

Abstract
The Soai reaction has profoundly impacted chemists’ perspective of chiral symmetry breaking, absolute asymmetric synthesis and its role in the origin of biological homochirality. Herein, we describe the unprecedented observation of asymmetry amplifying autocatalysis in the alkylation of 5-(trimethylsilylethynyl)pyridine-3-carbaldehyde using diisopropylzinc. Kinetic studies with a surrogate substrate and spectroscopic analysis of a series of zinc-alkoxides that incorporate specific structural mutations reveal a ‘pyridine-assisted cube escape’. The new tetrameric cluster functions as a catalyst that activates the substrate through a two point binding mode and poises a coordinated diisopropylzinc moiety for alkyl group transfer. Transition-state models leading to both the homochiral and heterochiral products were validated by density functional theory calculations. Moreover, experimental and computational analysis of the heterochiral complex provides a definitive explanation for the non-linear behavior of this system. Our deconstruction of the Soai system reveals the structural logic for autocatalyst evolution, function and substrate compatibility – a central mechanistic aspect of this iconic transformation.
The autocatalytic, asymmetry amplifying alkylation of pyrimidine-5-carbaldehydes with diisopropylzinc, crowned as the ‘Soai reaction’, occupies a venerable position in organic chemistry (Figure 1a).1,2 The transformation is peerless in its efficiency for asymmetric autocatalysis and sensitivity to initial chiral imbalances, making it a sui generis example of a reaction predisposed to evolve toward homochirality. Enantiopure products are obtained in three reaction cycles even with calculated autocatalyst e.e. as low as 5 × 10−5 %.3 In the absence of added catalyst, symmetry breaking can yield non-racemic products,4–7 thus categorizing the transformation as an example of spontaneous absolute asymmetric synthesis.8 A large variety of chiral additives2,9–15 and even circularly polarized light16 and isotopic chirality17–19 can influence the outcome of the reaction by biasing an initial imbalance toward one of the enantiomers. Soai’s seminal discoveries have received widespread attention in diverse chemical fields and have revived discussions regarding absolute asymmetric synthesis, chiral symmetry breaking, and the origin of biological homochirality.20–23
Figure 1:
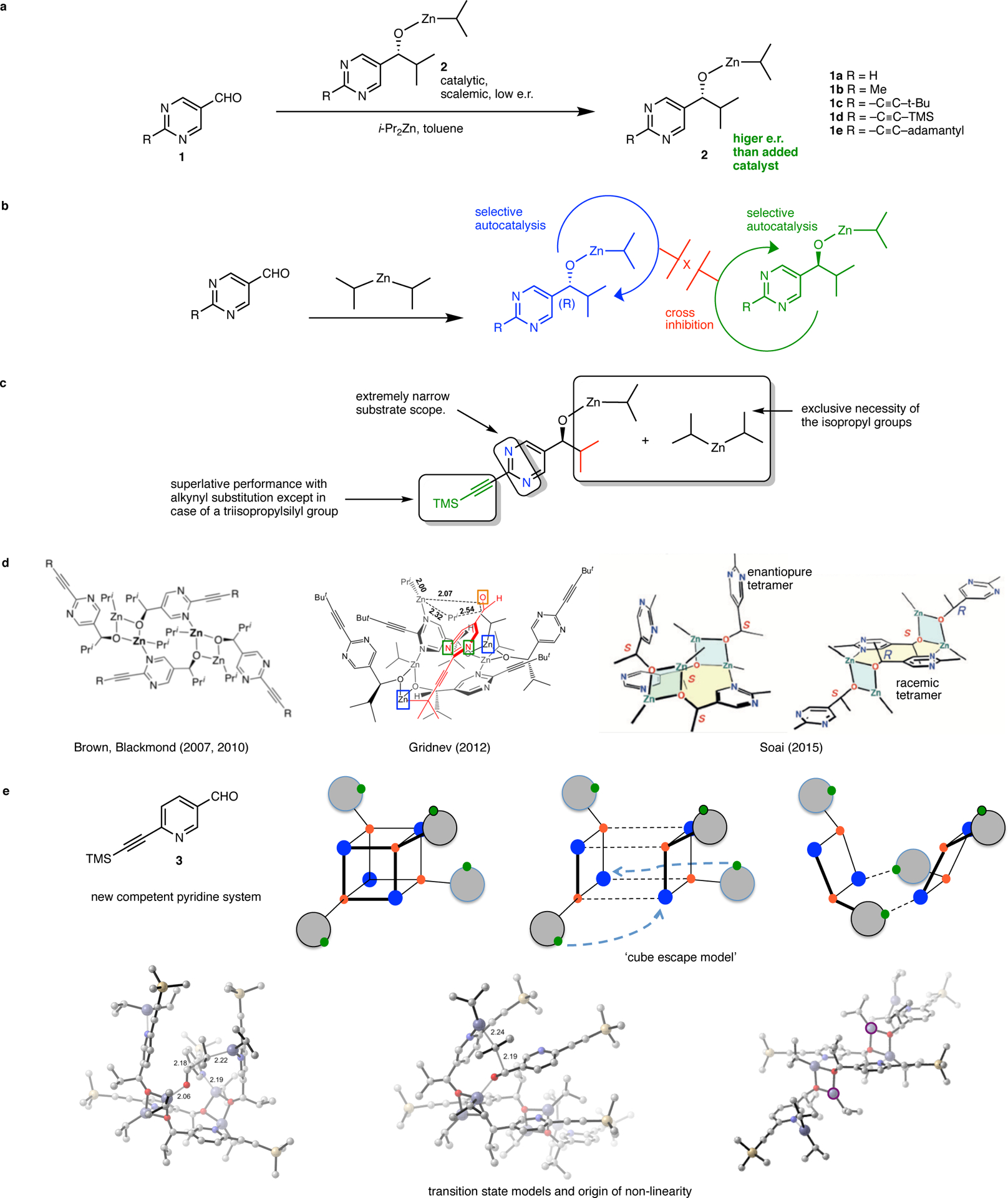
The Soai reaction system, current mechanistic understanding and salient contributions from this work. a. The diisopropylzinc alkylation of pyrimidine-5-carbaldehydes, termed the ‘Soai reaction’ is characterized by highly efficient autocatalysis and chiral amplification. The product zinc alkoxide is a selective autocatalyst for its own formation. Following Soai’s pioneering report with substrate 1a, competent activity has been subsequently described for other derivatives. In particular, the akynyl-substituted substrates 1c-1e demonstrate superior reactivity and chiral amplification. The transformation is exquisitely sensitive to initial chiral imbalances including perturbations caused by isotopic chirality. The reaction demonstrates spontaneous symmetry breaking and qualifies as an example of absolute asymmetric synthesis. b. Overall kinetic scheme for amplifying asymmetric autocatalysis (Frank, 1953). Accordingly, the product alkoxide enantiomers are self-catalytic while also being mutually inhibitory. Outstanding questions regarding the molecular details include the identity and mode of operation of the autocatalyst, a compelling transition-state hypothesis, an explanation for the origin of the non-linear effect, and a justification for the puzzling, idiosyncratic substrate constraints. c. Poorly understood structural constraints in the Soai reaction. d. The state of the art in mechanistic elucidation. From left – the SMS tetramer connectivity as first described by Brown and Blackmond, the Gridnev TS model of catalysis by the SMS tetramer and, crystal structures of the homochiral and heterochiral SMS tetramers. The crystal structure depicts the homochiral tetramer as a bowl shaped assembly with a central macrocycle (shaded yellow) and two ‘pyrimidine arms’ oriented in the same direction (in this case, the top face). The heterochiral tetramer possesses an identical connectivity but with the arms oriented on opposing faces of the central macrocycle. e. Contributions from this work include the discovery of a new amplifying autocatalytic pyridine system that leads to a structural simplification, the ‘cube escape’ model which elucidates the structural logic for the evolution of the Soai tetramer and the presentation of a new floor-to-floor TS model, fundamentally distinct from the Gridnev proposal, based on experimental and computational studies. The new model also provides insights into the structural constraints of the reactants and a compelling explanation for the origin of non-linearity.
The general kinetic scheme for amplifying autocatalysis, articulated by F.C. Frank in 1953 (Figure 1b) as a possible hypothesis for evolution of biological homochirality,24 was spectacularly brought to life by Soai’s seminal results. The product alkoxide, in some manner, acts as a highly enantioselective (auto)catalyst for its own production while concomitantly inhibiting catalysis (a positive non-linear effect) by its enantiomeric counterpart. This allows for the continuous autocatalytic enrichment of the major enantiomer with reaction progression. How this process actually transpires at the molecular level, is a question that has intrigued and challenged investigators for more than two decades. For a substantive understanding, the principle issues that need to be addressed are: (1) an elucidation of the precise identity and modus operandi of the autocatalyst, (2) a transition state structure rationalizing the basis of asymmetric autocatalysis, (3) an explanation for the origin of positive, non-linear (auto)catalysis, and, (4) a justification of the puzzling, restrictive and idiosyncratic substrate requirements that allow successful amplifying autocatalysis (Figure 1c).
Because catalyst aggregation is a predominant contributor to non-linear effects, establishing the identity of the product alkoxide (the autocatalyst) is of critical importance and has been the central focus of prior mechanistic investigations. Early studies indicating a dimeric autocatalyst model (see Supplementary Figure 1 for details)25–32 have since been superseded by the suggestion of a tetrameric alkoxide aggregate as the autocatalyst (Figure 1d). In 2007, Klankermayer et al. first suggested the square-macrocycle-square (SMS) tetramer, as a possible end product species.33 In 2010, Brown and Blackmond reported an intriguing inverse dependence of reaction rate on temperature and provided further support for the SMS tetramer as a viable species in the reaction cycle on the basis of further kinetic and spectroscopic investigations34 In 2012, DFT studies by Gridnev presented a candidate transition state model for alkyl transfer by the tetrameric autocatalyst. Finally in 2015, Soai et al. disclosed the first X-ray crystal structures of the enantiopure and racemic alkoxides which exist as tetramers or oligomers depending on crystallization conditions.35 The homochiral and heterochiral tetramers possess a 12-member macrocycle with a connectivity resembling the SMS tetrameric structure proposed earlier.34 The assembly can be considered as a concatamer of two Zn–O square dimers, ligated through pyrimidine–Zn coordination. In the homochiral tetramer, the unbound pyrimidine units (referred to as the tetramer ‘arms’) are oriented on the same face of the macrocycle. The racemic, heterochiral tetramer possesses a similar overall connectivity but the arms are placed on opposite sides of the macrocycle. In both the homochiral and heterochiral tetramers, a pair of three-coordinate, unsaturated (alkoxy)zinc atoms are present as part of the pyrimidinyl arms (boxed blue). The orientation of these unsaturated zinc centers follows the pattern of the arms. Finally, for both the tetramers, the crystal structure also showed that except for the two macrocycle residing nitrogen atoms, all the remaining six pyrimidinyl nitrogen atoms are bound by diisopropylzinc molecules (not shown in Figure 1d).
