

Graphical abstract
A small-molecule inhibitor of SARS-CoV, exhibiting an EC50 of 23 μM in cell-based assays, was identified by virtual screening against a computer model of a SARS-CoV cysteine proteinase.
Keywords: In silico screening, Anti-SARS agents, Antiviral agents, Small-molecule inhibitor leads, Docking, Chemical databases, Drug lead identification
Abstract
Virtual screening, a fast, computational approach to identify drug leads [Perola, E.; Xu, K.; Kollmeyer, T. M.; Kaufmann, S. H.; Prendergast, F. G. J. Med. Chem.2000, 43, 401; Miller, M. A. Nat. Rev. Drug Disc.2002, 1 220], is limited by a known challenge in crystallographically determining flexible regions of proteins. This approach has not been able to identify active inhibitors of the severe acute respiratory syndrome-associated coronavirus (SARS-CoV) using solely the crystal structures of a SARS-CoV cysteine proteinase with a flexible loop in the active site [Yang, H. T.; Yang, M. J.; Ding, Y.; Liu, Y. W.; Lou, Z. Y. Proc. Natl. Acad. Sci. U.S.A.2003, 100, 13190; Jenwitheesuk, E.; Samudrala, R. Bioorg. Med. Chem. Lett.2003, 13, 3989; Rajnarayanan, R. V.; Dakshanamurthy, S.; Pattabiraman, N. Biochem. Biophys. Res. Commun.2004, 321, 370; Du, Q.; Wang, S.; Wei, D.; Sirois, S.; Chou, K. Anal. Biochem.2005, 337, 262; Du, Q.; Wang, S.; Zhu, Y.; Wei, D.; Guo, H. Peptides2004, 25, 1857; Lee, V.; Wittayanarakul, K.; Remsungenen, T.; Parasuk, V.; Sompornpisut, P. Science (Asia)2003, 29, 181; Toney, J.; Navas-Martin, S.; Weiss, S.; Koeller, A. J. Med. Chem.2004, 47, 1079; Zhang, X. W.; Yap, Y. L. Bioorg. Med. Chem.2004, 12, 2517]. This article demonstrates a genome-to-drug-lead approach that uses terascale computing to model flexible regions of proteins, thus permitting the utilization of genetic information to identify drug leads expeditiously. A small-molecule inhibitor of SARS-CoV, exhibiting an effective concentration (EC50) of 23 μM in cell-based assays, was identified through virtual screening against a computer-predicted model of the cysteine proteinase. Screening against two crystal structures of the same proteinase failed to identify the 23-μM inhibitor. This study suggests that terascale computing can complement crystallography, broaden the scope of virtual screening, and accelerate the development of therapeutics to treat emerging infectious diseases such as SARS and Bird Flu.
Severe acute respiratory syndrome (SARS), an emerging infectious disease with severe mortality, is a viral respiratory illness caused by a human coronavirus called SARS-associated coronavirus (SARS-CoV).11 The sequencing of the SARS-CoV genome just 31 days after the outbreak of SARS was a testimony to the scientific community’s ability to rapidly respond to emerging diseases.12, 13 However, the use of this genome information to develop clinical treatments for SARS has slowed down. The SARS-CoV genome encodes a chymotrypsin-like cysteine proteinase (CCP, also known as Mpro or 3CLpro) that proteolytically processes polypeptides required for viral replication and transcription,14 representing an ideal drug target for treating SARS. Although small-molecule inhibitors of CCP have been identified,15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27 the development of these inhibitors as clinical drugs for treating SARS has not yet been achieved. New inhibitor leads of CCP are still highly desirable.
To identify new inhibitor leads of CCP, using the genomic information before any crystal structure of the enzyme was made available, a three-dimensional model in complex with a substrate fragment (ATVRLQp1Ap1′) was predicted by 200 molecular dynamics simulations (4.0 ns for each simulation with a 1.0-fs time step and different initial velocities) performed on terascale computers to predict conformations of the flexible loop (residues 45–48) according to a published protocol.28 An average structure of these simulations that represents CCP in the bound state was deposited to Protein Data Bank (PDB code: 2AJ5) and used as a drug target in virtual screening for small-molecule inhibitors, using a computer docking program, EUDOC.2, 29
Screening of 361413 small molecules against the 4.0-ns model of CCP identified 3958 compounds with total and van der Waals interaction energies lower than −40 and −25 kcal/mol, respectively. The use of such energy cut-offs was based on the observations that all experimentally confirmed micromolar inhibitors identified by EUDOC had total and van der Waals interaction energies lower than these cutoffs.1, 30 Twelve of them were selected for testing, after triaging compounds commercially unavailable and compounds with many chiral centers, poor solubility, or poor cell permeability.
