

Abstract
The secretory pathway in cells possesses an elaborate set of endoproteolytic enzymes that carry out a crucial step in protein precursor maturation. This step is proteolytic activation by cleavage at specific pairs of basic residues. These enzymes, named pro-protein convertases (PCs), are responsible for generating bioactive peptides and activating several enzymes and growth factors that are implicated in many important physiological events. PCs have roles in several pathologies including viral infections and cancers and, thus, are promising targets for therapeutic applications. Recent structural and homology-modeling studies demonstrate more similarity than expected at the catalytic site of the seven PCs, which makes the development of selective drugs to target individual PCs frustrating. Based on this information, we review the latest strategies to inhibit PCs, which might lead to the development of specific compounds.
Physiological functions of pro-protein convertases
The biology of pro-protein convertases (PCs) is fundamental to the cell infrastructure and to the proper coordination of the entire mammalian physiology. Through their endoproteolytic actions on inactive precursor proteins in the secretory pathway, PCs generate essential bioactive peptides, including hormones and neuropeptides (e.g. adrenocorticotropic hormone and insulin), that have vital roles in homeostatic balance and the regulation of life functions. They also activate growth factors and differentiation factors, proteins in the extracellular matrix and plasma, enzymes, receptors, viral-coat proteins and bacterial toxins. The PC family of enzymes is encoded by seven genes (see Box 1 and Table 1 for nomenclature), of which the archetype furin is the most studied (reviewed in 1, 2). Mammalian PCs are related to the yeast kexin and bacterial subtilisins, and Figure 1 presents the structural features of PCs. PCs are Ca2+-dependent serine proteases that were identified originally based on their ability to cleave precursor proteins at the peptide bond C-terminal to paired basic residues, such as K-R↓ and R-R↓. However, the cleavage-recognition motifs are now known to extend N-terminal to the cleavage site and often include additional arginine residues (e.g. R-x-[K/R]-R↓).
Box 1. The newly adopted PC nomenclature.
The seven members of the PC family are classified as MEROPS clan SB family S8 (http://merops.sanger.ac.uk). They are related to the bacterial family of degradative subtilisins (IUBMB EC 3.4.21.62) and, to some extent, are structurally and functionally analogous to the yeast kexin (EC 3.4.21.61). The family of PCs has several confusing nomenclatures. A consensus nomenclature was agreed at the Gordon Research Conference: Pro-protein Processing, Trafficking and Secretion (2004) and the officially adopted nomenclature of the pro-protein convertases is now furin (EC 3.4.21.75), PC2 (EC 3.4.21.93), PC1/3 (EC 3.4.21.94), PACE4, PC4, PC5/6 and PC7. The corresponding genes that encode these PCs are designated officially as PCSK type 1–PCSK type 7. Table 1 presents the names attributed to each PC and the corresponding official nomenclature.
Table 1.
PC nomenclaturea
| Gene designation (human) | Enzyme | Alternative enzyme names |
|---|---|---|
| PCb | SPC | |
| PCSK3 | Furin | PACE, PC1, SPC1 |
| PCSK2 | PC2 | SPC2 |
| PCSK1 | PC1/3 | PC1, PC3, SPC3 |
| PCSK6 | PACE4 | PACE4, SPC4 |
| PCSK4 | PC4 | SPC5 |
| PCSK5 | PC5/6 | PC5, PC6, SPC6 |
| PCSK7 | PC7 | PC8, LPC, SPC7 |
Abbreviations: LPC, lymphoma pro-protein convertase; PACE, paired basic amino acids-converting enzyme; PCSK, pro-protein convertase subtilisin/kexin type; SPC, subtilisin-like (or subtilase-like) pro-protein convertase.
‘Pro-hormone convertase’ is also used in the literature but is an inadequate appellation because pro-proteins other than pro-hormones are also matured by PCs.
Figure 1.
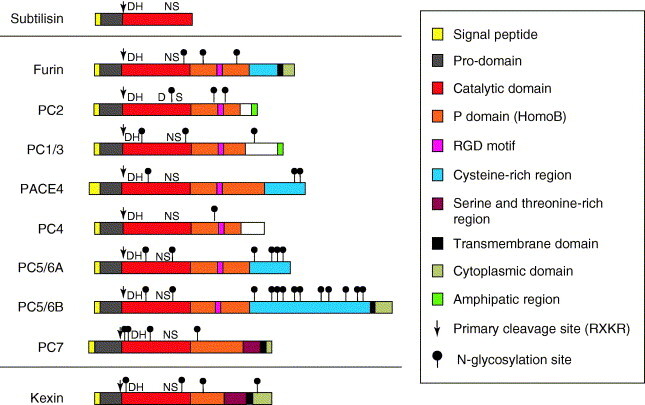
PC architecture. The PCs have a highly homologous architecture that is composed of: (i) an N-terminal signal peptide, which is responsible for directing proteins into the secretory pathway; (ii) a pro-domain, which acts as a putative intramolecular chaperone for facilitated transportation, folding and regulation of enzymatic activity; (iii) a catalytic domain, which contains an active site that is responsible for substrate-specific interactions and cleavage; (iv) a P domain, which is an independent folding moiety with the conformation of a barrel-like roll that is essential for structural cohesion of the enzyme and activity by regulating stability, and Ca2+- and pH-dependence; and (v) a divergent C-terminal domain that can contain membrane attachment sequences, Cys-rich regions and intracellular sorting signals. Additionally, furin contains two functional groups for internal ionic interactions (Ca2+-binding sites), one of which is beneath the active site and provides stability. D (aspartate), H (histidine) and S (serine) are part of the catalytic triad. N (asparagine) is located in the oxyanion hole and stabilizes, through its NH2 group, the negative charge that is developed in the peptide bond as a result of the nucleophilic attack from the catalytic serine. At least two isoforms of PC5/6 (A and B) have been identified, both of which are encoded by the same gene.
