

Abstract
Background
There was a causal relationship between elevated lipoprotein(a) [Lp(a)] levels and increased risk of calcific aortic valve stenosis (CAVS) in whites and blacks. The present study aimed to investigate whether Lp(a) levels were associated with aortic stenosis (AS) severity and clinical events in Chinese patients.
Methods
Levels of serum Lp(a) were measured in 652 patients with CAVS, whom all underwent baseline echocardiographic examination. The clinical endpoint was defined as a composite of aortic valve replacement (AVR) and cardiac death.
Results
Patients in the tertile 3 of Lp(a) had a higher percentage of severe AS compared with those in the tertile 1 and 2 of Lp(a) (46.2% vs. 33.9%, P = 0.005). Moreover, the top tertile of Lp(a) was an independent predictor of severe AS (OR = 1.78, 95% CI: 1.18–2.66, P = 0.006). However, there was no significant association between tertile 3 of Lp(a) and clinical events (hazard ratio: 0.73; 95% CI: 0.43–1.24; P = 0.239) in the multivariate Cox regression analysis during a mean follow-up time of 3.16 ± 2.74 years.
Conclusions
Elevated Lp(a) level was an independent predictor of severe AS by echocardiography in the Chinese population, but was not associated with the increased risk of AVR and cardiac death, suggesting that Lp(a) levels might be helpful in the risk stratification of patients with CAVS.
Keywords: Aortic stenosis, Calcification, Severity, Lipoprotein(a), Prognosis
1. Introduction
Currently, calcific aortic valve stenosis (CAVS) presented the third commonest cardiovascular disease in western countries, which affected 0.4% of the general population.[1]–[3] With the coming of an aging society, CAVS would become a major disease burden in China. As statins therapy failed to show any beneficial effect on the development or progression of aortic stenosis (AS),[4]–[7] Surgical or transcatheter aortic valve replacement (AVR) remained to be the only optimal choice for the treatment of severe symptomatic stenosis.[8]
Lipoprotein(a) [Lp(a)] is a lipoprotein subclass, which composed of a low-density lipoprotein cholesterol (LDL-C) and an apolipoprotein B100 (apoB100) molecule, covalently connected to apolipoprotein(a), a novel glycoprotein homologous with plasminogen.[9] In a body of studies, it was found that Lp(a), the major lipoprotein carrier of oxidized phospholipids (OxPLs),[10],[11] acting as a causal risk factor for CAVS characterized by progressive calcification and remodeling.[12] A genome-wide association study had demonstrated that a genetic variation in the Lp(a) (LPA) locus (rs10455872) which determined plasma Lp(a) concentration, to be associated with the presence of CAVS.[13] Subsequent large-scale Mendelian randomization studies had validated the causal relationship of raised Lp(a) levels and a higher risk of aortic stenosis.[14],[15] However, more prospective studies should be needed to understand the association of Lp(a) and AS progression rate for the purpose of finding a new therapeutic target and pushing a new frontier in the battle against CAVS. Recently studies indicated that increased Lp(a) levels were significantly related to the faster progression rate of AS evaluated by echocardiography and increased the progression of valvular calcification by CT.[16],[17] Nevertheless, the majority of studies at present were performed in whites and blacks, whether that relationship still existed in the Chinese population had not been validated considering the genetic differences among diverse ethnic groups.
The present study prospectively analyzed a cohort of Chinese patients with CAVS to explore whether Lp(a) levels were associated with the severity and prognosis of CAVS.
2. Methods
2.1. Study design and population
The present study was a prospective, observational cohort study. Between June 2013 and July 2014, a total of 652 patients (age > 18 years) with CAVS were consecutively recruited in Fuwai hospital (Beijing, China). Inclusion criteria was defined as following: peak aortic valve velocity (Vpeak) > 2.5 m/s and thickening of the aortic valve, limitation of valve mobility, and localized hyperechoic calcification of aortic valve confirmed by echocardiography. Exclusion criteria included severe aortic regurgitation, planned aortic valve replacement, the use of lipid-lowing medications, symptomatic or severe coronary heart disease, congestive heart failure, significant liver and kidney dysfunction, apparent hematological abnormalities, malignant tumors, thyroid dysfunction, gout, congenital heart and valvular disease, and infective endocarditis (Figure 1). This study was performed based on the Declaration of Helsinki and was permitted by the hospital's ethical review board (Fu Wai Hospital & National Center for Cardiovascular Diseases, Beijing, China). All participants gave written informed consent.
