect on evoked iGluSnFR responses.
Figure 2.
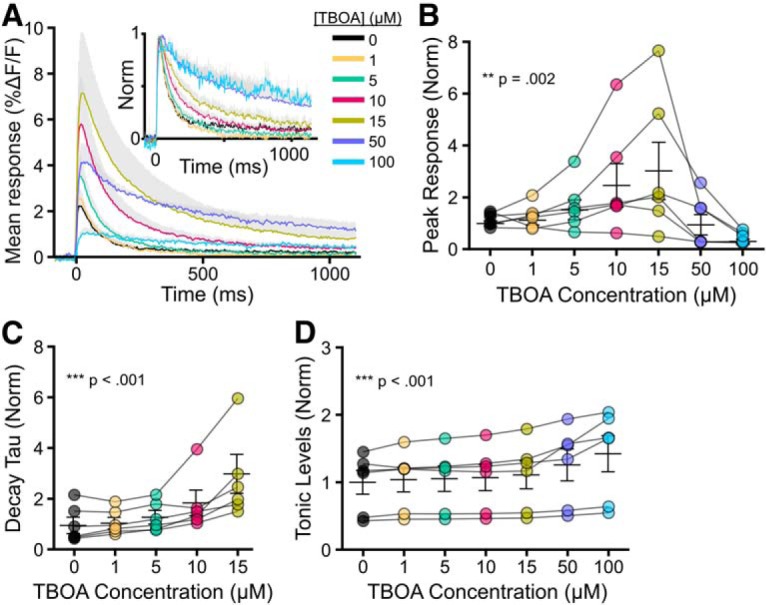
Effect of increasing concentrations of TBOA on evoked iGluSnFR responses. A, Grouped data show mean iGluSnFR responses to a single pulse of stimulation in the presence of 0, 1, 5, 10, 15, 50, and 100 μm TBOA. Gray shading represents SEM. Inset, Responses normalized to peak to help visualize the effect of TBOA on iGluSnFR decay. B–D, Peak (B), decay tau (C), and tonic levels (D) were normalized to the average value in the absence of TBOA. Lines connect paired data from the same slice.
To determine the effect of TBOA on glutamate dynamics during LTP induction (i.e., TBS), we used TBS to evoke iGluSnFR responses in slices bathed in regular ACSF and then, in the same slice, bath-applied either 5 or 15 μm TBOA for 10 min before evoking a second iGluSnFR response with TBS. This experimental design resulted in paired comparisons for each TBOA concentration. As a control, we found that the iGluSnFR response to the second TBS was not significantly different from the response to the first TBS when the slice was continuously bathed in ACSF (n = 5, RM two-way ANOVA, p(μmtreatment) = 0.149). TBOA, applied at 5 μm, resulted in a significant increase in the iGluSnFR peaks and decay taus associated with each burst of the TBS (Fig. 3A–D; peak: n = 6, RM two-way ANOVA, p(treatment) < 0.001, p(burst#) < 0.001, p(interaction) < 0.001; decay tau: n = 6, RM two-way ANOVA, p(treatment) < 0.001, p(burst#) = 0.706, p(interaction) = 0.700). Similar effects but of greater magnitude were observed following application of 15 μm TBOA (Fig. 3E–H; peak: n = 6, RM two-way ANOVA, p(treatment) < 0.001, p(burst#) < 0.001, p(interaction) = 0.001; decay tau: n = 6, RM two-way ANOVA, p(treatment) < 0.001, p(burst#) < 0.001, p(interaction) = 0.003). The peak ratio, calculated by dividing the peak iGluSnFR response after TBOA to the peak iGluSnFR response before TBOA for each burst, was larger for 15 μm TBOA than for 5 μm TBOA (Fig. 3I; n = 6 per concentration, RM two-way ANOVA, p(treatment) < 0.001, p(burst#) = 0.005, p(interaction) = 0.996). The decay tau ratio was also higher for 15 μm TBOA compared with 5 μm TBOA (Fig. 3J; n = 6 per concentration, RM two-way ANOVA, p(treatment) = 0.001, p(burst#) = 0.102, p(interaction) = 0.206). These data indicate that TBOA induces a concentration-dependent slowing of glutamate clearance and increase in extracellular accumulation during a standard LTP-inducing TBS protocol.
Figure 3.

Nonselective glutamate transporter inhibition slows glutamate clearance and impairs LTP. A, Mean (±SEM in gray) iGluSnFR response to TBS before (black) and after (orange) 5 μm TBOA application. B, Same data as shown in A, but normalized to the first peak to help visualize the effect of TBOA on iGluSnFR decay following each burst. C, D, Grouped data show the effect of 5 μm TBOA on iGluSnFR peak (C) and decay tau (D) values for each burst associated with TBS. E, Mean (±SEM) iGluSnFR response to TBS before (black) and after (blue) 15 μm TBOA application. F, Same data as shown in E, but normalized to the first peak to help visualize the effect of TBOA on the clearance rate following each burst. G, H, Grouped data show the effect of 15 μm TBOA on iGluSnFR peak (G) and decay tau (H) values for each burst associated with TBS. I, J, Peak (I) and decay (J) ratios for 5 and 15 μm TBOA. K–M, Representative traces (K), fEPSP slope (L), and fEPSP half-width (M) before and after 15 μm TBOA application. N, LTP in control (CTL), 5 μm TBOA, and 15 μm TBOA-treated slices. LTP is induced by TBS at time = 0. Representative traces show fEPSPs before (gray traces) and 55–60 min after TBS for each condition. O, Percent potentiation to analyze the effect of 5 and 15 μm TBOA on LTP. P, Percent potentiation plotted against the fold increase in the iGluSnFR decay tau during TBS for control and TBOA (5 and 15 μm) experiments at 25 and 32°C. Error bars indicate SEM. A, B, E, F, Gray shading represents SEM. *p < 0.05.
We then used conventional electrophysiology to quantify TBS-LTP in slices bathed in control ACSF or ACSF containing 5 or 15 μm TBOA. fEPSPs were recorded in CA1 stratum radiatum, and LTP was induced by TBS applied to the Shaffer collaterals. TBOA was bath-applied for a minimum of 10 min before TBS and remained in the bath for the duration of the recording. During baseline recordings, TBOA had no significant effect on fEPSP slope in response to single pulses (Fig. 3K,L; paired t test, n = 8, p = 0.987), and resulted in a nonsignificant trend to increase fEPSP half-width observed (Fig. 3K,M; paired t test, n = 8, p = 0.077). TBOA application resulted in a concentration-dependent impairment of TBS-LTP (Fig. 3N,O; control n = 10, 5 μm TBOA n = 6, 15 μm TBOA n = 6, one-way ANOVA, p = 0.028). The mean % LTP was not significantly reduced in 5 μm TBOA (Dunnett test vs control, p = 0.361), and post hoc significance was only observed for 15 μm TBOA (Dunnett test vs control, p = 0.016). As glutamate uptake as well as glutamate transporter surface diffusion are highly temperature-dependent (Wadiche and Kavanaugh, 1998; Murphy-Royal et al., 2015), we performed a subset of experiments at the near-physiological temperature of 32°C. When we plotted LTP strength against the magnitude of TBOA-induced glutamate slowing during TBS, including the experiments performed at 32°C (n = 6 for CTL iGluSnFR, n = 8 for CTL LTP, n = 6 for TBOA iGluSnFR, n = 9 for TBOA LTP), we found a clear relationship between LTP and glutamate dynamics (Fig. 3P); slower glutamate clearance rates significantly correlated with poor LTP consolidation (r = −0.483, p = 0.002). Together, these data indicate that nonselective inhibition of glutamate transporters promotes extracellular glutamate accumulation during TBS and results in a significant LTP impairment.
