

Abstract
Viruses are enormously efficient infectious agents that have been implicated in causing human disease for centuries. Transmission of these pathogens continues to be from one life form to another in the form of isolated cases, epidemics, and pandemics. Each infection requires entry into a susceptible host, replication, and evasion of the immune system. Viruses are successful pathogens because they target specific cells for their attack, exploit the cellular machinery, and are efficient in circumventing and/or inhibiting key cellular events required of survival. This article reviews some of the advances that have taken place in human virology in the past 50 years, emphasizing mechanisms that contribute to, and are involved with, virus survival and persistence.
History and progress
The field of virology was barely half a century old in 1948 when Dr Thomas Francis wrote 2 articles, “Viruses as Agents of Disease” and “The Prevention of Virus Disease,” that were published in this journal. Early leadership by Mayer, Ivanovsky, Loeffler, Frosch, Walter Reed and others, allowed progress from “contagium vivum fluidum”1 and a simple understanding of the existence and predatory nature of viruses to the characterization of viruses with regard to size, resistance to chemical and physical agents, host and tissue selectivity, and pathogenic and immunologic effects. These investigations made it clear that viruses were a very diverse group of pathogens. However, our knowledge of viral-cell interactions and the effect of viruses on the immune system was rudimentary. We held the understanding that a recovered individual is not susceptible to reinfection with the same virus, and that serum contained components that when mixed with virus and injected into a susceptible animal that animal was protected. These basic concepts served as the basis for classic studies of active and passive immunization. But several important advances that occurred between 1948 and 1957 jump-started the field of modern virology. These included the development of cultures of single animal cells,2, 3 Watson and Crick's identification of DNA and the genetic code,4 the development of optimal medium for growing cells, and the development of the viral plaque assay.5 By the early 1950s, Max Theiler and Jonas Salk (killed virus) had developed vaccines for yellow fever and polio, respectively, and through the benefits of growing the viruses in cell culture, shortly thereafter Sabin developed the oral (live attenuated virus) vaccine. Introduction of these vaccines into the human masses remains to this day one of the greatest accomplishments of preventive medicine.
In the 1960s, the transition from basic virology to molecular biology began. Viruses and components of viral infections were analyzed using gel electrophoresis, protein-antibody interactions, and biochemical assays to answer basic biologic questions. As a result, greater knowledge of virus replication, viral and cellular receptors, and immunologic interactions was achieved. Specifically, research during this time led to an understanding of the regulation of gene expression including transcription factors, enhancer elements, promoters, aspects of RNA polymerase, and reverse transcriptase, as well as the discovery of proto-oncogenes and tumor suppressor proteins. Scientists tagged viruses to identify intracellular locations of viral proteins, understand nuclear and cytoplasmic shuttling, and map neural circuitry. Complete genomic sequences of viruses have been recorded and entered into public databases. The benefits of databases such as FASTA and BLAST have led to searches in homology between motifs characteristic for specific gene products and the identification of novel viral genes and their functions.
Our knowledge also has advanced from the use of positive and negative selection procedures. Positive selection being the method whereby genomic fragments or single candidate genes are expressed in a suitable cell system and tested for functionality (ie, infection phenotype). In contrast, negative selection is based on the construction of viral mutants that lack specific genes and the implementation of studies that identify a change in phenotype when the viral mutant infects a particular cell or animal. Use of these techniques has led to the identification of virulence factors, novel mechanisms of regulation of cell surface receptors, and signal transduction pathways. Within the last decade, the emerging fields of genomics and proteomics have allowed for the functional analysis of a large number of transcripts and protein sequences that are expressed during viral infection that provide new clues as to the regulation of acute, chronic, and persistent viral infections, as well as reactivation and malignant transformation. Excitingly, the last decade has demonstrated that scientists have the knowledge and skill to harness unique features of viruses (eg, adenoviruses, retroviruses, herpesviruses) in implementing gene therapy and targeting processes important in chronic disease and cancer. However, mastery of this field remains to be seen.
Despite the exponential growth in virology during the last 30 years, mankind still suffers from transmission and disease when humans serve as hosts to viruses. Moreover, the outcomes are often severe when humans serve as novel hosts to emerging virus infection (eg, avian flu virus, Ebola virus, equine hemorrhagic fever viruses, Hanta virus, human immunodeficiency virus (HIV), Hendra virus, Nipah virus, sudden acute respiratory syndrome (SARS) coronavirus, and West Nile virus).6 Of great importance is the fact that the oral cavity continues to be the source of transmission of many viruses, the site of replication and asymptomatic shedding of viruses, and a site where persistent viral infections exist, the latter being a prerequisite for virally induced malignant transformation. Clearly, the field of virology has grown to the extent that a “state-of-the-art” paper would be exhaustive in length. Accordingly, this review focuses on specific viral cell interactions that allow the virus to survive the cellular attack and evade the immune system, establish persistent infections, and cause chronic disease. Additional topics will be covered in future reviews.
Viral counter defenses
Viruses have developed numerous strategies for subverting the host defenses that are launched during infection. The first innate defense encountered is cellular selectivity during the entry process. Viral attachment proteins bind to specific cell receptors (proteins, carbohydrates, or glycolipids) and coreceptors. The absence of a specific receptor shields the cell from attack. If this level of defense is foiled, upon binding receptors can sense microbial infection and trigger a multitude of antimicrobial and inflammatory responses. The toll-like receptor (TLR) family which consists of 10 to 15 members are well characterized in their ability to detect bacterial components (ie, lipoproteins and lipoteichoic acids, flagellin) as well as unmethylated CpG motif DNA of bacteria and viruses (detected by TLR9), double-stranded RNA (detected by TLR3) and single-stranded viral RNA (detected by TLR7).7 In particular, TLRs 3, 7, 8, and 9 specialize in viral detection and recognition of nucleic acids within the intracellular compartments which results in defensive signaling.
After receptor binding, entry is modulated by either direct fusion with the plasma membrane or clathrin- or nonclathrin-mediated endocytosis.8 Viruses that gain entry uncoat and deliver their genetic material and undergo a permissive or nonpermissive infection. A permissive cell permits virus replication and ultimate lysis of the host cell. In contrast, a nonpermissive cell downregulates virus replication and lytic gene expression resulting in little to no viral progeny. Nonpermissive infections can be abortive or persistent, and persistent infections can be active or latent. Latent infections are characterized by silencing of gene transcription, intermittent reactivation, or rarely oncogenic transformation.