Figure 1. The flow chart of the current study.

AS: aortic stenosis; AVR: aortic valve replacement; CAVS: calcific aortic valve stenosis.
For each patient, medical history was collected, and comorbidities and the cardiovascular risk profile were assessed. The morphology and structure of the aortic valve and the degree of aortic stenosis were observed by transthoracic two-dimensional echocardiography.
Hypertension was diagnosed by systolic blood pressure (SBP) ≥ 140 mmHg and/or diastolic blood pressure (DBP) ≥ 90 mmHg at least three times consecutively or receiving antihypertensive drugs therapy.
2.2. Echocardiography examination
The severity of AS was confirmed by an appointed echocardiographer on dedicated equipment (Phillips Epic7C, Netherlands) based on a formal protocol. 2-dimensional, M-mode and continuous wave Doppler examinations were performed with the use of an S5-1 pure wave probe. Continuous wave Doppler velocities were measured from the right sternal edge, apex, and suprasternal notch with the use of a D2 probe. Vpeak serves as a significant indicator for evaluating the severity of AS according to American Heart Association and American College of Cardiology criteria.[18] Peak and mean aortic valve pressure gradients were assessed by the modified Bernoulli equation. According to the recommendations on the Echocardiographic Assessment of Aortic Valve Stenosis from the European Association of Cardiovascular Imaging and the American Society of Echocardiography,[19] the definition of severe AS as follows: Vpeak ≥ 4 m/s or mean transvalvular gradient ≥ 40 mmHg.
2.3. Study outcome
All patients in the present study received telephone interviews by trained doctors who were blinded to the results of echocardiography and previous medical histories, and clinical outcomes of those patients were prospectively collected. The clinical endpoint was defined as a composite of AVR and cardiac death.
2.4. Laboratory tests
Plasma samples of all participants were collected in the morning after overnight fasting. Plasma levels of total cholesterol (TC), triglyceride (TG), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), apoA, apoB were measured using automatic biochemistry analyzer (Hitachi 7150, Tokyo, Japan). The serum Lp(a) levels were measured through an immune-turbidimetry assay (LASAY Lp(a) auto; SHIMA laboratories, Tokyo, Japan).
2.5. Statistical analysis
Statistical analyses were performed using SPSS version 22.0 software (SPSS Inc., Chicago, IL). A P value < 0.05 was considered statistically significant. Data were tested for normality and homogeneity with Levene variance distribution test. Continuous data were expressed as mean ± SD with normal distribution, or median (interquartile range) with the abnormal distribution. The differences in continuous variables between the two groups were assessed by Student's t-test or Mann-Whitney U tests as appropriate. Categorical variables were presented as n (%) and compared with the chi-square test or Fisher exact tests. The multivariate Logistic regression analyse was performed to confirm whether the tertile 3 of Lp(a) predicted severe AS. The event-free survival rates among tertiles of Lp(a) were measured by the Kaplan-Meier method and compared by the log-rank test.
3. Results
3.1. Baseline characteristics
A total of 652 patients with AS were enrolled in this study. Baseline demographic, laboratory and two-dimensional Doppler echocardiographic characteristics of all participants were shown on the basis of top Lp(a) tertile (Lp(a) > 38.15 mg/dL, n = 235) versus middle and bottom Lp(a) tertiles (Lp(a) ≤ 38.15 mg/dL, n = 417) in Table 1. The distribution of Lp(a) in the total population was shown in Figure 2.
