

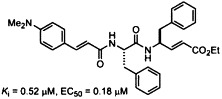
Graphical abstract
The dipeptide-conjugated ester derived from Phe-Phe dipeptide and 4-(dimethylamino)cinnamic acid shows a potent anti-SARS activity by inhibition of the 3CL protease.
Keywords: Severe acute respiratory syndrome; 3CL protease inhibitors; Peptides; α,β-Unsaturated esters; Cell-based assay; Antiviral agent
Abstract
The proteolytic processing of polyproteins by the 3CL protease of severe acute respiratory syndrome coronavirus is essential for the viral propagation. A series of tripeptide α,β-unsaturated esters and ketomethylene isosteres, including AG7088, are synthesized and assayed to target the 3CL protease. Though AG7088 is inactive (IC50 > 100 μM), the ketomethylene isosteres and tripeptide α,β-unsaturated esters containing both P1 and P2 phenylalanine residues show modest inhibitory activity (IC50 = 11–39 μM). The Phe-Phe dipeptide inhibitors 18a–e are designed on the basis of computer modeling of the enzyme–inhibitor complex. The most potent inhibitor 18c with an inhibition constant of 0.52 μM is obtained by condensation of the Phe-Phe dipeptide α,β-unsaturated ester with 4-(dimethylamino)cinnamic acid. The cell-based assays also indicate that 18c is a nontoxic anti-SARS agent with an EC50 value of 0.18 μM.
1. Introduction
Severe acute respiratory syndrome (SARS), which first occurred in Guandong (China) in November 2002, has spread over many countries in 2003. This illness is caused by infection with a novel human coronavirus (SARS-CoV).1 The fatality rate of SARS-CoV infection is rather high, estimated to be 14–15%. In only a few months nearly 1000 lives were claimed.2 The natural source of SARS-CoV is unclear, though studies on the molecular evolution of SARS-CoV indicate that the virus may have emerged from wild animals.3 At present, no efficacious therapy for SARS is available. Therefore, a search for effective antivirals for the SARS-CoV is of current interest.
The SARS-CoV belongs to Coronaviridae family,4 which includes porcine transmissible gastroenteritis virus (TGEV), human coronavirus (HCoV) 229E, mouse hepatitis virus, bovine coronavirus, and infectious bronchitis virus. The SARS-CoV is a positive-strand RNA virus that encodes two replicase polyproteins, pp1a and pp1b.4 Extensive proteolytic processing of these nonstructural polyproteins is required to provide the functional proteins for viral propagation. These processes are mediated primarily by the main protease (Mpro), which is also known as dimeric chymotrypsin-like protease (3CLpro).5 The active site of SARS-CoV 3CL protease contains Cys145 and His41 to constitute a catalytic dyad, in which cysteine functions as the common nucleophile in the proteolytic process.5 The 3CL protease cleaves pp1a at no less than 11 conserved sites with a sequence of (Leu, Met, Phe)-Gln ↓ (Ser, Ala, Gly), in which a P1 glutamine residue invariably occupies the S1 site.5, 6 The 3CL protease is essential for the propagation of the virus, and thus serves as a key target for the discovery of anti-SARS drugs.
So far, only a few inhibitors have been validated by in vitro protease assays. These protease inhibitors include C 2 symmetric peptidomimetic compounds,7 zinc-conjugated compounds,8 bifunctional aryl boronic acids,9 a quinolinecarboxylate derivative,10 a thiophenecarboxylate,11 and phthalhydrazide-substituted keto-glutamine analogs.12
AG7088, a ketomethyl isostere of a tripeptide-conjugated ester (compound 1a in Scheme 1 ), is a potent inhibitor against human rhinovirus (HRV) 3C protease,13 which is also a cysteine protease that mediates the maturation cleavages of replicase polyproteins.14 The structure of AG7088 incorporates a γ-lactam moiety to mimic the amido side chain of the P1 glutamine residue. To improve the cell membrane permeability, the P2 phenylalanine residue is replaced by a methylene isostere bearing a 4-fluorophenyl substituent. The previous pharmacophore models,5, 15 by analogy to the structure of TGEV main proteinase, indicated that AG7088 can partially fit into the putative binding pocket of the SARS-CoV 3CL protease, and the moiety of conjugated ester in AG7088 may function as an acceptor of Cys145. Thus, AG7088 is proposed to be a model for the design of inhibitors against the SARS-CoV 3CL protease.
Scheme 1.
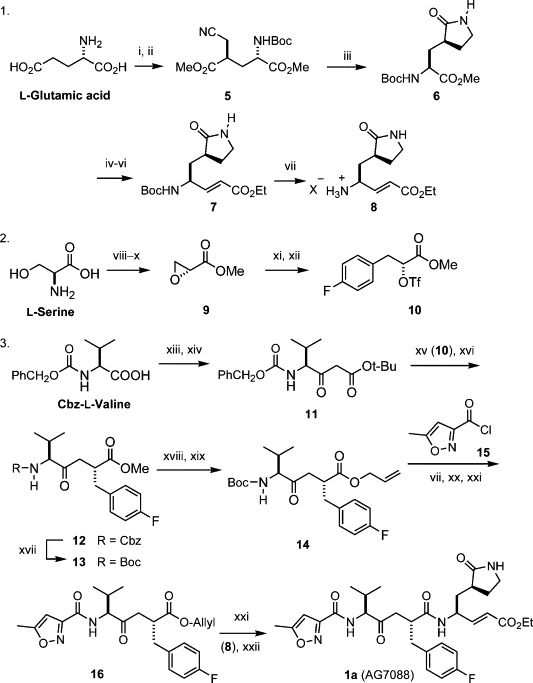
Synthesis of AG7088. Reagents and conditions: (i) Me3SiCl, MeOH, 0 °C, 18 h; then Boc2O, Et3N, 0–25 °C, 4 h; 96%. (ii) LiN(SiMe3)2, THF, −78 °C, 3 h; then BrCH2CN, 3.5 h; 82%. (iii) H2, cat. PtO2, MeOH, CHCl3, 25 °C, 12 h; then NaOAc, reflux, 12 h; 81%. (iv) NaBH4, LiCl, THF, EtOH, 25 °C, 18 h; 89%. (v) Pyridine-SO3, Me2SO, CH2Cl2, Et3N, −10 °C, 3 h. (vi) [EtO2CCHPO(OEt)2]−Na+, THF, −78 °C, 1 h; 75% yield for two steps. (vii) HCl, 1,4-dioxane, rt, 2 h. (viii) HBr, NaNO2, KBr, H2O, −10 °C, 12 h. (ix) KOH, EtOH, 0 °C, 12 h. (x) Me2SO4, CH2Cl2, (PhCH2)Et3N+Cl−, rt, 24 h; 70% for three steps, (xi) 4-FC6H4MgBr, CuBr–Me2S, THF, −35 °C, 1 h; 86%. (xii) (CF3SO2)2O, 2,6-lutidine, CH2Cl2, 0 °C, 40 min. (xiii) 1,1′-carbonyldiimidazole, THF, rt, 1 h. (xiv) CH3CO2-t-Bu, LiN(i-Pr)2, THF, −78 °C, 1 h; 65%. (xv) NaH, THF, 0 °C, 30 min; then triflate 10, THF, 0 °C to rt, 24 h. (xvi) CF3CO2H, CH2Cl2, rt, 24 h; 71% for two steps, (xvii) H2, Pd/C, Boc2O, MeOH, rt, 10 h; 83%. (xviii) LiOH (1.1 equiv), H2O, 1 h, 0 °C; 90%. (xix) allyl iodide, Cs2CO3, DMF, 45 °C, 5 h; 85%. (xx) N-methylmorpholine (NMM), CH2Cl2, 0–25 °C, 2 h; 88%. (xxi) Pd(PPh3)4, morpholine, THF, 25 °C, 3 h; 85%. (xxii) HOBt, EDCI, NMM, DMF, 0–25 °C, 20 h; 70%.
In this paper, we describe the synthesis, the inhibition of SARS-CoV 3CL protease and anti-SARS activity of AG7088, the related ketomethylene isosteres and the peptidomimetic α,β-unsaturated esters. Although AG7088 turns out to be inactive, the α,β-unsaturated ester 18c is found to be a potent antiviral agent with K i of 0.52 μM and EC50 of 0.18 μM.
2. Results and discussion
2.1. Synthesis of AG7088 and the related compounds
On the basis of the previously reported methods,(a), 16 an improved synthesis of AG7088 was accomplished by using the natural glutamic acid, serine, and valine as the building blocks (Scheme 1). In this approach, the dianionic alkylation of N-Boc glutamic acid dimethyl ester (step ii) occurred in a highly stereoselective manner, giving 5 as the exclusive product (82% yield).16b Reduction of the cyano group in 5 by catalytic hydrogenation, followed by in situ cyclization, afforded γ-lactam 6 in 81% yield. The ester group in 6 was effectively reduced by NaBH4 in the presence of LiCl. The alcohol product was then oxidized to an aldehyde (Parikh–Doering method), which was subsequently condensed with a Wittig reagent (Emmons method) to give the conjugated ester 7 in 75% yield (from 6).