Selective blockade of GLT-1 slows glutamate clearance but does not impair LTP
At the 5 and 15 μm concentrations used, TBOA would be expected to partially block GLT-1, the neuronal EAAT3, and, to a lesser extent, GLAST (IC50 for GLT-1, EAAT3, and GLAST is 6, 6, and 70 μm, respectively) (Shimamoto et al., 1998). Next, we asked whether a similar relationship between glutamate dynamics and synaptic plasticity could be observed following the selective blockade of GLT-1, the brain's most abundant glutamate transporter (Danbolt, 2001). The selective GLT-1 blocker DHK resulted in a concentration-dependent increase in the iGluSnFR peak and decay tau in response to a single pulse (Fig. 4A–C; peak: n = 6, RM one-way ANOVA, p < 0.001; decay tau: n = 6, RM one-way ANOVA, p < 0.001). Similar to our observations with TBOA, the concentration-dependent increase in iGluSnFR peak began to reverse at higher concentrations; in the case of DHK, concentrations >100 μm had a negative impact on the peak response (Fig. 4A,B). Unlike TBOA, and in agreement with a previous study from our laboratory (Pinky et al., 2018), DHK had no significant effect on tonic glutamate levels, as measured by the relative changes in the fluorescence intensity of basal (unstimulated) iGluSnFR (Fig. 4D; n = 6, RM ANOVA, p = 1.00). From these concentration-response experiments, we chose to further explore the effects of 50 and 100 μm DHK on glutamate dynamics during TBS and the magnitude of TBS-LTP.
Figure 4.
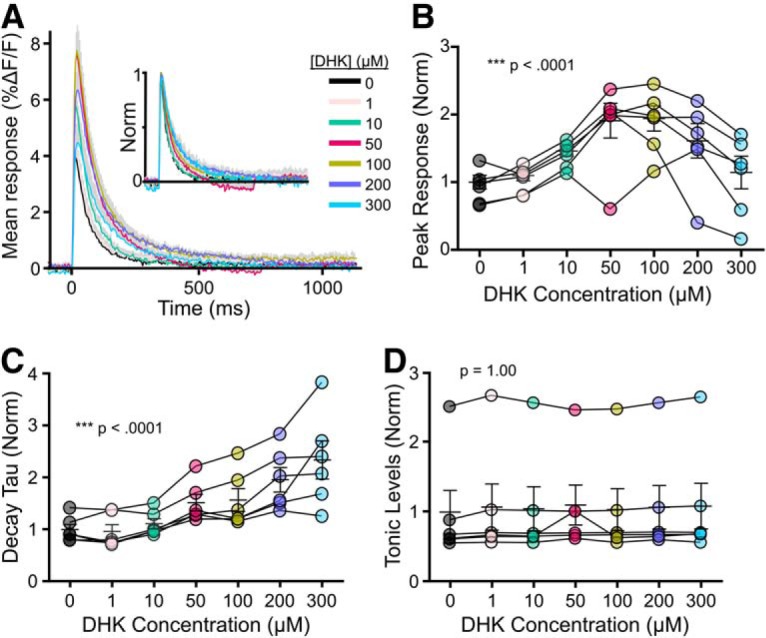
Effect of increasing concentrations of DHK on evoked iGluSnFR responses. A, Grouped data show mean iGluSnFR responses to a single pulse of stimulation (at time 0) in the presence of 0, 1, 10, 50, 100, 200, and 300 μm DHK. Gray shading represents SEM. Inset, Responses normalized to peak to help visualize the effect of DHK on iGluSnFR decay. B–D, Peak (B), decay tau (C), and tonic levels (D) were normalized to the average value in the absence of DHK. Lines connect paired data from the same slice.
When bath-applied at 50 μm, DHK resulted in a significant increase in the iGluSnFR peaks and decay taus associated with each burst of the TBS (Fig. 5A–D; peak: n = 6, RM two-way ANOVA, p(treatment) < 0.001, p(burst#) < 0.001, p(interaction) = 0.010; decay tau: n = 6, RM two-way ANOVA, p(treatment) < 0.001, p(burst#) = 0.002, p(interaction) = 0.170). Similar effects were observed following application of 100 μm DHK (Fig. 5E–H; peak response: n = 6, RM two-way ANOVA, p(treatment) < 0.001, p(burst#) = 0.044, p(interaction) = 0.063; decay tau: n = 6, RM two-way ANOVA, p(treatment) < 0.001, p(burst#) = 0.059, p(interaction) = 0.700). The peak ratio was not significantly different for 50 and 100 μm DHK (Fig. 5I; n = 6 per concentration, RM two-way ANOVA, p(treatment) = 0.720, p(burst#) = 0.130, p(interaction) = 1.00), although the decay tau ratio was significantly higher following 100 μm DHK compared with 50 μm DHK (Fig. 5J; n = 6 per concentration, RM two-way ANOVA, p(treatment) < 0.001, p(burst#) = 0.690, p(interaction) = 0.140). Thus, like TBOA, selective blockade of GLT-1 results slows glutamate clearance and enhances the amount of extracellular glutamate accumulation during a standard TBS protocol. Surprisingly, and in contrast to our TBOA results, we found that these concentrations of DHK did not affect the magnitude of TBS-LTP (Fig. 5K,L; control n = 10, 50 μm DHK n = 10, 100 μm DHK n = 6, one-way ANOVA, p = 0.796). When we plotted LTP strength against the magnitude of DHK-induced glutamate slowing, including a subset of experiments performed at near-physiological temperatures (32°C; n = 6 for CTL iGluSnFR, n = 8 for CTL LTP, n = 6 for DHK iGluSnFR, n = 6 for DHK LTP), we found no relationship between LTP and glutamate dynamics (Fig. 5M); slower glutamate clearance rates following selective GLT-1 inhibition were without effect on LTP consolidation (r = 0.054, p = 0.752). The only way we were able to observe significant LTP inhibition following selective GLT-1 blockade was when we further enhanced extracellular glutamate by combining 100 μm DHK with a TBS stimulus intensity 1.5-fold larger than the stimulus intensity used to evoke single fEPSPs (Fig. 5N; n = 5, p = 0.018, t test). Thus, LTP is much more sensitive to changes in glutamate dynamics induced by nonselective glutamate transporter blockade compared with selective GLT-1 blockade.
Figure 5.

Selective GLT-1 inhibition slows glutamate clearance but does not impair LTP. A, Mean (±SEM) iGluSnFR response to TBS before (black) and after (light green) 50 μm DHK application. B, Same data as shown in A, but normalized to the first peak. C, D, Grouped data show the effect of 50 μm DHK on iGluSnFR peak (C) and decay tau (D) values for each burst associated with TBS. E, Mean (±SEM) iGluSnFR response to TBS before (black) and after (dark green) 100 μm DHK application. F, Same data as shown in E, but normalized to the first peak. G, H, Grouped data show the effect of 100 μm DHK on iGluSnFR peak (G) and decay tau (H) values for each burst associated with TBS. I, J, Peak (I) and decay (J) ratios for 50 and 100 μm DHK. K, LTP in control, 50 μm DHK-treated, and 100 μm DHK-treated slices. LTP is induced by TBS at time = 0. Representative traces show fEPSPs before (gray traces) and 55–60 min after TBS for each condition. L, Percent potentiation to analyze the effect of 50 and 100 μm DHK on LTP. M, Percent potentiation plotted against the fold increase in the iGluSnFR decay tau during TBS for control and DHK (50 and 100 μm) experiments at 25°C and 32°C. N, Percent potentiation following a combination of DHK (100 μm) and a 1.5× stimulation intensity used during LTP induction. Error bars indicate SEM. A, B, E, F, Gray shading represents SEM. *p < 0.05.