Do these studies answer the four basic questions about the reaction mechanism? Brown and coworkers’ proposal of the SMS tetramer species was highly prescient.33,34 Soai’s seminal crystal structure elucidation35 proves that this aggregate is physically viable and strengthens the hypothesis of it also existing as a solution phase species. However, although the identity of the autocatalyst (as the SMS tetramer) may be now stated with some certainty, its mode of action remains highly speculative. Gridnev’s 2012 transition-state model, based on DFT studies, did not possess the benefit of Soai’s crystal structure connectivity to define the starting geometry of the SMS tetramer. The new observations and discoveries presented here draw a different conclusion than that of the Gridnev model. Our work present a TS model that is drastically distinct, where the substrate nitrogen atoms and the unsaturated zinc atoms (boxed green and blue respectively in Figure 1d) are involved in critical interactions necessary for catalysis, as well as a fundamentally different alkyl transfer configuration.
The origin of non-linearity, a defining hallmark of the reaction can be explained only when the differential activities of homochiral and heterochiral aggregates are delineated. Since a compelling transition-state hypothesis itself is lacking, the origin of non-linearity cannot be articulated. Our investigations, culminating into the new TS model presents a simple resolution of this issue.
Finally, experimental studies to rationalize the remarkable substrate constraints of the reactants are entirely absent.36 Although systematic studies with other dialkylzinc reagents are lacking, asymmetric amplification is observed exclusively with diisopropylzinc. 2-Alkynylpyrimidine-5-carbaldehydes37 remain the workhorse substrates and only scant reports with other competent aldehydes have been presented.38,39 Pyridine-3-carbaldehyde, for example does not demonstrate amplifying autocatalysis.40 Note that an inspection of the crystal structure (Figure 1d) does not reveal the significance of the second pyrimidine nitrogen in assembling the macrocyclic connectivity, nor the role played by the diisopropylzinc bound to this nitrogen with respect to catalysis. Similarly, the defining contributions of the alkyl groups towards product aggregation are not apparent. Moreover, the reasons for superlative performance of the alkynyl-substituted substrates remain unclear. Another idiosyncrasy is the bewildering failure of 2-(triisopropylsilylethynyl)pyrimidine-5-carbaldehyde to display amplifying autocatalysis.37 For a reaction demonstrating such striking characteristics, the extremely narrow substrate scope is perhaps unsurprising. Nevertheless, a fundamental rationalization of these structural constraints that allow exclusive access to the requisite catalytic intermediates remains obscure, highlighting our lack of understanding of this reaction. Brown and coworkers have described computational studies that suggest the importance of the isopropyl group in diverting aggregates away from the well-established cubic tetrameric species.33,41 However, no experimental studies on any of the Soai reaction substrates support or discredit this hypothesis. In particular, the precise role of the pyrimidine core and the alkynyl substituent remains enigmatic.
To summarize, although the current consensus of a tetrameric alkoxide catalyst represents a significant milestone in the identification of product aggregation, this knowledge has not yet enabled the formulation of a compelling transition state hypothesis nor provided deeper insights into the structural contributions from various parts of the molecule that give rise to asymmetric autocatalysis and non-linearity. The manner in which structural aspects of the reactants and the autocatalyst enable the realization of Frank’s scheme remains unclear. Two decades since Soai’s pioneering publication, a holistic comprehension of the reaction remains an open challenge with fundamental implications on our understanding of chiral symmetry breaking and absolute asymmetric synthesis. Our work addresses these questions (vida supra) that lie at the heart of unmasking the mechanism of the Soai reaction.
Results
Discovery of a New Amplifying Autocatalytic System
The first stage of our investigation was to deconstruct the Soai system and understand the contributions to catalysis and asymmetric amplification from its various components: (1) the dialkylzinc reagent, (2) the carbinol substituent, (3) the aromatic core and (4) the alkynyl substituent. Orienting studies began with the well-known pyrimidine-5-carbaldehyde 1d and different dialkylzinc reagents (these results will be disclosed in a full account of this work in the future). The next series of experiments probed the role of the aromatic core, which led us to the startling discovery that 5-(trimethylsilylethynyl)pyridine-3-carbaldehyde (3) displayed efficient amplifying asymmetric autocatalysis in the reaction with diisopropylzinc (Figure 2a). Product carbinol 4, when included in this reaction as a scalemic catalyst, afforded newly-generated product with higher enantioenrichment. This result was entirely unexpected and surprising because diisopropylzinc alkylation of a very similar substrate, 5-(t-butylethynyl)pyridine-3-carbaldehyde has been reported in literature (see Supplementary Information of Amedjkouh et. al., 2015) to show chiral erosion.42 We have been unable to reproduce the observation reported by Amedjkouh et al. Instead, we have observed amplifying autocatalysis for both substrates – diisopropylzinc alkylation of 5-(t-butylethynyl)pyridine-3-carbaldehyde also demonstrates amplifying autocatalysis (See Supplementary Figure 2).
Figure 2:
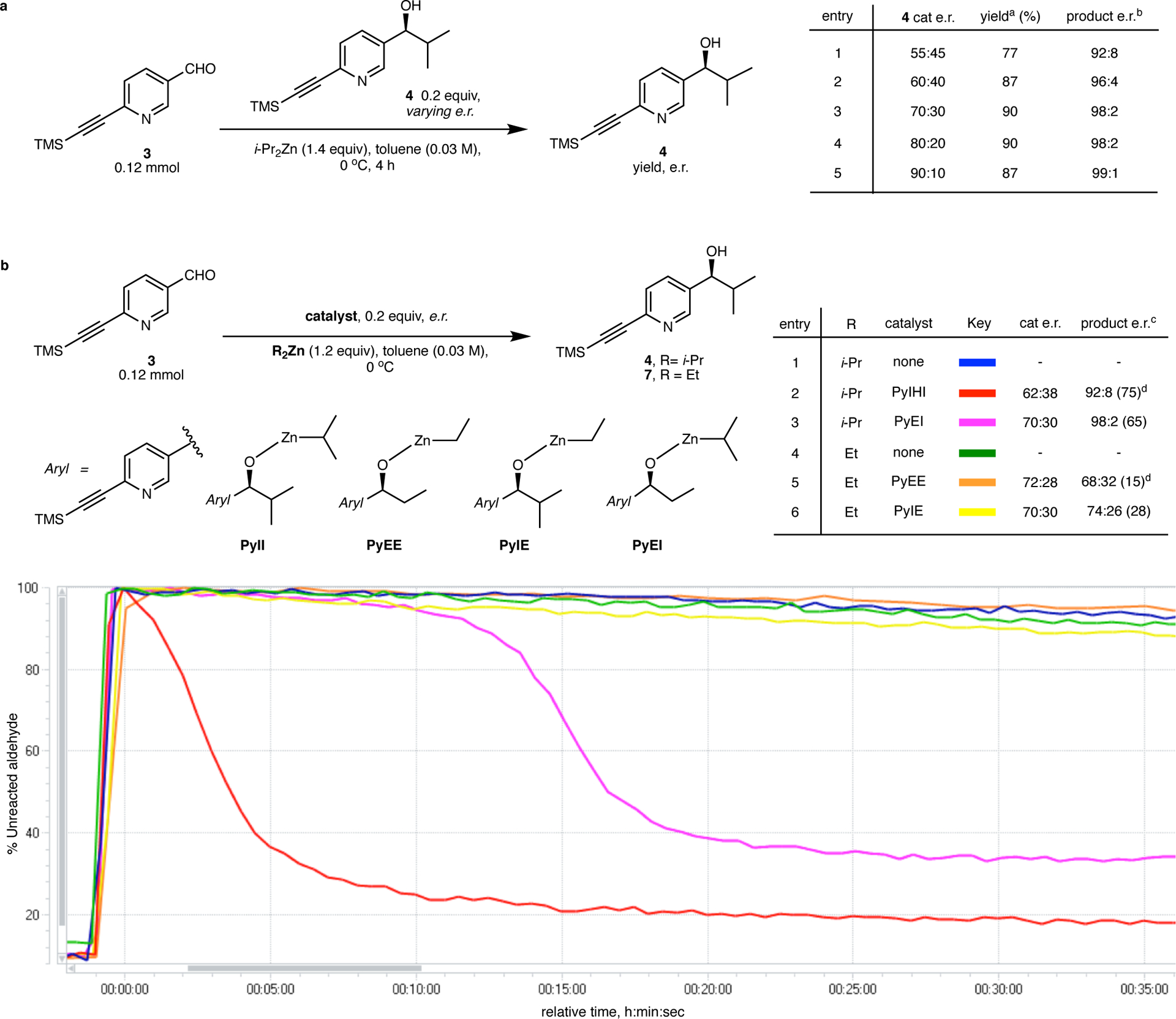
A new asymmetry amplifying, autocatalytic, pyridine system and a structure-activity relationship of the (auto)catalyst. a. Asymmetric autocatalysis with chiral amplification in alkylation of 3 with diisopropylzinc. Inclusion of the product carbinol results in autocatalytic generation of new product with higher enantioenrichment. The new system shares other striking characteristics of the original Soai system (see text). b. Qualitative comparison of catalytic activities and non-linear effect of alkoxides PyXX in dialkylzinc addition to 3 by in-situ IR monitoring of aldehyde consumption. Scalemic alkoxides PyXX were tested for activity and non-linear effect. Rapid conversion (red curve) and a strong non-linear effect is noted for the autocatalyst PyII. The other alkoxides show no initial rate enhancement and are poor catalysts – removal of a single methyl group from the PyII structure abrogates catalytic activity. a Yield of isolated, newly formed prouct; b e.r. of newly formed product excluding initial catalyst; c at specified conversion in parenthesis; d includes added catalyst. Product e.r. was analyzed by quenching a reaction aliquot at specified time, Supplementary Information, section 4.