On the other hand, the epoxyester 9 with (R)-chirality was obtained from l-serine via diazotization, bromide substitution, and base treatment.17 By the assistance of CuBr, the Grignard reagent 4-FC6H4MgBr reacted with 9 in a regioselective manner to give an alcohol product, which was then activated to the corresponding triflate 10. The SN2 reaction between triflate 10 and the valine-derived malonate 11 was followed by removal of the tert-butyloxycarbonyl (Boc) group under acidic conditions to provide the desired ketomethylene isostere 12. Catalytic hydrogenation of 12 in the presence of di-tert-butyl carbonate furnished a direct conversion of the benzyloxycarbonyl (Cbz) group to the Boc group in 13. Some ketomethylene isosteres are known to be susceptible to epimerization in alkaline conditions.13a In our case, hydrolysis of 13 was realized by using 1.1 equiv of LiOH (0 °C, 1 h), without complication of epimerization, and the acid product was then treated with allyl iodide in the presence of Cs2CO3 to give the allyl ester 14 as a single product. An oxidative condensation of 2,5-hexadione with aqueous HNO3 gave 5-methyl-3-isoxazolecarboxylic acid,18 and the corresponding acyl chloride 15 reacted with 14, after removal of the Boc group, to give amide 16 in the presence of N-methylmorpholine. The allyl group in 16 was smoothly removed by the catalysis of Pd(PPh3)4, and the resulting acid was coupled with the amine derived from 7 to culminate in the synthesis of AG7088. The spectroscopic properties of our prepared sample are in agreement with those reported for AG7088.13
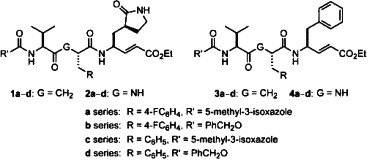
For the investigation of the structure–activity relationship, we also applied a combinatorial approach to synthesize other ketomethylene isosteres (e.g., 1b–d) having various R and R′ substituents at the P2 and N-terminal sites. In addition, the corresponding tripeptidomimetic α,β-unsaturated esters 2a–d were prepared by the procedures generally used in peptide synthesis. By using phenylalanine to replace the P1 residue of lactam-glutamate, a series of ketomethylene isosteres 3a–d and tripeptidomimetic α,β-unsaturated esters 4a–d were also synthesized for comparison study.
2.2. Inhibition assay against the SARS-CoV 3CL protease
According to the previously described fluorometric method,7, 19 the conjugated esters of 1–4 series were subjected to the inhibition assay against the SARS-CoV 3CL protease. The initial velocities of the inhibited reactions using 50 nM of SARS-CoV 3CL protease and 6 μM of the fluorogenic substrate were plotted against the different inhibitor concentrations to obtain the IC50 values. In comparison (Table 1 ), most of the tripeptidomimetic α,β-unsaturated esters (2 and 4 series) tend to be more active than the corresponding ketomethylene isosteres (1 and 3 series).
Table 1.
IC50 values of AG7088 (1a) and the related compounds for inhibition of SARS-CoV 3CL protease21
| Compound | G | R | R′ | IC50 (μM)a |
|---|---|---|---|---|
| 1a | CH2 | 4-FC6H4 | 5-Me-isoxazol-3-yl | >100 |
| 1b | CH2 | 4-FC6H4 | PhCH2O | >100 |
| 1c | CH2 | Ph | 5-Me-isoxazol-3-yl | >100 |
| 1d | CH2 | Ph | PhCH2O | >100 |
| 2a | NH | 4-FC6H4 | 5-Me-isoxazol-3-yl | >100 |
| 2b | NH | 4-FC6H4 | PhCH2O | >100 |
| 2c | NH | Ph | 5-Me-isoxazol-3-yl | 80 |
| 2d | NH | Ph | PhCH2O | 85 |
| 3a | CH2 | 4-FC6H4 | 5-Me-isoxazol-3-yl | 39 |
| 3b | CH2 | 4-FC6H4 | PhCH2O | 31 |
| 3c | CH2 | Ph | 5-Me-isoxazol-3-yl | 13 |
| 3d | CH2 | Ph | PhCH2O | 38 |
| 4a | NH | 4-FC6H4 | 5-Me-isoxazol-3-yl | 21 |
| 4b | NH | 4-FC6H4 | PhCH2O | 11 |
| 4c | NH | Ph | 5-Me-isoxazol-3-yl | 30 |
| 4d | NH | Ph | PhCH2O | 11 |
The highest concentration of samples used in tha assays is 100 μM.
Although AG7088 has been predicted to be a good inhibitor of SARS 3CL protease,5, 15 it turned out to be inactive at a concentration of 100 μM in our enzymatic assay. This result is in agreement with a cell-based assay that indicates AG7088 is inactive to the SARS-CoV.7, 20 However, the structure–activity relationship study on AG7088 analogs indicated that the inhibitory activity was improved by replacing the lactam-glutamine isostere with l-phenylalanine. For example, compounds 3a–d and 4a–d with a phenyl group replacing the P1 lactam moiety showed better inhibitory activity (IC50 = 11–39 μM) than AG7088 (1a) and its analogs (lb–d and 2a–d; IC50 ⩾80 μM).
2.3. Molecular modeling for the enzyme–inhibitor complexes
The observation of enhanced inhibitory activities of the compounds with phenylalanine replacement at the P1 positions prompted us to conduct the computational modeling of the enzyme in complexation with AG7088 or the inhibitor 3c. The target structure used for modeling is deduced from the complex structure of the SARS-CoV 3CL protease mutant (C145A) and the hexapeptide substrate, Ser-Gly-Val-Thr-Phe-Gln.21 Accordingly, the apoenzyme B-chain containing Cys145 was applied in the molecular simulation with the designated inhibitor.
From the computer docking, AG7088 cannot bind properly to the active site of SARS-CoV 3CL protease (Fig. 1 ). Though AG7088 can position its P2 fluorophenyl side chain into the S2 site, its P1 lactam moiety cannot fit to the S1 site of the protease without interfering with the proper disposition of the terminal conjugated ester group in the S1′ site. Unlike the effective binding of AG7088 with the Thr142 of Rhinovirus 3CL protease,5 the conjugated ester in AG7088 is prevented from suitable interaction with the Asn142 of the SARS-CoV 3CL protease, thereby forcing it to adopt a position outside the pocket defined by Cys145, Thr25, and Leu27. The possible steric effects are also exerted by the fluorophenyl group against Arg188 in the S2 pocket and by the isoxazoyl moiety against the hydrophobic residues in the S4 pocket.
Figure 1.
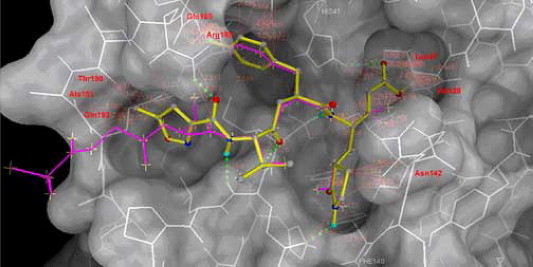
Structure of AG7088 (yellow) is modeled into structure of SARS-CoV 3CLpro by superimposing on the cocrystal structure of hexapeptide Ser-Gly-Val-Thr-Phe-Gln (magenta, the P6–P1 fragment). The close contact distances are computed in MGLTOOLS with a van der Waals radius factor of 1.0. Those residues that can contribute major steric effects (d < 2.0 Å) to AG7088 model are labeled in red.
In contrast, compound 3c having two phenyl groups at the P1 and P2 residues can fit, respectively, in the S2 and S3 pockets of the SARS-CoV 3CL protease (Fig. 2 ). The overlay of Ser-Gly-Val-Thr-Phe-Gln hexapeptide upon 3c shows a conformational similarity. The simulation model also reveals the key hydrogen bondings of 3c with the Glu166, Gln189, and Gln192 residues in the active sites of protease. It is noted that the isoxazolyl moiety in 3c adopts a conformation different from that in AG7088, and thus accommodates a favorable hydrogen bonding with Gin 192 in the S4 pocket. The binding mode of 3c thus contributes to a better inhibitory activity (IC50 = 13 μM) than that of AG7088 (IC50 > 100 μM). As the P1 and P2 residues in 3c move to the S2 and S3 pockets, the moiety of conjugated ester in 3c may occupy the S1 site to exert interactions with His 163 and Gly143. However, the conjugated ester is still unreachable (>4.5 Å) by Cys145 to render a Michael reaction for the covalent C–S bond formation.
Figure 2.
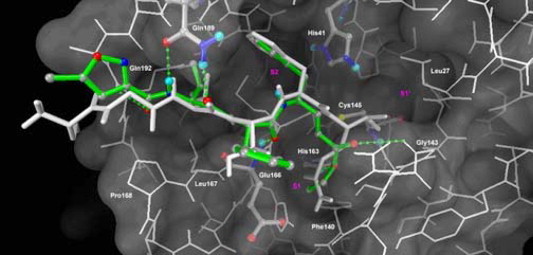
A modeled complex of SARS-CoV 3CL protease with the inhibitor 3c. The protease is shown in white line model, whereas the catalytic dyad (His41 and Cys145), His163 in S1 site, Glu166, Gln189 and compound 3c are highlighted in atom-colored ball-and-stick model. The stick of compound 3c is colored green. Individual atoms are displayed in gray (carbon), blue (nitrogen), red (oxygen), and yellow (sulfur). The hexapeptide Ser-Gly-Val-Thr-Phe-Gln (P6-P1), in white stick model, is overlaid for a comparison. The displayed structure of compound 3c is elected from the docked cluster with the lowest binding free energy. Potential hydrogen-bonding interactions of compound 3c with residues in the binding pocket are shown in green broken lines.
2.4. Design of dipeptide inhibitors having double Michael acceptors at both C- and N-terminals
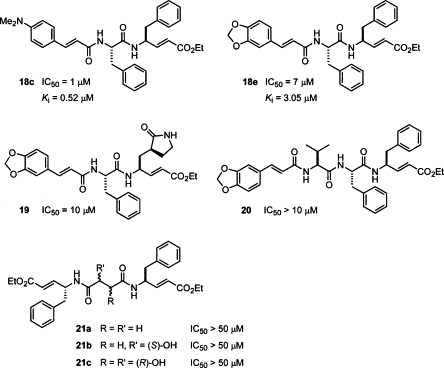
As the molecular modeling reveals that compounds of 3 and 4 series show better inhibitory activity than the compounds of 1 and 2 series by placing their P1 and P2 phenylalanine residues in the S2 and S3 pockets of the SARS-CoV 3CL protease. We surmise that the Phe-Phe dipeptides containing conjugated amido moieties at the N-terminals, such as compounds 18a–e with virtually pseudo-C 2 symmetry, may exhibit greater inhibitory activities.