Of the 12 compounds tested in cell-based inhibition assays, one compound, CS11 (Fig. 1 ), inhibited the human SARS-CoV Toronto-2 strain with an EC50 of 23 μM. CS11 was not toxic to normal cells at 23 μM. Four additional compounds showed 13–17% inhibition at a drug concentration of 32 μM.
Figure 1.
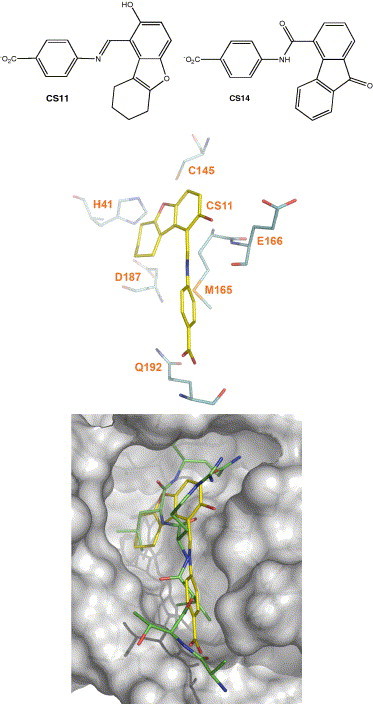
CS11 in complex with the active site of CCP. (Top) Chemical structures and protonation states of small-molecule inhibitors (CS11 and CS14) of severe acute respiratory syndrome coronavirus chymotrypsin-like cysteine proteinase (CCP); (middle) key intermolecular interactions of CS11 with the active-site residues of CCP; (bottom) overlay of CS11 with a reported substrate fragment (ATVRLQp1Ap1′) bound in the active site of CCP (PDB codes: 1P76 and 2AJ5).
The result of the cell-based assay for CS11 agrees with the EUDOC-generated CS11-bound CCP complex. In the complex model (Fig. 1), the cyclohexenyl and phenyl rings of CS11 occupy two hydrophobic regions of the active site, with the methylene and phenyl groups of CS11 mimicking the side chains of LeuP2 and ValP4 bound in a reported CCP complex;28 the carboxylate and hydroxyl groups of CS11 have hydrogen bonds with the amide proton of Gln192 and the carboxylate oxygen of Glu166, respectively. This model suggests that the potency of CS11 can be improved by minor structural modifications. For example, replacing the 4-aminobenzoic acid moiety of CS11 by a 4-amino-3-methylbenzoic acid would better mimic the side chain of ValP4. CS11 is easy to synthesize and derivatize, and it is therefore an excellent inhibitor lead of CCP.
These results demonstrate that, given the SARS-CoV genome only, one can identify a small molecule that is able to penetrate cells and rescue them from viral infection, leapfrogging the experimentally determined structures of CCP. This fact is supported by a recent report that Cinanserin was, according to cell-based assays, identified as an inhibitor of SARS-CoV by a small-scale virtual screen using a different computer model of CCP.27 Together, the two screens exemplify a genome-to-drug-lead approach that enables the direct utilization of available genetic information in drug lead identification.