This reaction (diisopropylzinc alkylation of 3) with the new substituted pyridine-system is homogenous throughout its course even at high concentrations and is conveniently monitored by tracking aldehyde consumption using in-situ IR spectroscopy. The transformation displays all the characteristics of an amplifying, autocatalytic process (Supplementary Figure 3) including sigmoidal reaction profiles and a higher catalytic rate with a homochiral autocatalyst than a heterochiral one (further highlighting the non-linear effect). Furthermore, it also demonstrates other non-obvious, idiosyncratic qualities typical to the Soai reaction,34,37 including absence of autocatalysis with diethylzinc (Figure 2b, entries 4–5), absence of autocatalysis with the triisopropylsilyl analog, as well as an inverse rate profile with respect to temperature (Supplementary Figure 4). These observations, bearing striking resemblance to the original Soai system establish that, just as is the case in the iconic pyrimidine-system, non-linear amplifying autocatalysis is also operative in the reaction of 3 and diisopropylzinc. Clearly, one of the nitrogen atoms in the pyrimidine ring is dispensable for this phenomenon.
Catalyst Structure-Activity Relationships
To evaluate contributions of the alkyl group on the carbinol center in the catalyst and the O–Zn–alkyl group, a qualitative comparison of catalysis by four different zinc alkoxides (PyII, PyEE, PyIE and PyEI) was made by in-situ IR monitoring of dialkylzinc additions to 3 in catalyzed and uncatalyzed reactions (Figure 2b) (nomenclature: Py/Ph = pyridine/phenyl indicating the aromatic core, I/E = isopropyl/ethyl indicating the carbinol alkyl group and I/E = isopropyl/ethyl indicating the alkylzinc group). In all cases, the corresponding scalemic carbinols were used for generating the zinc alkoxides (see Supplementary Information, Sections 3.1–3.3 for details) and the product enantiomeric composition was analyzed to assess any non-linear effects. Under these conditions, the uncatalyzed reactions (entries 1 and 4) are sluggish, show low conversion and ultimately stall. Inclusion of either PyEE (entry 5) or PyIE (entry 6) shows no appreciable rate enhancement. In the case of PyEE, the product carbinol has a lower e.r. than the added catalyst whereas a minor positive non-linear effect is seen in case of PyIE. Entry 2 represents the autocatalytic reaction with diisopropylzinc and reiterates the high catalytic efficiency and strong non-linear behavior of catalyst PyII. Finally, with alkoxide PyEI (entry 3), a sigmoidal aldehyde decay with high final product e.r. is achieved. The absence of an initial rate enhancement is noteworthy. This behavior is interpreted to arise from a low catalytic activity of PyEI giving rise to an initial production of PyII followed by strong asymmetric autocatalysis from a buildup of PyII. Taken together, these observations indicate that in this alkoxide series, only PyII possesses the requisite structural requirements for efficient, amplifying catalysis and replacement of either or both isopropyl groups results in alkoxides (PyEE, PyIE and PyEI) that possess markedly inferior catalytic efficiencies.
Spectroscopy of Zinc Alkoxides and the Structural Uniqueness of PyII
To gain insights into the structural requirements for efficient catalysis, occurring only in case of PyII, a spectroscopic study to characterize the solution state structures of phenyl (PhEE, PhEI, PhIE and PhII) and pyridyl (PyEE, PyEI, PyIE and PyII) zinc alkoxides in toluene was initiated (Figures 3 and 4). The high solubility of these species along with their stability in toluene for extended periods represented a significant advantage over their pyrimidine analogs and facilitated detailed 1D and 2D NMR spectroscopic characterization.
Figure 3:
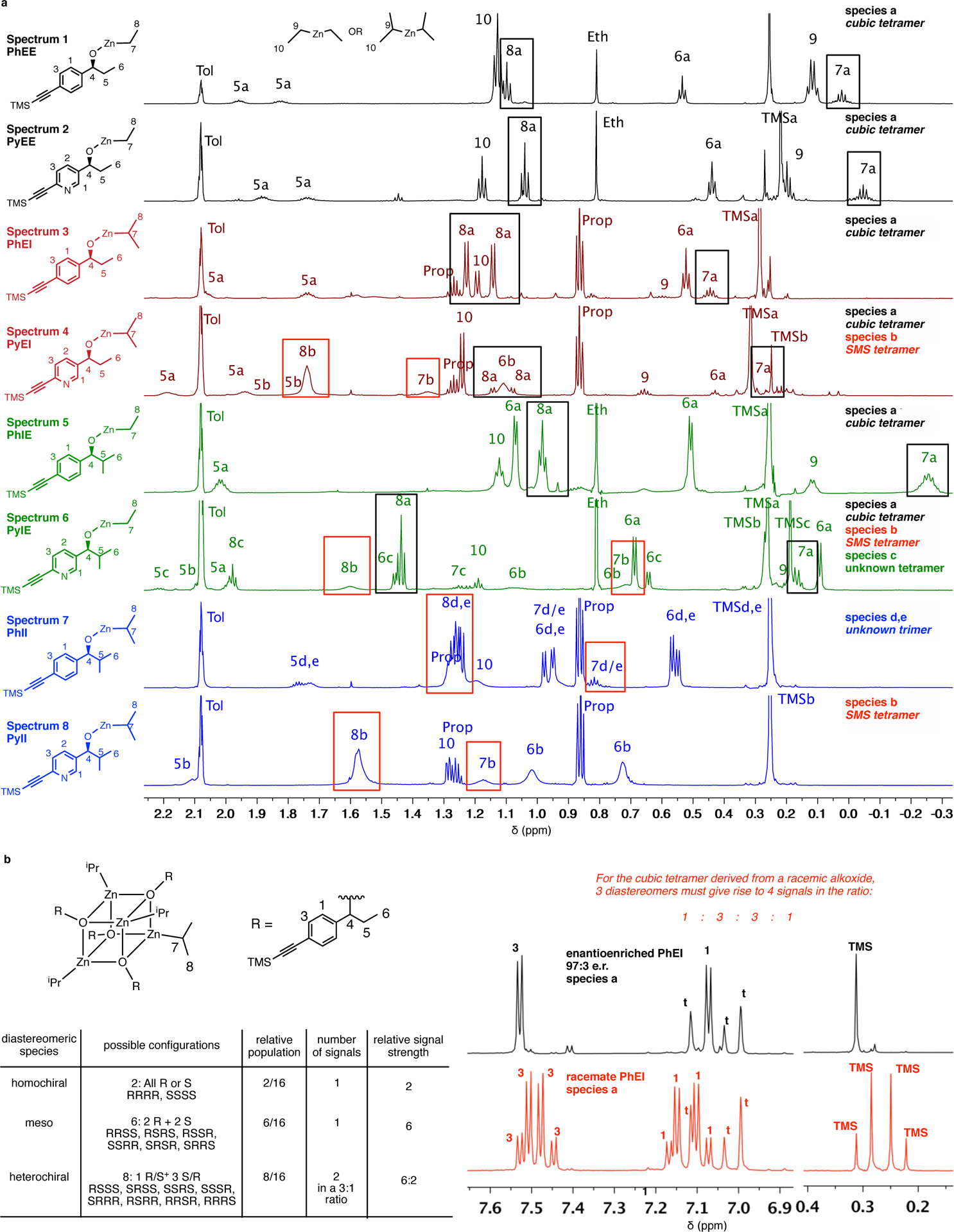
Spectroscopic characterization of zinc alkoxides reveals structure-dependent solution state aggregation. Partial 1H-NMR spectra (750 MHz) in toluene-d8 at 23 °C. tol/t = residual toluene, prop = propane, eth = ethane. Enantioenriched zinc alkoxides (alkoxides differing by only a nitrogen atom in the aromatic core are assigned the same color and are arranged successively) with signature chemical shifts for species a (black boxed) and species b (red boxed). Additional DOSY experiments (not shown here) and racemate analysis (see panel b) assist in characterizing aggregate species. Species a, observed for PhEE/EI/IE and PyEE/EI/IE, is a cubic tetramer and is characterized by sharp peaks and upfield chemical shifts for protons H(7) and H(8). In contrast, PyII exists exclusively as species b (the SMS tetramer) whose peaks are highly broadened, with downfield chemical shifts for protons H(7) and H(8). PyEI and PyIE exist as a mixture of species a and b. A third species, c (an uncharacterized tetrameric aggregate) is observed as a component of PyIE. PhII exists as a mixture of two uncharacterized trimers (species d, e)
Figure 4:
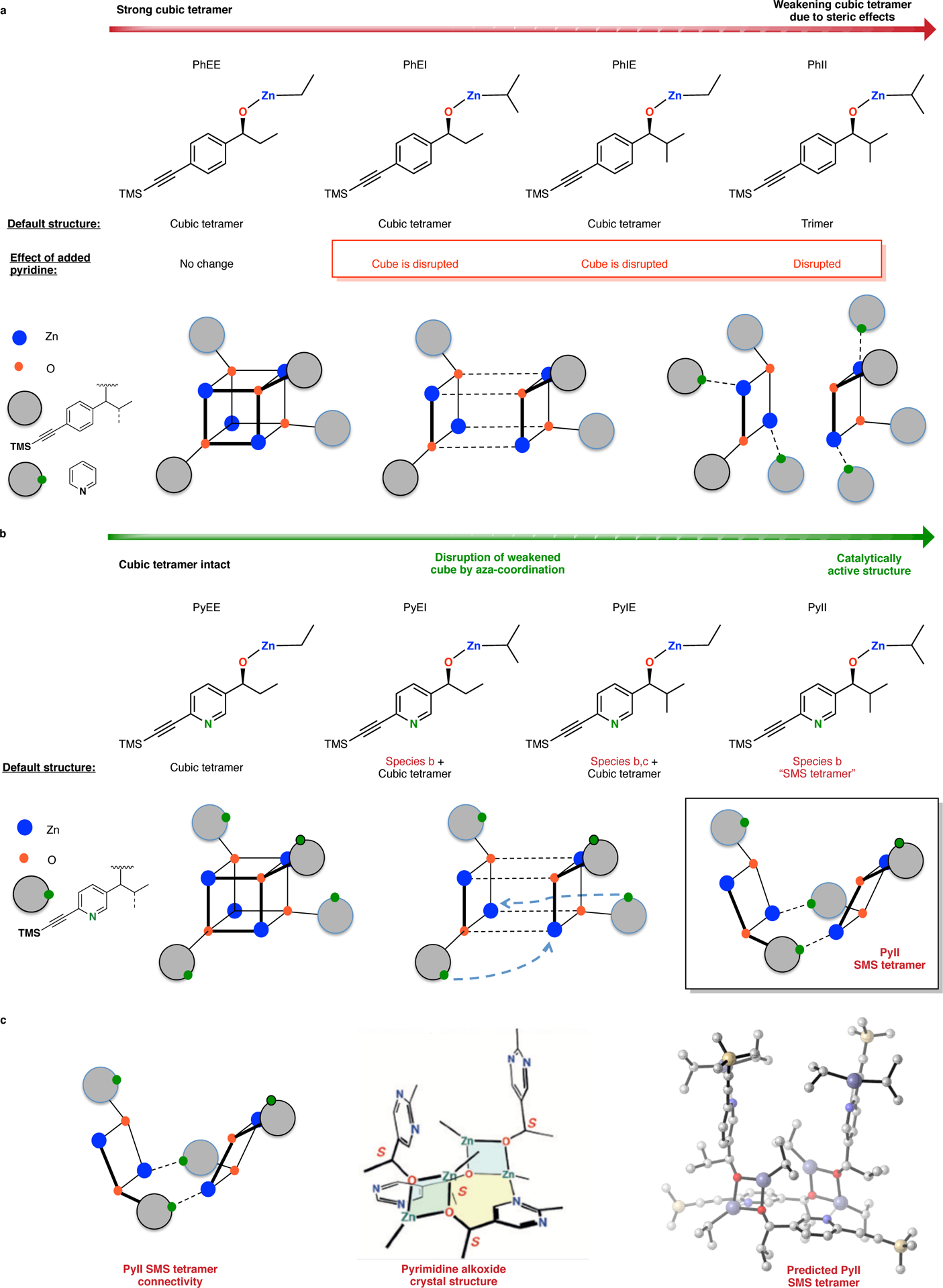
Diastereomeric species possible in a racemic cubic tetramer with their relative peak distributions. In the racemic cubic tetramer, three diastereomeric aggregates (homochiral, meso and heterochiral) can exist. The table lists all possible configurations of these species along with the expected, relative peak signal strengths. Note that the heterochiral aggregate alone will give rise to two peaks in the ratio 3:1. Thus, an overall quadruplet pattern (four peaks) arising from a mixture of the three aggregates is predicted. 1H NMR of the aryl and TMS region for racemic PhEI displays this predicted pattern.