Table 1. Baseline characteristics of all participants according to Lp(a) levels.
| Lp(a) ≤ 38.15 mg/dL Tertiles 1 and 2 (n = 417) | Lp(a) > 38.15 mg/dL Tertiles 3 (n = 235) | P-value | |
| Age, yrs | 60 ± 18 | 66 ± 15 | 0.009 |
| Male | 242 (58.1%) | 137 (58.6%) | 0.934 |
| BMI, kg/m2 | 23.30 ± 3.79 | 23.92 ± 3.69 | 0.213 |
| Hypertension | 226 (54.3%) | 155 (66.3%) | 0.006 |
| Diabetes mellitus | 39 (9.5%) | 53 (22.8%) | < 0.001 |
| Smoke | 139 (33.5%) | 95 (40.6%) | 0.233 |
| TC, mmol/L | 4.34 ± 1.12 | 4.68 ± 1.15 | 0.016 |
| TG, mmol/L | 1.42 ± 0.76 | 1.31 ± 0.66 | 0.215 |
| LDL-C, mmol/L | 2.68 ± 0.90 | 2.95 ± 0.95 | 0.016 |
| HDL-C, mmol/L | 1.17 ± 0.39 | 1.22 ± 0.35 | 0.259 |
| Lp(a), mg/dL | 11.65 (6.16–20.94) | 68.26 (47.49–95.19) | < 0.001 |
| ApoA, mg/dL | 1.34 ± 0.36 | 1.38 ± 0.35 | 0.355 |
| ApoB, mg/dL | 0.87 ± 0.28 | 0.98 ± 0.31 | 0.004 |
| Vpeak, m/s | 3.43 ± 1.14 | 3.70 ± 1.12 | 0.012 |
| Peak transvalvular gradient, mmHg | 52.22 ± 33.27 | 57.85 ± 34.38 | 0.064 |
| Mean transvalvular gradient, mmHg | 35.58 ± 20.50 | 36.99 ± 21.03 | 0.511 |
| Diameter of aortic valve ring, mm | 22.48 ± 2.78 | 22.27 ± 2.78 | 0.410 |
| Anteroposterior diameter of aortic valve sinus, mm | 32.02 ± 4.86 | 31.64 ± 4.41 | 0.383 |
| Diameter of ascending aorta, mm | 35.58 ± 6.97 | 35.20 ± 6.19 | 0.531 |
Data are expressed as mean ± SD or median (interquartile range) or n (%). ApoA: apolipoprotein A; ApoB: apolipoprotein B; BMI: body mass index; HDL-C: high density lipoprotein cholesterol; LDL-C: low density lipoprotein cholesterol; Lp(a): lipoprotein(a); TC: total cholesterol; TG: triglyceride; Vpeak: peak aortic valve velocity.
Figure 2. Distribution of Lp(a) levels in the total population.
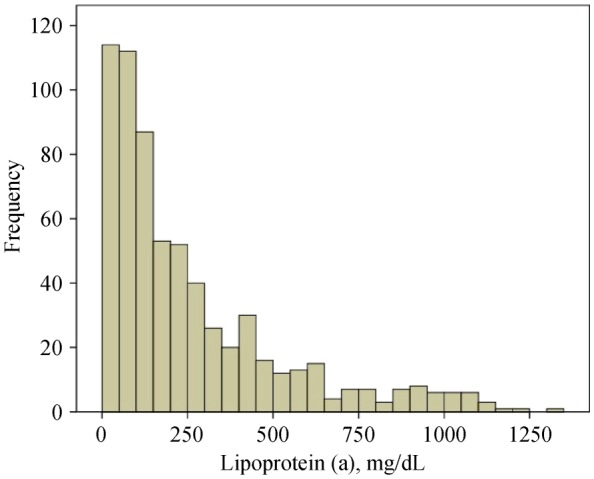
AS: aortic stenosis; CAVS: calcific aortic valve stenosis; Lp(a): lipoprotein(a); Vpeak: peak aortic valve velocity.