At the onset of infection, for a virus to survive within a cell, the virus must balance its own growth with death of the host and circumvention of the immune response. Strategies for survival involve regulating apoptosis, inhibiting interferon production, modulating the major histocompatibility complex (MHC) class I function that ultimately affects the cytotoxic lymphocyte (CTL) and natural killer (NK) response, and limiting cytokine and chemokine production/function. Long-term survival (ie, latency) requires downregulation of lytic gene expression, inhibition of apoptosis, and minimizing the inflammatory response.
Apoptosis
Apoptosis, or programmed cell death, is a highly regulated and conserved series of sequential cellular events that results from receptor- or mitochondrial-mediated pathways in response to a variety of stimuli, including viral infection and the appearance of double-stranded RNA. The process is regulated ( Fig 1) by a family of aspartate-specific cysteinyl proteases, or caspases, that converge at a number of downstream points resulting in proteolytic cleavage and enzyme activation.9 Caspases are segregated into 2 distinct subfamilies. The “apoptotic” caspases (2, 3, 6, 7, 8, 9, and 10) are involved in the cascade that results in protease production, chromatin condensation, and cellular degradation. The “inflammatory” caspases (1, 4, and 5) provide a second round of defense against viral infection.10 The inflammatory caspases are involved in the proteolytic maturation of key cytokines (ie, interleukin (IL)-1β and IL-18). Cytokine IL-18, also known as interferon (IFN)-inducing factor, directs the production of IFN-γ.11 In turn, IFN-γ induces expression of proteolytic active subunits that lead to proteolysis and antigenic processing by TAP proteins. TAP proteins are critical for displaying viral antigens on the cell surface (see Cellular Immunity, below).12
Fig 1.
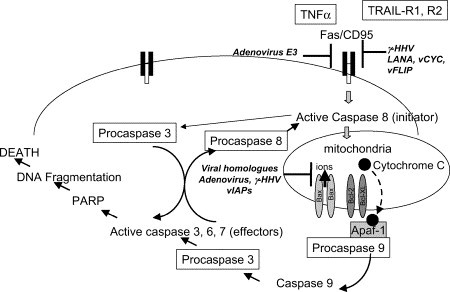
Viral mechanisms involved in subverting the apoptotic pathway. PARP, Poly (ADP-ribose) polymerase.
Clearly, apoptosis is an important target of virus defense, because early destruction of an infected cell could greatly reduce replication and the number of viral progeny produced. Interestingly, viruses have evolved several methods for suppressing or delaying apoptosis as well as encoding proteins that function as inducers of apoptosis. This apparent yin-yang relationship with apoptosis is important to prolong the life of the cell yet facilitate the release and spread of viral progeny at the appropriate time.13, 14
Viruses regulate apoptosis by several mechanisms including the targeting of the tumor suppressor gene product p53, the Fas death receptor, and by producing caspase inhibitors and viral Bcl-2 homologs.15 Adenovirus, for example, encodes several gene products that influence apoptosis. The E1A gene product stabilizes p53 and induces p53-dependent apoptosis.14, 16 In contrast, the adenovirus E3 gene product promotes degradation of Fas, and the adenovirus E1B proteins antagonize p53 function. Viral homologs of Bcl-2, an apoptosis suppressor that binds with Bax, are produced by adenovirus, Epstein-barr virus (EBV, BHRF1 protein), and other viruses.17, 18
There are several classes of caspase inhibitors encoded by viruses. These include the serine proteinase inhibitors (serpins: CrmA/SPI-2), viral inhibitors of apoptosis (vIAPs), p35, and inhibitors of procaspase 8 protease (also known as FLICE).19 CrmA and p35 block caspase 1, previously termed IL-1β–converting enzyme (ICE).20 Caspase 1 functions primarily as an activator of proinflammatory cytokines, but also has apoptosis-inducing ability in select mammalian cells, such as neurons. The vIAPs appear to inhibit Bax-mediated apoptosis in human cells rather than directly inhibiting caspases.21
Several human herpesviruses encode FLICE-inhibitory proteins (FLIPs) that block TRAIL-mediated cell death by interfering with procaspase 8 protease (FLICE) activation.
For example, the β-herpesviruses (cytomegalovirus (CMV)) encode a viral inhibitor of caspase activation (vICA) which inhibits caspase 8 (FLICE) activation,22 and γ-herpesviruses encode vFLIPs (eg, K13) which inhibit activation of caspases by molecular mimicry.23 CMV also encodes a viral mitochondrial inhibitor of apoptosis (vMIA) (encoded by the UL37 gene) which inhibits activation of mitochondrial pores in a manner similar to members of the antiapoptotic Bcl family.24, 25 The alpha herpesvirus HSV-1 encodes several antiapoptotic gene products (ie, ICP4, ICP27, γ34.5, Us3, gJ)26, 27, 28, 29, 30 that modulate apoptosis at several levels, including antagonism of double-stranded RNA-activated protein kinase (PKR), a downstream induction molecule of the interferon signaling pathway31, 32 Of note, all γ-herpesviruses express viral homologues of cellular antiapoptotic genes, including 1 or 2 Bcl-2 homologues.33
Interferon
Interferon, discovered in the late 1950s when scientists observed that virus-infected cells secrete a factor that mediates the transfer of a viral-resistant state,34 is a family of regulatory glycoprotein cytokines that modulate both innate and adaptive antimicrobial immunity. They are products of an infected cell genome and one of the key factors in the host response against viral infection. IFNs serve as an early defense system that precedes the onset of the immune response and are triggered by envelope glycoproteins, CpG DNA, or double-stranded RNA. In recombinant formulations, they have been used in medicine and dentistry to combat various viral infections.35, 36
Human IFNs are classified based on the sequence of amino acids into 3 main groups – α, β, and γ – and 3 that are less extensively studied (ω, κ, and τ, not discussed further in this review). IFN-α and -β are produced rapidly when viral factors interact with cellular pattern-recognition receptors such as TLRs and cytosolic receptors. Historically, synthesis of IFN-α has been attributed to macrophages and B cells, and IFN-β has been considered to be produced by fibroblasts. More recently, plasmacytoid dendritic cells have been shown to produce IFN-α preferentially to IFN-β. Both IFN-α and -β prevent the replication of viruses by inducing formation of secondary messengers which include IFN regulatory factor (IRF) 3, IRF-5, IRF-7, c-Jun/ATF-2, and NF-κB.37 IFN-γ is synthesized by activated T lymphocytes and natural killer (NK) cells following receptor-mediated stimulation or in response to cytokines produced by macrophages or antigen-presenting cells (ie, primarily IL-12, IL-18, and IFN-α/β) or by stimulation through T cell receptors (TCRs) or NK cell receptors. It is a powerful activator of mononuclear phagocytes, thus enhancing their ability to destroy intracellular microorganisms and tumor cells. IFNs mediate their antiviral action through IFN-stimulated genes (ISG), which number in hundreds. IFNs also regulate the cell cycle and have antiproliferative effects.
