

Abstract
The emerging disease SARS is caused by a novel coronavirus that encodes several unusual RNA-processing enzymes, including non-structural protein 15 (Nsp15), a hexameric endoribonuclease that preferentially cleaves at uridine residues.1., 2., 3. How Nsp15 recognizes and cleaves RNA is not well understood and is the subject of this study. Based on the analysis of RNA products separated by denaturing gel electrophoresis, Nsp15 has been reported to cleave both 5′ and 3′ of the uridine.1., 2. We used several RNAs, including some with nucleotide analogs, and mass spectrometry to determine that Nsp15 cleaves only 3′ of the recognition uridylate, with some cleavage 3′ of cytidylate. A highly conserved RNA structure in the 3′ non-translated region of the SARS virus was cleaved preferentially at one of the unpaired uridylate bases, demonstrating that both RNA structure and base-pairing can affect cleavage by Nsp15. Several modified RNAs that are not cleaved by Nsp15 can bind Nsp15 as competitive inhibitors. The RNA binding affinity of Nsp15 increased with the content of uridylate in substrate RNA and the co-factor Mn2+. The hexameric form of Nsp15 was found to bind RNA in solution. A two-dimensional crystal of Nsp15 in complex with RNA showed that at least two RNA molecules could be bound per hexamer. Furthermore, an 8.3 Å structure of Nsp15 was developed using cyroelectron microscopy, allowing us to generate a model of the Nsp15-RNA complex.
Abbreviations used: SARS, severe acute respiratory syndrome; MALDI-TOF, matrix-assisted laser desorption/ionization time-of-flight; TLC, thin-layer chromatography
Keywords: SARS, coronavirus, endoribonuclease, modified RNAs, hexamer
Introduction
Severe acute respiratory syndrome (SARS) is an economically important emerging health threat. SARS is caused by a novel coronavirus, SARS-CoV, which has a ∼30 kb, capped and polyadenylated positive-strand RNA genome.4 , 5 The genome of coronaviruses code for several proteins not usually found in RNA viruses, including enzymes that putatively function in RNA processing.6 These proteins include: Nsp14, an exonuclease and Nsp15, which has similarities to the U16 SnoRNA processing endonuclease, XendoU.7 , 8
The roles of these putative RNA processing proteins in SARS-CoV infection remain to be elucidated. Bhardwaj et al. and Ivanov et al. demonstrated independently that the recombinant Nsp15 protein of the SARS-CoV produced in Escherichia coli has endoribonuclease activity that preferentially cleaves RNAs at uridylate in a reaction stimulated by Mn2+.1 , 2 Furthermore, a single-nucleotide mutation in the Nsp15 gene of the human coronavirus prevented viral RNA accumulation, suggesting that Nsp15 is required for successful viral infection.1 A more thorough analysis of mutations in the Nsp15 ortholog of the related Arterivirus revealed that several mutations, including those in the putative active site, reduced viral plaque formation, but did not abolish infection in cultured cells.9 In contrast, several other mutations prevented infection and reduced subgenomic RNA accumulation. It is thus likely that Nsp15 may have more than one role in the infection cycles of coronaviruses and its relatives.
A number of mechanistic questions remain concerning how Nsp15 recognizes and cleaves RNA. First, it is not clear what features in the RNA can direct higher affinity recognition of the RNA substrate. While Bhardwaj et al. observed that single-stranded RNA (ssRNA) is preferred for cleavage, Ivanov et al. proposed that the preferred substrate is double-stranded RNA (dsRNA).1 , 2 Second, Guarino et al. demonstrated that Nsp15 exists as a hexameric structure (a dimer of trimers), whose formation is correlated with endoribonuclease activity.3 This raises the question of whether hexamers are required for RNA binding. Third, whether Mn2+ participates in cleavage or RNA binding has not been determined. Lastly, there is a discrepancy in the literature about the activity of Nsp15. Ivanov et al. concluded that Nsp15 cleaves both 3′ and 5′ of a uridine base in structured RNA, while Bhardwaj et al. reported cleavage 5′ of the uridine.1 , 2 In both of these reports, the claim of 5′ cleavage was based primarily on the migration of the RNAs relative to molecular mass markers in denaturing polyacrylamide gels. Additional analysis of RNA recognition and cleavage by Nsp15 is thus needed to define its mechanism of action.
In this work, we used a series of RNAs, including some with specific modifications to probe RNA binding and RNA cleavage by Nsp15. We demonstrated that Nsp15: (1) cleaves 3′ of target uridylate, and 3′ of cytidylate in the appropriate sequence context; (2) preferentially cleaves at uridylate bases that do not participate in base-pairing; (3) has higher affinity for RNAs that have consecutive uridylate bases; (4) uses Mn2+ to enhance RNA binding; (5) binds RNA at the interface of the dimers of trimers; (6) contains grooves that could accommodate RNAs at the interfaces of the trimers of dimers. Lastly, we used a real time cleavage assay to examine substrate recognition by Nsp15 in vitro and identified several competitive inhibitors of Nsp15.
Results
Ribose-modified RNAs
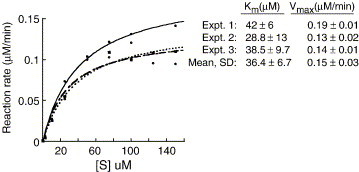
We used modified RNAs to examine the mechanism of RNA recognition and cleavage by Nsp15. The 16 nt RNA named R16.4 was used as a prototype, since it contains only one uridylate and its cleavage product can be resolved easily on a denaturing gel (Figure 1(a)). Radiolabeled R16.4 was readily cleaved by wild-type Nsp15, but not by a catalytically inactive mutant, H234A (Figure 1(a)). Notably, the cleavage product migrated as a 12 nt RNA in a denaturing gel with single nucleotide resolution. Furthermore, analysis of the kinetics of cleavage revealed that its K m was 36.4(±6.7) uM and the V max was 0.15(±0.03) μM/min in three independent experiments (Figure 1(b) and Supplementary Data Figure 1).
Figure 1.
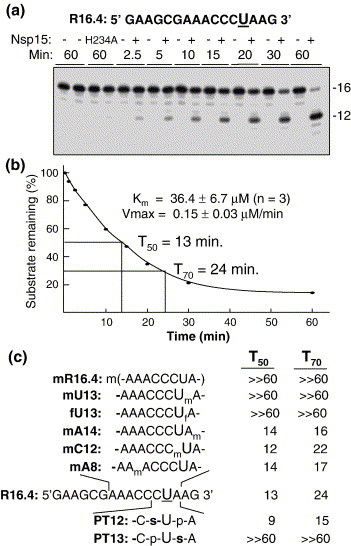
RNA cleavage by the SARS-CoV Nsp15. (a) The image from a denaturing 20% polyacrylamide gel shows radiolabeled R16.4 treated over time with Nsp15 or with the inactive mutant H234A. At the time-point designated above the appropriate lane, an aliquot of the reaction was removed and stored in a sample dye containing urea until the entire set had been collected. The sequence of RNA R16.4 is shown above the gel image. The only uridylate, at position 13, is in a larger font and underlined. We note that the major cleavage product migrated in the gel as a 12 nt RNA in this gel electrophoresis system. (b) Quantification of the amount of uncleaved RNA over a period of 1 h. The results from the gel image were quantified by use of a phosphorimager and plotted. T50 and T70 represent the time required to cleave 50% and 70% of substrate, respectively. (c) Analysis of the effects of ribose and phosphodiester modifications on the kinetics of Nsp15 cleavage. The sequence of unmodified R16.4 is shown in its entirety. For the other RNAs, only the residues surrounding the modifications are shown. m denotes a 2′-methoxy group and f denotes a 2′-fluoro group. p denotes a phosphodiester and s denotes a phosphorothioate. T50 and T70 values for each RNA were derived from plots of the amount of products formed over a 1 h time-course.
