

Publisher Summary
This chapter discusses the chemical and biological aspects of Narcissus alkaloids. Numerous alkaloids have been isolated from Narcissus speciesasaresult of the continuing search for novel alkaloids with pharmacological activity in the Amaryllidaceae family. The alkaloids isolated from this genus, classified in relation to the different skeleton types. The different Narcissus wild species and intersectional hybrids, grouped into subgenera and sections, with their corresponding alkaloids, arranged according to their ring system are listed. The biosynthetic pathways of Narcissus alkaloids includes: (1) enzymatic preparation of the precursors, (2) primary cyclization mechanisms, (3) enzymatic preparation of intermediates, (4) secondary cyclization, diversification, and restructuring. The chapter discusses proton nuclear magnetic resonance (1H NMR), carbon nuclear magnetic resonance (13C NMR), and mass spectrometry (MS) for Narcissus alkaloids. A list of the different Narcissus alkaloids, their spectroscopic properties, and literature with the most recent spectroscopic data is given. Several Narcissus extracts shows the following activities: antiviral, prophage induction, antibacterial, antifungal, antimalarial, insecticidal, cytotoxic, antitumor, antimitotic, antiplatelet, hypotensive, emetic, acetylcholine esterase inhibitory, antifertility, antinociceptive, chronotropic, pheromone, plant growth inhibitor, and allelopathic.
I. Introduction
A. Geographical Distribution and Taxonomical Aspects
The genus Narcissus L. belongs to the Narcisseae, one of the 15 tribes of the Amaryllidaceae, a widely distributed monocotyledonous family of 59 genera and about 850 species (1). The Amaryllidaceae are richly represented in the tropics and have pronounced centers of diversity in South Africa and the Andean region. Some genera are also found in the Mediterranean area and the temperate regions of Asia. The family's phylogenetic relationships closely follow geographic distribution, with much regional endemism, which adds credence to a Gondwana origin for the family at a time when the continents were much closer together (1). The hypothesis that the family evolved in Africa and subsequently spread to other continents, further suggesting that South America is the center of secondary diversification is supported by the matK sequence data (2).
Narcissus comprises approximately 80–100 wild species of perennial geophytes, geographically concentrated in southwestern Europe, with a center of diversity in the Iberian Peninsula and North Africa, which substantiates the hypotheses that landmasses of Europe and Africa were once joined. A few species extend into France and Italy, and even fewer are found in the Balkans and the eastern Mediterranean. Records outside this area, such as N. tazetta variants in China and Japan, are almost certainly ancient introductions 3., 4.. The native habitats of Narcissus species are very varied, ranging from lowland to mountain sites, including grassland, scrub, woods, river banks, and rocky crevices (3). Most of the species flower in late winter and spring, although a few flower in the autumn (5). All species are insect pollinated, with the majority possessing showy flowers, some of which are highly scented (6). The major pollinators are bees, butterflies, flies, and hawkmoths 5., 7., 8..
The taxonomy of the genus Narcissus is complex and unsettled because of its very varied wild populations, the ease with which hybridization occurs naturally, accompanied by extensive cultivation, breeding, selection, escape, and naturalization, and also because many descriptions of taxons have been based on garden specimens, several of which were probably of hybrid origin. However, although the status bestowed upon the individual groups may vary somewhat from author to author, the basic content of each group is similar in the various classifications 3., 4., 5.. The most recent classification Scheme 4, which incorporates elements of those of Fernandes and Webb 9., 10., 11., also notes that substantial work of revision is needed for some taxa in the genus. The inferred plastid-based phylogeny, on the other hand, reveals that floral morphology has been remarkably dynamic during the history of Narcissus, indicating extensive and repeated diversification and convergent evolution. Polymorphic sexual systems in Narcissus have evolved independently from the ancestral monomorphic state on at least six occasions 5., 12.. As these morphologies are associated with particular suites of pollinators, the recurring transitions from one floral design to another have probably accompanied pollinator transitions. Additionally, the remarkable resemblance of flower features between unrelated species indicates that pollinator activity might have driven flower convergence (13).
Scheme 4.
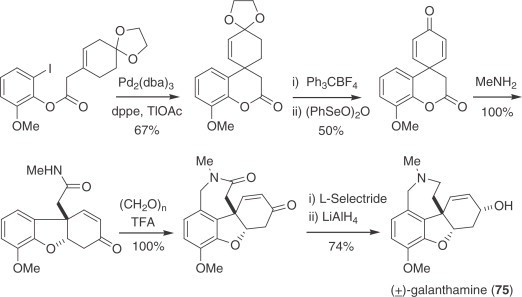
Palladium-mediated synthesis of galanthamine (75).
Most of the species of Narcissus will hybridize but, significantly, there is great variation in the fertility of the offspring, depending upon the degree of relationship between the parents (4). Hybridization has become very popular, resulting in thousands of commercial Narcissus cultivars that are in most cases larger and more robust than their wild parents (3). Moreover, large garden varieties of daffodils have recently been crossed with many of the small wild species to produce delightfully graceful blossoms. Unfortunately, the ability to produce new kinds of miniature daffodils is hampered by the disappearance of many of the tiny wild species (14). Thus, the survival of a number of Narcissus species is under threat because of over-collection and habitat destruction. There is a need to maintain vigilance in the conservation of wild species and of their many variants (15).
Narcissus bulbs have been an important floricultural crop in Western Europe since the late nineteenth century, although the bulbs have been grown in the Netherlands since the sixteenth century. At the start of the twenty-first century, the daffodil remains one of the major ornamental spring-flowering bulb crops grown in temperate regions, with large areas of field-grown crops providing both bulbs and flowers 15., 16.. Rees (17) estimated that the area of Narcissus grown in gardens, parks, cemeteries, etc., is five times the area grown commercially.
The development of the horticultural classification of Narcissus cultivars in 13 divisions was described by Kington (18) and, in order to avoid confusion due to the re-use of earlier names, since 1998, the International Daffodil Register and Classified List is updated annually by supplements of newly registered names (4).
B. The Amaryllidaceae Alkaloids
A particular characteristic of the Amaryllidaceae is a consistent presence of an exclusive group of alkaloids, which have been isolated from the plants of all the genera of this family. The Amaryllidaceae alkaloids represent a large, and still expanding, group of isoquinoline alkaloids, the majority of which are not known to occur in any other family of plants. Since the isolation of the first alkaloid, lycorine, from Narcissus pseudonarcissus in 1877, substantial progress has been made in examining the Amaryllidaceae plants, although they still remain a relatively untapped phytochemical source (1). At present, over 300 alkaloids have been isolated from plants of this family (19) and, although their structures vary considerably, these alkaloids are considered to be biogenetically related.
The large number of structurally diverse Amaryllidaceae alkaloids are classified mainly into nine skeleton types, for which the representative alkaloids are norbelladine, lycorine, homolycorine, crinine, hemanthamine, narciclasine, tazettine, montanine, and galanthamine (Fig. 1 ). With the aim of unifying the numbering system of the different skeleton types, Ghosal's model will be used in this review (20).
Figure 1.
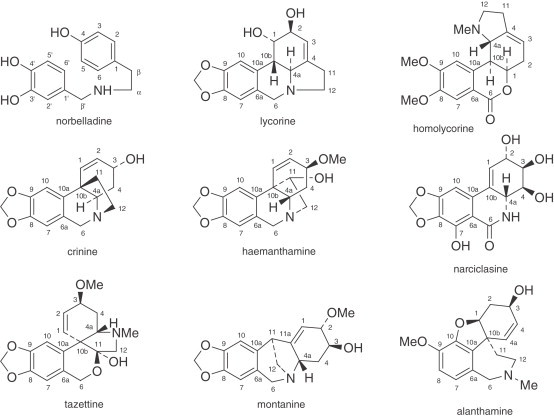
Amaryllidaceae alkaloid types.
As the alkaloids of the Amaryllidaceae family species fall mainly into one of these subgroups, they can serve as a classifying tool for including genera and species in this family. Thus, the genus Behria, in spite of having been classified as Amaryllidaceae (21), does not have any Amaryllidaceae alkaloids (22) and should therefore be included in the Alliaceae family. Furthermore, although it is unusual to find other types of alkaloids in this family, if present, they are always accompanied by typical Amaryllidaceae alkaloids. The classical example is the reported presence of the mesembrane (Sceletium) alkaloids, generally found in the Aizoaceae family 23., 24., in a few species of Amaryllidaceae such as Hymenocallis arenicola, Crinum oliganthum, N. pallidulus, and N. triandrus 25., 26., 27.. In turn, the unexpected isolation of (−)-capnoidine and (+)-bulbocapnine from Galanthus nivalis subsp. cilicicus is the first report of the occurrence of classical isoquinoline alkaloids in a typical member of the Amaryllidaceae (28). On the contrary, the isolation of the Amaryllidaceae alkaloid crinamine from the wild yam Dioscorea dregeana (Dioscoreaceae) has also proven that Amaryllidaceae alkaloids may well be encountered in other plant families (29). Both of these results suggest that the definition of the Amaryllidaceae alkaloids should be reconsidered, and also the alkaloidal profile of this plant family.
The presence of alkaloids in plants is believed to be a protective adaptation, which in Amaryllidaceae is connected with the seasonal cycle of development, many species grow in early spring when other genera are only just starting to grow (30). Thus, Amaryllidaceae plants present an ontogenic variation of alkaloids, and the effects of stress, such as incisional injuries or insect attacks, on alkaloid metabolism causes almost complete hydrolysis of the alkaloidal conjugates and also produces oxidized metabolites (31). Regarding co-evolutionary adaptation, a notable example is the elaboration of a new conjugate alkaloid (telastasine) by the insect Polytela gloriosa Fab. (Noctuidae), a smokey-gray moth that is adapted to toxic Amaryllidaceae plants 32., 33.. Plants and insects use the same detoxification mechanism. These alkaloids, essential for plant survival, have a wide range of interesting physiological effects, including antitumor, antiviral, acetylcholinesterase inhibitory, immunostimulatory, and antimalarial activities, some of them being of particular interest because of their potential use in clinical therapy (34).
Plants of the Amaryllidaceae family have been used for thousands of years as herbal remedies. The alkaloids from their extracts have been the object of active chemical investigation for nearly 200 years. Over the past two decades many have been isolated, screened for different biological activities, and synthesized by a number of research groups. An important handicap is the availability of these alkaloids, which are obtained only in minute quantities from natural sources. Since isolation in larger quantities is not practical, there is a strong case for the development of syntheses or semisyntheses of these alkaloids and their derivatives as potential prodrugs (35).
The history of the Amaryllidaceae alkaloids, their structural elucidation, and their biological profiles, as well as their synthesis, have been summarized on several occasions 20., 33., 36., 37., 38., 39., 40., 41., 42., 43., 44., 45., 46., 47., 48. which, together with the regular publications in the journal Natural Product Reports 34., 49., 50., 51., 52., 53., 54., 55., 56., 57., 58., 59., 60., 61., 62., 63., 64., 65., 66., represent a valuable source of information.
The present review provides coverage of the isolation, spectroscopy, biological activity, and chemical synthesis of the Amaryllidaceae alkaloids present in the genus Narcissus up to July 2005.
II. Narcissus Alkaloids and their Occurrence
Numerous alkaloids have been isolated from Narcissus species as a result of the continuing search for novel alkaloids with pharmacological activity in the Amaryllidaceae family. The alkaloids isolated from this genus, classified in relation to the different skeleton types, are shown in Table I, Table II, Table III, Table IV, Table V, Table VI, Table VII . Table VIII lists the different Narcissus wild species and intersectional hybrids, grouped into subgenera and sections, with their corresponding alkaloids, arranged according to their ring system. The occurrence of alkaloids in Narcissus cultivars is shown in Table IX .
Table I.
Narcissus Alkaloid Structures. Lycorine Type
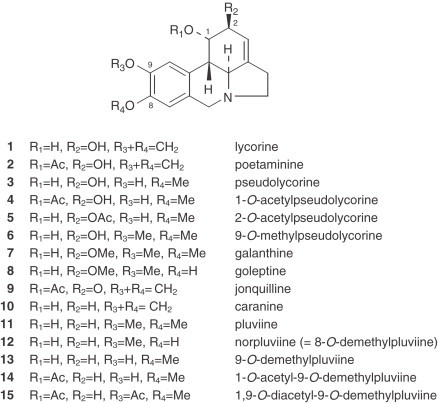 |
 |
 |
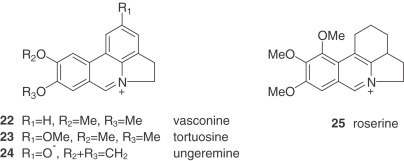 |
Table II.
Narcissus Alkaloid Structures. Homolycorine Type
 |
 |
Table III.
Narcissus Alkaloid Structures. Hemanthamine Type
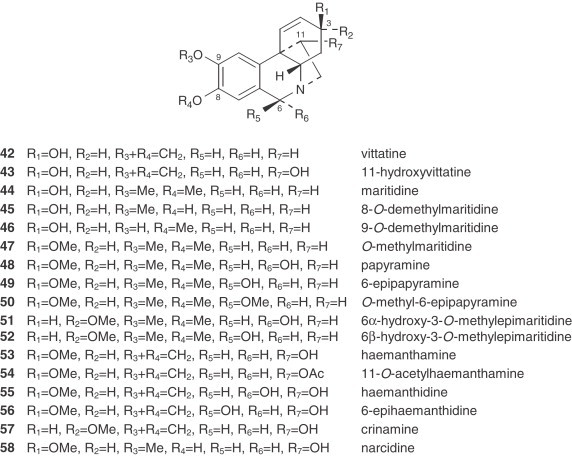 |
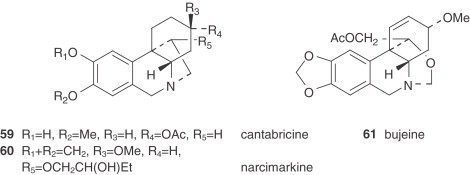 |
Table IV.
Narcissus Alkaloid Structures. Tazettine Type
 |
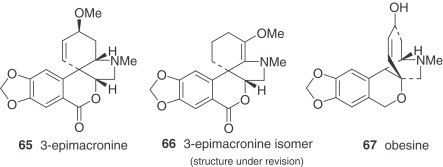 |
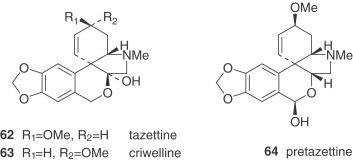
Table V.
Narcissus Alkaloid Structures. Narciclasine and Montanine Types
 |
 |
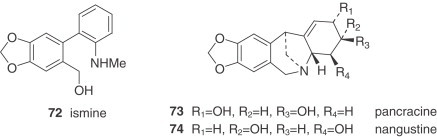 |
Table VI.
Narcissus Alkaloid Structures. Galanthamine Type
 |
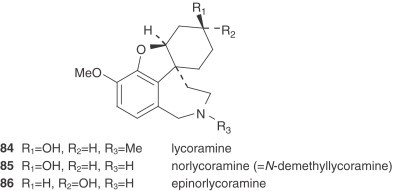 |
Table VII.
Narcissus Alkaloid Structures. Other Types
 |
 |
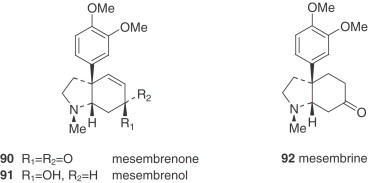 |
Table VIII.
Occurrence of Narcissus Alkaloids in Wild Species and Intersectional Hybrids
| Speciesa | Skeleton types: (alkaloids)b | References |
|---|---|---|
| 1. Subgenus Narcissus | ||
| a- Section Narcissus L. | ||
| N. angustifolius Curtis ex Haw. | GA: (75) | (88) |
| N. angustifolius Curtis subsp. transcarpathicus Kricsfalusy | LY: (3, 22, 24); HL: (27); | |
| MN: (73, 74); OA: (88) | (80) | |
| N. poeticus L. | LY: (1, 2, 6, 7, 11, 16, 18); | 89., 90., 91., 92., 93., 94., 95. |
| HL: (26, 35); MN: (73); GA: (75, 79) | ||
| N. poeticus L. var. ornatus Hort. | LY: (1, 2, 7); HL: (26, 35, 41); | 89., 96., 97., 98. |
| HT: (53); TZ: (62); GA: (75) | ||
| b- Section Pseudonarcissi DC. | ||
| Group A [Plant small, usually less than 15 cm] | ||
| N. asturiensis (Jordan) Pugsley | HT: (53, 55) ; TZ: (62, 65); | (99) |
| NC: (70, 72) | ||
| N. cyclamineus DC | NC: (68); GA: (83) | 100., 101. |
| N. jacetanus Fdez. Casas | LY: (1, 3, 20, 21) | (102) |
| N. lobularis Hort. | HT: (53); GA: (75) | (103) |
| N. muñozii-garmendiae Fdez. Casas | HL: (26, 35, 36) | (104) |
| N. vasconicus Fdez. Casas | LY: (1, 22); HL: (26, 28) | (70) |
| Group B [Plant often 15–60 cm or more and/or with large flowers] | ||
| N. bicolor L. | LY: (22); HL: (29); TZ: (64, 65); | (79) |
| NC: (71, 72) | ||
| N. bujei (Fdez. Casas) Fdez. Casas | HL: (26, 27, 30, 35, 36, 38); | 74., 93. |
| HT: (53, 54, 57, 61); TZ: (62); GA: (75) | ||
| N. confusus Pugsley | HL: (26, 29); HT: (53); TZ: (64); | 82., 83., 93. |
| GA: (75, 79, 80) | ||
| N. eugeniae Fdez. Casas | HL: (26, 35); GA: (75) | 93., 105., 106. |
| N. leonensis Pugsley | LY: (1); GA: (78, 79, 86) | 93., 107. |
| N. pallidiflorus Pugsley | HL: (26, 27); HT: (53); TZ: (64); | (86) |
| NC: (72); OA: (89) | ||
| N. perez-chiscanoi Fdez. Casas | GA: (75, 79) | (93) |
| N. primigenius (Laínz) Fdez. Casas & Laínz | HL: (26, 27); HT: (45, 53) | (108) |
| N. pseudonarcissus L. | LY: (1); HL: (26, 30); HT: (53); | 95., 109., 110. |
| OA: (87) | ||
| N. radinganorum Fdez. Casas | HL: (26, 27); HT: (46) | (111) |
| N. tortuosus Haworth | LY: (1, 23) | (68) |
| c- Section Ganymedes Salisbury ex Schultes and Schultes | ||
| N. pallidulus Graells | LY: (25); OA: (90) | 26., 67. |
| N. triandrus L. | OA: (90, 91, 92) | (112) |
| d- Section Jonquillae De Candolle | ||
| N. assoanus Léon-Dufour=N. requienii Roemer | LY: (3, 4, 5, 20, 21) | 87., 113., 114. |
| N. jonquilla L. | NC: (68); GA: (75, 84) | (115) |
| 2. Subgenus Hermione (Salisbury) Spach | ||
| a- Section Hermione | ||
| i. Subsection Hermione [syn. Section Tazettae De Candolle] | ||
| A. Series Hermione | ||
| N. aureus Loiseleur=N. tazetta L. ssp. aureus (Loisel.) Baker | LY: (1) | (116) |
| N. canaliculatus Guss. | HT: (53); TZ: (62); NC: (68) | 89., 101. |
| N. tazetta L. | LY: (1, 3, 7, 16, 18); | 89., 117., 118., 119., 120., 121., 122., 123., 124., 125., 126., 127., 128., 129. |
| HL: (26, 27, 30, 34); | ||
| HT: (47, 53, 55, 56); | ||
| TZ: (62, 63, 64); NC: (68, 71) | ||
| GA: (75, 82, 83) | ||
| N. tazetta L. ssp. tazetta | LY: (1); HT: (43, 53); | (130) |
| TZ: (62); GA: (78) | ||
| N. tazetta L. var. chinensis Roem | LY: (1, 3, 11); HL: (26, 35); | 131., 132., 133. |
| HT: (44, 47, 48, 49, 51, 52, 55, 56); | ||
| TZ: (62, 64); GA: (75, 76, 84) | ||
| B. Series Albiflorae | ||
| N. dubius Gouan | LY: (3); HL: (32, 33) | 72., 134. |
| N. panizzianus Parlatore | LY: (7); HL: (26); | (135) |
| HT: (48, 49); TZ: (64) | ||
| N. papyraceus Ker-Gawler. | LY: (1, 3); HL: (26, 27, 40); | 91., 136., 137. |
| HT: (44, 47, 48, 49, 50, 60); | ||
| TZ: (62); GA: (75, 84) | ||
| N. tortifolius Fdez. Casas | HL: (26, 27, 32, 33); GA: (75) | (138) |
| ii. Subsection Serotini [syn. Section Serotini Parlatore] | ||
| N. serotinus Löfl. ex L. | TZ: (66); NC: (68) | 101., 139. |
| 3. Subgenus Corbularia (Salisb.) Pax [syn. Section Bulbocodii De Candolle] | ||
| N. bulbocodium L. | HT : (53); TZ : (62, 64); | (112) |
| NC: (72); GA: (75, 81) | ||
| N. cantabricus DC. | HT: (42, 57, 59); TZ: (62) | (140) |
| N. nivalis Graells | LY: (6); GA: (75, 78) | (141) |
| N. obesus Salisbury | HT: (53); TZ: (64, 65, 67); | (76) |
| NC: (71, 72); GA: (75) | ||
| Intersectional hibrids | ||
| Intersection [Jonquillae DC. x Narcissus L.] | ||
| N. x gracilis Sabine | LY: (1); TZ: (62); GA: (75) | (142) |
| Intersection [Narcissus L. x Tazettae DC.] | ||
| N. x biflorus Curtis | LY: (1) | (116) |
| Intersection [Jonquillae DC. x Pseudonarcissii DC.] | ||
| N. x odorus L. var. rugulosus | LY: (1); HL: (26, 34, 37); | 101., 142. |
| TZ: (62); NC: (68); GA: (75) | ||
| Other Narcissus | ||
| Narcissus sp. | LY: (2); NC: (68, 69); GA: (75) | 95., 125., 143., 144. |
Wild Narcissus species and hybrids are grouped into Subgenera and Sections according to the Narcissus classification of Mathew (4). Some taxonomical aspects are also based on the works of Barra and López-González 145., 146., Dorda and Fernández-Casas 147., 148., 149., 150., 151., 152., Fernandes 10., 153. and Fernández-Casas 154., 155., 156., 157..
Skeleton types: LY, lycorine; HL, homolycorine; HT, hemanthamine; TZ, tazettine; NC, narciclasine; MN, montanine; GA, galanthamine; OA, other alkaloids. Alkaloid names: see Tables I–VII.
Table IX.
Occurrence of Alkaloids in Narcissus Cultivars
| Narcissus cultivarsa | Skeleton types: (alkaloids)b | References |
|---|---|---|
| Division 1 – Trumpet Daffodils | ||
| Narcissus ‘Celebrity’ | NC: (68) | (101) |
| Narcissus ‘Covent Garden’ | LY: (1, 7, 11); HL: (35); | 103., 158. |
| HT: (53); GA: (75, 78, 85) | ||
| Narcissus ‘Dutch Master’ | GA: (75) | (81) |
| Narcissus ‘Early Glory’ | LY: (1, 7); HT: (53); GA: (75) | (103) |
| Narcissus ‘Flower Carpet’ | NC: (68) | (101) |
| Narcissus ‘Golden Harvest’ | NC: (68); GA: (75) | 81., 101. |
| Narcissus ‘Grand Maître’ | LY: (1, 7); HL: (26); HT: (53); | (103) |
| TZ: (62); GA: (75) | ||
| Narcissus ‘Imperator’ | LY: (1, 7, 11); HL: (26, 35); | (103) |
| HT: (53); GA: (75) | ||
| Narcissus ‘King Alfred’ | LY: (1, 6, 7, 11, 16, 20); | 92., 101., 103., 125., 158., 159., 160., 161. |
| HL: (26, 27, 34, 35, 37); | ||
| HT: (53, 58); NC: (68); | ||
| GA: (75, 78, 85) | ||
| Narcissus ‘Magnet’ | LY: (7, 11); HT: (53) | (103) |
| Narcissus ‘Magnificience’ | LY: (1, 7, 11); HT: (53); | 103., 158. |
| GA: (75, 78, 85) | ||
| Narcissus ‘Mount Hood’ | HL: (26, 34); NC: (68); | 101., 162., 163., 164. |
| GA: (75, 84, 85) | ||
| Narcissus ‘Mrs. Ernst H. Krelage’ | LY: (1, 7, 11); HT: (53); | 103., 163. |
| NC: (68); GA: (75) | ||
| Narcissus ‘Music Hall’ | LY: (1, 7); HT: (53) | (103) |
| Narcissus ‘Oliver Cromwell’ | LY: (7, 11); HT: (53); GA: (75) | (103) |
| Narcissus ‘President Lebrun’ | NC: (68) | 101., 163. |
| Narcissus ‘Queen of Bicolors’ | LY: (1, 7, 11); HT: (53) | (103) |
| Narcissus ‘Rembrandt’ | LY: (1); HT: (53); NC: (68); | 101., 103. |
| GA: (75, 78, 85) | ||
| Narcissus ‘Rockery Beauty’ | LY: (7, 16); HT: (53); GA: (78, 85) | (103) |
| Narcissus ‘Romaine’ | LY: (7, 11); HL: (35); HT: (53) | (103) |
| Narcissus ‘Spring Glory’ | LY: (1, 7, 11); HT: (53); | (103) |
| GA: (78, 85) | ||
| Narcissus ‘Unsurpassable’ | LY: (11); HL: (35); HT: (53); | 81., 103. |
| GA: (75) | ||
| Narcissus ‘Victoria’ | LY : (1, 7, 11); HT: (53); | (103) |
| GA: (78, 85) | ||
| Narcissus ‘Wrestler’ | LY: (7, 11); HT: (53); GA: (75) | (103) |
| Division 2 – Large-Cupped Daffodils | ||
| Narcissus ‘Carabiniere’ | NC: (68) | (101) |
| Narcissus ‘Carlton’ | LY: (13, 14, 15); | 101., 165., 166., 167., 168. |
| HL: (26, 30, 31, 34, 35, 36, 37, 38); | ||
| HT: (42, 53); NC: (68); | ||
| GA: (75, 77, 78, 83, 84, 86) | ||
| Narcissus ‘Clamor’ | NC: (68) | 101., 163. |
| Narcissus ‘Daisy Schäffer’ | LY: (7, 11); GA: (75) | (103) |
| Narcissus ‘Deanna Durbin’ | LY: (1, 7, 11, 16); HT: (53) | 103., 158. |
| Narcissus ‘Flower Record’ | LY: (1, 7, 11); HT: (53); GA: (75) | 81., 103. |
| Narcissus ‘Folly’ | LY: (1); TZ : (62) | (118) |
| Narcissus ‘Fortune’ | LY: (19); HL: (34, 37); | 81., 103., 169., 170. |
| HT: (53); GA: (75) | ||
| Narcissus ‘Gigantic Star’ | GA: (75) | (81) |
| Narcissus ‘Helios’ | LY: (7, 11); HL: (26, 35); | 101., 103., 125. |
| HT: (53); NC: (68); GA: (75) | ||
| Narcissus ‘Ice Follies’ | LY: (1, 7, 10, 13, 14); HL: (34); | 81., 164., 171., 172. |
| HT: (53); GA: (75, 84, 85) | ||
| Narcissus ‘John Evelyn’ | LY: (1, 7,11); HT: (53); GA: (75) | (103) |
| Narcissus ‘Marion Cran’ | LY: (1, 7); HT: (53); GA: (75) | (103) |
| Narcissus ‘Mercato’ | NC: (68) | (101) |
| Narcissus ‘Mrs. R.O. Backhouse’ | NC: (68) | (101) |
| Narcissus ‘Nova Scotia’ | LY: (1, 7); HT: (53); GA: (75) | (103) |
| Narcissus ‘Oranje Bruid’ | NC: (68) | (101) |
| Narcissus ‘Pluvius’ | LY: (1, 7, 11, 16); HT: (53); | 103., 158. |
| GA: (75) | ||
| Narcissus ‘Salome’ | LY: (3, 22, 23); HL: (34, 39); | 71., 81. |
| HT: (57); GA: (75, 78) | ||
| Narcissus ‘Scarlet Elegance’ | NC: (68) | 101., 163. |
| Narcissus ‘Sempre Avanti’ | LY: (1, 7, 11, 16); HT: (53); | 101., 103., 125., 163. |
| NC: (68) | ||
| Narcissus ‘Suda’ | LY: (7, 11); HL: (35); | (103) |
| HT: (53); GA: (75) | ||
| Narcissus ‘Toronto’ | LY: (1, 7); HT: (53) | (103) |
| Narcissus ‘Tunis’ | NC: (68) | 101., 163. |
| Narcissus ‘Walt Disney’ | NC: (68) | 101., 163. |
| Narcissus ‘Yellow Sun’ | GA: (75) | (81) |
| Division 3 – Small-Cupped Daffodils | ||
| Narcissus ‘Barrett Browning’ | GA: (75) | (81) |
| Narcissus ‘Daphne’ | LY: (1); HL: (26, 35); GA: (75) | (103) |
| Narcissus ‘Verger’ | NC: (68); GA: (75) | 81., 101., 163. |
| Division 4 – Double Daffodils | ||
| Narcissus ‘Cheerfulness’ | NC: (68); GA: (75) | 101., 164. |
| Narcissus ‘Dick Wilden’ | GA: (75) | (81) |
| Narcissus ‘Flower Drift’ | GA: (75) | (81) |
| Narcissus ‘Inglescombe’ | LY: (1, 11); HL: (26, 34, 35); | 103., 162. |
| HT: (53); GA: (75, 84, 85) | ||
| Narcissus ‘Insulinde’ | LY: (1, 7, 11); HT: (53) | (103) |
| Narcissus ‘Irene Copeland’ | LY: (1); GA: (83) | (103) |
| Narcissus ‘Livia’ | LY: (1, 7, 10, 11); HT: (53) | (103) |
| Narcissus ‘Sir Winston Churchill’ | LY: (17) | (173) |
| Narcissus ‘Texas’ | LY: (1, 11, 12); HT: (53); | 101., 103., 163., 174., 175., 176. |
| NC: (68); GA: (75, 83) | ||
| Narcissus ‘Twink’ | LY: (1, 7, 11); HT: (53); GA: (75) | 103., 158. |
| Narcissus ‘Van Sion’ | LY: (1, 7, 11); HL: (26, 35); | 81., 103., 158. |
| HT: (53); TZ: (62); GA: (75, 78, 85) | ||
| Division 5 – Triandrus Daffodils | ||
| Narcissus ‘Thalia’ | LY: (1); HL: (26, 35); | 101., 125., 142. |
| HT: (53); NC: (68) | ||
| Narcissus ‘Tresamble’ | HL: (35); HT: (53); | 101., 125., 142. |
| NC: (68); GA: (75) | ||
| Division 6 – Cyclamineus Daffodils | ||
| Narcissus ‘Beryl’ | LY: (1, 7, 16) | (142) |
| Narcissus ‘Cairhays’ | LY: (7, 11); HT: (55, 56) | (142) |
| Narcissus ‘February Gold’ | HL: (26, 35); GA: (75, 84) | 81., 142. |
| Narcissus ‘Peeping Tom’ | LY: (11); HL: (35); TZ: (62) | (142) |
| Narcissus ‘Tête-a-Tête’ | GA: (75) | (81) |
| Division 7 – Jonquilla and Apodanthus Daffodils | ||
| Narcissus ‘Golden Sceptre’ | LY: (1, 7, 8, 9); | 142., 177., 178., 179., 180. |
| HL : (26, 30, 34, 35, 37); | ||
| HT: (53); TZ: (62); GA: (75) | ||
| Narcissus ‘Pipit’ | GA: (75) | (168) |
| Narcissus ‘Trevithian’ | LY: (1); HL: (35); TZ: (62); | 101., 142. |
| NC: (68); GA: (75) | ||
| Division 8 – Tazetta Daffodils | ||
| Narcissus ‘Cragford’ | LY: (1); HL: (26); HT: (53); | (89) |
| TZ: (62) | ||
| Narcissus ‘Early Perfection’ | LY: (1, 11); HL: (26, 34); | (89) |
| HT: (53); TZ: (62) | ||
| Narcissus ‘Geranium’ | LY: (1, 16); HL: (26); HT: (53); | 89., 122., 143., 164. |
| TZ: (62); NC: (68); GA: (75) | ||
| Narcissus ‘La Fiancée’ | LY: (1, 7); HL: (26); | (89) |
| HT: (53); TZ: (62) | ||
| Narcissus ‘Laurens Koster’ | LY: (1); HL: (26); HT: (53); | (89) |
| TZ: (62) | ||
| Narcissus ‘L’innocence’ | LY: (1, 16); HL: (26); | (89) |
| HT: (53); TZ: (62) | ||
| Narcissus ‘Minnow’ | GA: (75) | (81) |
| Narcissus ‘Scarlet Gem’ | LY: (1, 7); HL: (26); | (89) |
| HT: (53); TZ: (62) | ||
| Narcissus ‘Silver Chimes’ | LY: (1); HT: (53); TZ: (62) | (142) |
| Narcissus ‘St. Agnes’ | LY: (1); HL: (26); HT: (53); | (89) |
| TZ: (62) | ||
| Narcissus ‘Totus Albus’ | NC: (68) | 101., 163. |
| Division 9 – Poeticus Daffodils | ||
| Narcissus ‘Actaea’ | LY: (1, 7, 16); HL: (35); | 89., 101. |
| NC: (68); GA: (75) | ||
| Narcissus ‘Sarchedon’ | LY: (1, 7, 16); HL: (35); GA: (75) | (89) |
| Division 12 – Other Daffodils | ||
| Narcissus ‘Brougshane’ | GA: (75) | (81) |
| Narcissus ‘Kristalli’ | LY: (1); TZ: (62); GA: (75, 83) | (118) |
Horticultural classification of Narcissus cultivars according to the International Daffodil Register and Classified List of the Royal Horticultural Society (18).
Skeleton types: LY, lycorine; HL, homolycorine; HT, hemanthamine; TZ, tazettine; NC, narciclasine; MN, montanine; GA, galanthamine. Alkaloid names: see Tables I–VII.
A. Alkaloid Types and Distribution
The presence of almost 100 alkaloids in this genus (Tables I–VII) means that approximately a third of the total number of alkaloids isolated from Amaryllidaceae have been found in the genus Narcissus. Amaryllidaceae alkaloids are present in all the species and varieties of Narcissus studied. In general, a series of related alkaloids is found in each plant: often a few major metabolites and several minor components, which differ in the position of their substitutents. Up to now, about 40 wild species and around 100 cultivars have been studied in relation to the presence of alkaloids (Table VIII, Table IX), which means that more than half of the Narcissus species or varieties have still to be explored in this aspect.
1. Lycorine and Homolycorine Types
The alkaloids of the lycorine and homolycorine groups are, on the whole, the most common alkaloids in this genus. Lycorine (1), galanthine (7), and pluviine (11) (lycorine type) and homolycorine (26) and lycorenine (35) (homolycorine type) are particularly frequent, lycorine being the most abundant. The presence of these alkaloids is very significant in the sections Narcissus (mainly lycorine type), Pseudonarcissi (mainly homolycorine type), and Tazettae of the wild species, and in the Divisions 1, 2, and 4 of cultivars.
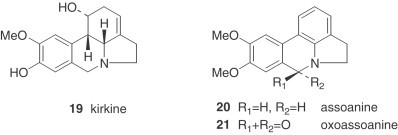
Almost all of the lycorine-type alkaloids isolated from Narcissus present a trans-union between B/C rings, making them especially vulnerable to oxidative processes. It is interesting to observe that four of these alkaloids, namely vasconine (22), tortuosine (23), ungeremine (24), and roserine (25) possess an unusual quaternary nitrogen 67., 68., 69., 70., 71.. The species N. pallidulus (section Ganymedes), unusual because it contains mesembrane-type alkaloids, is even more exceptional due to the presence of roserine.
The alkaloids of the homolycorine series, formed by a restructuring of lycorine-type alkaloids, are absent from some tribes of the Amaryllidaceae, such as the Amaryllideae or Hemantheae (44). For that reason, the presence of these alkaloids is a distinctive feature of the Narcisseae tribe. Moreover, all the Narcissus alkaloids of the homolycorine series display a B/C ring junction with a cis stereochemistry. An exceptional homolycorine-type alkaloid is dubiusine (33), which has an unusual hydroxybutyryl substituent (72).