Viral evasion of IFN occurs by several strategies. In the majority of infections, viruses encode products that antagonize either the IFN signal transduction pathway or cellular proteins induced by IFN that are responsible for inhibiting virus replication ( Fig 2).38 Adenovirus, EBV, papillomavirus, and members of the Paramyxovirinae subfamily encode proteins that inhibit the JAK-STAT (Janus kinase–signal transducer and activator of transcription) signaling pathways that are required for IFN production. Specifically, adenovirus encodes the oncoprotein E1A which inhibits the activation of ISG factor 3 (ISGF3).39, 40 Paramyxovirinae reduce the effectiveness of the IFN response by targeting STAT1 for degradation or by interference with STAT phosphorylation or stability.41, 42 Kaposi's sarcoma–associated herpesvirus (KSHV) encodes the pleiotropic gene product latency-associated nuclear antigen (LANA) that acts downstream of ISGF3 and inhibits p53.43, 44 In an alternate approach, HPV encodes 2 proteins, E6 and E7, that bind to IRF-3 and IRF-1, respectively, both of which inhibit the transactivation functions of the bound IRF.45, 46
Fig 2.
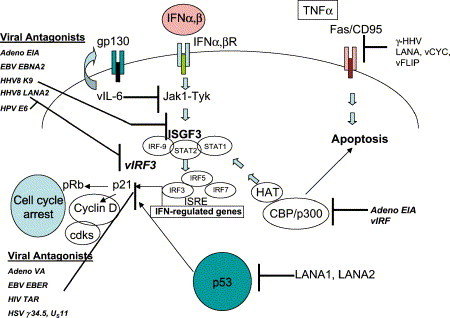
Viral mechanisms involved in subverting the interferon response. IFN, Interferon; IFNR, IFN receptor; IRF, INF regulatory factor; ISRE, IFN stimulable response element; HAT, histone acetyl transferase.
Viruses also encode proteins that mimic cellular components of the IFN signal transduction pathway, including homologs of the IFN receptors, a viral ISRE-like promoter element, and viral homolog of IRF (vIRF). For example, Poxviruses antagonize IFN signals by encoding soluble IFN receptor homologs.47, 48 EBV encodes a viral ISRE and HHV8 encodes vIRF from the K9 ORF that functions as a repressor of transcriptional activation induced by IFN-α, -β, and -γ.49 In addition, several viruses have developed strategies to inhibit IFN-inducible, RNA-dependent protein kinase (PKR). PKR, when antagonized, leads to phosphorylation of eIF-2α which results in inhibition of the IFN-induced antiviral response of the host.50 Adenovirus, herpesviruses, influenza, and SV40 antagonize PKR by different mechanisms involving degradation of PKR, prevention of PKR activation, and resistance to downstream kinase activation.51, 52, 53
Cellular immunity
The MHC class I–restricted T cell response can result in a lethal hit before virus replication. Thus, many viruses have developed strategies for interfering with antigen presentation to MHC class I molecules and intracellular trafficking of MHC molecules. Viruses target the MHC-I at almost all steps of its trafficking: in the endoplasmic reticulum (ER), in the cytoplasm on its way to the surface, and after the MHC reaches the cell surface ( Fig 3).
Fig 3.
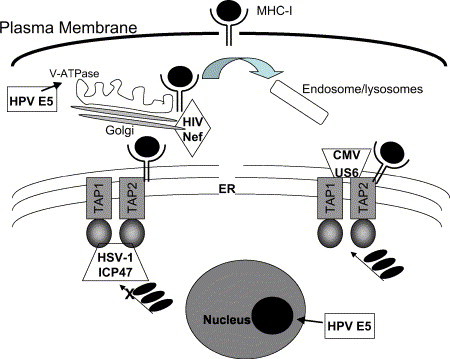
Viral mechanisms involved in subverting the MHC-1 response.
One key target in the viral defense against the cellular arm of the immune system is attack of the transporter protein associated with antigen processing (TAP). TAP loads short antigenic peptides to the MHC which stabilize the class I complexes and allows their migration to the cell surface. Without the peptide cargo, MHC class I molecules are unstable and dissociate. HSV-1 and HSV-2 encode infected cell polypeptide (ICP)-47, an immediate early gene product, that interacts with the TAP protein in the cytosol to prevent peptide binding to TAP.54 Human cytomegalovirus (HCMV) encodes US6, a 183–amino acid glycoprotein, that blocks peptide transport by binding to TAP in the endoplasmic reticulum.55 Although efficient at both retaining MHC-I molecules and preventing CTL recognition, HCMV also uses additional viral proteins (US2, US3, US6, and US11) to evade the immune system.56 A different approach is taken by EBV. This human γ-herpesvirus encodes a glycine-alanine repeat (GAr) domain on EBV-encoded nuclear antigen (EBNA) 1 that inhibits ubiquitin/proteasome-dependent proteolysis of EBV antigens.57 Thus, processing (ie, degradation) of viral proteins into antigenic peptides is restricted.58 KSHV, a lymphotropic γ-herpesvirus, interferes with MHC-I antigen presentation by ubiquitinating the cytosolic domain of the MHC-I.59 Herpesviruses also produce proteins that target MHC class I molecules for degradation in lysosomal compartments60 and downregulate expression of major histocompatibility complex molecules by shutting off host cell protein synthesis by the gene known as virion host shutoff (VHS, UL41).61
In contrast, HIV with its simple genome encodes fewer proteins but accomplishes similar immune evasion by pluripotent accessory proteins. For example, Nef, 1 of the 6 regulatory proteins encoded by HIV, has multiple functions. In addition to enhancing virion infectivity, Nef binds to and inhibits the surface expression of the major MHC-I,62 downregulates the cell surface expression of CD4 (the main HIV receptor),63 and facilitates CD4 receptor endocytosis.64 Through the function of Nef in redirecting the trafficking of immune receptors, infected T lymphocytes are able to hide from the immune system allowing for viral spread.