A 12 nt product corresponds to cleavage 5′ of the uridylate by Nsp15 (Figure 1(a)). However, since XendoU cleaves only 3′ of the uridylate,10 we examined whether ribose C2′ modifications would affect cleavage. A version of R16.4 with every ribose methylated at the C2′ position, named mR16.4, was not cleaved significantly over the 1 h course of the experiment, while the time required to cleave half of the unmodified R16.4 (T 50) was 14 min (Figure 1(c)). Furthermore, RNAs with U13 modified with a 2′-methoxy or a 2′-fluoro group (RNAs mU13 and fU13, respectively), were not cleavage by Nsp15 (Figure 1(c)). Next, we determined whether methylation of the neighboring nucleotides (mC12 and mA14) affected cleavage. The T 50 for mC12, and mA14 was 12 min and 14 min., respectively, which is similar to that for R16.4 (Figure 1(c)). These results indicate that only the ribose in the target uridylate influences cleavage at that nucleotide.
Phosphodiester modifications
We further investigated whether phosphorothioates adjacent to the target uridine would affect cleavage. RNA PT12, with a phosphorothioate immediately 5′ of U13, was cleaved at a rate similar to R16.4, while PT13, with a phosphorothioate immediately 3′ of U13, was resistant to cleavage (Figure 1(c)). Phosphorothioates involving non-bridging sulfur atoms could exist as two chiral forms and, since PT13 was not noticeably cleaved, both isomers inhibited cleavage. The addition of cadmium to 1 mM did not rescue cleavage of PT13, suggesting that the defect was not due to the inability of the 3′ phosphorothioate to bind Mn2+ (data not shown). The interaction of non-cleavable RNAs with Nsp15 will be addressed below.
The reduced cleavage observed with PT13 and RNAs with modifications of the C2′ ribose in U13 suggests that cleavage occurs 3′ of uridylate, as proposed by Ivanov et al.1 However, the claim that cleavage can occur 5′ of the uridylate was not supported by the data obtained with mC12 and PT12.1 , 2 Because this conclusion was based solely on the mobility of the bands in denaturing gels, which could have been affected by RNA conformation, we decided to re-evaluate whether cleavage can occur 5′ of the uridylate.
The 3′ cleavage by Nsp15
To further analyze whether Nsp15 cleavage occurred 5′ or 3′ of the uridylate, or both, we subjected the cleavage products of R16.4 to mass spectrometry. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry analysis of the untreated R16.4 revealed a peak at 5162 Da and its doubly charged species showed a peak at 2582.9 Da (Figure 2(a), upper panel). When treated with Nsp15, the peak of 5162 Da was reduced and peaks at 4239 Da appeared that would correspond to a 13 nt cleavage product containing a 3′ phosphate group (Figure 2(a), lower panel). No significant peak could be seen in the region below 900 Da (data not shown). The additional ion of 2120.9 Da is likely a doubly charged version of the 4239 Da ion. The mass spectrometry results demonstrate that Nsp15 cleaves only 3′ of the uridylate. We did not observe an ion that would correspond to the 13 nt cleavage product containing a cyclic phosphodiester. However, the treatments of the RNA to ensure ionization could have affected the integrity of the cyclic phosphodiester.
Figure 2.
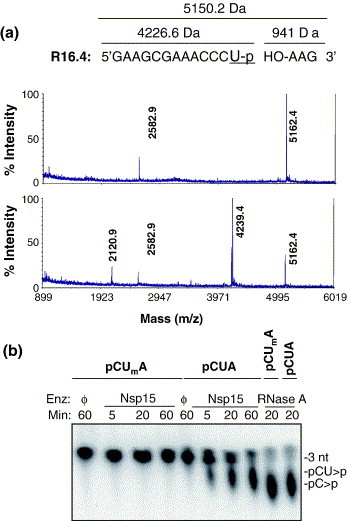
Cleavage 3′ of uridylate. (a) R16.4, untreated (upper panel) and treated with Nsp15 (lower panel). The spectra show both the singly ionized product and the doubly ionized product, which is half of the mass of the former. The expected mass of untreated R16.4 is 5150.2 Da and cleavage 3′ of the U13 position should generate products of 4210.6 Da and 939.6 Da. (b) Cleavage of pCpUpA is inhibited by a modification in the uridine ribose 2′-OH. The oligonucleotide substrates were radiolabeled at its 5′ phosphate group using phage T4 polynucleotide kinase reactions containing [γ-32P]ATP. The time when the reaction was terminated is given in minutes. The presence of no enzyme (ϕ), Nsp15 or RNase A is noted above the corresponding reaction on the image of the TLC plate. The identities of the cleavage products are labeled to the right of the TLC image.
To confirm that Nsp15 cleavage occurred 3′ of the target uridylate, we analyzed Nsp15 cleavage using short oligonucleotide substrates and thin-layer chromatography (TLC). This strategy has been used to detect cleavage products generated by endoribonucleases.11 RNase A, which cleaves 3′ of uridylate and cytidylate and leaves a 2′-3′ cyclic phosphodiester, was used as a control.12 The trinucleotide substrate pCpUpA was radiolabeled at the 5′ end. Cleavage of this substrate by Nsp15 3′ of the only U should release a radiolabeled 2 nt product, while the 5′ cleavage would generate a radiolabeled 1 nt product that should co-migrate with the 1 nt radiolabeled product of RNase A (Figure 2(b)). The product released by Nsp15 migrated closer to the input 3-mer than the RNase A product, consistent with cleavage only 3′ of uridylate (Figure 2(b)). Additionally, modification of the uridine C2′with a methoxy group prevented cleavage of CUA by Nsp15, while RNase A still generated the expected 1 nt product (Figure 2(b)). Also, cleavage of another trinucleotide, pGpUpA, with either Nsp15 or RNaseA generated a 2 nt product but pGpU remained unaltered, demonstrating that both RNase A and Nsp15 cleaved 3′ of uridylate (data not shown). Thus, analysis of multiple substrates demonstrated that Nsp15 cleaves only 3′ of the target uridine.
Neighboring residues
Ribonucleases often prefer certain cleavage sites based on the context of the target nucleotide.13 Therefore, we examined whether nucleotides adjacent to uridylate could influence cleavage. RNA R16.4 (with the sequence of CUA from nucleotides 12 to 14) and its derivatives were all predicted by mfold to lack a stable structure (data not shown).14 Substitution of the 5′ neighbor of the target uridylate with G or A had only modest effects on Nsp15 cleavage (underlined residue in Figure 3(a)). A ∼4-fold reduction in cleavage was observed when the base 3′ of the target uridylate was changed from the original A to a G (Figure 3(a)). Furthermore, C at position 14 (relevant sequence of -CUC-) resulted in two cleavage products, suggesting that two cleavages took place at the positions indicated by the backslash: -C/ UC- and -CU/ C (Figure 3(a)). These cleavages were not observed with a target sequence of -GUC- or -AUC- (Figure 3(b)). MALDI-TOF mass spectrometry was used to confirm the Nsp15 cleavage site in the RNA with the -CUC- target sequence. The spectrum of untreated RNA showed a single peak of 5126.3 Da that corresponds well to the calculated mass of the input RNA (5126.214 Da; Figure 3(c); upper panel). The Nsp15-treated samples contained additional peaks of 4209.1 Da and 3902.1 Da, which would correspond to the 13 nt and 12 nt cleavage products, respectively. Lastly, ions of 1223.9 Da and 917.8 Da, are good matches with a 4 nt and a 3 nt cleavage product, respectively (Figure 3(c); lower panel). All of these results demonstrate that the identity of the nucleotide immediately 3′ of the uridylate can influence cleavage by Nsp15. Also, Nsp15 could cleave at a cytidylate in a certain sequence context. It is important to note, however, where Nsp15 did cleave at a cytidylate, it did so 3′ of the cytidylate. Determination of the kinetic parameters for cleavage at C12 and U13 indicates that these cleavages occurred at similar rates, with K m = 15 μM and V max = 0.05 μM/min for cleavage at C12 and K m = 26.9 μM and V max = 0.08 μM/min for cleavage at U13.
Figure 3.