2. Hemanthamine, Tazettine, Narciclasine, and Montanine Types
The main groups of alkaloids originating from a para–para′ oxidative phenolic coupling of O-methylnorbelladine (87) (hemanthamine, tazettine, and narciclasine types) are frequently represented in this genus by the model alkaloid of each group, hemanthamine (53), tazettine (62), and narciclasine (68), respectively. Their presence is significant in the Pseudonarcissi, Tazettae, and Bulbocodii sections of wild species, and in the Divisions 1, 2, and 8 of cultivars. The hemanthamine-type alkaloids are the most abundant, being the biogenetic source of the other types.
The lack of crinine-type alkaloids (β-5,10b-ethano bridge configuration) in this genus, which only has representatives of the α-5,10b-ethano bridge series (hemanthamine type), is a very significant taxonomic feature. Additionally, in Narcissus, the hemanthamine-type alkaloids never show any substitution in the aromatic ring at position 7, which is usual in crinine-type alkaloids from tribes such as the Amaryllideae or Hemantheae (44). The pairs of alkaloids with a hydroxyl substituent at C-6, like papyramine/6-epipapiramine (48/49) or hemanthidine/6-epihemanthidine (55/56), always appear as a mixture of epimers in solution (73). It is also interesting to mention the unusual structure of bujeine (61), an alkaloid of the hemanthamine type that has a modified bridge with a heteroatom between C-11 and C-12 and an acetoxymethyl substituent at the 11 endo position (74). Ismine (72) is considered to be a catabolic product from the hemanthamine series.
Tazettine (62) is a widely reported alkaloid in the Amaryllidaceae family, particularly in Narcissus (see Table VIII, Table IX), although it is known to be an extraction artifact of pretazettine (64) (75). All the alkaloids of the tazettine type that are isolated from Narcissus species show the typical methylenedioxy group between the C-8 and C-9 positions. Obesine (67) is an exceptional tazettine-type alkaloid with a seven-membered ring (76).
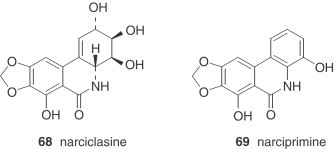
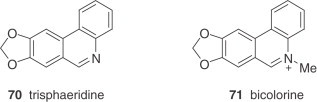
Narciclasine (68) is reasonably abundant in some Narcissus spp. and has served as a very useful intermediate for synthetic conversion into (+)-pancratistatin (see Section IV) and to conduct a series of structure–activity relationship studies 77., 78.. Bicolorine (71), another member of the narciclasine series, is an unusual, completely aromatized quaternary alkaloid with an N-methyl group (79).
The montanine-type alkaloids, also deriving from the hemanthamine series, are very unusual. Only two, namely pancracine (73) and nangustine (74), have been isolated from two species, both belonging to the Narcissus section of this genus. Nangustine has a unique substitution pattern (80), being the first 5,11-methanomorphanthridine alkaloid with a C-3/C-4 substitution, instead of a typical C-2/C-3.
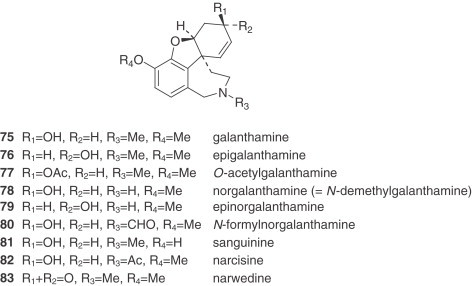
3. Galanthamine Type
The galanthamine-type alkaloids are the only group among the Amaryllidaceae alkaloids showing two ortho aromatic protons in ring A. This type of alkaloid often shows an N-methyl group, or occasionally N-formyl or N-acetyl derivatives. They are frequent in Narcissus, galanthamine (75) being the most usual and representative of them 30., 81.. The concentration of galanthamine in Narcissus has been found to vary widely between species and varieties, from trace amounts to 2.5% (30). The potential of some Narcissus cultivars (‘Carlton’, ‘Gigantic Star’, Ice Follies’, and ‘Fortune’) and wild species (N. confusus) as sources of galanthamine and related alkaloids has been recognized 81., 82., 83., 84., and research has been initiated into the agronomic factors that affect their content. Narcissus has two important advantages over Leucojum aestivum: first, bulbs of many Narcissus cultivars are available in commercial quantities, offering the possibility of establishing large-scale cultivation in a short time; second, a comprehensive body of information already exists regarding narcissus propagation, physiology, breeding, and cultivation (85).
4. Other Alkaloids
The mesembrane (Sceletium)-type alkaloids are also present in Narcissus plants. It is noteworthy that the alkaloids of this group (mesembrenone (90), mesembrenol (91), and mesembrine (92)) isolated from Narcissus are restricted to the species of the Ganymedes section, which is therefore of chemotaxonomic interest because they are generally found in the Aizoaceae, a dicotyledonous family 23., 24.. Both the Amaryllidaceae and Sceletium-type alkaloids have common biogenetic precursors, although their biosyntheses are fundamentally different (33).
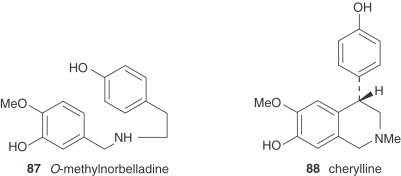
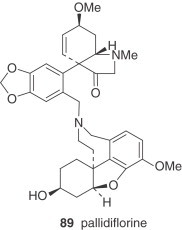
There are also two unclassified alkaloids, namely cherylline (88) and pallidiflorine (89). Cherylline, a 4-substituted tetrahydroisoquinoline derivative, is a unique representative of rare phenolic Amaryllidaceae alkaloids. Pallidiflorine, a heterobis alkaloid isolated from N. pallidiflorus (86), is the only bis-alkaloid present in this genus isolated up to now. This alkaloid is formed from two directly joined moieties, one of them being the alkaloid lycoramine (galanthamine type), and the other being the reverse form of the hemiacetal at C-11 of tazettine.
B. Ontogenic Variations
It is well established that profiles of alkaloids vary with time, location, and developmental stage. In many instances, the site of biosynthesis is restricted to a single organ, but accumulation of the corresponding products can be detected in several other plant tissues. Long-distance transport must take place in these instances. There are only a few data on the ontogenic variations and distribution of alkaloids in species of the Amaryllidaceae family, and some results have been obtained in Narcissus species, such as N. assoanus (with only lycorine-type alkaloids) or N. confusus (with alkaloids of the homolycorine, hemanthamine, tazettine, and galanthamine types) 84., 87..
During the vegetative period, the bulb of N. assoanus has a lower concentration of alkaloids than the aerial part. Pseudolycorine (3), the major alkaloid of both the aerial parts and the bulb, represents around 50% of the total alkaloids. The synthesis of alkaloids might take place mainly in the aerial part, where they accumulate. Once flowers have been fecundated, they are transported to the bulb. It seems that, in order to avoid autotoxicity, pseudolycorine is stored in the bulbs as the 1-O-Ac (4) and 2-O-Ac (5) derivatives (87). The specific localization of alkaloids make sense if their role as defense and/or signal compounds is accepted. As a rule, vulnerable tissues are defended more than old, senescent tissues.
Galanthamine (75) and four other alkaloids of N. confusus were found to be present in all of the organs at every stage, with the exception of hemanthamine (53), which does not occur in senescent flowers. The main alkaloid is galanthamine, followed by N-formylnorgalanthamine (80) or tazettine (62), depending on the sample, while homolycorine (26) is in general the least common. The organ with the highest total alkaloid accumulation throughout the ontogenic cycle is the bulb, except at the end of the cycle, when the alkaloids accumulate mainly in the flower stem, reaching a concentration of up to 2.5% by dry weight (84).
III. Biosynthetic Pathways
Most of the biosynthetic research done on Amaryllidaceae alkaloids was carried out in the 1960s and early 1970s. Since then, the only noteworthy study has been the biosynthesis of galanthamine (75) and related alkaloids (181). As in most alkaloid biosyntheses, that of the Amaryllidaceae follows a sequential pattern.
A. Enzymatic Preparation of the Precursors
Although l-phenylalanine (l-phe) and l-tyrosine (l-tyr) are closely related in their chemical structure, they are not interchangeable in plants. In the Amaryllidaceae alkaloids, l-phe serves as a primary precursor of the C6–C1 fragment, corresponding to ring A and the benzylic position (C-6), and l-tyr is the precursor of ring C, the two-carbon side chain (C-11 and C-12) and nitrogen, C6–C2–N. The conversion of l-phe to the C6–C1 unit requires the loss of two carbon atoms from the side chain as well as the introduction of at least two oxygenated substituents into the aromatic ring, which is performed via cinnamic acids. The presence of the enzyme phenylalanine ammonia lyase (PAL) has been demonstrated in Amaryllidaceae plants (182), and the elimination of ammonia mediated by this enzyme is known to occur in an antiperiplanar manner to give trans-cinnamic acid, with loss of the β-pro-S hydrogen (183). Thus, it may be expected that l-phe would be incorporated into Amaryllidaceae alkaloids with retention of the β-pro-R hydrogen. However, feeding experiments in Narcissus ‘King Alfred’ showed that tritium originally present at C-β of l-phe, whatever the configuration, was lost in the formation of several hemanthamine- and homolycorine-type alkaloids, which led to the conclusion that fragmentation of the cinnamic acids involves the oxidation of C-β to the ketone or acid level, the final product being protocatechuic aldehyde or its derivatives (Fig. 2 ). On the other hand, l-tyr is degraded no further than tyramine before incorporation into the Amaryllidaceae alkaloids.
Figure 2.
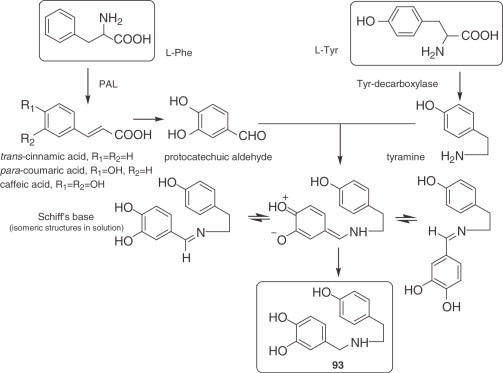
Biosynthetic pathway to norbelladine (93).
B. Primary Cyclization Mechanisms
Tyramine and protocatechuic aldehyde or its derivatives are logical components for the biosynthesis of the precursor norbelladine (93). This pivotal reaction represents the entry of primary metabolites into a secondary metabolic pathway. The junction of the amine and the aldehyde results in a Schiff's base, two of which have been isolated up to now from several Crinum species: craugsodine (184) and isocraugsodine (185). The existence of Schiff's bases in nature, as well as their easy conversion into the different ring systems of the Amaryllidaceae alkaloids, suggest that the initial hypothesis about this biosynthetic pathway was correct.
C. Enzymatic Preparation of Intermediates
In 1957, Barton and Cohen (186) proposed that norbelladine (93) or related alkaloids could undergo oxidative coupling in Amaryllidaceae plants, once ring A had been suitably protected by methylation, resulting in the different skeletons of the Amaryllidaceae alkaloids (Fig. 3 ). The key intermediate in most of cases is O-methylnorbelladine (87).
Figure 3.
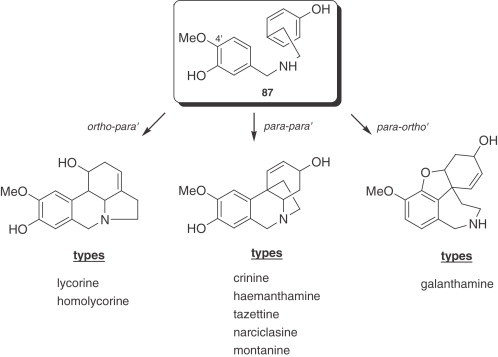
Phenol oxidative coupling in Amaryllidaceae alkaloids.
D. Secondary Cyclization, Diversification, and Restructuring
Secondary cyclization is produced by an oxidative coupling of O-methylnorbelladine.
1. Lycorine and Homolycorine Types
The alkaloids of this group are derivatives of the pyrrolo[de]phenanthridine (lycorine type) and the 2-benzopirano-[3,4-g]indole (homolycorine type) skeletons, and both types originate from an ortho–para′ phenol oxidative coupling (Fig. 4 ).
Figure 4.
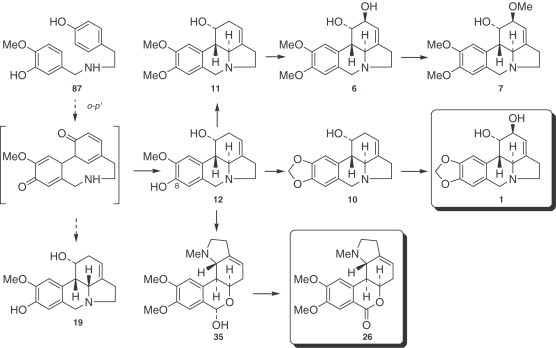
Alkaloids proceeding from an ortho–para′ coupling.
The biological conversion of cinnamic acid via hydroxylated cinnamic acids into the C6–C1 unit of norpluviine (12) has been used in a study of hydroxylation mechanisms in higher plants (187). When [3-3H, β-14C] cinnamic acid was fed to Narcissus ‘Texas’ a tritium retention in norpluviine (12) of 28% was observed, which is very close to the predicted value resulting from para-hydroxylation with hydrogen migration and retention.
In the conversion of O-methylnorbelladine (87) into lycorine (1), the labeling position [3-3H] on the aromatic ring of l-tyr afterward appears at C-2 of norpluviine (12), which is formed as an intermediate, the configuration of the tritium apparently being β (176). This tritium is retained in subsequently formed lycorine (1), which means that hydroxylation at C-2 proceeds with an inversion of configuration (188) by a mechanism involving an epoxide, with ring opening followed by allylic rearrangement of the resulting alcohol (Fig. 5 ). Supporting evidence comes from the incorporation of [2β-3H]caranine (10) into lycorine (1) in Zephyranthes candida (189). However, hydroxylation of caranine (10) in Clivia miniata occuring with retention of configuration was also observed (190). Further, [2α-3H,11-14C]caranine (10) was incorporated into lycorine (1) with high retention of tritium at C-2, indicating that no 2-oxo-compound can be implicated as an intermediate.
Figure 5.
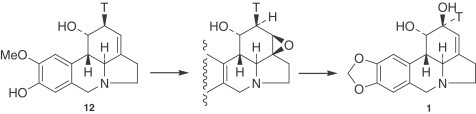
Biosynthesis of lycorine (1) with inversion of the configuration.
The conversion of the O-methoxyphenol to the methylenedioxy group may occur late in the biosynthetic pathway. Tritiated norpluviine (12) is converted to tritiated lycorine (1) by Narcissus ‘Deanna Durbin’, which demonstrates the previously mentioned conversion and also indicates that the C-2 hydroxyl group of lycorine (1) is derived by allylic oxidation of either norpluviine (12) or caranine (10) (191).
Regarding the conversion of [2β-3H,8-OMe-14C]pluviine (11) into galanthine (7), in Narcissus ‘King Alfred’, the retention of 79% of the tritium label confirms that hydroxylation of C-2 may occur with inversion of configuration (92).
It was considered (192) that another analogous epoxide 94 could give narcissidine (16) in the way shown by loss of the pro-S hydrogen from C-11, galanthine (7) being a suitable substrate for epoxidation. Labeled [α-14C, β-3H]-O-methylnorbelladine (87), when fed to Narcissus ‘Sempre Avanti’, afforded galanthine (7) (98% tritium retention) and narcissidine (16) (46% tritium retention). Loss of hydrogen from C-11 of galanthine (7) was therefore stereospecific. In the 1990s, Kihara et al. (193) isolated a new alkaloid, incartine (94), from the flowers of Lycoris incarnata, which could be considered as the biosynthetic intermediate of this pathway (Fig. 6 ).
Figure 6.
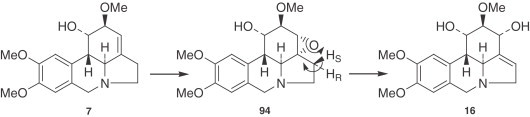
Conversion of galanthine (7) to narcissidine (16) via epoxide (94).
The biological conversion of protocatechuic aldehyde into lycorenine (35), which proceeds via O-methylnorbelladine (87) and norpluviine (12), first involves a reduction of the aldehyde carbonyl, and afterward, in the generation of lycorenine (35), oxidation of this same carbon atom. The absolute stereochemistry of these processes has been elucidated in subsequent experiments (194), and the results show that hydrogen addition and removal take place on the re-face of the molecules concerned (195), the initially introduced hydrogen being the one later removed (196). It is noteworthy that norpluviine (12), unlike pluviine (11), is converted in Narcissus ‘King Alfred’ primarily to alkaloids of the homolycorine type. Benzylic oxidation of position 6 would afford 95, followed by ring opening to form an amino aldehyde. Hemiacetal formation and methylation could provide lycorenine (35) (92), and, on subsequent oxidation, could afford homolycorine (26), as shown in Fig. 7 .
Figure 7.
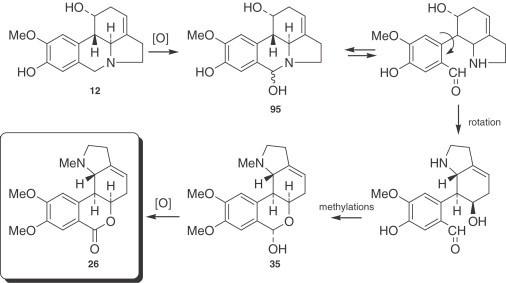
Conversion of norpluviine (12) to homolycorine-type alkaloids.
2. Crinine, Hemanthamine, Tazettine, Narciclasine, and Montanine Types
This group includes the alkaloids derived from 5,10b-ethanophenanthridine (crinine and hemanthamine types), 2-benzopyrano[3,4-c]indole (tazettine type), phenanthridine (narciclasine type), and 5,11-methanomorphanthridine (montanine type) skeletons, originating from a para–para′ phenol oxidative coupling (Fig. 8 ).
Figure 8.
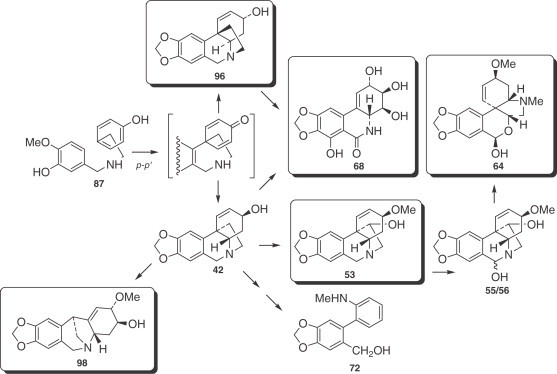
Alkaloids proceeding from a para–para′ coupling.
Results of experiments with labeled crinine (96), and less conclusively with oxovittatine, indicate that the two, naturally occurring enantiomeric series, represented in Fig. 8 by crinine (96) and vittatine (42), are not interchangeable in Nerine bowdenii (197).
Incorporation of O-methylnorbelladine (87), labeled in the methoxy carbon and also in positions [3,5-3H], into the alkaloid hemanthamine (53) was without loss of tritium, half of which was sited at C-2 of 53. Consideration of the possible mechanisms involved in relation to tritium retention led to the suggestion that the tritium which is expected at C-4 of 53 might not be stereospecific (198). The conversion of O-methylnorbelladine (87) into hemanthamine (53) involves loss of the pro-R hydrogen from the C-β of the tyramine moiety as well as the further entry of a hydroxyl group at this site (199). The subsequent benzylic oxidation results in a 55/56 epimeric mixture that even HPLC cannot separate. The epimeric forms were proposed to be interchangeable through 97. The biosynthetic conversion of the 5,10b-ethanophenanthridine alkaloids to the 2-benzopyrano[3,4-c]indole was demonstrated by feeding tritium-labeled alkaloids to Sprekelia formosissima. It was shown that this plant converts hemanthamine (53) to hemanthidine/epihemanthamine (55/56), and subsequently to pretazettine (64), in an essentially irreversible manner (200). This transformation was considered to proceed through 97 or the related alkoxide anion, although intermediate 97 have never been detected by spectral methods (201) (Fig. 9 ).
Figure 9.
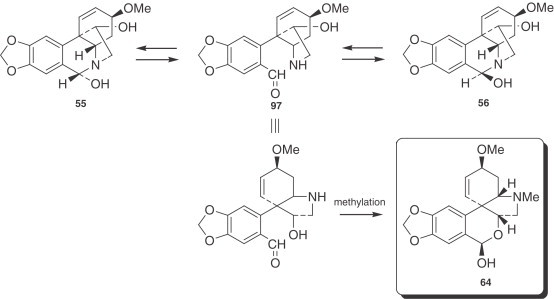
Biosynthesis of pretazettine (64).
It has also been proved that formation of the alkaloid narciclasine (68) proceeds from the pathway for crinine- and hemanthamine-type alkaloids, and not through norpluviine (12) and lycorine (1) derivatives. In fact, in view of its structural affinity to both hemanthamine (53) and lycorine (1), narciclasine (68) could be derived by either of the pathways. When O-methylnorbelladine (87), labeled in the methoxy carbon and in both protons of positions 3 and 5 of the tyramine aromatic ring, was administered to Narcissus plants, all four alkaloids incorporated activity. The isotopic ratio [3H:14C] for norpluviine (12) and lycorine (1) was, as expected, 50% that of the precursor, because of its ortho–para′ coupling. On the contrary, in hemanthamine (53) the ratio was unchanged. These results show clearly that the methoxy group of 87 is completely retained in the alkaloids mentioned, providing a satisfactory internal standard and showing that the degree of tritium retention is a reliable guide to the direction of phenol coupling. Narciclasine (68) showed an isotopic ratio (75%) higher than that of lycorine (1) or norpluviine (12), though lower than that of hemanthamine (53). However, the fact that more than 50% of tritium is retained suggests that O-methylnorbelladine (87) is incorporated into narciclasine (68) via para–para′ phenol oxidative coupling.
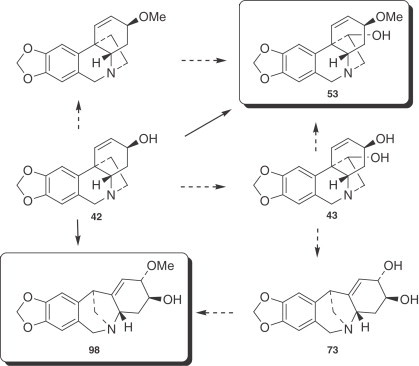
O-methylnorbelladine (87) and vittatine (42) are implicated as intermediates in the biosynthesis of narciclasine (68) 202., 203., 204., and the loss of the ethane bridge from the latter could occur by a retro-Prins reaction on 11-hydroxyvittatine (43). Strong support for this pathway was obtained by labeling studies. 11-Hydroxyvittatine (43) has also been proposed as an intermediate in the biosynthesis of hemanthamine (53) and montanine (98) (a 5,11-methanomorphanthridine alkaloid) following the observed specific incorporation of vittatine (42) into the two alkaloids in Rhodophiala bifida (197) (Fig. 10 ).
Figure 10.
Proposed biosynthetic pathways to hemanthamine (53) and montanine (98).
Fuganti and Mazza 203., 204. concluded that in the late stages of narciclasine (68) biosynthesis, the two-carbon bridge is lost from the oxocrinine skeleton, passing through intermediates bearing a pseudoaxial hydroxy-group at C-3 position and further hydrogen removal from this position does not occur. Noroxomaritidine was also implicated in the biosynthesis of narciclasine (68) and further experiments (205) showed that it is also a precursor for ismine (72).
The alkaloid ismine (72) has also been shown (206) to be a transformation product of the crinine-hemanthamine series. The precursor, oxocrinine labeled with tritium in the positions 2 and 4, was administered to Sprekelia formosissima plants and the radioactive ismine (72) isolated was shown to be specifically labeled at the expected positions.
3. Galanthamine Type
These alkaloids have a dibenzofuran nucleus (galanthamine type) and are obtained from a para–ortho′ phenol oxidative coupling.
The initial studies of this pathway suggested that the para–ortho′ coupling does not proceed from O-methylnorbelladine (87) but from N,O-dimethylnorbelladine (99) to finally give galanthamine (75) (207). N,O-dimethylnorbelladine (99) was first isolated from Pancratium maritimum (208), a species that also contains galanthamine (75).
However, the most recent study seems to contradict the evidence set forth above. Experiments carried out with application of 13C-labeled O-methylnorbelladine (87) to organs of field-grown L. aestivum have shown that the biosynthesis of galanthamine (75) involves the phenol oxidative coupling of O-methylnorbelladine (87) to a postulated dienone, which undergoes spontaneous closure of the ether bridge to yield N-demethylnarwedine (1 0 0), giving norgalanthamine (78) after stereoselective reduction. Furthermore, it was shown that norgalanthamine (78) is N-methylated to galanthamine (75) in the final step of biosynthesis (181) (Fig. 11 ). In contrast to the literature, N,O-dimethylnorbelladine (99) was metabolized to a lesser extent in L. aestivum and incorporated into galanthamine (75) as well as norgalanthamine (78) at about one-third of the rate of O-methylnorbelladine (87).
Figure 11.
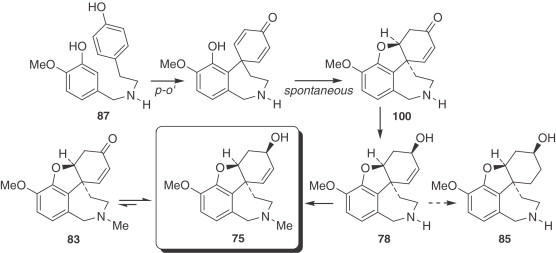
Biosynthesis of galanthamine (75) and derivatives.
According to Eichhorn et al. (181), narwedine (83) is not the direct precursor of galanthamine (75), and could possibly exist in equilibrium with galanthamine (75), a reaction catalyzed by a hypothetically reversible oxido-reductase.
Chlidanthine (1 0 1), by analogy with the known conversion of codeine to morphine, might be expected to arise from galanthamine (75) by O-demethylation. This was shown to be true when both galanthamine (75) and narwedine (83), with tritium labels, were incorporated into chlidanthine (1 0 1) (209).
IV. Synthetic Studies
The synthesis of Narcissus alkaloids has been the subject of intense efforts over the past few years. An exhaustive report is beyond the scope of this chapter, and the reader is referred to recent and excellent reviews in the field for a more thorough account 34., 60., 61., 62., 63., 64., 65., 66., 210., 211.. Several approaches to the total synthesis of these alkaloids have demonstrated their efficiency. We describe herein some representative examples, classified according to mechanistic criteria.
A. Oxidative Couplings
Biomimetic oxidative phenolic coupling still stands as a practical method for the preparation of these alkaloids. New syntheses of (−)-narwerdine (83) and (−)-galanthamine (75) have been disclosed, the former having been prepared in its enantiopure form from the corresponding racemate through a crystallization-induced chiral transformation (212). Derivatives of 1-methylgalanthamine (213), as well as galanthamine analogues having the N atom at altered positions in the azepine ring (214), have also been reported. A significant improvement to the oxidation protocol is the use of PIFA as the oxidant on a symmetrical substrate, allowing efficient synthesis of racemic narwedine and galanthamine (215) (Scheme 1 ).
Scheme 1.
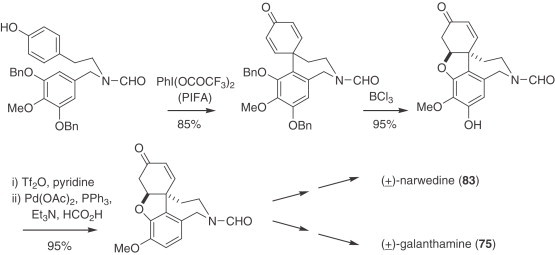
Synthesis of racemic narwedine (83) and galanthamine (75).
The same group later reported an efficient route to (−)-galanthamine (75) by a variation of the method whereby remote asymmetric induction was used (216) as well as the total syntheses of siculine, oxocrinine, epicrinine, and buflavine (217).
A solid-phase version of this biomimetic method has been developed in the context of diversity-oriented synthesis to generate a library of galanthamine-like molecules, which allowed the identification of compound A, a potent new blocker of protein trafficking from the Golgi apparatus to the plasma membrane. Interestingly, galanthamine (75) does not exhibit this biological activity (218) (Scheme 2 ).
Scheme 2.
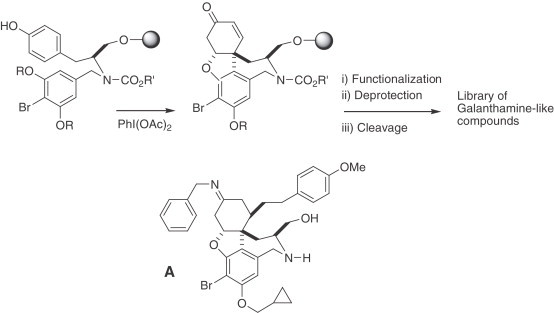
Diversity-oriented synthesis of galanthamine-like compounds.
A new oxidative rearrangement has led to the synthesis of (±)-lycoramine (219), and a related Lewis-acid-promoted transformation has allowed the preparation of mesembrine (92) and crinane in racemic form (220).
An impressive demonstration of the use of solid-supported reagents and scavengers in the total synthesis of natural products was provided by the group of Ley (for previous work, see ref. 221). As an application of this new methodology (intended to expedite synthetic routes, as the only work up required in these protocols involves filtration, followed by evaporation of the solvent), (+)-plicamine was synthesized without the need for chromatography. The sequence starts with p-hydroxyphenylglycine which, upon functionalization, undergoes a reductive amination with piperonal. The resulting product is subjected to biomimetic phenol oxidative coupling with solid-supported iodonium diacetate. An acid-promoted conjugate addition closed the lactam ring, and subsequent functional group transformations led to the desired product. Every single step used solid-supported reagents or solid-supported scavengers (Scheme 3 ) 222., 223.. The same group later disclosed the syntheses of (−)-obliquine and (+)-plicane (224).
Scheme 3.
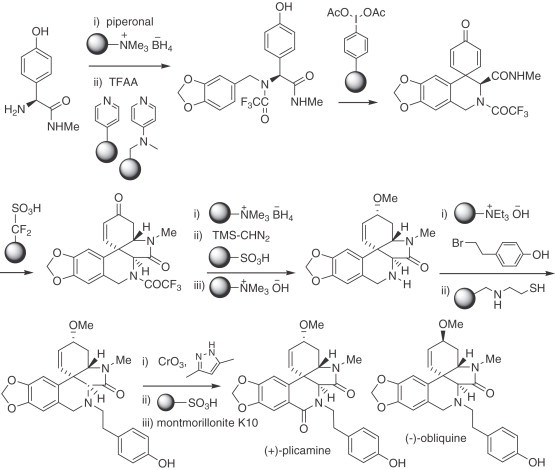
Synthesis of plicamine and obliquine using solid-supported reagents.
B. Organometallic-Promoted Reactions
Transition metals, especially palladium, have been used for the stereoselective synthesis of these alkaloids with remarkable success. Since Overman's approach to (±)-tazettine (62) using an intramolecular Heck reaction (225), this methodology for the construction of the critical quaternary stereogenic center has been implemented for the preparation of many alkaloids. Grigg later reported access to (R,R)-crinane via regiospecific palladium-mediated cyclization (226). An efficient total synthesis of racemic galanthamine (75) following this approach has been recently disclosed (227). After the key intramolecular Heck reaction, deprotection of the acetal moiety, followed by oxidation and aminolysis of the lactone, promoted the spontaneous conjugate addition of the phenolic hydroxyl, to afford the tricyclic intermediate. A Pictet–Spengler reaction was then used to close the azepine ring. Subsequent functional group transformations allowed the completion of the total synthesis (Scheme 4 ).
Related strategies were used to prepare racemic maritidine (44) (228) and lycoramine (84) 229., 230.. An elegant and unified approach to synthesize (−)-pancracine (73), (−)-montanine (98), (−)-coccinine, and (−)-brunsvigine used an intramolecular Heck reaction for the ring closure of the central azepine ring, connecting the bromoarene and terminal alkene moieties. The key precursor was prepared via stereospecific intramolecular allenylsilane addition to an imine (231). Enders recently reported an asymmetric synthesis of the 1-epi aglycon of cripowellins A and B, based on an intramolecular Heck reaction of a functionalized bromoaryl-azacyclononene, which in turn was prepared via ring closing metathesis of an open-chain precursor (232). Buflavine was prepared in a very short route by assembling a biphenyl intermediate through a Suzuki coupling (233).
Recently, palladium-catalyzed asymmetric allylic substitution of an activated cyclohexenol derivative has allowed two enantioselective syntheses of (−)-galanthamine (75) 234., 235.. Both approaches rely on the enantioselective preparation of the same tricyclic intermediate, which is subsequently converted to the alkaloid via stereocontrolled transformations; the most efficient of which comprised stereoselective allylic oxidation of the cyclohexene moiety (Scheme 5 ). The same methodology allowed the synthesis of (−)-codeine and (−)-morphine (236). The same group had earlier reported the synthesis of (+)-pancratistatin following a related strategy (237). Use of a tosylamide as the nucleophile in the displacement of an activated aryl-cyclohexenol derivative enabled the preparation of a chiral intermediate which was sequentially converted into (+)-crinamine (57), (−)-hemanthidine (55), and (+)-pretazettine (64) (238).
Scheme 5.
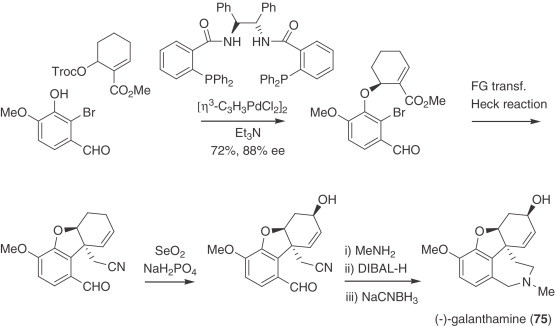
Enantioselective synthesis of (−)-galanthamine (75).
Regioselective C–H activation is often used for intramolecular biaryl preparations in Pd-mediated transformations. Thus concise syntheses of a large collection of alkaloids have been reported using variations of this approach 239., 240., 241. which differ mainly in terms of the functionalization of the precursor, palladium catalysts, and ligands. An efficient synthesis of racemic γ-lycorane was recently disclosed whereby the key step is the intramolecular α-arylation of a cyclohexanone derivative (242). Some heterocyclic synthetic precursors (e.g. substituted indoles, quinolines, quinolones, phenanthridines, and phenanthridinones) have also been prepared en route to the alkaloids [hippadine, trisphaeridine (70), and crinasiadine] via Pd- and Cu-mediated reactions 243., 244..
The use of main group metals has also facilitated alkaloid synthesis, exemplified in the preparation of (−)-brunsvigine by intramolecular anionic cyclization (the addition of an organolithium derivative to an amide) (245). Also, a Meyers biaryl coupling has been used as the key step in a total synthesis of buflavine (246). The connection of directed ortho metalations with cross-coupling reactions is the basis of very elegant syntheses of buflavine, 8-O-demethylbuflavine, pratosine, and hippadine 247., 248.. Synthesis of (−)-mesembrine has been achieved through a route involving stereoselective ring opening of an aryl-substituted epoxide with a Grignard reagent (249).
C. Enzymatic Dihydroxylation
The development of enzymatic dihydroxylation of aromatics has enabled synthetic access to a large collection of cyclohexadiene diols in enantiopure form. These compounds have become invaluable synthetic materials and are pivotal intermediates in the total syntheses of structurally diverse natural products (250). In particular, the group of Hudlicky which developed the methodology has devised short, efficient, and enantioselective total syntheses of alkaloids and related compounds to meet quantity demands for materials used in biological studies. Lycoricidine was prepared in only nine steps from bromobenzene (251), and the same starting material was used in straightforward syntheses of pancratistatin and its 7-deoxy derivative (252). Recently, the Hudlicky group published the synthesis of narciclasine (68), based on the whole-cell fermentation of 1,3-dibromobenzene by a recombinant Escherichia coli (Scheme 6 ). The resulting dienediol was subjected to a hetero-Diels–Alder reaction with a nitroso derivative and the corresponding adduct underwent Suzuki coupling followed by reductive opening of the oxazine N–O bond. Stereoselective reduction of the resulting ketone, exchange of the acetonide moiety with acetate groups, a Bischler–Napieralski-type ring closure and the removal of the ester and phenolic methyl protecting groups led to the desired product (253).
