

Graphical abstract
By using structure-based virtual screening approach in conjunction with chemical synthesis and bioassay, four potent CypA inhibitors have been discovered. Compound 16h is active with very close potency to CsA in inhibiting the proliferation of spleen cells, demonstrating that this compound may be a good lead for discovering new immunosuppressive agents.

Keywords: Cyclophilin A, Inhibitor, Drug design, Virtual screening
Abstract
Cyclophilin A (CypA) is a member of cyclophilins, a family of the highly homologous peptidyl prolyl cis–trans isomerases (PPIases), which can bind to cyclosporin A (CsA). CypA plays critical roles in various biological processes, including protein folding, assembly, transportation, regulation of neuron growth, and HIV replication. The discovery of CypA inhibitor is now of a great special interest in the treatment of immunological disorders. In this study, a series of novel small molecular CypA inhibitors have been discovered by using structure-based virtual screening in conjunction with chemical synthesis and bioassay. The SPECS_1 database containing 85,000 small molecular compounds was searched by virtual screening against the crystal structure of human CypA. After SPR-based binding affinity assay, 15 compounds were found to show binding affinities to CypA at submicro-molar or micro-molar level (compounds 1–15). Seven compounds were selected as the starting point for the further structure modification in considering binding activity, synthesis difficulty, and structure similarity. We thus synthesized 40 new small molecular compounds (1–6, 15, 16a–q, 17a–d, and 18a–l), and four of which (compounds 16b, 16h, 16k, and 18g) showed high CypA PPIase inhibition activities with IC50s of 2.5–6.2 μM. Pharmacological assay indicated that these four compounds demonstrated somewhat inhibition activities against the proliferation of spleen cells.
1. Introduction
Cyclophilins were discovered originally for their high affinity against cyclosporin A (CsA), an immunosuppressive drug used to prevent allograft rejection.1 The general biological function of cyclophilins is to catalyze the cis–trans isomerization of peptide bonds preceding proline in protein and peptides.2 Of the 15 known human cyclophilins, cyclophilin A (hCypA) is one of the most important members and has been widely studied for mapping its biological functions.3 In addition to a binding target of CsA, hCypA is able to enhance the rate of folding (or unfolding) of proteins via its peptidyl-prolyl isomerase (PPIase) activity.4 hCypA performs an essential function in HIV-1 replication by binding specifically with the capsid domain (CA) of the Gag polyprotein.5 hCypA may also render neuroprotective/neurotrophic effects6 when presented at high levels in the brain.7 Recently, it was discovered that the nucleocapsid (N) protein of SARS coronavirus (SARS-CoV) can bind to hCyPA, which may be associated with SARS-CoV infection.8 CypA has attracted considerable attention not only for its novel catalytic activity, but also for its therapeutic significance. It is reported that CypA–CsA binding might inhibit the serine/threonine phosphatase activity of calcineurin, thereby blocking the production of cytokines including interleukin-29 and interferon γ.10
The discovery of CsA more than two decades ago heralded a new era in the field of organ transplantation. Since then, the number of transplanted organs has grown continuously and the search for novel immunosuppressants has intensified.11 However, inhibitors of CypA are mainly derived from the natural sources (such as FK506,12 rapamycin,13 and sanglifehrin A14) and peptide analogues,15 which are all large molecules, and little has been reported regarding the small molecule CypA inhibitors.
Although immunosuppressants like CsA have improved the rates of transplant success and prolonged patient survivals, some side effects with this agent have been sequentially found such as nephrotoxicity,16 hypertension17, and cardiotoxicity.18 This thereby prompted us to discover novel small molecule CypA inhibitors in order to decrease the side effects. Structure-based ligand design has led to the identification of compounds that are currently in clinical trials or into the market.19 The wealth of structural and functional information of CypA15 has offered a solid starting point for the rational structure-based design of inhibitors. By using docking-based virtual screening approach in conjunction with surface plasmon resonance (SPR) determination, 15 novel small molecule CypA binders (hits) (compounds 1–15) have been discovered. According to the binding potency, structural similarity, and synthetic complexity, seven hits (compounds 1–6 and 15) were selected as the starting points for further structural optimization. Totally, 40 new compounds including 1–6 and 15 (the preparation methods for these seven compounds have not been reported) and their 33 analogues (16a–q, 17a–d, and 18a–l) have been synthesized and tested with biological assay. Finally, four compounds (16b, 16h, 16k, and 18g) were found to show high CypA PPIase (peptidyl-prolyl isomerase) inhibition activities and one compound (16h) is active in inhibiting the proliferation of spleen cells.
2. Materials and methods
2.1. Small molecular database for virtual screening
SPECS database contains the structural information of 280,000 small molecules (http://www.specs.net). SPECS Company supplies all the compound samples collected from difference sources. First, SPECS database was evaluated using our own filter of druglikeness.20 Non-druglike molecules were removed from the database, and finally 85,000 potentially druglike molecules were selected out for docking screening. These molecules were saved in the SPECS_1 database.
2.2. Virtual screening by molecular docking
The crystal structure of CypA in complex with sanglifehrin macrolide (SFM) (PDB entry 1NMK)14 recovered from the Brookhaven Protein Data Bank (http://www.rcsb.org/pdb) was used as a target for virtual screening on the SPECS_1 database (http://www.specs.net). In the present study, the DOCK4.0 program21 was employed for the primary screening. Residues of CypA around SFM at a radius of 6.5 Å were isolated for constructing the grids of the docking screening. The resulting substructure included all residues of the binding pocket. During the docking calculation, Kollman-all-atom charges22 were assigned to the protein and Gasteiger–Marsili partial charges23 were assigned to the small molecules in the SPECS_1 database. Conformational flexibility of the compounds from the database was considered in the docking search.
The DOCK suite of programs is designed to find possible orientations of a ligand in a ‘receptor’ site.21 The orientation of a ligand is evaluated with a shape scoring function and/or a function approximating the ligand–receptor binding energy. The shape scoring function is an empirical function resembling the van der Waals attractive energy. The ligand–receptor binding energy is taken to be approximately the sum of the van der Waals and electrostatic interaction energies. After the initial orientation and scoring evaluation, a grid-based rigid body minimization is carried out for the ligand to locate the nearest local energy minimum within the receptor binding site. The position and conformation of each docked molecule were optimized using single anchor search and torsion minimization method of DOCK4.0.21 Thirty configurations per ligand building a cycle and 50 maximum anchor orientations were used in the anchor-first docking algorithm. All docked configurations were energy minimized using 100 maximum iterations and 1 minimization cycle.
The 5000 molecules with the highest score as obtained by DOCK search were re-scored by using the Consensus Score (CScore)24 method encoded in Sybyl6.8.25 The binding free energies and the binding modes of the top 1301 molecules with CScore ⩾4 to CypA were calculated and modeled using the FlexX26 program encoded in Sybyl6.8.25 FlexX is a flexible docking method that uses an incremental construction algorithm to place ligand into an active site. By dividing the ligand into small fragments and building incrementally, the conformational flexibility of the ligand can be considered in reasonable times. The Kollman-all-atom charge22 and Gasteiger-Marsili partial charge23 were used for cyclophilin A and candidate molecules, respectively, in the FlexX simulations.
Finally 82 compounds were distinguished and selected for bioassay on the basis of score by virtual screening and druglikeness analysis. The virtual screening was performed on a 128-processor SGI Origin 3800 supercomputer.
2.3. Chemistry
2.3.1. Design of analogues of compounds 1–6, 15
Compounds1–6 and 15 (Fig. 1 ) bearing the highest binding affinities with CypA as obtained by the surface plasmon resonance (SPR) determination (see Section 4) for the 82 candidate compounds selected by virtual screening were used as lead compounds for designing new CypA inhibitors. Based on the structural and enzyme binding activity features of compounds 1–6 and 15, 33 new analogues (16a–q, 17a–d, and 18a–l) (Table 1, Table 2 ) were designed and synthesized for the first round. Keeping the key groups of compounds 1–4—2-substituted quinoline ring and sulfanilamide moiety—we used various setric, electronic, and hydrophobic groups to substitute at position 2 of the quinoline ring in compounds 1–4 (Fig. 1) and obtained 10 analogues (16a–j) (Table 1). Replacing the quinoline ring with a pyridine ring, we obtained seven compounds (16k–q) (Table 1). Keeping the key groups of compounds 5 and 6—phenyl alkyl acylamide and sulfanilamide moiety—we changed the length of the alkyl linkers, coupled with the sulfisoxazole of compound 1, and obtained four compounds (17a–d) (Table 1). On the basis of the structural features of compound 15, we designed compounds 18a–j (Table 2), maintained dibenzoic acid or dibenzoic acid ester moiety, and replaced 3,5-dimethoxybenzamido group in compound 15 with other electronic and hydrophobic substituted benzamido groups. Substituting the methylene group of compound 15 with an ethylene group, we obtained compounds 18k–l (Table 2).
Figure 1.
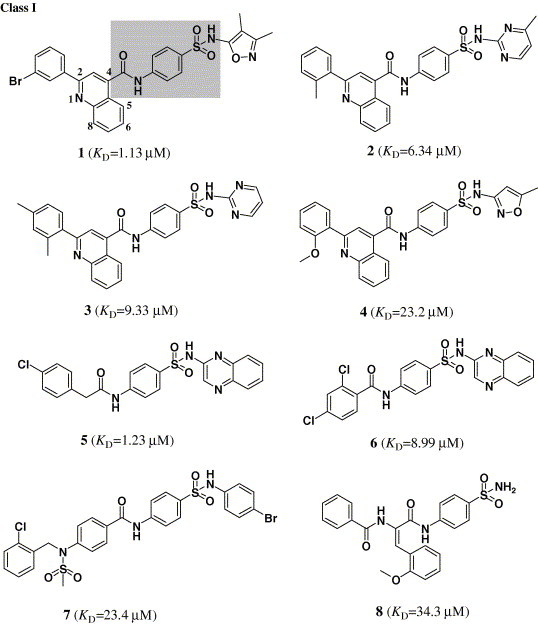
The structures of 15 binders (or hits) of CypA selected from the candidates by virtual screening and SPR-based binding affinity assay.
Table 1.