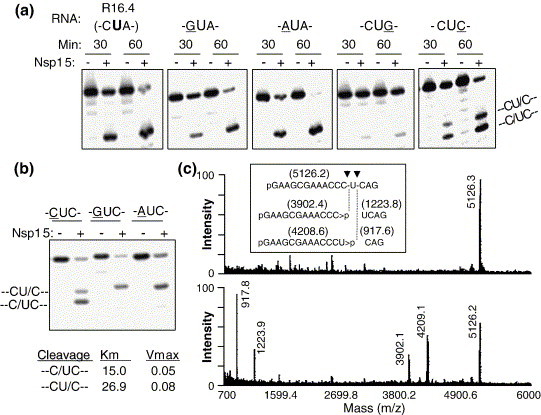
Effects of nucleotides adjacent to U13 on Nsp15 activity. (a) Gel images of the products generated by Nsp15 from RNA R16.4 and RNAs with altered residues immediately adjacent to U13. The sequences of nucleotides 12–14 of the RNAs tested are shown above the gel images. The residues altered from R16.4 are underlined. (b) Gel image showing an effect of nucleotides immediately upstream of –UC- on RNA cleavage by Nsp15. R16.4 with C14 was further modified to contain C, G or an A at the 12th position, indicated above the panel. Kinetic parameters, Km and Vmax, are indicated at the bottom of the image. (c) MALDI-TOF spectra of untreated (upper panel) and Nsp15 treated (lower panel) R16.4 containing the –CUC- sequence at positions 12–14.
Base modifications
How Nsp15 can recognize the target base is not understood. We used uracil analogs in the context of R16.4 to examine how Nsp15 specifically recognizes the target uridine basse. We first tested the RNA named ψU, which has a pseudouridine base at position 13, which should retain all of the uridine hydrogen bonding moieties, but has an additional N1 imino group (Figure 4(a)). ψU was cleaved by Nsp15 at a rate indistinguishable from that of R16.4 (Figure 4(a)), indicating that the non-hydrogen bonding face of the uracil is not important for cleavage. Next, we replaced the C4 carbonyl group of U13 with a thio group in RNA 4SU and found that the T 50 was increased from 13 min to 33 min (Figure 4(a)). Lastly, we replaced the imino group of the uracil with an N3 methyl group (N3-Me) and found that cleavage was abolished (Figure 4(a)). Thus, moieties in the target uracil are required for cleavage by Nsp15. Since these moieties would be blocked in a base-paired portion of the RNA, we hypothesize that Nsp15 will preferentially cleave only sites within unpaired RNA.
Figure 4.
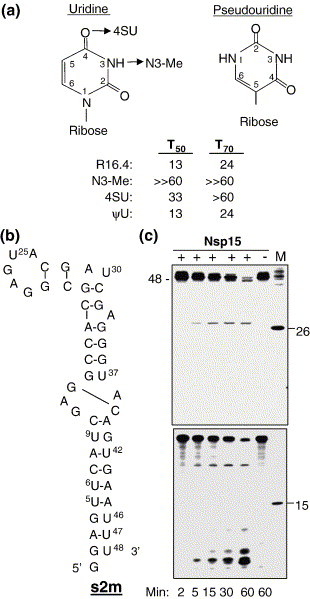
Effects of base modifications on Nsp15 cleavage. (a) A summary of the uracil analogs tested in the context of RNA R16.4. The arrows denote the names of the RNAs with a modified uracil. T50 and T70 values of several RNAs with modified uridine bases at position 13 were derived from kinetic assays as described for Figure 1. (b) A structure of a highly conserved RNA present at the 3′-untranslated region of the SARS-CoV RNA named s2m. The schematics of the RNA are drawn from the information given by Robertson et al.15 (c) Cleavage of S2m by Nsp15 over a time-course. RNAs of 26 nt and 15 nt radiolabeled with phage T4 kinase and [32P]ATP were used as molecular mass markers. The denaturing 15% polyacrylamide gel on the top was electrophoresed using conditions that better resolve the longer RNAs while the denaturing 25% polyacrylamide gel at the bottom retained the smaller molecular cleavage products generated from the 5′ end of s2m.
RNA structure and Nsp15 cleavage
We wanted to analyze Nsp15 cleavage in a more complex, structured RNA molecule. Robertson et al. recently solved the crystal structure of a highly conserved 48 nt RNA from the 3′ non-translated region of the SARS-CoV.15 This RNA, called s2m, contains several interesting features, including a right-angle turn and a 5 nt loop that mimics a tetraloop (Figure 4(b)). s2m contains a potential cleavage site in the 5 nt loop at position 25 and in the right-angle bend at nucleotide 30 (Figure 4(b)). When s2m was labeled at the 5′ terminus and subjected to cleavage by Nsp15, a 30 nt product accumulated over time, indicating preferential cleavage at U30 (Figure 4(c)). A fragment corresponding to U25 was not detected, perhaps due to other interactions within the tetraloop, which would protect it. Larger fragments of ∼40-48 nt were detected, which could correspond to RNAs that were cleaved at U residues near the 3′ end. When the products were resolved in a 25% polyacrylamide gel, fragments of 5 nt or 6 nt in length were detected. These products likely arose from cleavage of the long helix when the ends frayed and the exposed uridylate bases became accessible to Nsp15 (Figure 4(c), lower panel). This observation explains the apparently non-stoichiometric conversion of the full-length s2m to the 30 nt product (Figure 4(c)). Significantly, we did not observe efficient cleavage at base-paired U42, U37 or U9. Altogether, the results in Figure 4 suggest that Nsp15 cleavage is influenced by RNA structure, and that only some uridylate bases in the loops of RNA are cleaved preferentially.
Nsp15–RNA analog interaction
To determine whether the non-cleavable RNA analogs could interact with Nsp15, we needed to establish a more convenient and quantitative assay to monitor its activity. Since Nsp15 has a cleavage mechanism similar to that of RNase A, we selected two fluorogenic RNase A substrates that have been developed for real time assays, named rU and rC. Both substrates contain three deoxyribonucleotides and either a uridylate or a cytidylate as the only ribonucleotide. In addition, rU and rC both contain a 5′ carboxyfluorescein (FAM) and a 3′ fluorescence resonance emergency transfer quencher, tetramethylrhodamine. Cleavage by ribonuclease is expected to relieve quenching and increase fluorescence (Figure 5(a)). Fluorescence from these substrates was recorded continuously at 518 nm, a wavelength suitable for FAM. As expected, no increase in fluorescence was observed in reactions containing rC and Nsp15, even in the presence of Mn2+, since Nsp15 does not cleave RNAs efficiently at cytidylate, except in a specific sequence context (Figure 3). Some fluorescence was observed with rU in the absence of Mn2+, but the addition of Mn2+ increased the rate of cleavage significantly (Figure 5(a)). The role of Mn2+ in Nsp15 activity will be addressed later. Furthermore, the addition of the inactive mutant H234A resulted in only a minimal change in the fluorescent emission over the same time-course (data not shown). These results demonstrate that the assay can be used to examine the effects of competitors.
Figure 5.

Examinations of Nsp15 interaction with RNA analogs. (a) A real time endoribonuclease assay for Nsp15. The sequence of the quenched fluorescent substrate named rU (IDT, Coralville, IA) is shown, with fluorophores FAM and tetramethylrhodamine. The only ribonucleotide in this construct, uridylate, is in a larger font and underlined. A second version in which the uridylate was replaced with ribocytidine (rC) is used as a control. The fluorescent outputs from the two substrates were determined in the presence and in the absence of Mn2+, as indicated to the right of the graph. (b) A graph demonstrating inhibition of fluorogenic rU substrate turnover in the presence of increasing concentration of unlabeled rU (no inhibitor), a 16 nt DNA(D16.4), or R16.4 analogs (PT16 and N3meU). The equation used for the curve fit is in the box where the effects of various treatments are summarized: m2 denotes the apparent Km. (c) Image of an SDS/polyacrylamide gel where kinased RNAs (the identities of which are indicated above each lane) were crosslinked at 1200 μJ for 3 min to 1 μg of Nsp15. BSA was present at the same concentration. The letter ϕ is used to indicate that no protein was added in that reaction.
Next, we wanted to determine whether the modified RNAs that are not cleaved efficiently by Nsp15 can act as inhibitors of rU cleavage by Nsp15. To allow a quantitative comparison of the results, we determined the rate of cleavage of rU in the presence of increasing concentrations of a non-fluorogenic version of rU and fitted the data to the equation in Figure 5(b), as was done for characterization of RNaseA activity.16 The apparent K m for rU can be determined from the value m2, which was 169 μM. This value was changed only minimally when the reaction was challenged with of a DNA (1 μM) version of R16.4, D16.4. When R16.4 was present at 50 nM in the assay (a value approximating the IC50), the amount of unlabeled rU required to decrease the turnover of fluorogenic rU substrate by 50% was reduced by ∼50-fold, to 2.4 μM (Figure 5(b)). Similar effects were seen with the poorly cleavable analogs PT13, mR16.4, and N2MeU (Figure 1, Figure 4, Figure 5); and data not shown).