Scheme 6.
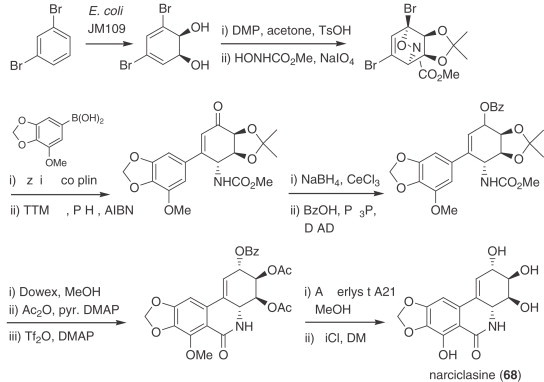
Synthesis of narciclasine (68).
The same group also disclosed the synthesis of epi-7-deoxypancratistatin via an aza-Payne rearrangement (254) (Scheme 7 ). Analogues of narciclasine (68), pancrastistatin, and 7-deoxypancratistatin have been synthesized using modifications of the reported procedures as well as new methodologies (e.g. addition of indoles to oxiranes and aziridines derived from cyclohexadiene diols) 255., 256., 257., 258..
Scheme 7.
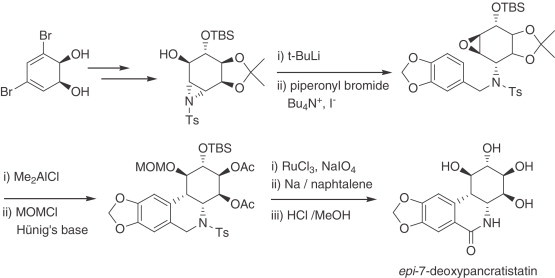
Synthesis of epi-7-deoxypancratistatin.
Pancratistatin analogues having a carbohydrate-derived structure in place of the polyhydroxycyclohexane moiety have been prepared for studying the minimum pharmacophore of the parent molecule (259).
D. Radical Reactions
The well-established radical reactions have been used to drive selective connectivity, often without the need of protecting groups normally required in polar approaches. Also the ‘living’ nature of some radical additions has triggered elegant and efficient reaction cascades for the formation of multiple C–C bonds.
The group of Magnus has continued to exploit the β-azidonation reaction of silyl enol ethers, which they used in the synthesis of pancratistatin (260), and has applied the same type of approach for the synthesis of a lycorane derivative. The pyrrolidine ring was closed via a cobalt-promoted intramolecular radical addition-dehydrogenation reaction (261). Parsons has reported model studies on the cyclization of diversely substituted 2-amino-cyclohex-2-enone derivatives to yield octahydroindole derivatives related to the Amaryllidaceae alkaloids. In these processes, radical cyclizations were promoted with tributyltin hydride, samarium (II) iodide, or manganese (III) acetate (262). Curran has reported a radical cyclization leading to spirocyclohexadienones, which includes the formal synthesis of aza-galanthamine (263). Keck reported an impressive radical cascade process as the crucial step in his total synthesis of 7-deoxypancratistatin (264). A modified approach led to the synthesis of (+)-lycoricidine, via 6-exo-cyclizations of substituted alkenyl radicals with oxime ethers (265) (Scheme 8 ). The precursor was prepared from d-gulonolactone, which was protected, selectively transformed, and coupled through a Sonogashira reaction with a substituted iodoarene to yield the key alkyne intermediate. PhSH was added under photochemical activation to generate a vinyl radical, which was then added to the oxime ether moiety to close the six-membered ring with high stereoselectivity. Treatment of the resulting compound with SmI2 allowed the cleavage of the N–O bond, the removal of the thioether, and the formation of the lactam. The final step was the deprotection of the acetonide group. Ent-lycoricidine, and (+)-narciclasine (68) have been similarly prepared.
Scheme 8.
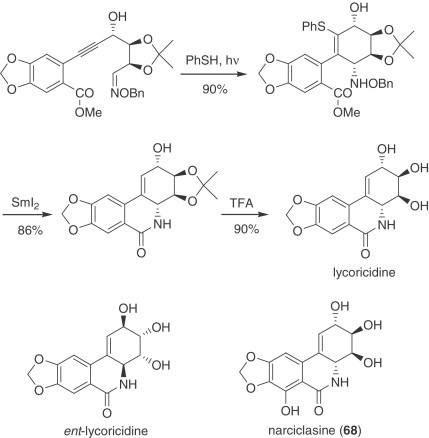
Radical synthesis of lycoricidine.
The formal synthesis of racemic pancracine (73) via radical cyclization of a N-(2-cyclohexenyl)-α-phenylthioacetamide has been reported (266). Racemic montanine (98), coccinine, and pancracine (73) have been prepared using a phenylthiotetrahydroisoquinoline derivative as the radical precursor (267). Recently, Zard has described the construction of the carbon skeleton of kirkine (19). In these model studies, thiosemicarbazide proradicals interact with lauroyl peroxide (a tin-free initiator) to trigger a reaction cascade that ultimately yields the desired tetracyclic system of kirkine (19) (268).
An interesting Csp 2–Csp 2 coupling via an aryl radical cyclization has been used as the key step in a unified strategy to synthesize vasconine (22), assoanine (20), oxoassoanine (21), and pratosine. Tin hydride cyclization of an N-(o-bromobenzyl)-aniline yielded a substituted phenanthridine which was subsequently transformed into vasconine (22), from which the remaining natural products were prepared (269) (Scheme 9 ).
Scheme 9.
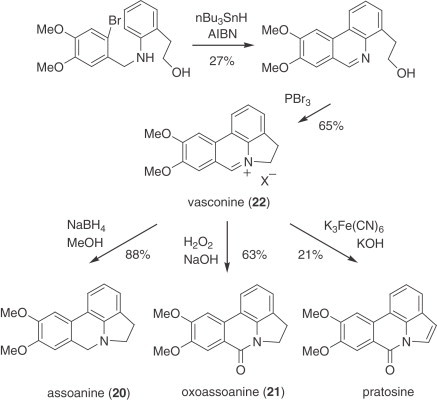
Radical synthesis of vasconine (22), assoanine (20), oxoassoanine (21), and pratosine.
E. Pericyclic Processes
The aza-Cope–Mannich cyclization protocol, developed by Overman has been strategically applied in the synthesis of racemic and enantiopure pancracine (73) (270). Later, Kim developed a synthetic approach based on a [3,3]-sigmatropic rearrangement as the key step in the stereoselective synthesis of racemic pancratistatin (271). Horner–Wasdsworth–Emmons reaction provided a reactive olefin that upon heating afforded the rearranged (aryl)cyclohexenylcarbaldehyde which, in turn, was further oxidized to the corresponding acid and subjected to a halolactonization and elimination protocol to yield the unsaturated lactone. This intermediate was converted into the hydroxyester, and subsequent functional-group transformations allowed the stereocontrolled installation of the remaining stereogenic centers and the final ring closure. The sequence ended with deprotection of the esters and methyl phenolic groups (Scheme 10 ).
Scheme 10.
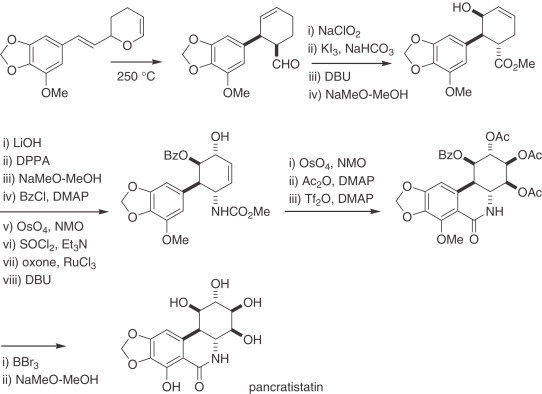
Synthesis of racemic pancratistatin.
Rigby's studies on the synthesis of alkenylisocyanates fostered the preparation of a suitable substituted aryl enamide, which on photocyclization yielded the polysubstituted pentacyclic system. Key to the success of this process is the hydrogen bond between the phenolic OH and the carbonyl group, which restricts the rotation around the aryl–amide bond and directs the cyclization. Further functionalization allowed the total synthesis of pancratistatin (272) and narciclasine (68) (273) (Scheme 11 ). The [4+1] cycloaddition of bis(alkylthio)carbenes with vinyl isocyanates was the key process in a recent synthesis of (±)-mesembrine (92) (274).
Scheme 11.
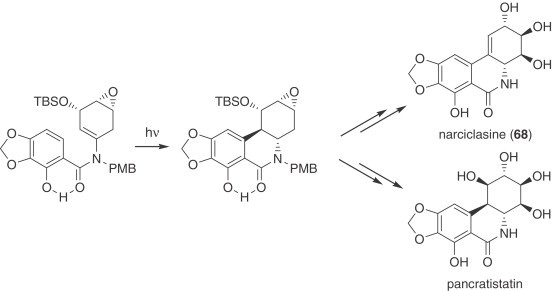
Synthesis of narciclasine (68) and pancratistain.
The Diels–Alder cycloaddition has played an important role in the synthesis of Narcissus alkaloids and related structures. Boger has prepared anhydrolycorinone using an intramolecular hetero-Diels–Alder reaction in which a 1,3,4-oxadiazole unit interacted with an electron-rich double bond to yield an intermediate furan adduct that underwent another intramolecular [4+2] cycloaddition with a second olefin moiety (275). Padwa has reported a conceptually attractive entry to this family of alkaloids based on intramolecular cycloadditions on substituted furanyl carbamates. The resulting adduct can be elaborated into diversely substituted hydroindolinones via nitrogen-assisted ring opening (276) or Rh(II)-promoted nucleophilic reaction (277). The former has provided compounds which on subsequent transformations yielded γ-lycorane and 1-deoxylycorine as racemates, whereas the latter afforded materials for the preparation of epi-zephyranthine (Scheme 12 ).
Scheme 12.
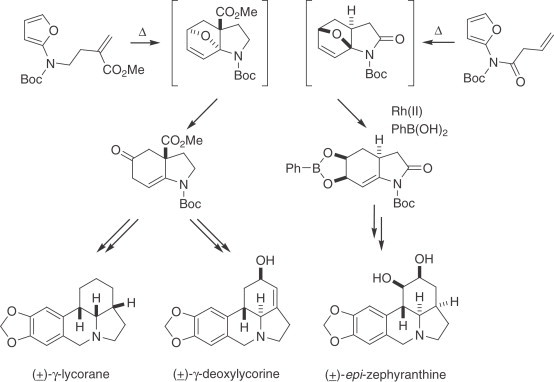
Synthesis of γ-lycorane, 1-deoxylycorine, and epi-zephyranthine.
F. Polar Reactions
Haseltine has described an enantioselective formal synthesis of pancratistatin in which the stereocontrol is driven by the acetonide of conduritol A. The enantioselective hydrolysis (desymmetrization) of this compound was achieved with a lipase, and the aryl–cyclohexane ring bond was formed through an intramolecular cyclization of the activated benzene ring with an allylic triflate (278). Plumet reported a total synthesis of (+)-7-deoxypancratistatin based on the conjugate addition of an aryl-lithium species to a bicyclic conjugated sulfone derived from furan, which enabled the efficient installation of the six stereogenic centers of the cyclohexane ring (279) (Scheme 13 ).
Scheme 13.
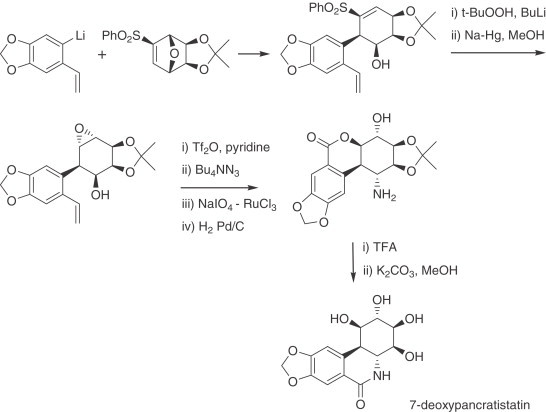
Synthesis of 7-deoxypancratistatin.
The next series of syntheses is based on conjugate additions. A 2-arylcyclohexanone was regio- and stereoselectively added to nitroethylene to access the octahydroindole core present in the alkaloids. This has enabled the total synthesis of (±)-γ-lycorane and (±)-crinane (280). Tomioka described a chemoselective conjugate addition – nitro Michael reaction sequence to prepare α- and β-lycoranes in their racemic form (281). The addition of an arylcuprate to a D-manitol-derived conjugate ester provided access to synthetic precursors of the Amaryllidaceae alkaloids (282). Similarly, the addition of arylcuprates to enantiomerically pure conjugate esters derived from d-xylose, allowed, after pertinent FG transformations and ring-closing metathesis, the preparation of novel 1-aryl-1-deoxyconduritols F. These compounds are advanced intermediates en route to pancratistatin derivatives (283). A 15-step synthesis of racemic mesembrine (92) featuring the intramolecular addition of an amine to a cyclohexenone moiety to close the octahydroindolone ring system was recently disclosed (284). Taber has described an elegant approach to (−)-mesembrine relying on a conjugate addition and an intramolecular alkylidene C–H insertion (285).
Intramolecular additions to N-acyliminium ions (generated by Pummerer reaction) were used to prepare highly functionalized tricyclic intermediates for the synthesis of the putative alkaloid jamtine (286). Synthesis of cherylline (88) in both of its enantiopure forms was achieved using a chiral auxiliary through a sequence involving reductive amination-acid-promoted cyclization (287).
Short syntheses of enantiomerically pure narciclasine (68) and lycoricidine based on the intramolecular acid-catalyzed arene-epoxide coupling have been described 288., 289.. Bromohydroxylation of a protected aminocyclohexenol afforded the corresponding bromohydrin as a mixture of two trans stereoisomers, which was subsequently transformed to link an arylmethyl moiety in basic medium with the concomitant formation of the epoxide ring, thereby setting the stage for the crucial carbon–carbon bond formation required for cyclization. Subsequent functional group transformations, exchange of protecting groups, oxidation of the benzylic position, and finally deprotection, afforded lycoricidine (Scheme 14 ). Narciclasine (68) has been prepared following a related pathway. The same group has also reported a convenient approach to chiral O-isopropylidene-protected 4-aminocyclohexenol (290).
Scheme 14.
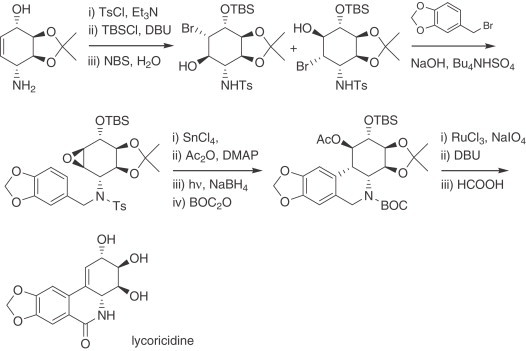
Total synthesis of lycoricidine.
G. Semisynthesis and Chiral Pool Approaches
Taking advantage of the availability of narciclasine (68) from plant extracts, Pettit used the compound as a starting material in an efficient synthesis of pancratistatin (77). The same group has also described related approaches for the preparation of a pancratistatin phosphate prodrug (291) as well as for the natural product 7-deoxy-trans-dihydronarciclasine and related derivatives (292). In another context, an improved protocol for the synthesis of (−)-galanthamine (75), based on the spontaneous resolution of either of the enantiomers of narwedine (83), has been reported (293).
A short route to enantiomerically pure lactone analogues of narciclasine and lycoricidine uses d-gulonolactone as the chiral pool source, as it contains all of the stereogenic centers of the products in their correct configuration. Connectivity with the functionalized aryl moiety arises from the addition of an organolithium reagent to the carbonyl group of the gulonolactone (294). The total syntheses of (−)-hemanthidine (55), (+)-pretazettine (64), and (+)-tazettine (62) were successfully achieved starting from the α-methyl-d-mannopyranoside (295). Vittatine (42) has been prepared from d-glucose. The sequence involved a Ferrier carbocyclization to yield a cyclohexenone derivative that, after functionalization and a Claisen rearrangement, gave an intermediate with the quaternary carbon atom and the required stereochemistry for the closure of the aryloctahydroindole ring system to provide the desired product (296).
d-(−)-quinic acid has been efficiently used as the synthetic precursor for the incorporation of the aminocyclohexanetriol moiety present in 2,7-dideoxypancratistatin (297) (Scheme 15 ).
Scheme 15.
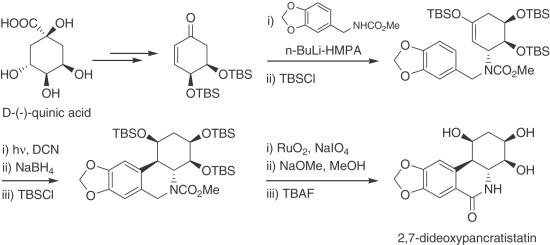
Synthesis of 2,7-dideoxypancratistatin from d-(−)-quinic acid.
V. Spectroscopy and Alkaloid Data
There follows a discussion of proton nuclear magnetic resonance (1H NMR), carbon nuclear magnetic resonance (13C NMR), and mass spectrometry (MS) of the Narcissus alkaloids. A list of the different Narcissus alkaloids, their spectroscopic properties, and literature with the most recent spectroscopic data is given in Table X .
Table X.
Narcissus Alkaloid Data
| Alkaloida | MF (MW)b | Spectroscopic data | References |
|---|---|---|---|
| 1 Lycorine | C16H17NO4 (287) | UV, IR, MS, 1H NMR, 13C NMR, CD, X-ray | 298., 299., 300., 301. |
| 2 Poetaminine | C18H19NO5 (329) | UV, IR | (302) |
| 3 Pseudolycorine | C16H19NO4 (289) | UV, IR, MS, 1H NMR, 13C NMR | (113) |
| 4 1-O-acetyl-pseudolycorine | C18H21NO5 (331) | UV, IR, MS, 1H NMR | (113) |
| 5 2-O-acetyl-pseudolycorine | C18H21NO5 (331) | UV, IR, MS, 1H NMR, 13C NMR | (113) |
| 6 9-O-methyl-pseudolycorine | C17H21NO4 (303) | UV, IR, MS, 1H NMR | 159., 303. |
| 7 Galanthine | C18H23NO4 (317) | UV, MS, 1H NMR, 13C NMR | 135., 304. |
| 8 Goleptine | C17H21NO4 (303) | IR | (179) |
| 9 Jonquilline | C18H17NO5 (327) | UV, IR | (180) |
| 10 Caranine | C16H17NO3 (271) | UV, IR, MS, 1H NMR, CD | 305., 306. |
| 11 Pluviine | C17H21NO3 (287) | UV, IR | 176., 307. |
| 12 Norpluviine | C16H19NO3 (273) | UV, IR | 176., 308. |
| 13 9-O-demethyl- pluviine | C16H19NO3 (273) | UV, MS, 1H NMR, 13C NMR | (167) |
| 14 1-O-acetyl-9-O-demethylpluviine | C18H21NO4 (315) | UV, MS, 1H NMR, 13C NMR | (167) |
| 15 1,9-O-diacetyl-9-O-demethylpluviine | C20H23NO5 (357) | UV, MS, 1H NMR, 13C NMR | (167) |
| 16 Narcissidine | C18H23NO5 (333) | UV, IR, MS, 1H NMR, X-ray | 309., 310., 311., 312., 313. |
| 17 Ungiminorine | C17H19NO5 (317) | UV, MS, 1H NMR, 13C NMR | 173., 314., 315. |
| 18 Nartazine | C20H23NO6 (373) | IR | (89) |
| 19 Kirkine | C16H19NO3 (273) | IR, MS, 1H NMR, 13C NMR | (316) |
| 20 Assoanine | C17H17NO2 (267) | UV, IR, MS, 1H NMR, 13C NMR | (114) |
| 21 Oxoassoanine | C17H15NO3 (281) | UV, IR, MS, 1H NMR, 13C NMR | (114) |
| 22 Vasconine | C17H16NO2 (266) | IR, MS, 1H NMR, 13C NMR | 70., 71. |
| 23 Tortuosine | C18H18NO3 (296) | IR, MS, 1H NMR, 13C NMR | 68., 69. |
| 24 Ungeremine | C16H11NO3 (265) | UV, IR, MS, 1H NMR, 13C NMR | 317., 318., 319., 320. |
| 25 Roserine | C18H22NO3 (300) | MS, 1H NMR, 13C NMR | (67) |
| 26 Homolycorine | C18H21NO4 (315) | UV, IR, MS, 1H NMR, 13C NMR, CD, X-ray | 82., 300., 321., 322. |
| 27 8-O-demethyl-homolycorine | C17H19NO4 (301) | UV, IR, MS, 1H NMR, 13C NMR, CD, X-ray | 111., 300., 323., 324. |
| 28 8-O-demethyl-8-O-acetylhomolycorine | C19H21NO5 (343) | IR, MS, 1H NMR, 13C NMR | (70) |
| 29 9-O-demethyl-homolycorine | C17H19NO4 (301) | UV, IR, MS, 1H NMR, 13C NMR | (82) |
| 30 Masonine | C17H17NO4 (299) | UV, IR, MS, 1H NMR, 13C NMR, CD | 165., 321. |
| 31 Normasonine | C16H15NO4 (285) | UV, IR, MS, 1H NMR, 13C NMR | (165) |
| 32 9-O-demethyl-2α-hydroxy-homolycorine | C17H19NO5 (317) | IR, MS, 1H NMR, 13C NMR | (138) |
| 33 Dubiusine | C23H27NO8 (445) | UV, IR, MS, 1H NMR, 13C NMR | (72) |
| 34 Hippeastrine | C17H17NO5 (315) | UV, IR, MS, 1H NMR, 13C NMR, CD | 71., 300., 325., 326. |
| 35 Lycorenine | C18H23NO4 (317) | UV, MS, 1H NMR, 13C NMR, X-ray | 106., 327., 328., 329. |
| 36O-methyllycorenine | C19H25NO4 (331) | IR, MS, 1H NMR, 13C NMR, X-ray | 74., 104. |
| 37 Oduline | C17H19NO4 (301) | UV, IR, MS, 1H NMR, 13C NMR | (165) |
| 38 6-O-methyloduline | C18H21NO4 (315) | UV, IR, MS, 1H NMR, 13C NMR | (165) |
| 39 2α-Hydroxy-6-O-methyloduline | C18H21NO5 (331) | IR, MS, 1H NMR, 13C NMR | (71) |
| 40 8-O-demethyl-homolycorine-N-oxide | C17H19NO5 (317) | UV, IR, MS, 1H NMR, 13C NMR | (137) |
| 41 Poetinatine | C20H23NO6 (373) | IR, MS, 1H NMR | (98) |
| 42 Vittatine | C16H17NO3 (271) | UV, IR, MS, 1H NMR, 13C NMR, CD | 208., 300., 330., 331. |
| 43 11-Hydroxyvittatine | C16H17NO4 (287) | UV, IR, MS, 1H NMR, 13C NMR, CD | 208., 300. |
| 44 Maritidine | C17H21NO3 (287) | UV, IR, MS, 1H NMR, 13C NMR, CD, X-ray | 136., 332., 333., 334., 335., 336. |
| 45 8-O-demethyl-maritidine | C16H19NO3 (273) | IR, MS, 1H NMR, 13C NMR | 108., 331., 337. |
| 46 9-O-demethyl-maritidine | C16H19NO3 (273) | IR, MS, 1H NMR | (111) |
| 47O-methylmaritidine | C18H23NO3 (301) | UV, IR, MS, 1H NMR, 13C NMR, CD | 133., 137. |
| 48 Papyramine | C18H23NO4 (317) | UV, IR, MS, 1H NMR, 13C NMR | 135., 137. |
| 49 6-Epipapyramine | C18H23NO4 (317) | UV, IR, MS, 1H NMR, 13C NMR | 135., 137. |
| 50O-methyl-6-epipapyramine | C19H25NO4 (331) | UV, IR, MS, 1H NMR, 13C NMR | (137) |
| 51 6α-Hydroxy-3-O-methylepimaritidine | C18H23NO4 (317) | UV, IR, MS, 1H NMR, CD | (133) |
| 52 6β-Hydroxy-3-O-methylepimaritidine | C18H23NO4 (317) | UV, IR, MS, 1H NMR, CD | (133) |
| 53 Hemanthamine | C17H19NO4 (301) | UV, IR, MS, 1H NMR, 13C NMR, CD, X-ray | 83., 331., 338., 339. |
| 54 11-O-acetyl- hemanthamine | C19H21NO5 (343) | IR, MS, 1H NMR, 13C NMR, CD | (74) |
| 55 Hemanthidine | C17H19NO5 (317) | UV, IR, MS, 1H NMR, 13C NMR, CD | 73., 300., 325., 331., 340., 341. |
| 56 6-Epihemanthidine | C17H19NO5 (317) | UV, IR, MS, 1H NMR, 13C NMR, CD | 73., 300., 325., 331., 340., 341. |
| 57 Crinamine | C17H19NO4 (301) | UV, IR, MS, 1H NMR, 13C NMR, CD, X-ray | 298., 342., 343., 344. |
| 58 Narcidine | C17H21NO4 (303) | UV, IR, MS, 1H NMR | (161) |
| 59 Cantabricine | C18H23NO4 (317) | IR, MS, 1H NMR, 13C NMR | (140) |
| 60 Narcimarkine | C21H29NO5 (375) | IR, MS | (91) |
| 61 Bujeine | C20H23NO6 (373) | IR, MS, 1H NMR, 13C NMR, CD | (74) |
| 62 Tazettine | C18H21NO5 (331) | UV, IR, MS, 1H NMR, 13C NMR, CD, X-ray | 300., 345., 346., 347., 348., 349., 350., 351. |
| 63 Criwelline | C18H21NO5 (331) | UV, MS, 1H NMR, 13C NMR, CD | 352., 353., 354., 355. |
| 64 Pretazettine | C18H21NO5 (331) | UV, IR, MS, 1H NMR, CD | 300., 347. |
| 65 3-Epimacronine | C18H19NO5 (329) | IR, MS, 1H NMR, 13C NMR, CD, X-ray | 79., 300., 337., 356. |
| 66 3-Epimacronine isomer | C18H19NO5 (329) | IR, MS, 1H NMR, 13C NMR | (139) |
| 67 Obesine | C16H17NO4 (287) | MS, 1H NMR, 13C NMR | (76) |
| 68 Narciclasine | C14H13NO7 (307) | UV, IR, MS, 1H NMR, 13C NMR, X-ray | 120., 122., 125. |
| 69 Narciprimine | C14H9NO5 (271) | UV, IR, MS, 1H NMR | 125., 357. |
| 70 Trisphaeridine | C14H9NO2 (223) | UV, IR, MS, 1H NMR, 13C NMR | 99., 358. |
| 71 Bicolorine | C15H12NO2 (238) | IR, MS, 1H NMR, 13C NMR | (79) |
| 72 Ismine | C15H15NO3 (257) | UV, IR, MS, 1H NMR, 13C NMR, X-ray | 99., 358., 359., 360. |
| 73 Pancracine | C16H17NO4 (287) | UV, MS, 1H NMR, 13C NMR, CD | 80., 361. |
| 74 Nangustine | C16H17NO4 (287) | IR, MS, 1H NMR, 13C NMR | (80) |
| 75 Galanthamine | C17H21NO3 (287) | UV, IR, MS, 1H NMR, 13C NMR, CD, X-ray | 83., 354., 362., 363. |
| 76 Epigalanthamine | C17H21NO3 (287) | UV, IR, MS, 1H NMR, CD | 354., 364., 365. |
| 77O-acetyl-galanthamine | C19H23NO4 (329) | UV, MS, 1H NMR, 13C NMR | (167) |
| 78 Norgalanthamine | C16H19NO3 (273) | UV, IR, MS, 1H NMR, 13C NMR, CD, X-ray | 141., 366., 367., 368. |
| 79 Epinorgalanthamine | C16H19NO3 (273) | IR, MS, 1H NMR, 13C NMR | (107) |
| 80N-formyl-norgalanthamine | C17H19NO4 (301) | UV, IR, MS, 1H NMR, 13C NMR | (83) |
| 81 Sanguinine | C16H19NO3 (273) | UV, IR, MS, 1H NMR, 13C NMR | (369) |
| 82 Narcisine | C18H21NO4 (315) | UV, IR, MS, 1H NMR, 13C NMR | (117) |
| 83 Narwedine | C17H19NO3 (285) | UV, IR, MS, 1H NMR | 364., 367. |
| 84 Lycoramine | C17H23NO3 (289) | IR, MS, 1H NMR, 13C NMR | 336., 367., 369. |
| 85 Norlycoramine | C16H21NO3 (275) | IR, MS, 1H NMR | (337) |
| 86 Epinorlycoramine | C16H21NO3 (275) | IR, MS, 1H NMR, 13C NMR | (107) |
| 87O-methyl-norbelladine | C16H19NO3 (273) | 1H NMR, 13C NMR | (181) |
| 88 Cherylline | C17H19NO3 (285) | IR, MS, 1H NMR, 13C NMR, X-ray | (370) |
| 89 Pallidiflorine | C34H40N2O7 (588) | IR, MS, 1H NMR, 13C NMR | (86) |
| 90 Mesembrenone | C17H21NO3 (287) | IR, MS, 1H NMR, 13C NMR | 26., 371. |
| 91 Mesembrenol | C17H23NO3 (289) | UV, IR, MS, 1H NMR, CD | (372) |
| 92 Mesembrine | C17H23NO3 (289) | IR, MS, 1H NMR, 13C NMR | 284., 285., 373. |
Alkaloids are listed in numerical order and grouped according to their ring system (see Tables I–VII).
MF (MW)=Molecular formula (Molecular weight).
A. Proton Nuclear Magnetic Resonance
1H NMR spectroscopy gives important information about the different types of Amaryllidaceae alkaloids. In the last two decades, the routine use of 2D NMR techniques has facilitated the structural assignments and the establishment of their stereochemistry. A compilation of the different 1H NMR spectra arranged according to the different skeleton types is shown in Table XI, Table XII, Table XIII, Table XIV, Table XV, Table XVI, Table XVII .
Table XI.
1H NMR Data of Lycorine-Type Alkaloids
| Alkaloid | 1 | 3 | 4 | 5 | 6 | 7 | 10 | 13 | 14 | 15 | 19 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H-1 | 4.27 br s | 4.85 br s | 5.58 m | 4.43 t | 4.53 dd | 4.55 s | 4.70 m | 4.26 dd | 5.54 dd | 5.51 m | 4.37 q |
| H-2α | 3.97 br s | 4.50 t | 4.15 dd | 5.29 dd | 4.18 m | 3.72 m | 2.59 m | 2.3–2.6 m | 2.3–2.5 m | 2.36 m | 2.46 ddd |
| H-2β | — | — | — | — | — | — | 2.59 m | 2.3–2.4 m | 2.3–2.5 m | 2.36 m | 2.34 dddd |
| H-3 | 5.37 br s | 5.60 t | 5.69 dd | 5.45 t | 5.55 m | 5.55 br s | 5.41 m | 5.54 br d | 5.39 br s | 5.32 br t | 5.87 br dt |
| H-4a | 2.60 d | 3.02 br d | 2.90 br s | 2.85 d | 2.87 dd | 2.65 s | 2.78 dd | 2.70 br d | 2.3–2.5 m | 2.2–2.3 m | 4.24 br d |
| H-6α | 3.32 d | 3.68 br d | 3.61 dd | 3.54 d | 3.55 dd | 3.40 br d | 3.52 dd | 3.31 d | 3.54 br d | 3.5–3.6 br s | 4.40 d |
| H-6β | 4.02 d | 4.16 d | 4.17 d | 4.14 d | 4.16 d | 4.05 d | 4.13 d | 3.93 d | 3.59 br d | 3.5–3.6 br s | 4.61 dt |
| H-7 | 6.68 s | 6.71 s | 6.70 s | 6.61 s | 6.75 s | 6.52 s | 6.58 s | 6.59 s | 6.64 s | 6.71 s | 6.60 s |
| H-10 | 6.81 s | 6.89 s | 6.74 s | 6.83 s | 6.96 s | 6.78 s | 6.82 s | 6.82 s | 7.08 s | 7.14 br s | 7.08 s |
| H-10b | 2.50 m | 2.74 br d | 2.90 br s | 2.69 d | 2.76 ddd | 2.65 s | 2.41 ddd | 2.98 dd | 3.33 dd | 3.27 dd | 3.52 dd |
| H-11 (2H) | 2.44 m | 2.6–2.7 m | 2.66 t | 2.63 m | 2.64 m | 2.4–2.6 m | 2.59 m | 2.5–2.6 m | 2.3–2.5 m | 2.2–2.4 m | 2.8–2.9 m |
| H-12α | 2.19 ddd | 2.6–2.7 m | 2.59 br t | 2.42 dt | 2.42 br q | 2.25 dd | 2.33 br q | 2.3–2.4 m | 2.79 m | 2.6–2.8 m | 3.71 ddd |
| H-12β | 3.19 dd | 3.37 dd | 3.33 m | 3.35 dt | 3.35 ddd | 3.25 ddd | 3.32 ddd | 3.32 m | 2.79 m | 2.6–2.8 m | 3.83 ddd |
| OCH2O | 5.95 s | — | — | — | — | — | 5.91 (2d) | — | — | — | — |
| OMe | — | 3.84 s | 3.84 s | 3.82 s | 3.85 s | 3.78 s | — | 3.89 s | 3.86 s | 3.74 s | 3.87 s |
| OMe | — | — | — | — | 3.80 s | 3.74 s | — | — | — | — | — |
| OMe | — | — | — | — | — | 3.40 s | — | — | — | — | — |
| OAc | — | — | 1.92 s | 2.06 s | — | — | — | — | 1.98 s | 2.23 s | — |
| OAc | — | — | — | — | — | — | — | — | — | 1.92 s | — |
| Solvent | a | b | b | b | c | d | d | d | d | d | c |
| MHz | 300 | 200 | 200 | 200 | 270 | 200 | 270 | 400 | 400 | 400 | 500 |
| Reference | (298) | (113) | (113) | (113) | (303) | (135) | (305) | (167) | (167) | (167) | (316) |
| Solvent a: DMSO-d6, b: CDC13-CDC3OD, c: CD3OD, d: CDCl3 | |||||||||||
| Alkaloid | 20 | 21 | 22 | 23 | 24 |
|---|---|---|---|---|---|
| H-1 | 7.31 dd | 7.85 br d | 8.32 d | 8.13 d | 7.54 s |
| H-2 | 6.75 t | 7.27 t | 7.84 t | — | — |
| H-3 | 6.99 dt | 7.33 br d | 7.70 d | 7.59 br s | 7.34 s |
| H-6 | 4.09 s | — | 10.42 s | 9.55 s | 9.19 s |
| H-7 | 6.64 s | 7.57 s | 8.09 s | 7.92 s | 7.59 s |
| H-10 | 7.17 s | 7.81 s | 7.86 s | 8.34 s | 7.88 s |
| H-11 (2H) | 3.00 br t | 3.45 br t | 3.76 t | 3.88 t | 3.70 t |
| H-12 (2H) | 3.32 t | 4.45 br t | 5.42 t | 5.35 t | 5.12 t |
| OMe | 3.93 s | 4.09 s | 4.19 s | 4.33 s | — |
| OMe | 3.87 s | 4.03 s | 4.08 s | 4.20 s | — |
| OMe | — | — | — | 4.17 s | — |
| OCH2O | — | — | — | — | 6.32 s |
| Solvent | d | b | b | c | e |
| MHz | 200 | 200 | 500 | 500 | 600 |
| Reference | (114) | (114) | (71) | (68) | (320) |
| Solvent a: DMSO-d6, b: CDC13-CDC3OD, c: CD3OD, d: CDCl3, e: D2O+0.01% | |||||
| Alkaloid | 16 | 17 | 25 |
|---|---|---|---|
| H-1α | — | — | 2.85 ddd |
| H-1β | 4.66 m | 4.69 br s | 3.13 dd |
| H-2α | 3.4–4.2 m | 3.69 m | 1.8–1.95 m |
| H-2β | — | — | 2.25–2.4 m |
| H-3α | — | — | 1.38 qd |
| H-3β | 4.66 m | 4.60 br s | 2.25–2.4 m |
| H-4 | — | — | 3.4–3.5 m |
| H-4a | 3.4–4.2 m | 4.09 m | — |
| H-6α | 3.54 d | 3.69 m | 9.50 s (1H) |
| H-6β | 4.09 d | 4.15 br d | |
| H-7 | 6.68 s | 6.78 s | 6.95 s |
| H-10 | 6.88 s | 6.93 s | — |
| H-10b | 2.70 d | 2.84 br d | — |
| H-11α | 5.56 br s (1H) | 5.75 br d (1H) | 2.05 ddd |
| H-11β | 2.70 dt | ||
| H-12α | 3.4–4.2 m (2H) | 3.69 m | 4.75–4.95 m (2H) |
| H-12β | 4.15 br d | ||
| OMe | 3.86 s | — | 4.15 s |
| OMe | 3.82 s | — | 4.05 s |
| OMe | 3.44 s | 3.44 s | 3.90 s |
| OCH2O | — | 5.92 br s | — |
| Solvent | d | c | d |
| MHz | 400 | 600 | 250 |
| Reference | (313) | (173) | (67) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3, e: D2O+0.01% TFA-d1.