Chemical structures of compounds 1–6 and 16–17, and their activities 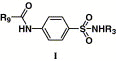
| Compound | R9 | R3 | KDa (μM) | % inhibition at 10 μMb |
|---|---|---|---|---|
| 1 | 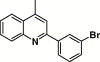 |
3,4-Dimethyl-5-isoxazolyl | 1.13 | 3.20 |
| 2 | 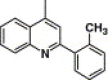 |
4-Methyl-2-pyrimidinyl | 6.34 | 11.2 |
| 3 | 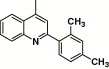 |
2-Pyrimidinyl | 9.33 | 10.2 |
| 4 | 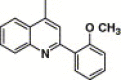 |
5-Methyl-3-oxazolyl | 23.2 | 26.5 |
| 16a | 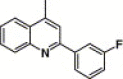 |
3,4-Dimethyl-5-isoxazolyl | 10.2 | 18.0 |
| 16b | 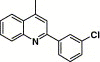 |
3,4-Dimethyl-5-isoxazolyl | 52.3 | 67.3 |
| 16c |  |
3,4-Dimethyl-5-isoxazolyl | 4.11 | 10.0 |
| 16d | 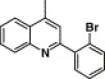 |
3,4-Dimethyl-5-isoxazolyl | 223 | 15.0 |
| 16e | 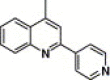 |
3,4-Dimethyl-5-isoxazolyl | 10.6 | 40.0 |
| 16f | 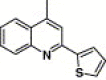 |
3,4-Dimethyl-5-isoxazolyl | 91.3 | 30.0 |
| 16g | 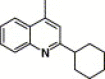 |
3,4-Dimethyl-5-isoxazolyl | 1.79 | NA |
| 16h | 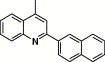 |
3,4-Dimethyl-5-isoxazolyl | 10.1 | 77.4 |
| 16i | 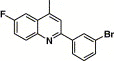 |
3,4-Dimethyl-5-isoxazolyl | 2.25 | 32.0 |
| 16j | 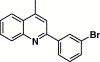 |
2-Quinoxalinyl | 3.13 | 36.0 |
| 16k | 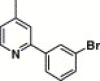 |
3,4-Dimethyl-5-isoxazolyl | 46.0 | 74.7 |
| 16l | 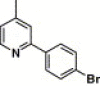 |
3,4-Dimethyl-5-isoxazolyl | 89.5 | NA |
| 16m | 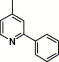 |
3,4-Dimethyl-5-isoxazolyl | 14.5 | NA |
| 16n | 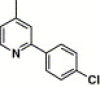 |
3,4-Dimethyl-5-isoxazolyl | 1.86 | NA |
| 16o | 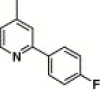 |
3,4-Dimethyl-5-isoxazolyl | 42.4 | 26.0 |
| 16p | 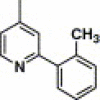 |
3,4-Dimethyl-5-isoxazolyl | 7.61 | NA |
| 16q | 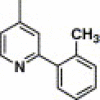 |
4-Methyl-2-pyrimidinyl | 340 | NA |
| 5 |  |
2-Quinoxalinyl | 1.23 | 3.2 |
| 6 | 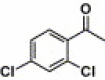 |
2-Quinoxalinyl | 8.99 | 7.5 |
| 17a | 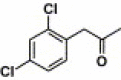 |
2-Quinoxalinyl | 37.7 | NA |
| 17b | 2-Quinoxalinyl | 23.4 | 5.6 | |
| 17c | 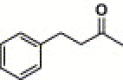 |
2-Quinoxalinyl | 1.06 | NA |
| 17d |  |
3,4-Dimethyl-5-isoxazolyl | 0.35 | NA |
Data obtained from SPR-based binding affinity assay. KD represents the equilibrium constant.
Data are means of three independent experiments.
Table 2.
Chemical structures of compounds 15 and 18 and their activities 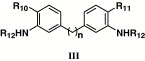
| Compound | n | R10 | R11 | R12 | KDa (μM) | % inhibition at 10 μMb |
|---|---|---|---|---|---|---|
| 15 | 1 | COOH | COOH | 3,5-Dimethoxy-benzoyl | 0.58 | 12.4 |
| 18a | 1 | CO2CH3 | CO2CH3 | 3,5-Dimethoxy-benzoyl | NA | NA |
| 18b | 1 | COOH | COOH | Benzoyl | NAc | NAc |
| 18c | 1 | CO2CH3 | CO2CH3 | Benzoyl | 32.3 | 11.2 |
| 18d | 1 | COOH | COOH | 4-Methoxy-benzoyl | NAc | NAc |
| 18e | 1 | CO2CH3 | CO2CH3 | 4-Methoxy-benzoyl | NA | NA |
| 18f | 1 | CO2CH3 | CO2CH3 | 4-Cl-Benzoyl | 140 | 10.2 |
| 18g | 1 | CO2CH3 | CO2CH3 | Benzyl | 414 | 86.0 |
| 18h | 1 | COOH | CO2CH3 | Benzyl | 0.23 | 6.5 |
| 18i | 1 | COOH | COOH | Benzyl | 184 | NA |
| 18j | 1 | — | — | 3,5-Dimethoxy-benzoyl | 3.3 | 43.0 |
| 18k | 2 | CO2CH3 | CO2CH3 | 3,5-Dimethoxy-benzoyl | 87.5 | 15.6 |
| 18l | 2 | COOH | COOH | 3,5-Dimethoxy-benzoyl | NAc | NAc |
Data obtained from SPR-based binding affinity assay. KD represents the equilibrium constant.
Data are means of three independent experiments.
These compounds cannot be assayed for poor solubility in DMSO.
2.3.2. Synthetic procedures
Scheme 1 depicts the sequence of reactions that led to the preparation of compounds 1–4 and 16a–j using isatins (19) as the starting material. In general, the substituted 4-quinolinecarboxylic acids 20 were prepared by isatins (19) condensation with ketones in ethanol. Compound 20 was converted to the corresponding acyl chlorides (21) by refluxing with SOCl2.27 Then 21 was substituted by using different sulfanilamides, giving the target compounds 1–4 and 16a–j (Table 1). The key intermediate 25 required by synthesizing compound 16j was prepared from compound 23 28 by sulfamation and deprotection.
Scheme 1.
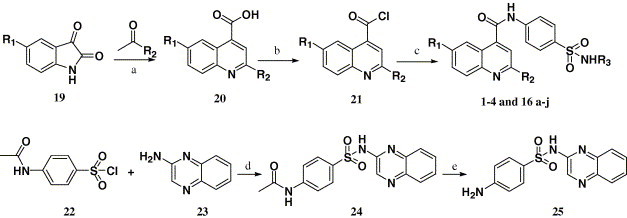
Reagents and conditions: (a) (i) KOH, C2H5OH, H2O, reflux, (ii) HCl, H2O; (b) SOCl2, reflux; (c) R3NHSO2C6H6NH2-p, pyridine, 25 °C; (d) pyridine, 25 °C; (e) (i) NaOH, H2O, reflux, (ii) HCl.
Compounds 16k–q were synthesized through the route outlined in Scheme 2 . 2-Bromo-1-arylethanone (26) was reacted by refluxing with ammonium formate in 98% HCOOH, which afforded 4-aryloxazole (27).29 Compound 28 was synthesized using the Usui Y. method30 by compound 27 reacting with maleic acid at 110 °C for 15 min. Then, the target compounds 16k–q were obtained after steps c–d of Scheme 1 were employed (Table 1).
Scheme 2.
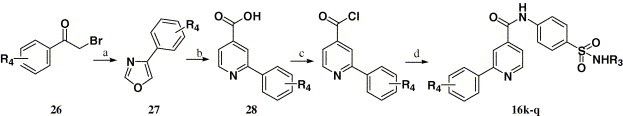
Reagents and conditions: (a) HCOOH (98%), HCOONH4, reflux; (b) maleic acid, 110 °C; (c) SOCl2, reflux; (d) R3NHSO2C6H6NH2-p, pyridine, 25 °C.
Compounds 5–6 and 17a–d were synthesized by using the same process as compounds 1–4, except compound 20 was replaced by compound 29. The synthetic route is outlined in Scheme 3 .
Scheme 3.
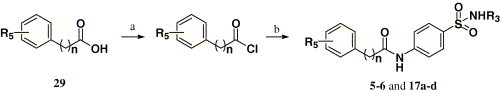
Reagents and conditions: (a) SOCl2, reflux; (b) R3NHSO2C6H6NH2-p, pyridine, 25 °C.
Scheme 4 depicts the sequence of reactions that led to the preparation of compounds 15 and 18a–i using anthranilic acid (30) as the starting material. Compound 31a 31 was synthesized by heating anthranilic acid (30) with HCHO in 36% HCl. Refluxing 31a with saturated HCl in methanol afforded compound 31b. Acylation 31a–b with various substituted benzoyl chlorides in pyridine afforded compounds 15 and 18a–f. Compound 31b reacted with benzyl bromide in refluxing NMP produced compound 18g, which was hydrolyzed using LiOH at 25 and 80 °C to afford monoester 18h and dicarboxylic acid 18i, respectively.
Scheme 4.
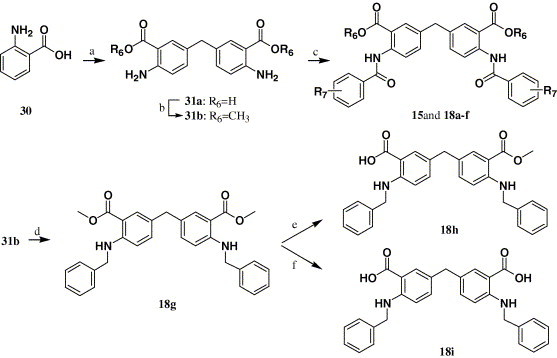
Reagents and conditions: (a) 3% HCHO, HCl, H2O; (b) CH3OH, HCl; (c) substituted benzoyl chlorides, pyridine, 25 °C; (d) benzyl bromide, NMP, Na2CO3, H2O; (e) LiOH, THF/CH3OH/H2O = 3:1:1, 25 °C; (f) LiOH, THF/CH3OH/H2O = 3:1:1, 80 °C.