HPV utilizes 2 early proteins (E5 and E7) to persist undetected within epithelial cells. The early gene product E7 of the oncogenic strains HPV-16 and -18 downregulates MHC-I expression at the transcriptional level by inhibiting the promoters of the MHC-I heavy chain, TAP-1, and LMP-2.65 E5 decreases MHC-I expression at the transcriptional level and causes retention of MHC-I in the Golgi apparatus. Within the Golgi, HPV E5 inactivates the ATPase proton pump system. As a result, acidification is blocked, local pH rises, and MHC-I trafficking is perturbed.66 Thus, it is clear that viruses have achieved ingenious methods for interference with MHC-I antigen presentation and inhibition of the cellular immune response.
Viral persistence
Viruses persist in cells because they are able to downregulate key processes that if left unattended would result in cell death. Originally attributed in part to the immune response, increasing evidence suggests regulation of key genes plays an important role in the process. Specifically, regulation of viral transcription and genomic replication allows for long-term viral stability and survival.
Many viruses, including those that cause persistent infections and chronic disease (ie, hepatitis C virus, hepatitis B virus, HIV, human herpesviruses, HPV, and JC virus), are successful because of their cell tropism and ability to autoregulate their replication efficiently within specific cells. Common features of autoregulation include sensors to the external environment, negative feedback loops, transcriptional enhancers specific for cells that host the persistent infection, and transcriptional silencers. In some cases autoregulation results in steady-state levels of virus replication; in other infections, the virus enters latency only to reactivate intermittently.
The importance of autoregulation is apparent from both in vivo and in vitro studies.67 For example, during lentivirus (HIV, simian immunodeficiency virus, and feline immunodeficiency virus) infection, viremia peaks early after infection then declines to a steady-state level. The effect is not altered by the presence of steroid-induced immunosuppression, and clearance of infected white blood cells is not associated with an earlier presence of antibody, cytokine response, or cytotoxic lymphocyte activity.68 In another common clinical example, successful antiviral therapy results in dramatic drops in viremia, often to undetectable levels.69 However, replication of virus often rebounds rapidly to pretreatment levels upon drug withdrawal, and in vitro studies indicate that cellular and immune functions are not contributory to the observed outcome. Even when antiviral therapy achieves a sustained virologic response (ie, absence of viremia 6 months after the end of treatment), highly sensitive assays (ie, polymerase chain reaction) detect residual viral genomes in most patients, indicating the ability of viruses to persist and autoregulate based on their environment.70, 71
Herpesviruses are well known to establish latency in a variety of cell types, and this family of viruses has the ability to autoregulate. This is illustrated by HSV-1 and HSV-2, which can undergo dichotomous life cycles: a lytic infection in epithelium and a latent infection in neurons. In fact, when neuronal cells are infected with HSV-1 or HSV-2 at low multiplicity of infection in vitro, the majority of cells can survive for many days even in the absence of immune cells and without the addition of antiviral drugs to the culture medium.72 Similarly, if the rare neuronal cells supporting replication are eliminated using acycloguanosine in the above mentioned system, over 95% of the remaining population harbors a quiescent infection for weeks after the antiviral drug is removed, again in the absence of immune cells.73, 74
Viruses regulate replication of their genome in a complex manner, but achieve the outcome through use of viral sensors, repressors, and effectors. Viral sensors “sense” perturbations in the viral equilibrium within the cell and signal change at the appropriate time. With many viruses (ie, HIV, hepatitis C virus, hepatitis B virus), envelope proteins play the role of sensors, because envelope proteins can influence virus replication in both a positive and a negative manner.75, 76, 77, 78 For HPV, regulation is through cellular factors that bind to the promoters of E1 and E2.79 For HSV-1, transcription of the immediate-early (IE) genes during the lytic infection is regulated by the binding of a tegument protein (VP16) with the cellular protein host cell factor (HCF) and Oct-1.80 Thus, VP16 would seem to be a logical choice for a sensor. However, latency can be established in the presence of VP1681 and reactivation occurs without the transactivating domain of VP16.82 Thus, downstream factors of VP16 (eg, ICP0, ICP4, or other unknown factors) serve as sensors of the environment and regulate the balance between latency and reactivation.
The “effectors” (transactivators and replicative enzymes such as RNA polymerase) modulate virus replication and are the targets of the sensors. Effectors are tightly regulated (ie, repressed at certain times) but dynamically modifiable, typically by proteins bound to critical regions of the genome. These proteins afford protection by limiting changes in conformation of, or enzymatic action on, the restricted gene. Histones are the most notable guardians of the effectors. Histones permit access of DNA to specific activators or repressors, general transcription factors, and RNA polymerase by posttranslational modification (acetylation, methylation and phosphorylation) of their amino terminal tails.83, 84 For example, hyperacetylation of histones is associated with an “open chromatin” conformation and transcriptional activation, whereas hypoacetylation of the histone complex is associated with condensed (hetero-) chromatin and gene silencing. Several human herpesviruses85, 86, 87 utilize these mechanisms for regulating latency and reactivation. In addition, the active regions of the latent α-herpesvirus genome appear to be segregated from the repressed gene regions by boundary or insulator elements, similar to that found on cellular chromosomes88 (D Bloom, personal communication). These chromatin insulators appear to be able to protect genes in one region from the regulatory influence of adjacent regions through conserved CTCF motifs.89 Herpesviruses also encode proteins such as LANA and latency-associated transcripts (LAT) that appear to regulate viral transcription during latency.44, 90
Integration, and the site of integration, into the host chromatin is another mechanism that can regulate viral gene transcription. For example, viruses that integrate (ie, HIV) preferentially select chromosomal sites where high-level transcription of key transactivators is maintained.91 This is accomplished by viruses preferentially integrating in chromatin regions characterized by an open structure (a hallmark of actively transcribed genes). The process by which this is regulated is not completely clear, but it has been suggested that cellular proteins may interact with integrase, the viral protein that catalyzes the integration reaction, in a manner that is site specific.