Table XII.
1H NMR Data of Homolycorine-Type Compounds: Lactone Alkaloids
| Alkaloid | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 40 |
|---|---|---|---|---|---|---|---|---|---|---|
| H-1 | 4.81 ddd | 4.78 m | 4.86 br d | 4.80 ddd | 4.75 m | 4.85 s | 4.59 br d | 4.63 dd | 4.58 br s | 4.83 m |
| H-2 (2H) | 2.49 m | 2.59 m | 2.5–2.7 m | 2.51 m | 2.60 m | 2.62 m | 4.28 br t (Hβ) | 5.46 dd (Hβ) | 4.38 br s (Hβ) | 2.64 m |
| H-3 | 5.50 m | 5.50 d | 5.63 br d | 5.55 m | 5.50 br d | 5.64 m | 5.68 br s | 5.62 m | 5.63 br s | 5.76 br d |
| H-4a | 2.72 dd | 2.73 dd | 2.96 br d | 2.71 br d | 2.74 m | 5.50 br d | 2.66 br d | 2.74 br d | 2.62 d | 3.87 br d |
| H-7 | 7.57 s | 7.60 s | 7.47 s | 7.54 s | 7.49 s | 7.52 s | 7.47 s | 7.52 s | 7.45 s | 7.44 s |
| H-10 | 6.99 s | 6.98 s | 7.13 s | 6.91 s | 6.96 s | 7.11 s | 6.95 s | 6.97 s | 6.92 s | 7.24 s |
| H-10b | 2.64 dd | 2.63 dd | 2.86 dd | 2.60 dd | 2.74 m | 2.66 d | 2.83 dd | 2.79 br d | 2.85 dd | 3.50 dd |
| H-11 (2H) | 2.6–2.7 m | 2.50 m | 2.5–2.7 m | 2.5–2.6 m | 2.50 m | 2.52 m | 2.5–2.6 m | 2.5–2.7 m | 2.48 m | 3.57 m |
| H-12α | 3.14 ddd | 3.14 ddd | 3.31 ddd | 3.15 ddd | 3.18 ddd | 3.15 ddd | 3.17 ddd | 3.19 ddd | 3.13 ddd | 2.88 m |
| H-12β | 2.24 ddd | 2.25 dd | 2.45 dd | 2.30 ddd | 2.27 dd | 2.89 dd | 2.31 dd | 2.37 dd | 2.23 ddd | 2.72 m |
| OCH2O | — | — | — | — | 6.07 (2d) | 6.05 d | — | — | 6.04 (2d) | — |
| OMe | 3.96 s | 3.95 s | 3.97 s | 3.94 s | — | — | 3.95 s | 3.94 s | — | 3.96 s |
| OMe | 3.95 s | — | — | — | — | — | — | — | — | — |
| NMe | 2.00 s | 2.00 s | 2.16 s | 2.01 s | 2.06 s | — | 2.08 s | 2.06 s | 2.03 s | 2.94 s |
| OAc | — | — | 2.00 s | — | — | — | — | 2.00 s | — | — |
| CHA | — | — | — | — | — | — | — | 2.53 dd | — | — |
| CHB | — | — | — | — | — | — | — | 2.65 dd | — | — |
| CHOH | — | — | — | — | — | — | — | 5.24 m | — | — |
| Me | — | — | — | — | — | — | — | 1.29 d | — | — |
| Solvent | d | d | b | b | d | d | b | b | d | b |
| MHz | 200 | 400 | 250 | 200 | 400 | 400 | 200 | 400 | 500 | 200 |
| Reference | (82) | (324) | (70) | (82) | (165) | (165) | (138) | (72) | (71) | (137) |
| Solvent a: DMSO-d6, b: CDC13-CDC3OD, c: CD3OD, d: CDCl3 | ||||||||||
| Alkaloid | 35 | 36 | 37 | 38 | 39 |
|---|---|---|---|---|---|
| H-1α | 4.35 br d | 4.29 br d | 4.35 d | 4.17 d | 4.15 br s |
| H-2α | 2.35 m | 2.35 m | 2.31 dm | 2.26 dd | — |
| H-2β | 2.65 m | 2.65 m | 2.62 dm | 2.53 dm | 4.21 br s |
| H-3 | 5.50 br s | 5.50 br d | 5.46 br d | 5.39 br s | 5.69 br s |
| H-4a | 2.78 br d | 2.77 dd | 2.72 br d | 2.69 br d | 2.87 d |
| H-6β | 5.93 s | 5.54 s | 5.99 s | 5.39 br s | 5.43 s |
| H-7 | 6.93 s | 6.80 s | 6.85 s | 6.67 s | 6.73 s |
| H-10 | 6.99 s | 6.93 s | 6.90 s | 6.77 s | 6.94 s |
| H-10b | 2.44 dd | 2.44 dd | 2.4–2.5 m | 2.35 dd | 2.85 d |
| H-11 (2H) | 2.4–2.6 m | 2.4–2.6 m | 2.4–2.5 m | 2.3–2.4 m | 2.54 m |
| H-12α | 3.15 ddd | 3.15 ddd | 3.14 ddd | 3.08 dd | 3.33 m |
| H-12β | 2.27 dd | 2.23 dd | 2.25 dd | 2.17 dd | 2.38 dt |
| OCH2O | — | — | 5.97 d | 5.83 d | 5.91 (2d) |
| OMe | 3.88 s | 3.89 s | — | 3.43 s | 3.51 s |
| OMe | 3.87 s | 3.88 s | — | — | — |
| OMe | — | 3.55 s | — | — | — |
| NMe | 2.08 s | 2.10 s | 2.11 s | 2.05 s | 2.22 s |
| Solvent | c | d | d | d | d |
| MHz | 250 | 200 | 400 | 400 | 500 |
| Reference | (106) | (104) | (165) | (165) | (71) |
Solvent a, DMSO-d6; b, CDCl3-CD3OD; c, CD3OD; d, CDCl3.
Table XIII.
1H NMR Data of Hemanthamine-Type Alkaloids
| Alkaloid | 42 | 43 | 44 | 45 | 46 | 47 | 48/49 | 53 | 54 | 55/56 | 57 | 58 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H-1 | 6.44 d | 6.41 d | 6.71 d | 6.65 d | 6.58 d | 6.49 d | 6.65 d/6.66 d | 6.36 d | 6.40 d | 6.33 d | 6.22 br s | 6.48 d |
| H-2 | 6.06 dd | 6.19 dd | 5.93 dd | 5.99 dd | 5.91 ddd | 6.08 dd | 6.00 dd | 6.25 dd | 6.21 dd | 6.27 dd | 6.22 br s | 6.35 dd |
| H-3 | 4.46 m | 4.28 ddd | 4.27 ddd | 4.36 m | 4.31 td | 3.85 m | 3.88 m/3.85 m | 3.82 m | 3.91 m | 3.85 m | 3.98 m | 3.86 m |
| H-4α | 1.85 ddd | 2.28 ddd | 1.81 ddd | 1.75 ddd | 1.73 td | 1.73 ddd | 1.77 ddd/1.60 ddd | 2.11 ddd | 2.07 ddd | 2.36 ddd/2.21 ddd | 2.08 m | 2.11 ddd |
| H-4β | 2.58 ddd | 1.85 ddd | 1.95 m | 2.03 ddd | 1.90 dddd | 2.95 ddd | 2.21 br d/2.09 m | 1.96 ddd | 1.98 ddd | 2.12 ddd/2.00 ddd | 2.08 m | 2.04 ddd |
| H-4a | 3.87 m | 3.47 dd | 3.48 dd | 3.37 m | 3.43 dd | 3.90 dd | 3.59 dd/3.90 m | 3.25 dd | 3.41 m | 3.56 dd/3.20 m | 3.22 dd | 3.38 m |
| H-6α | 4.07 d | 3.84 d | 3.84 d | 3.79 d | 3.81 d | 4.25 d | —/5.16 s | 3.72 d | 3.77 d | —/5.02 s | 3.66 d | 3.70 d |
| H-6β | 4.71 d | 4.35 d | 4.39 d | 4.39 d | 4.41 d | 4.80 d | 5.86 s/— | 4.25 d | 4.40 d | 5.69 s/— | 4.28 d | 4.31 d |
| H-7 | 6.53 s | 6.56 s | 6.66 s | 6.56 s | 6.53 s | 6.57 s | 7.03 s/6.89 s | 6.41 s | 6.52 s | 6.94 s/6.79 s | 6.47 s | 6.53 s |
| H-10 | 6.87 s | 6.94 s | 6.97 s | 6.82 s | 6.87 s | 6.81 s | 6.80 s/6.83 s | 6.74 s | 6.95 s | 6.70 s/6.73 s | 6.79 s | 6.78 s |
| H-11 endo | 2.15 m | 3.98 dd | 1.95 m | 1.95 m | 1.8–2.0 m | 2.22 ddd | 2.00 m | 3.96 dd | 5.05 dd | 3.92 m | 3.92 m | 3.99 ddd |
| H-11 exo | 2.32 m | — | 2.17 m | 2.15 m | 2.19 ddd | 2.37 ddd | 2.00 m | — | — | — | — | — |
| H-12 endo | 3.17 m | 3.20 dd | 2.93 ddd | 2.95 m | 2.91 ddd | 3.27 ddd | 3.02 ddd/2.84 ddd | 3.30 dd | 3.41 m | 4.20 dd/3.30 m | 3.40 m | 3.39 m |
| H-12 exo | 3.87 m | 3.50 dd | 3.34 m | 3.37 m | 3.31 ddd | 4.08 ddd | 3.73 ddd/3.39 m | 3.19 dd | 3.41 m | 2.96 dd/3.20 m | 3.40 m | 3.26 dd |
| OCH2O | 5.95 s | 5.89 s | — | — | — | — | — | 5.81 (2d) | 5.95 s | 5.83 (2d)/5.86 (2d) | 5.86 s | — |
| OMe | — | — | 3.81 s | 3.89 s | 3.82 s | 3.86 s | 3.88 s | — | — | — | — | 3.86 s |
| OMe | — | — | 3.77 s | — | — | 3.82 s | 3.88 s | — | — | — | — | — |
| OMe | — | — | — | — | — | 3.34 s | 3.37 s/3.31 s | 3.36 s | 3.41 s | 3.32 s/3.28 s | 3.40 s | 3.35 s |
| OAc | — | — | — | — | — | — | — | — | 2.03 s | — | — | — |
| Solvent | d | c | c | d | d | d | d | d | d | d | d | d |
| MHz | 360 | 270 | 300 | 360 | 200 | 200 | 200 | 360 | 500 | 360 | 300 | 360 |
| Reference | (331) | (376) | (336) | (331) | (111) | (137) | (135) | (331) | (74) | (331) | (298) | (161) |
| Solvent a: DMSO-d6, b: CDC13-CDC3OD, c: CD3OD, d: CDCl3 | ||||||||||||
| Alkaloid | 59 | 61 |
|---|---|---|
| H-1ax | 1.77 ddd | 6.38 d (1H) |
| H-1eq | 2.39 dt | |
| H-2ax | 1.61 dddd | 6.08 dd (1H) |
| H-2eq | 2.06 m | |
| H-3 | 4.67 tt (β) | 3.89 ddd (α) |
| H-4ax | 1.44 ddd | 2.43 ddd |
| H-4eq | 2.23 m | 2.06 ddd |
| H-4a | 3.18 dd | 3.29 dd |
| H-6α | 3.91 d | 4.04 d |
| H-6β | 4.50 d | 4.45 d |
| H-7 | 6.48 s | 6.57 s |
| H-10 | 6.74 s | 6.81 s |
| H-11 exo | 2.30 ddd | 3.93 dd |
| H-11 endo | 1.83 ddd | — |
| H-12 exo | 3.60 m | 4.82 d |
| H-12 endo | 2.94 m | 4.51 d |
| OMe | 3.82 s | 3.36 s |
| OCH2O | — | 5.91 s |
| CHA | — | 4.27 dd |
| CHB | — | 3.58 dd |
| OAc | 2.02 s | 2.00 s |
| Solvent | d | d |
| MHz | 500 | 500 |
| Reference | (140) | (74) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
Table XIV.
1H NMR Data of Tazettine-Type Alkaloids
| Alkaloid | 62 | 63 | 65 | 67 |
|---|---|---|---|---|
| H-1 | 5.60 ddd | 5.78 d | 5.44 ddd | 5.86 d |
| H-2 | 6.15 ddd | 6.20 dd | 5.97 d | 6.13 d |
| H-3 | 4.13 m | 3.89 ddd | 4.12 m | 4.3–4.4 m |
| H-4α | 2.20 m | 1.93 ddd | 1.70 m | 2.39 br d |
| H-4β | 1.60 m | 2.09 ddd | 2.55 m | 1.68 ddd |
| H-4a | 2.83 m | 2.95 t | 3.10 m | 3.15 br s |
| H-6 | 4.65 d | 4.68 d | — | 4.02 d |
| H-6′ | 4.95 dd | 4.94 d | — | 4.38 d |
| H-7 | 6.50 br s | 6.55 s | 7.52 s | 6.66 s |
| H-10 | 6.85 s | 6.52 s | 6.73 s | 6.70 s |
| H-10b | — | — | — | 2.81 br d |
| H-11 | — | — | 4.43 dd | — |
| H-12 | 2.65 d | 2.83 d | 2.76 dd | 3.01 d |
| H-12′ | 3.30 d | 3.30 d | 3.16 dd | 3.10 d |
| OCH2O | 5.90 s | 5.92 s | 6.01 s | 5.99 s |
| OMe | 3.45 s | 3.45 s | 3.40 s | — |
| NMe | 2.40 s | 2.38 s | 2.50 s | — |
| Solvent | d | d | d | c |
| MHz | 90 | 300 | 100 | 250 |
| Reference | (347) | (355) | (337) | (76) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
Table XV.
1H NMR Data of Narciclasine-Type Alkaloids
| Alkaloid | 68 | 70 | 71 | 72 |
|---|---|---|---|---|
| H-1 | 6.17 ddd | 8.36 dddd | 8.73 dd | 6.98 dd |
| H-2 | 4.23 ddd | 7.61 ddd | 8.02 td | 6.81 ddd |
| H-3 | 3.92 ddd | 7.67 ddd | 8.09 td | 7.28 ddd |
| H-4 | 3.90 dd | 8.11 ddd | 8.31 dd | 6.73 dd |
| H-4a | 4.35 ddd | — | — | — |
| H-6 | — | 9.06 s | 10.42 s | 4.26 d |
| H-6′ | — | — | — | 4.20 d |
| H-7 | — | 7.32 s | 7.97 s | 7.00 s |
| H-10 | 6.75 s | 7.89 s | 8.16 s | 6.67 s |
| OCH2O | 6.01 (2d) | 6.15 s | 6.41 s | 5.99 s |
| NMe | — | — | 4.73 s | 2.73 s |
| Solvent | c | d | b | d |
| MHz | 500 | 200 | 200 | 200 |
| Reference | (122) | (358) | (79) | (358) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
Table XVI.
1H NMR Data of Montanine-Type Alkaloids
| Alkaloid | 73 | 74 |
|---|---|---|
| H-1 | 5.37 br dddd | 5.52 dt |
| H-2α | — | 2.05 ddt |
| H-2β | 3.73 dddd | 2.57 dddd |
| H-3 | 3.65 ddddd (β) | 3.62 ddd (α) |
| H-4α | 1.36 ddd | 3.31 t |
| H-4β | 1.83 ddd | — |
| H-4a | 3.20 ddd | 3.16 br d |
| H-6α | 4.16 br d | 4.32 d |
| H-6β | 3.63 br d | 3.83 d |
| H-7 | 6.60 br s | 6.51 s |
| H-10 | 6.67 br s | 6.56 s |
| H-11 | 3.25 br s | 3.33 br d |
| H-12ax | 2.86 br s | 3.03 d |
| H-12eq | 2.86 br s | 2.94 dd |
| OCH2O | 5.90 (2d) | 5.86 (2d) |
| Solvent | A | c |
| MHz | 400 | 500 |
| Reference | (361) | (80) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
Table XVII.
1H NMR Data of Galanthamine-Type Alkaloids
| Alkaloid | 75 | 76 | 77 | 78 | 79 | 80 | 81 | 83 | 84 | 86 |
|---|---|---|---|---|---|---|---|---|---|---|
| H-1 | 4.73 m | 4.58 m | 4.58 m | 4.67 m | 4.62 br s | 4.50 m | 4.52 br s | 4.72 m | 4.28 m | 4.37 t |
| H-2α | 2.12 ddd | 1.69 ddd | 2.10 ddd | 2.24 ddd | 2.04 ddd | 1.95 ddd | 2.10 ddd | 2.73 dd | 1.4–1.9 m | 1.88 m |
| H-2β | 2.80 ddt | 2.77 dddd | 2.69 ddd | 2.60 m | 2.68 dd | 2.61 ddt | 2.49 dm | 3.14 m | 2.40 br d | 2.50 ddd |
| H-3 | 4.25 m | 4.61 dddd | 5.34 dd | 4.28 m | 4.15 m | 4.07 m | 4.15 br t | — | 3.98 m | 4.09 m |
| H-4 | 6.10 ddd | 6.05 dt | 5.92 d | 6.06 ddd | 5.98 d | 6.00 ddd | 5.91 dd | 6.02 d | 1.4–1.9 m (2H) | 1.5–1.7 m (2H) |
| H-4a | 6.21 dd | 5.79 dt | 6.28 d | 6.23 dt | 6.05 d | 5.88 dd | 6.11 d | 6.92 d | 1.4–1.9 m (2H) | 1.7–1.9 m (2H) |
| H-6 | 3.79 dd | 3.61 d | 3.77 d | 4.21 d | 3.93 d | 3.94 dd | 3.64 d | 3.76 d | 3.54 d | 3.98 s |
| H-6′ | 4.21 d | 4.06 d | 4.22 d | 4.44 d | 4.04 d | 4.80 d E | 4.06 d | 4.12 d | 3.92 d | 3.98 s |
| 5.10 d Z | ||||||||||
| H-7 | 6.74 d | 6.55 d | 6.61 d | 6.84 d | 6.62 d | 6.60 d | 6.57 d | 6.64 d | 6.51 d | 6.65 d |
| H-8 | 6.78 d | 6.61 d | 6.68 d | 6.90 d | 6.68 d | 6.76 d | 6.52 d | 6.68 d | 6.57 d | 6.62 d |
| H-11α | 2.20 ddd | 2.16 dt | 2.18 dd | 2.08 m | 1.88 dt | 1.71 ddd | 1.65 dm | 2.26 dt | 1.4–1.9 m | 1.7–1.9 m |
| H-11β | 1.69 ddd | 1.63 ddd | 1.65 dd | 2.08 m | 1.80 td | 1.81 ddd | 2.05 m | 1.87 d | 1.4–1.9 m | 1.7–1.9 m |
| H-12α | 3.16 dt | 3.04 br d | 3.14 br d | 2.42 m | 3.22 m | 3.82 ddd E | 3.01 dm | 3.14 m | 2.96 t | 3.19 dt |
| 3.15 ddd Z | ||||||||||
| H-12β | 3.39 ddd | 3.25 dt | 3.40 br t | 3.57 m | 3.38 dt | 3.61 ddd | 3.22 br t | 3.27 t | 3.12 t | 3.43 m |
| OMe | 3.95 s | 3.82 s | 3.85 s | 3.91 s | 3.84 s | 3.84 s | — | 3.82 s | 3.76 s | 3.85 s |
| NMe | 2.52 s | 2.56 s | 2.45 s | — | — | — | 2.38 s | 2.45 s | 2.29 s | — |
| NCHO | — | — | — | — | — | 8.02 s | — | — | — | — |
| — | — | — | 8.07 s | — | — | — | — | |||
| OAc | — | — | 2.04 s | — | — | — | — | — | — | |
| Solvent | d | d | d | c | d | d | c | d | d | d |
| MHz | 200 | 400 | 400 | 200 | 200 | 200 | 250 | 400 | 400 | 200 |
| Reference | (83) | (365) | (167) | (141) | (107) | (83) | (377) | (367) | (367) | (107) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
1. Lycorine Type
This group has been subject to several 1H NMR studies and lycorine (1) as well as its main derivatives has been completely assigned. The general characteristics of the 1H NMR spectra are
-
a.
Two singlets for the para-oriented aromatic protons, together with a unique olefinic proton.
-
b.
Two doublets as an AB system corresponding to the benzylic protons of C-6. The deshielding observed in the β-protons of positions 6 and 12 in relation to their α-homologues is due to the effect of the cis-lone pair of the nitrogen atom.
-
c.
Like almost all other lycorine-type examples, the alkaloids isolated from the Narcissus genus show a trans B/C ring junction, the coupling constant being J 4a,10b ∼11 Hz. Only kirkine (19) shows a cis B/C ring junction, with a smaller coupling constant J 4a,10b 8 Hz.
In the plant, the alkaloid lycorine (1) is particularly vulnerable to oxidation processes, giving several ring-C aromatized products.
2. Homolycorine Type
This group includes lactone, hemiacetal, or the more unusual cyclic ether alkaloids. The general traits for this type of alkaloids can be summarized as follows:
-
a.
Two singlets for the para-oriented aromatic protons. In lactone alkaloids, the deshielding of H-7 is caused by the peri-carbonyl group.
-
b.
The hemiacetal alkaloids always show the substituent at C-6 in an α-disposition.
-
c.
The majority of alkaloids belong to a single enantiomeric series containing a cis B/C ring junction, which is congruent with the small size of the coupling constant J 1,10b. In the Narcissus genus, no exception to this rule has been observed.
-
d.
The large coupling constant between H-4a and H-10b (J 4a,10b∼10 Hz) is only consistent with a trans-diaxial relationship.
-
e.
In general, ring C presents a vinylic proton. If position 2 is substituted by an OH, OMe, or OAc group, it always displays an α-disposition.
-
f.
The singlet corresponding to the N-methyl group is in the range of δ 2.0–2.2 ppm, its absence being very unusual.
-
g.
The H-12α is more deshielded than H-12β as a consequence of the cis-lone pair of the nitrogen atom.
Homolycorine-type alkaloids with a saturated ring C have been studied by Jeff and coworkers (374). They describe empirical correlations of N-methyl chemical shifts with stereochemical assignments of the B/C and C/D ring junction.
3. Hemanthamine Type
The absolute configuration of these alkaloids is determined through the circular dichroism spectrum. The alkaloids of the Narcissus genus are exclusively of the hemanthamine type, while in genera such as Brunsvigia, Boophane, etc., the crinine-type alkaloids are predominant. It is also noteworthy that the alkaloids isolated from the Narcissus genus do not show additional substitutions in the aromatic ring, apart from those of C-8 and C-9. On the contrary, in the genera where crinine-type alkaloids predominate, the presence of alkaloids with a methoxy substituent at C-7 is quite common. Thus, hemanthamine-type alkaloids show the following characteristics:
-
a.
Two singlets for the para-oriented aromatic protons.
-
b.
Using CDCl3 as the solvent, the magnitude of the coupling constants between each olefinic proton (H-1 and H-2) and H-3 gives information about the configuration of the C-3 substituent. Thus, in those alkaloids in which the two-carbon bridge (C-11 and C-12) is cis to the substituent at C-3, H-1 shows an allylic coupling with H-3 (J 1,3∼1–2 Hz) and H-2 shows a smaller coupling with H-3 (J 2,3∼0–1.5 Hz), as it occurs in crinamine (57). On the contrary, in the corresponding C-3 epimeric series, e.g. hemanthamine (53), a larger coupling between H-2 and H-3 (J 2,3 5 Hz) is shown, the coupling between H-1 and H-3 not being detectable.
-
c.
There is frequently an additional coupling of H-2 with the equatorial H-4β in a W-mechanism, while the proton H-4α shows a large coupling with H-4a (J 4 α ,4a∼13 Hz) due to their trans-diaxial disposition, characteristic of the hemanthamine series.
-
d.
Two doublets for an AB system corresponding to the benzylic protons of position C-6.
-
e.
The pairs of alkaloids with a hydroxy substituent at C-6, like papyramine/6-epipapyramine (48/49), hemanthidine/6-epihemanthidine (55/56), etc., appear as a mixture of epimers not separable even by HPLC.
-
f.
Also, in relation to position C-6, it is interesting to note that ismine (72), a catabolic product from the hemanthamine series, shows a restricted rotation around the biarylic bond, which makes the methylenic protons at the benzylic position magnetically non-equivalent.
4. Tazettine Type
Although tazettine (62) is one of the most widely reported alkaloids in the Amaryllidaceae family, it was found to be an extraction artifact from pretazettine (64) (75).
The presence of an N-methyl group (2.4–2.5 ppm) in tazettine-type alkaloids immediately distinguishes them from the hemanthamine type, from which they proceed biosynthetically. Moreover, the 1H NMR spectrum always shows the signal corresponding to the methylenedioxy group.
We have also included the alkaloid obesine (67) in this group, although it exhibits some structural differences with the skeleton type.
5. Galanthamine Type
Among the Amaryllidaceae alkaloids, only the galanthamine type shows an ortho-coupling constant between both the aromatic protons of ring A. The general characteristics of their 1H NMR spectra are
-
a.
Two doublets for the two ortho-oriented aromatic protons with a coupling constant of J 7,8 ∼8 Hz.
-
b.
The assignment of the substituent stereochemistry at C-3 is made in relation with the coupling constants of the olefinic protons H-4 and H-4a. When the coupling constant J 3,4 is about 5 Hz, the substituent is pseudoaxial, while if it is ∼0 Hz this indicates that the substituent at C-3 is pseudoequatorial.
-
c.
Two doublets as an AB system corresponding to the benzylic protons of C-6.
-
d.
The existence of the furan ring results in a deshielding effect in H-1.
-
e.
This type of alkaloid often shows an N-methyl group, but occasionally an N-formyl group has been reported.
B. Carbon13 Nuclear Magnetic Resonance
13C NMR spectroscopy has been extensively used for determining the carbon framework of Amaryllidaceae alkaloids, and there are several major contributions 330., 346., 375.. The preliminary assignments are made on the basis of chemical shifts and multiplicities of the signals (by DEPT experiment). The use of 2D NMR techniques, such as HMQC and HMBC, allow the assignments to be corroborated. Table XVIII, Table XIX, Table XX, Table XXI, Table XXII, Table XXIII, Table XXIV show a compilation of the different 13C NMR spectra classified according to the different types.
Table XVIII.
13C NMR Data of Lycorine-Type Alkaloids
| Alkaloid | 1 | 3 | 5 | 7 | 13 | 14 | 15 | 19 | 21 | 22 | 23 | 24 | 25 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C-1 | 70.2 | 70.7 | 68.5 | 68.3 | 69.8 | 70.3 | 69.9 | 65.4 | 119.4 | 120.0 | 102.2 | 105.4 | 31.7 |
| C-2 | 71.7 | 71.8 | 73.9 | 81.0 | 33.5 | 28.8 | 28.8 | 34.7 | 123.9 | 131.2 | 164.5 | 163.8 | 21.8 |
| C-3 | 118.5 | 118.6 | 114.0 | 115.1 | 116.2 | 115.0 | 114.6 | 123.2 | 123.9 | 125.6 | 118.0 | 119.1 | 23.0 |
| C-4 | 141.7 | 141.7 | 144.6 | 143.9 | 139.6 | 139.2 | 139.4 | 133.9 | 124.3 | 123.2 | 139.7 | 140.9 | 40.0 |
| C-4a | 60.8 | 61.4 | 60.9 | 60.9 | 59.5 | 60.4 | 60.2 | 71.2 | 131.3 | 136.0 | 133.0 | 133.2 | 162.4 |
| C-6 | 56.7 | 56.4 | 56.7 | 56.6 | 55.3 | 51.9 | 51.8 | 66.2 | 157.5 | 144.7 | 142.6 | 143.1 | 138.5 |
| C-6a | 129.7 | 127.3 | 127.9 | 129.3 | 126.0 | 125.0 | 129.2 | 124.8 | 129.0 | 121.3 | 122.9 | 125.3 | 119.3 |
| C-7 | 107.0 | 110.5 | 110.4 | 110.8 | 112.4 | 113.4 | 111.5 | 115.3 | 108.6 | 110.5 | 111.0 | 109.2 | 96.8 |
| C-8 | 145.2 | 146.2 | 146.0 | 147.8 | 146.0 | 145.4 | 149.4 | 149.1 | 149.8 | 151.7 | 153.1 | 153.1 | 150.5 |
| C-9 | 145.6 | 145.1 | 146.0 | 147.6 | 144.4 | 144.0 | 137.5 | 150.2 | 153.3 | 157.9 | 159.1 | 158.6 | 146.8 |
| C-10 | 105.1 | 111.3 | 111.3 | 108.0 | 110.2 | 110.2 | 121.2 | 111.9 | 103.3 | 102.4 | 104.6 | 103.1 | 142.0 |
| C-10a | 129.6 | 126.5 | 127.2 | 126.6 | 128.8 | 129.2 | 132.5 | 123.7 | 120.3 | 130.6 | 131.4 | 134.3 | 127.1 |
| C-10b | 40.2 | 39.4 | 41.1 | 41.5 | 41.6 | 38.0 | 38.2 | 42.1 | 117.0 | 136.0 | 126.1 | 127.6 | 135.5 |
| C-11 | 28.1 | 28.3 | 28.8 | 28.5 | 27.3 | 28.4 | 28.6 | 26.2 | 27.5 | 27.4 | 27.8 | 29.4 | 26.8 |
| C-12 | 53.3 | 53.9 | 53.9 | 53.8 | 52.1 | 52.4 | 52.3 | 67.5 | 46.8 | 55.4 | 57.0 | 58.0 | 57.4 |
| OCH2O | 100.6 | — | — | — | — | — | — | — | — | — | — | 106.4 | — |
| OMe | — | 56.1 | 56.0 | 57.3 | 55.9 | 56.0 | 55.9 | 56.2 | 56.3 | 56.7 | 57.8 | — | 62.5 |
| OMe | — | — | — | 56.0 | — | — | — | — | 56.2 | 56.4 | 57.4 | — | 62.4 |
| OMe | — | — | — | 55.9 | — | — | — | — | — | — | 57.0 | — | 56.8 |
| OOCMe | — | — | 170.9 | — | — | 170.6 | 170.6 | — | — | — | — | — | — |
| OOCMe | — | — | 21.3 | — | — | 21.4 | 20.6 | — | — | — | — | — | — |
| OOCMe | — | — | — | — | — | — | 169.2 | — | — | — | — | — | — |
| OOCMe | — | — | — | — | — | — | 21.3 | — | — | — | — | — | — |
| Solvent | a | b | b | d | d | d | d | c | b | b | c | c | d |
| MHz | 75 | 50 | 50 | 50 | 100 | 100 | 100 | 50 | 50 | 50 | 50 | 50 | 62 |
| Reference | (298) | (113) | (113) | (135) | (167) | (167) | (167) | (316) | (114) | (71) | (68) | (319) | (67) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
Table XIX.
13C NMR Data of Homolycorine-Type Alkaloids
| Alkaloid | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 | 40 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C-1 | 77.7 | 79.2 | 77.5 | 77.9 | 77.3 | 75.6 | 82.5 | 79.7 | 82.2 | 67.6 | 66.6 | 66.7 | 66.4 | 72.8 | 78.8 |
| C-2 | 31.3 | 32.1 | 30.9 | 31.3 | 31.1 | 31.2 | 66.2 | 69.2 | 66.2 | 32.6 | 31.3 | 31.7 | 31.3 | 68.3 | 31.4 |
| C-3 | 115.2 | 117.1 | 116.5 | 116.1 | 115.6 | 115.1 | 118.5 | 115.1 | 119.4 | 117.2 | 115.7 | 115.7 | 115.6 | 119.4 | 121.0 |
| C-4 | 140.9 | 141.3 | 138.7 | 140.0 | 140.2 | 139.4 | 143.3 | 147.4 | 142.4 | 140.8 | 140.7 | 140.6 | 140.3 | 144.0 | 135.6 |
| C-4a | 66.6 | 68.0 | 66.8 | 66.9 | 66.8 | 59.1 | 66.4 | 67.5 | 67.1 | 68.5 | 67.3 | 67.5 | 67.5 | 68.3 | 79.3 |
| C-6 | 165.9 | 167.9 | 166.3 | 167.0 | 165.4 | 165.1 | 165.7 | 165.4 | 165.0 | 92.3 | 98.2 | 91.8 | 98.2 | 98.5 | 167.1 |
| C-6a | 116.9 | 117.9 | 116.5 | 115.6 | 118.5 | 118.0 | 118.5 | 115.4 | 118.0 | 131.3 | 130.5 | 132.0 | 131.8 | 126.9 | 117.6 |
| C-7 | 111.9 | 116.9 | 115.9 | 112.7 | 109.7 | 109.9 | 112.0 | 115.0 | 109.8 | 111.9 | 109.7 | 107.4 | 107.3 | 107.6 | 117.1 |
| C-8 | 148.9 | 147.9 | 146.3 | 148.1 | 151.8 | 152.2 | 147.6 | 148.2 | 148.0 | 149.6 | 148.3 | 147.0 | 146.8 | 147.1 | 147.9 |
| C-9 | 153.1 | 153.8 | 152.2 | 152.0 | 147.8 | 147.8 | 151.8 | 152.0 | 151.9 | 149.6 | 148.3 | 147.0 | 146.7 | 147.1 | 153.4 |
| C-10 | 110.8 | 112.3 | 110.9 | 115.0 | 108.6 | 107.4 | 114.7 | 112.7 | 108.5 | 114.0 | 112.4 | 109.8 | 109.5 | 109.8 | 112.6 |
| C-10a | 137.8 | 137.6 | 135.7 | 138.0 | 139.7 | 138.7 | 137.2 | 136.7 | 138.8 | 128.6 | 125.6 | 128.2 | 126.9 | 131.2 | 134.9 |
| C-10b | 43.8 | 43.7 | 42.2 | 43.0 | 43.8 | 43.0 | 38.2 | 39.9 | 38.4 | 44.7 | 44.0 | 44.2 | 43.9 | 39.2 | 37.4 |
| C-11 | 28.1 | 28.6 | 27.4 | 27.9 | 27.9 | 29.6 | 27.0 | 28.0 | 27.3 | 28.7 | 27.8 | 28.1 | 27.9 | 27.7 | 26.2 |
| C-12 | 56.6 | 57.3 | 56.0 | 56.5 | 56.3 | 44.1 | 55.8 | 56.3 | 55.9 | 57.6 | 56.6 | 56.7 | 56.5 | 56.3 | 70.7 |
| OCH2O | — | — | — | — | 102.0 | 102.0 | — | — | 102.1 | — | — | 101.0 | 100.8 | 101.1 | — |
| OMe | 56.4 | 56.7 | 56.0 | 56.2 | — | — | 55.2 | 56.2 | — | 56.5 | 55.8 | — | 55.2 | 55.5 | 56.8 |
| OMe | 56.2 | — | — | — | — | — | — | — | — | 56.5 | 55.5 | — | — | — | — |
| OMe | — | — | — | — | — | — | — | — | — | — | 55.1 | — | — | — | — |
| NMe | 44.2 | 44.1 | 42.9 | 43.3 | 43.5 | — | 42.2 | 43.3 | 42.9 | 44.3 | 43.8 | 44.3 | 44.0 | 43.8 | 56.2 |
| OOCMe | — | — | 174.9 | — | — | — | — | 169.4 | — | — | — | — | — | — | — |
| OOCMe | — | — | 21.0 | — | — | — | — | 21.1 | — | — | — | — | — | — | — |
| OOCCH2CHOHMe | — | — | — | — | — | — | — | 170.8 | — | — | — | — | — | — | — |
| OOCCH2CHOHMe | — | — | — | — | — | — | — | 40.8 | — | — | — | — | — | — | — |
| OOCCH2CHOHMe | — | — | — | — | — | — | — | 66.7 | — | — | — | — | — | — | — |
| OOCCH2CHOHMe | — | — | — | — | — | — | — | 19.9 | — | — | — | — | — | — | — |
| Solvent | d | c | b | b | d | d | b | b | d | c | d | d | d | d | b |
| MHz | 63 | 50 | 62 | 50 | 100 | 100 | 50 | 50 | 50 | 62 | 50 | 100 | 100 | 50 | 50 |
| Reference | (321) | (324) | (70) | (82) | (165) | (165) | (138) | (72) | (71) | (106) | (104) | (165) | (165) | (71) | (137) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
Table XX.