Compounds 18j–l were synthesized through the route outlined in Scheme 5 . Refluxing 5-methyl-2-nitrobenzoic acid (32) with saturated HCl in methanol afforded compound 33, which was then brominated by NBS in chloroform to afford compound 34.32 Compound 34 was coupled with compound 33, giving the key intermediate 35 in the presence of n-BuLi at −78 °C, which was then reduced by Pd-C. Compound 18j was obtained by N-acylation of 37 with 3,5-dimethoxybenzoyl chloride in pyridine. Compound 18k was obtained under the same conditions as compound 18a, and compound 18l was quantitatively obtained by hydrolysis of 18k using LiOH at room temperature.
Scheme 5.
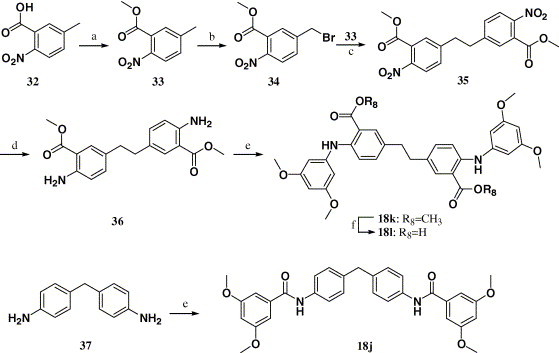
Reagents and conditions: (a) CH3OH, HCl; (b) NBS, CHCl3, BzooBz; (c) n-BuLi, THF, −78 °C; (d) Pd-C, H2, THF; (e) 3,5-dimethoxybenzoyl chloride, pyridine, 25 °C; (f) LiOH, THF/CH3OH/H2O = 3:1:1, 25 °C.
2.4. Biological assay
2.4.1. Binding assay
CypA was expressed and purified according to our previously reported procedure.8 The binding affinities of the virtual screening candidates to CypA in vitro were determined by employing the surface plasmon resonance (SPR) technology. The measurement was performed using the dual flow cell Biacore 3000 instrument (Biacore AB, Uppsala, Sweden). Immobilization of the CypA to the hydrophilic carboxymethylated dextran matrix of the sensor chip CM5 (Biacore) was carried out by the standard primary amine coupling reaction wizard. The CypA to be covalently bound to the matrix was diluted in 10 mM sodium acetate buffer (pH 6.0) to a final concentration of 0.035 mg/mL. Equilibration of the baseline was completed by a continuous flow of HBS-EP running buffer (10 mM Hepes, 150 mM NaCl, 3 mM EDTA, and 0.005% (v/v) surfactant P20, pH 7.4) through the chip for 1–2 h. All the Biacore data were collected at 25 °C with HBS-EP as running buffer at a constant flow rate of 30 μL/min. All the sensorgrams were processed by using automatic correction for non-specific bulk refractive index effects. The equilibrium constants (K D values) evaluating the protein–ligand binding affinities were determined by the steady state affinity fitting analysis of the Biacore data.
2.4.2. Enzymatic activity assay
CypA PPIase activity was measured at 4 °C by using the standard chymotrypsin-coupled assay.33 The assay buffer (50 mM Hepes, 100 mM NaCl, pH 8.0) and CypA (500 nM stock solution) were pre-cooled to 4 °C, to which then was added 15 μL of 3 mg/mL chymotrypsin in 1 mM HCl. The reaction was initiated by adding 12 μL of 3.8 mM peptide substrate (Suc-Ala-Ala-cis-Pro-Phe–pNA) in LiCl/THF solution with rapid inversion. After a delay from the onset of mixing (usually 6 s), the absorbance of p-nitroaniline was followed at 390 nM until the reaction was complete (1 min). The final concentration of LiCl in the assay was 9.6 mM; THF was present at a concentration of 2% (v/v). Absorbance readings were collected every 0.1 s by a U-2010 spectrophotometer. The progress curves were analyzed by non-linear least-squares fit.
The inhibition assays of compounds were performed in the same manner as mentioned above. A 0.6 μL aliquot of the compounds in DMSO was added to the CypA solution in the assay buffer. After being pre-incubated for 1 h at 4 °C, the assay was started by the addition of chymotrypsin and the substrate. To calculate the half-maximal inhibitory concentration (IC50), the percent of remaining PPIase activity is plotted against the common logarithm of the compound concentration, and the data were fitted using the sigmoidal fitting model by the Origin7.0 software.
2.4.3. T cell viability assay
Female ICR strains of mice, 6–8 weeks old and 20 ± 2 g, were purchased from the Experimental Animal House of China Pharmaceutical University (Nanjing, China) and were maintained in plastic cages with free access to pellet food and water (12 h light/dark cycle, 21 ± 2 °C). This study complied with the current ethical regulations on animal research in Nanjing University and the mice used were treated humanely.
The spleen was aseptically taken from mice, crushed gently, and separated into single cells by squeezing in 5 mL d-Hanks’ solution (Gibco-BRL). The cells obtained were passed through eight layers of gauze and centrifuged at 1000 rev/min for 5 min at 4 °C. Pellets were added into 10 mL of sterile 0.17 M Tris-(hydroxymethyl) aminomethane containing 0.75% NH4Cl (pH 7.5) followed by centrifugation to remove erythrocytes. After washing twice with RPMI 1640 containing 100 U mL−1 penicillin, 100 U mL−1 streptomycin, and 10% fetal calf serum (FCS) (RPMI 1640 medium), they were re-suspended in the RPMI 1640 medium and used for cell culture.
Spleen cells were cultured in 96-well flat-bottomed microplates (Falcon) at a density of 5 × 105 cells per well in RPMI 1640 medium (0.2 mL) and stimulated with 5 μg mL−1 Con A for 72 h at 37 °C in 5% CO2-air in the presence or absence (control group) of various concentrations of compounds. Then the cell growth was evaluated with modified MTT assay.34 Briefly, 20 μL of 5 mg mL−1 MTT in RPMI 1640 was added for a further 4 h incubation. After removing the supernatant, 200 μL of DMSO was added to dissolve the formazan crystals. The plate was shaken for 10 min and then read on an ELISA reader (Sunrise Remote/Touch Screen; TECAN, Austria) at 540 nm. All assays were run in triplicate and the effect of compounds on the proliferation of mouse spleen cells induced by Con A was calculated by Eq. 1
| (1) |
Data were expressed as means ± SD (significance of differences between groups). Statistical analysis was evaluated by one-way analysis of variance, followed by Student’s two-tailed t test for the evaluation of the difference between two groups and Dunnett’s t test between control group and multiple dose groups. One-way analysis of variance revealed a significant effect at P < 0.05.
3. Results and discussion
3.1. Identification of binders (hits) by virtual screening
Targeting the crystal structure of CypA (PDB entry 1NMK),14 we searched the SPECS_1 database by using DOCK4.0.21 The small molecules were ranked according to their scores calculated by using the scoring function of DOCK. The top 5000 candidate molecules were obtained with the best scores by a shape complementarity scoring function in DOCK.21 These compounds were re-estimated using Cscore.24 A total of 1301 compounds with Cscore = 4 or 5 (the best score of Cscore is 5) were subsequently docked and ranked using the FlexX26 program. Finally, 82 compounds were selected according to the score of FlexX for biological assay. Because enzymatic assay is time-consuming, therefore, surface plasmon resonance (SPR) measurements were used for the primary screening, determining the binding affinity of these 82 candidate molecules to CypA. Immobilization of CypA resulted in a resonance signal at about 5370 resonance units (RUs). Among the 82 compounds, the biosensor RUs of 15 compounds were concentration-dependent. The collected data indicated that these 15 compounds can bind to CypA in vitro and the binding affinities to cyclophilin A are in the submicro- or micro-molar range (K D = 53.4–0.58μM). These compounds could be designated as binders (or hits) of CypA, their chemical structures and binding affinities shown in Figure 1.
3.2. Analogue design and synthesis
Structurally, the 15 hits can be divided into three classes: class I includes eight compounds containing sulfanilamide moiety (compounds 1–8); class II compounds are acylhydrazone derivatives (compounds 9–12); class III is comprised by compounds 13–15, which have no distinct structural character (Fig. 1). Considering the structural similarity and the binding affinity with CypA, we took compounds 1–6 in class I and compound 15 in class III as leaders for structural optimization. Totally, 40 compounds (1–6, 15, 16a–q, 17a–d, and 18a–l) were designed and synthesized, and their chemical structures are shown in Table 1, Table 2. These compounds were synthesized through the routes outlined in Scheme 1, Scheme 2, Scheme 3, Scheme 4, Scheme 5, and the details for synthetic procedures and structural characterizations are described in the Experimental section.
3.3. Biological activities
3.3.1. The PPIase activity of CypA
For the primary assay, the percent inhibitions of the compounds at 10 μM were measured. The results are listed in Table 1, Table 2. Four compounds, i.e., 16b, 16h, 16k, and 18g, can remarkably inhibit the PPIase activity of CyPA (percent inhibition at 10 μM > 50%), indicating that these four compounds are CypA inhibitors. Therefore, we determined their IC50 values, which are, respectively, 4.88, 2.84, 2.48, and 6.23 μM (Fig. 2 ). In addition, the binding affinities of the 33 analogues were determined by using the SPR technology, the results also listed in Table 1, Table 2. The PPIase activities of the compounds do not consistently correlate with the SPR binding affinities. The reason may be that the protein was immobilized to a sensor chip in the SPR assay, which affects the conformational flexibility of the protein. Accordingly, good binders identified by the SPR assay may not be good inhibitors. To find CypA inhibitors rather than binders, the effects of compounds on the PPIase activity of CypA have to be determined.
Figure 2.
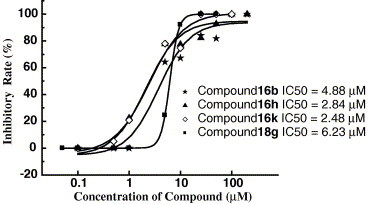
Concentration dependence of PPIase activity by 16b, 16h, 16k, and 18g, the concentration of CyPA was kept constant at 25 nM, while the concentration of compounds ranged from 0.1 to 200 μM.