During the past few years, several important pathologies have been linked with PC-like activity in the cell-secretory pathway and at the cell membrane. The aim is to develop inhibitors that target individual PCs because not every PC is implicated in a given disease state. In this article, we review the involvement of PCs in various pathologies, which justifies their targeting for therapeutic applications. Although increasing evidence of similarity between the various PCs raises the issue of whether specific compounds that target individual PCs can be developed, five major approaches that are described in this article appear to make this possible.
PCs as pharmacological targets
There is increasing interest in PCs as novel targets for drug design because they are proteolytically selective enzymes, the tissue and cellular localization patterns of which have been studied extensively [3]. In addition, they have well-defined cellular tasks and are crucial for the initiation and progress of many important diseases, most prominently in several viral infections and cancers. For example, furin and PC7 are required for the activation of HIV-1 viral coat protein glycoprotein 160 (gp160) to gp120 and gp41 to produce virions that are effective in receptor binding and membrane fusion during infection [4]. A similar PC-mediated processing requirement for viral spread and cytopathicity also occurs with the Epstein-Barr virus gB glycoprotein [5], the hepatitis B virus e antigen [6] and the severe acute respiratory syndrome coronavirus spike glycoprotein [7]. These roles make drugs that target PCs potential antiviral agents. In the area of cancer, ongoing work is clarifying the pivotal role of PCs in growth regulation and tumor progression by activating cancer-promoting factors such as stromelysin-3 [8], vascular endothelial growth factor c (VEGF-C) [9], insulin-like growth factor 1 receptor (IGFr1) 10, 11, transforming growth factor β (TGF-β) [12] and membrane type 1 matrix metalloproteinase (MMP-1) [13]. In addition, PC-inhibition studies are beginning to provide direct evidence of their roles in tumorigenesis and invasiveness 13, 14, 15.
Evidence of the diverse roles of PCs in other pathologies is also accumulating. Bacterial pathogens such as Pseudomonas aeruginosa exotoxin A and the protective antigen of Bacillius anthracis tripartite toxin require furin-mediated cleavage for cytotoxicity, which can be reduced and blocked by PC inhibitors 16, 17. PCs have a role in neurodegenerative and aging maladies such as Alzheimer's disease through the activation of pro-β site amyloid-precursor-protein-cleaving enzyme (BACE1) [18] and its novel binding partner BRI3 [19], and in arthritis through the processing of aggrecanase-1 [20] and tumor necrosis factor α-converting enzyme (TACE) [21]. Coordinated interplay between PC2 and PC1/3 has been uncovered recently in the production of peptides derived from cocaine and amphetamine-regulated transcript precursor (proCART) [22], which are believed to potentiate anorexia.
Thus, we conclude that the potential of PC inhibitors as novel therapeutic agents is considerable and warrants further analysis. To date, no PC-oriented therapies are available. To achieve this goal, new insights are needed into the molecular determinants that are responsible for the proteolytic activities of individual PCs.
The rationale for the development of PC inhibitors
Strategically, the mainstream of PC inhibitor development is to target their catalytic site. This is based on the fact that PCs are highly selective proteinases that cleave an uncommon sequence motif in proteins (e.g. R-x-K-R). However, the most potent inhibitors known to date, which target the catalytic site, are not significantly specific for a single PC. Comparing inhibitors of PCs shows that many have high potency but are not specific for one PC (Figure 2 ). In addition, PCs are often coexpressed within cells, so that all cells express a ‘cocktail’ of at least two PCs. Thus, poorly selective PC inhibitors will affect multiple cellular functions, and not only the pathological processes that are targeted.
Figure 2.
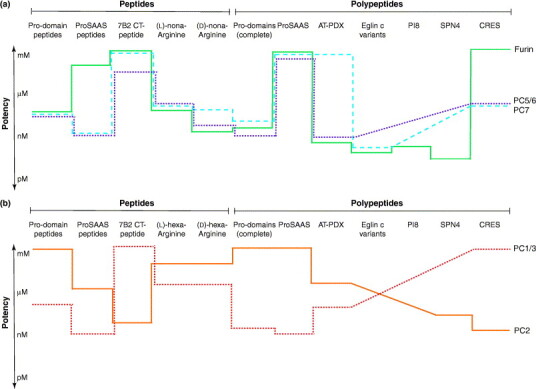
Potency and specificity of PC inhibitors. Schematic representation of the inhibitory potency of peptide and polypeptide inhibitors of PCs. Both PCs of the constitutive secretory pathway (furin, PC5/6 and PC7) (a) and PCs of the regulated secretory pathway (PC2 and PC1/3) (b) are shown. Inhibitory potency (logarithmic scale) is represented by the average of the Ki values determined in vitro in studies cited in this review. For example, most pro-domain peptides inhibit furin, PC1/3, PC5/6 and PC7 at mid-nanomolar concentrations, but are ineffective at PC2. The lower the value, the higher the inhibition potency. Diagonal traces mean that inhibition potency is undetermined.