Variations in glutamate dynamics do not correlate with LTP magnitude in control conditions
As our transporter blocker experiments above suggest that considerable transporter dysfunction is required to exert a negative impact on TBS-LTP, we reasoned that the slice-to-slice variability in glutamate dynamics observed in control conditions would be too insignificant to have any influence over LTP magnitude. To determine whether the natural (i.e., control) variation in glutamate clearance rates correlates with LTP magnitude, we combined iGluSnFR imaging with conventional electrophysiological recordings in the same experiments. First, a stable baseline was established by the fEPSP slope. The iGluSnFR response to a single pulse was then captured before LTP induction, and we also captured the iGluSnFR response during LTP induction with TBS. fEPSPs were then recorded every 20 s for 60 min after induction in the same slices (Fig. 6A,B). If synaptic plasticity exhibits a high sensitivity to glutamate clearance kinetics during TBS, we should observe the weakest LTP in slices that cleared glutamate the slowest during LTP induction. Importantly, iGluSnFR expression itself had no impact whatsoever on the magnitude of LTP observed (Fig. 6B, inset; n = 10 without iGluSnFR, n = 15 with iGluSnFR, t test, p = 0.996). In response to a single pulse, iGluSnFR decay tau varied from 30 to 75 ms; however, there was no significant correlation between iGluSnFR decay tau and LTP magnitude (Fig. 6C; n = 15, p = 0.870). Similarly, LTP magnitude did not correlate with glutamate dynamics during TBS under control conditions, as quantified by either the decay tau after the first burst (Fig. 6D; n = 15, p = 0.630) or the area under the curve (AUC) of the iGluSnFR response to the full TBS (Fig. 6E; n = 15, p = 0.099). This area measurement reflects the relative amount of extracellular glutamate accumulation throughout the entire TBS; and while slices with larger iGluSnFR areas tended to have weaker LTP, this trend did not reach statistical significance. These experiments indicate that LTP strength is not significantly affected by the 2.5-fold variation in iGluSnFR decay tau we observed in control conditions. Together with our findings with TBOA and DHK, our data indicate that considerable glutamate slowing induced by nonselective transporter inhibition is required before a detrimental effect on TBS-LTP can be observed.
Figure 6.
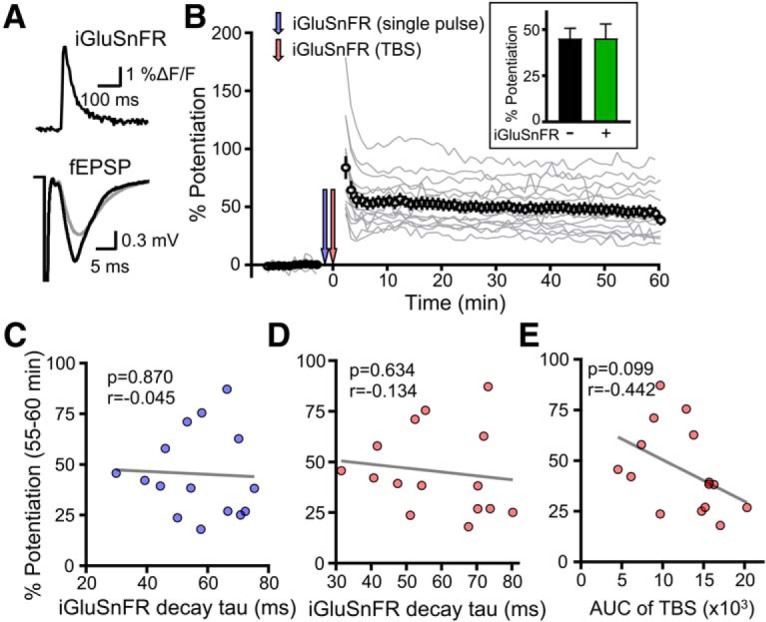
Lack of relationship between glutamate dynamics and LTP in control conditions. A, Top, Representative iGluSnFR response to a single pulse of electrical stimulation. Bottom, fEPSP responses before (gray) and 1 h after TBS (black). B, Experimental design showing the combination of LTP and iGluSnFR. Gray traces represent the electrophysiological responses for individual experiments. Inset, Bar graph represents percent potentiation in slices with (+) and without (−) iGluSnFR expression. C, Plot of the iGluSnFR decay tau to a single pulse and the resultant magnitude of LTP for each experiment. D, Plot of the iGluSnFR decay tau to the first burst of TBS and the resultant magnitude of LTP for each experiment. E, Plot of the iGluSnFR AUC to TBS and the resultant magnitude of LTP for each experiment.
Glutamate transporter inhibition does not impair LTP through excessive activation of NMDARs or recruitment of common LTD pathways.
Next, we sought to determine the precise mechanism by which glutamate transporter dysfunction impairs LTP. By altering the spatiotemporal dynamics of extracellular glutamate during LTP-inducing stimuli, it is possible that TBOA promotes the simultaneous activation of intracellular signaling associated with LTP and LTD that oppose each other. To determine whether the excessive glutamate accumulation during TBOA-TBS activated an NMDAR-independent LTD pathway to oppose LTP consolidation, we blocked NMDARs with the competitive antagonist d-APV (50–100 μm). In the presence of d-APV, TBS-LTP was significantly impaired as expected (Larson and Lynch, 1988). However, LTD was not revealed in the presence of both d-APV and TBOA, suggesting that TBOA is unlikely to recruit NMDAR-independent LTD during TBS (Fig. 7A,B; control n = 10, d-APV n = 6, d-APV+TBOA n = 6; one-way ANOVA, p = 0.005; post hoc Tukey tests: control vs d-APV p = 0.021, control vs d-APV+TBOA p = 0.010, d-APV vs d-APV+TBOA p = 0.947). In agreement, the TBOA-induced LTP impairment was not due to the activation of Group 1 mGluR-mediated LTD (Lüscher and Huber, 2010), as the mGluR1 antagonist AIDA (200 μm) was unable to restore TBOA-LTP to control levels (Fig. 7C,D; control n = 10, AIDA n = 7, AIDA+TBOA n = 6; one-way ANOVA, p = 0.009; post hoc Tukey tests: control vs AIDA p = 0.992, control vs AIDA+TBOA p = 0.011, AIDA vs AIDA+TBOA p = 0.023). Paired-pulse ratio experiments indicate that TBOA-TBS did not result in a lasting decrease in release probability that could account for the LTP deficit (Fig. 7E,F; n = 7; paired t test; p = 0.169). In addition, LTP was not impaired when TBOA was washed from the slice 30 min before LTP induction, suggesting that TBOA during the single test pulses does not cause an inhibitory metaplasticity that impairs subsequent LTP (CTL % LTP = 52.96 ± 7.18, n = 10; TBOA wash % LTP = 43.03 ± 9.76, n = 5, t test, p = 0.434). Similar results to our d-APV experiments were obtained with the NMDAR pore-blocker MK-801 (100 μm; Fig. 7G,H; control n = 10, MK-801 n = 6, MK-801+TBOA n = 6, one-way ANOVA, p < 0.001; post hoc Tukey tests: control vs MK-801 p < 0.001, control vs MK-801+TBOA p < 0.001, MK-801 vs MK-801+TBOA p = 0.765). As MK-801 preserves the glutamate binding domain of NMDARs, this result also indicates that the TBOA-LTP impairment does not result from the recruitment of metabotropic NMDAR LTD (Nabavi et al., 2013). Likewise, inhibition of GSK-3, a kinase known to play a critical role in NMDAR-dependent LTD (Peineau et al., 2007; Jo et al., 2011; Bradley et al., 2012), was also unable to restore TBOA-LTP to control levels (Fig. 7I,J; control n = 10, SB n = 7, SB+TBOA n = 7; one-way ANOVA p < 0.001; post hoc Tukey tests: control vs SB p = 0.990, control vs SB+TBOA p < 0.001, SB vs SB+TBOA p < 0.001). Indeed, while SB415286 (10 μm) had no effect on LTP on its own, TBS induced a clear LTD when GSK-3 was inhibited in the presence of TBOA. Excessive activation of NMDARs can have a detrimental effect on LTP (Katagiri et al., 2001). However, to our surprise, we were unable to restore TBOA-LTP to control levels by using subsaturating concentrations (1–10 μm) of d-APV. No differences were observed between d-APV concentrations within this range, and the data were pooled (Fig. 7K,L; control n = 10, d-APV(1–10 μm) n = 14, d-APV(1–10 μm)+TBOA n = 23; one-way ANOVA, p < 0.001; post hoc Tukey tests; control vs d-APV(1–10 μm) p = 0.220, control vs d-APV(1–10 μm)+TBOA p < 0.001, d-APV(1–10 μm) vs d-APV(1–10 μm)+TBOA p = 0.043). Ifenprodil (3–10 μm) was also unable to restore TBOA-LTP to control levels (Fig. 7M,N; ifenprodil n = 10; ifenprodil + TBOA n = 12, one-way ANOVA, p < 0.001; post hoc Tukey tests; control vs ifenprodil p = 0.783, control vs ifenprodil+TBOA p < 0.001, ifenprodil vs ifenprodil+TBOA p = 0.002), suggesting that excessive activation of GluN2B-containing NMDARs cannot account for the LTP impairment observed in the present study.