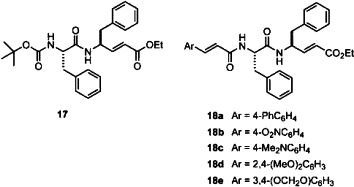
A strategy of parallel synthesis in a microtiter plate22 is applied for the construction of 18a–e and analogs. Thus, the Phe-Phe dipeptidyl α,β-unsaturated ester 17 was prepared by the amidation of N-Boc-phenylalanine with ethyl 4-amino-5-phenyl-2-pentenoate,23 and then treated with HCl in 1,4-dioxane to remove the Boc group. The resulting amine was reacted with a library of α,β-unsaturated carboxylic acids to give 18a–e and analogs, which were subjected, without isolation, to the inhibition assay against the SARS-CoV 3CL protease. On the basis of the preliminary results of assays, some of the most promising inhibitor candidates (18a–e) were selected for the scale-up synthesis. The IC50 values and inhibition constants (K i) of the pure samples were then measured to validate their activities (Table 2 ).
Table 2.
Protease inhibition, anti-SARS, and cytotoxicity properties of the dipeptide inhibitors 18a–e
| Compound | IC50 (μM)a | Ki (μM) | EC50 (μM) | CC50 (μM)b | S.I.c |
|---|---|---|---|---|---|
| 1a | >100 | NDd | —e | ND | |
| 3c | 13 | NDd | —e | ND | |
| 18a | 10 | 6.44 ± 0.8 | 18.86 | >200 | >10 |
| 18b | 5 | 2.48 ± 0.89 | 9.45 | >200 | >20 |
| 18c | 1 | 0.52 ± 0.024 | 0.18 | >200 | >1000 |
| 18d | 10 | 9.049 ± 2.35 | 0.11 | >200 | >1000 |
| 18e | 7 | 3.046 ± 0.61 | 0.16 | >200 | >1000 |
The highest concentration of samples is 10 μM in the assay against the SARS-CoV 3CL protease.
The highest concentration of sample is 200 μM in the assay of cytotoxicity on Vero E6 cells.
Selectivity index, the ratio of CC50 to EC50.
Not determined.
No protective effect against infection of Vero E6 cells at the concentration of 10 μM.
It is fortunate to find that the dipeptidomimetic α,β-unsaturated esters 18a–e bearing the cinnamyl moieties at the N-terminals showed good inhibitory activity against the SARS-CoV 3CL protease. Among them, compound 18c (JMF1521) derived from Phe-Phe dipeptide α,β-unsaturated ester and 4-(dimethylamino)cinnamic acid is the most potent inhibitor with an IC50 value of 1 μM and K i value of 0.52 μM. The double reciprocal plots of the initial rate versus substrate concentration indicate that all these compounds (18a–e) are competitive inhibitors. A representative example for the inhibition of the SARS-CoV 3CL protease by 18c is shown in Figure 3 .
Figure 3.
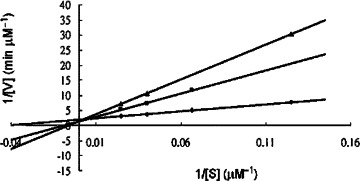
Lineweaver–Burk plot for inhibition of the SARS-CoV 3CL protease by the dipeptide-conjugated ester 18c.
The computer model for the complex of SARS-CoV 3CL protease with 18c (Fig. 4 ) shows that the P1 and P2 phenyl groups occupy the S2 and S3 pockets, respectively. The conjugated (dimethylamino)cinnamyl group disposes a rather rigid coplanar structure in the N-terminal and renders effective hydrogen bondings with the Glu166, Gln189, and Gln192 residues of the enzyme.
Figure 4.
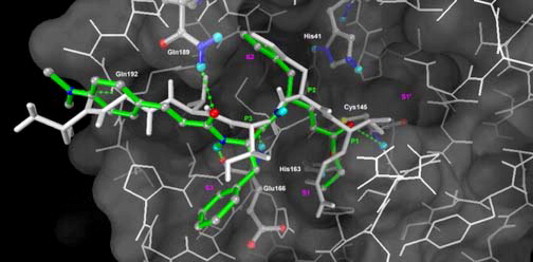
A modeling complex of SARS-CoV 3CL protease with the inhibitor 18c (colored green). The notations are the same as those shown in Figure 2.
For comparison, compounds 19, 20, and 21a–c were also synthesized and subjected to the inhibition assay. The dipeptide derivative 19 contains a γ-lactam moiety at the P1 site in lieu of the phenyl group in 18e. The tripeptide derivative 20 is a homolog of dipeptide 18e by the introduction of an additional P3 valine residue. The dimeric peptide conjugated esters 21a–c were prepared by using a diacid, e.g., succinic acid, S-malic acid, and (2R,3R)-tartaric acid, as the core structure to link with the phenylalanine α,β-unsaturated ester. Compound 18e derived from Phe-Phe dipeptide and 3,4-(methylenedioxy)cinnamic acid showed an IC50 value of 7 μM and K i value of 3.05 μM. The assays indicated that none of 19, 20, and 21a–c were active against the SARS-CoV 3CL protease.
2.5. Cell-based assays
The antiviral activities of 1a (AG7088), 3c, and 18a–e were evaluated by the protection from cytopathogenic effect (CPE) on Vero E6 cells.7 While 1a and 3c are inactive, 18a is active as an anti-SARS agent at a concentration of 3.3 μM, and 18b–e are even more potent with anti-SARS activities at 1 μM. The anti-SARS compounds 18a–e were further evaluated at multiple concentrations to deduce the concentrations for 50% inhibition of the host growth (CC50)24 and the 50% inhibitory concentrations of virus replication (EC50) by the enzyme-linked immunosorbent assay (ELISA).7 None of 18a–e are toxic to the growth of host cells at 200 μM, the highest concentration used in this assay. The EC50 values of compounds 18c, 18d, and 18e deduced by ELISA measurements of the viral spike protein expressions are 0.11–0.18 μM (Table 2). Thus, the ratio of CC50 to EC50, the selectivity index (SI), for compounds 18c–e is very high (>1000). The antiviral activity of 18c was confirmed by Western blotting assay using antispike protein monoclonal antibody (Fig. 5 ). A significant suppression of viral expression of 3CL protease was also observed on treatment with 18c at a concentration of 1 μM (data not shown).
Figure 5.
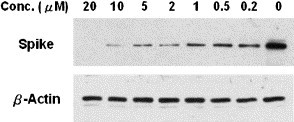
Western blot analysis of the inhibitory effect of compound 18c on the SARS-CoV spike protein synthesis.
Although the real reasons for smaller EC50 than K i are unclear, we speculate that the environment and the physical state of the 3CL protease may be very different between the SARS-infected cells and the recombinant 3CL protease used in the cell-free assays. We tried assay conditions using a low concentration (5 nM) of the SARS 3CL protease in an extended reaction time (1 h) and showed no sign of slow inhibition due to the irreversible bond formation with compounds 18c–e. The assay with a prolonged period is infeasible due to depletion of the substrate. It is uncertain whether the actual inhibitor binding may be tighter through a slow conformational change. On the other hand, the inhibitor may partially block the process for polyprotein maturation.21 Previous studies suggested that dimeric 3CL protease is the active form.5 Studies using cell-free systems also suggested that the N- and C-terminal tagged forms can be autoproteolyzed, and the autoproteolytic process is susceptible to protease inhibitors.21 Furthermore, 3CL protease can also proteolyze polyproteins in trans for their maturation.21 The cell-free studies therefore suggested the presence of multiple forms of proteolytic activities, and the physical states of the 3CL protease and their reactivity in SARS-infected cells are likely to be even more complicated. Like 3CL protease, the HIV protease is synthesized as part of the polyprotein that is autoproteolyzed for the protease maturation. It is thought that the dimerization of the HIV protease takes place while the enzyme is still in the form of a polyprotein that is synthesized and migrated to the membrane budding sites. Both the location and the timing of the autoproteolysis are crucial for the HIV viral infectivity.25, 26 Very little is known on the physical state and the enzyme reactivity of the 3CL protease in SARS-infected cells. It is thus impossible to surmise, at this time, the rate-limiting target of the protease inhibitors on SARS infectivity. More studies are needed to understand the regulation, proteolytic activity, and physical states of the enzyme present in the SARS-infected cells for the control of SARS virus using protease inhibitors.
3. Conclusion
We have devised an improved method for the preparation of AG7088 in an efficient manner. Although AG7088 and the analogs bearing P1 lactam-glutamine residue (1a–d and 2a–d) show no inhibitory activity against the SARS-CoV 3CL protease, the related conjugated esters 3a–d and 4a–d are found to exhibit improved activities. The Phe-Phe dipeptide inhibitors 18a–e are designed on the basis of computer modeling of the enzyme–inhibitor complex. The most potent inhibitor 18c, with an inhibition constant of 0.52 μM, is obtained by condensation of the Phe-Phe dipeptide α,β-unsaturated ester with 4-(dimethylamino)cinnamic acid. The cell-based assays indicate that 18c is a nontoxic anti-SARS agent with an EC50 value of 0.18 μM. In addition to valinomycin with an EC50 of 0.85 μM,7 to our knowledge compounds 18c–e represent the most potent anti-SARS agents known to date. Our computational study of structure–activity relationships shows that hydrogen bonding with the main chain Glu166 and the side chain Gln189 are crucial to the inhibitory potency. This interaction is supported by a modeling complex of SARS-CoV 3CL protease with the inhibitor 18c. We are currently investigating the cocrystal structure, which can give an insight into the proposed complexation mode for further design of new anti-SARS compounds.
