

Graphical abstract
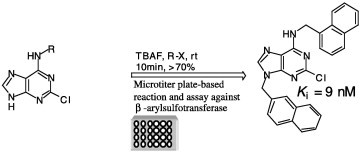
Combinatorial reactions in microtiter plates followed by screening in situ have been developed with the use of tetrabutylammonium fluoride to accelerate the N9-alkylation of purine derivatives with organic halides as demonstrated in the discovery of the most potent arylsulfotarnsferase inhibitor to date.
Keywords: TBAF, Purine-based libraries, N-alkylation, Sulfotransferase inhibitors
Abstract
Tremendous efforts have been invested in the synthesis of purine libraries due to their importance in targeting various enzymes involved in different diseases and cellular processes. The synthesis of N9-alkylated purine scaffolds relied mostly on Mitsunobu conditions with a variety of alcohols or strong basic conditions with different organic halides. A more reliable and efficient way for the synthesis of N9-alkylated purine scaffolds is reported. This method uses tetrabutylammonium fluoride (TBAF) to assist such chemistry. In many cases, the reactions were completed within 10 min and gave the desired product in high yield and selectivity. Moreover, these mild reaction conditions permitted its use in combinatorial reactions in microtiter plates followed by in situ screening for the discovery of potent sulfotransferase inhibitors.
1. Introduction
Purine derivatives have been used to target various enzymes involved in different diseases and cellular processes, most notably the kinase family.1 Thus, tremendous efforts have been invested to develop an efficient chemistry toward the synthesis of purine libraries that could be used as a source for both drug leads and probes for analyzing complex cellular processes.1, 2 Previously, synthesis of a N9-alkylated purine library relied on using either a Mitsunobu condition with a variety of alcohols3 or a strong basic condition (NaH, K2CO3) with a variety of alkyl and benzyl halides.4 These reactions require long reaction times (10–48 h), low temperatures for a Mitsunobu condition or elevated temperatures for the basic condition, and an inert atmosphere due to the sensitivity of the reagents in order to give the product in high yield and higher selectivity for the N9 versus the N7-alkylated product. Herein, we report that tetrabutylammonium fluoride (TBAF) at room temperature remarkably accelerates the N9-alkylation of the purine ring with a variety of organic halides. Moreover, these mild reaction conditions and the efficiency of this chemistry permit its use in combinatorial reactions in microtiter plates followed by in situ screening for the discovery of potent sulfotransferase inhibitors.
2. Results and discussion
TBAF, in addition to its wide use as a reagent for desilylation reaction,5 has also been used as an activator for organic halides to assist the reaction of palladium-catalyzed cross-coupling,6 non-Sonogashira-type palladium-catalyzed coupling of terminal alkynes,7 alkylation of phenols,8 and more recently in the catalysis of the synthesis of 5-substituted 1H-tetrazoles.9 Considering the role of TBAF in these reactions, as well as the basic nature of the fluoride ion,10 we wanted to examine the effect of TBAF alone in the N9-alkylation of purine scaffolds, as this type of chemistry would be accelerated if both a base and activator for the organic halides are present in the reaction. This mild condition, if achieved, is ideal for reactions in a microtiter plate for the high-throughput screening against several important enzymes. Initially, we investigated the alkylation reaction of 2,6-dichloropurine with benzyl bromide and methyl iodide. In both cases, the reaction was completed within 10 min upon addition of 2 equiv of the organic halide and 2 equiv of TBAF. The reactions were analyzed by LC–MS and gave a mixture of the two isomers N9/N7 in 7/3 ratio and excellent yields (isolated) (entries 1 and 2, Table 1 ). The reaction proceeded well in different solvents such as THF, DMF, and DMSO, and was carried out in open air. Similar results were obtained with 2-amino-6-chloropurine (entry 4, Table 1). Interestingly, using methyl bromoacetate gave better selectivity with excellent yield. Similarly, we also found that in the case of 1-bromodecane with 2-amino-6-chloropurine a minor amount of the N7-alkylated product was observed; however, the reaction in this case required a longer time for completion (entry 5, Table 1).