The first approaches to developing specific PC inhibitors were based on our understanding of the molecular determinants of selectivity of cleavage by PCs. The cleavage sites of numerous furin substrates were aligned to assess these molecular determinants [23]. However, subsequent cleavage studies show that many substrates are also processed by other PCs. This has been emphasized recently by studies in transgenic inducible-knockout mice, which lack furin in the liver [24]. Known substrates of furin in the liver (e.g. albumin, α5 integrin, lipoprotein-receptor-related protein, vitronectin, α1-microglobulin and bikunin) are either processed or partially processed in these mice, which indicates redundancy with other PCs in the liver. These data also underline the potential undesirable effects of nonspecific PC inhibitors (that inhibit several PCs), which would block the processing of more proteins than desired if applied therapeutically. Uniquely specific PC inhibitors have the potential advantage that they can be used in a combination therapy approach, should simultaneous blocking of two PCs be required.
Currently, the development of PC inhibitors is based on: (i) newly acquired structural and modeling information about PCs; (ii) peptides that are optimized by combinatorial methods; (iii) engineering polypeptide protease inhibitors that have not co-evolved with PCs but are used as stable scaffolds on which to graft the appropriate modifications to specifically target PCs; (iv) endogenous, co-evolutionary inhibitors; and (v) small non-peptidyl compounds and derivatives. Each of these approaches is discussed below.
The structural and molecular modeling approach
Understanding of the molecular basis of pro-protein processing by PCs and the design of specific inhibitors have both benefited from the recent elucidation of the three-dimensional conformation of the kinetically trapped furin [Protein Data Bank (PDB) (http://www.rcsb.org/pdb/) entry 1P8J, molecule A] [25] and of the yeast homolog kexin (PDB entry 1ot5, chain A) [26]. The crystal structure of furin provides crucial information on the spatial arrangement of the two domains that are implicated in PC activity: the catalytic domain, which contains the active site that is responsible for substrate-specific recognition and processing; and the P domain, which is responsible for enzyme stability (Figure 3 a). The crystal structure of furin in a covalently bound complex with the suicide inhibitor decanoyl-RVKR-chloromethylketone (dec-RVKR-cmk) [25] has led to the identification of the reactive amino acids that make up the S1–S4 subsite pockets* . This structural information permits modeling and docking analysis 27, 28, which has led to the extrapolation of further subsites, including the S5, S6 and S1′ subsite pockets (Figure 3b, Table 2 ). Apart from S3 and S1′, all subsite pockets of the active site of furin contain several protophilic carboxylates and carboxamides, which explains the stringent preference for either arginyl or lysyl functionality groups in substrates and inhibitors (Table 2).
Figure 3.

Structure aspects of PCs. (a) Ribbon representation of the furin crystal structure complexed with the permanent inhibitor dec-RVKR-cmk showing the three-dimensional arrangement of the catalytic and P domains, which are essential for PC activity. Dec-RVKR-cmk is shown in ball and stick rendering where green is carbon, blue is nitrogen and red is oxygen. The catalytic triad (Asp153, His194 and Ser368) is shown in yellow. α-Helical regions are shown in red, β-sheets are shown in blue, and the two Ca2+-binding sites are shown as purple spheres. The image is rendered using iMol 0.3 (http://www.pirx.com/iMol/). (b) Representation of the electrostatic surface potential of the active site of furin with a modeled sequence of six residues from the C-terminal of the furin pro-domain (KRRTKR). White annotations indicate the inhibitor residues and black annotations show the catalytic triad and the subsite residues (S1–S6). (c,d) Representation of the S1 subsite with a P1 arginine residue for furin (c) and PC7 (d). The amino acids of the S1 subsite (furin: Asp258 and Asp306; and PC7: Asp292 and Asp340) and those in close proximity (within 3.2 Å) are shown in a ball-and-stick representation, whereas the P1 arginine is rendered as both ball and stick and a mesh surface. This comparison demonstrates the close identity of the S1 subsites, with the exception of Thr309 for furin and Ala343 for PC7. Similar analysis of the S1 subsite of all other PCs reveals perfect identity with furin (not shown).
Table 2.