Viral persistence increases the likelihood of chronic infection and replication, but under certain circumstances also contributes to increased risk of oncogenic transformation. This can occur through chromosomal instability and virus integration,92 and the ability of several specific viral proteins to bind and inactivate p53 or less frequently pRb ( Fig 4).93 p53 is a checkpoint protein that interacts with CDK/cyclin inhibitors and p16, p27, and p21 to arrest the cell cycle in the G1 phase and can send signals for apoptosis through the regulatory proteins Bax, Bcl-2, and c-myc.94 The clinical importance of p53 inactivation is exemplified in that persistent HPV infection is associated with an increased risk of developing cervical cancer in young women, and recent findings suggest that the persistence of HPV DNA in treated tissue after cancer therapy is highly predictive of local recurrence.95
Fig 4.
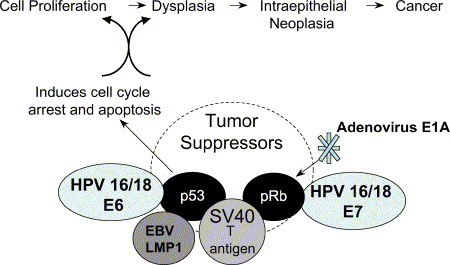
Viral mechanisms involved in dysregulation of apoptosis and increased risk for progression to neoplasia.
Conclusion
In this brief review, examples of mechanisms that contribute to survival and persistence of viruses within their host were presented. Emphasis was placed on mechanisms that permit survival of host defenses, evasion of the immune system, and establishment of chronic infections. Detailed knowledge of these processes has led to many therapeutic successes. However, additional knowledge is required for us to make strides in eliminating human suffering caused by these intracellular pathogens. It is hoped that 50 years from now when another review may appear in this journal on this topic, we will have a better understanding of how to eliminate persistent viral infections and identify patients at risk for virally induced complications of acute and chronic infections and will have harnessed the power of viruses to undergo selective lytic replication in tumor cells and modulate chronic disease.
Lexington, Ky UNIVERSITY OF KENTUCKY
Footnotes
This work was supported in part by NIH grant DE14142.
References
- 1.Levine A.J. The origins of virology. In: Knipe D.M., Howley P.M., editors. Fields virology. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 1–18. [Google Scholar]
- 2.Likely G.D., Sanford K.K., Earle W.R. Further studies on the proliferation in vitro of single isolated tissue cells. J Natl Cancer Inst. 1952;13:177–184. [PubMed] [Google Scholar]
- 3.Sanford K.K., Likely G.D., Bryan W.R., Earle W.R. The infection of cells in tissue culture with Rous sarcoma virus. J Natl Cancer Inst. 1952;12:1317–1343. [PubMed] [Google Scholar]
- 4.Watson J.D., Crick F.H. The structure of DNA. Cold Spring Harb Symp Quant Biol. 1953;18:123–131. doi: 10.1101/sqb.1953.018.01.020. [DOI] [PubMed] [Google Scholar]
- 5.Bachrach H.L., Callis J.J., Hess W.R., Patty R.E. A plaque assay for foot-and-mouth disease virus and kinetics of virus reproduction. Virology. 1957;4:224–236. doi: 10.1016/0042-6822(57)90060-0. [DOI] [PubMed] [Google Scholar]
- 6.Geisbert T.W., Jahrling P.B. Exotic emerging viral diseases: progress and challenges. Nat Med. 2004;10(12 Suppl):S110–S121. doi: 10.1038/nm1142. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y., Huang C.T., Huang X., Pardoll D.M. Persistent toll-like receptor signals are required for reversal of regulatory T cell–mediated CD8 tolerance. Nat Immunol. 2004;5:508–515. doi: 10.1038/ni1059. [DOI] [PubMed] [Google Scholar]
- 8.Earp L.J., Delos S.E., Park H.E., White J.M. The many mechanisms of viral membrane fusion proteins. Curr Top Microbiol Immunol. 2005;285:25–66. doi: 10.1007/3-540-26764-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dery O., Corvera C.U., Steinhoff M., Bunnett N.W. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am J Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- 10.Martinon F., Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–574. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 11.Gracie J.A., Robertson S.E., McInnes I.B. Interleukin-18. J Leukoc Biol. 2003;73:213–224. doi: 10.1189/jlb.0602313. [DOI] [PubMed] [Google Scholar]
- 12.Hisamatsu H., Shimbara N., Saito Y., Kristensen P., Hendil K.B., Fujiwara T. Newly identified pair of proteasomal subunits regulated reciprocally by interferon gamma. J Exp Med. 1996;183:1807–1816. doi: 10.1084/jem.183.4.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Everett H., McFadden G. Apoptosis: an innate immune response to virus infection. Trends Microbiol. 1999;7:160–165. doi: 10.1016/s0966-842x(99)01487-0. [DOI] [PubMed] [Google Scholar]
- 14.Shen Y., Shenk T.E. Viruses and apoptosis. Curr Opin Genet Dev. 1995;5:105–111. doi: 10.1016/s0959-437x(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 15.White E. Life, death, and the pursuit of apoptosis. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- 16.Teodoro J.G., Branton P.E. Regulation of p53-dependent apoptosis, transcriptional repression, and cell transformation by phosphorylation of the 55-kilodalton E1B protein of human adenovirus type 5. J Virol. 1997;71:3620–3627. doi: 10.1128/jvi.71.5.3620-3627.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bilbao G., Contreras J.L., Zhang H.G., Pike M.J., Overturf K., Mikheeva G. Adenovirus-mediated gene expression in vivo is enhanced by the antiapoptotic Bcl-2 gene. J Virol. 1999;73:6992–7000. doi: 10.1128/jvi.73.8.6992-7000.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grinnell B.W., Wagner R.R. Inhibition of DNA-dependent transcription by the leader RNA of vesicular stomatitis virus: role of specific nucleotide sequences and cell protein binding. Mol Cell Biol. 1985;5:2502–2513. doi: 10.1128/mcb.5.10.2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richter B.W., Duckett C.S. The IAP proteins: caspase inhibitors and beyond. Sci STKE. 2000;2000:PE1. doi: 10.1126/stke.2000.44.pe1. [DOI] [PubMed] [Google Scholar]