3.3.2. Inhibition activity against the proliferation of spleen cells
To test the immunosuppressive effects of 16a, 16h, 16k, and 18g, anti-proliferation activities on spleen cells in vitro of the four inhibitors have been evaluated. The result is listed in Table 3 , indicating that compounds 16a, 16h, and 16k show partial influence on the viability of the naïve spleen cells. It is remarkable that the effective concentration of compound 16h is ∼10 μM in inhibition of the proliferation of mouse spleen cells induced by Con A, which is slightly lower than that of CsA. This indicates that compound 16h is a good lead for designing more potent immunosuppressive agents.
Table 3.
Effect of compounds 16b, 16h, 16k, 18l, and CsA on the proliferation of mouse spleen cells induced by Con A in vitro
| Dose (μM) | Inhibitory rate (%)a |
||||
|---|---|---|---|---|---|
| CsA | 16b | 16h | 16k | 18g | |
| 100 | 84.31 ± 4.88b | 47.35 ± 4.98b | 46.74 ± 5.39b | 49.05 ± 4.69b | 28.83 ± 3.39b |
| 10 | 82.57 ± 5.20b | 29.78 ± 5.00 | 34.26 ± 5.60b | 22.71 ± 7.06 | 7.46 ± 4.13 |
| 1 | 56.52 ± 1.60 | 14.13 ± 9.97 | 20.76 ± 8.41 | 15.33 ± 7.81 | 1.20 ± 6.41 |
Each value indicates the mean ± SD of three experiments using three mice with triplicate sets in each assay.
One-way analysis of variance revealed a significant effect at P < 0.05.
3.4. Binding models
To gain structural information for further structural optimization, the 3D binding models of the designed compounds to CypA were generated based on the docking simulation, Figure 3 shows the interaction models of four potent inhibitors (16a, 16h, 16k, and 18g) to CypA. From Figure 3, one can see that there are two binding sites in the surface binding groove of CypA: site A is a small binding pocket formed by Lys82, Met100, Ala101, Asn102, Ala103, Thr107, Asn108, Gly109, Ser110, Gln111, and Phe112; near site A, there is a larger pocket, which is called site B, formed by Phe60, Met61, Gln63, Ala101, Asn102, Ala103, Phe113, Leu122, and His126; there is a ‘saddle’ between the two sites. The binding models (Figs. 3a–c) indicate that class I compounds (Table 1) interact with CypA in a similar way: the common sulfanilamide moiety occupies the polar binding site (site B), forming three to four hydrogen-bonds (H-bonds) with residues Gln63, Gly75, Asn102, Thr107, Ser110, and Gln111 (Fig. 4 ); the 2-substituents of quinoline rings (1–4 and 16a–j) and pyridine rings (16k–q) or aryl rings (5, 6 and 17a–d) fill in the hydrophobic pocket (site A); and the quinoline moiety (1–4 and 16a–j), the pyridine moiety (16k–q) or alkyl groups (5, 6 and 17a–d) act as linkers interacting with the saddle part between sites A and B. The interacting models for class III compounds to CypA are mostly similar to those for class I compounds, except that compounds 15, 18a, 18d–f, and 18j–l extend from site B due to their longer linkers and/or substituents.
Figure 3.
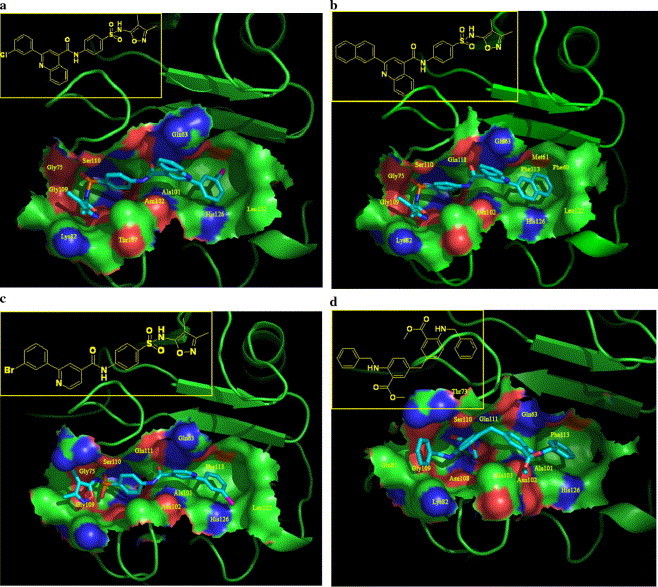
Three-dimensional structural modes of inhibitors 16b (a), 16h (b), 16k (c), and 18g (d) to CypA derived from the docking simulations. These four images were generated using the Pymol program (http://www.pymol.org/). The CypA surface was colored by electrostatic potential.
Figure 4.
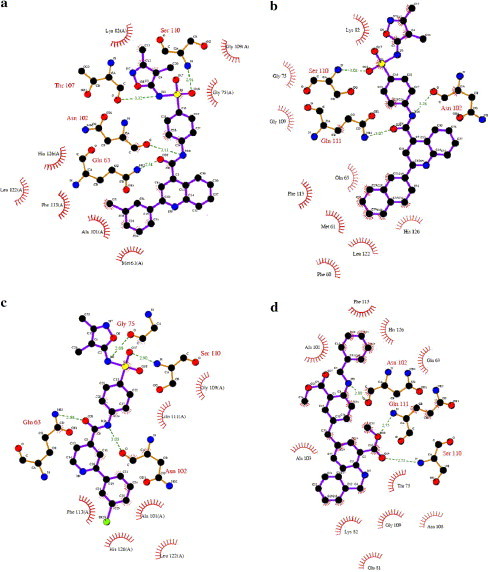
Two-dimensional representation for the interacting mode of compounds 16b (a), 16h (b), 16k (c), and 18g (d) with CypA. It is drawn using the LIGPLOT program.35 A distance between donor and acceptor of less than 3.4 Å is considered as a hydrogen-bond, and a 4.1 Å distance between two hydrophobic atoms is considered a hydrophobic interaction.
In summary, we have discovered four potent CypA inhibitors (16a, 16h, 16k, and 18g) by using a structure-based virtual screening approach in conjunction with chemical synthesis and bioassay. One of them (compound 16h) is active with very close potency to CsA in inhibiting the proliferation of spleen cells, demonstrating that this compound may be a good lead for discovering new immunosuppressive agents. Discovering small molecule CypA inhibitors is an urgent need, so the new chemical structures produced in this study are of significance.
4. Experimental
The reagents (chemicals) were purchased from Lancaster, Acros, and Shanghai Chemical Reagent Company, and used without further purification. Analytical thin-layer chromatography (TLC) was HSGF 254 (150–200 μm thickness, Yantai Huiyou Company, China). Yields were not optimized. Melting points were measured in capillary tube with a SGW X-4 melting point apparatus without correction. Nuclear magnetic resonance (NMR) spectra were given on a Brucker AMX-400 NMR (IS as TMS). Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Low- and high-resolution mass spectra (LRMS and HRMS) were given with electric, electrospray, and matrix-assisted laser desorption ionization (EI, ESI, and MALDI) produced by Finnigan MAT-95, LCQ-DECA spectrometer, and IonSpec 4.7 T.
4.1. 4-Amino-N-quinoxalin-2-yl benzenesulfonamide (25)
A mixture of 4-acetylamino-benzenesulfonyl chloride 22 (2.34 g, 10 mmol) and compound 15 28 (1.45 g, 10 mmol) in pyridine (20 mL) was stirred overnight at 25 °C, poured into H2O (50 mL), and extracted with EtOAc. The combined organic layer was washed, dried, filtered, and condensed. The residue was purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:2, v/v), to afford 24 (1.08 g, 31.6%) as a pale yellow solid; 1H NMR (DMSO): δ 2.03 (s, 3H), 7.61 (t, 1H), 7.78 (m, 4H), 7.90 (d, 1H), 8.02 (d, 2H), 8.62 (s, 1H); EI-MS m/z 342 (M+), 277 (100%).
A mixture of compound 24 (1 g, 3 mmol) and NaOH (1.5 g, 37.5 mmol) in H2O (15 mL) was refluxed for 2 h to generate a yellow solution, which was acidified to pH 5.5 at 70–80 °C. After cooling to room temperature, the precipitate was collected, washed with H2O, and dried, to afford compound 25 (0.8 g, 91.2%) as a yellow solid: mp 258–260 °C; EI-MS m/z 300 (M+), 236 (100%).
4.2. General procedures for preparations of 2027 are described as those for 2-(3-fluorophenyl)-quiniline-4-carboxylic acid (20a)
As illustrated in Scheme 1, isatin 19 (34.0 mmol), 3-fluoroacetophenone (40.8 mmol), and 85% KOH pellets (102 mmol) were dissolved in EtOH (40 mL), and the reaction mixture was stirred at 80 °C for 24 h. The mixture was condensed, diluted with water, and acidified with concd HCl, and the precipitate was collected, washed, and dried to afford 20a; yield 80%; mp 232–234 °C; 1H NMR (DMSO): δ 7.33–7.42 (m, 3H), 7.55 (d, 1H), 7.75 (m, 1H), 7.87 (m, 1H), 8.02 (s, 1H), 8.13 (d, 1H), 8.71 (d, 1H); EI-MS m/z 266 (M+−1), 222 (100%).
4.3. General procedures for preparations of 28 are described as those for 2-phenylpyridine-4-carbonic acid (28m)
A mixture of α-bromoacetophenone 26m (10 g, 50 mmol), HCOONH4 (11 g, 174 mmol), and 98% HCOOH (53 mL) was refluxed under stirring for 2.5 h. The reaction mixture was poured into H2O, alkalinized with concentrated NaOH, and extracted with Et2O. The combined organic layer was washed, dried, filtered, and condensed. The crude product obtained was purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:10, v/v), to afford 27m (1.2 g, 16.7%) as a yellow oil; 1H NMR (CDCl3): δ 7.33 (d, 1H), 7.42 (d, 2H), 7.75 (d, 2H), 7.94 (m, 2H).
A mixture of 4-phenyloxazole 27m (1 g, 6.9 mmol) and maleic acid (0.83 g, 7.1 mmol) was heated at 110 °C until melting for 2 h. The solid obtained after cooling was triturated with Et2O, collected by suction filtration. The filter cake was recrystallized with 95% EtOH to yield 28m (0.2 g, 14.6%) as a yellow solid: mp 263–264 °C; 1H NMR (DMCO): δ 7.56 (m, 3H), 7.85 (d, 1H), 8.20 (d, 2H), 8.38 (s, 1H), 8.88 (d, 1H). EI-MS m/z 199 (M+) (100%), 155.