Important PC subsite residuesa
| PC | S6 | S5 | S4 | S3 | S2 | S1 | Triad | S1′ |
|---|---|---|---|---|---|---|---|---|
| Furin | E230, D233 | E257, D264 | E236, D264 | L227, T254 | D154, D191 | D258, D306 | D153, H194, S368 | R193, R197, H364 |
| PC2 | P245, D249 | T273, D280 | E252, D280 | L242, W270 | D169, F206 | D274, D321 | D168, H209, S385 | S208, R212, H381 |
| PC1/3 | I244, D247 | N271, E278 | E250, E278 | L241, W268 | D168, E205 | D272, D320 | D167, H208, S382 | K207, R211, H378 |
| PACE4 | D283, D286 | D310, D317 | E289, D317 | L280, W307 | D207, D244 | D311, D359 | D206, H247, S421 | K256, R250, H417 |
| PC4 | A233, D236 | E260, D267 | E239, D267 | L220, W257 | D157, D193 | D271, D309 | D156, H197, S371 | R196, R200, H367 |
| PC5/6 | D249, D252 | D276, D283 | E255, D283 | L246, W273 | D173, E210 | D277, D325 | D172, H213, S385 | K212, R216, H381 |
| PC7 | P264, D267 | D291, D298 | E270, D298 | L261, W288 | D188, G225 | D292, D340 | D187, H228, S406 | H227, R231, H402 |
Numbering of the amino acids starts at the first amino acid (methionine) of the signal peptide sequence. Triad residues and S1–S4 subsites residues are deduced from the crystal structure of furin complexed with dec-RVKR-cmk. S5, S6 and S1′ are extrapolated by sequence-alignment analysis and confirmed by homology modeling. Residues in bold and underlined contrast with the active site of furin. Refer to the footnote for subsite nomenclature.
Based on homology modeling, the catalytic domains of the seven PCs are highly homologous (50–70%) [28] and the level of identity is even greater for the residues of the catalytic-cleft subsites (Table 2). For example, for all PCs, the S1 subsite that interacts with the P1 arginine residue of a substrate is composed of two acidic aspartic acid residues (Table 2) and three residues of the catalytic triad (aspartic acid, histidine and serine). In addition, nine additional amino acids that provide the steric environment to the P1 lateral chain are identical in all PCs, as illustrated by the comparison of furin and PC7 (Figure 3c,d).
The subsite identities go beyond the S1 subsite, and are also found from S2 to S5 (Table 2). However, differences increase away from the catalytic triad, at the S6 subsite pocket and beyond. These observations lower expectations for the possible development of short peptide inhibitors of PCs (four–five residues) that are specific for individual PCs. However, the differences observed at the S6 subsite indicate that specific, short-peptide inhibitors require at least six residues.
Beyond the S6 subsite, further differences might be exploited to design specific inhibitors. As yet, these differences are not well identified but some attempts have been made. For example, a recent study demonstrates that exploiting the catalytic-site proximity of so-called ‘adventitious contacts’ can lead to more-specific inhibitors [29]. The mapping of proximal sites is also aided by solution-structure studies of the PC1/3 pro-domain [30] and subsequent docking analysis with PCs. Unlike dec-RVKR-cmk, which maps only four subsite positions [28], pro-domain-docking analysis demonstrates distinctions between furin and the other PCs at the S5-subsite and S6-subsite pockets (Table 2). Further structural studies of the crystallization of furin and other PCs complexed with different inhibitors are needed to provide a better understanding of the catalytic site of PCs.
The combinatorial approach
The inhibitory properties of a small-peptide inhibitor can be optimized in vitro using positional scanning of synthetic peptide combinatorial libraries (PS-SPCLs) 31, 32. An optimal sequence, determined by screening PS-SPCLs can then be embedded into polypeptides to achieved higher specificity and potency. Such combinatorial libraries of degenerate mixtures of small-peptide inhibitors have been used successfully to compile the most potent sequence for inhibiting furin (nona-arginine) [33], and to identify a previously unknown sequence, LLRVKR, which potently inhibits PC1/3 31, 34. This latter sequence was identified in a novel, endogenous protein precursor, proSAAS, that has a tissue-distribution pattern that resembles closely that of PC1/3 35, 36. The development of nona-arginine and LLRVKR as PC inhibitors demonstrates the strength of PS-SPCLs in identifying potent inhibitory peptide sequences.
In the case of the polyarginine sequence, peptides ranging from four to nine residues are potent inhibitors of furin in both enantiomeric configurations [33]. The most potent polyarginine peptide is d-nona-arginine (K i=1 nM) [27]. Interestingly, all l- and d-polyarginine peptides are also potent inhibitors of PC5/6 and PC7 (M. Fugère et al., unpublished), which highlights the difficulty in achieving substantial specificity using the active site exclusively. From a therapeutic perspective, d-polyarginines are interesting as lead compounds because they cannot be cleaved by PCs and are thus more stable. d-Peptide therapeutics are also less prone to other proteolytic activities such as carboxypeptidases. d-hexa-arginine blocks the activation of P. aeruginosa exotoxin A [16] and d-nona-arginine partially protects macrophages and mice against anthrax toxemia 17, 27. l-Hexa-arginine is an efficient inhibitor of the processing of gp160 (env) at the REKR551 sequence in vitro and ex vivo, and effectively suppresses HIV-1 infection of T cells and macrophages [4]. Both furin and PC7, the two principal PCs that are expressed by HIV-host cells, are the best candidates for processing the precursor of HIV-1 gp160 [37]. If polyarginine peptides do not exhibit undesirable interactions with other biological processes they might become key components for a combined therapy that targets furin, PC5/6 and PC7 simultaneously. Conversely, introducing these polyarginine peptide sequences into engineered polypeptides might achieve greater specificity while retaining high potency.