Figure 7.

TBOA-induced LTP impairment is not mediated by NMDAR overactivation or recruitment of common LTD pathways. A, B, LTP in control conditions and in the presence of a saturating concentration of the NMDAR antagonist d-APV (50–100 μm) alone and in combination with 15 μm TBOA. Percent potentiation shown in B. C, D, LTP in control conditions and in the presence of the Group I mGluR antagonist AIDA (200 μm) alone and in combination with 15 μm TBOA. Percent potentiation shown in D. E, Paired pulse ratio (normalized to baseline) before and after TBS in the presence of 15 μm TBOA. F, Paired pulse ratio before and 30 min after TBS in the presence of 15 μm TBOA. G, H, LTP in control conditions and in the presence of the use-dependent NMDAR pore blocker MK-801 (100 μm) alone and in combination with 15 μm TBOA. Percent potentiation shown in H. I, J, LTP in control conditions and in the presence of the GSK inhibitor SB 412586 (10 μm) alone and in combination with 15 μm TBOA. Percent potentiation shown in J. K, L, LTP in control conditions and in the presence of subsaturating concentrations of d-APV (1–10 μm) alone and in combination with 15 μm TBOA. Percent potentiation shown in L. M, N, LTP in control conditions and in the presence of the GluN2B-containing NMDAR antagonist ifenprodil (3–10 μm) alone and in combination with 15 μm TBOA. Percent potentiation shown in N. Error bars indicate SEM. *p < 0.05; **p < 0.01; ***p < 0.0001.
These results were surprising given the previously reported links between glutamate spillover, exNMDAR overactivation, and synapse weakening (Katagiri et al., 2001; S. Li et al., 2009, 2011; Scimemi et al., 2009; Varga et al., 2015). Therefore, to further probe the possible involvement of excessive NMDAR activity in the TBOA-LTP impairment, we fixed acute slices 10 min following TBS, resectioned them to 16 μm on a cryostat, and quantified pERK immunofluorescence levels in CA1 stratum radiatum. pERK is a kinase essential for TBS-LTP (Zhu et al., 2015) but is inactivated by high levels of exNMDAR (Ivanov et al., 2006) or GluN2B-containing NMDAR (Kim et al., 2005) stimulation. Thus, if exNMDAR and/or GluN2B-containing NMDAR overactivation mediates the TBOA-LTP impairment, we would expect to see a reduction in the TBS-induced recruitment of pERK in slices treated with TBOA. The mean pERK fluorescence intensity was transiently elevated by TBS as previously reported (Wang et al., 2014); however, pERK levels increased, even higher following TBS in TBOA-treated slices, with significant post hoc differences identified both 10 and 30 min after TBS (Fig. 8A–C; n = 4–6 slices per time per condition; two-way ANOVA, p(treatment) = 0.005, p(time) < 0.001, p(interaction) = 0.106; post hoc Bonferroni tests comparing TBS vs TBS+TBOA; 0 min p > 0.05, 10 min p = 0.023, 20 min p > 0.05, 30 min p = 0.040). We also stained separate slices with an antibody against total ERK. We found that total ERK levels were unchanged by TBS or by TBOA-TBS (Fig. 8C; n = 4–6 slices per condition, two-way ANOVA, p(treatment) = 0.724, p(time) = 0.747, p(interaction) = 0.273). Therefore, TBS and TBOA-TBS increased ERK phosphorylation rather than total ERK expression levels. This pERK hyperactivation argues against the overactivation of exNMDARs or GluN2B-containing NMDARs in the present conditions.
Figure 8.

Glutamate transporter inhibition promotes pERK hyperactivity and reduces the postsynaptic population response to TBS. A, Representative images of pERK (red) and DAPI (pseudocolored green for clarity) in slices without TBS, with TBS, and with TBS in the presence of 15 μm TBOA. B, Representative image shows dendritic pERK staining in the stratum radiatum. C, Grouped data show pERK (left) and total ERK (right) intensity, normalized to background fluorescence in a nonstimulated region of the slice. D, Representative fEPSP responses to TBS in control (CTL) conditions and in the presence of 15 μm TBOA. E, Grouped data show the AUC of the fEPSP response to each burst during TBS. F, Representative fEPSP responses to TBS in the presence of d-APV (50–100 μm) alone and in combination with 15 μm TBOA. G, Grouped data show the AUC of the fEPSP response to each burst during TBS. Error bars indicate SEM. *p < 0.05.
As extracellular glutamate accumulation during TBS is enhanced in the presence of TBOA, we next asked whether the postsynaptic population response to TBS was also enhanced in the presence of TBOA. We calculated the AUC of the fEPSP response to each of the 10 bursts associated with the TBS train. Compared with control slices, TBOA resulted in a nonsignificant increase in the mean fEPSP area to the first burst, but this was followed by a significant reduction in fEPSP area to subsequent bursts (Fig. 8D,E; control n = 14, TBOA n = 9; RM two-way ANOVA, p(treatment) = 0.008, p(burst#) < 0.001, p(interaction) < 0.001; post hoc Bonferroni tests for control vs TBOA: burst #2 p = 0.017, burst #3 p = 0.001, burst #4 p = 0.005, burst #5 p = 0.026, all other bursts p > 0.05). With the exception of the first burst, the dampening effect of TBOA on the postsynaptic response to TBS was both mimicked and occluded by pretreatment with 50–100 μm d-APV (Fig. 8F,G; d-APV n = 6, d-APV+TBOA n = 6, RM two-way ANOVA, p(treatment) = 0.530, p(burst#) < 0.001, p(interaction) = 0.561; post hoc Bonferroni tests for d-APV vs d-APV+TBOA: all bursts p > 0.05). These data indicate that, despite the increase in extracellular glutamate, TBOA reduces, rather than enhances, the overall population response to TBS. Thus, it is unlikely that glutamate receptors are overactivated throughout the course of the TBS paradigm when glutamate uptake is inhibited with TBOA.
Glutamate transporter inhibition impairs LTP by excess calcium influx through L-type VGCCs
Calcium influx through NMDARs is required for TBS-LTP at the CA3-CA1 synapse. Considering our finding that the fEPSP response to TBS is reduced by glutamate transporter inhibition, we next quantified the intracellular calcium response to TBS in control conditions and in the presence of 15 μm TBOA. These experiments were paired, in that each slice was initially subjected to TBS in ACSF, followed by a 10 min bath application of TBOA (15 μm), and a second TBS was evoked. To quantify the effect of TBOA, the AUC of the GCaMP6f response to the second TBS was divided by that to the first TBS; these data are expressed in Figure 9 as “GCaMP6f AUC (TBS2/TBS1).” As we reasoned that the calcium influx associated with the second TBS may be influenced by the synaptic changes induced by the first TBS, these data were compared with control slices that also received two TBS trains separated by 10 min, but ACSF alone (i.e., no TBOA) was bathed continuously throughout the experiment. Indeed, in control slices, the AUC of the GCaMP6f response the second TBS was 1.35 (± 0.16)-fold greater than the response to the first TBS. However, in TBOA-treated slices, the fold GCaMP6f response increased to 4.51 (± 0.97), a value significantly higher than control slices. The TBOA-induced increase in the calcium response to TBS was not prevented by ifenprodil (10 μm) but was completely prevented by d-APV (50–100 μm; Fig. 9A–C; n = 6 or 7 per condition; two-way ANOVA; p(TBOA) < 0.001; p(antagonists) = 0.001; p(interaction) = 0.016, post hoc Bonferroni tests for ACSF vs TBOA p = 0.034, with ifenprodil p < 0.001, with d-APV p = 0.999). These data suggest that the excess calcium response during TBOA-TBS is at least initially dependent on NMDAR activation, but not on ifenprodil-sensitive (i.e., GluN1/GluN2B diheteromeric) NMDARs.