Table 1.
TBAF assisted N9-alkylation
| Entry | Scaffolds | R–X | Yield (%) | N9/N7 | Reaction time |
|---|---|---|---|---|---|
| 1 | 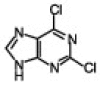 |
95 | 70/30 | 10 min | |
| 2 | 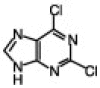 |
CH3I | 96 | 70/30 | 10 min |
| 3 | 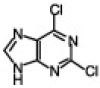 |
98 | 86/14 | 10 min | |
| 4 | 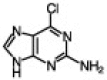 |
90 | 70/30 | 10 min | |
| 5 | 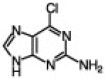 |
CH3(CH3)8–CH2–Br | 85 | 95/5 | 2 h |
| 6 | 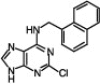 |
 |
97 | 98/2 | 10 min |
| 7 | 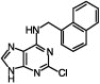 |
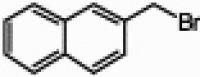 |
95 | 99/1 | 10 min |
| 8 | 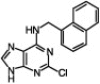 |
95 | 98/2 | 10 min | |
| 9 | 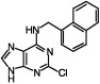 |
70 | 98/2 | 3 h | |
| 10 | 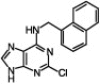 |
50 | 98/2 | 12 h |
It has been observed that substitution at the C6 position affects the N9/N7 alkylation ratio.1d Indeed, in all the cases that we tested with the 2-chloro-6-naphthylpurine scaffold and a variety of organic halides, only a very minute amount of the N7-alkylated product was detected (entries 6–10, Table 1). However, sterically hindered primary alkyl halides (entry 9, Table 1) and secondary alkyl halides (entry 10, Table 1) required longer reaction times and higher temperatures (60 °C) and the reaction gave the product with high selectivity, but with lower yield.
Next, we examined whether this chemistry could be applied to reactions in microtiter plates followed by in situ screening of the product for the discovery of potent inhibitors against a chosen target. To be useful, the chemistry should be efficient, selective, and free of protecting group manipulations so that the product could be used directly for screening in situ without isolation. Since its introduction,11 this approach was found to be very successful for the discovery of potent inhibitors against different enzymes, such as HIV protease,11, 12 β-arylsulfotransferase,13 SARS coronavirus protease,14 α-1,3-fucosyltransferase,15 and α-fucosidase.16 As a model system, we have selected the enzyme β-arylsulfotransferase-IV (β-AST-IV), which represents an effective model for studying sulfotransferase inhibition.17
Our laboratory had previously screened a library of 35,000 purine and pyrimidine analogues against β-AST-IV, which resulted in the inhibitor 2-chloro-6-naphthylpurine, 1, (K i = 96 nM, Scheme 1 ) binding to the aryl site of the enzyme.18 Thirty organic halides were reacted in microtiter plates with the 2-chloro-6-naphthylpurine core and the reactions were analyzed by TLC and by LC–MS for completion and selectivity ratio (Scheme 1). Most of these reactions were completed within 20 min and gave the N9-alkylated product in high yield (>95%). Wells were diluted to 100 nM and assayed directly against β-AST-IV, as was previously described.19 Two wells gave better inhibition activity than the original core. These two inhibitors were synthesized, purified, and characterized, and their K i values were determined to be 15 and 9 nM for compounds 3 and 2, respectively (Scheme 1).
Scheme 1.
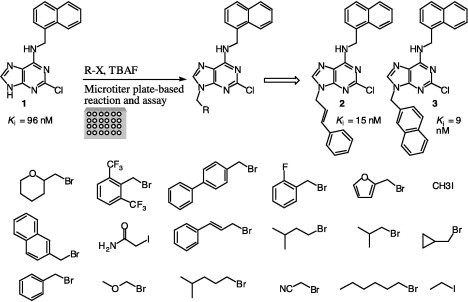
Combinatorial reactions in microtiter plate for the discovery of potent sulfotransferase inhibitors.