- 20.Gagliardini V., Fernandez P.A., Lee R.K., Drexler H.C., Rotello R.J., Fishman M.C. Prevention of vertebrate neuronal death by the crmA gene. Science. 1994;263:826–828. doi: 10.1126/science.8303301. [DOI] [PubMed] [Google Scholar]
- 21.Wilkinson J.C., Wilkinson A.S., Scott F.L., Csomos R.A., Salvesen G.S., Duckett C.S. Neutralization of Smac/Diablo by inhibitors of apoptosis (IAPs). A caspase-independent mechanism for apoptotic inhibition. J Biol Chem. 2004;279:51082–51090. doi: 10.1074/jbc.M408655200. [DOI] [PubMed] [Google Scholar]
- 22.Skaletskaya A., Bartle L.M., Chittenden T., McCormick A.L., Mocarski E.S., Goldmacher V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci U S A. 2001;98:7829–7834. doi: 10.1073/pnas.141108798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Q., Zachariah S., Chaudhary P.M. The human herpes virus 8–encoded viral FLICE-inhibitory protein induces cellular transformation via NF-kappaB activation. J Biol Chem. 2003;278:52437–52445. doi: 10.1074/jbc.M304199200. [DOI] [PubMed] [Google Scholar]
- 24.Goldmacher V.S. vMIA, a viral inhibitor of apoptosis targeting mitochondria. Biochimie. 2002;84:177–185. doi: 10.1016/s0300-9084(02)01367-6. [DOI] [PubMed] [Google Scholar]
- 25.Goldmacher V.S., Bartle L.M., Skaletskaya A., Dionne C.A., Kedersha N.L., Vater C.A. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc Natl Acad Sci U S A. 1999;96:12536–12541. doi: 10.1073/pnas.96.22.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koyama A.H., Adachi A. Induction of apoptosis by herpes simplex virus type 1. J Gen Virol. 1997;78:2909–2912. doi: 10.1099/0022-1317-78-11-2909. [DOI] [PubMed] [Google Scholar]
- 27.Zhou G., Galvan V., Campadelli-Fiume G., Roizman B. Glycoprotein D or J delivered in trans blocks apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. J Virol. 2000;74:11782–11791. doi: 10.1128/jvi.74.24.11782-11791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benetti L., Roizman B. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci U S A. 2004;101:9411–9416. doi: 10.1073/pnas.0403160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogg P.D., McDonell P.J., Ryckman B.J., Knudson C.M., Roller R.J. The HSV-1 Us3 protein kinase is sufficient to block apoptosis induced by overexpression of a variety of Bcl-2 family members. Virology. 2004;319:212–224. doi: 10.1016/j.virol.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 30.Sanfilippo C.M., Chirimuuta F.N., Blaho J.A. Herpes simplex virus type 1 immediate-early gene expression is required for the induction of apoptosis in human epithelial HEp-2 cells. J Virol. 2004;78:224–239. doi: 10.1128/JVI.78.1.224-239.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diaz-Guerra M., Rivas C., Esteban M. Full activation of RNaseL in animal cells requires binding of 2-5A within ankyrin repeats 6 to 9 of this interferon-inducible enzyme. J Interferon Cytokine Res. 1999;19:113–119. doi: 10.1089/107999099314252. [DOI] [PubMed] [Google Scholar]
- 32.Zhou A., Paranjape J., Brown T.L., Nie H., Naik S., Dong B. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polster B.M., Pevsner J., Hardwick J.M. Viral Bcl-2 homologs and their role in virus replication and associated diseases. Biochim Biophys Acta. 2004;1644:211–227. doi: 10.1016/j.bbamcr.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 34.Isaacs A., Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147:258–267. [PubMed] [Google Scholar]
- 35.Davis G.L. Treatment of acute and chronic hepatitis C. Clin Liver Dis. 1997;1:615–630. doi: 10.1016/s1089-3261(05)70325-1. [DOI] [PubMed] [Google Scholar]
- 36.Grubman M.J. New approaches to rapidly control foot-and-mouth disease outbreaks. Expert Rev Anti Infect Ther. 2003;1:579–586. doi: 10.1586/14787210.1.4.579. [DOI] [PubMed] [Google Scholar]
- 37.Malmgaard L. Induction and regulation of IFNs during viral infections. J Interferon Cytokine Res. 2004;24:439–454. doi: 10.1089/1079990041689665. [DOI] [PubMed] [Google Scholar]
- 38.Knipe D.M., Samuel C.E., Palese P. Virus-host cell interactions. In: Knipe D.M., Howley P.M., Griffine D.E., Lamb R.A., Martin M.A., Roizman B., editors. Fields Virology. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 133–170. [Google Scholar]
- 39.Ackrill A.M., Foster G.R., Laxton C.D., Flavell D.M., Stark G.R., Kerr I.M. Inhibition of the cellular response to interferons by products of the adenovirus type 5 E1A oncogene. Nucleic Acids Res. 1991;19:4387–4393. doi: 10.1093/nar/19.16.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burgert H.G., Ruzsics Z., Obermeier S., Hilgendorf A., Windheim M., Elsing A. Subversion of host defense mechanisms by adenoviruses. Curr Top Microbiol Immunol. 2002;269:273–318. doi: 10.1007/978-3-642-59421-2_16. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez J.J., Horvath C.M. Host evasion by emerging paramyxoviruses: Hendra virus and Nipah virus V proteins inhibit interferon signaling. Viral Immunol. 2004;17:210–219. doi: 10.1089/0882824041310568. [DOI] [PubMed] [Google Scholar]
- 42.Precious B., Young D.F., Andrejeva L., Goodbourn S., Randall R.E. In vitro and in vivo specificity of ubiquitination and degradation of STAT1 and STAT2 by the V proteins of the paramyxoviruses simian virus 5 and human parainfluenza virus type 2. J Gen Virol. 2005;86:151–158. doi: 10.1099/vir.0.80263-0. [DOI] [PubMed] [Google Scholar]
- 43.Fukushi M., Higuchi M., Oie M., Tetsuka T., Kasolo F., Ichiyama K. Latency-associated nuclear antigen of Kaposi's sarcoma–associated herpesvirus interacts with human myeloid cell nuclear differentiation antigen induced by interferon alpha. Virus Genes. 2003;27:237–247. doi: 10.1023/a:1026391715071. [DOI] [PubMed] [Google Scholar]