4. Experimental
4.1. General
Melting points are uncorrected. 1H NMR spectra were recorded at 400 MHz; 13C NMR spectra were recorded at 100 MHz. Chemical shifts (δ) are given in parts per million (ppm) relative to residual solvent [δ 7.24 (s) for CHCl3 and δ 2.49 (m) for DMSO-d 6]. The splitting patterns are reported as s (singlet), d (doublet), t (triplet), and multiplet (m). Chemical shifts of 13C NMR spectra are reported relative to CDCl3 (δ 77.0 for the central line of triplet) and DMSO-d 6 [δ 39.5 (m)]. Mass spectra were recorded at an ionizing voltage of 70 or 20 eV. Merck silica gel 60F sheets were used for analytical thin-layer chromatography (TLC). Merck silica gel 60F glass plates (20 cm × 20 cm with 2 mm thickness) were used for preparative TLC. Column chromatography was performed on silica gel (70–230 mesh) using gradients of EtOAc/hexane as eluents.
4.2. Chemistry
Reactions requiring dry conditions were carried out under an inert atmosphere using standard techniques. All the reagents and solvents were of reagent grade and were used without further purification unless otherwise specified. THF was distilled from sodium benzophenone ketyl under N2. Tripeptide ketomethylene isosteres lb–d and 3a–d were prepared by the procedure similar to that for 1a (AG7088). Peptide α,β-unsaturated esters 2a–d and 4a–d were prepared by the similar procedure. Compounds 5–13 were prepared according to the previously described procedures.(a), 16
4.2.1. Allyl (2R,5S)-5-(tert-butoxycarbonyl)amino-2-(4-fluorobenzyl)-6-methyl-4-oxo-heptanoate (14)
Lithium hydroxide (3.3 mmol, 3.3 mL of 1 M aqueous solution) was added to a solution of methyl ester 13 (1.186 g, 3 mmol) in MeOH (20 mL) at 0 °C. The mixture was stirred for 1 h at 0 °C, 1 N HCl (20 mL) was added, and the mixture was extracted with EtOAc (30 mL, 4×). The combined organic layers were dried over MgSO4 and concentrated under reduced pressure to provide a crude carboxylic acid (1.028 g, 90%).
The crude product was dissolved in DMF (20 mL) and treated with allyl iodide (0.36 mL, 4.0 mmol) in the presence of Cs2CO3 (1.14 g, 3.5 mmol) at 45 °C for 5 h. The mixture was diluted with water (20 mL) and extracted with EtOAc (30 mL, 3×). The combined organic layers were washed with water (20 mL, 3×), dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography (EtOAc/hexane, 1:1) to afford allyl ester 14 (839 mg, 84%) as a colorless oil.
Compound 14: oil; TLC (EtOAc/hexane, 1:1) R f = 0.33; IR (neat) 3101, 2983, 1721, 1605, 1451, 1111 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.15–7.12 (2H, m), 7.02–6.90 (2H, m), 5.85–5.69 (1H, m), 5.27–5.19 (2H, m), 5.10 (1H, d, J = 8.6 Hz), 4.53–4.49 (2H, m), 4.25–4.13 (1H, m), 3.16–3.10 (1H, m), 3.08–2.95 (1H, m), 2.91–2.69 (2H, m), 2.55–2.38 (1H, m), 2.17–2.00 (1H, m), 1.49 (9H, s), 0.93 (3H, d, J = 6.8 Hz), 0.70 (3 H, d, J = 6.8 Hz); 13C NMR (CDCl3, 100 MHz): δ 204.4 (C), 171.1 (C), 148.5 (C), 137.6 (C), 135.0 (CH), 129.1 (CH, 2×), 128.8 (CH, 2×), 126.6 (C), 114.1 (CH2), 71.8 (CH2), 63.1 (C), 42.5 (CH), 38.9 (CH2), 32.7 (CH2), 28.1 (CH), 28.0 (CH3, 3×), 19.7 (CH), 16.3 (CH3, 2×); FAB-MS 422.2 (M++H); HRMS calcd for C23H33FNO5 (M++H), 422.2343; found, 422.2341.
4.2.2. Allyl (2R,5S)-2-(4-fluorobenzyl)-6-methyl-5-[(5-methyl-3-isoxazolyl)carbonyl)]amino-4-oxoheptanoate (16)
A solution of HCl in 1,4-dioxane (4.0 M, 5 mL) was added to a solution of allyl ester 14 (210 mg, 0.5 mmol) in 1,4-dioxane (5 mL) at room temperature. The mixture was stirred for 2 h, and then concentrated under reduced pressure to give a crude aminium salt. This material was dissolved in CH2Cl2 (10 mL) and cooled to 0 °C. 4-Methylmorpholine (0.17 mL, 1.5 mmol) and 5-methylisoxazole-3-carbonyl chloride (88 mg, 0.6 mmol) were added sequentially. The mixture was removed from the ice bath, stirred for 2 h at 25 °C, and diluted with CH2Cl2 (20 mL). The mixture was washed with 10% aqueous citric acid (20 mL) and brine (20 mL). The organic phase was dried over Na2SO4, concentrated, and purified by flash column chromatography (EtOAc/hexane, 1:4) to give the desired product 16 (187 mg, 88%).
Compound 16: oil; TLC (EtOAc/hexane, 1:1) R f = 0.28; IR (neat) 3071, 2944, 1714, 1700, 1612, 1466 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.13–7.09 (2H, m), 6.98–6.87 (2H, m), 6.44 (1H, s), 5.86–5.61 (1H, m), 5.23–5.09 (3H, m), 4.61–4.52 (2H, m), 4.23 (1H, dd, J = 8.5, 3.7 Hz), 3.13–3.07 (1H, m), 2.97–2.86 (2H, m), 2.72–2.61 (1H, m), 2.51–2.40 (1H, m), 2.49 (3H, s), 2.21–2.03 (1H, m), 0.95 (3H, d, J = 8.1 Hz), 0.71 (3H, d, J = 8.1 Hz); 13C NMR (CDCl3, 100 MHz): δ 204.4 (C), 171.9 (C), 169.8 (C), 160.8 (C), 160.2 (C), 157.9 (C), 138.1 (C), 128.5 (CH, 2×), 128.2 (CH, 2×), 126.1 (CH), 112.1 (CH2), 103.8 (CH), 72.6 (CH2), 64.5 (CH), 39.0 (CH2), 34.5 (CH2), 28.6 (CH), 20.9 (CH), 20.3 (CH3), 16.6 (CH3, 2×); FAB-MS m/z 431.2 (M++H); HRMS calcd for C23H28FN2O5 (M++H), 431.1982; found, 431.1983.
4.2.3. Ethyl 4-[2-(4-fluorobenzyl)-6-methyl-5-(5-methyl-3-isoxazolyl)carbonylamino-1,4-dioxoheptylamino]-5-(2-oxo-3-pyrrolidinyl)-2-pentenoate (1a, AG7088)
Compound 16 (129 mg, 0.3 mmol) in anhydrous THF (10 mL) was stirred with Pd(PPh3)4 (36 mg, 0.03 mmol) and morpholine (0.25 mL, 3.0 mmol) for 3 h at 25 °C. The mixture was concentrated under reduced pressure, diluted with CH2Cl2 (30 mL), and washed with 2 N HCl (10 mL) and water (20 mL). The organic phase was extracted with saturated NaHCO3 aqueous solution (30 mL, 3×). The combined aqueous extracts were acidified to pH 2 with 5% aqueous KHSO4 at 0 °C, and then extracted with Et2O (30 mL, 5×). The ethereal extract was dried (MgSO4), filtered, and the filtrate was concentrated under reduced pressure to give the corresponding acid of 16 (99 mg, 85%).
Compound 7 (81 mg, 0.25 mmol) was treated with HCl in 1,4-dioxane, by a procedure similar to that for 14, to give aminium salt 8. This material and the carboxylic acid derived from 16 (99 mg, 0.25 mmol) were dissolved in DMF (5 mL) and cooled to 0 °C, followed by the addition of 4-methylmorpholine (0.08 mL, 0.75 mmol), HOBt (41 mg, 0.3 mmol), and EDCI (58 mg, 0.3 mmol). The mixture was removed from the ice bath, stirred for 20 h at 25 °C, diluted with CH2Cl2 (15 mL), and washed with 10% aqueous citric acid (8 mL) and water (10 mL, 3×). The organic phase was dried over Na2SO4, concentrated, and purified by flash column chromatography (MeOH/CH2Cl2, 1:99) to provide 105 mg of 1a (70% yield).
Compound 1a: white solid; mp 180–182 °C (lit.13a mp 178–181 °C); TLC (CH3OH/CH2Cl2, 1:9) R f = 0.50; IR (KBr) 3299, 1678, 1521, 1325, 1180 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.34 (1H, d, J = 8.7 Hz), 7.11–7.08 (2H, m), 6.94–6.90 (2H, m), 6.58 (1H, dd, J = 11.7, 5.2 Hz), 6.38 (1H, s), 6.05 (1H, br s, NH), 5.42 (1H, dd, J = 11.6, 1.4 Hz), 4.63 (1H, dd, J = 8.7, 4.4 Hz), 4.50–4.37 (1H, m), 4.14 (2H, q, J = 6.8 Hz), 3.31–3.20 (2H, m), 3.05–2.99 (1H, m), 2.98–2.89 (1H, m), 2.88–2.79 (1H, m), 2.70–2.51 (3H, m), 2.45 (3H, s), 2.25–2.00 (3H, m), 1.85–1.75 (1H, m), 1.74–1.60 (1H, m), 1.50–1.42 (1H, m), 1.25 (3H, t, J = 6.8 Hz), 0.85 (3H, d, J = 6.8 Hz), 0.79 (3H, d, J = 6.8 Hz) ; 13C NMR (CDCl3, 100 MHz): δ 206.7 (C), 173.4 (C), 171.1 (C), 166.0 (C), 162.6 (C), 160.2 (C), 158.9 (C), 158.1 (C), 147.1 (CH), 134.0 (C), 130.3 (CH, 2×), 120.5 (CH), 115.1 (CH), 114.8 (CH, 2×), 101.3 (CH), 62.8 (CH), 60.4 (CH2), 49.0 (CH), 43.9 (CH), 42.0 (CH2), 40.5 (CH2), 38.3 (CH2), 34.9 (CH2), 30.4 (CH), 28.7 (CH2), 19.9 (CH3), 17.1 (CH3), 14.3 (CH2), 12.4 (CH3); FAB-MS 599.3 (M++H); HRMS calcd for C31H40FN4O7, 599.2801 (M++H); found, 599.2811. Anal. calcd for C31H39FN4O7: C 62.19, H 6.57, N 9.36. Found: C 62.12, H 6.60, N 9.37.