In general, for all alkoxides, spectra obtained from racemic samples showed greater multiplicity of signals and additional peaks compared to spectra obtained from enantioenriched samples, proving that none of these alkoxides exist as monomers (Supplementary Information, Section 3). Figure 3 presents the upfield region of the eight, enantioenriched alkoxides to allow comparison of important spectral characteristics. Different colors highlight four possible combinations of the carbinol–alkyl and O–Zn–alkyl groups. For the enantioenriched phenyl alkoxides PhEE (spectrum 1), PhEI (spectrum 3) and PhIE (spectrum 5), a single set of peaks corresponding to a predominant species was observed (Supplementary Information, Section 3 for complete spectral comparisons). The signature characteristics of this species are the sharp, clearly resolved peaks with only minor perturbation over a wide temperature range (–20 – 50 °C, Supplementary Information, Section 3) and the atypical upfield chemical shifts of the alkylzinc protons H(7) and H(8) (Figure 3, black boxes) in comparison to the free dialkylzinc protons H(9) and H(10).
For alkoxide PhEI, a remarkable quadruplet splitting pattern in the racemic aggregate is observed in comparison to the enantiopure aggregate (Figure 4). Such a pattern is consistent with a symmetric, cubic tetramer structure, which finds ample precedent.43,44 The racemic cubic tetramer can exist as a mixture of three diastereomers depending on the configurations at the four constituent alkoxide stereogenic centers: (1) chiral (D2), (2) meso (S4) and (3) heterochiral (C1) (Figure 4). On the basis of the statistical populations of these three species, the ideal 1:3:3:1 peak pattern seen here provides compelling evidence for the structure of PhEI as a racemic, cubic tetramer.
The atypical upfield shift of the alkylzinc protons is believed to arise from shielding by the aryl groups and is also observed in the ethylzinc alkoxide of (S)-1-phenylethanol (Supplementary Table 5 and Figures 127–130), the methylzinc alkoxide of which has been proposed to form a cubic tetramer.45 On the basis of these observations, species PhEE, PhEI and PhIE are believed to form cubic tetramers (species a, Figure 3) in solution and the upfield chemical shifts of protons H(7) and H(8) (black boxes) are a diagnostic feature of this structure.
The spectrum of pyridylzinc alkoxide PyEE (spectrum 2) shows a predominant species matching the spectral characteristics of a cubic tetramer. Clearly, presence of the pyridine nitrogen in PyEE does not disrupt the natural preference to form a cubic tetramer. Increasing the bulk of the alkyl groups in the pyridyl series in species PyEI (spectrum 4) and PyIE (spectrum 6) still maintained the cubic tetramer assembly (species a, black boxed diagnostic peaks) but also showed the presence of other species (species b, c), the peaks of which are labeled with the suffix ‘b’ or ‘c’ in Figure 3 (c.f. H(7b), H(6c)). Broad peaks with downfield shifts of protons H(7) and H(8) (red boxes) characterize species b. A third species (c) with sharp peaks and downfield alkylzinc protons can be identified in the spectrum of PyIE (spectrum 6). In the spectra for the bulkiest derivatives of both phenyl and pyridyl complexes PhII (spectrum 7) and PyII (spectrum 8) respectively, diagnostic peaks for the cubic tetramer are completely absent. Two components can be detected in the spectrum of PhII (species d, e) whereas the single species with broad peaks seen in the spectrum of PyII resembles species b (in the spectra of PyEI and PyIE) in chemical shift patterns (red boxed). Species b and c also show diagnostic peak differences in the aryl region in comparison to species a (Supplementary Figure 105).
Diffusion Ordered Spectroscopy (DOSY) studies of the alkoxides for molecular weight estimations (Supplementary Figure 5, also see Supplementary Information Section 3 for detailed DOSY data) established that all of these compounds except PhII are tetrameric and are unchanged over a temperature range of 0 to 50 °C, highlighting the stability of these aggregates. DOSY analysis showed that PhII exists as a mixture of trimers. This evidence provides further support to the hypothesis that species a are cubic tetramers and reveals that species b and c represent distinct aggregate structures that are also tetrameric.
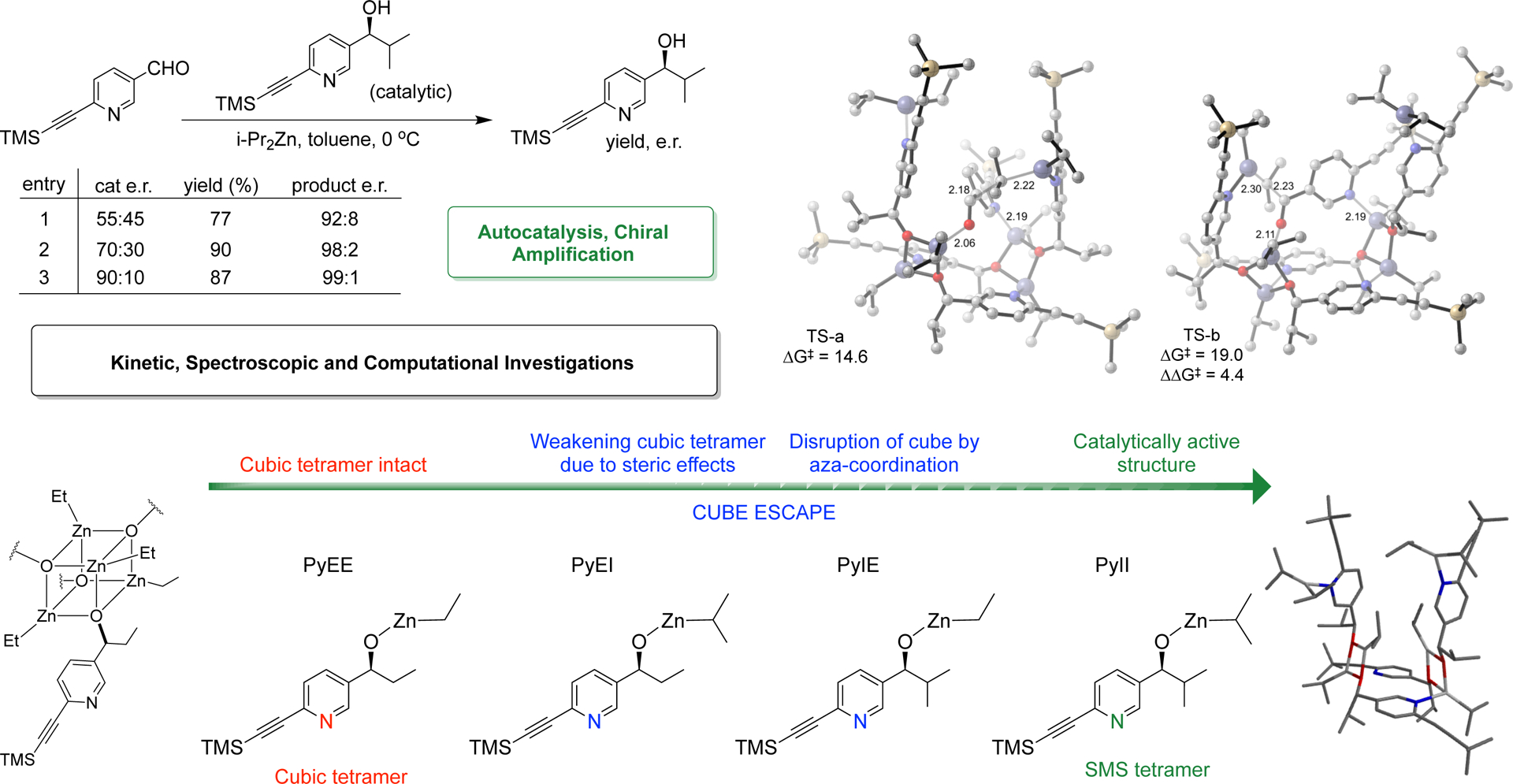
Rationalizing the effect of structure on alkoxide aggregation (Figure 5) begins with the assumption that a cubic tetramer is the preferred constitution for these alkylzinc alkoxides. However, replacement of ethyl groups around the cube core by bulkier isopropyl groups results in destabilization of the cube structure by steric repulsion to the point at which the cube is no longer viable as in cases of PhII and PyII. A weakened cube can be further disrupted by a pyridine nitrogen through coordination to the zinc alkyls to assist in the formation of alternative tetrameric aggregates. Indeed, addition of pyridine to solutions of PhEI, PhIE and PhII results in disruption of the aggregate structure whereas PhEE remains unaffected (Figure 5a) (Supplementary Figure 108). With a pyridine nitrogen available for coordination to zinc, alkoxides PyEE, PyEI, PyIE and PyII are poised to escape the cube only if the core is sufficiently weakened. The resulting continuum of structures displayed by these alkoxides represents this phenomenon. Whereas PyEE maintains a cubic tetramer constitution because its core is sufficiently stable to resist pyridine disruption, PyEI and PyIE exhibit formation of other tetrameric species owing to partial cube escape facilitated by nitrogen coordination. Finally, zinc alkoxide PyII represents the end point with complete cube escape and access to an exclusive, alternative tetrameric aggregate structure. The simplest way in which such a cube-escape can occur is an intramolecular coordination of the pyridyl nitrogen to expand the cube to a square-macrocycle-square (SMS) connectivity (Figure 5b). This connectivity is identical to the crystal structure described by Soai. Clearly, only a single nitrogen is required in the aromatic core to build this tetramer. In light of the striking similarity of PyII to the structure and properties of the original Soai system 2d, as well as the rationale for the assembly of the SMS tetramer, the proposal that its identity is analogous to that found in the recently disclosed X-ray crystal structure of 2c (the ‘Soai tetramer’) seems compelling (Figure 5c).