A crosslinking assay was used to confirm that Nsp15 could bind non-cleavable RNAs. Nsp15 was crosslinked individually to kinased RNAs by UV irradiation and then electrophoresed in a denaturing polyacrylamide gel. Radiolabeled bands corresponding to the Nsp15–RNA complex were detected at varying abundances with all of the analog-containing RNAs (Figure 5(c)). No crosslinking was observed with BSA present at the same concentration in a parallel reaction. Together with the results from the fluorogenic assays, these results confirm that the derivatives of R16.4 that cannot be cleaved efficiently retain the ability to interact with Nsp15, albeit to different extents.
Nsp15–RNA interaction
Since Nsp15 prefers to cleave RNAs containing uridylate, we examined whether the number of uridines in an RNA affect binding by Nsp15. First, we compared the ability of 16-mer oligonucleotides, composed entirely of adenylate (rA16) or uridylate (rU16), to inhibit Nsp15 activity in the fluorogenic assay. The kinetics of inhibition was biphasic for both RNAs, but rU16 was 3.5-fold more effective in inhibiting Nsp15 cleavage (IC50 of 15 nM and K i of 2.3 nM) than rA16 (IC50 of 53 nM and Ki of 7.6 nM) (Figure 6(a)), indicating that Nsp15 has a higher affinity for RNA containing uridylate. We examined the ability of wild-type and a catalytically defective Nsp15, H234A, to be crosslinked to radiolabeled rU16 and/or rA16. When the crosslinked products were resolved by SDS-PAGE, we observed that mutant H234A crosslinked well with rU16, but not to rA16, suggesting that binding to rU16 was preferred even in the absence of RNA cleavage (Figure 6(b)). We note that in this assay it is more useful to compare the crosslinking results obtained with H234A, since wild-type Nsp15 would digest rU16, hence resulting in a lack of signal with rU16 (Figure 6(b)).
Figure 6.
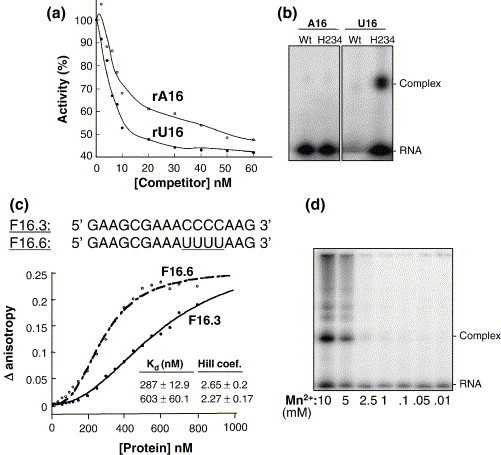
Nsp15-RNA interaction. (a) Inhibition of rU cleavage assay by RNAs rA16 or rU16. Increasing amounts of competitor RNAs (rA16 or rU16) were added to the reactions, allowed to equilibrate for 10 s and monitored for rate of cleavage. (b) Image of an SDS/polyacrylamide gel where kinased RNAs (identities of which are indicated above the lanes) were UV-crosslinked as described above to 500 ng of either WT or H234A mutant proteins. The mix contained an equal amount of BSA as a control for non-specific crosslinking. (c) Analysis of H234A-RNA interaction using fluorescence anisotropy. The anisotropy change was measured with increasing concentrations of H234A. Each datum point represents the average of ten anisotropy values. The Kd for the interaction and Hill coefficient (H.C) were derived from the binding isotherm using the Hill equation, as described in Materials and Methods. (d) SDS-PAGE image of radiolabeled rU16 crosslinked to H234A in the presence of increasing concentrations of MnCl2.
We used fluorescence spectroscopy to determine the affinity of Nsp15 to RNA containing or lacking uridylate. Fluorescein-labeled RNAs F16.3 and F16.6 containing four cytidylates or four uridylates, respectively, were synthesized for anisotropy measurements in the presence of increasing concentrations of H234 (Figure 6(c)). We observed a significant anisotropy change in response to increasing concentrations of H234A (Figure 6(c)). The dissociation constant derived by fitting the data to the Hill equation shows Nsp15 has a K d of 287 nM for F16.6 (with four uridylate bases) and of 603 nM for F16.3 (with four cytidylate bases). Both RNAs were bound with Hill coefficients in excess of 2.0, indicating cooperativity (Figure 6(c)). Please note that in these calculations, we did not assume that Nsp15 exists as a hexamer.3
Role of Mn2+ in Nsp15 activity
Given that ribonucleases that act through the formation of a 2′-3′ cyclic phosphodiester do not require divalent metal for cleavage, and that Nsp15 endoribonuclease activity is stimulated by Mn2+, we hypothesize that Mn2+ affects Nsp15 binding to the substrate RNA. Radiolabeled rU16 was crosslinked to the H234 mutant in the presence of increasing concentrations of Mn2+. Crosslinking was increased dramatically with concentrations of Mn2+ of 5 mM or higher (Figure 6(d)), consistent with our previous observation that the K d for Mn2+ was ∼5 mM.2 These results indicate that Mn2+ likely affects Nsp15 binding to RNA.
The hexameric form of Nsp15 binds RNA
Nsp15 can exist in solution in equilibrium as a monomer, a trimer, and a hexamer.3 The hexameric state is the one that correlates with enzymatic activity.3 We wanted to address which form of Nsp15 binds RNA. To facilitate detection, a 5′ biotinylated RNA containing ten uridylate bases (U10) was used. The catalytically inactive mutant, H249A, was mixed with U10 at a molar ratio of 1:1.5 and subjected to gel-filtration chromatography. The elution profile of H249A revealed peaks at the expected hexamer, trimer, and monomer locations in fractions 23–25, 26–29, and 30–33, respectively (Figure 7(a)).3 The presence of Nsp15 in these fractions was confirmed by SDS-PAGE (Figure 7(b)). RNA U10 subjected to gel-filtration chromatography in the absence of H249A eluted primarily in fractions 36–41 (Figure 7c)), the position expected for a short RNA oligonucleotide. However, in the presence of H249A, U10 was enriched in fractions 22–26, which contain Nsp15 hexamers, with some of it trailing in the fractions containing trimers. U10 was absent from the fractions containing monomeric Nsp15 (Figure 7(d)). These results suggest that the hexameric form of Nsp15 is the one that binds RNA.
Figure 7.
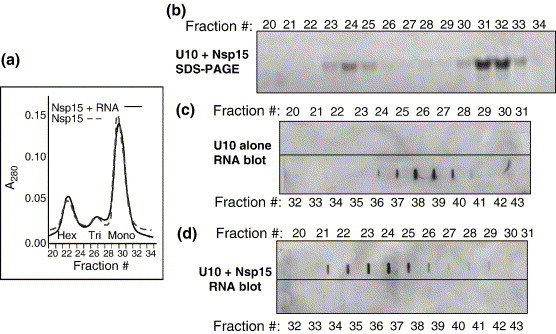
Analysis of Nsp15-RNA complex. (a) Gel-filtration profile of the catalytically inactive Nsp15 mutant, H249A in the presence (unbroken line) or in the absence (broken line) of biotin-labeled RNA U10. The positions where hexamer, trimer and monomer elute were characterized by Guarino et al.3 (b) Individual gel-filtration fractions on SDS-PAGE. (c) Slot blot of gel filtration fractions obtained when Nsp15 and RNA subjected to UV crosslinking were passed through the column. (d) Slot blot analysis of gel-filtration fractions performed with only RNA. Fraction numbers are indicated above the gel and blots. The RNAs in the blots were detected by probing with horse-radish peroxidase-conjugated streptavidin and developed using enhanced chemiluminiscence.