Compared with patients in middle and bottom Lp(a) tertiles, those in top tertile of Lp(a) tended to be older (66 ± 15 vs. 60 ± 18 years, P = 0.009), had higher prevalence of diabetes (22.8% vs. 9.5%, P < 0.001) and hypertension (66.3% vs. 54.3%, P = 0.006). Besides, those in tertile 3 of Lp(a) had higher TC levels (4.68 ± 1.15 vs. 4.34 ± 1.12 mmol/L, P = 0.016), higher LDL-C levels (2.95 ± 0.95 vs. 2.68 ± 0.90 mmol/L, P = 0.016), and higher Vpeak(3.70 ± 1.12 vs. 3.43 ± 1.14 mmol/L, P = 0.012). However, the differences in peak transvalvular gradient, mean transvalvular gradient, anteroposterior diameter of aortic valve sinus, the diameter of ascending aorta between two groups did not reach statistical significance.
3.2. Lp(a) levels and the severity of AS
Compared with those in bottom and middle tertiles of Lp(a), patients in top tertile of Lp(a) had higher percentage of severe AS (46.2% vs. 33.9%, P = 0.005, Figure 2). Moreover, after adjusting for the traditional risk factors, including age, sex, body mass index (BMI), hypertension, diabetes, smoking, and LDL-C, multivariate Logistic regression analysis showed that the tertile 3 of Lp(a) was a independent predictor of severe AS evaluated by echocardiography (OR = 1.78, 95% CI: 1.18–2.66, P = 0.006) (Table 2). Moreover, hypertension (OR = 2.40, 95% CI: 1.52–3.80, P < 0.001) and diabetes (OR 2.74, 95% CI 1.54–4.87, P = 0.001) were also independently associated with severe AS.
Table 2. Logistic regression analysis for predictors of severe AS.
| Univariate logistic regression |
Multivariate logistic regression |
|||
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| Age | 1.01 (0.99–1.02) | 0.483 | 1.00 (0.98–1.01) | 0.697 |
| Male | 1.23 (0.86–1.74) | 0.254 | 1.26 (0.79–2.00) | 0.332 |
| BMI | 1.04 (0.99–1.09) | 0.162 | 1.03 (0.98–1.09) | 0.286 |
| Hypertension | 1.92 (1.33–2.77) | 0.001 | 2.40 (1.52–3.80) | < 0.001 |
| Diabetes | 2.35 (1.44–3.82) | 0.001 | 2.74 (1.54–4.87) | 0.001 |
| Smoke | 1.20 (0.84–1.72) | 0.327 | 1.12 (0.70–1.79) | 0.652 |
| LDL-C | 0.89 (0.74–1.08) | 0.239 | 0.84 (0.68–1.04) | 0.108 |
| Tertile 3 of Lp(a) | 1.68 (1.17–2.41) | 0.005 | 1.78 (1.18–2.66) | 0.006 |
AS: aortic valve stenosis; BMI: body mass index; LDL-C: low density lipoprotein-cholesterol; Lp(a): lipoprotein(a).
3.3. Lp(a) and clinical events
During a mean follow-up time of 3.16 ± 2.74 years, 293 patients with severe AS who received planned AVR were excluded, patients with mild to moderate AS (> 2.5 m/s < Vpeak < 4 m/s) were followed-up for the composite endpoint of unplanned AVR and cardiac death (n = 359). Among 359 patients with mild to moderate AS, 75 patients received unplanned AVR operation and four patients died of cardiac cause. After adjustment for age, sex, BMI, hypertension, diabetes, smoke and LDL-C, no significant association was found between tertile 3 of Lp(a) and higher rate of clinical events (hazard ratio = 0.73; 95% CI: 0.43–1.24; P = 0.239) in the multivariate Cox regression analysis (Figure 3). As depicted in Figure 4, the Kaplan-Meier survival curve showed no significant difference in the event-free survival rates between tertile 3 level of Lp(a) and tertile 1 or 2 level of Lp(a).
Figure 3. The percentages of severe AS according to plasma Lp(a) levels.

AS: aortic valve stenosis; BMI: body mass index; LDL-C: low density lipoprotein-cholesterol; Lp(a): lipoprotein(a).