Figure 5:
Structural logic for the evolution of the catalytically active, PyII SMS tetramer. a. Sterically compromised PhXX cubic tetramers are disrupted by pyridine coordination. Note that PhEE is unaffected due to a strong cubic core. b. A conceptually similar ‘cube escape’ occurs in the PyXX alkoxides with intramolecular pyridine coordination yielding the SMS tetramer in case of PyII. PyEE, with minimal steric encumbrance, maintains the cubic tetrameric assembly. Partial cube escape in case of PyEI/IE results in a mixture of species. c. The predicted identity of the cube-escaped PyII SMS tetramer by analogy with the Soai pyrimidine alkoxide crystal structure. The PyII structure, with a central macrocycle and two ‘pyridine arms’ (each bound with diisopropylzinc molecules) facing the top face of the macrocycle, is identical in connectivity to the pyrimidine crystal structure.
Reactant Constraints of PyII and Kinetics with a Surrogate Substrate
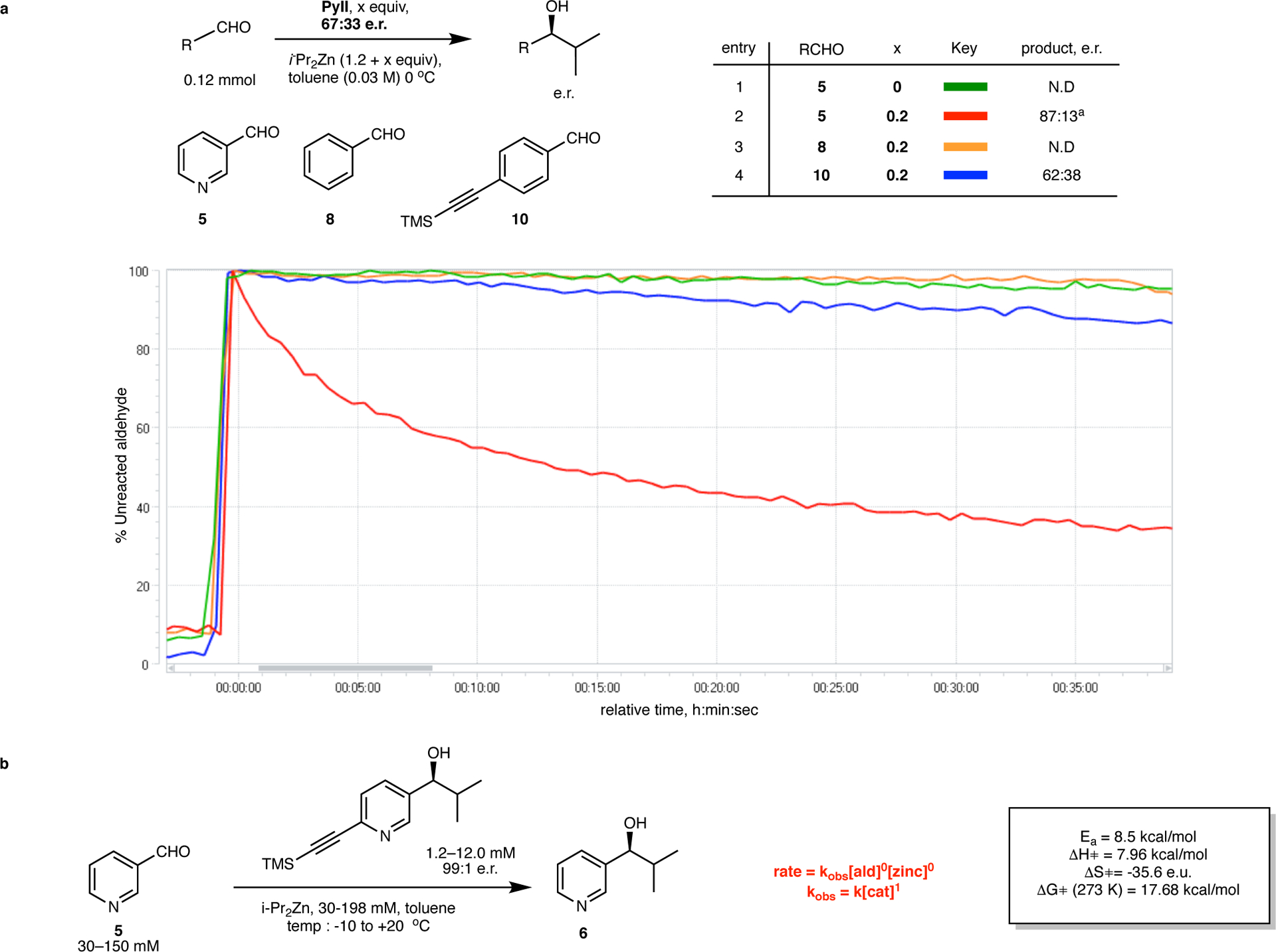
We discovered that PyII can maintain its critical attributes of enantioselective catalysis and positive non-linear effect in the non-autocatalytic alkylation of pyridine-3-aldehyde (5) with diisopropylzinc (Figure 6, entries 1 and 2). However, the aldehyde decay profile is reminiscent of a conventional catalyst-substrate reaction because the newly formed product contributes minimally to catalysis (Supplementary Figure 167)40. This reaction, in which pyridine-3-aldehyde acts as a surrogate substrate, is thus representative of the alkyl transfer step in the autocatalytic reaction, and was employed to provide insights into the mode of action of the PyII catalyst. In-situ IR monitoring of aldehyde consumption was employed for a comprehensive initial rate kinetic analysis of the diisopropylzinc alkylation of 5 catalyzed by PyII (Figure 6b, see Supplementary Information, section 2 for details). The initial rate displayed a first order dependence on catalyst concentration and zeroth order dependence on both 5 and diisopropylzinc (Supplementary Figure 6), indicating that saturation binding of both reactants to the catalyst aggregate precedes the rate limiting alkyl transfer. We believe that the decrease in conversion rate over time arises from catalyst poisoning and/or product inhibition by the product alkoxide that can mimic the PyII monomer and incorporate into the PyII aggregate. In theory, this hypothesis can be confirmed by the observation of a decreased initial rate in presence of the isopropylzinc alkoxide of 6 (the reaction product). However, such an experiment was precluded by the insolubility of this species when independently prepared. The insolubility of the isopropylzinc alkoxide of 6 should not be confused with the autocatalytic reaction of 3, which is completely homogenous; the surrogate system is simply an investigative tool to interrogate the alkyl transfer step. Activation parameters were determined assuming a tetrameric catalyst aggregate. A significant observation which highlights the substrate specificity of the PyII catalyst was the poor reactivity of benzaldehyde 8 and 4-(trimethylsilylethynyl)benzaldehyde 10 under identical catalytic conditions (Figure 6, entries 3 and 4).
Figure 6:
Substrate constraints and kinetic studies for the enantioselective, positive non-linear alkyl transfer activity of PyII a. In-situ IR monitoring profiles for aldehyde consumption in diisopropylzinc alkylations catalyzed by scalemic PyII. No reactivity is seen for substrates 8 and 10 whereas rapid reaction with a positive non-linear effect is observed with 5, suggesting that nitrogen coordination to a catalyst site is necessary for reactivity. Note that the reaction with 5 is not autocatalytic (since the product – the isopropylzinc alkoxide of 6 is an incompetent autocatalyst under these conditions) and demonstrates conventional catalyst-substrate kinetic profiles. 5 is thus utilized as a ‘competent surrogate’ to study the alkyl transfer activity of PyII. a measured at ~70% conversion. b. Substrate Rate law and Arrhenius parameters for the non-autocatalytic reaction with surrogate substrate 5, as measured using initial rate kinetic analysis. The reaction rate was found to be first order in catalyst concentration and zero order in both reactant concentrations.
Substrate Binding and Catalysis
For the hypothesis that PyII emulates the ‘Soai tetramer’ to be valid, it must also provide a clear explanation for the observed saturation kinetic behavior (with 5) and production of homochiral products from both 3 and 5. Catalysis must be facilitated by a reactant-binding model that would account for activation of the carbonyl group as well as an incoming diisopropylzinc prior to alkyl transfer. The critical demonstration of poor reactivity of aldehydes 8 and 10 (Figure 6a) indicates that pyridine-3-carbaldehydes are a privileged substrate class for the SMS tetramer owing to nitrogen coordination to a favorable site on the catalyst. Independent spectroscopic studies revealed that: (1) the dialkylzinc binds to the pyridine nitrogens of PyII (Supplementary Figure 107) as well as substrates 3 and 5 (Supplementary Figure 113), (2) the carbonyl group in 5 has an inherently poor affinity to diisopropylzinc and binding is exclusively at the ring nitrogen with diagnostic chemical shift changes (Supplementary Figure 112, 113) and, (3) the carbonyl group in 5 can coordinate to an unsaturated zinc in the alkoxide core (Supplementary Figure 110, 111).