4.2.4. Ethyl 4-(tert-butoxycarbonyl)amino-5-phenyl-2-pentenoate (17)
Ethyl (2E,4S)-4-(tert-butoxycarbonyl)amino-5-phenyl-2-pentenoate23 was prepared from N-Boc-l-Phe by a sequence of esterification (CH3I, KHCO3, rt, 8 h, 94%), reduction (NaBH4, LiCl, THF, EtOH, rt, 12 h, 89%), oxidation (pyridine·SO3, DMSO, Et3N, −10 °C to rt, 1 h), and olefination [EtO2CCH2PO(OEt)2, NaN(TMS)2, THF, −78 °C to rt, 2 h, 84% for two steps], and then treated with HCl in 1,4-dioxane to remove the Boc group. The resulting amine was reacted with N-Boc-l-Phe in the presence of EDCI, HOBt, and 4-methylmorpholine, by a procedure similar to that for compound 1a, to give the desired product 17 in 82% overall yield.
Compound 17: white solid; mp 129–131 °C; TLC (EtOAc/hexane, 3:7) R f = 0.27; IR (KBr) 3214, 1711, 1645 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.30–7.15 (8H, m), 7.05 (2H, d, J = 6.7 Hz), 6.77 (1H, dd, J = 15.5, 4.3 Hz), 6.00 (1H, d, J = 8.1 Hz), 5.59 (1H, d, J = 15.5 Hz), 4.99–4.87 (2H, m), 4.29–4.25 (1H, m), 4.16 (2H, q, J = 7.1 Hz), 2.99–2.97 (2H, m), 2.81 (2H, d, J = 6.7 Hz), 1.40 (9H, s), 1.27 (3H, t, J = 7.1 Hz); 13C NMR (CDCl3, 100 MHz): δ 170.6 (C), 166.0 (C), 155.3 (C), 146.2 (C), 136.4 (C), 136.0 (CH), 129.3 (CH, 2×), 129.2 (CH, 2×), 128.8 (CH, 2×), 128.6 (CH, 2×), 127.1 (CH), 126.9 (CH), 121.5 (CH), 80.3 (C), 60.4 (CH2), 56.0 (CH), 50.6 (CH), 40.4 (CH2), 38.4 (CH2), 28.2 (CH3, 3×), 14.6 (CH3); FAB-MS 467.57 (M++H); HRMS calcd for C27H35N2O5, 467.5771 (M++H); found, 467.5775.
4.2.5. Dipeptidomimetic α,β-unsaturated esters 18a–e
The Phe-Phe dipeptide α,β-unsaturated ester 17 (235 mg, 0.5 mmol) was treated with HCl in 1,4-dioxane, by a procedure similar to that for 14, to give the corresponding aminium salt, which was then subjected to coupling reactions with appropriate (substituted) cinnamic acids (0.55 mmol) in DMF (10 mL) by promotion of HBTU (0.6 mmol) and i-Pr2NEt (2.5 mmol), by a procedure similar to that for 1a, to afford the dipeptide conjugated esters 18a–e (69–83% yields).
4.2.5.1. Ethyl 5-phenyl-4-[2-(4-phenylcinnamyl)amino-1-oxo-3-phenyl]propylamino-2-pentenoate (18a)
Colorless solid; mp 220–223 °C; TLC (CH3OH/CH2Cl2, 1:19) R f = 0.45; IR (KBr) 3211, 1709, 1629, 1521, 1259 cm−1; 1H NMR (DMSO-d 6, 400 MHz): δ 8.36 (2H, d, J = 8.2 Hz), 7.73–7.69 (4H, m), 7.63 (2H, d, J = 8.2 Hz), 7.46–7.42 (4H, m), 7.22–7.15 (10H, m), 6.78 (1H, dd, J = 15.7, 5.2 Hz), 6.75 (1H, d, J = 15.7 Hz), 5.67 (1H, d, J = 15.7 Hz), 4.71–4.65 (2H, m), 4.09 (2H, q, J = 7.1 Hz), 2.97–2.77 (4H, m), 1.19 (3H, t, J = 7.1 Hz); 13C NMR (DMSO-d 6, 100 MHz): δ 171.0 (C), 165.9 (C), 165.1 (C), 148.5 (C), 141.4 (C), 139.8 (C), 138.9 (C), 138.1 (C), 138.0 (CH), 134.4 (CH), 129.7 (CH, 2×), 129.6 (CH, 2×), 129.5 (CH, 2×), 128.6 (CH, 2×), 128.5 (CH, 2×), 128.3 (CH, 2×), 127.6 (CH, 2×), 127.1 (CH, 2×), 126.8 (CH, 2×), 126.7 (CH), 122.4 (CH), 120.7 (CH), 60.4 (CH2), 54.9 (CH), 51.4 (CH), 38.4 (CH2, 2×), 14.6 (CH3); FAB-MS m/z 573.28 (M++H); HRMS calcd for C37H37N2O4, 573.2753 (M++H); found, 573.2755. Anal. calcd for C37H36N2O4: C 77.60, H 6.34, N 4.89. Found: C 77.58, H 6.34, N 4.90.
4.2.5.2. Ethyl 4-[2-(4-nitrocinnamyl)amino-1-oxo-3-phenyl]propylamino-5-phenyl-2-pentenoate (18b)
Colorless solid; mp 247–250 °C; TLC (EtOAc/hexane, 1:1) R f = 0.27; IR (KBr) 3291, 1711, 1637, 1533, 1421, 1259 cm−1; 1H NMR (DMSO-d 6, 400 MHz): δ 8.49 (1H, d, J = 8.5 Hz), 8.38 (1H, d, J = 8.5 Hz), 8.25 (2H, d, J = 8.7 Hz), 7.80 (2H, d, J = 8.7 Hz), 7.49 (1H, d, J = 15.8 Hz), 7.29–7.14 (10H, m), 6.88 (1H, d, J = 15.8 Hz), 6.78 (1H, dd, J = 15.8, 5.2 Hz), 5.66 (1H, d, J = 15.8 Hz), 4.67–4.63 (2H, m), 4.09 (2H, q, J = 7.1 Hz), 2.96–2.65 (4H, m), 1.19 (3H, t, J = 7.1 Hz); 13C NMR (DMSO-d 6, 100 MHz): δ 171.8 (C), 165.9 (C), 164.3 (C), 148.5 (C), 147.9 (C), 141.9 (C), 138.1 (C), 137.9 (CH), 137.1 (CH), 129.7 (CH), 129.5 (CH), 129.0 (CH), 128.6 (CH, 2×), 128.5 (CH, 2×), 126.8 (CH, 2×), 126.7 (CH, 2×), 126.6 (CH, 2×), 124.6 (CH, 2×), 120.1 (CH), 60.4 (CH2), 54.7 (CH), 51.4 (CH), 39.3 (CH2), 31.2 (CH2), 14.6 (CH3); FAB-MS m/z 542.23 (M++H); HRMS calcd for C31H32N3N3O6, 542.2291 (M++H); found, 542.2289. Anal. calcd for C31H31N3O6: C 68.75, H 5.77, N 7.76. Found: C 68.77, H 5.56, N 7.76.
4.2.5.3. Ethyl 4-{2-[4-((dimethylamino)cinnaniyl]-amino-1-oxo-3-phenyl}propylamino-5-phenyl-2-pentenoate (18c)
Colorless solid; mp 225–227 °C; TLC (EtOAc/hexane, 1:1) R f = 0.24; IR (KBr) 3213, 1707, 1521, 1459, 1255 cm−1; 1H NMR (DMSO-d 6, 400 MHz): δ 8.32 (1H, d, J = 8.2 Hz), 8.16 (1H, d, J = 8.2 Hz), 7.36 (2H, d, J = 8.6 Hz), 7.29–7.14 (11H, m), 6.79 (1H, dd, J = 15.8, 5.1 Hz), 6.70 (2H, d, J = 8.6 Hz), 6.41 (1H, d, J = 15.7 Hz), 5.66 (1H, d, J = 15.7 Hz), 4.65–4.60 (2H, m), 4.09 (2H, q, J = 7.1 Hz), 2.94 (6H, s), 2.90–2.64 (4H, m), 1.18 (3H, t, J = 7.1 Hz); 13C NMR (DMSO-d 6, 100 MHz): δ 171.2 (C), 166.0 (C), 165.9 (C), 151.5 (C), 148.6 (C), 139.8 (C), 138.2 (C), 129.7 (CH, 2×), 129.5 (CH, 2×), 129.3 (CH, 2×), 128.6 (CH, 2×), 128.5 (CH, 2×), 126.7 (CH, 2×), 122.7 (CH), 126.8 (CH, 2×), 120.6 (CH), 116.8 (CH), 112.4 (CH), 60.4 (CH2), 54.6 (CH), 51.4 (CH), 40.5 (CH2, 2×), 38.4 (CH2, 2×), 14.6 (CH3); FAB-MS m/z 540.06 (M++H); HRMS calcd for C33H38N3O4, 540.0641 (M++H); found, 540.0639. Anal. calcd for C33H37N3O4: C 73.44, H 6.91, N 7.79. Found: C 73.47, H 6.93, N 7.78.