Figure 4. Multivariate Cox regression analysis for AVR and cardiac death.

AS: aortic stenosis; AVR: aortic valve replacement; BMI: body mass index; LDL-C: low density lipoprotein-cholesterol.
Figure 5. The event-free Survival analysis according to lipoprotein(a) [Lp(a)] levels for aortic valve replacement and cardiac death.
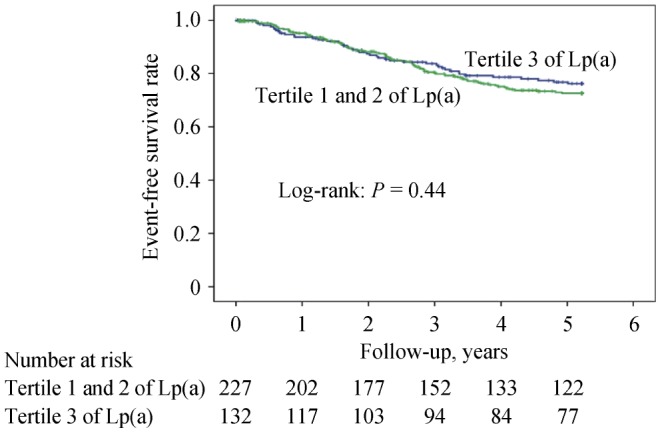
Lp(a): lipoprotein(a).
4. Discussion
The present study confirmed that increased levels of Lp(a) was an independent predictor of severe AS in the Chinese patients with CAVS assessed by echocardiography. However, in our study, no association was found between increased levels of Lp(a) and clinical events, including AVR and cardiac death during the follow-up period.
Recent studies suggested the increased plasma levels of Lp(a) determined by gene were closely associated with the aortic valve calcification (AVC) and stenosis in the general population.[13]–[15],[20] Lp(a) was firstly reported as a risk factor for AS by an epidemiological survey in 1995,[21] which was then confirmed by several cross-sectional and retrospective studies.[22]–[24] Then, Thanassoulis, et al.[13] found that one SNP in the LPA locus encoding Lp(a) (rs10455872) was associated with AVC detected by CT or clinical AS. The Copenhagen City Heart Study (1991 to 2011; n = 10,803) and the Copenhagen General Population Study (2003 to 2011; n = 66,877)[14] demonstrated the significant relationship between increased Lp(a) levels, corresponding genotypes and elevated risk of AS. Patients with Lp(a) > 90 mg/dL had a 2.9-fold risk of AS. In the EPIC-Norfolk study[15] which included 17,553 participants, Mendelian randomization study and replication in a case-control cohort indicated an increased risk of AS in participants with rs10455872 and Lp(a) > 50 mg/dL, and Lp (a) ≥ 50 mg/dL was associated with an HR of 1.98 for risk of AS. MESA study explored the relationship between risk of Lp(a) and CAVS in 4678 multi-ethnic participants,[20] the results showed that Lp(a) ≥ 30 mg/dL was associated with AVC in white subjects (RR = 1.56; 95% CI: 1.24–1.96); while in black subjects, that relationship only reached the verge of statistical difference (RR = 1.55, 95% CI: 0.98–2.44, P = 0.059). The prevalence of AVC increased significantly in whites with Lp(a) ≥ 50 mg/dL (RR = 1.72, 95% CI: 1.36–2.17). In a recent study, it was also found the plasma Lp(a) levels were positively correlated with the severity of calcification in patients with the bicuspid aortic valve.[25] These results revealed a significant association between the level of Lp(a) and the degree of AVC in white and black individuals. However, Thanassoulis, et al.[13] suggested no significant association between Lp(a) levels and AVC in 774 Chinese Americans. In the MESA study,[20] no significant association was also found between Lp(a) and AVC in 559 Chinese Americans. Therefore, it is necessary to further verify the causal relationship between Lp(a) and AVC in the Chinese population.