An 8.3 Å structure of Nsp15
Previously, we published a ∼21 Å structure of negatively stained Nsp15, but we sought to obtain a higher resolution structure of Nsp15 using cryo-electron microscopy (EM), and to integrate the biochemical information concerning Nsp15-RNA interaction with a model for the structure of Nsp15. Cryo-EM is advantageous for structure determination, since it does not involve staining with specific dyes that could reflect a bias for dye accumulation.
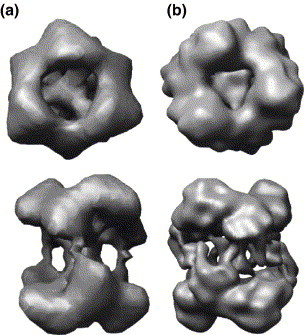
Cryo-EM images of Nsp15 of corresponding areas but with different degrees of defocus were taken. The defocus in Figure 8(a) is 1 μm, which has very low contrast. The contrast of the particles is higher when subjected to a greater (3 μm) defocus (Figure 8(b)), and this view is useful for selecting individual particles. A total of 30,000 individual particles were used for contrast transfer function correction. After eight cycles of refinement using the 3D reconstruction of negatively stained Nsp15 as an initial model, the structures converged as judged from Fourier shell correlation (FSC, Figure 8(c)). An EOTEST (thick line in Figure 8(c)) shows the resolution is 8.3 Å. There was good correlation between the class averages and reprojections from the final 3D reconstruction (Figure 8(e)). Importantly, the primary structure is that of a hexamer arranged in the form of a dimer of trimers, corroborating the structures observed previously.3 However, this data set is more complete than that used for image construction by Guarino et al.3 and revealed significantly more features of the hexamer; nearly all of the potential particle orientations were observed, as represented by the asymmetric triangle in Figure 8(d). (A comparison of the reconstructed 21 Å and 8.3 Å structures is provided as Supplementary Data Figure 2.) Class averages of different orientations of the Nsp15 hexamer were obtained to allow for the reconstruction of the 3D model for Nsp15 (Figure 8(e)). The final reconstructed model is shown in Figure 8(f), with each subunit represented in a different color. We note that each subunit contains two larger domains and one or two smaller domains. The two trimers are apparently connected in a tail-to-tail orientation through one of the smaller domains. Also, the channel visible in the top view of the hexamer is smaller in the higher resolution structure (Supplementary Data Figure 2). Finally, the Nsp15 hexamer apparently lacks long α-helices, since these secondary structures should be apparent at 8.3 Å resolution.17
Figure 8.
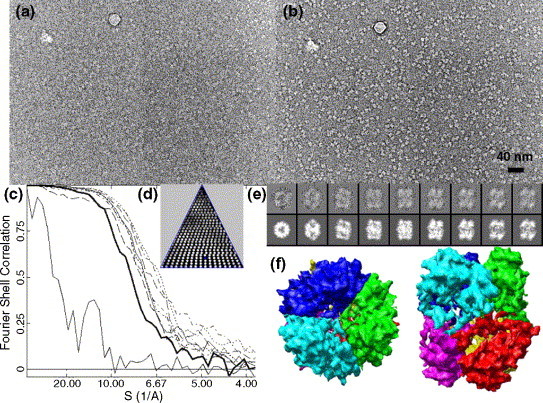
Cryo-EM 3D reconstruction of Nsp15 K289A. (a) Close to focus (1.5 μm) image of Nsp15. (b) Far-defocus (5.5 μm) image of the same area. (c) Refinement process monitored by Fourier shell correction. EOTEST is shown as a thick line. (d) The asymmetric triangle shows the orientation of the particles. (e) Upper row: class average of the particles; bottom row: reprojection from the final 3D model. (f) Surface display of the 3D structure, showing the six subunits in different colors. The figures to the left and right are the top and side views of the hexamer.
RNA substrate binding at the inter-trimer interface
The structure of Nsp15 alone suggests that it could either bind RNA through the top of the trimer, where there is a depression, or between the interfaces of the dimer of trimers, where a number of grooves exist. To determine how Nsp15 recognizes RNA, we used the lipid monolayer method to produce 2D crystals of Nsp15 K289A in the presence or in the absence of RNA. K289A was selected because it is catalytically inactive and forms hexamers efficiently.3 For ease of manipulation, these 2D crystals were stained with uranyl acetate, which will obscure some features of Nsp15. However, we were able to detect the location of three different RNAs in complex with Nsp15. The first is Stem-loop T, which has tightly compacted structure that was solved by nuclear magnetic resonance spectroscopy and demonstrated to interact with Nsp15.2 , 18 The second is a 40 bp poly(I:C) synthetic double-stranded oligonucleotide (Supplementary Data Figure 3). The third RNA is U16, which binds well to Nsp15 (Figure 9(a), (b) and (d)). In all of the crystallization attempts, the RNA was at a tenfold higher stoichiometry than Nsp15.
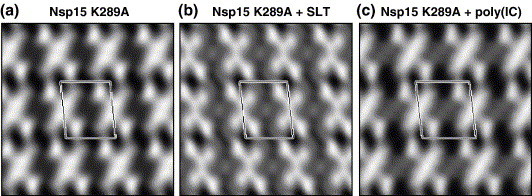
Figure 9.
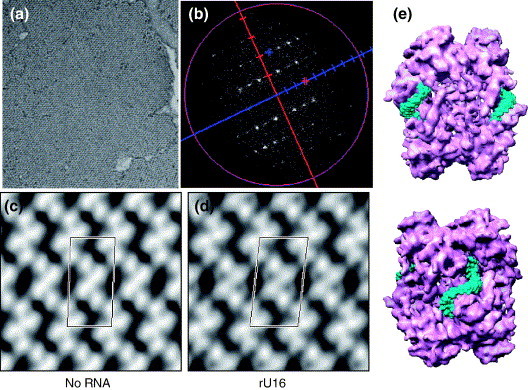
RNA binding by Nsp15 K289A. (a) Electron micrograph of negatively stained 2D crystal of Nsp15. (b) Fourier transform of the 2D crystal. (c) The 2D projection of Nsp15 with P2 symmetry. One unit cell is labeled;(a = 92.9 Å, b = 186.7 Å, γ = 87.6°). (d) The 2D projection of Nsp15- in complex with U16 (P2 symmetry, a = 95.8 Å, b = 179.8 Å, γ = 83.8°). (e) Two rU16s are fitted into the grooves present in the interface between the dimers of trimers of a hexameric Nsp15. The bottom image is rotated by 90° relative to the top image.
Electron crystallographic analysis of Nsp15 lacking RNA shows a homogeneous structure with two X-shaped Nsp15 hexamers per unit cell (Figure 9(a)–(c)). The unit cell has the dimension of a = 9.3 nm, b = 18.7 nm, with γ = 87.6°. This projection closely resembles the side-on view of the 3D structure of Nsp15 that was determined by single-particle analysis.3 However, Nsp15 in complex with U16 produced lattices with a slightly different unit cell dimensions and distinct additional densities at the inter-trimer interface (Figure 9(d); a = 9.6 nm, b = 18 nm, with γ = 83.8°). The difference in the unit cell dimensions suggests that RNA binding may induce a conformational change in Nsp15. Similar changes in the density distribution were observed in the presence of Stem-loop T and poly(I:C) (Supplementary Data Figure 3). It is possible that not all bound RNA molecules are visible in the orientation we are using, since there is no differentiation of information along the z-axis in the projection. In any case, the RNA-related densities are located exclusively at the inter-trimer interface, suggesting that one binding site is shared between two trimers.
To put the crystal data into a 3D perspective, we modeled the approximate location of the RNA into the 3D reconstruction of the cryo-EM structure of Nsp15 K289A. Nsp15 hexamers contain distinct grooves. One particularly pronounced groove that resides between two opposing subunits is related by 2-fold symmetry, and resembles the letter S. This groove is of sufficient size to accommodate one molecule of rU16. Figure 10(e) shows two views of two U16 molecules fitted into this groove. It is possible that even longer RNA molecules could pass through the groove between the trimers.