13C NMR Data of Hemanthamine-Type Alkaloids
| Alkaloid | 42 | 43 | 44 | 45 | 47 | 48 | 49 | 50 | 53 | 54 | 55 | 56 | 57 | 59 | 61 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C-1 | 132.1 | 133.2 | 132.3 | 130.3 | 128.2 | 132.4 | 132.0 | 132.4 | 128.0 | 127.5 | 126.7 | 126.3 | 123.6 | 25.4 | 131.4 |
| C-2 | 127.4 | 127.6 | 128.6 | 127.6 | 127.3 | 125.6 | 125.9 | 125.2 | 127.2 | 129.5 | 132.3 | 132.8 | 136.0 | 26.4 | 129.1 |
| C-3 | 64.0 | 64.6 | 64.5 | 63.3 | 70.7 | 72.4 | 72.4 | 72.4 | 73.0 | 72.4 | 72.5 | 72.5 | 76.0 | 69.9 | 72.5 |
| C-4 | 32.7 | 32.8 | 33.5 | 31.6 | 25.6 | 28.7 | 28.1 | 28.5 | 29.5 | 28.2 | 27.8 | 27.6 | 30.1 | 30.3 | 26.7 |
| C-4a | 62.3 | 64.0 | 64.1 | 62.8 | 64.8 | 62.2 | 56.3 | 57.1 | 62.7 | 62.8 | 61.6 | 56.2 | 66.1 | 66.0 | 53.4 |
| C-6 | 62.8 | 63.7 | 62.6 | 60.9 | 57.1 | 87.0 | 88.9 | 96.4 | 63.3 | 61.0 | 85.8 | 88.4 | 63.4 | 59.1 | 57.6 |
| C-6a | 126.3 | 126.3 | 125.8 | 121.1 | 116.5 | 127.4 | 126.5 | 125.5 | 126.9 | 126.2 | 129.2 | 127.8 | 126.5 | 118.4 | 131.8 |
| C-7 | 106.9 | 107.9 | 111.9 | 109.8 | 110.0 | 111.0 | 112.5 | 112.4 | 106.9 | 106.6 | 108.2 | 109.5 | 106.9 | 109.0 | 105.7 |
| C-8 | 145.6 | 147.9 | 149.2 | 144.8 | 148.9 | 147.8 | 147.8 | 147.3 | 146.5 | 146.5 | 146.5 | 146.4 | 146.2 | 146.3 | 146.2 |
| C-9 | 146.0 | 148.4 | 149.1 | 146.0 | 149.0 | 148.5 | 148.7 | 148.2 | 147.0 | 146.5 | 147.4 | 147.7 | 146.5 | 145.4 | 145.5 |
| C-10 | 102.7 | 104.4 | 107.8 | 109.2 | 106.2 | 105.4 | 105.4 | 105.0 | 103.3 | 103.4 | 102.7 | 102.9 | 103.2 | 109.6 | 105.4 |
| C-10a | 138.3 | 136.9 | 138.7 | 136.5 | 138.0 | 136.4 | 137.4 | 138.1 | 135.0 | 134.2 | 134.7 | 135.8 | 135.3 | 137.1 | 129.1 |
| C-10b | 44.2 | 51.4 | 45.4 | 44.0 | 44.9 | 44.7 | 44.1 | 43.7 | 50.0 | 49.2 | 50.7 | 50.3 | 50.3 | 42.4 | 36.9 |
| C-11 | 44.2 | 80.8 | 45.0 | 42.8 | 40.6 | 42.3 | 40.9 | 41.1 | 80.0 | 80.2 | 79.2 | 78.3 | 80.0 | 35.5 | 75.3 |
| C-12 | 53.5 | 61.5 | 54.0 | 52.8 | 52.3 | 42.0 | 47.9 | 48.4 | 61.5 | 60.4 | 52.0 | 57.8 | 61.2 | 50.8 | 81.0 |
| OCH2O | 100.6 | 102.4 | — | — | — | — | — | — | 101.0 | 100.8 | 101.0 | 101.0 | 100.9 | — | 100.8 |
| OMe | — | — | 56.8 | 55.8 | 56.1 | 56.6 | 56.6 | 56.8 | 56.0 | 56.5 | 56.8 | 56.5 | 55.8 | 55.8 | 56.5 |
| OMe | — | — | 56.5 | — | 56.1 | 56.6 | 56.6 | 56.0 | — | — | — | — | — | — | — |
| OMe | — | — | — | — | 56.1 | 56.1 | 56.0 | 55.8 | — | — | — | — | — | — | — |
| OMe | — | — | — | — | — | — | — | 55.7 | — | — | — | — | — | — | — |
| OOCMe | — | — | — | — | — | — | — | — | — | 170.0 | — | — | — | 170.5 | — |
| OOCMe | — | — | — | — | — | — | — | — | — | 21.2 | — | — | — | 21.0 | — |
| CH2OOCMe | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 64.6 |
| CH2OOCMe | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 170.8 |
| CH2OOCMe | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 20.9 |
| Solvent | d | c | c | b | d | d | d | d | d | d | d | d | d | d | d |
| MHz | 20 | 67.5 | 75 | 50 | 50 | 50 | 50 | 50 | 20 | 50 | 125 | 125 | 75 | 50 | 50 |
| Reference | (330) | (376) | (336) | (108) | (137) | (135) | (135) | (137) | (330) | (74) | (73) | (73) | (298) | (140) | (74) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
Table XXI.
13C NMR Data of Tazettine-Type Alkaloids
| Alkaloid | 62 | 63 | 65 | 67 |
|---|---|---|---|---|
| C-1 | 130.5 | 130.1 | 131.3 | 132.4 |
| C-2 | 128.4 | 128.9 | 126.0 | 136.5 |
| C-3 | 72.4 | 72.1 | 72.7 | 63.6 |
| C-4 | 26.6 | 25.4 | 29.8 | 34.4 |
| C-4a | 69.9 | 68.2 | 63.3 | 68.5 |
| C-6 | 65.0 | 62.6 | 168.5 | 62.2 |
| C-6a | 127.8 | 126.2 | 118.6 | 131.0 |
| C-7 | 103.7 | 108.5 | 103.8 | 107.3 |
| C-8 | 146.3 | 146.6 | 147.1 | 148.4 |
| C-9 | 146.3 | 146.2 | 152.3 | 147.3 |
| C-10 | 109.1 | 104.2 | 111.0 | 111.0 |
| C-10a | 125.4 | 130.9 | 142.2 | 125.0 |
| C-10b | 50.1 | 50.0 | 46.2 | 50.3 |
| C-11 | 101.7 | 102.6 | 80.1 | 82.7 |
| C-12 | 61.7 | 64.5 | 53.5 | 55.7 |
| OCH2O | 100.6 | 100.9 | 102.1 | 101.9 |
| OMe | 55.6 | 56.7 | 56.2 | — |
| NMe | 41.9 | 40.6 | 42.8 | — |
| Solvent | d | d | d | c |
| MHz | 16 | 75 | 50 | 62 |
| Ref. | (346) | (355) | (79) | (76) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c; CD3OD, d: CDCl3.
Table XXII.
13C NMR Data of Narciclasine-Type Alkaloids
| Alkaloid | 68 | 70 | 71 | 72 |
|---|---|---|---|---|
| C-1 | 124.7 | 122.0 | 120.2 | 129.9 |
| C-2 | 69.1 | 126.7 | 125.9 | 118.0 |
| C-3 | 72.3 | 128.1 | 131.0 | 129.1 |
| C-4 | 68.8 | 129.9 | 133.0 | 110.7 |
| C-4a | 52.8 | 143.8 | 136.5 | 146.7 |
| C-6 | 172.1 | 151.8 | 152.8 | 63.5 |
| C-6a | 129.2 | 123.1 | 126.6 | 134.0 |
| C-7 | 168.9 | 105.5 | 108.3 | 109.7 |
| C-8 | 152.3 | 148.1 | 159.2 | 147.5 |
| C-9 | 144.8 | 148.2 | 152.0 | 147.4 |
| C-10 | 95.7 | 99.9 | 102.2 | 110.2 |
| C-10a | 132.1 | 130.3 | 122.0 | 131.2 |
| C-10b | 133.3 | 124.3 | 135.3 | 127.2 |
| OCH2O | 102.0 | 101.9 | 105.7 | 101.3 |
| NMe | — | — | 45.9 | 30.8 |
| Solvent | a | d | c | d |
| MHz | 68 | 50 | 50 | 50 |
| Reference | (122) | (99) | (79) | (79) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
Table XXIII.
13C NMR Data of Montanine-Type Alkaloids
| Alkaloid | 73 | 74 |
|---|---|---|
| C-1 | 115.6 | 114.7 |
| C-2 | 68.2 | 35.5 |
| C-3 | 70.4 | 72.3 |
| C-4 | 29.3 | 75.5 |
| C-4a | 58.5 | 70.0 |
| C-6 | 59.2 | 62.0 |
| C-6a | 122.2 | 125.2 |
| C-7 | 106.2 | 107.8 |
| C-8 | 146.8 | 148.3 |
| C-9 | 146.3 | 147.6 |
| C-10 | 106.8 | 108.3 |
| C-10a | 131.1 | 133.6 |
| C-11 | 44.7 | 46.4 |
| C-11a | 150.0 | 147.5 |
| C-12 | 54.5 | 56.9 |
| OCH2O | 100.7 | 102.1 |
| Solvent | c | c |
| MHz | 50 | 50 |
| Reference | (80) | (80) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
Table XXIV.
13C NMR Data of Galanthamine-Type Alkaloids
| Alkaloid | 75 | 77 | 78 | 79 | 80 | 81 | 82 | 83 | 84 | 86 |
|---|---|---|---|---|---|---|---|---|---|---|
| C-1 | 88.1 | 86.2 | 88.2 | 88.4 | 87.9/88.7 | 88.8 | 88.7 | 88.0 | 89.8 | 90.0 |
| C-2 | 30.0 | 27.7 | 30.3 | 29.9 | 29.8 | 31.3 | 29.2 | 37.3 | 31.5 | 31.7 |
| C-3 | 62.2 | 63.2 | 61.7 | 61.9 | 61.4 | 62.6 | 61.4 | 194.4 | 65.2 | 65.5 |
| C-4 | 126.8 | 123.3 | 129.2 | 127.6 | 128.0/128.2 | 128.6 | 128.2 | 127.1 | 27.6 | 27.8 |
| C-4a | 126.0 | 121.8 | 125.9 | 127.1 | 125.8/126.2 | 128.1 | 125.1 | 144.3 | 31.7 | 37.5 |
| C-6 | 60.5 | 59.9 | 51.6 | 53.5 | 41.0/52.8 | 61.6 | 58.4 | 60.7 | 60.4 | 53.8 |
| C-6a | 129.5 | 127.2 | 123.6 | 132.1 | 127.4 | 128.0 | 127.1 | 129.4 | 129.1 | 127.2 |
| C-7 | 121.6 | 130.2 | 122.8 | 120.9 | 119.9/121.6 | 123.1 | 121.2 | 122.0 | 121.6 | 120.8 |
| C-8 | 110.5 | 111.7 | 112.2 | 111.3 | 111.4 | 116.7 | 111.3 | 111.8 | 111.3 | 110.9 |
| C-9 | 145.5 | 146.7 | 146.9 | 146.2 | 146.2/146.5 | 146.8 | 146.8 | 147.0 | 146.2 | 146.6 |
| C-10 | 144.0 | 144.4 | 145.3 | 144.1 | 144.3/144.5 | 142.6 | 144.5 | 144.0 | 144.0 | 144.2 |
| C-10a | 132.7 | 131.9 | 133.1 | 133.1 | 131.8/131.9 | 134.2 | 131.2 | 130.5 | 136.3 | 136.6 |
| C-10b | 48.2 | 47.8 | 48.2 | 48.5 | 48.0/48.1 | 47.9 | 48.1 | 49.0 | 46.7 | 47.4 |
| C-11 | 34.0 | 33.7 | 35.6 | 39.7 | 46.6/46.7 | 35.5 | 35.1 | 33.2 | 23.9 | 24.2 |
| C-12 | 54.3 | 53.4 | 45.9 | 46.8 | 35.7/39.1 | 55.2 | 35.2 | 54.1 | 54.1 | 47.4 |
| OMe | 55.5 | 56.0 | 56.2 | 55.9 | 55.8 | — | 55.6 | 56.0 | 55.9 | 56.1 |
| NMe | 42.2 | 40.9 | — | — | — | 43.1 | — | 42.4 | 41.9 | — |
| NCHO | — | — | — | — | 162.1/162.7 | — | — | — | — | — |
| OCMe | — | 170.9 | — | — | — | — | 161.2 | — | — | — |
| OCMe | — | 21.4 | — | — | — | — | 21.4 | — | — | — |
| Solvent | d | d | b | d | b | c | d | d | d | d |
| MHz | — | 100 | 50 | 50 | 50 | 75 | 50 | 90 | 25 | 50 |
| Reference | (362) | (167) | (141) | (107) | (83) | (377) | (117) | (181) | (367) | (107) |
Solvent a: DMSO-d6, b: CDCl3-CD3OD, c: CD3OD, d: CDCl3.
The 13C NMR spectra of Narcissus alkaloids can be divided in two regions. The low-field region (>90 ppm) contains signals of the carbonyl group, the olefinic and aromatic carbons, as well as that of the methylenedioxy group. The other signals corresponding to the saturated carbon resonances are found in the high-field region, the N-methyl being the only characteristic group, easily recognizable by a quartet signal between 40 and 46 ppm.
The effect of a substituent (OH, OMe, OAc) on the carbon resonances is of considerable importance in localizing the position of the functional groups.
The analysis of the spectra allows conclusions to be drawn about the following aspects:
-
•
The number of methine olefinic carbons.
-
•
The presence and nature of the nitrogen substituent.
-
•
The existence of a lactonic carbonyl group.
-
•
The presence of a quaternary carbon signal assignable to C-10b in the chemical shift range of 42–50 ppm.
C. Mass Spectrometry
Extensive studies on the mass spectrometry of Amaryllidaceae alkaloids by electron impact were reported in the 1960s and 1970s 94., 310., 328., 353., 378., 379., 380., 381., 382., 383., 384., 385.. The fragmentation patterns in the electron impact mass spectrometry (EIMS) of various skeletal types are fairly well documented and have considerable diagnostic value.
1. Lycorine Type
The molecular ion appears as a quite intense peak, and generally suffers the loss of water, as well as C-1 and C-2 and their substituents, by a retro-Diels–Alder fragmentation (Fig. 12 ). The loss of water is not present in the spectra of acetyl derivatives.
Figure 12.
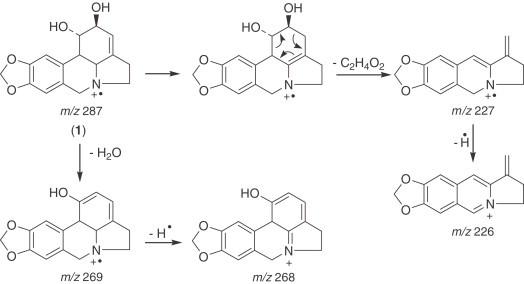
Mass fragmentation pattern of lycorine (1).
The ease of the loss of water from the molecular ion was found to be greatly dependent on the stereochemistry of the C-2 hydroxyl group. Thus, in the mass spectrum of lycorine (1) the relative intensity is low, while in 2-epilycorine it is the base peak (310).
2. Homolycorine Type
In this type of structure, the cleavage of the labile bonds in ring C by a retro-Diels–Alder reaction is dominant, generating two fragments: one, the most characteristic, represents the pyrrolidine ring (together with the substituents in position 2), and the other (a less-abundant fragment) encompasses the aromatic lactone or hemilactone moiety (Fig. 13 ). A further general and noteworthy feature is the low abundance of the molecular ion in all alkaloids with a double bond Δ3,4 (378).
Figure 13.
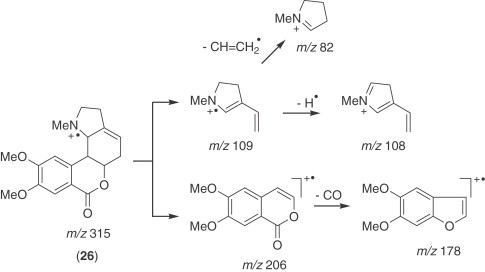
Mass fragmentation pattern of homolycorine (26).
3. Hemanthamine Type
The following observations about this type of alkaloids should be considered:
-
(i)
In most cases, the molecular ion is the base peak.
-
(ii)
The aromatic ring plays an important role in the stabilization of the ions, which is retained in all fragments of high mass, while the nitrogen atom is often lost.
-
(iii)
The fragmentation mechanisms are initiated by the rupture of a bond β to the nitrogen atom, which implies the opening of the C-11/C-12 bridge 382., 383..
-
a.
Alkaloids with a saturated ring C and no bridge substituent. The configuration of the ring C substituent plays a minor role in the fragmentation process.
-
b.
Alkaloids with a double bond (Δ 1,2 ) in ring C and no bridge substituent. The fragmentation pattern involves ruptures of C-4a/C-10b and C-3/C-4 bonds. A characteristic feature is the loss of a nitrogen-containing moiety, C3H5N [M+-55].
-
c.Alkaloids with a double bond (Δ 1,2 ) in ring C and a hydroxyl substituent at C-11. The presence of a hydroxyl group on C-11 is responsible for dramatic changes in the fragmentation pattern (Fig. 14 ), and it is profoundly influenced by the stereochemistry. There are three fundamental patterns of fragmentation:
-
•Loss of CH3OH: it is more favorable when the two-carbon bridge and the substituent on C-3 are on the same side of the molecule.
-
•Loss of C2H6N: the relative significance of the loss of this neutral nitrogen moiety is governed by the ease with which the methanol is eliminated.
-
•Loss of CHO: A peak at m/z [M+-29] due to the loss of an aldehyde radical is present in all alkaloids of this type.
-
•
Figure 14.
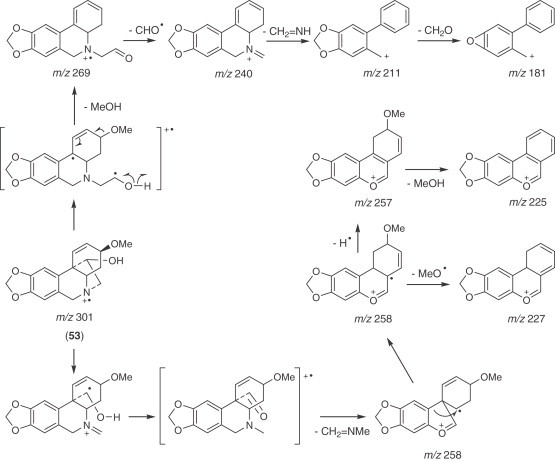
Mass fragmentation pattern of hemanthamine (53).
4. Tazettine Type
Minor changes in stereochemistry are sufficient to cause appreciable differences in the stereoisomers in this kind of structure. Thus, in the MS of tazettine (62), with a β-configuration of the methoxyl group at C-3, the dominant ion occurs at m/z [M+-84], following a C-ring fragmentation by a retro-Diels–Alder process. In contrast, the mass spectrum of its epimer criwelline (63) contains a peak of low abundance at m/z [M+-84] (Fig. 15 ). Ions occur in both stereoisomers owing to the successive loss of a methyl radical and water from the molecular ion (353).
Figure 15.
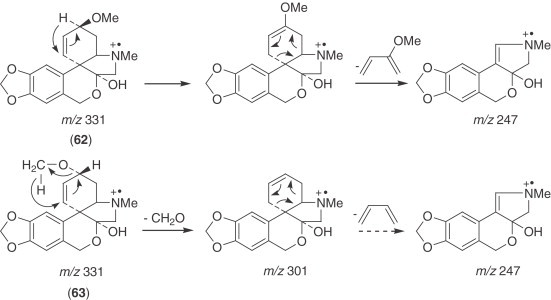
Mass fragmentation pattern of tazettine (62) and criwelline (63).
5. Montanine Type
The mass spectral fragmentation patterns observed for alkaloids containing the 5,11-methanomorphanthridine nucleus greatly depend on the nature and particular configuration of the substituents at C-2 and C-3. Thus, all the alkaloids that possess a methoxyl group give rise to an M+-31 ion.
The configuration of the C-2 substituent has a considerable effect on the extent to which the retro-Diels–Alder fragmentation ion is observed (Fig. 16 ). There is a definite enhancement of this fragmentation when the C-2 has an α-configuration (94).
Figure 16.
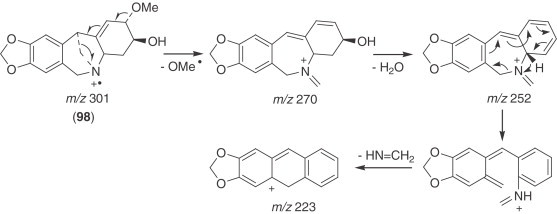
Mass fragmentation pattern of montanine (98).
6. Galanthamine Type
In this type of structure, the intense molecular ion as well as the [M+-1] peak, the breaking of ring C (losing a C4H6O fragment), and the elimination of elements of ring B (including the nitrogen atom) are characteristic (Fig. 17 ). This behavior is similar for the dihydro derivatives (380).
Figure 17.
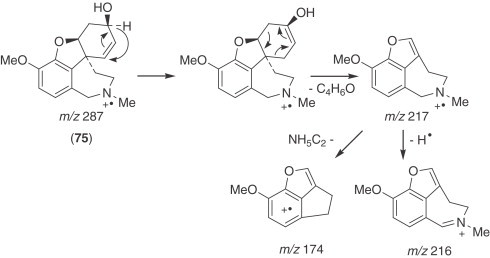
Mass fragmentation pattern of galanthamine (75).
VI. Biological and Pharmacological Activities
A. Traditional Uses
1. Traditional Medicinal Usage
Considering that the Narcissus species are a rich source of alkaloids, it is not surprising that, despite their lethal potential, plants of this genus have been used throughout history in traditional medicine to treat a variety of medicinal problems (386). N. poeticus, for example, is described in the Bible as a well-established treatment for symptoms that would now be defined as cancer (387). In the fourth century bc, Hippocrates of Cos (the ‘Father of Medicine’) recommended a pessary prepared from Narcissus oil (probably N. poeticus) for the management of uterine tumors (388). In the first century ad, Pliny the Elder also recorded the topical use of N. poeticus and N. pseudonarcissus for this purpose. It is now known that N. poeticus contains 0.012% of the antineoplastic agent narciclasine (68) in the fresh bulb 14., 101.. Arabian, North African, and Chinese medical practitioners of the Middle Ages continued using Narcissus oil in cancer treatment (389). For example, bulbs of N. tazetta L. var. chinensis, cultivated in China as a decorative plant, were used topically for the treatment of tumors in folk medicine. In this case, pretazettine (64) was established to be one of the antitumor active compounds 133., 390.. The bulbs of N. tazetta continue to be used in Turkey as a home remedy for the treatment of abscesses because of their antiphlogistic and analgesic properties (391).
Narcissus species have also been used as applications for wounds, hard imposthumes, strained sinews, stiff or painful joints, and other local ailments, being the basis of an ancient ointment called ‘Narcissimum’(14). The powdered flowers have been used as an emetic, and, in the form of syrup or infusions, have been considered useful for relieving the congestive bronchial catarrh in children, and also epidemic dysentery (392). In France, narcissus flowers have been used as an antispasmodic (14). The Arabians commended the oil as a cure for baldness and as an aphrodisiac (393). In John K’Eogh's Irish Herbal, the roots pounded with honey were recommended to treat burns, bruised sinews, dislocations and old aches, remove freckles, heal abscesses and sores, and draw out thorns and splinters 14., 394.. The bulbs of N. tazetta have also been used as a contraceptive. The influence of the daffodil on the nervous system has led to the use of its flowers and bulbs for hysterical affections and even epilepsy. A homoeopathic medicine is also made from the bulbs and used for respiratory disease, particularly bronchitis and whooping cough (14).
2. Toxic and Hallucinogenic Effects
Plants of this genus have been used throughout history as a stimulant to induce trance and hallucinations, and as an agent in suicide. Socrates called the narcissus the ‘Chaplet of the infernal Gods’, because of its narcotic effects. Pliny, in turn, describes the narcissus as ‘narce narcissum dictum, non a fabuloso puero’, which translated means ‘named narcissus from narce, not from the fabulous boy’. The Greek narkao, meaning to be numb, originates in the narcotic properties of the plant (14).
It has been known for a long time that daffodil ingestion is very dangerous, resulting in toxic symptoms in both man and warm-blooded animals such as cattle, goats, cats, and pigs 393., 395., 396.. After ingestion of Narcissus species such as N. pseudonarcissus or N. jonquilla (115), the first visible symptoms are salivation, acute abdominal pains, nausea, vomiting, and diarrhea, followed by neurological (trembling, convulsions, paralysis, etc.) and cardiac sequels, and sometimes resulting in death, if eaten in larger quantities. There have been many cases of poisoning or death when the bulbs have been cooked by mistake in the place of leeks or onions (393). Recovery, however, is usually complete within a few hours without any treatment being necessary 14., 397., but in cases of massive ingestion, activated charcoal, salts, and laxatives are administered. When symptoms are severe, atropine sulphate is given by intravenous injection and it may be necessary to induce vomiting or remove stomach contents (398).
The good news is that the bulb tastes awful, making it highly unlikely that anyone could even keep down one bite. In an experiment performed with several plant species that are not consumed by animals, the plant with the most repellent activity was the daffodil, specifically N. pseudonarcissus (399). As a consequence, an animal repellent containing alkaloids isolated from members of the genus Narcissus has been designed to repel animals from vegetation by rendering it unpalatable, being also effective against fungi, molds, and bacteria 400., 401..
Not all Narcissus species are equally dangerous. The bulbs of N. poeticus, for example, are more dangerous than those of N. pseudonarcissus. Neither do all plant tissues have the same concentration or profile of alkaloids. Thus, the alkaloid content of N. papyraceus is five times higher in the aerial part than in the bulbs, being toxic for herbivorous mammals (137). The distribution of the alkaloids in the plant tissues can be related with the plant defense mechanism.
Some Narcissus species, such as N. pseudonarcissus, can produce harmful effects without being swallowed. Thus, those who pick and pack the flowers are liable to develop dermatitis, probably due to the irritant effects of the sap or an allergic reaction 397., 402., 403., 404., 405., 406.. The compounds responsible for the irritation are not known, but alkaloids are thought to be involved (110). When extracts of the bulbs are applied to open wounds they can produce staggering, numbness of the whole nervous system, and paralysis of the heart (393). Furthermore, the scent of flowers of species such as N. bulbocodium can produce headaches and even vomiting if they are placed in confined spaces. Indeed, many people refuse to have daffodils in their house, considering them to be unlucky for the way they hang their heads, which suggests tears and unhappiness (14).
The mucilage secreted by bulbs can also produce harmful effects in plant species such as rose, rice, and cabbage, inhibiting seed germination and seedling growth 120., 407..
3. Other Uses
The olfactory qualities of the Narcissus flower have made it a valuable component of luxury perfumes since time immemorial, although the main components of the volatile part of narcissus absolute are not of alkaloidal origin. However, alkaloids are present, together with essential oils, in some Narcissus-derived perfumes, such as jonquil absolute 408., 409., 410..
B. Biological Activities of Plant Extracts
Several Narcissus extracts have shown the following activities: antiviral 390., 411., 412., 413., 414., 415., 416., 417., prophage induction (418), antibacterial 418., 419., 420., antifungal 419., 421., 422., antimalarial 419., 423., insecticidal (419), cytotoxic 390., 411., 419., 424., antitumor 390., 395., 412., 413., 415., 425., antimitotic (426), antiplatelet (419), hypotensive (427), emetic (395), acetylcholine esterase inhibitory (93), antifertility (428), antinociceptive (391), chronotropic (427), pheromone (429), plant growth inhibitor, and allelopathic 120., 143., 407., 427..
C. Biological and Pharmacological Activities of Alkaloids
The alkaloids from the genus Narcissus are the compounds responsible for the majority of the above-mentioned activities, although the mannosa-binding lectins have also received much interest recently 430., 431., 432., 433., 434., 435..
In spite of the great variety of pharmacological and/or biological properties exhibited by these alkaloids, only some of the activities of a reduced number have been reported, and the most extensively studied effect is that of non-specific inhibition. The relationship of chemical structure and biological activity is largely unknown, and further studies are needed to explore the full therapeutic potential of these alkaloids. The most-studied alkaloids in this group are galanthamine (75), lycorine (1), narciclasine (68), and pretazettine (64), which possess a diversity of pharmacological activities.
1. Lycorine Type
Lycorine (1), the most frequent and characteristic of the Amaryllidaceae alkaloids, has been reported to be a powerful inhibitor of ascorbic acid (l-Asc) biosynthesis 436., 437., and thus has proved to be a useful tool in studying Asc-dependent metabolic reactions in l-Asc-synthesizing organisms 438., 439.. Lycorine is actually a powerful inhibitor of the activity of l-galactono-γ-lactone dehydrogenase, the terminal enzyme of l-Asc biosynthesis 440., 441., 442., 443., which appears to be localized in the mitochondrial membrane 444., 445.. Galanthine (7) also has a high capacity to inhibit ascorbic acid biosynthesis (437).
It is well documented that lycorine (1) is a powerful inhibitor of cell growth, cell division, and organogenesis in higher plants, algae, and yeasts, inhibiting the cell cycle during interphase, which seems to be related with the l-Asc levels 438., 446., 447., 448., 449., 450.. In plants, it also inhibits cyanide-insensitive respiration, peroxidase activity, and protein synthesis 451., 452., 453.. The effects of lycorine on l-Asc biosynthesis have been reported to occur at concentrations below those at which protein synthesis is affected, but it seems difficult to completely rule out non-specific effects of this alkaloid since it has been reported that, at least in yeasts, lycorine is able to interact directly with mitocondrial DNA. Thus, differing sensitivity to the alkaloid among cells devoid of mitochondrial DNA (rho 0) and cells with mitochondrial DNA either rho + or rho − has been found in yeasts 440., 448., 454., 455., rho 0 cells being resistant to high concentrations of the drug 450., 456., 457., 458.. Some strains can even adapt to the presence of lycorine, because they are able to degrade the alkaloid and use its biotransformation products as growth-stimulating factors (458).
Lycorine-1-O-β-d-glucoside, on the other hand, promotes cell growth, seed germination, and the rate of development of root and root hairs in higher plants. The glucosyloxy derivatives of lycorine (1) and pseudolycorine (3) and their aglycones form stable complexes with phytosterols and with divalent metal ions, and are able to translocate them from the rhizosphere to the aerial part (347). Palmilycorine and some acylglucosyloxy conjugates of lycorine, in turn, are frequently encountered among the phytosterols exhibiting membrane-stabilizing action. Additionally, lycorine-1-O-β-d-glucoside and acylglucosyloxy conjugates of lycorine are used by plants for recognition to reject the vast majority of microorganisms and parasites (459).
In animals, lycorine (1) shows antitumor activity 460., 461., reported to inhibit the in vivo and in vitro growth of a variety of tumor cells, such as BL6 mouse melanoma, Lewis lung carcinoma, murine ascites, or HeLa cells 20., 298., 459., 462., 463., 464., 465.. It induces flat morphology in K-ras-NRK cells (transformed fibroblasts) (466), and reduces the cellular activity in femoral bone marrow tissue that results in granulocytic leucopenia and a decrease in the number of erythrocytes. This alkaloid's mechanism of action is thought to be through the inhibition of protein synthesis at the ribosomal level, even though the cytotoxic effects of calprotectin can also be suppressed using lycorine 424., 460., 461., 467., 468.. Lycorine also inhibits murine macrophage production of tumor necrosis factor alpha (TNF-α) (469), and shows inhibitory effects on nitric oxide production and induction of inducible nitric oxide synthase (NOS) in lipopolysaccharide-activated macrophages (470). The molecular mechanism of lycorine against leukemia (human cell line HL-60) shows that it can suppress cell growth and reduce cell survival by arresting the cell cycle at the G2/M phase and inducing apoptosis of tumor cells (471). It displays pronounced cell growth inhibitory activities against both parental and multidrug resistant L5178 mouse lymphoma cell lines, but is almost inactive in inhibiting the glycoprotein responsible for the efflux-pump activity of tumor cells. Assays for interactions with tRNA revealed that the antiproliferative effects of lycorine result from its complex formation with tRNA (73). Interaction of lycorine (1), pseudolycorine (3), and 2-O-acetylpseudolycorine (5) with DNA has also been observed 472., 473..
Some other alkaloids of this series, such as caranine (10), galanthine (7), pseudolycorine (3), and 2-O-acetylpseudolycorine (5), are also active against a variety of tumor cells 415., 463., 474.. Pseudolycorine inhibits the protein synthesis in tumor cells at the step of peptide bond formation, but it has a different binding site than lycorine 467., 475.. Ungeremine (24), a natural metabolite of lycorine (1), is responsible, at least in part, for the growth-inhibitory and cytotoxic effects of lycorine, being active against leukemia 476., 477.. Lycorine-1-O-β-d-glucoside, in turn, has the reverse effect of lycorine, and may produce mitogenic activity in animal cells (478).
Lycorine (1) and pseudolycorine (3) exert antiviral effects on several RNA- and DNA-containing viruses (479). Antiviral activity has been observed in tests with flaviviruses, and to a slightly lesser degree, bunyaviruses. Lycorine and pseudolycorine also show inhibitory activity against the Punta Toro and Rift Valley fever viruses, but with low selectivity 480., 481.. Lycorine, in turn, acts as an anti-SARS-CoV (Severe Acute Respiratory Syndrome-associated Coronavirus) and shows pronounced activity against poliomyelitis, coxsackie, and herpes type 1 viruses 20., 482.. It possesses high antiretroviral activity accompanied by low therapeutic indices (483). The relationship between its structure and the mechanism of activity has been studied in the Herpes simplex virus, suggesting that alkaloids that may eventually prove to be antiviral agents have a hexahydroindole ring with two functional hydroxyl groups (484). The activity was found to be due to the inhibition of multiplication, and not to the direct inactivation of extracellular viruses, and the mechanism of the antiviral effect was partially explained as a blocking of viral DNA polymerase activity 33., 413., 479., 485..
Lycorine (1) has appreciable inhibitory activity against acetylcholinesterase (486). Cholinesterase activity appears to be associated with the two free hydroxyl groups present in some of the alkaloids of this structural type (487). The higher cholinesterase activity of assoanine (20) and oxoassoanine (21) with respect to the other lycorine-type alkaloids could be explained by an aromatic ring C, which gives a certain planarity to those molecules (93). Another alkaloid, galanthine (7), exhibits powerful cholinergic activity and has therefore attracted much interest in the treatment of myasthenia gravis, myopathy, and diseases of the central nervous system (488). Caranine (10), pseudolycorine (3), ungiminorine (17), and in particular, ungeremine (24), also show an inhibitory effect on acetylcholinesterase 93., 173., 320..