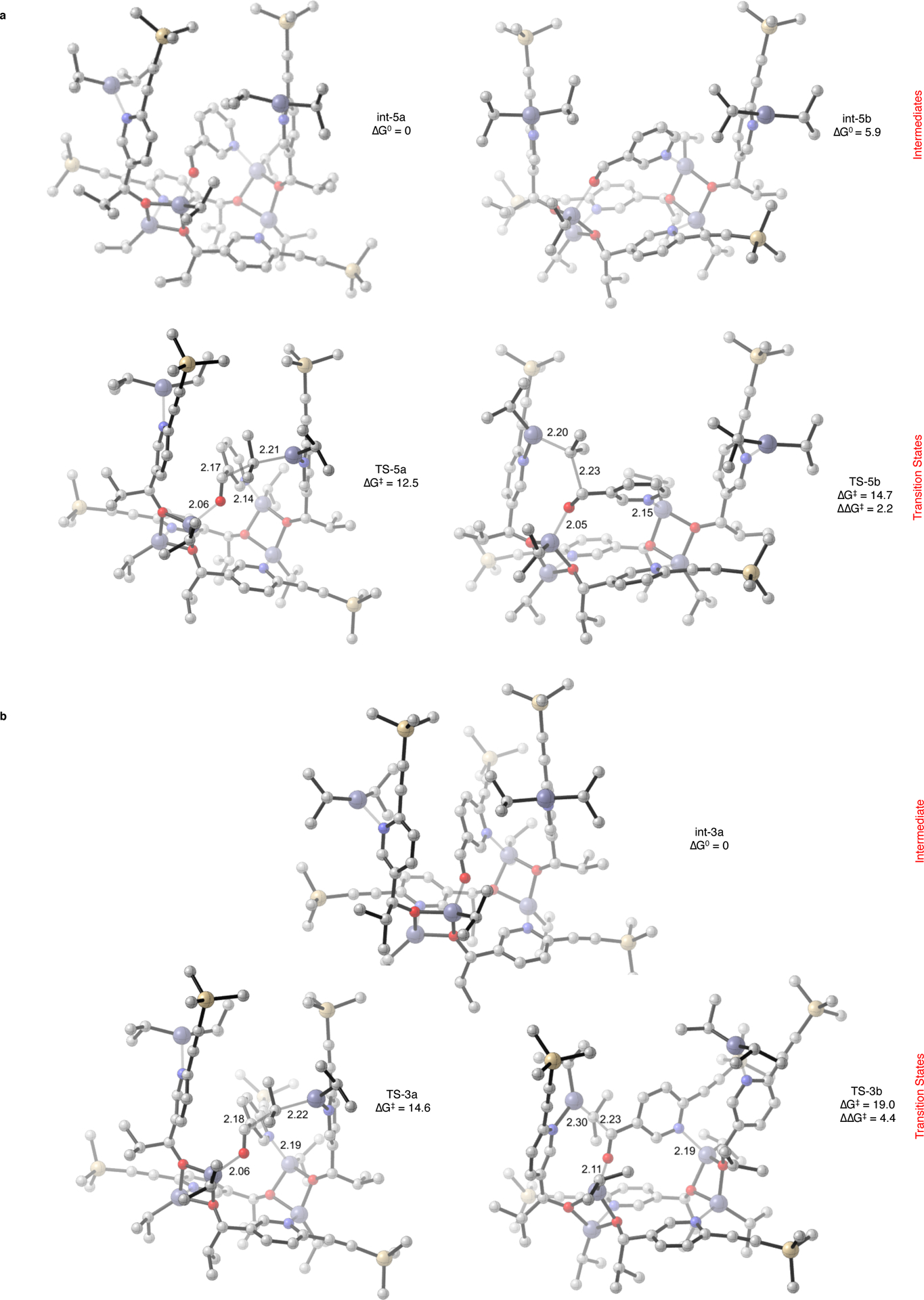
In light of these considerations, the energetic preferences of two proposed models (floor-to-floor and floor-to-arm) describing reactant docking prior to the alkyl transfer were interrogated with density functional theory (DFT) calculations (Figure 7). The crystal structure for 2e was employed as the starting geometry for the homochiral PyII tetramer after the replacement of pyrimidine cores and tert-butyl groups by pyridines and trimethylsilyl groups, respectively. In the floor-to-floor model, the substrate, through a two point binding, bridges the two distal unsaturated zinc alkoxide centers in the Zn–O squares whereas in the floor-to-arm model, the substrate through a two point binding, bridges an unsaturated zinc alkoxide atom in the Zn–O square and a dialkylzinc atom on the opposite arm. Note that in both models, the reactants are electronically activated toward alkyl transfer – the aldehyde (rendered more electrophilic) by coordination to the Lewis acidic zinc center and diisopropylzinc (rendered more nucleophilic) by coordination to the Lewis basic nitrogen. Computational evaluation of the binding of aldehyde 5 to homochiral PyII tetramer reveals a preference for the floor-to-floor model (int-5a, Figure 7a) (for the floor-to-arm model calculations, refer Supplementary Information, Section 5, int 5f-5h). Alkyl group transfer by diisopropylzinc is primed for delivery to the Si face of the aldehyde in int-5a, which would lead to the observed (S) stereoisomer. Conversely, the Re face is accessible only through a rotation of both the substrate and one arm of the tetramer as shown in int-5b, which is unfavorable by 5.9 kcal/mol. The stereocontrolling transition structures TS-5a and TS-5b arising from int-5a and int-5b, and having activation free energy barriers of 12.5 kcal/mol and 14.7 kcal/mol respectively are shown in Figure 7a. This energy difference is in agreement with the observed stereoselectivity of PyII with pyridine-3-carbaldehyde 5 (95:5 e.r., ΔΔG‡ = 1.7 kcal/mol, see Supplementary Table 1, Exp 5a-c). Although of no direct stereochemical consequence, it is interesting to note that the isopropyl group transfers (SE2 reactions) occur with “inversion of configuration” at the methine carbon (vide infra).
Figure 7:
Catalysis by the PyII SMS tetramer – DFT studies for floor-to-floor substrate docking and alkyl transfer. a. Catalysis with 5 as substrate. The carbonyl oxygen and aryl nitrogen of substrate 5 coordinate to the two unsaturated zinc centers across the macrocycle floor (int-5a/5b), thus the nomenclature ‘floor-to-floor’. These two bound intermediates respectively give rise to the competing transition state structures TS-5a/5b. The favored TS-5a involves delivery of the isopropyl group from the ‘right arm’ of the tetramer to produce the homomorphic product alkoxide. TS-5b, leading to the heteromorphic product via isopropyl delivery from the ‘left arm’, is disfavored by 2.2 kcal/mol. b. (Auto)catalysis with 3 as substrate. The only identified, floor-to-floor bound stable intermediate int-3a (analogous to int-5a) leads to the competing transition state structures TS-3a and TS-3b. The former is similar to TS-5a, where isopropyl delivery from the ‘right arm’ leads the observed homomorphic product. In contrast, TS-5b is highly disfavored (by 4.4 kcal/mol) because of the distortion required to accommodate the substrate for isopropyl delivery from the ‘left arm’. Note that the presence of the alkynyl substituent in substrate 3 precludes a geometry similar to TS-5b, thus contributing to increased selectivity in case of the autocatalytic reaction. Transition structures TS-3a–3b and TS-5a–5b, and optimized minima int-3a and int-5a–5b are calculated at the M06–2X/def2-TZVPP–SMD (toluene)//B3LYP/6–31G(d) level of theory. Energies reported in kcal/mol. Purple, zinc; yellow, silicon; red, oxygen; blue, nitrogen; gray, carbon. Hydrogens are hidden for clarity. ΔG0, free energy of activation. ΔΔG‡, relative free energy activation barrier.
The transition structures and minima for the autocatalytic reaction of aldehyde 3 and tetramer PyII were also investigated computationally (Figure 7b). The ground state geometry int-3a is quite similar to int-5a, N.B. the bulky aldehyde 3 is conformationally restricted in the tetramer. The lowest-energy transition structure TS-3a leads to the alcohol (S)-4, and the next higher energy transition structure TS-3b (4.4 kcal/mol) leads to the (R)-4 product. This large difference in energy comports with an enantioselective autocatalytic mechanism. TS-3a is analogous to TS-5a, in which facile alkyl transfer (with “inversion”) takes place from diisopropylzinc in the ground state geometry int-3a. Contrariwise, TS-3b, represents an SE2-type mechanism with “retention of configuration” at the methine of the transferring isopropyl group. This higher-energy pathway occurs because the left arm of the tetramer does not rotate for alkyl transfer to the Re-face of the aldehyde as in int-5b. This conformation is inaccessible for aldehyde 3 because to arrange this substrate to receive the isopropyl group would require that the trimethylsilylethynyl substituent be in the same space as the opposite arm (N.B., the C5 position of aldehyde 5 in both int-5b and TS-5b, which corresponds to the trimethylsilylethynyl-substituted position of 3). A similar type of transition structure was located for aldehyde 5, i.e. without the trimethylsilyethynyl group (TS-5e, Supplementary Figure 174), which was 5.2 kcal/mol higher in energy. This observation reveals that the constrained SE2 substitution with retention of configuration (TS-3b and TS-5e) is highly disfavored compared to the invertive SE2 pathway seen in TS-5a and TS-3a. Thus, the stereoselectivity arises from the steric environment of the chiral tetramer, which favors Si-face addition to the aldehyde when bound in the preferred floor-to-floor arrangement.