Discussion
The goals for this study were to clarify the mechanism of cleavage by the SARS-CoV Nsp15 and to better understand how it recognizes RNA. We used several RNAs, including some with nucleotide analogs, to demonstrate that Nsp15 cleaved 3′ of the specifically recognized uridine, that the site and rate of cleavage are affected by the nucleotide 3′ of the uridylate, and that unpaired uridylate bases are preferred for cleavage. RNAs that are not cleaved by Nsp15 can retain the ability to interact with Nsp15 and act as competitive inhibitors. Furthermore, RNAs with multiple uridine bases had a higher affinity of binding to Nsp15, and Mn2+ enhanced RNA–Nsp15 interaction. Lastly, RNA is apparently recognized at the interface between the dimer of trimers in the hexameric Nsp15 structure, and this complex was modeled into an 8.3 Å structure of Nsp15 determined by cryo-EM and image reconstruction.
The SARS-CoV Nsp15 protein has been claimed to cleave both 5′ and 3′ of uridylate residues in RNA through the formation of a 2′-3′cyclic phosphodiester.1 Analysis of the cleavage products by mass spectrometry and TLC shows that Nsp15 cleavage occurs only 3′ of uridylate. RNAs containing modifications 3′, but not 5′, of the cognate uridylate were not cleaved by Nsp15. The cleavage mechanism is likely through the formation of 2′-3′ cyclic phosphodiester formation, as predicted by Ivanov et al.1 We have evidence for a 2′-3′ cyclic phosphodiester, because we could not radiolabel the 3′ end of the cleaved product unless we first treated it with kinase and alkaline phosphatase to break the cyclic phosphodiester (data not shown). The previous claims of the 5′ cleavage activity were likely based on the effects of the 2′-3′ cyclic phosphodiester on migration of the cleavage products in polyacrylamide gels.1 , 2 We also found that Nsp15 could cleave 3′ of cytidylate when it was present immediately 5′ of -UC-.
Ribonucleases are generally classified into two groups on the basis of whether they leave a 5′ or 3′ phosphate at the cleavage site.19 The former group includes RNases that cleave base-paired RNAs, such as the RNase H family and RNases III, while the latter includes members of the RNase A family that preferentially cleave unpaired residues, and includes barnase, RNase T1, and RNase T2.20., 21., 22., 23., 24., 25., 26. The two classes of ribonucleases have very different mechanisms. Those that leave a 5′ phosphate possess a metal ion at the active site.19 In contrast, the ribonucleases that cleave 3′ of the target residue use two side-chains at the active site to catalyze acid–base hydrolysis, leaving a 2′-3′ cyclic phosphodiester.
Our data suggest that Nsp15 has a cleavage mechanism highly similar to that of RNase A, XendoU, and RNAse T1. However, unlike these endoribonucleases, Mn2+ can enhance RNA binding to Nsp15 (Figure 6(d)). This requirement contrasts with XendoU, which does not depend on Mn2+ for RNA binding.8 RNase A and T1 are also known to not require a co-factor. The concentration of Mn2+ needed for binding in vitro is at least one logarithmic unit higher than the concentration of Mn2+ present in cells.27 , 28 This suggests that the availability of Mn2+ may regulate Nsp15. Alternatively, the environment within the cell may reduce the concentration of Mn2+ needed for activity.
Our analysis of the cleavage of the s2m RNA, and RNAs with analogs in the uracil base, in ribose C2′ positions indicate that Nsp15 the target uridine must be exposed in single-stranded portions of an RNA. This is consistent with our previous claim that Nsp15 preferred unpaired uridylate and is in conrast to the interpretations offered by Ivanov et al.1 Furthermore, specificity for the uracil is apparently conferred by recognition of moieties in uracil that participate in Watson–Crick base pairing. The nucleotide immediately 3′ of the uracil may modify this interaction to influence the rate of cleavage, and possibly the site of cleavage (Figure 4(a)). More efficient cleavage of the target uridylate was observed when the immediate 3′ nucleotide is an adenylate.
In addition to conferring specificity for cleavage, uridylate bases in RNAs appear to contribute to binding by Nsp15, as measured by fluorescence spectroscopy and UV crosslinking studies. We observed also that co-crystals of Nsp15 with a highly structured RNA resulted in RNA binding to only one side of the hexamer, while co-crystals of Nsp15 with RNA U16 can result in at least two RNA molecules per hexamer, perhaps due to higher-affinity binding.
The 3D structure as determined by cryo-EM agrees well with the 3D reconstruction obtained from negatively stained specimens (Supplementary Data Figure 2). The overall size and shape of the molecule, and the relative positions of the two larger and the two smaller domains of each subunit are very similar. However, the cryo-EM reconstruction offers additional detail at the trimer–trimer interface and displays six connecting protein densities between the two trimers instead of three (not counting the central density) in the negatively stained structure. This difference may be due to preferential staining.
At first glance, it is curious that the SARS-CoV, a virus with one of the longest RNA genomes known, would encode an endoribonuclease. While an endoribonuclease may be useful in the processing of the SARS-CoV RNAs and/or in attacking cellular RNAs, its presence raises the question of how can Nsp15 be regulated to prevent unintended cleavage of the viral genome. Some precedence is provided by the pestivirus endoribonuclease Erns, which is also a structural glycoprotein.29 Erns could be kept in an inactive form until is it assembled outside of the viral capsid. We note that the Nsp15 hexamer is the form that binds RNA efficiently (Figure 6), and that all three Nsp15-RNA co-crystals have the RNA positioned at the interface between the dimers of trimers (Figure 9 and Supplemental Data Figure 3), suggesting that hexamer formation needs to precede RNA binding, and that the active site is on the surface of the hexamer. Also, the specificity required for RNA cleavage could protect the SARS-CoV RNA, since not all uridylate bases within an RNA molecule are equally sensitive to cleavage, as seen with s2m (Figure 4(c) and (d)D). The complex of Nsp15 and RNA awaits further structural and biochemical analysis in order to confirm the proposed model of interaction (Figure 9(e)).
The complex requirements for Nsp15 to interact with RNA should allow the use of RNA mimetics as specific inhibitors of Nsp15. Since several mutations in Nsp15 orthologs of human coronavirus and arterivirus resulted in viruses that are negatively affected in plaque formation and could lead to abolituon of infection altogether, it should be possible to inhibit the infection of SARS-CoV.9 The demonstration that the cleavage mechanism of Nsp15 is similar to RNaseA should allow the application of technologies adapted for RNaseA. A fluorogenic assay used for RNase A has already allowed the continuous monitoring of Nsp15 activity and the characterization of potential inhibitors.
Materials and Methods
RNAs
Most RNAs were chemically synthesized by Dharmacon, Inc. (Boulder, CO). Stem–loop 2 motif RNA (s2m) was transcribed by phage T7 RNA polymerase as described.15 All RNAs were purified from denaturing polyacrylamide gels, quantified by spectrophotometry, and checked for quality on a denaturing polyacrylamide gel. Recombinant Nsp15 protein was expressed and purified as described.2
Endoribonuclease assays
The standard RNA cleavage assay used 1 × 104 CPM of 5′-end radiolabeled RNA substrate (1 μM final RNA concentration) and 0.026 μM Nsp15 in 50 mM Tris–HCl (pH 7.5), 50 mM KCl, 1 mM dithiothreitol, 5 mM MnCl2 at 30 °C. Labeling of the Nsp15 substrates was performed as described.2 The endoribonuclease reactions were terminated by the addition of a gel-loading buffer that contained 7.5 M urea. Products were separated by electrophoresis in 20% polyacrylamide gels containing 7.5 M urea , unless stated otherwise. Gels were wrapped in plastic and exposed to a PhosphorImager screen for quantification using Molecular Dynamics software. Each result shown was reproduced in at least one, and usually two independent experiments.
For kinetic analysis, the assay mixture contained 50 mM Tris –HCl (7.2), 50 mM KCl, 5 mM MnCl2 and 1 mM DTT. The reaction was started by addition of Nsp15 at 30 °C. The cleavage product generated at various substrate concentrations was quantified at time-points ranging between 2 min and 20 min. Data were fit by linear least-squares regression analysis to determine activity in units of μM/min. Values of K m and V max were determined by non-linear least-squares regression analysis of data fit to the Michaelis–Menten equation.