3. Conclusion
In conclusion, the new method described here represents a useful way toward rapidly accessing N9-alkylated purine analogues. The reaction of the purine scaffold with organic halides in the presence of TBAF is rapid, reliable, and gives the product in high yield and selectivity. The chemistry is also amenable to combinatorial reactions in microtiter plates followed by in situ screening, as was demonstrated in the discovery of two potent sulfotransferase inhibitors. We believe that this chemistry should ease the preparation of purine-based libraries and its combination with the in situ screening approach could represent a useful method for selecting potent inhibitors against other targets. The scope, limitation, and further application of the TBAF-assisted N-alkylation are currently under investigation.
4. Experimental
4.1. General procedure (entry 3, Table 1)
2,6-Dichloropurine (100 mg, 0.53 mmol) was dissolved in 500 μL THF at room temperature and to this was added 1 mL (1.0 mmol) TBAF (1.0 M solution in THF, Aldrich). To the clear mixture was added methyl bromo acetate (100 mL, 1.0 mmol). The reaction was kept for 10 min at room temperature, as at this time TLC analysis (CHCl3/MeOH, 10:1) indicated complete consumption of the purine. The reaction was directly loaded into the column and the product was eluted with a solution of hexane/ethyl acetate, 1:1 to yield 120 mg product as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.19 (s, 1H), 5.06 (s, 2H), 3.84 (s, 3H); 13C NMR 166.57, 153.3, 153.1, 152.0, 146.02 130.3, 53.3, 44.39; ESI-MS Calcd for (C8H6Cl2N4O2)H+ 260.99. Found 261.0 (entry 1, Table 1): 1H NMR (500 MHz, CDCl3) δ 8.06 (s, 1H), 7.27–7.3 (m, 5H), 5.4 (s, 2H); 13C NMR 153.07, 151.75, 145.53, 133.92, 133.91, 130.55, 129.32, 129.29, 128.98, 128.04, 128.00, 47.95; ESI-MS Calcd for (C12H8Cl2N4)H+ 279.01. Found 279.0, (entry 7, Table 1). 1H NMR (500 MHz, DMSO) δ 8.9 (br s, 1H), 8.3 (s, 1H), 8.28 (d, 8 Hz, 2H), 7.96–7.79 (m, 7H), 7.56–7.43 (m, 8H), 5.54 (s, 2H), 5.14 (d, 2H, 5.9 Hz); 13C NMR 155.97, 154.18, 150.89, 135.04, 134.15, 133.64, 133.26 132.26, 131.69, 129.4 128.63, 128.26, 127.35, 127.12, 127.07, 126.94, 126.64, 126.44, 125.93, 124.33, 119.15, 47.39, 42.20; ESI-MS Calcd for (C27H20ClN5)H+ 450.14. Found 450.0.
4.2. Library preparation for in situ screening
Thirty milligrams of 2-chloro-6-naphthylpurine was dissolved in 195 μL of 1 M solution of TBAF in DMF and to this was added 105 μL anhydrous DMF. From this stock solution 10 μL was taken and added to 30 wells of the microtiter plate. To each well was added 2 equiv of different alkyl bromide and the plate was kept at room temperature. The reactions were analyzed by TLC and LC–MS (C8 column). Most of the reactions at this time were completed. The wells were then diluted to 100 nM ready for the assay.