The protein-bioengineering approach
Astute bioengineering of protease inhibitors that did not co-evolve with PCs has also led to the development of potent PC inhibitors. The first successful example is α1-antitrypsin, which has been transformed into a potent furin inhibitor by the insertion of a furin-like recognition sequence (Arg355-Ile-Pro-Arg358) into its reactive-site loop. This bioengineered protein, named antitrypsin-portland (α1-AT-PDX), inhibits furin and PC5/6 without altering its serpin-like mechanism of inhibition (reviewed in [32]). The potential therapeutic value of α1-AT-PDX is demonstrated by its ability, following exogenous application, to prevent processing of HIV type 1 gp160 and maturation ex vivo of pro-gB, the precursor of human cytomegalovirus envelope glycoprotein of β-herpesviridae. More recently, α1-AT-PDX has been tested in cancer-related applications. Whereas overexpression of furin and other PCs is associated with a significant increase in metastatic spreading potential and tumor-cell proliferation 8, 38, α1-AT-PDX decreases the invasiveness of head and neck squamous-cell carcinoma by 50% in vitro, and by up to 80% when inoculated in tracheal xenotransplants [15]. Expression of α1-AT-PDX from a DNA vector in transfectant tumor xenotransplants of astrocytoma cells also suppresses efficiently tumorigenicity and invasiveness [13]. Although α1-AT-PDX represents a strong case for targeting PCs in pathophysiological processes, the large size of this protein (45 kDa) limits its practical application. Nonetheless, α1-AT-PDX might be used effectively to block processing events that occur at the outer surface of the cell membrane.
A second example of a successful bioengineering approach has been achieved using eglin c, a potent inhibitor of elastase, cathepsin G and degradative subtilisins. Initially, arginine residues were inserted into the reactive site loop of eglin c to target furin and kexin [39]. Then, randomization of strategic eglin c residues that make contact with PC residues outside the catalytic cleft, namely ‘adventitious contacts’, proved extremely efficient in designing two variants of eglin c that target either furin or PC7 with significant specificity [29]. These two variants, Val33-Arg42-Arg45-Asp49-eglin c (K i=270 pM for furin and 11 nM for PC7) and Arg42-Arg45-Trp49-eglin c (K i=10 nM for furin and 470 pM for PC7) demonstrate that optimizing flanking contacts by in vitro molecular evolution provides a powerful technique to generate specific inhibitors for each PC. These two derivatives of eglin c show that differences outside but near the active site of PCs favor both binding affinity and specificity of the inhibitor. These eglin c variants might serve as biotechnological tools to dissect the functions of furin and PC7 in the cell and at the membrane, for co-crystalization processes, and as lead compounds to design potent, specific and smaller therapeutic drugs.
The endogenous approach
Endogenous molecules that inhibit PCs in vivo represent a major lead for designing pharmacological inhibitors of PCs. Although some are large polypeptides, core inhibitory domains can be identified and used as lead compounds. Examples of endogenous inhibitors are PC pro-domains, the neuroendocrine 7B2 protein, the proSAAS precursor and the proteinase inhibitor 8 (reviewed in [32]). Each contains a PC-recognition sequence and they act as substrate analogs, real substrates or ‘false baits’ for PCs. Recently, the inhibitory effects on PCs of two new candidate endogenous inhibitors, the cystatin-related epididymal spermatogenic (CRES) protein [40] and the serine-protease inhibitor 4 SPN4A/4.1 41, 42, 43, have been described.
PC pro-domains are autoprocessed in cis by their cognate PC, but remain bound to the active site through their C-terminal PC-recognition sequence until the complex reaches the compartment of zymogen activation. Thus, pro-domains are dual-function molecules, being the first substrate and first inhibitor encountered by PCs in cells. Evaluation in vitro of the inhibitory capacity of the seven complete pro-domains and of derived, short C-terminal dodecapeptides demonstrates their nanomolar potency and some specificity [44]. However, none is a specific inhibitor of a unique PC. The lack of pro-domain specificity is also observed in inhibition experiments in cell culture. Recently, overexpression in trans in Chinese hamster ovary (CHO) cells of the wild-type PC5/6 pro-domain and a selection of key residue mutants efficiently inhibited the processing of an overexpressed substrate, proVEGF-C [45]. However, maturation of proVEGF-C is also decreased significantly by expression of the pro-domains of furin, PACE4 and PC7 in CHO cells. To date, there is no indication that specificity of inhibition can be achieved without significant structural modification of pro-domains. Nevertheless, because pro-domains have multiple interactions with PCs outside the catalytic site, bioengineering might result in more specific pro-domains. Co-crystalization of a pro-domain–PC complex might provide the information required to map these interactions.
Another endogenous candidate is CRES, a protein that has divergent functions. CRES is related to the family 2 cystatins, a family of cysteine-protease inhibitors of the larger cystatin superfamily. The inability of CRES to inhibit C1-type cysteine proteases and the presence of two accessible paired Lys-Lys basic-residue sites has prompted the assumption that CRES might inhibit PC2. Assays in vitro demonstrate that CRES inhibits PC2 but not furin and PC1/3 [40]. Inhibition of PC2 occurs at low nanomolar concentrations and is competitive, which indicates interaction with the catalytic site.