Lycorine (1) is an analgesic, more so than aspirin, and a hypotensive 489., 490., as are caranine (10) and galanthine (7). The analgesic activity exhibited by the Amaryllidaceae alkaloids is attributed to their resemblance to the morphine and codeine skeletons. Lycorine also has antiarrhythmic action, and lycorine hydrochloride is a strong broncholytic (30). In fact, lycorine shows a relaxant effect on an isolated epinephrine-precontracted pulmonary artery and increases contractility and the rate of an isolated perfused heart. These effects are mediated by stimulation of β-adrenergic receptors (491).
Lycorine (1) also has a strong inhibitory effect on parasite (Encephalitozoon intestinalis) development (492) and antifungal activity against Candida albicans (326). Additionally, lycorine has antifeedant (493), antimalarial 423., 494., emetic (495), anti-inflammatory (496), antiplatelet (419), as well as antifertility (490) activities. Galanthine (7), in turn, shows mild in vitro activity against Tripanosoma brucei rhodesiense and Plasmodium falciparum (497).
2. Homolycorine Type
It is reported that some alkaloids of this series, such as homolycorine (26), 8-O-demethylhomolycorine (27), 9-O-demethyl-2α-hydroxyhomolycorine (32), dubiusine (33), hippeastrine (34), lycorenine (35), or O-methyllycorenine (36), present cytotoxic effects against non-tumoral fibroblastic LMTK cells (463), also being moderately active in inhibiting the in vivo and in vitro growth of a variety of tumor cells, such as Molt 4 lymphoma, HepG2 human hepatoma, LNCaP human prostate cancer, or HT 341., 463., 490.. Dubiusine, lycorenine, 8-O-demethylhomolycorine and 9-O-demethyl-2α-hydroxyhomolycorine also show DNA binding activity comparable to that of vinblastine (472). Homolycorine possesses high antiretroviral activity, accompanied by low therapeutic indices (483). Hippeastrine, in turn, displays antiviral activity against Herpes simplex type 1 (484).
Dubiusine (33), homolycorine (26), 8-O-demethylhomolycorine (27), and lycorenine (35) have a hypotensive effect on the arterial pressure of normotensive rats (498). Lycorenine also shows a vasodepressor action ascribed to the maintenance of its α-adrenergic blocking action, and produces bradycardia by modifying vagal activity (499). Another feature of lycorenine is its analgesic activity (20).
Homolycorine (26) and masonine (30) are other inductors of delayed hypersensitivity in animals (110). Hippeastrine (34), in turn, shows antifungal activity against C. albicans and it also possesses a weak insect antifeedant activity (326).
3. Hemanthamine Type
Hemanthamine (53), hemanthidine (55), crinamine (57), maritidine (44), and papyramine (48) display pronounced cell growth inhibitory activities against a variety of tumor cells, such as Rauscher viral leukemia, Molt 4 lymphoma, BL6 mouse melanoma, HepG2 human hepatoma, HeLa, LNCaP human prostate cancer, or HT 298., 336., 341., 424., 462., 463., 500.. Some of these alkaloids, namely crinamine, hemanthamine, and papyramine, also present a cytotoxic effect against non-tumoral fibroblastic LMTK cells (463). The mechanism of action of hemanthamine is thought to be through the inhibition of protein synthesis, blocking the peptide bond formation step on the peptidyl transferase center of the 60S ribosomal subunit 467., 475.. Hemanthamine and hemanthidine also display the same pronounced cell growth inhibitory activities against both parental and multidrug resistant L5178 mouse lymphoma cell lines as described above for lycorine (1) (73). Crinamine, in turn, shows inhibitory effects on nitric oxide (NO) production and induction of inducible nitric oxide synthase (NOS) in lipopolysaccharide-activated macrophages (470).
The antimalarial activity against strains of chloroquine-sensitive P. falciparum observed in hemanthamine (53) and hemanthidine (55) can be attributed to the methylenedioxybenzene part of the molecule and the tertiary nitrogen without methyl (423). Crinamine (57) also exhibits moderate antimalarial activity 494., 501.. Hemanthidine also works in vitro against Trypanosoma brucei rhodesiense, and to a lesser extent against Trypanosoma cruzi (497). Vittatine (42) has antibacterial activity against the Gram-positive Staphylococcus aureus and the Gram-negative E. coli (326), and the alkaloid crinamine shows strong activity against Bacillus subtilis and S. aureus (502).
Like lycorine (1), hemanthidine (55) has stronger analgesic and anti-inflammatory activity than aspirin 489., 496., and vittatine has been found to potentiate the analgesic effect of morphine (60). Moreover, some alkaloids of this series, such as hemanthamine (53) or papiramine (48) have a hypotensive effect 41., 498., and hemanthamine shows strong antiretroviral activity (483).
4. Tazettine Type
Tazettine (62) is mildly active against certain tumor cell lines 341., 424., 503., with a slight cytotoxicity when tested on fibroblastic LMTK cell lines (463). Tazettine also displays weak hypotensive and antimalarial activities and interacts with DNA 419., 472., 498.. Its chemically labile precursor, pretazettine (64), is far more interesting due to its antiviral and anticancer activities. In fact, when pretazettine is stereochemically rearranged to tazettine, the biological activity of the precursor is to a large extent reduced 504., 505..
Pretazettine (64) shows cytotoxicity against fibroblastic LMTK cell lines and inhibits HeLa cell growth, being therapeutically effective against advanced Rauscher leukemia, Ehrlich ascites carcinoma, spontaneous AKR lymphocytic leukemia, and Lewis lung carcinoma 412., 503., 506., 507., 508., 509., 510.. It is one of the most active of the Amaryllidaceae alkaloids against Molt4 lymphoid cells (463), and is used in combination with DNA-binding and alkylating agents in treating the Rauscher leukemia virus 412., 503.. In fact, pretazettine strongly inhibits the activity of reverse transcriptase from various oncogenic viruses by binding to the enzyme (20). It inhibits both the growth of the Rauscher virus and cellular protein synthesis in eukaryotic cells by a mechanism that does not affect DNA and RNA synthesis, even though it has a pronounced DNA-binding activity 415., 424., 467., 472., 481., 507., 511.. Pretazettine has also been shown to be active against selected RNA-containing flavoviruses (Japanese encephalitis, yellow fever, and dengue) and bunyaviruses (Punta Toro and Rift Valley fever) in organ culture (481). It also possesses pronounced activity against Herpes simplex type 1 virus (484). This activity may reflect a general ability to inhibit protein synthesis during viral replication (512).
5. Narciclasine Type
Narciclasine (68), an antimitotic and antitumoral alkaloid (143), affects cell division at the metaphase stage and inhibits protein synthesis in eukaryotic ribosomes by directly interacting with the 60S subunit and inhibiting peptide bond formation by preventing binding of the 3′ terminal end of the donor substrate to the peptidyl transferase center 467., 475., 513., 514., 515.. It also retards DNA synthesis (516) and inhibits calprotectin-induced cytotoxicity at a more than 10-fold lower concentration than lycorine (1) (468). The peculiar effects of narciclasine seem to arise from the functional groups and conformational freedom of its C-ring (122), with the 7-hydroxyl group believed to be important in its biological activity (253). This alkaloid, related to pancratistatin (516), is one of the most important antineoplastic Amaryllidaceae alkaloids (460) and shows some promise as an anticancer agent. It inhibits HeLa cell growth, has antileukemic properties and is active against a variety of tumor cells, such as human and murine lymphocytic leukemia, larynx, and cervix carcinomas, and Ehrlich tumor cells 33., 77., 386., 388., 389., 516.. No effect has been observed toward solid tumors. Narciclasine-4-O-β-d-glucopiranoside shows very similar cytotoxic and antitumoral activity to narciclasine (517).
Narciclasine (68) has a prophylactic effect on the adjuvant arthritis model in rats, significantly suppressing the degree of swelling of adjuvant-treated, as well as untreated, feet (468). This alkaloid is also active against Corynebacterium fascians, inhibits the pathogenic yeast Cryptococcus neoformans, and modifications, like 2,3,4,7-tetra-O-acetylnarciclasine inhibit, the growth of the pathogenic bacterium Neisseria gonorrhoeae (78). Antiviral activity has been observed against RNA-containing flaviviruses and bunyaviruses (481).
At the plant level, narciclasine (68) is a potent inhibitor, showing a broad range of effects, including the ability to inhibit seed germination and seedling growth of some plants in a dose-dependent manner, interacting with hormones in some physiological responses (518). Thus, indole-3-acetic acid cannot overcome the inhibition of elongation of wheat coleoptile sections caused by narciclasine. Additionally, narciclasine suppresses the gibberellin-induced α-amylase production in barley seeds and cytokinin-induced expansion and greening of excised radish cotyledons (120). Like lycorine (1), narciclasine also inhibits ascorbic acid biosynthesis (305). Narciclasine, present in daffodil mucilage, can delay tepal senescence in cut Iris flowers by attenuation of protease activity, which, in turn, is apparently related to the inhibition of the protein synthesis involved in senescence (519). At the organelle level, narciclasine inhibits both isocitrate lyase (ICL) activity in glyoxysomes and hydroxypyruvate reductase (HPR) activity in peroxysomes. It also blocks the formation of chloroplasts, markedly reducing the chlorophyll content of light-grown wheat seedlings, probably due to the inhibition of the formation of 5-aminolevulinic acid, an essential chlorophyll precursor (520). The formation of light-harvesting chlorophyll a/b binding protein (LHCP) is also inhibited by this alkaloid (521).
Some alkaloids of this series, such as trisphaeridine (70), possess high antiretroviral activities, accompanied by low therapeutic indices (483). Ismine (72), in turn, shows a significant hypotensive effect on the arterial pressure of normotensive rats (498) and is cytotoxic against Molt 4 lymphoid and LMTK fibroblastic cell lines (463).
6. Montanine Type
There is little information about the montanine-type alkaloids, only some data about pancracine (73), which shows antibacterial activity against S. aureus and Pseudomonas aeroginosa (326), as well as weak activity against Tripanosoma brucei rhodesiense, T. cruzi, and P. falciparum (80).
7. Galanthamine Type
Galanthamine (75), originally isolated from Galanthus nivalis L. in the 1940s, is a long-acting, selective, reversible and competitive inhibitor of acetylcholinesterase. This enzyme is responsible for the degradation of acetylcholine at the neuromuscular junction, in peripheral and central cholinergic synapses and in parasympathetic target organs 522., 523., 524.. Galanthamine has the ability to cross the blood–brain barrier and act within the central nervous system 525., 526.. It binds at the base of the active site gorge of acetylcholinesterase, interacting with both the choline-binding site and the acyl-binding pocket, having a number of moderate-to-weak interactions with the protein 527., 528., 529.. In addition, galanthamine stimulates pre- and postsynaptic nicotinic receptors which can, in turn, increase the release of neurotransmitters, thus directly stimulating neuronal function 524., 530.. It is also suggested that the stimulation of nicotinic receptors protects against apoptosis induced by β-amyloid toxicity 524., 531., 532.. Its dual mode of action (527), coupled with the evidence that galanthamine has reduced side effects, make it a promising candidate for the treatment of nervous diseases, paralysis syndrome, schizophrenia, and other forms of dementia, as well as Alzheimer's disease 524., 527., 528..
Galanthamine (75) has other noteworthy pharmacological actions, including an ability to amplify the nerve-muscle transfer (20), affecting membrane ionic processes (533). It is also known to cause bradycardia or atrioventricular conduction disturbances (41), has long been used as a reversal agent in anesthetic practice (181), inhibits traumatic shock, and has been patented for use in the treatment of nicotine dependence. Besides this, galanthamine acts as a mild analeptic, shows an analgesic power as strong as morphine, compensates for the effects of opiates on respiration, relieves jet lag, fatigue syndrome, male impotence, and alcohol dependence, and when applied in eye drops, reduces the intraocular pressure 20., 81., 85., 534.. It also acts as a hypotensive and has a weak antimalarial activity 419., 498..
At present, there is no preventative or curative treatment available for Alzheimer's disease, leaving the symptomatic relief offered by AChEI therapy as the only approved therapeutic option. Owing to the relative lack of alternative treatment, galanthamine (75) is a reasonable approximation of the ideal concept of symptomatic Alzheimer's disease therapy 523., 535.. Galanthamine hydrobromide (a third-generation cholinesterase inhibitor used against Alzheimer's disease) offers superior pharmacological profiles and increased tolerance compared to the original acetylcholinesterase inhibitors, physostigmine or tacrine 536., 537., 538., 539., 540.. Galanthamine is effective and well tolerated, resulting in short-term improvements in cognition, function, and daily life activities in patients with mild to moderate symptoms 530., 541., 542.. However, long-term benefits beyond 6 months are in question (543), because persistent elevation of acetylcholine may lead to over-stimulation of both nicotinic and muscarinic acetylcholine receptors, the former causing receptor desensitization and the latter potentially causing an increased frequency of cholinergic side effects 524., 530., 544.. The safety profile of galanthamine, as well as its clinical effectiveness, will only be demonstrated after large-scale clinical trials 544., 545., 546..
Broadly speaking, the development of galanthamine (75) into a widely used Alzheimer's drug can be divided into three main periods: (1) the early development in Eastern Europe for its use in the treatment of poliomyelitis; (2) the pre-clinical development in the 1980s; and (3) the clinical development in the 1990s (544). Galanthamine hydrobromide was first used by Bulgarian and Russian researchers in the 1950s and exploited for a variety of clinical purposes. It has been used clinically for postsurgery reversal of tubocurarine-induced muscle relaxation and for treating post-polio paralysis, myasthenia gravis, and other neuromuscular diseases, as well as traumatic brain injuries 547., 548.. As early as 1972, Soviet researchers demonstrated that galanthamine could reverse scopolamine-induced amnesia in mice, a finding that was demonstrated in man 4 years later. However, this alkaloid was not applied to Alzheimer's disease until 1986, long after the widely accepted cholinergic hypothesis had been first postulated, when researchers in Western Europe switched their attention to galanthamine because of its ability to penetrate the blood–brain barrier and specifically to augment the central cholinergic function 544., 549.. This led to clinical trials of galanthamine in the treatment of Alzheimer's disease. In 1996, Sanochemia Pharmazeutika in Austria first launched galanthamine as ‘NIVALIN®’, but its strictly limited availability meant the international pharmaceutical community adopted a cautions approach 181., 550., until Sanochemia Pharmazeutika developed a method to synthetically produce the compound in 1997 (551). Later, galanthamine was co-developed by Shire Pharmaceuticals (Great Britain) and the Janssen Research Foundation (Belgium), who have launched galanthamine as ‘REMINIL®’ in many countries 524., 544.. This renewed interest is reflected in the increasing number of scientific reviews concerned exclusively with galanthamine and its derivatives 522., 525., 526., 530., 538., 539., 547., 552., 553., 554., 555..
Sanguinine (81) has a more potent acetylcholinesterase inhibitory activity than galanthamine (75) due to an extra hydroxyl group available for potential interaction with acetylcholinesterase 93., 555.. Sanguinine, in turn, is 10-fold more selective than galanthamine for acetylcholinesterase (AChE) vs. butyrylcholinesterase (BuChE) (556). The lack of AChE inhibitory activity of lycoramine (84) and epinorlicoramine (86) could be due to the occurrence of a double bond in ring C, which does not allow these alkaloids to have the same spatial configuration as the active alkaloids of this series (93).
Narwedine (83), the biogenetic precursor of galanthamine (75), has been studied as a respiratory stimulator. It increases the amplitude and decreases the frequency of cardiac contractions and would therefore be of value in reducing blood loss during surgery (41). It also inhibits the action of narcotics and hypnotics, and increases the analgesic effect of morphine (60) as well as the pharmacological effects of caffeine, carbazole, arecoline, and nicotine (30).
8. Other Alkaloids
Cherylline (88) is a 4-arylisoquinoline derivative, a group with several potential medicinal properties (80), including a weak acetylcholinesterase inhibitory activity (486). Mesembrenone (90), in turn, is mildly active against Molt 4 lymphoid and non-tumoral fibroblastic LMTK cells (463), has a moderate hypotensive effect on arterial pressure, and interacts slightly with DNA 472., 498..
References
- 1.A. W. Meerow and D. A. Snijman, in “The Families and Genera of Vascular Plants” (K. Kubitzki, ed.), vol. III, p. 83. Springer, Berlin, 1998.
- 2.Ito M., Kawamoto A., Kita Y., Yukawa T., Kurita S. J. Plant Res. 1999;112:207. [Google Scholar]
- 3.G. R. Hanks, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks, ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 1. Taylor & Francis, London, 2002.
- 4.B. Mathew, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks, ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 30. Taylor & Francis, London, 2002.
- 5.Graham S.W., Barrett S.C.H. Am. J. Bot. 2004;91:1007. doi: 10.3732/ajb.91.7.1007. [DOI] [PubMed] [Google Scholar]
- 6.Dodson H.E.M., Arroyo J., Bergström G., Groth I. Biochem. Syst. Ecol. 1997;25:685. [Google Scholar]
- 7.Arroyo J., Barrett S.C.H., Hidalgo R., Cole W.W. Am. J. Bot. 2002;89:1242. doi: 10.3732/ajb.89.8.1242. [DOI] [PubMed] [Google Scholar]
- 8.Herrera C.M. Ecology. 1995;76:218. [Google Scholar]
- 9.A. Fernandes, Daffodil and Tulip Year Book (Royal Horticultural Society) 33, 37 (1968).
- 10.Fernandes A. An. Inst. Bot. A. J. Cavanilles. 1975;32:843. [Google Scholar]
- 11.Webb D.A. In: Tutin T.G., Heywood V.H., Burges N.A., Moore D.M., Valentine D.H., Walters S.M., Webb D.A., Chater A.O., Richardson B.K., editors. vol. 5. Cambridge University Press; Cambridge: 1980. p. 75. (Flora Europaea). [Google Scholar]
- 12.Barrett S.C.H., Harder L.D. New Phytol. 2005;165:45. doi: 10.1111/j.1469-8137.2004.01183.x. [DOI] [PubMed] [Google Scholar]
- 13.Pérez R., Vargas P., Arroyo J. New Phytol. 2003;161:235. [Google Scholar]
- 14.A. C. Dweck, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks, ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 19. Taylor & Francis, London, 2002.
- 15.G. R. Hanks, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks, ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 53. Taylor & Francis, London, 2002.
- 16.Blanchard J.W. Alpine Garden Society, Woking; Surrey: 1990. Narcissus: A Guide to Wild Daffodils. [Google Scholar]
- 17.A. R. Rees, The Garden (Royal Horticultural Society) 118, 404 (1993).
- 18.Kington S. Royal Horticultural Society; London: 1998. The International Daffodil Register and Classified List 1998. [Google Scholar]
- 19.Dictionary of Natural Products (Net Database). Chapman & Hall/CRC Press, London, 2005.
- 20.Ghosal S., Saini K.S., Razdan S. Phytochemistry. 1985;24:2141. [Google Scholar]
- 21.Wiggins I.L. Stanford University Press; Stanford: 1980. Flora of Baja California. [Google Scholar]
- 22.Bastida J., Sellés M., Codina C., Viladomat F., León de la Luz J.L. Planta Med. 1996;62:575. doi: 10.1055/s-2006-957979. [DOI] [PubMed] [Google Scholar]
- 23.Jeffs P.W. In: Manske R.H.F., Rodrigo R.G.A., editors. vol. 19. Academic Press; New York: 1981. p. 1. (The Alkaloids). [Google Scholar]
- 24.Smith M.T., Crouch N.R., Gericke N., Hirst M. J. Ethnopharmacol. 1996;50:119. doi: 10.1016/0378-8741(95)01342-3. [DOI] [PubMed] [Google Scholar]
- 25.Döpke W., Sewerin E., Trimiño Z. Z. Chem. 1980;20:298. [Google Scholar]
- 26.Bastida J., Viladomat F., Llabrés J.M., Ramirez G., Codina C., Rubiralta M. J. Nat. Prod. 1989;52:478. [Google Scholar]
- 27.J. A. Seijas, M. P. Vázquez-Tato, M. T. Linares, P. Ramil-Rego, and M. I. Buján, 8th International Electronic Conference on Synthetic Organic Chemistry (ECSOC-8), p. 625 (2004).
- 28.Kaya G.I., Unver N., Gözler B., Bastida J. Biochem. Syst. Ecol. 2004;32:1059. [Google Scholar]
- 29.Mulholland D.A., Crouch N., Decker B., Smith M.T. Biochem. Syst. Ecol. 2002;30:183. [Google Scholar]
- 30.O. A. Cherkasov and O. N. Tolkachev, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks, ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 242. Taylor & Francis, London, 2002.
- 31.Ghosal S., Singh S.K., Unnikrishnan S. Phytochemistry. 1990;29:805. [Google Scholar]
- 32.S. Ghosal, K. Datta, S. K. Singh, and Y. Kumar, J. Chem. Res.-S 334 (1990)
- 33.Hoshino O. In: Cordell G.A., editor. vol. 51. Academic Press; New York: 1998. p. 323. (The Alkaloids: Chemistry and Biology). [Google Scholar]
- 34.Jin Z. Nat. Prod. Rep. 2003;20:606. doi: 10.1039/b304144c. [DOI] [PubMed] [Google Scholar]
- 35.Hudlicky T., Rinner U., Gonzalez D., Akgun H., Schilling S., Siengalewicz P., Martinot T.A., Pettit G.R. J. Org. Chem. 2002;67:8726. doi: 10.1021/jo020129m. [DOI] [PubMed] [Google Scholar]
- 36.Grundon M.F. Alkaloids 1979;9:137. [Google Scholar]
- 37.Grundon M.F. Alkaloids 1981;10:135. [Google Scholar]
- 38.Grundon M.F. Alkaloids 1981;11:131. [Google Scholar]
- 39.Grundon M.F. Alkaloids 1982;12:151. [Google Scholar]
- 40.Grundon M.F. Alkaloids 1983;13:187. [Google Scholar]
- 41.Martin S.F. In: Brossi A., editor. vol. 30. Academic Press; New York: 1987. p. 251. (The Alkaloids). [Google Scholar]
- 42.Polt R. In: Hudlicky H., editor. vol. 3. JAI Press; Greenwich, CT: 1996. p. 109. (Organic Synthesis: Theory and Applications). [Google Scholar]
- 43.Lewis J.R. In: Sainsbury M., editor. vol. 4. Elsevier; Amsterdam: 1997. p. 165. (Rodd's Chemistry of Carbon Compounds (2nd Edition)). [Google Scholar]
- 44.Viladomat F., Bastida J., Codina C., Nair J.J., Campbell W.E. In: Pandalai S.G., editor. vol. 1. Research Signpost; Trivandrum: 1997. p. 131. (Recent Research Developments in Phytochemistry). [Google Scholar]
- 45.J. Bastida, F. Viladomat, and C. Codina, lskd asdkjf alskd, in “Studies in Natural Products Chemistry,” (Atta-ur-Rahman, ed.), vol. 20, p. 20, Elsevier, Amsterdam, 1998.
- 46.Quayle P. Annu. Rep. Prog. Chem., Sect. B: Org. Chem. 2000;96:259. [Google Scholar]
- 47.Prabhakar S., Tavares M.R. In: Pelletier S.W., editor. vol. 20. Pergamon Press; Oxford: 2001. p. 433. (Alkaloids: Chemical and Biological Perspectives). [Google Scholar]
- 48.J. Bastida and F. Viladomat, ksd lakdj, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks, ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 141. Taylor & Francis, London, 2002.
- 49.Grundon M.F. Nat. Prod. Rep. 1984;1:247. [Google Scholar]
- 50.Grundon M.F. Nat. Prod. Rep. 1985;2:249. doi: 10.1039/np9850200393. [DOI] [PubMed] [Google Scholar]
- 51.Grundon M.F. Nat. Prod. Rep. 1987;4:89. doi: 10.1039/np9870400089. [DOI] [PubMed] [Google Scholar]
- 52.Grundon M.F. Nat. Prod. Rep. 1989;6:79. doi: 10.1039/np9890600523. [DOI] [PubMed] [Google Scholar]
- 53.Lewis J.R. Nat. Prod. Rep. 1990;7:549. doi: 10.1039/np9900700549. [DOI] [PubMed] [Google Scholar]
- 54.Lewis J.R. Nat. Prod. Rep. 1992;9:183. doi: 10.1039/np9920900081. [DOI] [PubMed] [Google Scholar]
- 55.Lewis J.R. Nat. Prod. Rep. 1993;10:291. doi: 10.1039/np9931000029. [DOI] [PubMed] [Google Scholar]
- 56.Lewis J.R. Nat. Prod. Rep. 1994;11:329. doi: 10.1039/np9941100329. [DOI] [PubMed] [Google Scholar]
- 57.Lewis J.R. Nat. Prod. Rep. 1995;12:339. doi: 10.1039/np9951200135. [DOI] [PubMed] [Google Scholar]
- 58.Lewis J.R. Nat. Prod. Rep. 1996;13:171. [Google Scholar]
- 59.Lewis J.R. Nat. Prod. Rep. 1997;14:303. [Google Scholar]
- 60.Lewis J.R. Nat. Prod. Rep. 1998;15:107. [Google Scholar]
- 61.Lewis J.R. Nat. Prod. Rep. 1999;16:389. [Google Scholar]
- 62.Lewis J.R. Nat. Prod. Rep. 2000;17:57. doi: 10.1039/a809403i. [DOI] [PubMed] [Google Scholar]
- 63.Lewis J.R. Nat. Prod. Rep. 2001;18:95. doi: 10.1039/a909077k. [DOI] [PubMed] [Google Scholar]
- 64.Lewis J.R. Nat. Prod. Rep. 2002;19:223. doi: 10.1039/b007741k. [DOI] [PubMed] [Google Scholar]
- 65.Jin Z., Li Z., Huang R. Nat. Prod. Rep. 2002;19:454. doi: 10.1039/b108923b. [DOI] [PubMed] [Google Scholar]
- 66.Jin Z. Nat. Prod. Rep. 2005;22:111. doi: 10.1039/b316106b. [DOI] [PubMed] [Google Scholar]
- 67.Bastida J., Codina C., Viladomat F., Rubiralta M., Quirion J.C., Weniger B. J. Nat. Prod. 1992;55:134. [Google Scholar]
- 68.Bastida J., Fernández J.M., Viladomat F., Codina C., de la Fuente G. Phytochemistry. 1995;38:549. [Google Scholar]
- 69.Stark L.M., Lin X.F., Flippin L.A. J. Org. Chem. 2000;65:3227. doi: 10.1021/jo991902p. [DOI] [PubMed] [Google Scholar]
- 70.Bastida J., Codina C., Viladomat F., Rubiralta M., Quirion J.C., Weniger B. J. Nat. Prod. 1992;55:122. [Google Scholar]
- 71.Almanza G.R., Fernández J.M., Wakori E.W.T., Viladomat F., Codina C., Bastida J. Phytochemistry. 1996;43:1375. [Google Scholar]
- 72.Bastida J., Llabrés J.M., Viladomat F., Codina C., Rubiralta M., Feliz M. Phytochemistry. 1988;27:3657. [Google Scholar]
- 73.Hohmann J., Forgo P., Molnár J., Wolfard K., Molnár A., Thalhammer T., Máthé I., Sharples D. Planta Med. 2002;68:454. doi: 10.1055/s-2002-32068. [DOI] [PubMed] [Google Scholar]
- 74.Labraña J., Choy G., Solans X., Font-Bardia M., de la Fuente G., Viladomat F., Codina C., Bastida J. Phytochemistry. 1999;50:183. [Google Scholar]
- 75.Wildman W.C., Bailey D.T. J. Am. Chem. Soc. 1967;89:5514. doi: 10.1021/ja00996a032. [DOI] [PubMed] [Google Scholar]
- 76.Viladomat F., Bastida J., Codina C., Rubiralta M., Quirion J.C. J. Nat. Prod. 1992;55:804. [Google Scholar]
- 77.Pettit G.R., Melody N., Herald D.L. J. Org. Chem. 2001;66:2583. doi: 10.1021/jo000710n. [DOI] [PubMed] [Google Scholar]
- 78.Pettit G.R., Melody N., Herald D.L., Schmidt J.M., Pettit R.K., Chapuis J.C. Heterocycles. 2002;56:139. [Google Scholar]
- 79.Viladomat F., Bastida J., Tribó G., Codina C., Rubiralta M. Phytochemistry. 1990;29:1307. [Google Scholar]
- 80.Labraña J., Machocho A.K., Kricsfalusy V., Brun R., Codina C., Viladomat F., Bastida J. Phytochemistry. 2002;60:847. doi: 10.1016/s0031-9422(02)00154-1. [DOI] [PubMed] [Google Scholar]
- 81.M. Kreh, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 256. Taylor & Francis, London, 2002.
- 82.Bastida J., Llabrés J.M., Viladomat F., Codina C., Rubiralta M., Feliz M. J. Nat. Prod. 1987;50:199. [Google Scholar]
- 83.Bastida J., Viladomat F., Llabrés J.M., Codina C., Feliz M., Rubiralta M. Phytochemistry. 1987;26:1519. [Google Scholar]
- 84.López S., Bastida J., Viladomat F., Codina C. Planta Med. 2003;69:1166. doi: 10.1055/s-2003-818013. [DOI] [PubMed] [Google Scholar]
- 85.R. M. Moraes-Cerdeira, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 273. Taylor & Francis, London, 2002.
- 86.Codina C., Viladomat F., Bastida J., Rubiralta M., Quirion J.C. Phytochemistry. 1990;29:2685. [Google Scholar]
- 87.Viladomat F., Llabrés J.M., Bastida J., Cusidó R.M., Codina C. Physiol. Plant. 1986;68:657. [Google Scholar]
- 88.Cherkasov O.A., Tokhtabaeva G.M., Margvelashvili N.N., Maisuradze N.I., Tolkachev O.N. Rastit. Resur. 1988;24:414. [Google Scholar]
- 89.Boit H.G., Döpke W. Chem. Ber. 1956;89:2462. [Google Scholar]
- 90.Boit H.G., Stender W. Chem. Ber. 1954;87:624. [Google Scholar]
- 91.Döpke W., Sewerin E. Z. Chem. 1981;21:71. [Google Scholar]
- 92.Harken R.D., Christensen C.P., Wildman W.C. J. Org. Chem. 1976;41:2450. [Google Scholar]
- 93.López S., Bastida J., Viladomat F., Codina C. Life Sci. 2002;71:2521. doi: 10.1016/s0024-3205(02)02034-9. [DOI] [PubMed] [Google Scholar]
- 94.Wildman W.C., Brown C.L. J. Am. Chem. Soc. 1968;90:6439. doi: 10.1021/ja01025a036. [DOI] [PubMed] [Google Scholar]
- 95.Willaman J.J., Li H.L. Lloydia. 1970;33S:1. [Google Scholar]
- 96.Boit H.G. Chem. Ber. 1954;87:681. [Google Scholar]
- 97.Döpke W. Naturwissenschaften. 1963;50:354. [Google Scholar]
- 98.Döpke W., Nguyen T.D. Z. Chem. 1974;14:57. [Google Scholar]
- 99.Viladomat F., Sellés M., Codina C., Bastida J. Planta Med. 1997;63:583. doi: 10.1055/s-2006-957781. [DOI] [PubMed] [Google Scholar]
- 100.E. D. Bazhenova, K. U. Aliev, and U. B. Zakirov, Farmakol. Alkaloidov Serdech. Glikozidov. 100 (1971).
- 101.Piozzi F., Marino M.L., Fuganti C., Di Martino A. Phytochemistry. 1969;8:1745. [Google Scholar]
- 102.Bastida J., Viladomat F., Llabrés C. Codina J.M., Codina C., Rubiralta M. Planta Med. 1988;54:362. doi: 10.1055/s-2006-962460. [DOI] [PubMed] [Google Scholar]
- 103.Boit H.G., Döpke W., Beitner A. Chem. Ber. 1957;90:2197. [Google Scholar]
- 104.Codina C., Bastida J., Viladomat F., Fernández J.M., Bergoñon S., Rubiralta M., Quirion J.C. Phytochemistry. 1993;32:1354. [Google Scholar]
- 105.C. Codina, F. Viladomat, J. Bastida, and M. Rubiralta, Abstracts of the 36th Annual Congress on Medicinal Plant Research, p. 49. Freiburg (1988).
- 106.Codina C., Viladomat F., Bastida J., Rubiralta M., Quirion J.C. Nat. Prod. Lett. 1992;1:85. [Google Scholar]
- 107.Bastida J., Viladomat F., Bergoñon S., Fernández J.M., Codina C., Rubiralta M., Quirion J.C. Phytochemistry. 1993;34:1656. [Google Scholar]
- 108.Bastida J., Bergoñon S., Viladomat F., Codina C. Planta Med. 1994;60:95. doi: 10.1055/s-2006-959421. [DOI] [PubMed] [Google Scholar]
- 109.Cook J.W., Loudon J.D., McCloskey P. J. Chem. Soc. 1954;4176 [Google Scholar]
- 110.Gude M., Hausen B.M., Heitsch H., König W.A. Contact Dermatitis. 1988;19:1. doi: 10.1111/j.1600-0536.1988.tb02860.x. [DOI] [PubMed] [Google Scholar]
- 111.Bastida J., Llabrés J.M., Viladomat F., Codina C., Rubiralta M., Feliz M. Planta Med. 1988;54:524. doi: 10.1055/s-2006-962537. [DOI] [PubMed] [Google Scholar]
- 112.J. A. Seijas, M. P. Vázquez-Tato, J. Seijo-Muras, P. Ramil-Rego, and M. I. Buján, 8th International Electronic Conference on Synthetic Organic Chemistry (ECSOC-8), p. 623 (2004).
- 113.Llabrés J.M., Viladomat F., Bastida J., Codina C., Serrano M., Rubiralta M., Feliz M. Phytochemistry. 1986;25:1453. [Google Scholar]
- 114.Llabrés J.M., Viladomat F., Bastida J., Codina C., Rubiralta M. Phytochemistry. 1986;25:2637. [Google Scholar]
- 115.Vigneau C., Tsao J., Ducluzeae R., Galzot J. J. Toxicol. Med. 1984;4:21. [Google Scholar]
- 116.Hung S.H., Chen Z.X., Li C.F. Yao Hsueh Hsueh Pao (Acta Pharm. Sin.) 1962;9:719. [PubMed] [Google Scholar]
- 117.Abdallah O.M. Phytochemistry. 1993;34:1447. [Google Scholar]
- 118.Abduazimov K.A., Yunusov S.Y. Khim. Prir. Soedin. 1967;3:64. [Google Scholar]
- 119.Abou-Donia A.H., Darwish F.A., Ghazy N.M. Alex. J. Pharm. Sci. 1989;3:122. [Google Scholar]
- 120.Bi Y.R., Yung K.H., Wong Y.S. Plant Sci. 1998;135:103. [Google Scholar]
- 121.Boit H.G., Döpke W. Naturwissenschaften. 1960;47:109. [Google Scholar]
- 122.Evidente A. Planta Med. 1991;57:293. doi: 10.1055/s-2006-960098. [DOI] [PubMed] [Google Scholar]
- 123.Evidente A., Lanzetta R., Abou-Donia A.H., Amer M.E., Kassem F.F., Harraz F.M. Arch. Pharm. 1994;327:595. [Google Scholar]
- 124.Furusawa E., Furusawa S., Tani S., Irie H., Kitamura K., Wildman W.C. Chem. Pharm. Bull. 1976;24:336. [Google Scholar]
- 125.Piozzi F., Fuganti C., Mondelli R., Ceriotti G. Tetrahedron. 1968;24:1119. [Google Scholar]
- 126.Späth E., Kahovec L. Chem. Ber. 1934;67:1501. [Google Scholar]
- 127.Späth E., Kondo H., Kuffner F. Chem. Ber. 1936;69:1086. [Google Scholar]
- 128.Tani S., Kobayashi N., Fujiwara H., Shingu T., Kato A. Chem. Pharm. Bull. 1981;29:3381. [Google Scholar]
- 129.Youssef D.T., Khalifa A.A. Pharmazie. 2001;56:818. [PubMed] [Google Scholar]
- 130.Orhan I., Sener B. Chem. Nat. Compd. 2003;39:383. [Google Scholar]
- 131.Hung S.H., Ma G.E., Chu M. Yao Hsueh Hsueh Pao (Acta Pharm. Sin.) 1966;13:499. [Google Scholar]
- 132.Laing M., Clark R.C. Tetrahedron Lett. 1974;583 [Google Scholar]
- 133.Ma G.E., Li H.Y., Lu C.E., Yang X.M., Hong S.H. Heterocycles. 1986;24:2089. [Google Scholar]
- 134.J. Bastida, C. Codina, F. Viladomat, and M. Rubiralta, Proceedings of the 18th IUPAC Symposium Chem. Nat. Prod., p. 189. Strasbourg (1992).