4.3. Enzymatic assay
A 2× stock solution of 1 M tris(hydroxymethyl)-aminomethane buffer (1 mL, 200 mM, pH 7.6), 250 mM β-mercaptoethanol (250 μL, 12.5 mM), 2 mM PAP (25 μL, 10 μM), enzyme (5 μL), and 3.72 mL water was formulated. Inhibitor and 4 MUS solutions were diluted to 10× the desired final concentration. Inhibitors were dissolved in DMSO for the studies, and a final assay volume of 200 μL was used. Enzyme-containing stock solution (100 μL), inhibitor (20 μL), and water (40 μL) were combined in 96-well microplates, mixed, and allowed to remain for 10 min. The reaction was initiated with 4 MUS solution (20 μL) and production of fluorescent 4-methylumbelliferone was followed for 5 min to calculate the rates. Measurements were performed using a Packard Fusion plate reader. Inhibitor concentrations were chosen such that enzymatic rates were linear. For K i measurements, inhibitors were studied at 0, 50, 100, 150, 200, and 250 nM concentrations. The dissociation constants were obtained as the absolute value of the x-axis intercept on plotting K/V versus inhibitor concentration. Multiple K i values were determined and the results were averaged to yield the final reported values. The reactions were completed after 10 min. Those that still show starting material were heated to 60 °C for several hours. The wells were then assayed at 100 nM, in which compounds 2 and 3 showed better inhibition activity. These two compounds were synthesized on a large scale and their K i values were determined (see Fig. 1 ).
Figure 1.
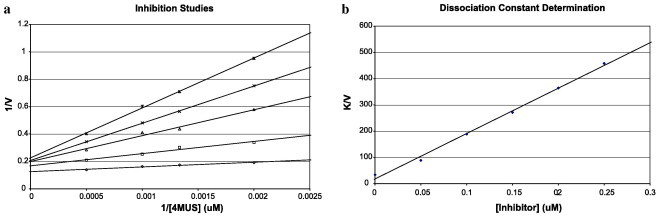
Inhibition of β-AST-IV with compound 2: (a) reciprocal rate versus reciprocal MUS concentration at 0, 50, 100, 150, 200, and 250 nM inhibitor; (b) slop replot.
Acknowledgments
We thank the National Institutes of Health and the Skaggs Institute for Chemical Biology for funding. We thank the National Science Council of Taiwan and the Genomic Research Center, Academia Sinica, for the financial support (C.-Y.W). We are also very thankful to Sheng-Kai Wang for the useful discussion.
References and notes
- 1.(a) Vesely J., Havlicek L., Strnad M., Blow J.J., Donella-Deana A., Pinna L., Letham D.S., Kato J., Detivaud L., Leclerc S. Eur. J. Biochem. 1994;224:771. doi: 10.1111/j.1432-1033.1994.00771.x. [DOI] [PubMed] [Google Scholar]; (b) Gray N.S., Wodicka L., Thunnissen A.-M.W.H., Norman T.C., Kwon S., Espinoza F.H., Morgan D.O., Barnes G., LeClerc S., Meijer L., Kim S.-H., Lockhart D.J., Schultz P.G. Science. 1998;281:533. doi: 10.1126/science.281.5376.533. [DOI] [PubMed] [Google Scholar]; (c) Chang Y.-T., Gray N.S., Rosania G.R., Sutherlin D.P., Kwon S., Norman T.C., Sarohia R., Leost M., Meijer L., Schultz P.G. Chem. Biol. 1999;6:361. doi: 10.1016/S1074-5521(99)80048-9. [DOI] [PubMed] [Google Scholar]; (d) Ding S., Gray N.S., Wu X., Ding Q., Schultz P.G. J. Am. Chem. Soc. 2002;124:1594. doi: 10.1021/ja0170302. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ding S., Gray N.S., Ding Q., Wu X., Schultz P.G. J. Comput. Chem. 2001;3:97. [Google Scholar]; (b) Ding S., Gray N.S., Ding Q., Schultz P.G. Tetrahedron Lett. 2001;42:8751. [Google Scholar]; (c) Ding S., Gray N.S., Ding Q., Schultz P.G. J. Org. Chem. 2001;66:8273. doi: 10.1021/jo016010f. [DOI] [PubMed] [Google Scholar]