4.4. General procedures for preparations of 4-substituted carboxamido sulfanilamides 1–6, 16a–q, and 17a–d are described as those for 1
4.4.1. 4-(2-(3-Bromophenyl)-quinoline-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl)benzene sulfonamide (1)
A suspension of 3-bromophenyl-quinoline-4-carboxylic acid (0.33 g, 1 mmol) in redistilled SOCl2 (6 mL) was refluxed gently for 3 h. A clear solution resulted. The SOCl2 was then evaporated under reduced pressure. The yellow solid thus obtained was used in the next step without further purification.
A mixture of yellow solid thus obtained, sulfisoxazole (0.27 g, 1 mmol), and pyridine (5 mL) was stirred overnight at 25 °C, poured into H2O (30 mL), and extracted with EtOAc. The combined organic layer was washed, dried, filtered, and condensed. The residue was purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:4, v/v), to afford 1 (120 mg, 20.8%) as a white solid: mp 254–256 °C; 1H NMR (DMSO): δ 1.71 (s, 3H), 2.10 (s, 3H), 7.57 (t, 1H), 7.75 (m, 2H), 7.84 (d, 2H), 7.90 (m, 1H), 8.03 (d, 2H), 8.22 (m, 2H), 8.40 (d, 1H), 8.48 (s, 1H), 8.58 (t, 1H); EI-MS m/z 578 (M++1), 203 (100%); HRMS (EI) m/z calcd C27H21BrN4O4S (M+) 576.0473, found 576.0467.
4.4.2. 4-(2-(2-Methylphenyl)-quinoline-4-carboxamido)-N-(4-methyl-pyrimidin-2-yl) benzenesulfonamide (2)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:4, v/v), yield 31.3%: mp 291–293 °C; 1H NMR (DMSO): δ 2.29 (s, 3H), 2.44 (s, 3H), 6.91 (d, 1H), 7.21 (m, 3H), 7.62 (d, 1H), 7.71 (t, 1H), 7.87 (t, 1H), 7.89 (s, 1H), 7.95 (d, 2H), 8.02 (d, 2H), 8.18 (q, 2H), 8.32 (d, 1H); EI-MS m/z 509 (M+), 336 (100%); HRMS (EI) m/z calcd C28H23N5O3S (M+) 509.1518, found 509.1521.
4.4.3. 4-(2-(2,4-Dimethylphenyl)-quinoline-4-carboxamido)-N-pyrimidin-2-yl benzenesulfonamide (3)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 40.0%: mp 240–241 °C; 1H NMR (DMSO): δ 2.36 (s, 3H), 2.42 (s, 3H), 7.05 (t, 1H), 7.21 (m, 2H), 7.57 (d, 1H), 7.71 (t, 1H), 7.87 (m, 2H), 7.96 (d, 2H), 8.02 (d, 2H), 8.17 (q, 2H), 8.52 (d, 2H); EI-MS m/z 509 (M+), 350 (100%); HRMS (EI) m/z calcd C28H23N5O3S (M+) 509.1515, found 509.1521.
4.4.4. 4-(2-(2-Methoxyphenyl)-quinoline-4-carboxamido)-N-(5-methyl-oxazol-2-yl) benzenesulfonamide (4)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 46.9%: mp 266–268 °C; 1H NMR (DMSO): δ 2.31 (s, 3H), 3.82 (s, 3H), 6.15 (s, 1H), 7.17 (t, 1H), 7.22 (d, 1H), 7.56 (t, 1H), 7.67 (t, 1H), 7.92 (m, 4H), 8.02 (d, 2H), 8.10 (s, 1H), 8.18 (t, 2H); EI-MS m/z 514 (M+), 234 (100%); HRMS (EI) m/z calcd C27H22N4O5S (M+) 514.1314, found 514.1311.
4.4.5. 4-(4-Chlorophenylacetamido)-N-quinoxalin-2-yl benzenesulfonamide (5)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 33.2%: mp 263–265 °C; 1H NMR (DMSO): δ 3.65 (s, 2H), 7.35 (m, 4H), 7.62 (t, 1H), 7.78 (m, 4H), 7.92 (d, 1H), 8.05 (d, 2H), 8.62 (br, 1H); EI-MS m/z 452 (M+), 387 (100%); HRMS (EI) m/z calcd C22H17ClN4O3S (M+) 452.0711, found 452.0710.
4.4.6. 4-(2,4-Dichlorobenzamido)-N-quinoxalin-2-yl benzenesulfonamide (6)
THis compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 43.8%: mp 217–219 °C; 1H NMR (DMSO): δ 7.51 (dd, 1H), 7.68 (m, 3H), 7.78 (t, 1H), 7.89 (d, 1H), 7.99 (m, 3H), 8.21 (d, 2H), 8.72 (s, 1H); EI-MS m/z 472 (M+), 173 (100%); HRMS (EI) m/z calcd C21H14Cl2N4O3S (M+) 472.0163, found 472.0164.
4.4.7. 4-(2-(3-Fluorophenyl)-quinoline-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16a)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 26.7%: mp 257–258 °C; 1H NMR (DMSO): δ 1.85 (s, 3H), 2.12 (s, 3H), 7.40 (t, 1H), 7.67 (q, 1H), 7.73 (t, 1H), 7.84 (d, 2H), 7.92 (t, 1H), 8.05 (d, 2H), 8.25 (m, 4H), 8.47 (s, 1H); EI-MS m/z 516 (M+), 222 (100%); HRMS (EI) m/z calcd C27H21FN4O4S (M+) 516.1261, found 516.1268.
4.4.8. 4-(2-(3-Chlorophenyl)-quinoline-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16b)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 30.5%: mp 237–238 °C; 1H NMR (DMSO): δ 1.70 (s, 3H), 2.10 (s, 3H), 7.62 (m, 2H), 7.72 (t, 1H), 7.86 (d, 2H), 7.89 (t, 1H), 8.04 (d, 2H), 8.22 (t, 2H), 8.35 (d, 1H), 8.45 (s, 1H), 8.48 (s, 1H); EI-MS m/z 532 (M+), 266 (100%); HRMS (EI) m/z calcd C27H21ClN4O4S (M+) 532.0969, found 532.0972.
4.4.9. 4-(2-(4-Bromophenyl)-quinoline-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16c)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 35.2%: mp 248–250 °C; 1H NMR (DMSO): δ 1.70 (s, 3H), 2.10 (s, 3H), 7.67 (t, 1H), 7.79 (d, 2H), 7.82 (d, 2H), 7.88 (t, 1H), 8.02 (d, 2H), 8.18 (d, 2H), 8.33 (d, 2H), 8.42 (s, 1H); EI-MS m/z 578 (M+), 203 (100%); HRMS (EI) m/z calcd C27H21BrN4O4S (M+) 576.0431, found 576.0467.
4.4.10. 4-(2-(2-Bromophenyl)-quinoline-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16d)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 40.3%: mp 243–244 °C; 1H NMR (DMSO): δ 1.74 (s, 3H), 2.15 (s, 3H), 7.47 (t, 1H), 7.57 (t, 1H), 7.75 (m, 2H), 7.81 (t, 3H), 7.90 (t, 1H), 8.00 (m, 3H), 8.17 (d, 1H), 8.22 (d, 1H); EI-MS m/z 578 (M+), 203 (100%); HRMS (EI) m/z calcd C27H21BrN4O4S (M+) 576.0464, found 576.0467.
4.4.11. 4-(2-Pyridin-4-yl-quinoline-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16e)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 37.7%: mp 238–239 °C; 1H NMR (DMSO): δ 2.04 (s, 3H), 2.35 (s, 3H), 7.97 (m, 1H), 8.14 (m, 3H), 8.32 (d, 2H), 8.47 (d, 1H), 8.52 (dd, 2H), 8.59 (d, 1H), 8.77 (s, 1H), 9.01 (dd, 2H); EI-MS m/z 499 (M+), 205 (100%); HRMS (EI) m/z calcd C26H21N5O4S (M+) 499.1316, found 499.1314.
4.4.12. 4-(2-Thiophen-2-yl-quinoline-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16f)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 24.3%: mp 234–235 °C; 1H NMR (DMSO): δ 1.70 (s, 3H), 2.10 (s, 3H), 7.25 (t, 1H), 7.64 (t, 1H), 7.76 (d, 1H), 7.82 (m, 3H), 8.11 (m, 5H), 8.34 (s, 1H); EI-MS m/z 504 (M+), 210 (100%); HRMS (EI) m/z calcd C25H20N4O4S2 (M+) 504.0916, found 504.0926.
4.4.13. 4-(2-Cyclohexanyl-quinoline-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16g)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 21.6%: mp 222–223 °C; 1H NMR (DMSO): δ 1.35 (m, 1H), 1.48 (m, 2H), 1.75 (m, 6H), 1.87 (m, 2H), 1.99 (m, 2H), 2.10 (s, 3H), 2.96 (m, 1H), 7.61 (t, 1H), 7.66 (s, 1H), 7.80 (m, 3H), 7.99 (d, 2H), 8.03 (d, 1H), 8.07 (d, 1H); EI-MS m/z 504 (M+), 210 (100%); HRMS (EI) m/z calcd C27H28N4O4S (M+) 504.1822, found 504.1831.
4.4.14. 4-(2-Naphthalen-2-yl-quinoline-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16h)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:3, v/v), yield 37.1%: mp 264–266 °C; 1H NMR (DMSO): δ 1.71 (s, 3H), 2.05 (s, 3H), 7.60 (m, 2H), 7.70 (t, 1H), 7.88 (m, 3H), 8.00 (m, 1H), 8.05 (d, 2H), 8.11 (d, 2H), 8.19 (d, 1H), 8.24 (d, 1H), 8.59 (m, 2H), 8.95 (s, 1H); EI-MS m/z 548 (M+), 254 (100%); HRMS (EI) m/z calcd C31H24N4O4S (M+) 548.1509, found 548.1518.