- 135.Bastida J., Codina C., Viladomat F., Rubiralta M., Quirion J.C., Husson H.P., Ma G.E. J. Nat. Prod. 1990;53:1456. [Google Scholar]
- 136.Hung S.H., Ma G.E., Sung S.Q. Acta Chim. Sin. 1981;39:529. [Google Scholar]
- 137.Suau R., Rico R., García A.I., Gómez A.I. Heterocycles. 1990;31:517. [Google Scholar]
- 138.Bastida J., Codina C., Viladomat F., Rubiralta M., Quirion J.C., Husson H.P. Phytochemistry. 1990;29:2683. [Google Scholar]
- 139.Vrondeli A., Kefalas P., Kokkalou E. Pharmazie. 2005;60:559. doi: 10.1002/chin.200546187. [DOI] [PubMed] [Google Scholar]
- 140.Bastida J., Contreras J.L., Codina C., Wright C.W., Phillipson J.D. Phytochemistry. 1995;40:1549. [Google Scholar]
- 141.Bastida J., Viladomat F., Llabrés J.M., Quiroga S., Codina C., Rubiralta M. Planta Med. 1990;56:123. doi: 10.1055/s-2006-960904. [DOI] [PubMed] [Google Scholar]
- 142.Boit H.G., Stender W., Beitner A. Chem. Ber. 1957;90:725. [Google Scholar]
- 143.Ceriotti G. Nature. 1967;213:595. doi: 10.1038/213595a0. [DOI] [PubMed] [Google Scholar]
- 144.Cherkasov O.A., Maisuradze N.I., Glyzina G.S., Gayevskii A.V. Khim.-Farm. Zh. 1989;23:621. [Google Scholar]
- 145.Barra A., López-González G. An. Jard. Bot. Madr. 1984;40:345. [Google Scholar]
- 146.Barra A., López-González G. An. Jard. Bot. Madr. 1984;40:369. [Google Scholar]
- 147.Dorda E., Fernández-Casas J. Fontqueria. 1984;5:15. [Google Scholar]
- 148.Dorda E., Fernández-Casas J. Fontqueria. 1984;6:7. [Google Scholar]
- 149.Dorda E., Fernández-Casas J. Fontqueria. 1989;27:103. [Google Scholar]
- 150.Dorda E., Fernández-Casas J. Fontqueria. 1990;30:235. [Google Scholar]
- 151.Dorda E., Fernández-Casas J. Fontqueria. 1994;39:69. [Google Scholar]
- 152.Dorda E., Rivas-Ponce M.A., Fernández-Casas J. Fontqueria. 1991;31:235. [Google Scholar]
- 153.Fernandes A. Fontqueria. 1991;31:141. [Google Scholar]
- 154.Fernández-Casas J. Fontqueria. 1983;3:23. [Google Scholar]
- 155.Fernández-Casas J. Fontqueria. 1984;5:35. [Google Scholar]
- 156.Fernández-Casas J. Fontqueria. 1984;6:35. [Google Scholar]
- 157.Fernández-Casas J. Fontqueria. 1986;11:15. [Google Scholar]
- 158.Boit H.G., Ehmke H. Chem. Ber. 1956;89:163. [Google Scholar]
- 159.Fales H.M., Giuffrida L.D., Wildman W.C. J. Am. Chem. Soc. 1956;78:4145. [Google Scholar]
- 160.C. Fuganti, D. Ghiringhelli, and P. Grasselli, Tetrahedron Lett. 2261 (1974).
- 161.Tojo E. J. Nat. Prod. 1991;54:1387. [Google Scholar]
- 162.Bastos J.K., Xu L., Nanayakkara N.P.D., Burandt C.L., Moraes-Cerdeira R.M., McChesney J.D. J. Nat. Prod. 1996;59:638. [Google Scholar]
- 163.Ceriotti G., Spandrio L., Gazzani G.A. Tumori. 1967;53:359. doi: 10.1177/030089166705300406. [DOI] [PubMed] [Google Scholar]
- 164.Moraes-Cerdeira R.M., Burandt C.L., Bastos J.K., Nanayakkara N.P.D., Mikell J., Thurn J., McChesney J.D. Planta Med. 1997;63:472. doi: 10.1055/s-2006-957740. [DOI] [PubMed] [Google Scholar]
- 165.Kreh M., Matusch R. Phytochemistry. 1995;38:1533. [Google Scholar]
- 166.Kreh M., Matusch R., Witte L. Phytochemistry. 1995;38:773. [Google Scholar]
- 167.Kreh M., Matusch R., Witte L. Phytochemistry. 1995;40:1303. [Google Scholar]
- 168.Mustafa N.R., Rhee I.K., Verpoorte R. J. Liq. Chromatogr. Relat. Technol. 2003;26:3217. [Google Scholar]
- 169.G. M. Gorbunova, V. I. Sheichenko, and O. N. Tolkachev, Khim. Prir. Soedin. 800 (1984).
- 170.G. M. Tokhtabaeva, V. I. Sheichenko, I. V. Yartseva, and O. N. Tolkachev, Khim. Prir. Soedin. 872 (1987).
- 171.Moraes-Cerdeira R.M., Bastos J.K., Burandt C.L., Nanayakkara N.P.D., Mikell J., McChesney J.D. Planta Med. 1997;63:92. doi: 10.1055/s-2006-957617. [DOI] [PubMed] [Google Scholar]
- 172.Abou-Donia A.H., Darwish F.A., Amer M.E., Kassem F.F., Hammoda H.M., Shin Y.G. Alex. J. Pharm. Sci. 2002;16:96. [Google Scholar]
- 173.Ingkaninan K., Hazekamp A., de Best C.M., Irth H., Tjaden U.R., van der Heijden R., van der Greef J., Verpoorte R. J. Nat. Prod. 2000;63:803. doi: 10.1021/np9905719. [DOI] [PubMed] [Google Scholar]
- 174.D. H. R. Barton and G. W. Kirby, J. Chem. Soc. 806 (1962).
- 175.G. W. Kirby and J. Michael, J. Chem. Soc.- Perkin Trans. I 115 (1973). [DOI] [PubMed]
- 176.G. W. Kirby and H. P. Tiwari, J. Chem. Soc.-C 676 (1966).
- 177.Döpke W. Naturwissenschaften. 1963;50:645. [Google Scholar]
- 178.Döpke W. Planta Med. 1963;11:154. [Google Scholar]
- 179.Döpke W. Arch. Pharm. 1964;297:39. doi: 10.1002/ardp.19642970105. [DOI] [PubMed] [Google Scholar]
- 180.Döpke W., Dalmer H. Naturwissenschaften. 1965;52:60. doi: 10.1007/BF00602926. [DOI] [PubMed] [Google Scholar]
- 181.Eichhorn J., Takada T., Kita Y., Zenk M.H. Phytochemistry. 1998;49:1037. [Google Scholar]
- 182.Suhadolnik R.J., Fischer A.G., Zulalian J. Biochem. Biophys. Res. Commun. 1963;11:208. doi: 10.1016/0006-291x(63)90335-8. [DOI] [PubMed] [Google Scholar]
- 183.R. H. Wightman, J. Staunton, A. R. Battersby, and K. R. Hanson, J. Chem. Soc., Perkin Trans. I 2355 (1972). [DOI] [PubMed]
- 184.S. Ghosal, Y. Kumar, S. K. Singh, and A. Shanthy, J. Chem. Res.-S 28 (1986).
- 185.Ghosal S., Shanthy A., Singh S.K. Phytochemistry. 1988;27:1849. [Google Scholar]
- 186.D.H.R. Barton and T. Cohen, Festschrift Arthur Stoll, p. 117. Birkhäuser Verlag, Basel, 1957.
- 187.W. R. Bowman, I. T. Bruce, and G.W. Kirby, J. Chem. Soc., Chem. Commun. 1075 (1969).
- 188.I. T. Bruce and G. W. Kirby, J. Chem. Soc., Chem. Commun. 207 (1968).
- 189.Wildman W.C., Heimer N.E. J. Am. Chem. Soc. 1967;89:5265. doi: 10.1021/ja00996a032. [DOI] [PubMed] [Google Scholar]
- 190.C. Fuganti and M. Mazza, J. Chem. Soc., Chem. Commun. 936 (1972)
- 191.A. R. Battersby, R. Binks, S. W. Breuer, H. M. Fales, W. C. Wildman, and R. J. Highet, J. Chem. Soc. 1595 (1964).
- 192.C. Fuganti, D. Ghiringhelli, and Grasselli, P. J. Chem. Soc., Chem. Commun. 350 (1974).
- 193.Kihara M., Xu L., Konishi K., Nagao Y., Kobayashi S., Shingu T. Heterocycles. 1992;34:1299. [Google Scholar]
- 194.C. Fuganti and M. Mazza, J. Chem. Soc., Perkin Trans. I 954 (1973).
- 195.Hanson K.R. J. Am. Chem. Soc. 1966;88:2731. [Google Scholar]
- 196.C. Fuganti and M. Mazza, J. Chem. Soc., Chem. Commun. 1196 (1971).
- 197.Feinstein A.I., Wildman W.C. J. Org. Chem. 1976;41:2447. [Google Scholar]
- 198.Fuganti C. Chim. Ind. (Milan) 1969;51:1254. [Google Scholar]
- 199.A. R. Battersby, J. E. Kelsey, and J. Staunton, J. Chem. Soc., Chem. Commun. 183 (1971).
- 200.Fales H.M., Wildman W.C. J. Am. Chem. Soc. 1964;86:294. [Google Scholar]
- 201.Wildman W.C., Bailey D.T. J. Am. Chem. Soc. 1969;91:150. [Google Scholar]
- 202.C. Fuganti, J. Staunton, and A. R. Battersby, J. Chem. Soc., Chem. Commun. 1154 (1971).
- 203.C. Fuganti and M. Mazza, J. Chem. Soc., Chem. Commun. 1388 (1971).
- 204.C. Fuganti and M. Mazza, J. Chem. Soc., Chem. Commun. 239 (1972).
- 205.C. Fuganti, Tetrahedron Lett. 1785 (1973).
- 206.C. Fuganti and M. Mazza, J. Chem. Soc., Chem. Commun. 1466 (1970).
- 207.D. H. R. Barton, G. W. Kirby, J. B. Taylor, and G. M. Thomas, J. Chem. Soc. 4545 (1963).
- 208.Vázquez-Tato M.P., Castedo L., Riguera R. Heterocycles. 1988;27:2833. [Google Scholar]
- 209.J. G. Bhandarkar and G. W. Kirby, J. Chem. Soc.-C 1224 (1970). [DOI] [PubMed]
- 210.Hudlicky T. J. Heterocycl. Chem. 2000;37:535. [Google Scholar]
- 211.U. Rinner and T. Hudlicky, Synlett 365 (2005).
- 212.Czollner L., Treu M., Froehlich J., Kueenburg B., Jordis U. ARKIVOC. 2001;2:191. [Google Scholar]
- 213.Hermetsberger M., Treu M., Jordis U., Mereiter K., Hametner C., Froehlich J. Monatsh. Chem. 2004;135:1275. [Google Scholar]
- 214.Lewin A.H., Szewczyk J., Wilson J.W., Carroll F.I. Tetrahedron. 2005;61:7144. [Google Scholar]
- 215.Node M., Kodama S., Hamashima Y., Baba T., Hamamichi N., Nishide K. Angew Chem., Int. Ed. 2001;40:3060. doi: 10.1002/1521-3773(20010817)40:16<3060::AID-ANIE3060>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 216.Kodama S., Hamashima Y., Nishide K., Node M. Angew. Chem., Int. Ed. 2004;43:2659. doi: 10.1002/anie.200353636. [DOI] [PubMed] [Google Scholar]
- 217.Kodama S., Takita H., Kajimoto T., Nishide K., Node M. Tetrahedron. 2004;60:4901. [Google Scholar]
- 218.Pelish H.E., Westwood N.J., Feng Y., Kirchhausen T., Shair M. J. Am. Chem. Soc. 2001;123:6740. doi: 10.1021/ja016093h. [DOI] [PubMed] [Google Scholar]
- 219.Fan C.-A., Tu Y.-Q., Song Z.-L., Zhang E., Shi L., Wang M., Wang B., Zhang S.-Y. Org. Lett. 2004;6:4691. doi: 10.1021/ol048105k. [DOI] [PubMed] [Google Scholar]
- 220.Song Z.L., Wang B.M., Tu Y.Q., Fan C.A., Zhang S.Y. Org. Lett. 2003;5:2319. doi: 10.1021/ol0346685. [DOI] [PubMed] [Google Scholar]
- 221.S. V. Ley, O. Schucht, A. W. Thomas, and P. J. Murray, J. Chem. Soc., Perkin Trans. I 1251 (1999).
- 222.Baxendale I.R., Ley S.V., Piutti C. Angew. Chem., Int. Ed. 2002;41:2194. doi: 10.1002/1521-3773(20020617)41:12<2194::aid-anie2194>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 223.Baxendale I.R., Ley S.V., Nessi M., Piutti C. Tetrahedron. 2002;58:6285. [Google Scholar]
- 224.Baxendale I.R., Ley S.V. Ind. Eng. Chem. Res. 2005;44:8588. [Google Scholar]
- 225.Abelman M.M., Overman L.E., Tran V.D. J. Am. Chem. Soc. 1990;112:6959. [Google Scholar]
- 226.Grigg R., Santhakumar V., Sridharan V., Thornton-pett M., Bridge A.W. Tetrahedron. 1993;49:5177. [Google Scholar]
- 227.Guillou C., Beunard J.-L., Gras E., Thal C. Angew. Chem., Int. Ed. 2001;40:4745. doi: 10.1002/1521-3773(20011217)40:24<4745::aid-anie4745>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 228.Bru C., Thal C., Guillou C. Org. Lett. 2003;5:1845. doi: 10.1021/ol0343358. [DOI] [PubMed] [Google Scholar]
- 229.Gras E., Guillou C., Thal C. Tetrahedron Lett. 1999;40:9243. [Google Scholar]
- 230.Liang P.-H., Liu J.-P., Hsin L.-W., Cheng C.-Y. Tetrahedron. 2004;60:11655. [Google Scholar]
- 231.Jin J., Weinreb S.M. J. Am. Chem. Soc. 1997;119:5773. [Google Scholar]
- 232.Enders D., Lenzen A., Raabe G. Angew. Chem., Int. Ed. 2005;44:3766. doi: 10.1002/anie.200500556. [DOI] [PubMed] [Google Scholar]
- 233.Sahakitpichan P., Ruchirawat S. Tetrahedron Lett. 2003;44:5239. [Google Scholar]
- 234.Trost B.M., Totse F.D. J. Am. Chem. Soc. 2000;122:11262. [Google Scholar]
- 235.Trost B.M., Tang W. Angew. Chem., Int. Ed. 2002;41:2795. doi: 10.1002/1521-3773(20020802)41:15<2795::AID-ANIE2795>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 236.Trost B.M., Tang W. J. Am. Chem. Soc. 2002;124:14542. doi: 10.1021/ja0283394. [DOI] [PubMed] [Google Scholar]
- 237.Trost B.M., Pulley S.R. J. Am. Chem. Soc. 1995;117:10143. [Google Scholar]
- 238.Nishimata T., Stao Y., Mori M. J. Org. Chem. 2004;69:1837. doi: 10.1021/jo030309b. [DOI] [PubMed] [Google Scholar]
- 239.Harayama T., Hori A., Abe H., Takeuchi Y. Tetrahedron. 2004;60:1611. [Google Scholar]
- 240.Torres J.C., Pinto A.C., Garden S.J. Tetrahedron. 2004;60:9889. [Google Scholar]
- 241.H.-J. Knölker and S. Filali, Synlett 1752 (2003).
- 242.Z. Shao, J. Chen, R. Huang, C. Wang, L. Li, and H. Zhang, Synlett 2228 (2003).
- 243.Hiroya K., Itoh S., Sakamoto T. J. Org. Chem. 2004;69:1126. doi: 10.1021/jo035528b. [DOI] [PubMed] [Google Scholar]
- 244.Banwell M.G., Lupton D.W., Ma X., Renner J., Sydnes M.O. Org. Lett. 2004;6:2741. doi: 10.1021/ol0490375. [DOI] [PubMed] [Google Scholar]
- 245.Sha C.-K., Hong A.-W., Huang C.-M. Org. Lett. 2001;3:2177. doi: 10.1021/ol016022n. [DOI] [PubMed] [Google Scholar]
- 246.Hoarau C., Couture A., Deniau E., Grandclaudon P. J. Org. Chem. 2004;67:5846. doi: 10.1021/jo0202379. [DOI] [PubMed] [Google Scholar]
- 247.Patil P.A., Snieckus V. Tetrahedron Lett. 1998;39:1325. [Google Scholar]
- 248.Hartung C.G., Fecher A., Chapell B., Snieckus V. Org. Lett. 2003;5:1899. doi: 10.1021/ol0344772. [DOI] [PubMed] [Google Scholar]
- 249.Taber D.F., He Y. J. Org. Chem. 2005;70:7711. doi: 10.1021/jo0511039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Hudlicky T., Gonzalez D., Gibson D.T. Aldrichim. Acta. 1999;32:35. [Google Scholar]
- 251.Hudlicky T., Olivo H.F., McKibben B. J. Am. Chem. Soc. 1994;116:5108. [Google Scholar]
- 252.Hudlicky T., Tian X., Königsberger K., Maurya R., Rouden J., Fan B. J. Am. Chem. Soc. 1996;118:10752. [Google Scholar]
- 253.Gonzalez D., Martinot T., Hudlicky T. Tetrahedron Lett. 1999;40:3077. [Google Scholar]
- 254.Rinner U., Siengalewicz P., Hudlicky T. Org. Lett. 2002;4:115. doi: 10.1021/ol0169877. [DOI] [PubMed] [Google Scholar]
- 255.Hudlicky T., Rinner U., Gonzalez D., Akgun H., Schilling S., Siengalewicz P., Martinot T.A., Pettit G. J. Org. Chem. 2002;67:8726. doi: 10.1021/jo020129m. [DOI] [PubMed] [Google Scholar]
- 256.Rinner U., Hudlicky T., Gordon H., Pettit G.R. Angew. Chem., Int. Ed. 2004;43:5342. doi: 10.1002/anie.200460218. [DOI] [PubMed] [Google Scholar]
- 257.Rinner U., Hillebrenner H.L., Adams D.R., Hudlicky T., Pettit G.R. Bioorg. Med. Chem. Lett. 2004;14:2911. doi: 10.1016/j.bmcl.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 258.Hudlicky T., Rinner U., Finn K.J., Ghiviriga I. J. Org. Chem. 2005;70:3490. doi: 10.1021/jo040292c. [DOI] [PubMed] [Google Scholar]
- 259.Phung A.N., Zannetti M.T., Whited G., Fessner W.-D. Angew. Chem., Int. Ed. 2003;42:4821. doi: 10.1002/anie.200352023. [DOI] [PubMed] [Google Scholar]
- 260.Magnus P., Sebhat I.K. Tetrahedron. 1998;54:15509. [Google Scholar]
- 261.Magnus P., Bailey J.M., Porter M.J. Tetrahedron. 1999;55:13927. [Google Scholar]
- 262.Parsons A.F., Williams D.A.J. Tetrahedron. 2000;56:7217. [Google Scholar]
- 263.González-López de Turiso F., Curran D.P. Org. Lett. 2005;7:151. doi: 10.1021/ol0477226. [DOI] [PubMed] [Google Scholar]
- 264.Keck G.E., Wager T.T., McHardy S.F. J. Org. Chem. 1998;63:9164. [Google Scholar]
- 265.Keck G.E., Wager T.T., Duarte-Rodríquez J.F. J. Am. Chem. Soc. 1999;121:5176. [Google Scholar]
- 266.M. Ikeda, M. Hamada, T. Yamashita, F. Ikegami, T. Sato, and H. Ishibashi, Synlett 1246 (1998).
- 267.M. Ishikazi, K. Kurihara, E. Tanazawa, and O. Hoshino, J. Chem. Soc., Perkin Trans. I 101 (1993).
- 268.Barclay G.L., Quiclet-Sire B., Sanchez-Jimenez G., Zard S.Z. Org. Biomol. Chem. 2005;3:823. doi: 10.1039/b416763e. [DOI] [PubMed] [Google Scholar]
- 269.Rosa A.M., Lobo A.M., Branco P.S., Prabhakar S., Sá-da-Costa M. Tetrahedron. 1997;53:299. [Google Scholar]
- 270.Overman L.E., Shim J. J. Am. Chem. Soc. 1993;58:4662. [Google Scholar]
- 271.Ko H., Kim E., Park J.E., Kim D., Kim S. J. Org. Chem. 2004;69:112. doi: 10.1021/jo035371n. [DOI] [PubMed] [Google Scholar]
- 272.Rigby J.H., Maharoof U.S.M., Mateo M.E. J. Am. Chem. Soc. 2000;122:6624. [Google Scholar]
- 273.Rigby J.H., Mateo M.E. J. Am. Chem. Soc. 1997;119:12655. [Google Scholar]
- 274.Rigby J.H., Dong W. Org. Lett. 2000;2:1673. doi: 10.1021/ol005706c. [DOI] [PubMed] [Google Scholar]
- 275.Wolkenberg S.E., Boger D.L. J. Org. Chem. 2002;67:7361. doi: 10.1021/jo020437k. [DOI] [PubMed] [Google Scholar]
- 276.Padwa A., Brodney M.A., Lynch S.M. J. Org. Chem. 2001;66:1716. doi: 10.1021/jo0014109. [DOI] [PubMed] [Google Scholar]
- 277.Wang Q., Padwa A. Org. Lett. 2004;6:2189. doi: 10.1021/ol049348f. [DOI] [PubMed] [Google Scholar]
- 278.Doyle T.J., Hendrix M., VanDerveer D., Javanmard S., Haseltine J. Tetrahedron. 1997;53:11153. [Google Scholar]
- 279.Aceña J.L., Arjona O., León M.L., Plumet J. Org. Lett. 2000;2:3683. doi: 10.1021/ol000268v. [DOI] [PubMed] [Google Scholar]
- 280.Gao S., Tu Y.Q., Song Z., Wang A., Fan X., Jiang Y. J. Org. Chem. 2005;70:6523. doi: 10.1021/jo0507690. [DOI] [PubMed] [Google Scholar]
- 281.Yasuhara T., Nishimura K., Yamashita M., Fukuyama N., Yamada K., Muraoka O., Tomioka K. Org. Lett. 2003;5:1123. doi: 10.1021/ol0341905. [DOI] [PubMed] [Google Scholar]
- 282.Manpadi M., Kornienko A. Tetrahedron Lett. 2005;46:4433. [Google Scholar]
- 283.Nadein O.N., Kornienko A. Org. Lett. 2004;6:831. doi: 10.1021/ol049942p. [DOI] [PubMed] [Google Scholar]
- 284.Chavan S.P., Khobragade D.A., Pathak A.B., Kalkote U.R. Tetrahedron Lett. 2004;45:5263. [Google Scholar]
- 285.Taber D.F., Neubert T.D. J. Org. Chem. 2001;66:143. doi: 10.1021/jo001237g. [DOI] [PubMed] [Google Scholar]
- 286.Padwa A., Danca M.D., Hardcastle K.I., McClure M.S. J. Org. Chem. 2003;68:929. doi: 10.1021/jo026471g. [DOI] [PubMed] [Google Scholar]
- 287.Lebrun S., Couture A., Deniau E., Grandclaudon P. Org. Biomol. Chem. 2003;1:1701. doi: 10.1039/b302168h. [DOI] [PubMed] [Google Scholar]
- 288.Elango S., Yan T.-H. J. Org. Chem. 2002;67:6954. doi: 10.1021/jo020155k. [DOI] [PubMed] [Google Scholar]
- 289.Elango S., Yan T.-H. Tetrahedron. 2002;58:7335. [Google Scholar]
- 290.Elango S., Wang Y.-C., Cheng C.-L., Yan T.-H. Tetrahedron Lett. 2002;43:3757. [Google Scholar]
- 291.Pettit G.R., Orr B., Ducki S. Anticancer Drug Des. 2000;15:389. [PubMed] [Google Scholar]
- 292.Pettit G.R., Melody N. J. Nat. Prod. 2005;68:207. doi: 10.1021/np0304518. [DOI] [PubMed] [Google Scholar]
- 293.Shieh W.-C., Carlson J.A. J. Org. Chem. 1994;59:5463. [Google Scholar]
- 294.Ibn-Ahmed S., Khaldi M., Chrétien F., Chapleur Y. J. Org. Chem. 2004;69:6722. doi: 10.1021/jo049153l. [DOI] [PubMed] [Google Scholar]
- 295.Baldwin S.W., Debenham J.S. Org. Lett. 2000;2:99. doi: 10.1021/ol9911472. [DOI] [PubMed] [Google Scholar]
- 296.M. Bohno, H. Imase, and N. Chida, Chem. Commun. 1086 (2004). [DOI] [PubMed]
- 297.G. Pandey, A. Murugan, and M. Balakrishnan, Chem. Commun. 624 (2002). [DOI] [PubMed]
- 298.Likhitwitayawuid K., Angerhofer C.K., Chai H., Pezzuto J.M., Cordell G.A. J. Nat. Prod. 1993;56:1331. doi: 10.1021/np50098a017. [DOI] [PubMed] [Google Scholar]
- 299.Spohn M., Brecht V., Frahm A.W. Arch. Pharm. 1994;327:123. [Google Scholar]
- 300.Wagner J., Pham H.L., Döpke W. Tetrahedron. 1996;52:6591. [Google Scholar]
- 301.Gopalakrishna E.M., Watson W.H., Pacheco P., Silva M. Cryst. Struct. Commun. 1976;5:795. [Google Scholar]
- 302.Döpke W., Dalmer H. Naturwissenschaften. 1965;52:61. doi: 10.1007/BF00602926. [DOI] [PubMed] [Google Scholar]
- 303.Evidente A., Iasiello I., Randazzo G. J. Nat. Prod. 1984;47:1003. [Google Scholar]
- 304.Kobayashi S., Ishikawa H., Kihara M., Shingu T., Hashimoto T. Chem. Pharm. Bull. 1977;25:2244. [Google Scholar]
- 305.Evidente A., Arrigoni O., Liso R., Calabrese G., Randazzo G. Phytochemistry. 1986;25:2739. [Google Scholar]
- 306.K. Kuriyama, T. Iwata, M. Moriyama, K. Kotera, Y. Hamada, R. Mitsui, and K. Takeda, J. Chem. Soc., Sec. B 46 (1967).
- 307.Boit H.G., Ehmke H., Uyeo S., Yajima H. Chem. Ber. 1957;90:363. [Google Scholar]
- 308.Sandberg F., Michel K.H. Acta Pharm. Suec. 1968;5:61. [PubMed] [Google Scholar]
- 309.Fales H.M., Wildman W.C. J. Am. Chem. Soc. 1958;80:4395. [Google Scholar]
- 310.T. H. Kinstle, W. C. Wildman, and C. L. Brown, Tetrahedron Lett. 4659 (1966).
- 311.Clardy J.C., Wildman W.C., Hauser F.M. J. Am. Chem. Soc. 1970;92:1781. [Google Scholar]
- 312.A. Immirzi and C. Fuganti, J. Chem. Soc.-B 1218 (1971).
- 313.Kihara M., Ozaki T., Kobayashi S., Shingu T. Chem. Pharm. Bull. 1995;43:318. [Google Scholar]
- 314.Richomme P., Pabuccuoglu V., Gözler T., Freyer A.J., Shamma M. J. Nat. Prod. 1989;52:1150. [Google Scholar]
- 315.Suau R., Gómez A.I., Rico R., Vazquez-Tato M.P., Castedo L., Riguera R. Phytochemistry. 1988;27:3285. [Google Scholar]
- 316.Bastida J., Codina C., Peeters P., Rubiralta M., Orozco M., Luque F.J., Chhabra S.C. Phytochemistry. 1995;40:1291. [Google Scholar]
- 317.Evidente A., Randazzo G., Surico G., Lavermicocca P., Arrigoni O. J. Nat. Prod. 1985;48:564. [Google Scholar]
- 318.Abou-Donia A.H., Abib A.A., El Din A.S., Evidente A., Gaber M., Scopa A. Phytochemistry. 1992;31:2139. [Google Scholar]
- 319.Bastida J., Codina C., Porras C.L., Paiz L. Planta Med. 1996;62:74. doi: 10.1055/s-2006-957808. [DOI] [PubMed] [Google Scholar]
- 320.Rhee I.K., Appels N., Hofte B., Karabatak B., Erkelens C., Stark L.M., Flippin L.A., Verpoorte R. Biol. Pharm. Bull. 2004;27:1804. doi: 10.1248/bpb.27.1804. [DOI] [PubMed] [Google Scholar]
- 321.Jeffs P.W., Abou-Donia A., Campau D., Staiger D. J. Org. Chem. 1985;50:1732. [Google Scholar]
- 322.Viladomat F., Bastida J., Codina C., Solans X., Font-Bardia M. Acta Crystallogr.-C. 1999;55:385. [Google Scholar]
- 323.Latvala A., Önür M.A., Gözler T., Linden A., Kivçak B., Hesse M. Phytochemistry. 1995;39:1229. [Google Scholar]
- 324.Latvala A., Önür M.A., Gözler T., Linden A., Kivçak B., Hesse M. Tetrahedron Asym. 1995;6:361. [Google Scholar]
- 325.H. Irie, Y. Tsuda, and S. Uyeo, J. Chem. Soc. 1446 (1959).
- 326.Evidente A., Andolfi A., Abou-Donia A.H., Touema S.M., Hammoda H.M., Shawky E., Motta A. Phytochemistry. 2004;65:2113. doi: 10.1016/j.phytochem.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 327.T. Kitagawa, W. I. Taylor, S. Uyeo, and H. Yajima, J. Chem. Soc. 1066 (1955).
- 328.T. Ibuka, H. Irie, A. Kato, S. Uyeo, K. Kotera, and Y. Nakagawa, Tetrahedron Lett. 4745 (1966).
- 329.Clardy J.C., Chan J.A., Wildman W.C. J. Org. Chem. 1972;37:49. [Google Scholar]
- 330.Frahm A.W., Ali A.A., Ramadan M.A. Magn. Reson. Chem. 1985;23:804. [Google Scholar]
- 331.Pabuççuoglu V., Richomme P., Gözler T., Kivçak B., Freyer A.J., Shamma M. J. Nat. Prod. 1989;52:785. [Google Scholar]
- 332.G. G. De Angelis and W. C. Wildman, Tetrahedron Lett. 729 (1969).
- 333.Tomioka K., Koga K., Yamada S.I. Chem. Pharm. Bull. 1977;25:2681. [Google Scholar]
- 334.Zabel V., Watson W.H., Pacheco P., Silva M. Cryst. Struct. Commun. 1979;8:371. [Google Scholar]
- 335.Ghosal S., Razdan A., Razdan S. Phytochemistry. 1985;24:635. [Google Scholar]
- 336.Youssef D.T.A., Frahm A.W. Planta Med. 1998;64:669. doi: 10.1055/s-2006-957549. [DOI] [PubMed] [Google Scholar]
- 337.Kihara M., Koike T., Imakura Y., Kida K., Shingu T., Kobayashi S. Chem. Pharm. Bull. 1987;35:1070. [Google Scholar]
- 338.Baudouin G., Tillequin F., Koch M. Heterocycles. 1994;38:965. [Google Scholar]
- 339.Watson W.H., Galloy J., Silva M. Acta Crystallogr.-C. 1984;40:156. [Google Scholar]
- 340.Trimiño Z., Castillo M., Spenglers I. Rev. Cubana Farm. 1989;23:147. [Google Scholar]
- 341.Antoun M.D., Mendoza N.T., Rios Y.R. J. Nat. Prod. 1993;56:1423. doi: 10.1021/np50098a030. [DOI] [PubMed] [Google Scholar]
- 342.Viladomat F., Bastida J., Codina C., Campbell W.E., Mathee S. Phytochemistry. 1994;35:809. [Google Scholar]
- 343.Viladomat F., Almanza G.R., Codina C., Bastida J., Campbell W.E., Mathee S. Phytochemistry. 1996;43:1379. [Google Scholar]
- 344.Roques R., Declercq J.P., Germain G. Acta Crystallogr.-B. 1977;33:3696. [Google Scholar]
- 345.Wildman W.C., Kaufman C.J. J. Am. Chem. Soc. 1954;76:5815. [Google Scholar]
- 346.Crain W.O., Wildman W.C., Roberts J.D. J. Am. Chem. Soc. 1971;93:990. doi: 10.1021/ja00733a035. [DOI] [PubMed] [Google Scholar]
- 347.Ghosal S., Kumar Y., Sinhg S. Phytochemistry. 1984;23:1167. [Google Scholar]
- 348.Trimiño Z., Iglesias C., Sánchez M., Spengler I. Rev. Cubana Quím. 1987;3:57. [Google Scholar]
- 349.Döpke W., Pham L.H., Gründemann E., Bartoszek M., Flatau S. Planta Med. 1995;61:564. doi: 10.1055/s-2006-959375. [DOI] [PubMed] [Google Scholar]
- 350.Pham L.H., Gründemann E., Wagner J., Bartoszek M., Döpke W. Phytochemistry. 1999;51:327. [Google Scholar]
- 351.Ide S., Sener B., Temizer H., Könükol S. Cryst. Res. Technol. 1996;31:617. [Google Scholar]
- 352.Hauth H., Stauffacher D. Helv. Chim. Acta. 1964;47:185. doi: 10.1002/hlca.19710540507. [DOI] [PubMed] [Google Scholar]
- 353.Duffield A.M., Aplin R.T., Budzikiewicz H., Djerassi C., Murphy C.F., Wildman W.C. J. Am. Chem. Soc. 1965;87:4902. doi: 10.1021/ja00949a038. [DOI] [PubMed] [Google Scholar]
- 354.De Angelis G.G., Wildman W.C. Tetrahedron. 1969;25:5099. [Google Scholar]
- 355.Razafimbelo J., Andriantsiferana M., Baudouin G., Tillequin F. Phytochemistry. 1996;41:323. [Google Scholar]
- 356.Linden A., Akineri G., Noyan S., Gozler T., Hesse M. Acta Crystallogr.-C. 1998;54:1653. [Google Scholar]
- 357.Spenglers I., Trimiño Z. Rev. Cubana Farm. 1989;23:151. [Google Scholar]
- 358.Suau R., Gómez A.I., Rico R. Phytochemistry. 1990;29:1710. [Google Scholar]
- 359.Youssef D.T.A. J. Nat. Prod. 2001;64:839. doi: 10.1021/np0005816. [DOI] [PubMed] [Google Scholar]
- 360.Viladomat F., Codina C., Bastida J., Solans X., Font-Bardia M. Acta Crystallogr.-C. 1998;54:81. [Google Scholar]
- 361.Ali A.A., Mesbah M.K., Frahm A.W. Planta Med. 1984;50:188. doi: 10.1055/s-2007-969669. [DOI] [PubMed] [Google Scholar]
- 362.Abdallah O.M., Ali A.A., Itokawa H. Phytochemistry. 1989;28:3248. [Google Scholar]
- 363.Peeters O.M., Blaton N.M., De Ranter C.J. Acta Crystallogr.-C. 1997;53:1284. [Google Scholar]
- 364.T. Kametani, K. Yamaki, H. Yagi, and K. Fukumoto, J. Chem. Soc.-C 2602 (1969). [DOI] [PubMed]
- 365.Vlahov R., Krikorian D., Spassov G., Chinova M., Vlahov I., Parushev S., Snatzke G., Ernst L., Kieslich K., Abraham W.R., Sheldrick W.S. Tetrahedron. 1989;45:3329. [Google Scholar]
- 366.Laiho S.M., Fales H.M. J. Am. Chem. Soc. 1964;86:4434. [Google Scholar]
- 367.Li H.Y., Ma G.E., Xu Y., Hong S.H. Planta Med. 1987;53:259. doi: 10.1055/s-2006-962697. [DOI] [PubMed] [Google Scholar]
- 368.Roques R., Lapasset J. Acta Crystallogr.-B. 1976;32:579. [Google Scholar]
- 369.Kobayashi S., Satoh K., Numata A., Shingu T., Kihara M. Phytochemistry. 1991;30:675. [Google Scholar]
- 370.Kobayashi S., Tokumoto T., Kihara M., Imakura K., Shingu T., Taira Z. Chem. Pharm. Bull. 1984;32:3015. [Google Scholar]
- 371.Jeffs P.W., Capps T., Johnson D.B., Karle J.M., Martin N.H., Rauckman B. J. Org. Chem. 1974;39:2703. [Google Scholar]
- 372.Jeffs P.W., Ahmann G., Campbell H.F., Farrier D.S., Ganguli G., Hawks R.L. J. Org. Chem. 1970;35:3512. [Google Scholar]
- 373.Denmark S.E., Marcin L.R. J. Org. Chem. 1997;62:1675. [Google Scholar]
- 374.Jeffs P.W., Mueller L., Abou-Donia A.H., Seif el-Din A.A., Campau D. J. Nat. Prod. 1988;51:549. [Google Scholar]
- 375.L. Zetta, G. Gatti, and C. Fuganti, J. Chem. Soc., Perkin Trans. II 1180 (1973).