Figure 9.
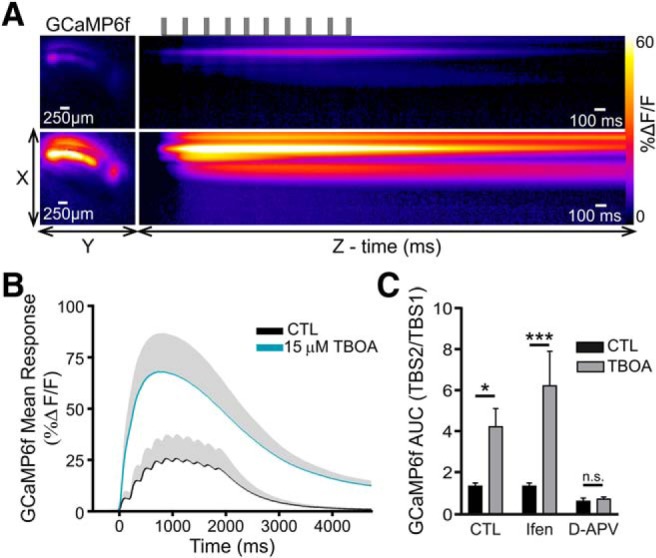
Glutamate transporter inhibition enhances the calcium response to TBS. A, Heat map represents the maximal GCaMP6f response frame (left) and the GCaMP6f response profile over time (right) in response to TBS, both in control conditions (top images) and in the presence of 15 μm TBOA (bottom images). Top, Black horizontal lines indicate the timing of the TBS protocol. B, Grouped data show the mean GCaMP6f response to TBS before (black) and after 15 μm TBOA application (blue). Gray shading represents SEM. C, Grouped data show the fold increase in the GCaMP6f AUC in response to the second TBS (TBS2) compared with the first (TBS1). For all experiments, the first TBS was applied in control conditions and the second TBS was applied in the presence of 15 μm TBOA. Experiments were performed both in the absence and the presence of either ifenprodil or d-APV as indicated. *p < 0.05; ***p < 0.001. n.s., non significant.
In the presence of TBOA, the NMDAR-mediated component of the fEPSP rapidly diminishes by the second burst of TBS (Fig. 8D–G). Thus, we reasoned that an additional source of calcium may contribute the dramatic effect of TBOA on the calcium response to TBS. We used dantrolene (20 μm), naspm (3 μm), and nifedipine (20 μm) to assess the putative contribution of intracellular calcium stores, GluA2-lacking AMPARs, and L-VGCCs to the excessive cytosolic calcium levels obtained during TBOA-TBS, respectively. Interestingly, all compounds were able to decrease the mean effect of TBOA on the total GCaMP6f area during TBS, suggesting the contribution of multiple calcium sources to TBOA-TBS (Fig. 10A–E; n = 6 per group, one-way ANOVA, p = 0.012, post hoc Dunnett tests vs TBOA alone: dantrolene+TBOA p = 0.013, naspm+TBOA p = 0.047, nifedipine+TBOA p = 0.009). Although DHK (100 μm) did not impair LTP (Fig. 5), we found that, like TBOA, DHK resulted in a robust increase in the calcium response to TBS (Fig. 10F). The DHK-induced increase in the calcium response to TBS was significantly reduced by dantrolene, naspm, and nifedipine (Fig. 10F; n = 5 or 6 per group, one-way ANOVA, p < 0.001, post hoc Dunnett tests vs DHK alone: dantrolene+DHK p < 0.001, naspm+DHK p < 0.001, nifedipine+DHK p < 0.001). We then assessed whether either of these compounds alone could prevent the inhibition of LTP induced by TBOA. Intriguingly, dantrolene had no effect on LTP in the absence of TBOA but resulted in robust LTD when coapplied with TBOA (Fig. 10G,H; control n = 10, dantrolene n = 6, dantrolene+TBOA n = 6; one-way ANOVA, p < 0.001; post hoc Tukey tests: control vs dantrolene p = 0.988, control vs dantrolene+TBOA p < 0.001, dantrolene vs dantrolene+TBOA p < 0.001). Naspm also had no effect on LTP when applied on its own but was unable to prevent the inhibitory effect of TBOA on LTP consolidation (Fig. 10I,J; control n = 10, naspm n = 4, naspm+TBOA n = 7; one-way ANOVA, p = 0.033, post hoc Tukey tests: control vs naspm p = 0.938, control vs naspm+TBOA p = 0.048, naspm vs naspm+TBOA p = 0.075). In contrast, nifedipine, which resulted in a nonsignificant increase in the magnitude of LTP when applied alone, completely prevented the degradative effect of TBOA on LTP (Fig. 10K,L; control n = 10, nifedipine n = 6, nifedipine+TBOA n = 8, one-way ANOVA, p = 0.166; post hoc Tukey tests: control vs nifedipine p = 0.219, control vs nifedipine+TBOA p = 0.271, nifedipine vs nifedipine+TBOA p = 0.965). Thus, while multiple calcium sources contribute to the calcium overload during TBOA-TBS, calcium entry through L-VGCCs is required for the plasticity impairment induced by glutamate transporter inhibition.
Figure 10.

Glutamate transporter inhibition impairs LTP by L-VGCC activation. A–D, Heat maps represent the maximal GCaMP6f response frame (left) and the GCaMP6f response profile over time (right) in response to TBS, both in control conditions (top images) and in the presence of 15 μm TBOA alone (A, bottom images) and in combination with dantrolene (Dan; 20 μm; B, bottom images), naspm (30 μm; C, bottom images), or nifedipine (Nif; 20 μm; D, bottom images). E, F, Grouped data show the fold increase in the GCaMP6f AUC in response to the second TBS (TBS2) compared with the first (TBS1). For all experiments, the first TBS was applied in control conditions and the second TBS was applied in the presence of 15 μm TBOA (E) or 100 μm DHK (F) alone or in combination with dantrolene, naspm, or nifedipine as indicated. G, H, LTP in control conditions and in the presence of the ryanodine receptor inhibitor, dantrolene, alone and in combination with 15 μm TBOA. Percent potentiation shown in H. I, J, LTP in control conditions and in the presence of the GluA2-lacking AMPAR blocker naspm alone and in combination with 15 μm TBOA. Percent potentiation shown in J. K, L, LTP in control conditions and in the presence of the L-VGCC blocker nifedipine alone and in combination with 15 μm TBOA. Percent potentiation shown in L. M, N, LTP in control conditions and in the presence of the calpain inhibitor MDL 28170 alone and in combination with 15 μm TBOA. MDL 28170 was applied 10 min after TBS. Percent potentiation shown in N. *p < 0.05; **p < 0.01; ***p < 0.001.
The calcium-dependent proteases calpain-1 and calpain-2 have received much recent attention for their opposing roles in synaptic plasticity (Baudry and Bi, 2016). Calpain-2 has been shown to act as a molecular brake that can limit the magnitude of TBS-LTP during the maintenance phase (Wang et al., 2014). Interestingly, calpain-2 can be activated by pERK (Glading et al., 2004; Chen et al., 2010) as well as millimolar concentrations of cytosolic calcium (Baudry and Bi, 2016). As TBOA-TBS resulted in both pERK hyperactivity and elevated cytosolic calcium, we asked whether calpain-2 recruitment could be acting as an LTP brake under these conditions. To test this hypothesis, we induced TBS-LTP in the presence or absence of TBOA, and bath-applied the calpain inhibitor MDL-28170 (10 μm) 10 min after LTP induction. This delayed application strategy was used in a previous study to inhibit calpain-2's effect on LTP consolidation without interfering with calpain-1's effect on LTP induction (Wang et al., 2014). MDL-28170 tended to result in a higher mean LTP magnitude than in control slices as expected (Wang et al., 2014), but TBOA still resulted in a significant LTP impairment compared with the MDL-28170-treated slices (Fig. 10M,N; control n = 10, MDL n = 7, MDL+TBOA n = 9, one-way ANOVA p = 0.038, post hoc Tukey tests: control vs MDL p = 0.168, control vs MDL+TBOA p = 0.597, MDL vs MDL+TBOA p = 0.031).