Last, the Spn4 gene in Drosophila melanogaster encodes the serpin SPN4A/4.1. SPN4A/4.1 has a consensus serpin sequence for the hinge region of the reactive-site loop (RSL), which indicates that it functions as a serpin, and includes a natural PC-recognition sequence (ArgP4-Arg-Lys-ArgP1↓) in its RSL. In vitro, SPN4A/4.1 inactivates human furin (K i=13 pM) and PC2 (K i=0.92 nM) by a slow-binding mechanism and the formation of a SDS-stable acyl–enzyme complex [41]. This kinetic trapping is typical of the serpin clade. That SPN4A/4.1 exerts potent inhibition on both furin and PC2 indicates a profound lack of selectivity between PCs in general. However, the removal of one amino acid in the RSL (AlaP6) generates a reduction of inhibition potency against PC2 (i.e. ID50 is 27-fold greater) but has no effect on furin. This change in specificity indicates that the RSL of SPN4A/4.1 is more amendable to modifications than other serpins such as α1-AT-PDX.
The chemical approach
Small non-peptidyl compounds represent a poorly explored area of PC inhibition. Because the activity of PCs is affected by metal ions, the first inhibitory chemical compounds investigated were 4′-(p-tolyl)-2,2′;6′,2″-terpyridine (TTP)-chelated metal salts such as Zn(TTP)Cl2 and Cu(TTP)Cl2. These bind slowly and competitively, and are irreversible inhibitors with low micromolar inhibition constants (IC50=5–10 μM), possibly by a chelate-coordinated interaction of the metal with the catalytic histidine of PCs [46] and Kex2 [47]. Although the free chelate alone is not inhibitory, the chelated metal salts are more potent than the solvated metal ions. Thus, the chelation of metal-based inhibitors to generate a prodrug serves to coordinate the delivery of the drug (either Zn2+ or Cu2+) to the active site of PCs. Modification and adjustment of the chelate might lead to greater inhibitory affinity and specificity for PCs. Also, as suggested by the authors, these metallodrugs might be developed further as wide-spectrum PC blockers for rapid intervention in aggressive viral infections and/or for bacterial detoxification.
Improving the specificity of inhibition of small chemical molecules that target the active site might prove difficult given the highly conserved amino acids of PC subsites. However, small chemical molecules, possibly obtained through library screening, might lead to the identification of compounds that can induce allosteric conformational changes that selectively affect PC activity. Presently, the only known PC allosteric sites are Ca2+-binding sites, but these appear to be unlikely sites for the design of specific PC inhibitors.
Allosteric inhibition of PCs might be a future avenue of research. It is mostly unexplored because of the difficulty in mapping allosteric sites and identifying ligands that cause conformational changes. However, screening large libraries of thiol-containing compounds has identified a regulatory site away from the active site for caspase-3 and caspase-7 that immobilizes the enzyme conformation when it forms a reversible disulfide bond with an accessible cysteine [48]. The link between this outer site and the active site is such that binding of the specific compounds blocks the interaction with substrates. We suggest that such strategies are also possible for inhibition of PCs.
Future perspectives
The design of unique, specific PC inhibitors represents a major challenge in light of the homology between the active sites of each family member. PC inhibitors have the potential to become important therapeutic agents, particularly in viral diseases and, possibly, in cancer. Additionally, studies to design specific PC inhibitors should: (i) identify the molecular determinants of selectivity between PCs; (ii) provide new pharmacological tools to probe normal PC functions; and (iii) define the true nature of compensating functions between PCs. The challenge is to identify small, but significant, differences away from the core active site and to exploit these through innovative and classical pharmacological drug design.
Acknowledgements
We thank Pierre Lavigne for helpful discussions of the structural considerations of PCs. This work was supported by a CIHR research grant to R.D. and CIHR studentship award to M.F.
Footnotes
The catalytic site of proteases, including PCs, is defined by subsite positions (e.g. S3, S2 and S1), which correspond to the residues of a binding substrate or inhibitor (P3-P2-P1). Thus, the substrate residue at position P1 binds the S1-subsite pocket of the protease. Each subsite can be composed of several amino acids. Substrate and inhibitor residues and subsites on the C-terminal side of the cleavage site are defined by primes (Pn′ and Sn′). The processing of substrates occurs at the P1-P1′ peptidyl bond (e.g. Pn-P3-P2-P1↓P1′-P2′-P3′-Pn′) [49].