4.2.5.4. Ethyl 4-[2-(2,4-dimethoxycinnamyl)amino-1-oxo-3-phenyl]propylamino-5-phenyl-2-pentenoate (18d)
Colorless solid; mp 203–205 °C; TLC (EtOAc/hexane, 1:1) R f = 0.27; IR (KBr) 3276, 2932, 1723, 1648, 1536, 1270, 1033 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.77 (1H, d, J = 15.7 Hz), 7.40 (2H, d, J = 8.5 Hz), 7.30–7.14 (7H, m), 7.06 (2H, d, J = 8.5 Hz), 6.80 (1H, dd, J = 15.7, 5.2 Hz), 6.50 (1H, d, J = 8.5 Hz), 6.47 (1H, s), 6.38 (1H, d, J = 15.7 Hz), 6.37 (1H, br s), 6.05 (1H, br s), 5.61 (1H, d, J = 15.7 Hz), 4.85–4.76 (2H, m), 4.18 (2H, q, J = 7.0 Hz), 3.88 (3H, s), 3.85 (3H, s), 3.12–3.06 (2H, m), 2.85–2.76 (2H, m), 1.30 (3H, t, J = 7.0 Hz); 13C NMR (CDCl3, 100 MHz): δ 170.6 (C), 166.9 (C), 166.1 (C), 162.4 (C), 159.8 (C), 146.3 (CH), 137.3 (C), 136.5 (C), 136.3 (CH), 130.7 (CH), 129.3 (CH), 129.2 (CH, 2×), 128.7 (CH, 2×), 128.5 (CH, 2×), 127.0 (CH, 2×), 126.7 (CH), 121.4 (CH), 118.2 (C), 116.6 (CH), 105.0 (CH), 98.4 (CH), 60.4 (CH2), 55.47 (CH3), 55.41 (CH3), 54.6 (CH), 51.1 (CH), 40.5 (CH2), 38.4 (CH2), 14.2 (CH3); FAB-MS m/z 557.26 (M++H); HRMS calcd for C33H37N2O6, 557.2651 (M++H); found, 557.2656. Anal. calcd for C33H36N2O6: C 71.20, H 6.52, N 5.03. Found: C 71.17, H 6.54, N 5.03.
4.2.5.5. Ethyl 4-{2-[3,4-(methylenedioxy)cinnamyl]amino-l-oxo-3-phenyl}propylamino-5-phenyl-2-pentenoate (18e)
Colorless solid; mp 192–194 °C; TLC (EtOAc/hexane, 1:1) R f = 0.33; IR (KBr) 3297, 1726, 1666, 1547, 1448, 1249 cm−1; 1H NMR (DMSO-d 6, 400 MHz): δ 8.32 (1H, d, J = 8.3 Hz), 8.18 (1H, d, J = 8.4 Hz), 7.32–7.10 (12H, m), 7.04 (1H, d, J = 8.0 Hz), 6.93 (1H, d, J = 8.0 Hz), 6.78 (1H, dd, J = 15.7, 5.2 Hz), 6.53 (1H, d, J = 15.7 Hz), 6.05 (2 H, s), 5.65 (1H, d, J = 15.7 Hz), 4.67–4.63 (2 H, m), 4.09 (2 H, q, J = 7.0 Hz), 2.93–2.75 (4H, m), 1.19 (3H, t, J = 7.0 Hz); 13C NMR (DMSO-d 6, 100 MHz): δ 171.0 (C), 165.9 (C), 165.3 (C), 148.9 (C), 148.5 (C), 148.4 (C), 139.2 (C), 138.8 (C), 138.1 (CH), 129.7 (CH), 129.5 (CH, 2×), 128.7 (CH), 128.6 (CH, 2×), 128.5 (CH, 2×), 126.8 (CH, 2×), 126.7 (CH), 123.7 (CH), 120.7 (CH), 120.4 (CH), 109.0 (CH), 106.2 (CH), 101.9 (CH2), 60.4 (CH2), 54.6 (CH), 51.4 (CH), 38.4 (CH2, 2×), 14.6 (CH3); FAB-MS m/z 541.23 (M++H); HRMS calcd for C32H33N2O6, 541.2338 (M++H); found, 541.2341. Anal. calcd for C32H32N2O6: C 71.09, H 5.97, N 5.18. Found: C 71.11, H 5.99, N 5.17.
4.2.6. Ethyl 4-{2-[3,4-(methylenedioxy)cinnamyl]amino-1-oxo-3-phenyl} propylamino-5-(2-oxo-3-pyrrolidyl)-2-pentenoate (19)
Condensation of l-Phe-OMe with 3,4-(methylenedioxy)cinnamic acid in the presence of EDCI, HOBt, and 4-methylmorpholine, followed by hydrolysis (LiOH, MeOH/H2O), afforded N-[3,4-(methylenedioxy)cinnamyl]phenylalanine in 64% yield. This product was reacted with the aminium salt 8 (HBTU, i-Pr2NEt, DMF, 0 °C to rt, 7 h), by a procedure similar to that for 18a–e, to give the title compound 19 in 57% yield.
Compound 19: white solid; mp 103–105 °C; TLC (MeOH/CH2Cl2, 1:99) R f = 0.24; IR (KBr) 3283, 2919, 1683, 1447, 1241 cm−1; 1 H NMR (CDCl3, 400 MHz): δ 7.81 (1H, d, J = 7.0 Hz), 7.50 (1H, d, J = 15.5 Hz), 7.29–7.20 (4H, m), 7.00–6.94 (2H, m), 6.79–6.67 (3H, m), 6.31–6.26 (2H, m), 5.99 (2H, s), 5.75 (1H, d, J = 15.5 Hz), 5.10–5.00 (1H, m), 4.57–4.40 (1H, m), 4.18 (2H, q, J = 7.1 Hz), 3.31–3.19 (3H, m), 3.08–3.06 (1H, m), 2.23–2.12 (2H, m), 2.10–1.81 (2H, m), 1.80–1.40 (2H, m), 1.30 (3H, t, J = 7.0 Hz); 13C NMR (CDCl3, 100 MHz): δ 171.2 (C), 166.2 (C), 165.8 (C), 165.7 (C), 149.2 (C), 148.2 (C), 146.8 (CH), 141.3 (C), 136.5 (CH), 129.7 (C), 129.1 (CH), 128.5 (CH, 2×), 126.9 (CH, 2×), 124.1 (CH), 121.2 (CH), 118.3 (CH), 108.5 (CH), 106.4 (CH), 101.4 (CH2), 60.5 (CH2), 54.8 (CH), 54.3 (CH), 49.5 (CH), 40.7 (CH2), 38.9 (CH2), 34.8 (CH2), 29.7 (CH2), 14.2 (CH3); FAB-MS 548.24 (M++H); HRMS calcd for C30H34N3O7, 548.2397 (M++H); found, 548.2399.
4.2.7. Ethyl 4-[N-(3,4-methylenedioxy)cinnamyl-valinyl-phenylalanyl]-5-phenyl-2-pentenoate (20)
By a procedure similar to that for 19, condensation of l-Val-OMe with 3,4-methylenedioxycinnamic acid gave N-(3,4-methylenedioxy)cinnamyl-valine, which was subjected to amidation with the amine derived from 17 to give compound 20 in 73% yield.
Compound 20: white solid; mp 207–209 °C; TLC (MeOH/CH2Cl2, 1:19) R f = 0.32; IR (KBr) 3284, 1678, 1437, 1255 cm−1; 1H NMR (DMSO-d 6, 400 MHz): δ 8.13 (2H, d, J = 7.1 Hz), 7.93 (1H, d, J = 7.1 Hz), 7.35–6.93 (14H, m), 6.93–6.60 (2H, m), 6.05 (2H, s), 5.62 (1H, d, J = 15.5 Hz), 4.66–4.60 (1H, m), 4.50–4.47 (1H, m), 4.24–4.21 (1H, m), 4.09 (2H, q, J = 7.1 Hz), 2.91–2.65 (4H, m), 2.02–1.87 (1H, m), 1.21–1.15 (9H, m); 13C NMR (DMSO-d 6, 100 MHz): δ 171.2 (C), 170.7 (C), 165.9 (C), 165.8 (C), 148.9 (C), 148.4 (C), 148.3 (CH), 139.2 (C), 138.1 (C), 137.9 (CH), 129.8 (C), 129.6 (CH, 2×), 129.5 (CH, 2×), 128.7 (CH, 2×), 128.6 (CH, 2×), 128.5 (CH), 126.7 (CH), 123.7 (CH), 120.7 (CH), 120.6 (CH), 109.1 (CH), 106.5 (CH), 101.9 (CH2), 60.2 (CH2), 58.4 (CH), 54.6 (CH), 51.2 (CH), 40.6 (CH2), 39.3 (CH2), 30.9 (CH), 19.6 (CH3), 19.4 (CH3), 14.3 (CH3); FAB-MS 640.30 (M++H); HRMS calcd for C37H42N3O7, 640.3023 (M++H); found, 640.3027.
4.2.8. N,N′-Bis-[2-(1-benzyl-3-ethoxycarbonyl)propenyl]1,4-butanediamide (21a)
Ethyl (2E,4S)-4-(tert-butoxycarbonyl)amino-5-phenyl-2- pentenoate23 was treated with HCl in 1,4-dioxane to remove the Boc group, and the resulting amine was reacted with succinic acid in the presence of EDCI, HOBt, and 4-methylmorpholine, by a procedure similar to that for 17, to give 21a in 80% yield.