The progression rate of AS was a crucial clinical aspect during the follow-up period. The ASTRONOMER trial[16] investigated the relationship between Lp(a) levels and the progression rate of valve stenosis by echocardiography, in which those in the top tertile of Lp(a) (Lp(a) ≥ 58.5 mg/dL) were found to have a 2-fold progression rate of AS than those in the middle and bottom tertiles of Lp(a) (Lp(a) < 58.5 mg/dL) after a mean follow-up of 3.5 ± 1.2 years. The ASTRONOMER study presented that raised Lp(a) levels associated with the fast progression of AS for the first time. A recent study, which combined the data from the Ring of Fire study and the SALTIRE study, showed that patients in the top tertile had more rapid -progression of AS by echocardiography as well as increased valvular calcification by CT.[17] Moreover, Capoulade, et al.[26] found that there was a linear relationship between plasma Lp(a) levels and AS progression rate in patients with mild to moderate CAVS, and that Lp(a) levels per 10-mg/dL increase was associated with a 1.10 OR for rapid AS progression.Moreover, the present study provide additional evidence that elevated Lp(a) levels (Lp(a) > 38.15 mg/dL) was an independent predictor of severe AS in Chinese population. In the ASTRONOMER study,[16] those patients in the top tertile of Lp(a) had a higher rate of clinical events including AVR and cardiac death. Zheng, et al.[17] analyzed results of the Ring of Fire study and the SALTIRE study, and found that elevated Lp(a) independently associated with an increased clinical event rate. However, we did not find a significant association between elevated plasma levels of Lp(a) and the need for AVR and death. The differences between the results of our study and those of the two prior studies might be attributed to the following reasons. Firstly, the mean age of patients in the present study (65 ± 14 years) was significant differ from those in the ASTRONOMER study (58 ± 13 years) and those in the Ring of Fire study and the SALTIRE study (70.3 ± 9.9 years). Secondly, the Lp(a) levels of participants in our study were lower than those in the ASTRONOMER study, and Lp(a) level was mainly dominated by the LPA gene. Thus, the ethnic diversities in gene variations may contribute to the difference of results. Therefore, whether the relationship between Lp(a) and severe AS or faster progression of AS is different among different ethnicity still needs further genetic extensive sample studies in the Chinese population.
The underlying mechanism that Lp(a) augmented the progression of CAVS may mainly through inducing the calcification of normal human aortic valve interstitial cells (HAVICs).[12] Lp(a) deposited in valvular tissue, increased ROS formation, promoted inflammatory response, and osteogenic differentiation.[27] A bound of evidence suggested that the plasma lipoprotein-associated phospholipase A2-phospholipase A2 [Phospholipase A2 (Lp-PLA2)] levels were significantly increased in patients with CAVS. Lp-PLA2 is also highly expressed in stenotic and calcified aortic valvular tissues, where it may lead to mineralization of HAVICs and the remodeling of the aortic valve.[28]–[30] Lp-PLA2 hydrolyzes oxidized phospholipids (oxPLs) carried by Lp(a) and produces lysophosphatidylcholine (LPC), a highly active proinflammatory and procalcifying factor[31] and play a role in inducing osteogenesis and mineralization of HAVICs.[29] Autotaxin (ATX), a lysophospholipase D enzyme, carried and transported by Lp(a) to the aortic valves. ATX breaks down LPC from oxPLs and generates free fatty acids and lysophosphatidic acid (LPA),[10],[32],[33] which is characterized by actively promoting inflammatory response and osteogenic transformation of HAVICs through LPAR1-RhoA-NF-κB pathway and inducing the expression of IL-6 and BMP-2.[32] In summary, the interaction of Lp(a), Lp-PLA2, ATX, LPA accelerated the progression of CAVS.
There were several limitations to the present study. Firstly, the relatively small sample size and the exclusion of patients who received planned AVR for severe AS might confine the generalizability of this study. Secondly, we did not perform annualized echocardiography examines for enrolled participants, so we cannot investigate the relationship of Lp(a) levels and rapid progression of AS in Chinese patients. Finally, we did not detect the LPA gene of the participants, which may be robust to demonstrate the results difference caused by ethnicity variation. Further investigations with a large number of Chinese patients and regular echocardiographic follow-up were needed.