Discussion
Prior research suggests a dichotomous role of NMDARs in LTP strength whereby synNMDAR activation promotes LTP while exNMDAR activation impairs LTP and/or promotes LTD (S. Li et al., 2011; Papouin et al., 2012; Liu et al., 2013; Varga et al., 2015). By combining real-time millisecond imaging of extracellular glutamate with conventional electrophysiology, we demonstrated a clear negative relationship between glutamate clearance times and LTP magnitude following nonselective inhibition of glutamate transporters. Unexpectedly, the same relationship was not observed following selective inhibition of GLT-1 with DHK. To our surprise, the ability for poor glutamate clearance to inhibit LTP was associated with excessive calcium influx through L-VGCCs during LTP induction.
The sensitivity of activity-dependent synaptic plasticity to glutamate dynamics
By plotting glutamate dynamics and LTP magnitude under different experimental conditions, it became apparent that LTP is much more sensitive to nonselective transporter inhibition compared with GLT-1-selective inhibition. The IC50 of TBOA for GLAST, GLT-1, and EAAT3 is 70, 6, and 6 μm, respectively (Shimamoto et al., 1998). Thus, it is possible that the 15 μm TBOA concentration used in the majority of our experiments exerts its effects primarily through inhibitory actions at GLT-1 and EAAT3, with a relatively minor contribution from GLAST inhibition. Interestingly, blocking the majority of GLT-1 with DHK concentrations upward of 100 μm (IC50 for GLT-1 = 23 μm) (Arriza et al., 1994) was unable to match the effect of 15 μm TBOA on both glutamate accumulation and LTP. While it is clear that complete GLT-1 loss of function can impact synaptic plasticity, as GLT-1 KO mice exhibit poor LTP induced by high-frequency stimulation (Katagiri et al., 2001), our DHK results suggest that the selective inhibition of a majority of GLT-1 transporters is insufficient to impair TBS-LTP. An unanswered question brought up by the present study is why DHK was unable to impair LTP. DHK increased extracellular glutamate and the population GCaMP6f response to TBS but was without effect on LTP magnitude. One possibility is that selective inhibition of GLT-1 increases glutamate and calcium levels at microdomains that differ from those evoked by TBOA. Indeed, while GLT-1 has been detected at presynaptic terminals as well as astrocytes, EAAT3 (which would also be largely inhibited by 15 μm TBOA) exists on neurons (Rimmele and Rosenberg, 2016). EAAT3 has been shown to buffer synaptically released glutamate, thereby restricting extrasynaptic receptor activation (Scimemi et al., 2009). It is of interest for future studies to explore the differing effects of selective and nonselective transporter blockade on subcellular glutamate and calcium dynamics.
It is important to note that our mechanistic results suggest that the sensitivity of LTP to glutamate dynamics may increase with age. We identified that the LTP impairment induced by glutamate transporter inhibition is dependent upon L-VGCCs. It is well established that L-VGCC expression and activity increase with age (Thibault and Landfield, 1996; Davare and Hell, 2003); thus, it is of interest for future studies to determine whether LTP becomes more sensitive to glutamate dynamics in the aging brain. The sensitivity of this relationship is likely to rely on numerous factors present at basal conditions, including, but not limited to, resting membrane potential, glutamate release probability, the tortuosity of the extracellular space, as well as the density and relative nanoscale distributions of glutamate transporters, glutamate receptors, and L-VGCCs.
Slow glutamate clearance impairs LTP: underlying mechanisms
We provide multiple lines of evidence to suggest that the TBS-LTP impairment induced by glutamate transporter inhibition is not due to NMDAR overactivation during TBS. First, low concentrations of d-APV were unable to prevent the TBOA-induced LTP impairment, suggesting that general, subunit-independent NMDAR overstimulation during TBS is unlikely to mediate the TBOA effect. Second, ifenprodil was also unable to prevent the TBOA-mediated calcium increase and LTP impairment, suggesting that selective overstimulation of GluN1/GluN2B diheteromeric NMDARs also does not mediate the TBOA effect. Third, if the LTP impairment resulted from excessive activation of exNMDARs, then we should have observed a reduction in pERK staining following TBS in TBOA-treated slices, as exNMDAR activation is linked to pERK inactivation (Ivanov et al., 2006). Rather, we observed clear evidence for pERK hyperactivity under our experimental conditions. Fourth, the postsynaptic population response to each burst of the TBS was rapidly degraded in TBOA-treated slices; this effect was not observed when NMDARs were blocked with d-APV, suggesting that the NMDAR component of the fEPSP response to TBS is decreased, rather than enhanced, in the presence of TBOA. On the other hand, it is possible that TBOA causes an NMDAR-dependent global depolarization, thereby depressing postsynaptic responses to TBS as a result of a reduced driving force. Indeed, such depolarization could also facilitate L-VGCC activation during TBS, which we found was responsible for the subsequent LTP impairment.
TBOA resulted in an exaggerated calcium response to TBS that was partially attributed to several sources, including intracellular stores, GluA2-lacking AMPARs, and L-VGCCs. Excessive calcium may help explain the reduction in the NMDAR component of the fEPSP during TBOA-TBS, as it is well known that high concentrations of cytosolic calcium can trigger calcium-dependent NMDAR inactivation (Legendre et al., 1993; Rosenmund and Stevens, 1996; Paoletti et al., 2013; Sibarov and Antonov, 2018). Interestingly, although inhibiting each of these calcium sources in isolation significantly reduced the excess calcium response to TBOA-TBS, only L-VGCC inhibition was able to prevent the subsequent LTP impairment. This selective dependency on L-VGCC supports the idea that the precise calcium source and resultant calcium nanodomains are likely essential mediators of the strength and direction of synaptic plasticity (Evans and Blackwell, 2015). In the striatum, where glutamate clearance is not as efficient as it is in the hippocampus (Pinky et al., 2018), L-VGCC activation represents an important switch that promotes synapse weakening in the form of LTD (Adermark and Lovinger, 2007). While the results of the present study suggest that NMDARs are not overactivated during TBS in the presence of TBOA, we cannot rule out a possible involvement of transient NMDAR overactivation in the TBOA-induced LTP deficit. The excess calcium observed during TBOA-TBS was completely prevented by d-APV; therefore, it is entirely possible that the glutamate spillover induced by TBOA reached NMDARs in close proximity to L-VGCCs. In this scenario, depolarization within the nanodomain of the L-VGCC would promote further calcium entry through L-VGCCs. Alternatively, the overactivation of L-VGCCs may stem largely from a TBOA-induced and NMDAR-dependent depolarization within the slice, facilitating L-VGCC activation during TBS. Arguing against an exNMDAR involvement is the fact that the GluN2B-containing NMDAR antagonist ifenprodil was unable to prevent the TBOA-induced calcium increase and LTP deficit. However, while GluN2B may be enriched extrasynaptically, GluN2A and GluN2B are far from exclusive to synaptic and extrasynaptic sites, respectively (Harris and Pettit, 2007; Petralia et al., 2010; Parsons and Raymond, 2014).