4.4.15. 4-(2-(3-Bromophenyl)-6-fluoro-quinoline-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16i)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 34.9%: mp 267–269 °C; 1H NMR (DMSO): δ 1.70 (s, 3H), 2.10 (s, 3H), 7.56 (t, 1H), 7.74 (d, 1H), 7.87 (m, 3H), 7.96 (d, 1H), 8.04 (d, 2H), 8.29 (q, 1H), 8.38 (d, 1H), 8.56 (m, 2H); EI-MS m/z 596 (M+), 221 (100%); HRMS (EI) m/z calcd C27H20FBrN4O4S (M+) 594.0376, found 594.0373.
4.4.16. 4-(2-(3-Bromophenyl)-quinoline-4-carboxamido)-N-quinoxalin-2-yl benzenesulfonamide (16j)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 38.2%: mp 285–286 °C; 1H NMR (DMSO): δ 7.53 (t, 1H), 7.67 (m, 2H), 7.76 (m, 2H), 7.86 (m, 2H), 7.95 (d, 1H), 8.00 (d, 2H), 8.14 (d, 2H), 8.18 (d, 2H), 8.35 (d, 1H), 8.40 (s, 1H), 8.53 (t, 1H), 8.72 (br, 1H); EI-MS m/z 611 (M+), 203 (100%); HRMS (EI) m/z calcd C30H20BrN5O3S (M+) 609.0452, found 609.0470.
4.4.17. 4-(2-(3-Bromophenyl)-pyridine-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16k)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 38.7%: mp 240–242 °C; 1H NMR (DMSO): δ 1.70 (s, 3H), 2.10 (s, 3H), 7.54 (t, 1H), 7.71 (d, 1H), 7.82 (d, 2H), 7.86 (d, 1H), 8.03 (d, 2H), 8.21 (d, 1H), 8.38 (t, 1H), 8.46 (s, 1H), 8.90 (d, 1H); EI-MS m/z 528 (M+), 260 (100%); HRMS (EI) m/z calcd C23H19BrN4O4S (M+) 526.0292, found 526.0311.
4.4.18. 4-(2-(4-Bromophenyl)-pyridine-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16l)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 35.2%: mp 248–249 °C; 1H NMR (DMSO): δ 1.67 (s, 3H), 2.08 (s, 3H), 7.76 (d, 2H), 7.84 (m, 3H), 8.02 (d, 2H), 8.16 (d, 2H), 8.41 (s, 1H), 8.89 (d, 1H); EI-MS m/z 526 (M+), 126 (100%); HRMS (EI) m/z calcd C23H19BrN4O4S (M+) 526.0308, found 526.0310.
4.4.19. 4-(2-Phenyl-pyridine-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16m)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 48.2%: mp 219–220 °C; 1H NMR (DMSO): δ 1.66 (s, 3H), 2.08 (s, 3H), 7.56 (m, 3H), 7.80 (m, 3H), 8.02 (d, 2H), 8.15 (d, 2H), 8.17 (s, 1H), 8.87 (d, 1H); EI-MS m/z 448 (M+), 57 (100%); HRMS (EI) m/z calcd C23H20N4O4S (M+) 448.1214, found 448.1206.
4.4.20. 4-(2-(4-Chlorophenyl)-pyridine-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16n)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 38.1%: mp 244–245 °C; 1H NMR (DMSO): δ 1.66 (s, 3H), 2.08 (s, 3H), 7.62 (d, 2H), 7.82 (m, 3H), 8.02 (d, 2H), 8.22 (d, 2H), 8.41 (s, 1H), 8.87 (d, 1H); EI-MS m/z 482 (M+), 216 (100%); HRMS (EI) m/z calcd C23H19ClN4O4S (M+) 482.0829, found 482.0816.
4.4.21. 4-(2-(4-Fluorophenyl)-pyridine-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16o)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 55.6%: mp 232–234 °C; 1H NMR (DMSO): δ 1.68 (s, 3H), 2.08 (s, 3H), 7.39 (t, 2H), 7.82 (m, 3H), 8.03 (d, 2H), 8.26 (m, 2H), 8.39 (s, 1H), 8.87 (d, 1H); EI-MS m/z 466 (M+), 200 (100%); HRMS (EI) m/z calcd C23H19FN4O4S (M+) 466.1107, found 466.1111.
4.4.22. 4-(2-(2-Methylphenyl)-pyridine-4-carboxamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (16p)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 25.1%: mp 202–203 °C; 1H NMR (DMSO): δ 1.66 (s, 3H), 2.08 (s, 3H), 2.35 (s, 3H), 7.36 (m, 3H), 7.48 (d, 1H), 7.79 (d, 2H), 7.84 (d, 1H), 8.01 (m, 3H), 8.87 (d, 1H); EI-MS m/z 462 (M+), 167 (100%); HRMS (EI) m/z calcd C24H22N4O4S (M+) 462.1345, found 462.1361.
4.4.23. 4-(2-(2-Methylphenyl)-pyridine-4-carboxamido)-N-(4-methyl-pyrimidin-2-yl) benzenesulfonamide (16q)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 28.9%: mp 242–243 °C; 1H NMR (DMSO): δ 2.31 (s, 3H), 2.35 (s, 3H), 6.90 (d, 2H), 7.35 (m, 3H), 7.47 (d, 1H), 7.82 (d, 1H), 8.01 (q, 4H), 8.32 (m, 1H), 8.87 (d, 1H); EI-MS m/z 459 (M+), 286 (100%); HRMS (EI) m/z calcd C24H21N5O3S (M+) 459.1369, found 459.1365.
4.4.24. 4-(2,4-Dichlorophenylacetamido)-N-quinoxalin-2-yl benzenesulfonamide (17a)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 35.2%: mp 235–236 °C; 1H NMR (DMSO): δ 3.83 (s, 2H), 7.38 (m, 1H), 7.42 (d, 1H), 7.55 (d, 1H), 7.62 (t, 1H), 7.79 (m, 4H), 7.92 (d, 1H), 8.04 (d, 2H), 8.62 (br, 1H); EI-MS m/z 486 (M+), 236 (100%); HRMS (EI) m/z calcd C22H16Cl2N4O3S (M+) 486.0309, found 486.0321.
4.4.25. 4-Benzamido-N-quinoxalin-2-yl benzenesulfonamide (17b)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 45.4%: mp 258–259 °C; 1H NMR (DMSO): δ 7.63 (m, 4H), 7.75 (t, 1H), 7.82 (m, 1H), 7.93 (m, 3H), 8.03 (d, 2H), 8.13 (d, 2H), 8.65 (br, 1H); EI-MS m/z 404 (M+), 105 (100%); HRMS (EI) m/z calcd C21H16N4O3S (M+) 404.0943, found 404.0943.
4.4.26. 4-(3-Phenyl-propionamido)-N-quinoxalin-2-yl benzenesulfonamide (17c)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 39.3%: mp 213–214 °C; 1H NMR (DMSO): δ 2.65 (t, 2H), 2.90 (t, 2H), 7.16 (t, 1H), 7.26 (m, 4H), 7.62 (t, 1H), 7.79 (m, 4H), 7.93 (d, 1H), 8.03 (d, 2H), 8.63 (br, 1H); EI-MS m/z 432 (M+), 367 (100%); HRMS (EI) m/z calcd C23H20N4O3S (M+) 432.1261, found 432.1256.
4.4.27. 4-(4-Chlorophenylacetamido)-N-(3,4-dimethyl-isoxazol-5-yl) benzenesulfonamide (17d)
This compound was purified by flash chromatography with EtOAc/petroleum ether (1:2, v/v), yield 49.3%: mp 193–194 °C; 1H NMR (DMSO): δ 1.60 (s, 3H), 2.07 (s, 3H), 3.69 (s, 2H), 7.39 (q, 4H), 7.69 (d, 2H), 7.78 (d, 2H); EI-MS m/z 419 (M+), 125 (100%); HRMS (EI) m/z calcd C19H18ClN4O3S (M+) 419.0704, found 419.0706.
4.4.28. 4,4′-Methylenebisanthranilic acid (31a)31
To a stirred mixture of anthranilic acid 30 (6.85, 50 mmol), H2O (63 mL), and 36% HCl (13 mL) at 50 °C was added 3% aqueous formaldehyde (18 mL). The resulting solution was heated at 70 °C and stirred for 4 h. Upon neutralization with ammonia, the precipitate was filtered, washed successively with H2O and hot acetic acid, followed by drying to afford 31a (4.8 g, 33.6%) as yellow solid: mp 258–260 °C.
4.4.29. 4,4′-Methylenebisanthranilic acid dimethyl ester (31b)
A mixture of 31a (1 g, 3.5 mmol) in methanol (30 mL) saturated with HCl was refluxed for 5 h, poured into crushed ice, and pH adjusted to 9 with 2 N NaOH. The precipitate was collected, dried, and purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:10, v/v), to afford 31b (0.30 g, 27.5%) as a white solid: mp 144–145 °C; 1H NMR (CDCl3): δ 3.80 (s, 2H), 3.88 (s, 6H), 5.60 (br, 2H), 6.62 (d, 2H), 7.08 (dd, 2H), 7.65 (d, 2H).
4.4.30. 6,6′-Bis-(3,5-dimethoxybenzamido)-3,3′-methanediyl-di-benzoic acid (15)
A mixture of 31a (0.2 g, 0.7 mmol), 3,5-dimethoxybenzoyl chloride (0.31 g, 1.54 mmol), and pyridine (5 mL) was stirred at 0 °C for 5 h. The resulting mixture was poured into H2O, and the precipitate was collected, dried, and purified by flash chromatography on silica gel, eluted with a mixture of CH3OH/CHCl3 (1:15, v/v), to afford 15 (70 mg, 16.3%) as a white solid: mp > 300 °C; 1H NMR (DMSO): δ 3.80 (s, 12H), 3.90 (s, 2H), 6.68 (s, 2H), 7.14 (s, 4H), 7.30 (d, 2H), 7.92 (s, 2H), 8.58 (d, 2H); MALDI-MS m/z 637 [M+Na]+; HRMS (MALDI) m/z calcd C33H30N2O10Na [M+Na]+ 637.17927, found 637.18080.