- 3.Toyota A., Katagiri N., Kaneko C. Heterocycles. 1993;36:1625. [Google Scholar]
- 4.(a) Montgomery J.A., Hewson K., Jr., Temple C. J. Med. Pharm. Chem. 1962;5:15. doi: 10.1021/jm01236a002. [DOI] [PubMed] [Google Scholar]; (b) Hamden M.R., Jarvest R.L. Tetrahedron Lett. 1985;26:4265. [Google Scholar]
- 5.Corey E.J., Venkateswarlu A.J. J. Am. Chem. Soc. 1972;94:6190. [Google Scholar]
- 6.Fugami K., Ohnuma S.-Y., Kameyama M., Saotome T., Kosugi M. Synlett. 1999:63. [Google Scholar]
- 7.Mori A., Kawashima J., Shimada T., Suguro M., Hirabayashi K., Nishihara Y. Org. Lett. 2000;2:2935. doi: 10.1021/ol0061586. [DOI] [PubMed] [Google Scholar]
- 8.Alauddin M.M., Miller J.M., Clark J.H. Can. J. Chem. 1984;62:263. [Google Scholar]
- 9.Amantini D., Beleggia R., Fringuelli F., Pizzo F., Vaccaro L. J. Org. Chem. 2004;69:2896. doi: 10.1021/jo0499468. [DOI] [PubMed] [Google Scholar]
- 10.Hayami J.-I., Uno N., Kaji A. Tetrahedron Lett. 1968;11:1385. [Google Scholar]
- 11.Brik A., Lin Y.-C., Elder J., Wong C.-H. Chem. Biol. 2002;9:891. doi: 10.1016/s1074-5521(02)00184-9. [DOI] [PubMed] [Google Scholar]
- 12.(a) Cheng T.-J., Brik A., Wong C.-H., Kan C.-C. Antimicrob. Agents Chemother. 2004;48:2437. doi: 10.1128/AAC.48.7.2437-2447.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brik A., Muldoon J., Lin Y.-C., Elder J.H., Goodsell D.S., Olson A.J., Fokin V.V., Sharpless K.B., Wong C.-H. ChemBioChem. 2003;4:1246. doi: 10.1002/cbic.200300724. [DOI] [PubMed] [Google Scholar]
- 13.Best M.D., Brik A., Chapman E., Lee L.V., Cheng W.-C., Wong C.-H. ChemBioChem. 2004;5:811. doi: 10.1002/cbic.200300841. [DOI] [PubMed] [Google Scholar]
- 14.Wu C.-Y., Jan J.-T., Ma S.-H., Kuo C.-J., Juan H.-F., Cheng Y.-S.E., Hsu H.-H., Huang H.-C., Wu D., Brik A., Liang F.-S., Liu R.-S., Fang J.M., Chen S.-T., Liang P.-H., Wong C.-H. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10012. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee L.V., Mitchell M.L., Huang S.-J., Fokin V.V., Sharpless K.B., Wong C.-H. J. Am. Chem. Soc. 2003;125:9588. doi: 10.1021/ja0302836. [DOI] [PubMed] [Google Scholar]
- 16.(a) Wu C.-Y., Chang C.-F., Chen J.S.-Y., Wong C.-H., Lin C.-H. Angew. Chem., Int. Ed. 2003;42:4661. doi: 10.1002/anie.200351823. [DOI] [PubMed] [Google Scholar]; (b) Chang C.F., Ho C.W., Wu C.Y., Chao T.A., Wong C.-H., Lin C.-H. Chem. Biol. 2004;11:1301. doi: 10.1016/j.chembiol.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 17.Burkart M.D., Izumi M., Chapman E., Lin C.-H., Wong C.-H. J. Org. Chem. 2000;65:5565. doi: 10.1021/jo000266o. [DOI] [PubMed] [Google Scholar]
- 18.Chapman E., Ding S., Schultz P.G., Wong C.-H. J. Am. Chem. Soc. 2002;124:14524. doi: 10.1021/ja021086u. [DOI] [PubMed] [Google Scholar]
- 19.Burkart M.D., Wong C.-H. Anal. Biochem. 1999;274:131. doi: 10.1006/abio.1999.4264. [DOI] [PubMed] [Google Scholar]