References
- 1.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rockwell N.C., Thorner J.W. The kindest cuts of all: crystal structures of Kex2 and furin reveal secrets of precursor processing. Trends Biochem. Sci. 2004;29:80–87. doi: 10.1016/j.tibs.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Bergeron F., et al. Subtilase-like pro-protein convertases: from molecular specificity to therapeutic applications. J. Mol. Endocrinol. 2000;24:1–22. doi: 10.1677/jme.0.0240001. [DOI] [PubMed] [Google Scholar]
- 4.Kibler K.V., et al. Polyarginine inhibits gp160 processing by furin and suppresses productive human immunodeficiency virus type 1 infection. J. Biol. Chem. 2004;279:49055–49063. doi: 10.1074/jbc.M403394200. [DOI] [PubMed] [Google Scholar]
- 5.Johannsen E., et al. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. U. S. A. 2004;101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Messageot F., et al. Proteolytic processing of the hepatitis B virus e antigen precursor. Cleavage at two furin consensus sequences. J. Biol. Chem. 2003;278:891–895. doi: 10.1074/jbc.M207634200. [DOI] [PubMed] [Google Scholar]
- 7.Bergeron E., et al. Implication of proprotein convertases in the processing and spread of severe acute respiratory syndrome coronavirus. Biochem. Biophys. Res. Commun. 2005;326:554–563. doi: 10.1016/j.bbrc.2004.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bassi D.E., et al. The proprotein convertases furin and PACE4 play a significant role in tumor progression. Mol. Carcinog. 2000;28:63–69. [PubMed] [Google Scholar]
- 9.Siegfried G., et al. The secretory proprotein convertases furin, PC5, and PC7 activate VEGF-C to induce tumorigenesis. J. Clin. Invest. 2003;111:1723–1732. doi: 10.1172/JCI17220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stawowy P., et al. Proprotein convertases regulate insulin-like growth factor 1-induced membrane-type 1 matrix metalloproteinase in VSMCs via endoproteolytic activation of the insulin-like growth factor-1 receptor. Biochem. Biophys. Res. Commun. 2004;321:531–538. doi: 10.1016/j.bbrc.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 11.Khatib A.M., et al. Inhibition of proprotein convertases is associated with loss of growth and tumorigenicity of HT-29 human colon carcinoma cells: importance of insulin-like growth factor-1 (IGF-1) receptor processing in IGF-1-mediated functions. J. Biol. Chem. 2001;276:30686–30693. doi: 10.1074/jbc.M101725200. [DOI] [PubMed] [Google Scholar]
- 12.Dubois C.M., et al. Evidence that furin is an authentic transforming growth factor-beta1-converting enzyme. Am. J. Pathol. 2001;158:305–316. doi: 10.1016/s0002-9440(10)63970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercapide J., et al. Inhibition of furin-mediated processing results in suppression of astrocytoma cell growth and invasiveness. Clin. Cancer Res. 2002;8:1740–1746. [PubMed] [Google Scholar]
- 14.Wick W., et al. BCL-2-induced glioma cell invasiveness depends on furin-like proteases. J. Neurochem. 2004;91:1275–1283. doi: 10.1111/j.1471-4159.2004.02806.x. [DOI] [PubMed] [Google Scholar]
- 15.Bassi D.E., et al. Furin inhibition results in absent or decreased invasiveness and tumorigenicity of human cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2001;98:10326–10331. doi: 10.1073/pnas.191199198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarac M.S., et al. The furin inhibitor hexa-D-arginine blocks the activation of Pseudomonas aeruginosa exotoxin A in vivo. Infect. Immun. 2002;70:7136–7139. doi: 10.1128/IAI.70.12.7136-7139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarac M.S., et al. Protection against anthrax toxemia by hexa-D-arginine in vitro and in vivo. Infect. Immun. 2004;72:602–605. doi: 10.1128/IAI.72.1.602-605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinnix I., et al. Convertases other than furin cleave beta-secretase to its mature form. FASEB J. 2001;15:1810–1812. doi: 10.1096/fj.00-0891fje. [DOI] [PubMed] [Google Scholar]
- 19.Wickham L., et al. beta-Amyloid protein converting enzyme 1 and brain-specific type II membrane protein BRI: binding partners processed by furin. J. Neurochem. 2005;92:93–102. doi: 10.1111/j.1471-4159.2004.02840.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang P., et al. Proprotein convertase furin interacts with and cleaves pro-ADAMTS4 (Aggrecanase-1) in the trans-Golgi network. J. Biol. Chem. 2004;279:15434–15440. doi: 10.1074/jbc.M312797200. [DOI] [PubMed] [Google Scholar]
- 21.Srour N., et al. TACE/ADAM-17 maturation and activation of sheddase activity require proprotein convertase activity. FEBS Lett. 2003;554:275–283. doi: 10.1016/s0014-5793(03)01159-1. [DOI] [PubMed] [Google Scholar]
- 22.Dey A., et al. Biological processing of the cocaine and amphetamine-regulated transcript precursors by prohormone convertases, PC2 and PC1/3. J. Biol. Chem. 2003;278:15007–15014. doi: 10.1074/jbc.M212128200. [DOI] [PubMed] [Google Scholar]
- 23.Nakayama K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J. 1997;327:625–635. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roebroek A.J., et al. Limited redundancy of the proprotein convertase furin in mouse liver. J. Biol. Chem. 2004;279:53442–53450. doi: 10.1074/jbc.M407152200. [DOI] [PubMed] [Google Scholar]