The real-time endoribonuclease assay used a substrate purchased from Integrated DNA Technologies, Inc, Coralville, IA. The substrate has a 5′ fluorophore carboxy fluorescein (FAM) and a 3′ tetramethylrhodamine that could quench FAM fluorescence. Of the four nucleotides, the uridylate or the cytidylate is the only ribonucleotide while the others are deoxyribonucleotides. The excitation wavelength was 492 nm, and the maximum emission of the substrate at 518 nm was determined in the buffer used for the gel-based assay. Changes in fluorescence over time were measured using an LS55 spectrometer (Perkin Elmer, Inc.).
Kinetic characterization of Nsp15 activity
Fluorogenic assays were performed at room temperature in 50 mM Hepes (pH 7.5), 50 mM KCl, 1 mM DTT and 5 mM MnCl2. Progress curves at a constant fluorogenic rU and increasing concentrations of unlabeled rU were monitored for 90 s with less than 10% substrate conversion. To determine the effect of RNA analogs, reactions were performed in the presence of 50 nM analogs. The data were fit by linear least-squares regression analysis to determine the rate of reaction. Values of apparent K m were determined by non-linear least-squares regression analysis of data fit to following equation:
where (ΔI/Δt)0 is the activity prior to the addition of unlabeled substrate.16
Mass spectrometry
For analyzing masses of products generated by Nsp15 endoribonuclease activity, R16.4 was incubated either with assay buffer or Nsp15 under standard assay conditions and then extracted with phenol/chloroform. The aqueous layer was passed through Zip Tip C18 (Millipore) in 200 mM triethanol ammonium acetate (TEAA), pH 7.0. The Zip Tip was washed twice with 200 mM TEAA (10 μl each wash) followed by two washes with 20 mM TEAA (10 μl each wash). Elution was done with 10 μl of 50% (v/v) methanol. Mass spectra were acquired on a Voyager-DE STR mass spectrometer (Applied Biosystems, Framingham, MA).
Thin-layer chromatography
Oligonucleotide products were analyzed using thin-layer chromatography. Briefly, from 1 to 2 μl of the endoribonuclease reactions were spotted on polyethylene imine (PEI) cellulose. Plates were wetted in water and then developed with a solution of 0.5 M potassium phosphate (pH 3.5).
Fluorescence anisotropy
Fluorescein-labeled RNA probes for the binding assays were diluted to 500 nM in 50 mM sodium acetate (pH 5.5), 50 mM KCl. Fluorescence measurements were made at room temperature with a Perkin-Elmer luminescence spectrometer LS55 and cuvettes with an optical path-length of 0.4 cm. Measurements were taken with an integration time of 1 s and a slit-width of 5 nm of both excitation (490 nm) and emission (520 nm). Anisotropy values were recorded 20 s after each protein addition to allow the sample to reach equilibrium. The anisotropy values are the average of ten measurements. Binding data were analyzed by non-linear, least-squares fitting using KaleidaGraph software (Synergy Software, Reading, PA). The Hill equation:
was used to determine the dissociation constant (K d). In this equation, ΔA is the anisotropy change caused by the ligand binding, B max is the maximum anisotropy change, × is the total concentration of the input protein and the exponential term n is the Hill coefficient.
Two-dimensional crystallization
His-tagged Nsp15 K289A was purified as described.3 The 2D crystals were obtained using the lipid monolayer method in conjunction with Ni-NTA-modified lipids.30 Briefly, 15 μl of Nsp15 K289A at 0.1 mg/ml (20 mM Hepes (pH 6.9), 100 mM NaCl, 10 mM MgCl2, 2% (w/v) PEG 3000, 1 mM NaN3) were added to a custom-designed Teflon well (4 mm wide and 1 mm deep), then 0.2 μl of mixed lipid solution of 0.1 mg/ml of Ni-NTA-dioleoylphosphatidylserine (DOPS) and 0.4 mg/ml of egg phosphatidyl choline (PC) in chloroform/hexane (1:1, v/v) was carefully layered on top of the protein solution and incubated overnight at 4 °C in a humid environment. Crystals were harvested using a carbon-coated copper grid, washed on a 50 μl droplet of distilled water and negatively stained with an aqueous solution of 1% (w/v) uranyl acetate (pH 4.25). A JEOL 1200EX transmission electron microscope (JEOL, Tokyo, Japan), operated at an acceleration voltage of 100 kV, was used to image the 2D crystals at a calibrated magnification of 39,900×. Co-crystallization of RNA substrate with Nsp15 K289A was performed using the same monolayer approach but with the addition of 3.5 μM Stem-loop T, or 0.12 μg/mg 40 bp poly(I:C), or 2.5 μM rU16. Electron micrographs were scanned using a Leafscan-45 microdensitometer with 5.14 Å/pixel at the specimen level.31 Crystallographic image processing was carried out with the CRISP software package.32
Cryo-EM and 3D image reconstruction
The cryo-EM data were collected at the National Center for Macromolecular Imaging at Houston.33 Briefly, 2.5 μl of purified Nsp15 K289 solution (0.2 mg/ml in 20 mM Hepes (pH 6.9), 100 mM NaCl, 1 mM NaN3) was applied to a glow-discharged holey carbon grid (Quantifoil R2-1), blotted with filter-paper, and flash-frozen with liquid ethane. A JEOL 2010F electron microscopy (JEOL, Tokyo Japan) was used for data collection with a Gatan model 626 cryostage (Gatan, Pleasanton CA). Each area was imaged twice on a 4K × 4K CCD camera (Gatan US4000) at a magnification of 60,000×, corresponding to 1.81 Å/pixel. A close-to-focus image was recorded first (1 to 2 nm); after 1 min, a far-defocus image (3 to 6 nm) was recorded.
The far-defocus image is used to select particles semi-automatically with the boxer program of the EMAN software package.34 , 35 The coordinates of the particles were then applied to the closed-to-focus image. The alignhuge program was used to align the two images. More than 30,000 particles were selected from 200 independently obtained images. The parameters for contrast transfer function correction of each particle set were determined with the ctfit program. After phase flipping, the amplitude correction was done automatically in refine command. A 3D model from negatively stained reconstruction of Nsp15 was used as the initial model for refinement.3 Then segment3D was used to separate the six subunits of the Nsp15 dimer of trimers and displayed in different colors with chimera software.36
Modeling RNA into the 3D structure of Nsp15
A 3 nt uridylate fragment from PDB (1WKS) was used to make the rU16 model. Alignments between the fragments were performed so that the orientation and distance of the bases were the same as within the fragment. There are several grooves within the Nsp15 hexamer. The grooves between the two subunits, which are related by a 2-fold axis, are big enough to fit the RNA. All the alignment and fitting were performed manually with chimera.36
Acknowledgements
We thank F. Walz and Ranjith Kumar for helpful discussions, and William K. Russell of the Laboratory for Biological Mass Spectrometry at TAMU for mass spectrometry results. Support by NSF grants MCB0332259 and MCB461511 is gratefully acknowledged. Cryo-EM data were collected at the National Center for Macromolecular Imaging at Houston with advice provided by Wah Chiu, Steve Ludtke, and Htet Khant.
Edited by D. E. Draper
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2006.06.021
Appendix A. Supplementary data
Supplementary Figure S1.
Kinetic analysis of cleavage at the uridylate in RNA R16.4 by Nsp15. Shown are the results from the three independent experiments in which the parameters were determined. Each experiment consisted of nine RNA concentrations analyzed over five time points. The mean Km and Vmax values derived from these experiments are summarized in Fig. 1B.
Supplementary Figure S2.
Comparison of the structures of the negatively stained Nsp15 hexamer and the cryo-EM structure filtered to 2 nm resolution. (A) Ttop and side views of the negatively-stained Nsp15 K289A. (B) Top and side views of the cryo-EM structure of Nsp15 K289A.
Supplementary Figure S3.
Co-crystals of Nsp15 K289A mutant protein without (A) and with RNAs (B, C). SLT is an tightly folded RNA of 35-nt (Kim et al., 2001) while poly(I:C) is a chemically synthesized RNA of 40 bps. The boxes in each panel define a unit cell. Note that the structures of each of the protein differed with the RNA bound, suggesting that Nsp15 could adjust its conformation in response to the RNA.