- 44.Renne R., Barry C., Dittmer D., Compitello N., Brown P.O., Ganem D. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma–associated herpesvirus. J Virol. 2001;75:458–468. doi: 10.1128/JVI.75.1.458-468.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koromilas A.E., Li S., Matlashewski G. Control of interferon signaling in human papillomavirus infection. Cytokine Growth Factor Rev. 2001;12:157–170. doi: 10.1016/s1359-6101(00)00023-x. [DOI] [PubMed] [Google Scholar]
- 46.Um S.J., Rhyu J.W., Kim E.J., Jeon K.C., Hwang E.S., Park J.S. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002;179:205–212. doi: 10.1016/s0304-3835(01)00871-0. [DOI] [PubMed] [Google Scholar]
- 47.Smith G.L., Symons J.A., Alcami A. Poxviruses: interfering with interferon. Semin Virol. 1998;8:409–418. [Google Scholar]
- 48.Alcami A., Symons J.A., Smith G.L. The vaccinia virus soluble alpha/beta interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J Virol. 2000;74:11230–11239. doi: 10.1128/jvi.74.23.11230-11239.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao S.J., Boshoff C., Jayachandra S., Weiss R.A., Chang Y., Moore P.S. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene. 1997;15:1979–1985. doi: 10.1038/sj.onc.1201571. [DOI] [PubMed] [Google Scholar]
- 50.Samuel C.E. Antiviral actions of interferon. Interferon-regulated cellular proteins and their surprisingly selective antiviral activities. Virology. 1991;183(1):1–11. doi: 10.1016/0042-6822(91)90112-o. [DOI] [PubMed] [Google Scholar]
- 51.Mathews M.B., Shenk T. Adenovirus virus-associated RNA and translation control. J Virol. 1991;65:5657–5662. doi: 10.1128/jvi.65.11.5657-5662.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samuel C.E. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desloges N., Rahaus M., Wolff M.H. Role of the protein kinase PKR in the inhibition of varicella-zoster virus replication by beta interferon and gamma interferon. J Gen Virol. 2005;86:1–6. doi: 10.1099/vir.0.80466-0. [DOI] [PubMed] [Google Scholar]
- 54.Tomazin R., Hill A.B., Jugovic P., York I., van Endert P., Ploegh H.L. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J. 1996;15:3256–3266. [PMC free article] [PubMed] [Google Scholar]
- 55.Lehner P.J., Karttunen J.T., Wilkinson G.W., Cresswell P. The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing–dependent peptide translocation. Proc Natl Acad Sci U S A. 1997;94:6904–6909. doi: 10.1073/pnas.94.13.6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farrell H., Degli-Esposti M., Densley E., Cretney E., Smyth M., Davis-Poynter N. Cytomegalovirus MHC class I homologues and natural killer cells: an overview. Microbes Infect. 2000;2:521–532. doi: 10.1016/s1286-4579(00)00315-4. [DOI] [PubMed] [Google Scholar]
- 57.Yin Y., Manoury B., Fahraeus R. Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus–encoded EBNA1. Science. 2003;301:1371–1374. doi: 10.1126/science.1088902. [DOI] [PubMed] [Google Scholar]
- 58.Dantuma N.P., Sharipo A., Masucci M.G. Avoiding proteasomal processing: the case of EBNA1. Curr Top Microbiol Immunol. 2002;269:23–36. doi: 10.1007/978-3-642-59421-2_2. [DOI] [PubMed] [Google Scholar]
- 59.Coscoy L., Ganem D. Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci U S A. 2000;97:8051–8056. doi: 10.1073/pnas.140129797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reusch U., Muranyi W., Lucin P., Burgert H.G., Hengel H., Koszinowski U.H. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 1999;18:1081–1091. doi: 10.1093/emboj/18.4.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trgovcich J., Johnson D., Roizman B. Cell surface major histocompatibility complex class II proteins are regulated by the products of the gamma(1)34.5 and U(L)41 genes of herpes simplex virus 1. J Virol. 2002;76:6974–6986. doi: 10.1128/JVI.76.14.6974-6986.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Collins K.L., Chen B.K., Kalams S.A., Walker B.D., Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 63.Garcia J.V., Miller A.D. Serine phosphorylation-independent downregulation of cell-surface CD4 by Nef. Nature. 1991;350:508–511. doi: 10.1038/350508a0. [DOI] [PubMed] [Google Scholar]
- 64.Piguet V., Chen Y.L., Mangasarian A., Foti M., Carpentier J.L., Trono D. Mechanism of Nef-induced CD4 endocytosis: Nef connects CD4 with the mu chain of adaptor complexes. EMBO J. 1998;17:2472–2481. doi: 10.1093/emboj/17.9.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Georgopoulos N.T., Proffitt J.L., Blair G.E. Transcriptional regulation of the major histocompatibility complex (MHC) class I heavy chain, TAP1 and LMP2 genes by the human papillomavirus (HPV) type 6b, 16 and 18 E7 oncoproteins. Oncogene. 2000;19:4930–4935. doi: 10.1038/sj.onc.1203860. [DOI] [PubMed] [Google Scholar]
- 66.Piguet V. Receptor modulation in viral replication: HIV, HSV, HHV-8 and HPV: same goal, different techniques to interfere with MHC-I antigen presentation. Curr Top Microbiol Immunol. 2005;285:199–217. doi: 10.1007/3-540-26764-6_7. [DOI] [PubMed] [Google Scholar]
- 67.Sallie R. Replicative homeostasis: a fundamental mechanism mediating selective viral replication and escape mutation. Virol J. 2005;2:10. doi: 10.1186/1743-422X-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Terwee J.A., Yactor J.K., Sondgeroth K.S., Vandewoude S. Puma lentivirus is controlled in domestic cats after mucosal exposure in the absence of conventional indicators of immunity. J Virol. 2005;79:2797–2806. doi: 10.1128/JVI.79.5.2797-2806.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neumann A.U., Lam N.P., Dahari H., Gretch D.R., Wiley T.E., Layden T.J. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 70.Feld J.J., Liang T.J. HCV persistence: cure is still a four letter word. Hepatology. 2005;41:23–25. doi: 10.1002/hep.20561. [DOI] [PubMed] [Google Scholar]
- 71.Radkowski M., Gallegos-Orozco J.F., Jablonska J., Colby T.V., Walewska-Zielecka B., Kubicka J. Persistence of hepatitis C virus in patients successfully treated for chronic hepatitis C. Hepatology. 2005;41:106–114. doi: 10.1002/hep.20518. [DOI] [PubMed] [Google Scholar]