4.4.31. 6,6′-Bis-benzamido-3,3′-methanediyl-di-benzoic acid (18b)
In the same manner as described for 15, 18b was prepared from benzoyl chloride, yield 20.0%: mp 288–289 °C; 1H NMR (CDCl3): δ 4.24 (s, 2H), 7.58 (m, 4H), 7.61 (m, 2H), 7.67 (s, 4H), 8.09 (s, 2H), 8.32 (m, 4H); EI-MS m/z 494 (M+), 105 (100%). HRMS (EI) m/z calcd C29H22N2O6 (M+) 494.1487, found 494.1479.
4.4.32. 6,6′-Bis-(4-methoxybenzamido)-3,3′-methanediyl-di-benzoic acid (18d)
In the same manner as described for 15, 18d was prepared from 4-methoxybenzoyl chloride; yield 20.0%: mp 280–282 °C;1H NMR (DMSO): δ 3.86 (s, 6H), 4.07 (s, 2H), 7.05 (d, 2H), 7.13 (d, 2H), 7.30 (d, 1H), 7.62 (d, 1H), 7.79 (d, 1H), 7.99 (m, 4H), 8.13 (d, 2H), 8.62 (d, 1H); EI-MS m/z 554 (M+), 135 (100%). HRMS (EI) m/z calcd C31H26N2O8 (M+) 554.1678, found 554.1690.
4.4.33. 6,6′-Bis-(3,5-dimethoxybenzamido)-3,3′-methanediyl-di-benzoic acid dimethyl ester (18a)
A mixture of 31b (50 mg, 0.15 mmol), 3,5-dimethoxybenzoyl chloride (68 mg, 0.34 mmol), and pyridine (5 mL) was stirred at 0 °C for 5 h. The resulting mixture was poured into H2O, and the precipitate was collected, dried, and purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:4, v/v), to afford 18a (23 mg, 22.5%) as a white solid: mp 194–196 °C; 1H NMR (DMSO): δ 3.85 (s, 12H), 3.88 (s, 6H), 4.05 (s, 2H), 6.78 (s, 2H), 7.07 (s, 4H), 7.59 (d, 2H), 7.88 (s, 2H), 8.44 (d, 2H); MALDI-MS m/z 665 [M+Na]+. HRMS (MALDI) m/z calcd C35H34N2O10Na [M+Na]+ 665.21057, found 665.21140.
4.4.34. 6,6′-Bis-benzamido-3,3′-methanediyl-di-benzoic acid dimethyl ester (18c)
In the same manner as described for 18a, 18c was prepared from benzoyl chloride, yield 20.3%: mp 214–216 °C; 1H NMR (DMSO): δ 3.88 (s, 6H), 4.06 (s, 2H), 7.65 (m, 8H), 7.90 (s, 2H), 7.97 (d, 4H), 8.50 (d, 2H); EI-MS m/z 522 (M+), 105 (100%); HRMS (EI) m/z calcd C31H26N2O6 (M+) 522.1781, found 522.1791.
4.4.35. 6,6′-Bis-(4-methoxybenzamido)-3,3′-methanediyl-di-benzoic acid dimethyl ester (18e)
In the same manner as described for 18a, 18e was prepared from 4-methoxybenzoyl chloride, yield 28.0%: mp 234–236 °C; 1H NMR (DMSO): δ 3.84 (s, 6H), 3.88 (s, 6H), 4.04 (s, 2H), 7.14 (d, 4H), 7.58 (d, 2H), 7.93 (m, 6H), 8.51 (d, 2H); MALDI-MS m/z 605 [M+Na]+. HRMS (MALDI) m/z calcd C33H30N2O8Na [M+Na]+ 605.18944, found 605.18960.
4.4.36. 6,6′-Bis-(4-chlorobenzamido)-3,3′-methanediyl-di-benzoic acid dimethyl ester (18f)
In the same manner as described for 18a, 18f was prepared from 4-chlorobenzoyl chloride, yield 26.0%: mp 241–242 °C; 1H NMR (CDCl3): δ 3.95 (s, 6H), 3.99 (s, 2H), 7.46 (d, 2H), 7.50 (d, 4H), 7.90 (s, 2H), 7.99 (d, 4H), 8.85 (d, 2H); EI-MS m/z 590 (M+), 139 (100%). HRMS (EI) m/z calcd C31H24Cl2N2O6 (M+) 590.1030, found 590.1011.
4.4.37. 6,6′-Bis-benzylamino-3,3′-methanediyl-di-benzoic acid dimethyl ester (18g)
To a stirred mixture of 31b (100 mg, 0.32 mmol), NaCO3 (80 mg, 0.75 mmol), and NMP (3 mL) was added benzyl bromide (0.09 mL, 0.75 mmol). The mixture was then heated at 90 °C for 10 h. The reaction mixture after cooling was transferred to a separatory funnel using a 1:1 mixture of H2O and EtOAc. The product was extracted with EtOAc. The combined organic phases were washed, dried, filtered, and concentrated. The residue was purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:20, v/v), to afford 18g (43 mg, 27.4%) as a white solid: mp 86–88 °C; 1H NMR (DMSO): δ 3.64 (s, 2H), 3.76 (s, 6H), 4.42 (d, 4H), 6.63 (d, 2H), 7.15 (d, 2H), 7.26 (m, 2H), 7.32 (m, 8H), 7.60 (d, 2H), 7.96 (t, 2H); MALDI-MS m/z 517 [M+Na]+. HRMS (MALDI) m/z calcd C31H30N2O4Na [M+Na]+ 517.20978, found 517.21090.
4.4.38. 6,6′-Bis-benzylamino-3,3′-methanediyl-di-benzoic acid monomethyl ester (18h)
A mixture of 18g (50 mg, 0.1 mmol) and LiOH (20 mg, 0.8 mmol) in THF/Methanol/H2O (3:1:1, v/v/v, 10 mL) was stirred at room temperature for 12 h. The resulting solution was acidified to pH 2 using 1 N HCl. The product was extracted into the organic phase using EtOAc (3 × 25 mL), dried, filtered, and concentrated. The residue was purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:4, v/v), to afford 18h (20 mg, 41.6%) as a white solid: mp 173–175 °C; 1H NMR (DMSO): δ 3.64 (s, 2H), 3.77 (s, 3H), 4.43 (t, 4H), 6.65 (q, 2H), 7.17 (q, 2H), 7.33 (m, 10H), 7.62 (q, 2H), 7.95 (t, 2H); MALDI-MS m/z 503 [M+Na]+. HRMS (MALDI) m/z calcd C30H28N2O4Na [M+Na]+ 503.19413, found 503.19610.
4.4.39. 6,6′-Bis-benzylamino-3,3′-methanediyl-di-benzoic acid (18i)
In the same manner as described for 18h but room temperature was changed into refluxing temperature, 18i was prepared as a white solid, yield 21.2%: mp 179–181 °C; 1H NMR (DMSO): δ 3.64 (s, 2H), 4.41 (s, 4H), 6.61 (d, 2H), 7.14 (d, 2H), 7.33 (m, 10H), 7.60 (s, 2H); MALDI-MS m/z 489 [M+Na]+. HRMS (MALDI) m/z calcd C29H26N2O4Na [M+Na]+ 489.17848, found 489.17930.
4.4.40. 5-Bromomethyl-2-nitro-benzoic acid methyl ester (34)32
A mixture of 5-methyl-2-nitro-benzoic acid 32 (1 g, 0.55 mmol) in methanol (30 mL) saturated with HCl was refluxed for 5 h, and most of solvent was evaporated, filtered, washed, and dried to afford 33 (0.91 g, 84.5%) as a white solid: mp 76–78 °C.
A mixture of 33 (0.9 g, 4.7 mmol), NBS (1.25 g, 7.0 mmol), benzoyl peroxide (0.05 g), and CH3Cl (30 mL) was stirred for 24 h. The resulting solution was washed successively with H2O, aqueous NaCO3, brine, dried, filtered, and concentrated. The residue was purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:10, v/v), to afford 34 (0.43 g, 33.8%) as a white solid: mp 58–61 °C.
4.4.41. 6,6′-Bis-nitro-3,3′-ethanediyl-di-benzoic acid dimethyl ester (35)
To a stirred mixture of 33 (0.5 g 2.5 mmol), n-BuLi (2.5 M in hexane) (2 mL, 5 mmol), and redistilled THF (10 mL) was added dropwise 34 (0.35 g, 1.3 mmol) in redistilled THF (5 mL) at −78 °C under N2. The resulting mixture was stirred for 3 h at −78 °C and then warmed to room temperature overnight. The solution was poured into aqueous NH4Cl, extracted with EtOAc. The combined organic phases were dried, filtered, and concentrated. The residue was purified by flash chromatography on silica gel, eluted with a mixture of EtOAc/petroleum ether (1:4, v/v), to afford 35 (0.11 g, 21.8%) as a yellow solid: 1H NMR (CDCl3): δ 3.07 (s, 4H), 3.93 (s, 6H), 7.37 (d, 2H), 7.50 (d, 2H), 7.89 (d, 2H); EI-MS m/z 388 (M+), 194 (100%).
4.4.42. 6,6′-Bis-amino-3,3′-ethanediyl-di-benzoic acid dimethyl ester (36)
A mixture of 35 (110 mg, 0.28 mmol), 10% palladium on charcoal (20 mg), and THF (20 mL) was stirred at 25 °C for 8 h in an atmosphere of hydrogen. The catalyst was filtered and the filtrate was concentrated to dryness. The residue was washed with a few methanol to afford 36 (90 mg) as a white solid: mp 216–218 °C.
4.4.43. 6,6′-Bis-(3,5-dimethoxybenzamido)-3,3′-ethanediyl-di-benzoic acid dimethyl ester (18k)
In the same manner as described for 18a, 18k was prepared from 36, yield 58.0%: mp 215–216 °C; 1H NMR (CDCl3): δ 2.93 (s, 4H), 3.88 (s, 12H), 3.94 (s, 6H), 6.64 (s, 2H), 7.18 (s, 4H), 7.44 (d, 2H), 7.87 (s, 2H), 8.83 (d, 2H); MALDI-MS m/z 679 [M+Na]+. HRMS (MALDI) m/z calcd C36H36N2O10Na [M+Na]+ 679.22622, found 679.22800.