References
- 1.Ivanov K.A., Hertzig T., Rozanov M., Bayer S., Thiel V., Gorbalenya A.E., Ziebuhr J. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Natl Acad. Sci. USA. 2004;101:12694–12699. doi: 10.1073/pnas.0403127101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhardwaj K., Guarino L., Kao C.C. The severe acute respiratory syndrome coronavirus Nsp15 protein is an endoribonuclese that prefers manganese as a cofactor. J. Virol. 2004;78:12218–12224. doi: 10.1128/JVI.78.22.12218-12224.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guarino L.A., Bhardwaj K., Dong W., Sun J., Holzenburg A., Kao C. Mutational analysis of the SARS virus Nsp15 endoribonuclease: identification of residues affecting hexamer formation. J. Mol. Biol. 2005;353:1106–1117. doi: 10.1016/j.jmb.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holmes K.V., Enjuanes L. Virology. The SARS coronavirus: a postgenomic era. Science. 2003;300:1377–1378. doi: 10.1126/science.1086418. [DOI] [PubMed] [Google Scholar]
- 5.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 6.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minskaia E., Hertzig T., Gorbalenya A.E., Campanacci V., Cambillau C., Canard B., Ziebuhr J. Discovery of an RNA virus 3′ → 5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl Acad. Sci. USA. 2006;103:5108–5113. doi: 10.1073/pnas.0508200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gioia U., Laneve P., Dlakic M., Arceci M., Bozzoni I., Caffarelli E. Functional characterization of XendoU, the endoribonuclease involved in small nucleolar RNA biosynthesis. J. Biol. Chem. 2005;280:18996–19002. doi: 10.1074/jbc.M501160200. [DOI] [PubMed] [Google Scholar]
- 9.Posthuma C.C., Nedialkova D.D., Zevenhoven-Dobbe J.C., Blokhuis J.H., Gorbalenya A.E., Snijder E.J. Site-directed mutagenesis of the Nidovirus replicative endoribonuclease NendoU exerts pleiotropic effects on the arterivirus life cycle. J. Virol. 2006;80:1653–1661. doi: 10.1128/JVI.80.4.1653-1661.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laneve P., Altieri F., Fiori M.E., Scaloni A., Bozzoni I., Caffarelli E. Purification, cloning, and characterization of XendoU, a novel endoribonuclease involved in processing of intron-encoded small nucleolar RNAs in Xenopus laevis. J. Biol. Chem. 2003;278:13026–13032. doi: 10.1074/jbc.M211937200. [DOI] [PubMed] [Google Scholar]
- 11.Walz F.G., Jr, Osterman H.L., Libertin C. Base-group specificity at the primary recognition site of ribonuclease T for minimal RNA substrates. Arch. Biochem. Biophys. 1979;195:95–102. doi: 10.1016/0003-9861(79)90330-8. [DOI] [PubMed] [Google Scholar]
- 12.Nogues M.V., Vilanova M., Cuchillo C.M. Bovine pancreatic ribonuclease A as a model of an enzyme with multiple substrate binding sites. Biochim. Biophys. Acta. 1995;1253:16–24. doi: 10.1016/0167-4838(95)00138-k. [DOI] [PubMed] [Google Scholar]
- 13.Fersht A.R. 2nd edit., W.H. Freeman; New York: 1985. Enzyme Structure and Mechanism. [Google Scholar]
- 14.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucl. Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson M.P., Igel H., Baertsch R., Haussler D., Ares M., Jr, Scott W.G. The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS Biol. 2005;3:86–94. doi: 10.1371/journal.pbio.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelemen B.R., Klink T.A., Behlke M.A., Eubanks S.R., Leland P.A., Raines R.T. Hypersensitive substrate for ribonucleases. Nucl. Acids Res. 1999;27:3696–3701. doi: 10.1093/nar/27.18.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiu W., Baker M.L., Jiang W., Dougherty M., Schmid M.F. Electron cryomicroscopy of biological machines at subnanometer resolution. Structure. 2005;13:363–372. doi: 10.1016/j.str.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 18.Kim C.H., Kao C.C. A mutant viral RNA promoter with an altered conformation retains efficient recognition by a viral RNA replicase through a solution-exposed adenine. RNA. 2001;7:1476–1485. [PMC free article] [PubMed] [Google Scholar]
- 19.Saida F., Uzan M., Bontems F. The phage T4 restriction endoribonuclease RegB: a cyclizing enzyme that requires two histidines to be fully active. Nucl. Acids Res. 2003;31:2751–2758. doi: 10.1093/nar/gkg377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deshpande R.A., Shankar V. Ribonucleases from T2 family. Crit. Rev. Microbiol. 2002;28:79–122. doi: 10.1080/1040-840291046704. [DOI] [PubMed] [Google Scholar]
- 21.Irie M., Ohgi K. Ribonuclease T2. Methods Enzymol. 2001;341:42–55. doi: 10.1016/s0076-6879(01)41144-x. [DOI] [PubMed] [Google Scholar]
- 22.Kanaya S. Prokaryotic type 2 RNases H. Methods Enzymol. 2001;341:377–394. doi: 10.1016/s0076-6879(01)41165-7. [DOI] [PubMed] [Google Scholar]
- 23.Lima W.F., Wu H., Crooke S.T. Human RNases H. Methods Enzymol. 2001;341:430–440. doi: 10.1016/s0076-6879(01)41168-2. [DOI] [PubMed] [Google Scholar]
- 24.Nicholson A.W. Escherichia coli ribonucleases: paradigms for understanding cellular RNA metabolism and regulation. In: D'Allessio G., Riordan J.F., editors. Ribonucleases: Structures and Functions. Academic Press; New York: 1997. pp. 1–49. [Google Scholar]
- 25.Mauguen Y., Hartley R.W., Dodson E.J., Dodson G.G., Bricogne G., Chothia C., Jack A. Molecular structure of a new family of ribonucleases. Nature. 1982;297:162–164. doi: 10.1038/297162a0. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida H. The ribonuclease T1 family. Methods Enzymol. 2001;341:28–41. doi: 10.1016/s0076-6879(01)41143-8. [DOI] [PubMed] [Google Scholar]
- 27.Quamme G.A., Dai L.J., Rabkin S.W. Dynamics of intracellular free Mg2+ changes in a vascular smooth muscle cell line. Am. J. Physiol. 1993;265:H281–H288. doi: 10.1152/ajpheart.1993.265.1.H281. [DOI] [PubMed] [Google Scholar]
- 28.Zhang R.Q., Ellis K.J. In vivo measurement of total body magnesium and manganese in rats. Am. J. Physiol. 1989;257:R1136–R1140. doi: 10.1152/ajpregu.1989.257.5.R1136. [DOI] [PubMed] [Google Scholar]
- 29.Hausmann Y., Roman-Sosa G., Thiel H.J., Rumenapf T. Classical swine fever virus glycoprotein E rns is an endoribonuclease with an unusual base specificity. J. Virol. 2004;78:5507–5512. doi: 10.1128/JVI.78.10.5507-5512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang W.H., Kornberg R.D. Electron crystal structure of the transcription factor and DNA repair complex, core TFIIH. Cell. 2000;102:609–613. doi: 10.1016/s0092-8674(00)00083-0. [DOI] [PubMed] [Google Scholar]
- 31.Mitsuoka K., Murata K., Kimura Y., Namba K., Fujiyoshi Y. Examination of the LeafScan 45, a line-illuminating micro-densitometer, for its use in electron crystallography. Ultramicroscopy. 1997;68:109–121. [Google Scholar]
- 32.Hovmoller S. CRISP – crystallographic image processing on a personal computer. Ultramicroscopy. 1992;41:121–136. [Google Scholar]
- 33.Booth C., Jiang W., Baker M., Zhou Z., Ludtke S., Chiu W. A 9 Å single particle reconstruction from CCD captured imaged on a 200 kV electron cryomicroscope. J. Struct. Biol. 2004;147:116–127. doi: 10.1016/j.jsb.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Ludtke S.J., Baldwin P.R., Chiu W. Semiautomated software for high resolution single-particle reconstructions. J. Struct. Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 35.Ludtke S.J., Chen D.H., Song J.L., Chuang D.T., Chiu W. Seeing GroEL at 6 Å resolution by single particle electron cryomicroscopy. Structure. 2004;12:1129–1136. doi: 10.1016/j.str.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 36.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]