In conclusion, this study demonstrated that elevated Lp(a) level was an independent predictor of the severe AS by echocardiography in the Chinese population, but was not associated with the increased risks of AVR and cardiac death, suggesting that Lp(a) levels might be helpful in the risk stratification of patients with CAVS.
Acknowledgments
We declare that we do not have any conflict of interest. We thanked patient advisers for the information they provided.
References
- 1.Nkomo VT, Gardin JM, Skelton TN, et al. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368:1005–1011. doi: 10.1016/S0140-6736(06)69208-8. [DOI] [PubMed] [Google Scholar]
- 2.d'Arcy JL, Coffey S, Loudon MA, et al. Large-scale community echocardiographic screening reveals a major burden of undiagnosed valvular heart disease in older people: the OxVALVE Population Cohort Study. Eur Heart J. 2016;37:3515–3522. doi: 10.1093/eurheartj/ehw229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindman BR, Clavel MA, Mathieu P, et al. Calcific aortic stenosis. Nat Rev Dis Primers. 2016;2:16006. doi: 10.1038/nrdp.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao Y, Nicoll R, He YH, et al. The effect of statins on valve function and calcification in aortic stenosis: a meta-analysis. Atherosclerosis. 2016;246:318–324. doi: 10.1016/j.atherosclerosis.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 5.Moura LM, Ramos SF, Zamorano JL, et al. Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J Am Coll Cardiol. 2007;49:554–561. doi: 10.1016/j.jacc.2006.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossebo AB, Pedersen TR, Boman K, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 7.Cowell SJ, Newby DE, Prescott RJ, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- 8.Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2438–2488. doi: 10.1016/j.jacc.2014.02.537. [DOI] [PubMed] [Google Scholar]
- 9.Garg V. The role of lipoprotein(a) in calcific aortic valve disease: insights from a large-cohort genetic study. JAMA Cardiol. 2018;3:24–25. doi: 10.1001/jamacardio.2017.4267. [DOI] [PubMed] [Google Scholar]
- 10.Torzewski M, Ravandi A, Yeang C, et al. Lipoprotein(a) associated molecules are prominent components in plasma and valve leaflets in calcific aortic valve stenosis. JACC Basic Transl Sci. 2017;2:229–240. doi: 10.1016/j.jacbts.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tselepis AD. Oxidized phospholipids and lipoprotein-associated phospholipase A2 as important determinants of Lp(a) functionality and pathophysiological role. J Biomed Res. 2016;32:13–22. doi: 10.7555/JBR.31.20160009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu B, Hafiane A, Thanassoulis G, et al. Lipoprotein(a) induces human aortic valve interstitial cell calcification. JACC Basic Transl Sci. 2017;2:358–371. doi: 10.1016/j.jacbts.2017.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63:470–477. doi: 10.1016/j.jacc.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 15.Arsenault BJ, Boekholdt SM, Dube MP, et al. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet. 2014;7:304–310. doi: 10.1161/CIRCGENETICS.113.000400. [DOI] [PubMed] [Google Scholar]
- 16.Capoulade R, Chan KL, Yeang C, et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J Am Coll Cardiol. 2015;66:1236–1246. doi: 10.1016/j.jacc.2015.07.020. [DOI] [PubMed] [Google Scholar]