4.4.44. 6,6′-Bis-(3,5-dimethoxybenzamido)-3,3′-ethanediyl-di-benzoic acid (18l)
A mixture of 18k (32 mg, 0.05 mmol) and LiOH (12 mg, 0.5 mmol) in THF/methanol/H2O (3:1:1, v/v/v, 20 mL) was stirred at room temperature for 12 h. The resulting solution was acidified to pH 2 using 1 N HCl. The product was extracted, dried, filtered, concentrated, and purified by flash chromatography using EtOAc/petroleum ether (2:1, v/v), to afford 18l (18 mg, 58.1%) as a pale red solid: mp > 300 °C; 1H NMR (DMSO): δ 2.91 (s, 4H), 3.86 (s, 12H), 6.74 (s, 2H), 7.07 (s, 4H), 7.54 (d, 2H), 7.90 (s, 2H), 8.59 (d, 2H); ESI-MS m/z 627 [M-H]+. HRMS (ESI) m/z calcd C34H31N2O10 [M−H]+ 627.1979, found 627.1989.
4.4.45. Bis-(4-(3,5-dimethoxybenzamido)-phenyl)-methane (18j)
In the same manner as described for 18a, 18j was prepared from 4,4′-methanediyl-bis-aniline 37, yield 56.0%; mp 185–187 °C; 1H NMR (CDCl3): δ 3.83 (s, 12H), 3.96 (s, 2H), 6.61 (t, 2H), 6.97 (d, 4H), 7.19 (d, 4H), 7.56 (d, 4H); ESI-MS m/z 525 [M−H]+. HRMS (ESI) m/z calcd C31H29N2O6 [M−H]+ 525.2026, found 525.2078.
Acknowledgments
We gratefully acknowledge financial support from the State Key Program of Basic Research of China (Grant 2002CB512802), the National Natural Science Foundation of China (Grants 20372069, 29725203, 20472094, and 20102007), the Basic Research Project for Talent Research Group from the Shanghai Science and Technology Commission, the Key Project from the Shanghai Science and Technology Commission (Grant 02DJ14006), the Key Project for New Drug Research from CAS, the Qi Ming Xing Foundation of Shanghai Ministry of Science and Technology (Grant 03QD14065), and the 863 Hi-Tech Programm (Grants 2002AA233061, 2002AA104270, 2002AA233011, and 2003AA235030).
Contributor Information
Hong Liu, Email: hliu@mail.shcnc.ac.cn.
Xu Shen, Email: xshen@mail.shcnc.ac.cn.
Hualiang Jiang, Email: hljiang@mail.shcnc.ac.cn.
References and notes
- 1.Handschumacher R.E., Harding M.W., Rice J., Drugge R.J., Speicher D.W. Science. 1984;226:544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- 2.(a) Fisher G., Whittman-Liebold B., Lang K., Kiefhaber T., Schmid F.X. Nature. 1989;337:476–478. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]; (b) Takahashi N., Hayano T., Suzuki M. Nature. 1989;337:473–475. doi: 10.1038/337473a0. [DOI] [PubMed] [Google Scholar]
- 3.Braaren D., Luban J. EMBO J. 2001;20:1300–1309. doi: 10.1093/emboj/20.6.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Galat A. Curr. Top. Med. Chem. 2003;3:1315–1347. doi: 10.2174/1568026033451862. [DOI] [PubMed] [Google Scholar]; (b) Dornan J., Taylor P., Walkinshaw M.D. Curr. Top. Med. Chem. 2003;3:1392–1409. doi: 10.2174/1568026033451899. [DOI] [PubMed] [Google Scholar]
- 5.Luban J., Bossolt K.L., Franke E.K., Kalpana G.V., Goff S.P. Cell. 1993;73:1067–1078. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- 6.(a) Capano M., Virji S., Crompton M. Biochem. J. 2002;363:29–36. doi: 10.1042/bj3630029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yoshimoto T., Siesjo B.K. Brain Res. 1999;832:283–291. doi: 10.1016/s0006-8993(99)01733-3. [DOI] [PubMed] [Google Scholar]; (c) Crompton M. J. Physiol. 2000;529:11–21. doi: 10.1111/j.1469-7793.2000.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawson T.M., Steiner J.P., Lyons W.E., Fotuhi M., Blue M., Snyder S.H. Neuroscience. 1994;62:569–580. doi: 10.1016/0306-4522(94)90389-1. [DOI] [PubMed] [Google Scholar]
- 8.(a) Luo C., Luo H.B. Biochem. Biophys. Res. Commun. 2004;321:557–565. doi: 10.1016/j.bbrc.2004.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen Z.N., Mi L., Xu J., Yu J. J. Infect. Dis. 2005;191:755–760. doi: 10.1086/427811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Friedman J., Weissman I. Cell. 1991;66:799–806. doi: 10.1016/0092-8674(91)90123-g. [DOI] [PubMed] [Google Scholar]; (b) Liu J., Farmer J.D., Lane W.S., Friedman J., Weissman I., Schreiber S.L. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 10.(a) Barbara E.B., Georg H., David F., Steven J.B. Curr. Opin. Immunol. 1993;5:763–773. [Google Scholar]; (b) Zou X.J., Matsumura Y.J., John P., Ryo S., Alberto M., Jack M., Stanley C. J. Transpl. Immunol. 1995;3:151–161. doi: 10.1016/0966-3274(95)80042-5. [DOI] [PubMed] [Google Scholar]
- 11.(a) Dumont F.J. Exp. Opin. Ther. Patents. 2001;11:377–404. [Google Scholar]; (b) Anderson, P. S., Kenyon, G. L., Marshall, G. R., Eds.; Perspectives in Drug Discovery and Design; ESCOM Science Publishers: Leiden, The Netherlands, 1994; Vol. 2, pp 3–248.
- 12.Siekierka J.J., Hung S.H., Poe M., Lin C.S., Sigal N.H. Nature. 1989;341:755–757. doi: 10.1038/341755a0. [DOI] [PubMed] [Google Scholar]
- 13.Clane R.Y., Lim S., Samaan A., Collier D.S.T. Lancet. 1989;334:227. doi: 10.1016/s0140-6736(89)90417-0. [DOI] [PubMed] [Google Scholar]
- 14.Rechard S., Jorg K., Luisa M.C., Charles D.P., Francesco S. J. Am. Chem. Soc. 2003;125:3849–3859. [Google Scholar]
- 15.Cui M., Huang X., Luo X., James M.B., Ji R., Chen K., Shen J., Jiang H. J. Med. Chem. 2002;45:5249–5259. doi: 10.1021/jm020082x. [DOI] [PubMed] [Google Scholar]
- 16.Su Q., Weber L., Le Hir M., Zenke G., Ryffel B. Ren. Physiol. Biochem. 1995;18:128–139. doi: 10.1159/000173910. [DOI] [PubMed] [Google Scholar]
- 17.Bennet W.M., Porter G.A. Am. J. Med. 1988;85:131–133. doi: 10.1016/s0002-9343(88)80330-9. [DOI] [PubMed] [Google Scholar]
- 18.Miller L.W. Am. J. Transplant. 2002;2:807–818. doi: 10.1034/j.1600-6143.2002.20902.x. [DOI] [PubMed] [Google Scholar]
- 19.(a) Kuntz I.D., Meng E.C., Shoichet B.K. Acc. Chem. Res. 1994;27:117–123. [Google Scholar]; (b) Wang J.L., Liu D.X., Zhang Z.J., Shan S., Han X.B., Srinivasula S.M., Croce C.M., Alnemri E.S., Huang Z.W. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7124–7129. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu H., Li Y., Song M., Tan X., Cheng F., Zheng S., Shen J., Luo X., Ji R., Yue J., Hu G., Jiang H., Chen K. Chem. Biol. 2003;10:1103–1113. doi: 10.1016/j.chembiol.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Zheng S., Luo X., Chen G., Zhu W., Jiang H. J. Chem. Inf. Model. 2005;45:856–862. doi: 10.1021/ci050031j. [DOI] [PubMed] [Google Scholar]
- 21.(a) Ewing T., Kuntz I.D. J. Comput. Chem. 1997;18:1175–1189. [Google Scholar]; (b) Kuntz I.D. Science. 1992;257:1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 22.Weiner S.J., Kollman P.A., Nguyen D.T., Case D.A. J. Comput. Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 23.Gasteiger J., Marsili M. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 24.(a) Muegge I., Rarey M. Small molecule docking and scoring. In: Lipkowitz K.B., Boyd D.B., editors. Reviews in Computational Chemistry. Wiley; New York: 2001. pp. 1–60. [Google Scholar]; (b) Charifson P.S., Corkery J.J., Murcko M.A., Walters W.P. J. Med. Chem. 1999;42:5100–5109. doi: 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- 25.Sybyl [molecular modeling package], version 6.8; Tripos Associates: St. Louis, MO, 2000.
- 26.Rarey M., Kramer B., Lengauer T., Klebe G. J. Mol. Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 27.Robert E.L., Philip S.B., Marion T.C., John F.C. J. Am. Chem. Soc. 1946;68:1813–1831. [Google Scholar]
- 28.Pfister K., Sullivan A.P., John W., Tishler M. J. Am. Chem. Soc. 1951;7:4955–4957. [Google Scholar]
- 29.Bredereck H., Gompper R. Chem. Ber. 1954;87:700–706. [Google Scholar]
- 30.Usui Y., Hara Y., Shimamoto N., Yurugi S., Masada T. Heterocycles. 1975;3:155–161. [Google Scholar]
- 31.Reddy L.M., Reddy P.P., Reddy P.S.N. Indian J. Chem. 2002;41:2405–2409. [Google Scholar]
- 32.Zrihen M., Labia R., Wakselman M. Eur. J. Med. Chem. Chim. Ther. 1983;18:307–314. [Google Scholar]
- 33.Kofron J.L., Kuzmic P., Kishore V., Colon-Bonilla E., Rich D.H. Biochemistry. 1991;30:6127–6134. doi: 10.1021/bi00239a007. [DOI] [PubMed] [Google Scholar]
- 34.Sargent J.M., Taylor C.G. Br. J. Cancer. 1989;60:206–210. doi: 10.1038/bjc.1989.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wallace A.C., Laskowski R.A., Thornton J.M. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]