It was previously shown that calcium influx through L-VGCCs is sufficient to induce persistent ERK activation (Dolmetsch et al., 2001); and while pERK is generally regarded as a positive regulator of LTP, numerous studies indicate that excessive levels of pERK, or “hyperactive ERK” (Guilding et al., 2007), can indeed suppress LTP and impair hippocampal-dependent learning and memory (Costa et al., 2002; Echeverria et al., 2004; Sweatt, 2004; Guilding et al., 2007; Cui et al., 2008). For example, ERK hyperactivity can impair LTP by increasing synapsin-1 phosphorylation and enhancing GABA release (Cui et al., 2008). In addition, pERK is known to activate calpain-2, a negative regulator of LTP (Baudry and Bi, 2016), via phosphorylation at Serine 50 (Glading et al., 2004; Zadran et al., 2009; Chen et al., 2010); however, the results of the present study suggest that excessive calpain-2 activation is unlikely to mediate the TBOA-induced LTP impairment. As L-VGCC activation is typically associated with enhanced LTP, the precise mechanism by which it is causing LTP impairment in the present study remains to be seen. Certain possibilities, including pERK hyperactivity, an enhancement of heterosynaptic LTD (Wickens and Abraham, 1991), and gene expression changes induced by L-VGCC conformational changes (B. Li et al., 2016), are of interest to explore in future studies.
In conclusion, here, we combined electrophysiology with real-time glutamate imaging to better understand the relationship between glutamate dynamics and activity-dependent synaptic plasticity. These experiments were performed in healthy control mice at a relatively young age. The reliance of the LTP deficit on L-VGCCs supports a critical importance of glutamate and calcium nanodomains in synaptic plasticity determination and suggests that the sensitivity of the glutamate dynamics/synaptic plasticity relationship may change with aging (Thibault and Landfield, 1996; Davare and Hell, 2003) as well as in neurodegenerative diseases where the nanoscale redistribution of synaptic proteins is observed. Both Alzheimer disease and Huntington disease are associated with reductions in GLT-1 mRNA and/or protein (Liévens et al., 2001; Behrens et al., 2002; Faideau et al., 2010; Zumkehr et al., 2015). The reported magnitude of GLT-1 reduction in these diseases, when considered in isolation, is unlikely to result in a clear LTP impairment. However, when considered together with increased L-VGCC density (Wang and Mattson, 2014) and/or the subcellular mislocalization of NMDARs (Milnerwood et al., 2010; Parsons and Raymond, 2014), it is possible that these factors combine to contribute to the poor LTP observed in animal models of these diseases. In all, the present study represents an essential step in understanding the relationship between the spatiotemporal dynamics of glutamate neurotransmission and activity-dependent synaptic plasticity, and exposes additional questions regarding the nature of this relationship during aging and in neurodegenerative disease.
Footnotes
This work was supported by Natural Sciences and Engineering Research Council of Canada Discovery Grant, Huntington Society of Canada Navigator Grant, and Alzheimer Society of Canada New Investigator Award. We thank Bimal Tennakoon, Vanessa Strong, and Jade Quirion for technical assistance; and the animal care staff at Memorial University for assistance with animal housing.
The authors declare no competing financial interests.
References
- Adermark L, Lovinger DM (2007) Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. J Neurosci 27:6781–6787. 10.1523/JNEUROSCI.0280-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14:5559–5569. 10.1523/JNEUROSCI.14-09-05559.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M, Bi X (2016) Calpain-1 and calpain-2: the yin and yang of synaptic plasticity and neurodegeneration. Trends Neurosci 39:235–245. 10.1016/j.tins.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB (2002) Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain 125:1908–1922. 10.1093/brain/awf180 [DOI] [PubMed] [Google Scholar]
- Bonde C, Sarup A, Schousboe A, Gegelashvili G, Zimmer J, Noraberg J (2003) Neurotoxic and neuroprotective effects of the glutamate transporter inhibitor dl-threo-beta-benzyloxyaspartate (dl-TBOA) during physiological and ischemia-like conditions. Neurochem Int 43:371–380. 10.1016/S0197-0186(03)00024-X [DOI] [PubMed] [Google Scholar]
- Bradley CA, Peineau S, Taghibiglou C, Nicolas CS, Whitcomb DJ, Bortolotto ZA, Kaang BK, Cho K, Wang YT, Collingridge GL (2012) A pivotal role of GSK-3 in synaptic plasticity. Front Mol Neurosci 5:13. 10.3389/fnmol.2012.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Libertini SJ, Wang Y, Kung HJ, Ghosh P, Mudryj M (2010) ERK regulates calpain 2-induced androgen receptor proteolysis in CWR22 relapsed prostate tumor cell lines. J Biol Chem 285:2368–2374. 10.1074/jbc.M109.049379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ (2002) Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 415:526–530. 10.1038/nature711 [DOI] [PubMed] [Google Scholar]
- Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, Parada LF, Mody I, Silva AJ (2008) Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 135:549–560. 10.1016/j.cell.2008.09.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. (2001) Glutamate uptake. Prog Neurobiol 65:1–105. 10.1016/S0301-0082(00)00067-8 [DOI] [PubMed] [Google Scholar]
- Davare MA, Hell JW (2003) Increased phosphorylation of the neuronal L-type Ca2+ channel Cav1.2 during aging. Proc Natl Acad Sci U S A 100:16018–16023. 10.1073/pnas.2236970100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME (2001) Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294:333–339. 10.1126/science.1063395 [DOI] [PubMed] [Google Scholar]
- Echeverria V, Ducatenzeiler A, Dowd E, Jänne J, Grant SM, Szyf M, Wandosell F, Avila J, Grimm H, Dunnett SB, Hartmann T, Alhonen L, Cuello AC (2004) Altered mitogen-activated protein kinase signaling, tau hyperphosphorylation and mild spatial learning dysfunction in transgenic rats expressing the β-amyloid peptide intracellularly in hippocampal and cortical neurons. Neuroscience 129:583–592. 10.1016/j.neuroscience.2004.07.036 [DOI] [PubMed] [Google Scholar]
- Evans RC, Blackwell KT (2015) Calcium: amplitude, duration, or location? Biol Bull 228:75–83. 10.1086/BBLv228n1p75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, Dufour N, Guillermier M, Brouillet E, Hantraye P, Déglon N, Ferrante RJ, Bonvento G (2010) In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum Mol Genet 19:3053–3067. 10.1093/hmg/ddq212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glading A, Bodnar RJ, Reynolds IJ, Shiraha H, Satish L, Potter DA, Blair HC, Wells A (2004) Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol 24:2499–2512. 10.1128/MCB.24.6.2499-2512.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilding C, McNair K, Stone TW, Morris BJ (2007) Restored plasticity in a mouse model of neurofibromatosis type 1 via inhibition of hyperactive ERK and CREB. Eur J Neurosci 25:99–105. 10.1111/j.1460-9568.2006.05238.x [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11:682–696. 10.1038/nrn2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL (2007) Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J Physiol 584:509–519. 10.1113/jphysiol.2007.137679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I (2006) Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol 572:789–798. 10.1113/jphysiol.2006.105510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G, Cho K (2011) Aβ1–42 inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat Neurosci 14:545–547. 10.1038/nn.2785 [DOI] [PubMed] [Google Scholar]
- Katagiri H, Tanaka K, Manabe T (2001) Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur J Neurosci 14:547–553. 10.1046/j.0953-816x.2001.01664.x [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M (2005) Differential roles of NR2A- and NR2B-containing NMDA receptors in ras-ERK signaling and AMPA receptor trafficking. Neuron 46:745–760. 10.1016/j.neuron.2005.04.031 [DOI] [PubMed] [Google Scholar]
- Koeglsperger T, Li S, Brenneis C, Saulnier JL, Mayo L, Carrier Y, Selkoe DJ, Weiner HL (2013) Impaired glutamate recycling and GluN2B-mediated neuronal calcium overload in mice lacking TGF-β1 in the CNS. Glia 61:985–1002. 10.1002/glia.22490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Lynch G (1988) Role of N-methyl-d-aspartate receptors in the induction of synaptic potentiation by burst stimulation patterned after the hippocampal θ-rhythm. Brain Res 441:111–118. 10.1016/0006-8993(88)91388-1 [DOI] [PubMed] [Google Scholar]