- 17.Zheng KH, Tsimikas S, Pawade T, et al. Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J Am Coll Cardiol. 2019;73:2150–2162. doi: 10.1016/j.jacc.2019.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg. 2014;148:e1–e132. doi: 10.1016/j.jtcvs.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 19.Baumgartner H, Hung J, Bermejo J, et al. Recommendations on the Echocardiographic Assessment of Aortic Valve Stenosis: A Focused Update from the European Association of Cardiovascular Imaging and the American Society of Echocardiography. J Am Soc Echocardiogr. 2017;30:372–392. doi: 10.1016/j.echo.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Cao J, Steffen BT, Budoff M, et al. Lipoprotein(a) levels are associated with subclinical calcific aortic valve disease in white and black individuals: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:1003–1009. doi: 10.1161/ATVBAHA.115.306683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gotoh T, Kuroda T, Yamasawa M, et al. Correlation between lipoprotein(a) and aortic valve sclerosis assessed by echocardiography (the JMS Cardiac Echo and Cohort Study) Am J Cardiol. 1995;76:928–932. doi: 10.1016/s0002-9149(99)80263-x. [DOI] [PubMed] [Google Scholar]
- 22.Stewart BF, Siscovick D, Lind BK, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol. 1997;29:630–634. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 23.Glader CA, Birgander LS, Soderberg S, et al. Lipoprotein(a), Chlamydia pneumoniae, leptin and tissue plasminogen activator as risk markers for valvular aortic stenosis. Eur Heart J. 2003;24:198–208. doi: 10.1016/s0195-668x(02)00385-8. [DOI] [PubMed] [Google Scholar]
- 24.Bozbas H, Yildirir A, Atar I, et al. Effects of serum levels of novel atherosclerotic risk factors on aortic valve calcification. J Heart Valve Dis. 2007;16:387–393. [PubMed] [Google Scholar]
- 25.Sticchi E, Giusti B, Cordisco A, et al. Role of lipoprotein (a) and LPA KIV2 repeat polymorphism in bicuspid aortic valve stenosis and calcification: a proof of concept study. Intern Emerg Med. 2019;14:45–50. doi: 10.1007/s11739-018-1925-8. [DOI] [PubMed] [Google Scholar]
- 26.Capoulade R, Yeang C, Chan KL, et al. Association of mild to moderate aortic valve stenosis progression with higher lipoprotein(a) and oxidized phospholipid levels: secondary analysis of a randomized clinical trial. JAMA Cardiol. 2018;3:1212–1217. doi: 10.1001/jamacardio.2018.3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu B, Khan K, Hamid Q, et al. Pathological significance of lipoprotein(a) in aortic valve stenosis. Atherosclerosis. 2018;272:168–174. doi: 10.1016/j.atherosclerosis.2018.03.025. [DOI] [PubMed] [Google Scholar]
- 28.Capoulade R, Mahmut A, Tastet L, et al. Impact of plasma Lp-PLA2 activity on the progression of aortic stenosis: the PROGRESSA study. JACC Cardiovasc Imaging. 2015;8:26–33. doi: 10.1016/j.jcmg.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 29.Mahmut A, Boulanger MC, El Husseini D, et al. Elevated expression of lipoprotein-associated phospholipase A2 in calcific aortic valve disease: implications for valve mineralization. J Am Coll Cardiol. 2014;63:460–469. doi: 10.1016/j.jacc.2013.05.105. [DOI] [PubMed] [Google Scholar]
- 30.Kolasa-Trela R, Fil K, Bazanek M, et al. Lipoprotein-associated phospholipase A2 is elevated in patients with severe aortic valve stenosis without clinically overt atherosclerosis. Clin Chem Lab Med. 2012;50:1825–1831. doi: 10.1515/cclm-2012-0015. [DOI] [PubMed] [Google Scholar]
- 31.Macphee CH, Nelson JJ, Zalewski A. Lipoprotein-associated phospholipase A2 as a target of therapy. Curr Opin Lipidol. 2005;16:442–426. doi: 10.1097/01.mol.0000174155.61307.5f. [DOI] [PubMed] [Google Scholar]
- 32.Nsaibia MJ, Boulanger MC, Bouchareb R, et al. OxLDL-derived lysophosphatidic acid promotes the progression of aortic valve stenosis through a LPAR1-RhoA-NF-kappaB pathway. Cardiovasc Res. 2017;113:1351–1363. doi: 10.1093/cvr/cvx089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouchareb R, Mahmut A, Nsaibia MJ, et al. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation. 2015;132:677–690. doi: 10.1161/CIRCULATIONAHA.115.016757. [DOI] [PubMed] [Google Scholar]