- 72.Miller C.S., Bhattacharjee P.S., Higaki S., Jacob R.J., Danaher R.J., Thompson H.W. Herpesvirus quiescence (QIF) in neuronal cells VI: correlative analysis demonstrates usefulness of QIF-PC12 cells to examine HSV-1 latency and reactivation and deregulated LAT ORF Expression. Curr Eye Res. 2003;26:239–248. doi: 10.1076/ceyr.26.3.239.14901. [DOI] [PubMed] [Google Scholar]
- 73.Danaher R.J., Jacob R.J., Miller C.S. Establishment of a quiescent herpes simplex virus type 1 infection in neurally differentiated PC12 cells. J Neurovirol. 1999;5:258–267. doi: 10.3109/13550289909015812. [DOI] [PubMed] [Google Scholar]
- 74.Danaher R.J., Jacob R.J., Chorak M., Freeman C.S., Miller C.S. Heat stress activates production of herpes simplex virus type 1 from quiescently infected neurally differentiated PC12 cells. J Neurovirol. 1999;5:374–383. doi: 10.3109/13550289909029478. [DOI] [PubMed] [Google Scholar]
- 75.Sakai H., Shibata R., Sakuragi J., Sakuragi S., Kawamura M., Adachi A. Cell-dependent requirement of human immunodeficiency virus type 1 Vif protein for maturation of virus particles. J Virol. 1993;67:1663–1666. doi: 10.1128/jvi.67.3.1663-1666.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wei Y., Tavis J.E., Ganem D. Relationship between viral DNA synthesis and virion envelopment in hepatitis B viruses. J Virol. 1996;70:6455–6458. doi: 10.1128/jvi.70.9.6455-6458.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iwatani Y., Kawano K., Ueno T., Tanaka M., Ishimoto A., Ito M. Analysis of dominant-negative effects of mutant Env proteins of human immunodeficiency virus type 1. Virology. 2001;286:45–53. doi: 10.1006/viro.2001.0944. [DOI] [PubMed] [Google Scholar]
- 78.Su L., Kaneshima H., Bonyhadi M.L., Lee R., Auten J., Wolf A. Identification of HIV-1 determinants for replication in vivo. Virology. 1997;227:45–52. doi: 10.1006/viro.1996.8338. [DOI] [PubMed] [Google Scholar]
- 79.Longworth M.S., Laimins L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004;68:362–372. doi: 10.1128/MMBR.68.2.362-372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spector D., Purves F., Roizman B. Role of alpha-transinducing factor (VP16) in the induction of alpha genes within the context of viral genomes. J Virol. 1991;65:3504–3513. doi: 10.1128/jvi.65.7.3504-3513.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sears A.E., Hukkanen V., Labow M.A., Levine A.J., Roizman B. Expression of the herpes simplex virus 1 alpha transinducing factor (VP16) does not induce reactivation of latent virus or prevent the establishment of latency in mice. J Virol. 1991;65:2929–2935. doi: 10.1128/jvi.65.6.2929-2935.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Steiner I., Spivack J.G., Deshmane S.L., Ace C.I., Preston C.M., Fraser N.W. A herpes simplex virus type 1 mutant containing a nontransinducing Vmw65 protein establishes latent infection in vivo in the absence of viral replication and reactivates efficiently from explanted trigeminal ganglia. J Virol. 1990;64:1630–1638. doi: 10.1128/jvi.64.4.1630-1638.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Iizuka M., Smith M.M. Functional consequences of histone modifications. Curr Opin Genet Dev. 2003;13:154–160. doi: 10.1016/s0959-437x(03)00020-0. [DOI] [PubMed] [Google Scholar]
- 84.Grewal S.I., Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301:798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- 85.Murphy J.A., Duerst R.J., Smith T.J., Morrison L.A. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J Virol. 2003;77:9337–9345. doi: 10.1128/JVI.77.17.9337-9345.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jenkins P.J., Binne U.K., Farrell P.J. Histone acetylation and reactivation of Epstein-Barr virus from latency. J Virol. 2000;74:710–720. doi: 10.1128/jvi.74.2.710-720.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu R., Misra V. Potential role for luman, the cellular homologue of herpes simplex virus VP16 (alpha gene trans-inducing factor), in herpesvirus latency. J Virol. 2000;74:934–943. doi: 10.1128/jvi.74.2.934-943.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Quinn J.P., McGregor R.A., Fiskerstrand C.E., Davey C., Allan J., Dalziel R.G. Identification of a novel multifunctional structural domain in the herpes simplex virus type 1 genome: implications for virus latency. J Gen Virol. 1998;79:2529–2532. doi: 10.1099/0022-1317-79-10-2529. [DOI] [PubMed] [Google Scholar]
- 89.West A.G., Gaszner M., Felsenfeld G. Insulators: many functions, many mechanisms. Genes Dev. 2002;16:271–288. doi: 10.1101/gad.954702. [DOI] [PubMed] [Google Scholar]
- 90.Garber D.A., Schaffer P.A., Knipe D.M. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J Virol. 1997;71:5885–5893. doi: 10.1128/jvi.71.8.5885-5893.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cereseto A., Giacca M. Integration site selection by retroviruses. AIDS Rev. 2004;6:13–21. [PubMed] [Google Scholar]
- 92.Wang S.S., Hildesheim A. Viral and host factors in human papillomavirus persistence and progression. J Natl Cancer Inst Monogr. 2003:35–40. doi: 10.1093/oxfordjournals.jncimonographs.a003480. [DOI] [PubMed] [Google Scholar]
- 93.Klein G. Perspectives in studies of human tumor viruses. Front Biosci. 2002;7:268–274. doi: 10.2741/A726. [DOI] [PubMed] [Google Scholar]
- 94.Collot-Teixeira S., Bass J., Denis F., Ranger-Rogez S. Human tumor suppressor p53 and DNA viruses. Rev Med Virol. 2004;14:301–319. doi: 10.1002/rmv.431. [DOI] [PubMed] [Google Scholar]
- 95.Nagai Y., Toma T., Moromizato H., Maehama T., Asato T., Kariya K. Persistence of human papillomavirus infection as a predictor for recurrence in carcinoma of the cervix after radiotherapy. Am J Obstet Gynecol. 2004;191:1907–1913. doi: 10.1016/j.ajog.2004.06.088. [DOI] [PubMed] [Google Scholar]