- Legendre P, Rosenmund C, Westbrook GL (1993) Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J Neurosci 13:674–684. 10.1523/JNEUROSCI.13-02-00674.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Xu H, Li Z, Wang Z, O'Malley TT, Zhang D, Walsh DM, Xu P, Selkoe DJ, Li S (2016) Soluble Aβ oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol Dis 85:111–121. 10.1016/j.nbd.2015.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Tadross MR, Tsien RW (2016) Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 351:863–867. 10.1126/science.aad3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D (2009) Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62:788–801. 10.1016/j.neuron.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31:6627–6638. 10.1523/JNEUROSCI.0203-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liévens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP (2001) Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis 8:807–821. 10.1006/nbdi.2001.0430 [DOI] [PubMed] [Google Scholar]
- Liu DD, Yang Q, Li ST (2013) Activation of extrasynaptic NMDA receptors induces LTD in rat hippocampal CA1 neurons. Brain Res Bull 93:10–16. 10.1016/j.brainresbull.2012.12.003 [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29:243–254. 10.1016/S0896-6273(01)00194-5 [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM (2010) Group 1 mGluR-dependent synaptic long-term depression (mGluR-LTD): mechanisms and implications for circuitry and disease. Neuron 65:445–459. 10.1016/j.neuron.2010.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen TW, Bargmann CI, Orger MB, Schreiter ER, Demb JB, Gan WB, Hires SA, Looger LL (2013) An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat Methods 10:162–170. 10.1038/nmeth.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI (2004) Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci 24:7821–7828. 10.1523/JNEUROSCI.1697-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA (2010) Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron 65:178–190. 10.1016/j.neuron.2010.01.008 [DOI] [PubMed] [Google Scholar]
- Murphy-Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J, Groc L, Oliet SH (2015) Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat Neurosci 18:219–226. 10.1038/nn.3901 [DOI] [PubMed] [Google Scholar]
- Nabavi S, Kessels HW, Alfonso S, Aow J, Fox R, Malinow R (2013) Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc Natl Acad Sci U S A 110:4027–4032. 10.1073/pnas.1219454110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, Eichenbaum H, Wiener SI, Wible CG (1991) Learning-related patterns of CA1 spike trains parallel stimulation parameters optimal for inducing hippocampal long-term potentiation. Hippocampus 1:181–192. 10.1002/hipo.450010206 [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14:383–400. 10.1038/nrn3504 [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH (2012) Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 150:633–646. 10.1016/j.cell.2012.06.029 [DOI] [PubMed] [Google Scholar]
- Parsons MP, Raymond LA (2014) Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82:279–293. 10.1016/j.neuron.2014.03.030 [DOI] [PubMed] [Google Scholar]
- Parsons MP, Vanni MP, Woodard CL, Kang R, Murphy TH, Raymond LA (2016) Real-time imaging of glutamate clearance reveals normal striatal uptake in Huntington disease mouse models. Nat Commun 7:11251. 10.1038/ncomms11251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL (2007) LTP inhibits LTD in the hippocampus via regulation of GSK3β. Neuron 53:703–717. 10.1016/j.neuron.2007.01.029 [DOI] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A (2009) Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32:421–431. 10.1016/j.tins.2009.05.001 [DOI] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA (2015) Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci 35:5187–5201. 10.1523/JNEUROSCI.4255-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Hua F, Yi Z, Zhou A, Ge L, Stephenson FA, Wenthold RJ (2010) Organization of NMDA receptors at extrasynaptic locations. Neuroscience 167:68–87. 10.1016/j.neuroscience.2010.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinky NF, Wilkie CM, Barnes JR, Parsons MP (2018) Region- and activity-dependent regulation of extracellular glutamate. J Neurosci 38:5351–5366. 10.1523/JNEUROSCI.3213-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimmele TS, Rosenberg PA (2016) GLT-1: the elusive presynaptic glutamate transporter. Neurochem Int 98:19–28. 10.1016/j.neuint.2016.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. 10.1016/S0896-6273(00)80146-4 [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW (1994) Localization of neuronal and glial glutamate transporters. Neuron 13:713–725. 10.1016/0896-6273(94)90038-8 [DOI] [PubMed] [Google Scholar]
- Scimemi A, Tian H, Diamond JS (2009) Neuronal transporters regulate glutamate clearance, NMDA receptor activation, and synaptic plasticity in the hippocampus. J Neurosci 29:14581–14595. 10.1523/JNEUROSCI.4845-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T (1998) dl-threo-β-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol 53:195–201. 10.1124/mol.53.2.195 [DOI] [PubMed] [Google Scholar]
- Shipton OA, Paulsen O (2014) GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos Trans R Soc Lond B Biol Sci 369:20130163. 10.1098/rstb.2013.0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibarov DA, Antonov SM (2018) Calcium-dependent desensitization of NMDA receptors. Biochem 83:1173–1183. 10.1134/S0006297918100036 [DOI] [PubMed] [Google Scholar]
- Soni N, Reddy BV, Kumar P (2014) GLT-1 transporter: an effective pharmacological target for various neurological disorders. Pharmacol Biochem Behav 127:70–81. 10.1016/j.pbb.2014.10.001 [DOI] [PubMed] [Google Scholar]
- Sweatt JD. (2004) Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14:311–317. 10.1016/j.conb.2004.04.001 [DOI] [PubMed] [Google Scholar]
- Thibault O, Landfield PW (1996) Increase in single L-type calcium channels in hippocampal neurons during aging. Science 272:1017–1020. 10.1126/science.272.5264.1017 [DOI] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ (2006) Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol 572:477–492. 10.1113/jphysiol.2005.103754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga E, Juhász G, Bozsó Z, Penke B, Fülöp L, Szegedi V (2015) Amyloid-β1–42 disrupts synaptic plasticity by altering glutamate recycling at the synapse. J Alzheimers Dis 45:449–456. 10.3233/JAD-142367 [DOI] [PubMed] [Google Scholar]
- Volianskis A, France G, Jensen MS, Bortolotto ZA, Jane DE, Collingridge GL (2015) Long-term potentiation and the role of N-methyl-d-aspartate receptors. Brain Res 1621:5–16. 10.1016/j.brainres.2015.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Kavanaugh MP (1998) Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J Neurosci 18:7650–7661. 10.1523/JNEUROSCI.18-19-07650.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mattson MP (2014) L-type Ca2+ currents at CA1 synapses, but not CA3 or dentate granule neuron synapses, are increased in 3xTgAD mice in an age-dependent manner. Neurobiol Aging 35:88–95. 10.1016/j.neurobiolaging.2013.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhu G, Briz V, Hsu YT, Bi X, Baudry M (2014) A molecular brake controls the magnitude of long-term potentiation. Nat Commun 5:1–12. 10.1038/ncomms4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby PJ, Rowan MJ, Anwyl R (2007) Beta-amyloid blocks high frequency stimulation induced LTP but not nicotine enhanced LTP. Neuropharmacology 53:188–195. 10.1016/j.neuropharm.2007.05.013 [DOI] [PubMed] [Google Scholar]
- Wickens JR, Abraham WC (1991) The involvement of L-type calcium channels in heterosynaptic long-term depression in the hippocampus. Neurosci Lett 130:128–132. 10.1016/0304-3940(91)90244-N [DOI] [PubMed] [Google Scholar]
- Yi JH, Hazell AS (2006) Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int 48:394–403. 10.1016/j.neuint.2005.12.001 [DOI] [PubMed] [Google Scholar]
- Zadran S, Qin Q, Bi X, Zadran H, Kim Y, Foy MR, Thompson R, Baudry M (2009) 17-beta-estradiol increases neuronal excitability through MAP kinase-induced calpain activation. Proc Natl Acad Sci U S A 106:21936–21941. 10.1073/pnas.0912558106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Liu Y, Wang Y, Bi X, Baudry M (2015) Different patterns of electrical activity lead to long-term potentiation by activating different intracellular pathways. J Neurosci 35:621–633. 10.1523/JNEUROSCI.2193-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumkehr J, Rodriguez-Ortiz CJ, Cheng D, Kieu Z, Wai T, Hawkins C, Kilian J, Lim SL, Medeiros R, Kitazawa M (2015) Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer's disease. Neurobiol Aging 36:2260–2271. 10.1016/j.neurobiolaging.2015.04.005 [DOI] [PubMed] [Google Scholar]