- 376.Evidente A. J. Nat. Prod. 1986;49:168. [Google Scholar]
- 377.Mary A., Renko D.Z., Guillou C., Thal C. Bioorg. Med. Chem. 1998;6:1835. doi: 10.1016/s0968-0896(98)00133-3. [DOI] [PubMed] [Google Scholar]
- 378.Schnoes H.K., Smith D.H., Burlingame A.L., Jeffs P.W., Döpke W. Tetrahedron. 1968;24:2825. [Google Scholar]
- 379.Fales H.M., Milne G.W.A., Vestal M.L. J. Am. Chem. Soc. 1969;91:3682. doi: 10.1021/ja01041a064. [DOI] [PubMed] [Google Scholar]
- 380.Razakov R., Bochkarev V.N., Abduazimov Kh.A., Vul’fson N.S., Yunusov S.Y. Khim. Prir. Soedin. 1969;5:280. [Google Scholar]
- 381.Fales H.M., Lloyd H.A., Milne G.W.A. J. Am. Chem. Soc. 1970;92:1590. doi: 10.1021/ja00709a028. [DOI] [PubMed] [Google Scholar]
- 382.Longevialle P., Smith D.H., Burlingame A.L., Fales H.M., Highet R.J. Org. Mass Spectrom. 1973;7:401. [Google Scholar]
- 383.Longevialle P., Fales H.M., Highet R.J., Burlingame A.L. Org. Mass Spectrom. 1973;7:417. [Google Scholar]
- 384.Samuel E.H.C. Org. Mass Spectrom. 1975;10:427. [Google Scholar]
- 385.Onyiriuka O.S., Jackson A.H. Isr. J. Chem. 1978;17:185. [Google Scholar]
- 386.Pettit G.R., Pettit G.R., III, Groszek G., Backhaus R.A., Doubek D.L., Barr R.J., Meerow A.W. J. Nat. Prod. 1995;58:756. doi: 10.1021/np50119a017. [DOI] [PubMed] [Google Scholar]
- 387.Pettit G.R., Cragg G.M., Singh S.B., Duke J.A., Doubek D.L. J. Nat. Prod. 1990;53:176. doi: 10.1021/np50067a026. [DOI] [PubMed] [Google Scholar]
- 388.Pettit G.R., Gaddamidi V., Herald D.L., Singh S.B., Cragg G.M., Schmidt J.M., Boettner F.E., Williams M., Sagawa Y. J. Nat. Prod. 1986;49:995. doi: 10.1021/np50048a005. [DOI] [PubMed] [Google Scholar]
- 389.Pettit G.R., Pettit G.R., III, Backhaus R.A., Boyd M.R., Meerow A.W. J. Nat. Prod. 1993;56:1682. doi: 10.1021/np50100a004. [DOI] [PubMed] [Google Scholar]
- 390.Furusawa E., Suzuki N., Tani S., Furusawa S., Ishioka G.Y., Motobu J. Proc. Soc. Exp. Biol. Med. 1973;143:33. doi: 10.3181/00379727-143-37247. [DOI] [PubMed] [Google Scholar]
- 391.Çakici I., Ulug H.Y., Inci S., Tunçtan B., Abacioglu N., Kanzik I., Sener B. J. Pharm. Pharmacol. 1997;49:828. doi: 10.1111/j.2042-7158.1997.tb06121.x. [DOI] [PubMed] [Google Scholar]
- 392.Grigson G. Helicon Publishing Ltd.; Oxford: 1996. The Englishman's Flora. [Google Scholar]
- 393.Grieve M. Tiger Books International; London: 1998. A Modern Herbal – the Medicinal, Culinary, Cosmetic and Economic Properties, Cultivation and Folklore of Herbs, Grasses, Fungi, Shrubs and Trees with All Their Modern Scientific Uses. [Google Scholar]
- 394.Scott M. The Aquarium Press; Wellingborough: 1986. An Irish Herbal. The Botananalogia Universalis Hibernica by JohnK’Eogh (1681–1754) [Google Scholar]
- 395.Wu T.C., Chen S.T., Wang T.Y., Hsu P. Yao Hsueh Hsueh Pao (Acta Pharm. Sin.) 1965;21:31. [Google Scholar]
- 396.Jaspersen-Schib R., Theus L., Guirguis-Oeschger M., Gossweiler B., Meier-Abt P.J. Schweiz. Med. Wochenschr. 1996;126:1085. [PubMed] [Google Scholar]
- 397.C. G. Julian and P. W. Bowers, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 399. Taylor & Francis, London, 2002.
- 398.Junko I., Akito T., Yumiko K., Noriyoshi O. Pharm. Mon. 1994;36:855. [Google Scholar]
- 399.Ries S., Baughan R., Nair M.G., Schutzki R. HortTechnology. 2001;11:302. [Google Scholar]
- 400.R. K. O’Leary, U.S. pat. Appl. 6565867 B1 20030520 (CA 138: 364205) (2003).
- 401.R. K. O’Leary, U.S. pat. Appl. 2003202998 A1 20031030 (CA 139: 303299) (2003).
- 402.Bruynzeel D.P., De Boer E.M., Brouwer E.J., De Wolff F.A., De Haan P. Contact Dermatitis. 1993;29:11. doi: 10.1111/j.1600-0536.1993.tb04529.x. [DOI] [PubMed] [Google Scholar]
- 403.Bruynzeel D.P. Contact Dermatitis. 1997;37:70. doi: 10.1111/j.1600-0536.1997.tb00042.x. [DOI] [PubMed] [Google Scholar]
- 404.De Jong N.W., Vermeulen A.M., Gerth van Wijk R., De Groot H. Allergy. 1998;53:204. doi: 10.1111/j.1398-9995.1998.tb03872.x. [DOI] [PubMed] [Google Scholar]
- 405.Gonçalo S., Freitas J.D., Sousa I. Contact Dermatitis. 1987;16:115. doi: 10.1111/j.1600-0536.1987.tb01400.x. [DOI] [PubMed] [Google Scholar]
- 406.Güneser S., Atici A., Cengizler I., Alparslan N. Allergol. Immunopathol. 1996;24:116. [PubMed] [Google Scholar]
- 407.Van Doorn W.G. J. Am. Soc. Hort. Sci. 1998;123:146. [Google Scholar]
- 408.Maurer B. Perfum. Flavor. 1994;19(19–22):24. [Google Scholar]
- 409.Reuveni M., Sgi Z., Evnor D., Hetzroni A. Plant Sci. 1999;147:19. [Google Scholar]
- 410.C. Remy, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 392. Taylor & Francis, London, 2002.
- 411.Abou-Karam M., Shier W.T. J. Nat. Prod. 1990;53:340. doi: 10.1021/np50068a011. [DOI] [PubMed] [Google Scholar]
- 412.Furusawa E., Suzuki N., Furusawa S., Lee J.Y.B. Proc. Soc. Exp. Biol. Med. 1975;149:771. doi: 10.3181/00379727-149-38896. [DOI] [PubMed] [Google Scholar]
- 413.Papas T.S., Sandhaus L., Chirigos M.A., Furusawa E. Biochem. Biophys. Res. Commun. 1973;52:88. doi: 10.1016/0006-291x(73)90957-1. [DOI] [PubMed] [Google Scholar]
- 414.Ramanathan S., Furusawa E., Kroposki M., Furusawa S., Cutting W. Chemotherapy. 1968;13:121. doi: 10.1159/000220538. [DOI] [PubMed] [Google Scholar]
- 415.Suzuki N., Tani S., Furusawa S., Furusawa E. Proc. Soc. Exp. Biol. Med. 1974;145:771. doi: 10.3181/00379727-145-37892. [DOI] [PubMed] [Google Scholar]
- 416.Vacik J.P., Davis W.B., Kelling C.S., Schermeister L.J., Schipper I.A. Adv. Ophtalmol. 1979;38:281. [PubMed] [Google Scholar]
- 417.Van den Bergue D.A., Ieven M., Mertens F., Vlietinck J., Lammens E. Lloydia. 1978;41:463. [PubMed] [Google Scholar]
- 418.Dornberger K., Lich H. Pharmazie. 1982;37:215. [PubMed] [Google Scholar]
- 419.Sener B., Koyuncu M., Bingöl F., Muhtar F. Phuket; Thailand: 1997. IUPAC International Conference on Biodiversity and Bioresources: Conservation and Utilization. [Google Scholar]
- 420.Ieven M., Van den Bergue D.A., Mertens F., Vlietinck A., Lammens E. Planta Med. 1979;36:311. doi: 10.1055/s-0028-1097277. [DOI] [PubMed] [Google Scholar]
- 421.Chaumont J.P., Senet J.M. Plant. Med. Phytother. 1978;12:186. [Google Scholar]
- 422.Chaumont H., Schemaker H., Rouseau J. Plant. Med. Phytother. 1978;12:157. [Google Scholar]
- 423.Sener B., Orham I., Satayavivad J. Phytother. Res. 2003;17:1220. doi: 10.1002/ptr.1346. [DOI] [PubMed] [Google Scholar]
- 424.Furusawa E., Irie H., Combs D., Wildman W.C. Chemotherapy. 1980;26:36. doi: 10.1159/000237881. [DOI] [PubMed] [Google Scholar]
- 425.Furusawa E., Suzuki N., Ramanathan S., Furusawa S., Cutting W. Proc. Soc. Exp. Biol. Med. 1972;140:1034. doi: 10.3181/00379727-140-36606. [DOI] [PubMed] [Google Scholar]
- 426.Ikram M. Fitoterapia. 1983;54:123. [Google Scholar]
- 427.Chiu K.W., Lee Y.C., Yung K.H. Phytother. Res. 1992;6:231. [Google Scholar]
- 428.Matsui A.D.S., Rogers J., Woo Y.K., Cutting W.C. Med. Pharmacol. Exp. 1967;16:414. doi: 10.1159/000137022. [DOI] [PubMed] [Google Scholar]
- 429.Keiser I., Harris E.J., Miyashita D.H., Jacobson M., Perdue R.E. Lloydia. 1975;38:141. [PubMed] [Google Scholar]
- 430.Barre A., Van Damme E.J.M., Peumans W.J., Rougé P. Plant Physiol. 1996;112:1531. doi: 10.1104/pp.112.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Van Damme E.J.M., Peumans W.J., Barre A., Rougé P. Crit. Rev. Plant Sci. 1998;17:575. [Google Scholar]
- 432.E. J. M. Van Damme and W. J. Peumans, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 380. Taylor & Francis, London, 2002.
- 433.López S., Codina C., Bastida J., Viladomat F., Davidson E., Stewart D. J. Agric. Food Chem. 2002;50:2507. doi: 10.1021/jf011459v. [DOI] [PubMed] [Google Scholar]
- 434.López S., Armand-Ugon M., Bastida J., Viladomat F., Este J.A., Stewart D., Codina C. Planta Med. 2003;69:109. doi: 10.1055/s-2003-37715. [DOI] [PubMed] [Google Scholar]
- 435.Ooi L.S.M., Liu F., Ooi V.E.C., Ng T.B., Fung M.C. Biochem. Cell Biol. 2002;80:271. doi: 10.1139/o01-240. [DOI] [PubMed] [Google Scholar]
- 436.Arrigoni O., Arrigoni-Liso R., Calabrese G. Nature. 1975;256:513. [Google Scholar]
- 437.Evidente A., Cicala M.R., Randazzo G., Riccio R., Calabrese G., Liso R., Arrigoni O. Phytochemistry. 1983;22:2193. [Google Scholar]
- 438.Arrigoni O., Calabrese G., De Gara L., Bitonti M.B., Liso R. J. Plant Physiol. 1997;150:302. [Google Scholar]
- 439.De Tullio M.C., De Gara L., Paciolla C., Arrigoni O. Plant Physiol. Biochem. 1998;36:433. [Google Scholar]
- 440.Davey M.W., Persiau G., De Bruyn A., Van Damme J., Bauw G., Van Montagu M. Anal. Biochem. 1998;257:80. doi: 10.1006/abio.1997.2544. [DOI] [PubMed] [Google Scholar]
- 441.De Gara L., Paciolla C., Tommasi F., Liso R., Arrigoni O. J. Plant Physiol. 1994;144:649. doi: 10.1104/pp.99.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Imai T., Karita S., Shiratori G., Hatturi M., Nunome T., Ôba K. Plant Cell Physiol. 1998;39:1350. doi: 10.1093/oxfordjournals.pcp.a029341. [DOI] [PubMed] [Google Scholar]
- 443.Kuzniak E. Acta Physiol. Plant. 2004;26:327. [Google Scholar]
- 444.Arrigoni O., Paciolla C., De Gara L. B. Soc. Ital. Biol. Sper. 1996;75:37. [PubMed] [Google Scholar]
- 445.Arrigoni O., De Gara L., Paciolla C., Evidente A., De Pinto M.C., Liso R. J. Plant Physiol. 1997;150:362. [Google Scholar]
- 446.Arrigoni O. J. Bioenerg. Biomembr. 1994;26:407. doi: 10.1007/BF00762782. [DOI] [PubMed] [Google Scholar]
- 447.Córdoba-Pedregosa M.C., González-Reyes J.A., Cañadillas M.S., Navas P., Córdoba F. Plant Physiol. 1996;112:1119. doi: 10.1104/pp.112.3.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Del Giudice A., Massardo D.R., Manna F., Koltovaya N., Hartings H., Del Giudice L., Wolf K. Curr. Microbiol. 1997;34:382. doi: 10.1007/s002849900200. [DOI] [PubMed] [Google Scholar]
- 449.Liso R., Calabrese G., Bitonti M.B., Arrigoni O. Exp. Cell Res. 1984;150:314. doi: 10.1016/0014-4827(84)90574-3. [DOI] [PubMed] [Google Scholar]
- 450.Onofri S., Poerio E., Serangeli P., Tosi S., Garuccio I., Arrigoni O. Antonie Leeuwenhoek. 1997;71:277. doi: 10.1023/a:1000161921389. [DOI] [PubMed] [Google Scholar]
- 451.Arrigoni O., Arrigoni-Liso R., Calabrese G. Science. 1976;194:332. doi: 10.1126/science.194.4262.332. [DOI] [PubMed] [Google Scholar]
- 452.Liso R., De Gara L., Tommasi F., Arrigoni O. FEBS Lett. 1985;187:141. [Google Scholar]
- 453.Kukhanova M., Victorova L., Krayevsky A. FEBS Lett. 1983;160:129. doi: 10.1016/0014-5793(83)80951-x. [DOI] [PubMed] [Google Scholar]
- 454.Del Giudice A., Massardo D.R., Manna F., Evidente A., Randazzo G., Wolf K. Curr. Genet. 1984;8:493. doi: 10.1007/BF00410435. [DOI] [PubMed] [Google Scholar]
- 455.Massardo D.R., Manna F., Schafer B., Wolf K., Del Giudice L. Curr. Genet. 1994;25:80. doi: 10.1007/BF00712972. [DOI] [PubMed] [Google Scholar]
- 456.Massardo D.R., Manna F., Del Giudice L., Wolf K. Curr. Genet. 1990;17:455. doi: 10.1007/BF00334527. [DOI] [PubMed] [Google Scholar]
- 457.Massardo D.R., Zweifel S.G., Gunge N., Miyakawa I., Sando N., Del Giudice A., Wolf K., Del Giudice L. Can. J. Microbiol. 2000;46:1058. doi: 10.1139/w00-096. [DOI] [PubMed] [Google Scholar]
- 458.Onofri S., Barreca D., Garuccio I. Antonie Leeuwenhoek. 2003;83:57. doi: 10.1023/a:1022903504795. [DOI] [PubMed] [Google Scholar]
- 459.Ghosal S., Shanthy A., Kumar A., Kumar Y. Phytochemistry. 1985;24:2703. [Google Scholar]
- 460.Hua D.H., Saha S., Takemoto D.J. Anticancer Res. 1997;17:2435. [PubMed] [Google Scholar]
- 461.Yui S., Mikami M., Kitahara M., Yamazaki M. Immunopharmacology. 1998;40:151. doi: 10.1016/s0162-3109(98)00040-x. [DOI] [PubMed] [Google Scholar]
- 462.Lin L.Z., Hu S.F., Chai H.B., Pengsuparp T., Pezzuto J.M., Codell G.A., Ruangrungsi N. Phytochemistry. 1995;40:1295. doi: 10.1016/0031-9422(95)00372-e. [DOI] [PubMed] [Google Scholar]
- 463.Weniger B., Italiano L., Beck J.P., Bastida J., Bergoñon S., Codina C., Lobstein A., Anton R. Planta Med. 1995;61:77. doi: 10.1055/s-2006-958007. [DOI] [PubMed] [Google Scholar]
- 464.Nair J.J., Campbell W.E., Gammon D.W., Albrecht C.F., Viladomat F., Codina C., Bastida J. Phytochemistry. 1998;49:2539. doi: 10.1016/s0031-9422(99)00575-0. [DOI] [PubMed] [Google Scholar]
- 465.Min B.S., Gao J.J., Nakamura N., Kim Y.H., Hattori M. Chem. Pharm. Bull. 2001;49:1217. doi: 10.1248/cpb.49.1217. [DOI] [PubMed] [Google Scholar]
- 466.Kushida N., Atsumi S., Koyano T., Umezawa K. Drug Exp. Clin. Res. 1997;23:151. [PubMed] [Google Scholar]
- 467.Jimenez A., Santos A., Alonso G., Vázquez D. Biochim. Biophys. Acta. 1976;425:342. doi: 10.1016/0005-2787(76)90261-6. [DOI] [PubMed] [Google Scholar]
- 468.Mikami M., Kitahara M., Kitano M., Ariki Y., Mimaki Y., Sashida Y., Yamazaki M., Yui S. Biol. Pharm. Bull. 1999;22:674. doi: 10.1248/bpb.22.674. [DOI] [PubMed] [Google Scholar]
- 469.Yui S., Mikami M., Mikami Y., Sashida Y., Yamazaki M. Yakugaku Zasshi. 2001;121:167. doi: 10.1248/yakushi.121.167. [DOI] [PubMed] [Google Scholar]
- 470.Abdel-Halim O.B., Morikawa T., Ando S., Matsuda H., Yoshikawa M. J. Nat. Prod. 2004;67:1119. doi: 10.1021/np030529k. [DOI] [PubMed] [Google Scholar]
- 471.Liu J., Hu W.-X., He L.-F., Ye M., Li Y. FEBS Lett. 2004;578:245. doi: 10.1016/j.febslet.2004.10.095. [DOI] [PubMed] [Google Scholar]
- 472.Schmeda-Hirschmann G., Astudillo L., Bastida J., Viladomat F., Codina C. Bol. Soc. Chil. Quím. 2000;45:515. [Google Scholar]
- 473.Karadeniz H., Gulmez B., Sahinci F., Erdem A., Kaya G.I., Unver N., Kivcak B., Ozsoz M. J. Pharm. Biomed. Anal. 2003;32:295. doi: 10.1016/s0731-7085(03)00283-8. [DOI] [PubMed] [Google Scholar]
- 474.Pettit G.R., Gaddamidi V., Goswami A., Cragg G.M. J. Nat. Prod. 1984;47:796. doi: 10.1021/np50035a007. [DOI] [PubMed] [Google Scholar]
- 475.Jimenez A., Sánchez L., Vázquez D. FEBS Lett. 1975;60:66. doi: 10.1016/0014-5793(75)80420-0. [DOI] [PubMed] [Google Scholar]
- 476.Ghosal S., Singh S.K., Kumar Y., Unnikrishnan S., Chattopadhyay S. Planta Med. 1988;54:114. doi: 10.1055/s-2006-962364. [DOI] [PubMed] [Google Scholar]
- 477.Zee-Cheng R.K.Y., Yan S.J., Cheng C.C. J. Med. Chem. 1978;21:199. doi: 10.1021/jm00200a011. [DOI] [PubMed] [Google Scholar]
- 478.Chattopadhyay U., Chaudhuri L., Das S., Kumar Y., Ghosal S. Pharmazie. 1984;39:855. [PubMed] [Google Scholar]
- 479.Ieven M., Van den Bergue D.A., Vlietinck A. Planta Med. 1983;49:109. [PubMed] [Google Scholar]
- 480.Ieven M., Vlietinck A., Van den Bergue D.A., Totte J., Dommisse R., Esmans E., Alderweireldt F. J. Nat. Prod. 1982;45:564. doi: 10.1021/np50023a009. [DOI] [PubMed] [Google Scholar]
- 481.Gabrielsen B., Monath T.P., Huggins J.W., Kefauver D.F., Pettit G.R., Groszek G., Hollingshead M., Kirsi J.J., Shannon W.M., Schubert E.M., Dare J., Ugarkar B., Ussery M.A., Phelan M.J. J. Nat. Prod. 1992;55:1569. doi: 10.1021/np50089a003. [DOI] [PubMed] [Google Scholar]
- 482.Li S.Y., Chen C., Zhang H.Q., Guo H.Y., Wang H., Wang L., Zhang X., Hua S.N., Yu J., Xiao P.G., Li R.S., Tan X. Antiviral Res. 2005;67:18. doi: 10.1016/j.antiviral.2005.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Szlávik L., Gyuris A., Minárovits J., Forgo P., Molnár J., Hohmann J. Planta Med. 2004;70:871. doi: 10.1055/s-2004-827239. [DOI] [PubMed] [Google Scholar]
- 484.Renard-Nozaki J., Kim T., Imakura Y., Kihara M., Kobayashi S. Res. Virol. 1989;140:115. doi: 10.1016/s0923-2516(89)80089-5. [DOI] [PubMed] [Google Scholar]
- 485.Vrijsen R., Van der Berghe D.A., Vlietinck A.J., Boeyé A. J. Biol. Chem. 1986;261:505. [PubMed] [Google Scholar]
- 486.Elgorashi E.E., Stafford G.I., van Staden J. Planta Med. 2004;70:260. doi: 10.1055/s-2004-818919. [DOI] [PubMed] [Google Scholar]
- 487.Houghton P.J., Agbedahunsi J.M., Adegbulugbe A. Phytochemistry. 2004;65:2893. doi: 10.1016/j.phytochem.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 488.Cordell G.A. Wiley Interscience; New York: 1981. Introduction to Alkaloids: A Biogenetic Approach. [Google Scholar]
- 489.Tanker M., Çitoglu G., Gumusel B., Sener B. Int. J. Pharmacogn. 1996;34:194. [Google Scholar]
- 490.Fennell C.W., van Staden J. J. Ethnopharmacol. 2001;78:15. doi: 10.1016/s0378-8741(01)00305-1. [DOI] [PubMed] [Google Scholar]
- 491.Abdalla S., Abu Zarga M., Sabri S. Fitoterapia. 1993;64:518. [Google Scholar]
- 492.M. Q. Amara, J. F. Franetich, N. Bouladoux, D. Mazier, G. Eisenbrand, D. Marko, L. Meijer, C. Doerig, and I. Desportes-Livage, J. Eukaryot. Microbiol.99S(Suppl.) (2001). [DOI] [PubMed]
- 493.Singh R.P., Pant N.C. Experientia. 1980;36:552. [Google Scholar]
- 494.Campbell W.E., Nair J.J., Gammon D.W., Codina C., Bastida J., Viladomat F., Smith P.J., Albrecht C.F. Phytochemistry. 2000;53:587. doi: 10.1016/s0031-9422(99)00575-0. [DOI] [PubMed] [Google Scholar]
- 495.Schultz A.G., Holoboski M.A., Smyth M.S. J. Am. Chem. Soc. 1996;118:6210. [Google Scholar]
- 496.Çitoglu G., Tanker M., Gümüsel B. Phytother. Res. 1998;12:205. [Google Scholar]
- 497.Herrera M.R., Machocho A.K., Brun R., Viladomat F., Codina C., Bastida J. Planta Med. 2001;67:191. doi: 10.1055/s-2001-11495. [DOI] [PubMed] [Google Scholar]
- 498.Schmeda-Hirschmann G., Rodriguez J.A., Loyola J.I., Astudillo L., Bastida J., Viladomat F., Codina C. Pharm. Pharmacol. Commun. 2000;6:309. [Google Scholar]
- 499.Miyasaka K., Hiramatsu Y. Jpn. J. Pharmacol. 1980;30:655. doi: 10.1254/jjp.30.655. [DOI] [PubMed] [Google Scholar]
- 500.Alarcón M., Cea G., Weigert G. Bull. Environ. Contam. Toxicol. 1986;37:508. doi: 10.1007/BF01607796. [DOI] [PubMed] [Google Scholar]
- 501.Viladomat F., Bastida J., Codina C., Campbell W.E., Mathee S. Phytochemistry. 1995;40:307. [Google Scholar]
- 502.Adesanya S.A., Olugbade T.A., Odebiyi O.O., Aladesanmi J.A. Int. J. Pharmacogn. 1992;30:303. [Google Scholar]
- 503.Furusawa E., Furusawa S., Lee J.Y.B., Patanavanich S. Proc. Soc. Exp. Biol. Med. 1976;152:186. doi: 10.3181/00379727-152-39357. [DOI] [PubMed] [Google Scholar]
- 504.Kobayashi S., Kihara M., Shingu T., Shingu K. Chem. Pharm. Bull. 1980;28:2924. [Google Scholar]
- 505.Rigby J.H., Cavezza A., Heeg M.J. J. Am. Chem. Soc. 1998;120:3664. [Google Scholar]
- 506.Furusawa E., Furusawa S. Chemotherapy. 1986;32:521. doi: 10.1159/000238462. [DOI] [PubMed] [Google Scholar]
- 507.Furusawa E., Furusawa S. Oncology. 1988;45:180. doi: 10.1159/000226558. [DOI] [PubMed] [Google Scholar]
- 508.Furusawa E., Lockwood R.H., Furusawa S., Lum M.K.M., Lee J.Y.B. Chemotherapy. 1979;25:308. doi: 10.1159/000237856. [DOI] [PubMed] [Google Scholar]
- 509.Furusawa E., Lum M.K.M., Furusawa S. Chemotherapy. 1981;27:277. doi: 10.1159/000237992. [DOI] [PubMed] [Google Scholar]
- 510.Furusawa E., Furusawa E., Sokugawa L. Chemotherapy. 1983;29:294. doi: 10.1159/000238212. [DOI] [PubMed] [Google Scholar]
- 511.Furusawa E., Furusawa S., Lee J.Y.B., Patanavanich S. Chemotherapy. 1978;24:259. doi: 10.1159/000237790. [DOI] [PubMed] [Google Scholar]
- 512.D. Brown, in “Narcissus and daffodil: the genus Narcissus” (G.R. Hanks, ed.), vol. 21 in the series: “Medicinal and Aromatic Plants – Industrial Profiles”, p. 332. Taylor & Francis, London, 2002.
- 513.Baez A., Vazquez D. Biochim. Biophys. Acta. 1978;518:95. doi: 10.1016/0005-2787(78)90119-3. [DOI] [PubMed] [Google Scholar]
- 514.Jimenez A., Sánchez L., Vázquez D. FEBS Lett. 1975;55:53. doi: 10.1016/0014-5793(75)80955-0. [DOI] [PubMed] [Google Scholar]
- 515.Rodríguez-Fonseca C., Amils R., Garret R.A. J. Mol. Biol. 1995;247:224. doi: 10.1006/jmbi.1994.0135. [DOI] [PubMed] [Google Scholar]
- 516.Pettit G.R., Pettit G.R., III, Backhaus R.A., Boettner F.E. J. Nat. Prod. 1995;58:37. doi: 10.1021/np50115a004. [DOI] [PubMed] [Google Scholar]
- 517.Abou-Donia A.A., De Giulio A., Evidente A., Gaber M., Habib A.A., Lanzetta R., Seif El Din A.A. Phytochemistry. 1991;30:3445. [Google Scholar]
- 518.Ghosal S., Singh S., Kumar Y., Srivastava S. Phytochemistry. 1989;28:611. [Google Scholar]
- 519.Van Doorn W.G., Sinz A., Tomassen M.M. Phytochemistry. 2004;65:571. doi: 10.1016/j.phytochem.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 520.Bi Y., Guo J., Zhang L., Wong Y. J. Plant Physiol. 2003;160:1041. doi: 10.1078/0176-1617-00911. [DOI] [PubMed] [Google Scholar]
- 521.Bi Y., Zhang L., Guo J.K., Yung K., Wong Y. New Zeal. J. Crop Hort. Sci. 2003;31:335. [Google Scholar]
- 522.Fulton B., Benfield P. Drug Aging. 1996;9:60. doi: 10.2165/00002512-199609010-00006. [DOI] [PubMed] [Google Scholar]
- 523.Wilcock G., Wilkinson D. In: Alzheimer's Disease: Biology, Diagnosis and Therapeutics. Iqbal K., Winblad B., Nishimura T., Takeda M., Wisniewski H.M., editors. Wiley; West Sussex: 1997. p. 661. [Google Scholar]
- 524.Sramek J.J., Frackiewicz E.J., Cutler N.R. Expert Opin. Inv. Drug. 2000;9:2393. doi: 10.1517/13543784.9.10.2393. [DOI] [PubMed] [Google Scholar]
- 525.Mucke H.A.M. Drug Today. 1997;33:251. [Google Scholar]
- 526.Mucke H.A.M. Drug Today. 1997;33:259. [Google Scholar]
- 527.Greenblatt H.M., Kriger G., Lewis T., Silman I., Sussman J.L. FEBS Lett. 1999;463:321. doi: 10.1016/s0014-5793(99)01637-3. [DOI] [PubMed] [Google Scholar]
- 528.Greenblatt H.M., Guillou C., Guenard D., Argaman A., Botti S., Badet B., Thal C., Silman I., Sussman J.L. J. Am. Chem. Soc. 2004;126:15405. doi: 10.1021/ja0466154. [DOI] [PubMed] [Google Scholar]
- 529.Guillou C., Mary A., Renko D.Z., Gras E., Thal C. Bioorg. Med. Chem. Lett. 2000;10:637. doi: 10.1016/s0960-894x(00)00059-7. [DOI] [PubMed] [Google Scholar]
- 530.Scott L.J., Goa K.L. Drugs. 2000;60:1095. doi: 10.2165/00003495-200060050-00008. [DOI] [PubMed] [Google Scholar]
- 531.Arias E., Ales E., Gabilan N.H., Cano-Abad M.F., Villarroya M., Garcia A.G., López M.G. Neuropharmacology. 2004;46:103. doi: 10.1016/s0028-3908(03)00317-4. [DOI] [PubMed] [Google Scholar]
- 532.Kihara T., Sawada H., Nakamizo T., Kanki R., Yamashita H., Maelicke A., Shimohama S. Biochem. Biophys. Res. Commun. 2004;325:976. doi: 10.1016/j.bbrc.2004.10.132. [DOI] [PubMed] [Google Scholar]
- 533.Radicheva N., Mileva K., Stoyanova N., Georgieva B. Method. Find. Exp. Clin. Pharmacol. 1999;21:5. doi: 10.1358/mf.1999.21.1.527011. [DOI] [PubMed] [Google Scholar]
- 534.Blacker C.V.R., Greenwood D.T., Wesnes K.A., Wilson R., Woosward C., Howe I., Ali T. J. Am. Med. Assoc. 2004;292:1195. doi: 10.1001/jama.292.10.1195. [DOI] [PubMed] [Google Scholar]
- 535.Rainer M. Drug Today. 1997;33:273. [Google Scholar]
- 536.Ezio G. Neurochem. Int. 1998;32:413. doi: 10.1016/s0197-0186(97)00124-1. [DOI] [PubMed] [Google Scholar]
- 537.Hermann A.M., Mucke P.D. Drug Today. 1997;33:251. [Google Scholar]
- 538.Nordberg A., Svensson A.L. Drug Saf. 1998;19:465. doi: 10.2165/00002018-199819060-00004. [DOI] [PubMed] [Google Scholar]
- 539.Kewitz H. Drug Today. 1997;33:265. [Google Scholar]
- 540.Rainer M. CNS Drugs. 1997;7:89. doi: 10.2165/00023210-199707020-00001. [DOI] [PubMed] [Google Scholar]
- 541.M. A. Raskind, E. R. Peskind, T. Wessel, W. Yuan, and the Galanthamine USA-1 Study Group, Neurology54, 2261 (2000). [DOI] [PubMed]
- 542.Zhao Q., Brett M., van Osselaer N., Huang F., Raoult A., van Peer A., Verhaeghe T., Hust R. J. Clin. Pharmacol. 2002;42:1002. [PubMed] [Google Scholar]
- 543.Patterson C.E., Passmore A.P., Crawford V.L.S. Int. J. Clin. Pract. 2004;58:144. doi: 10.1111/j.1368-5031.2004.0107.x. [DOI] [PubMed] [Google Scholar]
- 544.Heinrich M., Teoh H.L. J. Ethnopharmacol. 2004;92:147. doi: 10.1016/j.jep.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 545.Jones R.W., Soininen H., Hager K., Aarsland D., Passmore P., Murthy A., Zhang R., Bahra R. Int. J. Geriatr. Psych. 2004;19:58. doi: 10.1002/gps.1038. [DOI] [PubMed] [Google Scholar]
- 546.Mannens G.S.J., A W. Snel C., Hendrickx J., Verhaeghe T., Le Jeune L., Bode W., van Beijsterveldt L., Lavrijsen K., Leempoels J., van Osselaer N., van Peer A., Meuldermans W. Drug Metab. Dispos. 2002;30:553. doi: 10.1124/dmd.30.5.553. [DOI] [PubMed] [Google Scholar]
- 547.Bores G.M., Kosley R.W. Drug Future. 1996;21:621. [Google Scholar]
- 548.Radicheva N., Vydevska M., Mileva K. Method. Find. Exp. Clin. Pharmacol. 1996;18:301. [PubMed] [Google Scholar]
- 549.Allain H., Bentué-Ferrer D., Belliard S., Derouesné C. In: Ellis G.P., Luscombe D.K., editors. vol. 34. Elsevier; Amsterdam: 1997. p. 1. (Progress in Medicinal Chemistry). [Google Scholar]
- 550.Hermann A.M., Mucke P.D. Drug Today. 1997;33:259. [Google Scholar]
- 551.Czollner L., Frantsits W., Kuenburg B., Hedenig U., Frohlich J., Jordis U. Tetrahedron Lett. 1998;39:2087. [Google Scholar]
- 552.Svensson A.L., Nordberg A. In: Alzheimer's Disease: Biology, Diagnosis and Therapeutics. Iqbal K., Winblad B., Nishimura T., Takeda M., Wisniewski H.M., editors. Wiley; West Sussex: 1997. p. 751. [Google Scholar]
- 553.Bullock R. Expert Rev. Neurotherapeutics. 2004;4:153. doi: 10.1586/14737175.4.2.153. [DOI] [PubMed] [Google Scholar]
- 554.Dengiz A.N., Kershaw P. CNS Spectr. 2004;9:377. doi: 10.1017/s1092852900009378. [DOI] [PubMed] [Google Scholar]
- 555.Bores G.M., Huger F.P., Petko W., Mutlib A.E., Camacho F., Rush D.K., Selk D.E., Wolf V., Kosley R.W., Davis L., Vargas H.M. J. Pharmacol. Exp. Ther. 1996;227:728. [PubMed] [Google Scholar]
- 556.Bachus R., Bickel U., Thomsen T., Roots I., Kewitz H. Pharmacogenetics. 1999;9:661. [PubMed] [Google Scholar]