Compound 21a: TLC (EtOAc/hexane, 1:4) R f = 0.21; IR (KBr) 3304, 3065, 2998, 1731, 1646, 1547, 1308, 1189 cm−1; 1H NMR (CDCl3, 300 MHz): δ 7.27–7.10 (10H, m), 6.86 (2H, dd, J = 13.6, 8.0 Hz), 6.48 (2H, d, J = 8.5 Hz), 5.81 (2H, dd, J = 13.6, 4.2 Hz), 4.87 (2H, dd, J = 13.2, 6.1 Hz), 4.10 (4H, q, J = 7.6 Hz), 2.93–2.77 (4H, m), 2.46 (4H, s), 1.23 (6H, t, J = 7.6 Hz); 13C NMR (CDCl3, 75 MHz): δ 171.5 (C, 2×), 166.0 (C, 2×), 146.7 (CH, 2×), 136.4 (C, 2×), 129.3 (CH, 4×), 128.5 (CH, 4×), 126.8 (CH, 2×), 121.3 (CH, 2×), 60.6 (CH, 2×), 51.2 (CH2, 2×), 40.3 (CH2, 2×), 31.4 (CH2, 2×), 14.1 (CH3, 2×); FAB-MS m/z 521.28 (M++H); HRMS calcd for C30H37N2O6, 521.2652 (M++H); found, 521.2655.
4.2.9. N,N′-Bis-[2-(1-benzyl-3-ethoxycarbonyl)propenyl]2-hydroxy-1,4-butanediamide (21b)
Using (S)-malic acid in lieu of succinic acid, compound 21b was prepared by a procedure similar to that for 21a.
Compound 21b: white solid; TLC (CH3OH/CH2Cl2, 1:19) R f = 0.23; IR (KBr) 3310, 3068, 1718, 1654, 1533, 1371, 1287, 1182 cm−1; 1H NMR (CDCl3, 300 MHz): δ 7.29–7.07 (10H, m), 6.92–6.77 (2H, m), 5.83–5.77 (2H, m), 5.81 (1H, br s), 4.85–4.78 (2H, m), 4.22 (2H, d, J = 6.7 Hz), 4.17–4.08 (4H, m), 2.93–2.78 (4H, m), 2.86–2.56 (2H, m), 2.19 (1H, dd, J = 15.6, 8.6 Hz), 1.25–1.17 (6H, m); 13C NMR (CDCl3, 75 MHz): δ 171.9 (C), 171.5 (C), 165.9 (C, 2×), 146.2 (CH), 146.1 (CH), 136.2 (C), 136.1 (C), 129.35 (CH, 2×), 129.25 (CH, 2×), 128.6 (CH, 2×), 128.5 (CH, 2×), 126.96 (CH), 126.92 (CH), 121.6 (CH), 121.5 (CH), 69.03 (CH), 60.6 (CH), 60.5 (CH), 51.0 (CH2), 50.6 (CH2), 40.5 (CH2), 40.2 (CH2), 38.9 (CH), 14.1 (CH3, 2×); FAB-MS m/z 537.3 (M++H); HRMS calcd for C30H37N2O7, 537.2601 (M++H); found, 537.2600.
4.2.10. N,N′-Bis-2-(1-benzyl-3-ethoxycarbonyl)propenyl]2,3-dihydroxy-1,4-butanediamide (21c)
Using (2R,3R)-tartaric acid in lieu of succinic acid, compound 21c was prepared by a procedure similar to that for 21a.
Compound 21c: TLC (CH3OH/CH2Cl2, 1:19) R f = 0.21; IR (neat) 3357, 2986, 2926, 1706, 1646, 1534, 1295, 1169 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.32–7.25 (6H, m), 7.16 (4H, d, J = 8.3 Hz), 6.87 (2H, dd, J = 15.7, 4.3 Hz), 5.74 (2H, dd, J = 15.7, 2.0 Hz), 5.01–4.93 (4H, m), 4.17–4.12 (4H, m), 2.89–2.87 (8H, m), 1.23 (6H, t, J = 7.1 Hz); 13C NMR (CDCl3, 100 MHz): δ 166.1 (C, 2×), 162.7 (C, 2×), 146.4 (C, 2×), 136.2 (CH, 2×), 129.3 (CH, 4×), 128.6 (CH, 4×), 126.9 (CH, 2×), 121.3 (CH, 2×), 71.5 (CH, 2×), 60.5 (CH2, 2×), 50.7 (CH, 2×), 40.3 (CH2, 2×), 14.1 (CH, 2×).
4.3. Inhibition assay against the SARS-CoV 3CL protease
A fluorometric assay19 was utilized to determine the inhibition constants of the prepared samples. Briefly, a fluorogenic peptide Dabcyl-KTSAVLQSGFRKME-Edans was used as the substrate; and the enhanced fluorescence due to cleavage of this substrate catalyzed by the protease was monitored at 538 nm with excitation at 355 nm. The IC50 value of individual sample was measured in a reaction mixture containing 50 nM of the SARS-CoV 3CL protease and 6 μM of the fluorogenic substrate in 20 mM Bis–Tris (pH 7.0). The enzyme stock solution was kept in 12 mM Tris–HCl (pH 7.5) containing 120 mM NaCl, 0.1 mM EDTA, and 1 mM DTT plus 7.5 mM β-ME before adding to the assay solution. The K i measurements were performed at two fixed inhibitor concentrations and various substrate concentrations.
4.4. Cell-based assay for the anti-SARS agents
Vero E6 cells (2 × 104/well) were cultured in a 96-well plate in FBS DMEM supplemented with 10% FBS. The culture medium was removed after 1-day incubation when the cells reached 80–90% confluence. A solution of 100 μL DMEM with 2% FBS containing the compound to be tested, was placed in three wells. Three wells containing cells in DMEM, with 2% FBS were used as the CPE-positive control. Cells were incubated in a CO2 incubator at 37 °C for 2 h, inoculated with SARS-CoV (H.K.) at a dose of 100 TCID50/well, and the cytopathic morphology of the cells was examined using an inverted microscope 72 h after infection.
4.5. Cytotoxicity study
Vero E6 cells were grown in DMEM supplemented with l-glutamine, nonessential amino acids, and 10% FBS. The cells were cultured in 75 cm2 flasks and incubated in a humidified 5% CO2 incubator at 37 °C. Cells were later seeded at 7 × 104 cells/mL onto a 96-well plate and left overnight before cytotoxicity study. MTS assay was performed using Cell Titer 96 nonRadioactive Cell Proliferation Assay Kits (Promega Co., Madison, WI, USA) to determine the population of living cells.23 This assay measures the amount of Formazan produced by metabolic conversion of Owen’s reagent, [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfo- phenyl)-2H-tetrazolium, inner salt; MTS], by a dehydrogenase present in the mitochondria of metabolically active cells and is directly proportional to the number of living cells. Briefly, after the incubations with the tested compounds at varied concentrations for 2 days, the culture medium was replaced with MTS/PMS in DMEM. After incubation for 2 h at 37 °C, the absorbance of the samples was measured with a plate reader at 490 nm. Data are expressed as percentage of control cells (as 100%) cultured in the absence of any tested compounds.
4.6. ELISA of anti-SARS agents
At the conclusion of the incubation, the SARS-virus-infected Vero E6 cells were rinsed with PBS, fixed in an ice-cold methanol/acetone (1:1) solution for 3 min at room temperature, and rinsed three times with PBS. The cells were blocked with 3% skimmed milk in PBS for 30 min at room temperature, and then incubated for 1 h at 37 °C with 1:2000 diluted monoclonal antibody (ascetic fluid) to the spike protein of SARS-CoV. All samples were washed with three changes of PBS-T buffer and twice with fresh changes of PBS buffer at room temperature, followed by incubation with HRP-labeled goat anti-mouse IgG for 30 min at room temperature. The plates were rinsed with PBS containing 0.05% Tween 20 between incubations. A substrate solution containing O-phenylenediamine dihydrochloride, citrate buffer (pH 5.0), and hydrogen peroxide was added to each well. The plates were covered and gently shaken at room temperature for 10 min. The reaction was stopped by the addition of sulfuric acid (3 N) and the plates were read immediately at 492 nm. The EC50 value for each tested agent was extrapolated from the linear regression plot of the agent concentration versus OD492.
4.7. Western blot analysis of anti-SARS agents
SARS-CoV-infected Vero E6 cells were treated with compounds at varied concentrations for 24 or 48 h then lysed in a lysis buffer for 3 min. The cell debris was spun down and all cell lysates were harvested for electrophoresis and Western blotting assay with SDS–PAGE and a Hybond-C Extra membrane (Amersham Pharmacia). The resulting membrane was blocked in 3% skimmed milk in PBS for 30 min at room temperature, and then treated with 1:1000 diluted antispike protein monoclonal antibody (ChemiconMAb 1501) for 1 h at room temperature. The membrane was rinsed by using two changes of PBS-T buffer, and then washed once for 15 min and twice for 5 min each with fresh PBS buffer at room temperature, followed by treatment with horseradish peroxidase-labeled goat anti-mouse IgG for 30 min and 1:2000 dilution for 1 h. The membrane was washed as above, and the mixed enhanced chemiluminescence detection reagent was added to the protein side of the membrane. The blot was placed with the protein side up, in the film cassette, to visualize the level of protein expression.