- 25.Henrich S., et al. The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nat. Struct. Biol. 2003;10:520–526. doi: 10.1038/nsb941. [DOI] [PubMed] [Google Scholar]
- 26.Holyoak T., et al. 2.4 A resolution crystal structure of the prototypical hormone-processing protease Kex2 in complex with an Ala-Lys-Arg boronic acid inhibitor. Biochemistry. 2003;42:6709–6718. doi: 10.1021/bi034434t. [DOI] [PubMed] [Google Scholar]
- 27.Kacprzak M.M., et al. Inhibition of furin by polyarginine-containing peptides: nanomolar inhibition by nona-D-arginine. J. Biol. Chem. 2004;279:36788–36794. doi: 10.1074/jbc.M400484200. [DOI] [PubMed] [Google Scholar]
- 28.Henrich S., et al. Proprotein convertase models based on the crystal structures of furin and kexin: explanation of their specificity. J. Mol. Biol. 2005;345:211–227. doi: 10.1016/j.jmb.2004.10.050. [DOI] [PubMed] [Google Scholar]
- 29.Komiyama T., et al. Optimization of protease-inhibitor interactions by randomizing adventitious contacts. Proc. Natl. Acad. Sci. U. S. A. 2003;100:8205–8210. doi: 10.1073/pnas.1032865100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tangrea M.A., et al. Solution structure of the pro-hormone convertase 1 pro-domain from Mus musculus. J. Mol. Biol. 2002;320:801–812. doi: 10.1016/s0022-2836(02)00543-0. [DOI] [PubMed] [Google Scholar]
- 31.Apletalina E., et al. Identification of inhibitors of prohormone convertases 1 and 2 using a peptide combinatorial library. J. Biol. Chem. 1998;273:26589–26595. doi: 10.1074/jbc.273.41.26589. [DOI] [PubMed] [Google Scholar]
- 32.Fugere M., Day R. Inhibitors of the subtilase-like pro-protein convertases (SPCs) Curr. Pharm. Des. 2002;8:549–562. doi: 10.2174/1381612023395736. [DOI] [PubMed] [Google Scholar]
- 33.Cameron A., et al. Polyarginines are potent furin inhibitors. J. Biol. Chem. 2000;275:36741–36749. doi: 10.1074/jbc.M003848200. [DOI] [PubMed] [Google Scholar]
- 34.Basak A., et al. Inhibitory specificity and potency of proSAAS-derived peptides toward proprotein convertase 1. J. Biol. Chem. 2001;276:32720–32728. doi: 10.1074/jbc.M104064200. [DOI] [PubMed] [Google Scholar]
- 35.Fricker L.D., et al. Identification and characterization of proSAAS, a granin-like neuroendocrine peptide precursor that inhibits prohormone processing. J. Neurosci. 2000;20:639–648. doi: 10.1523/JNEUROSCI.20-02-00639.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanoue E., Day R. Coexpression of proprotein convertase SPC3 and the neuroendocrine precursor proSAAS. Endocrinology. 2001;142:4141–4149. doi: 10.1210/endo.142.9.8386. [DOI] [PubMed] [Google Scholar]
- 37.Moulard M., Decroly E. Maturation of HIV envelope glycoprotein precursors by cellular endoproteases. Biochim. Biophys. Acta. 2000;1469:121–132. doi: 10.1016/s0304-4157(00)00014-9. [DOI] [PubMed] [Google Scholar]
- 38.Taylor N.A., et al. Curbing activation: proprotein convertases in homeostasis and pathology. FASEB J. 2003;17:1215–1227. doi: 10.1096/fj.02-0831rev. [DOI] [PubMed] [Google Scholar]
- 39.Komiyama T., Fuller R.S. Engineered eglin c variants inhibit yeast and human proprotein processing proteases, Kex2 and furin. Biochemistry. 2000;39:15156–15165. doi: 10.1021/bi001907c. [DOI] [PubMed] [Google Scholar]
- 40.Cornwall G.A., et al. The cystatin-related epididymal spermatogenic protein inhibits the serine protease prohormone convertase 2. Endocrinology. 2003;144:901–908. doi: 10.1210/en.2002-220997. [DOI] [PubMed] [Google Scholar]
- 41.Richer M.J., et al. The Spn4 gene of Drosophila encodes a potent furin-directed secretory pathway serpin. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10560–10565. doi: 10.1073/pnas.0401406101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osterwalder T., et al. Drosophila serpin 4 functions as a neuroserpin-like inhibitor of subtilisin-like proprotein convertases. J. Neurosci. 2004;24:5482–5491. doi: 10.1523/JNEUROSCI.5577-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oley M., et al. Inhibition of furin by serpin Spn4A from Drosophila melanogaster. FEBS Lett. 2004;577:165–169. doi: 10.1016/j.febslet.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 44.Fugere M., et al. Inhibitory potency and specificity of subtilase-like pro-protein convertase (SPC) prodomains. J. Biol. Chem. 2002;277:7648–7656. doi: 10.1074/jbc.M107467200. [DOI] [PubMed] [Google Scholar]
- 45.Nour N., et al. Structure-function analysis of the prosegment of the proprotein convertase PC5A. J. Biol. Chem. 2003;278:2886–2895. doi: 10.1074/jbc.M208009200. [DOI] [PubMed] [Google Scholar]
- 46.Podsiadlo P., et al. Furin inhibition by compounds of copper and zinc. J. Biol. Chem. 2004;279:36219–36227. doi: 10.1074/jbc.M400338200. [DOI] [PubMed] [Google Scholar]
- 47.Brinkerhoff C.J., et al. Protease inhibitors formed in situ from copper and tridentate chelates: a generalized approach towards metal-based pharmaceuticals. ChemBioChem. 2002;3:1141–1143. doi: 10.1002/1439-7633(20021104)3:11<1141::AID-CBIC1141>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 48.Hardy J.A., et al. Discovery of an allosteric site in the caspases. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schechter I., Berger A. On the active site of proteases. 3. Mapping the active site of papain; specific peptide inhibitors of papain. Biochem. Biophys. Res. Commun. 1968;32:898–902. doi: 10.1016/0006-291x(68)90326-4. [DOI] [PubMed] [Google Scholar]