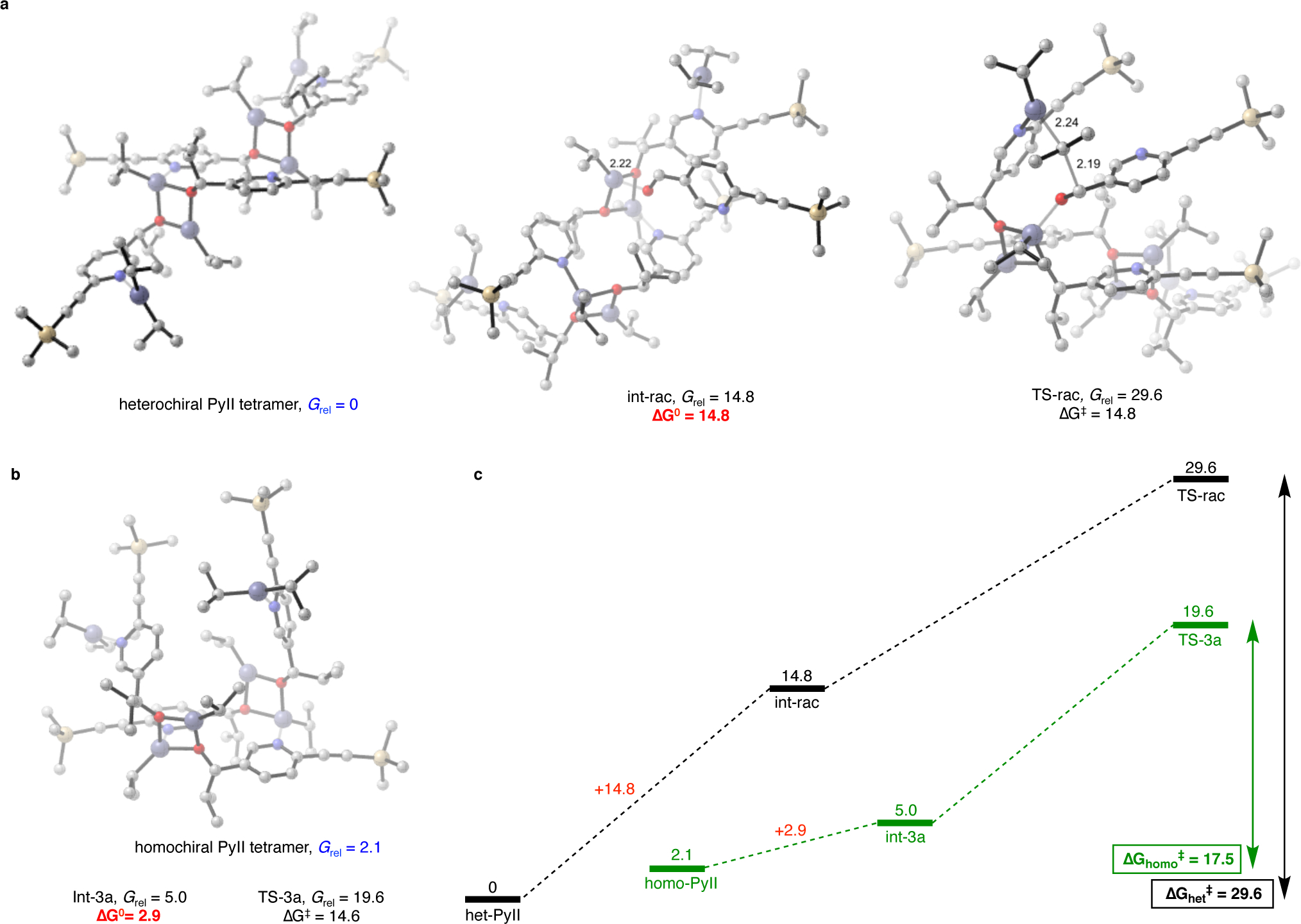
Finally, the catalytic competence of the heterochiral (racemic) PyII tetramer was computationally investigated (Figure 8). The heterochiral aggregate was modeled according to the Soai crystal structure for racemic 2c (Figure 1). The heterochiral tetramer is energetically preferred to the homochiral PyII tetramer by 2.1 kcal/mol (Grel blue). From inspection of this structure, it is immediately apparent that substrate binding in a two-point, floor-to-floor arrangement is impossible because the requisite, unsaturated zinc atoms are positioned on opposite sides of the macrocycle. Indeed, the only stable binding mode located for substrate 3 to the heterochiral PyII tetramer was a single-point binding leading to int-rac (Figure 8a red, ΔG0 = 14.8 kcal/mol). This complex is highly disfavored in comparison to the ‘floor-to-floor’ binding to the homochiral PyII tetramer leading to int-3a (Figure 8b red, ΔG0 = 2.9 kcal/mol). Interestingly, the activation barrier for the alkyl transfer int-rac to TS-rac (ΔG‡ = 14.8 kcal/mol) is nearly identical to the barrier for the homochiral assembly int-3a to TS-3a (ΔG‡ = 14.6 kcal/mol). Thus, the heterochiral PyII tetramer is catalytically incompetent not because the alkyl transfer is unfavorable, but rather because substrate binding is energetically inaccessible (Figure 8c). Therefore, because the heterochiral PyII tetramer is energetically preferred and kinetically incompetent compared to the homochiral PyII tetramer (ΔGhet‡ – ΔGhomo‡ = 12.1 kcal/mol), a mechanism for non-linearity in the asymmetric autocatalytic action of the homochiral PyII SMS tetramer is now established.
Figure 8.
Origin of non-linearity in the amplifying, autocatalytic reaction – DFT studies for comparison of catalysis by the heterochiral (panel a and black graphic in panel c) and homochiral (panel b and green graphic in panel c) PyII tetramers. The heterochiral tetramer is thermodynamically preferred to the homochiral tetramer by 2.1 kcal/mol (Grel numbers in blue). A floor-to-floor substrate binding to the heterochiral tetramer is impossible, because the requisite, unsaturated zinc atoms are located on opposite sides of the macrocycle. Alkyl transfer can thus occur only through a single-point binding pathway (int-rac and TS-rac), which is overall disfavored. Panel c summarizes the comparison in the form of a relative energy diagram. The disfavored binding to the heterochiral SMS tetramer (red, 14.8 kcal/mol) as compared to the homochiral SMS tetramer (red, 2.9 kcal/mol) results in the disparate activities of the two tetramers – catalysis by the latter (green pathway) is highly preferred over the former (black pathway). This thermodynamic preference for the heterochiral tetramer, along with its catalytic incompetence gives rise to the non-linear effect in the Soai reaction. Energies are calculated at the M06–2X/def2-TZVPP–SMD (toluene)//B3LYP/6–31G(d) level of theory and reported in kcal/mol. Purple, zinc; yellow, silicon; red, oxygen; blue, nitrogen; gray, carbon. Hydrogens are hidden for clarity. ΔG0, free energy of binding. ΔG‡, free energy activation barrier.
Discussion
The Soai reaction represents a fortuitous confluence of three phenomena in catalysis: autocatalysis, enantioselective catalysis and non-linear effects (asymmetric amplification). The pyridine-based autocatalytic system 3 provides a conceptual simplification because omission of a nitrogen atom in the aromatic core of the pyrimidine system in turn translates to four fewer coordinating sites on the SMS tetramer. The new system is amenable to detailed kinetic and spectroscopic investigations and presents decisive answers to critical mechanistic questions highlighted at the beginning of this article. The experimentally verified pyridine-coordination-assisted ‘cube escape’ model provides a compelling hypothesis to explain how the SMS macrocycle tetramer is a natural evolutionary endpoint of the pyridinyl (and presumably pyrimidyl) alkoxides. Conversely, the reaction of diethylzinc generates a catalytically inactive cubic tetramer product and fails to display asymmetric autocatalysis. The ring nitrogen atom with a 1,3-relationship to the alkoxide oxygen is indispensable for the assembly of the catalyst by bridging two dimeric units and providing binding sites for reactants on the catalyst arms. Complementarily, this 1,3-relationship in the substrate facilitates a two-point binding mode for aldehyde activation.
In other words, we suggest that the SMS tetramer allows catalysis with an ‘expansion’ of the cubic tetramer, made possible by replacing two Zn–O cube bonds by the aromatic pyridine linker (Figure 5). Such a structural change provides two unsaturated zinc atoms for reactant coordination, at a distance defined by the aromatic pyridine linkers, whose lengths precisely match the requisite dimensions for a ‘floor-to-floor’ substrate binding. The resulting bound complex is poised to produce the homochiral product alkoxide due to the constrained geometry of the tetramer, which favors alkyl delivery from only one diisopropylzinc-bound arm. This model also elucidates how inclusion of the alkynyl substituent naturally improves selectivity by further disfavoring the competing pathway through an unfavorable steric interaction. Likewise, benzaldehydes, incapable of such a two-point binding are incompetent substrates.
The floor-to-floor model presented here can be contrasted with the Gridnev proposal (Supplementary Figure 7). We have observed that the pyrimidine autocatalyst shares identical reactant constraints as the PyII tetramer – no reaction is seen with benzaldehydes 8 and 10. The Gridnev model presents no interaction of either the pyrimidine-nitrogen atoms or the unsaturated zinc atoms when in fact (based on the substrate constraints), just as is the case with PyII, the docking of these centers is clearly critical for successful catalysis. Furthermore, our computational studies indicate that the four-center cyclic, Noyori like alkyl transfer configuration proposed in the Gridnev model is prohibitively high in energy in comparison to the direct transfer seen in the floor-to-floor model (see Supplementary Information, Section 5, TS 5g-5h).
The articulation of the floor-to-floor model now allows for a simple resolution of another central issue – how does asymmetric amplification occur in the reaction? The geometry of the energetically preferred heterochiral racemic tetramer precludes such a floor-to-floor binding, making this adduct catalytically inactive. The phenomenological interpretation of this conclusion is illustrated in Supplementary Figure 8. Consider a scalemic (60:40, S/R e.r.) mixture of the PyII alkoxide. The racemic constituent of this mixture will preferentially form the heterochiral SMS tetramer, which is catalytically inactive. The excess, enantiopure component (in this case the S stereoisomer) will assemble into the catalytically active homochiral PyII aggregate. Owing to the highly selective catalysis by the homochiral autocatalyst, such a bifurcation of the scalemic alkoxide will give rise to a strong positive non-linear effect – in this case, formally manifesting as autocatalytic, asymmetric amplification as exemplified in the Frank scheme.
In conclusion, these results contribute to addressing longstanding mechanistic questions regarding the extraordinary Soai reaction, provide an opportunity to further broaden the reaction scope to other substituted pyridine-3-carbaldehydes, and will serve as a platform for further studies and explorations in the fascinating area of asymmetric autocatalysis.
Supplementary Material
Acknowledgments
We are grateful for generous financial support from the University of Illinois. S.V.A. is grateful to the University of Illinois for Graduate Fellowships. A.S. thanks the NIH Chemistry-Biology Interface Research Training Grant (T32GM008496). We are also grateful for the support services of the NMR, mass spectrometry and microanalytical laboratories of the University of Illinois at Urbana-Champaign.
Footnotes
Competing interests
The authors declare no competing interests.
Data Availability
All data mentioned in this manuscript is available as part of the main article or as Supplementary Information (Supplementary Figures 1–177, Tables 1 to 6). Initial rate kinetics data and DFT computation data is included in separate files.