4.8. Computer modeling
Docking experiments were conducted by using Autodock 3.0.5 with a Lamarckian Genetic Algorithm (LGA).27 The crystal structure (PDB code 1c145a) for the complex of a mutated (C145A) SARS-CoV 3CL protease with a hexapeptide substrate of Ser-Gly-Val-Thr-Phe-Gln was refined using Cys at residue 145 and only the apo chain B of the dimer in this simulation. The structures of inhibitory compounds were built in CAChe (Fujitsu, Japan) and refined by performing an optimized geometry calculation in Mechanics using augmented MM2 parameters and stored in PDB format. MGLTOOLS (MGL, Scripps Institute) was used for structure preparation and parameter creation to meet the input requirements of Autodock. Briefly, essential hydrogen atoms were added to the structure model of 3CLpro followed by assigning Kollman united atom charges and solvation parameters. Compound molecules were assigned Gasteiger–Marsili charges,28 merge nonpolar H atoms, and defined torsions. Autogrid tool in Autodock 3.0.5 was applied to compute energy grids (60 × 60 × 70 in xyz directions with 0.375 Å spacing) of various types of compound atoms. These grip maps were centered at the active site. During docking experiments, each compound was kept flexible (except for their rings and amide bonds) and the protein was rigid. Solis and Wets’ local search method with LGA was applied to generate available conformations of compound structures within the active site. The conformational search was conducted utilizing 0.2 Å quaternion and 2° torsion steps. For each compound structure, 5 × 106 energy was evaluated and 80 poses were selected from 5 × 105 generations per run. Plausible docking modes were elected by collecting binding free energies, clustering all simulated poses structures within 2.0 Å root mean square deviations, and selecting the most abundant cluster with highest affinity energy. The conserved hydrogen bond interactions with Glu166 and Gln189, which observed in crystal complex, were considered as the additional factors when electing the binding mode. Pictures of the final simulated docking modes were generated in MGLTOOLS.
Acknowledgment
We thank National Science Council for financial support.
References and notes
- 1.(a) Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]; (b) Drosten C., Günther S., Preiser W., van der Werf S., Brodt H.-R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A.M. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]; (c) Peiris J.S.M., Lai S.T., Poon L.L.M., Guan Y., Yam L.Y.C., Lim W., Nicholls J., Yee W.K.S., Yan W.W., Cheung M.T. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Helth Organization, Communicable Disease Surveillance & Response, website: http://www.who.int/csr/sars/archive/2003_05_07a/en and http://www.who.int/csr/sars/country/en/country2003_08_15.pdf. Summary table of SARS cases by country (1 November, 2002, to 7 August, 2003).
- 3.He J.-F., Peng G.-W., Min J., Yu D.-W., Liang W.-L., Zhang S.-Y., Xu R.-H., Zheng H.-Y., Wu X.-W., Xu J. Science. 2004;303:1666–1669. [Google Scholar]
- 4.(a) Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.-H. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]; (b) Marra M.A., Jones S.J.M., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S.N., Khattra J., Asano J.K., Barber S.A., Chan S.Y. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]; (c) Ruan Y.J., Wei C.L., Ee L.A., Vega V.B., Thoreau H., Yun S.T.S., Chia J.M., Ng P., Chiu K.P., Lim L. Lancet. 2003;361:1779–1785. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]; (b) Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chou K., Wei D., Zhong W. Biochem. Biophys. Res. Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang C., Wei P., Fan K., Liu Y., Lai L. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]; (c) Du Q.-S., Wang S.-Q., Zhu Y., Wei D.-Q., Guo H., Sirois S., Chou K.-C. Peptides. 2004;25:1857–1864. doi: 10.1016/j.peptides.2004.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu C.-Y., Jan J.-T., Ma H.-H., Kuo C.-J., Juan H.-F., Cheng Y.-S.E., Hsu H.-H., Huang H.-C., Wu D., Brik A., Liang F.-S., Liu R.-S., Fang J.-M., Chen S.-T., Liang P.-H., Wong C.-H. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu J.T.-A., Kuo C.-J., Hsieh H.-P., Wang Y.-C., Huang K.-K., Lina C.P.-C., Huang P.-F., Chen X., Liang P.-H. FEBS Lett. 2004;574:116–120. doi: 10.1016/j.febslet.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Biochemsitry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 10.Kao R.Y., Tsui W.H.W., Lee T.S.W., Tanner J.A., Watt R.M., Huang J.-D., Hu L., Chen G., Chen Z., Zhang L., He T., Chan K.-H., Tse H., To A.P.C., Ng L.W.Y., Wong B.C.W., Tsoi H.-W., Yang D., Ho D.D., Yuen K.-Y. Chem. Biol. 2004;11:1293–1299. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain R.P., Pettersson H.I., Zhang J., Aull K.D., Fortin P.D., Huitema C., Eltis L.D., Parrish J.C., James M.N.G., Wishart D.S., Vederas J.C. J. Med. Chem. 2004;47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 13.(a) Dragovich P.S., Prins T.J., Zhou R., Webber S.E., Marakovits J.T., Fuhrman S.A., Patick A.K., Matthews D.A., Lee C.A., Ford C.E., Burke B.J., Rejto P.A., Hendrickson T.F., Tuntland T., Brown E.L., Meador J.W., III, Ferre R.A., Harr J.E.V., Kosa M.B., Worland S.T. J. Med. Chem. 1999;42:1213–1224. doi: 10.1021/jm9805384. [DOI] [PubMed] [Google Scholar]; (b) Patick A.K., Binford S.L., Brothers M.A., Jackson R.L., Ford C.E., Diem M.D. Antimicrob. Agents Chemother. 1999;43:2444–2450. doi: 10.1128/aac.43.10.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Matthews D.A., Dragovich P.S., Webber S.E., Fuhrman S.A., Patick A.K., Zalman L.S., Hendrickson T.F., Love R.A., Prins T.J., Marakovits J.T., Zhou R., Tikhe J., Ford C.E., Meador J.W., Ferre R.A., Brown E.L., Binford S.L., Brothers M.A., Delisle D.M., Worland S.T. Proc. Natl. Acad. Sci. U.S.A. 1999;96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Kräusslich H.G., Wimmer E. Annu. Rev. Biochem. 1988;57:701–754. doi: 10.1146/annurev.bi.57.070188.003413. [DOI] [PubMed] [Google Scholar]; (b) Kay J., Dunn B.M. Biochem. Biophys. Acta. 1990;1048:1–18. doi: 10.1016/0167-4781(90)90015-t. [DOI] [PubMed] [Google Scholar]; (c) Lawson M.A., Semler B.L. Curr. Top. Microbiol. Immunol. 1990;161:49–87. [PubMed] [Google Scholar]
- 15.Sirois S., Wei D.-Q., Du Q., Chou K.-C. J. Chem. Inf. Comput. Sci. 2004;44:1111–1122. doi: 10.1021/ci034270n. [DOI] [PubMed] [Google Scholar]
- 16.(a) Tian, Q.; Nayyar, N. K.; Babu, S.; Tao, J.; Moran, T. J.; Dagnino, R.; Mitchell, L. J.; Remarchuk, T. P.; Melnick, M. J.; Bender, S. L. U.S. Pat. Appl. Publ. 2002.; (b) Tian Q., Nayyar N.K., Babu S., Chen L., Tao J., Lee S., Tibbetts A., Moran T., Liou J., Guo M., Kennedy T.P. Tetrahedron Lett. 2001;42:6807–6809. [Google Scholar]
- 17.(a) Larchevêque M., Petit Y. Tetrahedron Lett. 1987;28:1993–1996. [Google Scholar]; (b) Pègorier L., Petit Y., Larchevêque M. Synthesis. 1994:1403–1405. [Google Scholar]
- 18.Street L.J., Sternfeld F., Jelley R.A., Reeve A.J., Carling R.W., Moore K.W., McKernan R.M., Bindi Sohal B., Cook S., Pike A., Dawson G.R., Bromidge F.A., Wafford K.A., Seabrook G.R., Thompson S.A., Marshall G., Pillai G.V., Castro J.L., Atack J.R., MacLeod A.M. J. Med. Chem. 2004;47:3642–3657. doi: 10.1021/jm0407613. [DOI] [PubMed] [Google Scholar]
- 19.Kuo C.-J., Chi Y.-H., Hsu T.-A., Liang P.-H. Biochem. Biophys. Res. Commun. 2004;318:862–867. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.An interview mentions that AG7088 may be inactive against the SARS-CoV 3CL protease. See: Clarke, T. Nature2003, 423 (May 15). http://www.nature.com/nature/focus/SARS/index.html.
- 21.Hsu, M.-F.; Kuo, C.-J.; Chang, K.-T.; Chang, H.-C.; Chou, C.-C.; Ko, T.-P.; Shr, H.-L.; Chang, G.-G.; Wu, Y.-T.; Andrew Wang, H.-J.; Liang, P.-H. J. Biol. Chem.2005, in press. [DOI] [PMC free article] [PubMed]
- 22.(a) Wu C.-Y., Chang C.-F., Chen J.S.-Y., Wong C.-H., Lin C.-H. Angew. Chem., Int. Ed. 2003;42:4661–4664. doi: 10.1002/anie.200351823. [DOI] [PubMed] [Google Scholar]; (b) Chang C.-F., Ho C.-W., Wu C.-Y., Chao T.-A., Wong C.-H., Lin C.-H. Chem. Biol. 2004;11:1301–1306. doi: 10.1016/j.chembiol.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Ohba T., Ikeda E., Wakayama J., Takei H. Bioorg. Med. Chem. Lett. 1996;6:219–224. [Google Scholar]
- 24.Buttke T.M., McCubrey J.A., Owen T.C. J. Immunol. Methods. 1993;157:233–240. doi: 10.1016/0022-1759(93)90092-l. [DOI] [PubMed] [Google Scholar]
- 25.Kohl N.E., Emini E.A., Schleif W.A., Davis L.J., Heimbach J.C., Dixon R.A.F., Scolnick E.M., Sigal I.S. Proc. Natl. Acad. Sci. U.S.A. 1988;85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kraüsslich H.-G. Proc. Natl. Acad. Sci. U.S.A. 1991;88:3213–3217. doi: 10.1073/pnas.88.8.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris G.M., Goodsell D.S., Halliday R.S., Heuy R., Hart W.E., Belew R.K., Olson A.J. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 28.Gasteiger J., Marsili M. Tetrahedron. 1980;36:3219–3288. [Google Scholar]