References
- 1.Soai K, Shibata T, Morioka H & Choji K Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature 378, 767–768 (1995). [Google Scholar]
- 2.Soai K, Kawasaki T & Matsumoto A Asymmetric Autocatalysis of Pyrimidyl Alkanol and Its Application to the Study on the Origin of Homochirality. Acc. Chem. Res 47, 3643–3654, doi: 10.1021/ar5003208 (2014) [DOI] [PubMed] [Google Scholar]
- 3.Sato I, Urabe H, Ishiguro S, Shibata T & Soai K Amplification of Chirality from Extremely Low to Greater than 99.5 % ee by Asymmetric Autocatalysis. Angewandte Chemie International Edition 42, 315–317, doi: 10.1002/anie.200390105 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Soai K et al. Asymmetric synthesis of pyrimidyl alkanol without adding chiral substances by the addition of diisopropylzinc to pyrimidine-5-carbaldehyde in conjunction with asymmetric autocatalysis. Tetrahedron: Asymmetry 14, 185–188, doi: 10.1016/S0957-4166(02)00791-7 (2003). [DOI] [Google Scholar]
- 5.Singleton DA & Vo LK Enantioselective Synthesis without Discrete Optically Active Additives. J. Am. Chem. Soc 124, 10010–10011, doi: 10.1021/ja027129o (2002). [DOI] [PubMed] [Google Scholar]
- 6.Singleton DA & Vo LK A Few Molecules Can Control the Enantiomeric Outcome. Evidence Supporting Absolute Asymmetric Synthesis Using the Soai Asymmetric Autocatalysis. Org. Lett 5, 4337–4339, doi: 10.1021/ol035605p (2003). [DOI] [PubMed] [Google Scholar]
- 7.Soai K, Shibata T, Kowata Y: Japan Kokai Tokkyo Koho JP 1997 9–268179. Application date: February 1 and April 18, 1996. [Google Scholar]
- 8.Mislow K Absolute Asymmetric Synthesis: A Commentary. Collect. Czech. Chem. Commun 68, 849–864 (2003). [Google Scholar]
- 9.Sato I et al. Highly enantioselective synthesis of organic compound using right- and left-handed helical silica. Tetrahedron Lett 44, 721–724, doi: 10.1016/S0040-4039(02)02707-7 (2003). [DOI] [Google Scholar]
- 10.Lutz F, Sato I & Soai K The Asymmetric Power of Chiral Ligands Determined by Competitive Asymmetric Autocatalysis. Org. Lett 6, 1613–1616, doi: 10.1021/ol049559k (2004). [DOI] [PubMed] [Google Scholar]
- 11.Kawasaki T et al. Highly enantioselective asymmetric autocatalysis using chiral ruthenium complex-ion-exchanged synthetic hectorite as a chiral initiator. Organic & Biomolecular Chemistry 7, 1073–1075, doi: 10.1039/B823282B (2009). [DOI] [PubMed] [Google Scholar]
- 12.Sato I et al. Determination of absolute configurations of amino acids by asymmetric autocatalysis of 2-alkynylpyrimidyl alkanol as a chiral sensor. J. Organomet. Chem 692, 1783–1787, doi: 10.1016/j.jorganchem.2006.11.042 (2007). [DOI] [Google Scholar]
- 13.Kawasaki T et al. Asymmetric Amplification Using Chiral Cocrystals Formed from Achiral Organic Molecules by Asymmetric Autocatalysis. Angew. Chem. Int. Ed 44, 2774–2777, doi: 10.1002/anie.200462963 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Sato I et al. Asymmetric Induction by Helical Hydrocarbons: [6]- and [5]Helicenes. Angew. Chem. Int. Ed 40, 1096–1098, doi: (2001). [DOI] [PubMed] [Google Scholar]
- 15.Kawasaki T et al. Chiral Discrimination of Cryptochiral Saturated Quaternary and Tertiary Hydrocarbons by Asymmetric Autocatalysis. J. Am. Chem. Soc 128, 6032–6033, doi: 10.1021/ja061429e (2006). [DOI] [PubMed] [Google Scholar]
- 16.Kawasaki T et al. Enantioselective Synthesis of Near Enantiopure Compound by Asymmetric Autocatalysis Triggered by Asymmetric Photolysis with Circularly Polarized Light. J. Am. Chem. Soc 127, 3274–3275, doi: 10.1021/ja0422108 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki T et al. Autocatalysis Triggered by Carbon Isotope (13C/12C) Chirality. Science 324, 492–495, doi: 10.1126/science.1170322 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Kawasaki T et al. Asymmetric Autocatalysis: Triggered by Chiral Isotopomer Arising from Oxygen Isotope Substitution. Angew. Chem. Int. Ed 50, 8131–8133, doi: 10.1002/anie.201102263 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto A et al. Asymmetric Induction by a Nitrogen 14N/15N Isotopomer in Conjunction with Asymmetric Autocatalysis. Angew. Chem. Int. Ed 55, 15246–15249, doi:doi: 10.1002/anie.201608955 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blackmond DG The Origin of Biological Homochirality. Cold Spring Harbor Perspectives in Biology 2, a002147, doi: 10.1101/cshperspect.a002147 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flügel RM Chirality and Life: A Short Introduction to the Early Phases of Chemical Evolution 1 edn, (Springer-Verlag; Berlin Heidelberg, 2011). [Google Scholar]
- 22.Weissbuch I & Lahav M Crystalline Architectures as Templates of Relevance to the Origins of Homochirality. Chem. Rev 111, 3236–3267, doi: 10.1021/cr1002479 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Cintas P in Top. Curr. Chem (Springer-Verlag; Berlin Heidelberg, 2013). [Google Scholar]
- 24.Frank F On spontaneous asymmetric synthesis. Biochim. Biophys. Acta 11, 459–463 (1953). [DOI] [PubMed] [Google Scholar]
- 25.Blackmond DG, McMillan CR, Ramdeehul S, Schorm A & Brown JM Origins of Asymmetric Amplification in Autocatalytic Alkylzinc Additions. J. Am. Chem. Soc 123, 10103–10104, doi: 10.1021/ja0165133 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Buono FG & Blackmond DG Kinetic Evidence for a Tetrameric Transition State in the Asymmetric Autocatalytic Alkylation of Pyrimidyl Aldehydes. J. Am. Chem. Soc 125, 8978–8979, doi: 10.1021/ja034705n (2003). [DOI] [PubMed] [Google Scholar]
- 27.Sato I et al. Relationship between the time, yield, and enantiomeric excess of asymmetric autocatalysis of chiral 2-alkynyl-5-pyrimidyl alkanol with amplification of enantiomeric excess. Tetrahedron: Asymmetry 14, 975–979, doi: 10.1016/S0957-4166(03)00164-2 (2003). [DOI] [Google Scholar]
- 28.Gridnev ID, Serafimov JM & Brown JM Solution Structure and Reagent Binding of the Zinc Alkoxide Catalyst in the Soai Asymmetric Autocatalytic Reaction. Angew. Chem. Int. Ed 43, 4884–4887, doi: 10.1002/anie.200353572 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Islas JR et al. Mirror-symmetry breaking in the Soai reaction: A kinetic understanding. Proceedings of the National Academy of Sciences of the United States of America 102, 13743–13748, doi: 10.1073/pnas.0503171102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schiaffino L & Ercolani G Unraveling the Mechanism of the Soai Asymmetric Autocatalytic Reaction by First-Principles Calculations: Induction and Amplification of Chirality by Self-Assembly of Hexamolecular Complexes. Angew. Chem. Int. Ed 47, 6832–6835, doi: 10.1002/anie.200802450 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Ercolani G & Schiaffino L Putting the Mechanism of the Soai Reaction to the Test: DFT Study of the Role of Aldehyde and Dialkylzinc Structure. The Journal of Organic Chemistry 76, 2619–2626, doi: 10.1021/jo102525t (2011). [DOI] [PubMed] [Google Scholar]
- 32.Buhse T, Noble-Terán ME, Cruz J-M, Micheau J-C & Coudret C in Advances in Asymmetric Autocatalysis and Related Topics (eds Pályi Gyula, Kurdi Róbert, & Zucchi Claudia) 71–110 (Academic Press, 2017). [Google Scholar]
- 33.Klankermayer J, Gridnev ID & Brown JM Role of the isopropyl group in asymmetric autocatalytic zinc alkylations. Chem. Commun, 3151–3153, doi: 10.1039/B705978G (2007). [DOI] [PubMed] [Google Scholar]
- 34.Quaranta M, Gehring T, Odell B, Brown JM & Blackmond DG Unusual Inverse Temperature Dependence on Reaction Rate in the Asymmetric Autocatalytic Alkylation of Pyrimidyl Aldehydes. J. Am. Chem. Soc 132, 15104–15107, doi: 10.1021/ja103204w (2010). [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto A et al. Crystal Structure of the Isopropylzinc Alkoxide of Pyrimidyl Alkanol: Mechanistic Insights for Asymmetric Autocatalysis with Amplification of Enantiomeric Excess. Angew. Chem. Int. Ed 54, 15218–15221, doi: 10.1002/anie.201508036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gehring T, Busch M, Schlageter M & Weingand D A concise summary of experimental facts about the Soai reaction. Chirality 22, E173–E182, doi: 10.1002/chir.20849 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Shibata T, Yonekubo S & Soai K Practically Perfect Asymmetric Autocatalysis with (2-Alkynyl-5-pyrimidyl)alkanols. Angew. Chem. Int. Ed 38, 659–661, doi: (1999). [DOI] [PubMed] [Google Scholar]
- 38.Tanji S et al. Asymmetric autocatalysis of 5-carbamoyl-3-pyridyl alkanols with amplification of enantiomeric excess. Tetrahedron: Asymmetry 11, 4249–4253, doi: 10.1016/S0957-4166(00)00420-1 (2000). [DOI] [Google Scholar]
- 39.Shibata T, Choji K, Hayase T, Aizu Y & Soai K Asymmetric autocatalytic reaction of 3-quinolylalkanol with amplification of enantiomeric excess. Chem. Commun, 1235–1236, doi: 10.1039/CC9960001235 (1996). [DOI] [Google Scholar]
- 40.Soai K, Niwa S & Hori H Asymmetric self-catalytic reaction. Self-production of chiral 1-(3-pyridyl)alkanols as chiral self-catalysts in the enantioselective addition of dialkylzinc reagents to pyridine-3-carbaldehyde. J. Chem. Soc., Chem. Commun, 982–983, doi: 10.1039/C39900000982 (1990). [DOI] [Google Scholar]
- 41.Brown JM, Gridnev I & Klankermayer J in Amplification of Chirality (ed Soai Kenso) 35–65 (Springer; Berlin Heidelberg, 2008). [Google Scholar]
- 42.Romagnoli C, Sieng B & Amedjkouh M Asymmetric Amplification Coupling Enantioselective Autocatalysis and Asymmetric Induction for Alkylation of Azaaryl Aldehydes. Eur. J. Org. Chem 2015, 4087–4092, doi: 10.1002/ejoc.201500508 (2015). [DOI] [Google Scholar]
- 43.Noltes JG & Boersma J Investigations on organozinc compounds IX. Coordination chemistry of organozinc compounds RZnX: organozinc derivatives of tert-butanol, some phenols, diethylhydroxylamine and some oximes. J. Organomet. Chem 12, 425–431, doi: 10.1016/S0022-328X(00)88695-7 (1968). [DOI] [Google Scholar]
- 44.Jana S, Berger RJF, Fröhlich R, Pape T & Mitzel NW Oxygenation of Simple Zinc Alkyls: Surprising Dependence of Product Distributions on the Alkyl Substituents and the Presence of Water. Inorg. Chem 46, 4293–4297, doi: 10.1021/ic062438r (2007). [DOI] [PubMed] [Google Scholar]
- 45.Kitamura M, Okada S, Suga S & Noyori R Enantioselective addition of dialkylzincs to aldehydes promoted by chiral amino alcohols. Mechanism and nonlinear effect. J. Am. Chem. Soc 111, 4028–4036, doi: 10.1021/ja00193a040 (1989). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.