To demonstrate the necessity of the genome-to-drug-lead approach, the virtual screen that identified CS11 was repeated with two crystal structures of CCP in its bound and unbound states (PDB codes: 1UK4 and 1UK2).3 Surprisingly, both screens failed to identify CS11 as a possible inhibitor. Further, the respective numbers of potential inhibitors (472 and 181) identified from the screens using the bound and unbound crystal structures are much smaller than the number (3958) identified using the computer model. The small number of potential inhibitors identified using the crystal structures may explain the fact that there has been no report of an experimentally confirmed SARS-CoV inhibitor identified by the virtual screening using the crystal structures of CCP only.4, 5, 6, 7, 8, 9, 10, 26
The different screening results could be explained by the difference primarily in the main-chain conformation of the flexible loop (residues 45–48) of CCP (Fig. 2 ). The flexible loop was not determined in the crystal structure solved at a resolution of 1.9 Å (PDB code: 1Q2W). In the bound and unbound crystal structures solved at resolutions of 2.5 and 2.2 Å, respectively, the flexible loop was determined with relatively low real-space correlation coefficients for residues 45–48 (PDB codes: 1UK4 and 1UK2).3 In the two crystal structures, the main-chain conformation of the flexible loop pushes Met49 towards His41 which in turn forces Gln189 to block the space above Asp187 and His41 in the active site, whereas the main-chain conformation of the flexible loop in the 4.0-ns model moves both Met49 and Gln189 away from His41 and thus removes the blockage of Gln189 seen in the crystal structures (Fig. 3 ). Although a flexible docking algorithm can adjust the receptor conformation to accommodate the docked ligand, there has been no literature report that it can adequately adjust the main-chain conformation of the receptor for complexation.
Figure 2.
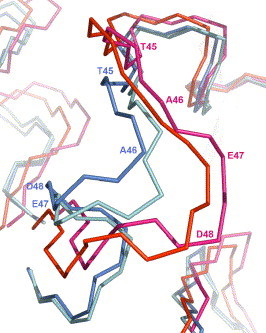
Comparison of the backbone conformations in the flexible loop (residues 45–48) of SARS-CoV CCP. Backbones (CA, C, and N) of the flexible loop (residues 45–48) in the active site of severe acute respiratory syndrome coronavirus chymotrypsin-like cysteine proteinase (CCP) in the overlaid models of the 2.0-ns computer model (magenta, PDB code: 1P76), the 4.0-ns computer model (red, PDB code: 2AJ5), the crystal structure in the bound state (blue, PDB code: 1UK4), and the crystal structure in the unbound state (cyan, PDB code: 1UK2).
Figure 3.
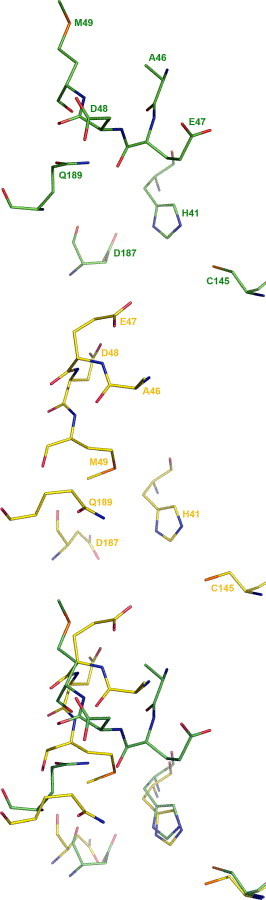
Comparison of residue conformations in the flexible loop (residues 45–48) of CCP. Residues in the flexible loop of severe acute respiratory syndrome coronavirus chymotrypsin-like cysteine proteinase (CCP) in the 4.0-ns computer model (top, PDB code: 2AJ5), the crystal structure in the bound state (middle, PDB code: 1UK4), and the overlay of the two (bottom).
To substantiate that the different screening results were caused by the structural differences of the drug target, one must prove that the identification of experimentally confirmed active inhibitors through virtual screening is in fact sensitive to regional structural differences of the target. A CCP model averaged from 200 2.0-ns simulations28 was used to repeat the virtual screening. The 2.0-ns average structure was purposefully contracted by omitting root-mean-square fit in averaging instantaneous structures. Consequently, relative to all atoms of residues 14–180 in the 4.0-ns model of CCP, the root mean square deviation (RMSD) of the 2.0-ns model (1.11 Å) is smaller than those of the bound (2.38 Å) and unbound (2.39 Å) crystal structures.
Interestingly, the virtual screening using the 2.0-ns model failed to identify CS11 as a potential inhibitor, and it resulted in a smaller number (2298) of potential inhibitors than the number obtained using the 4.0-ns structure (3985) but much larger than those obtained using the bound (472) and unbound (181) crystal structures. These numbers are proportional to regional structural differences estimated by the aforementioned RMSDs.
To evaluate the effect of regional structural changes experimentally, 17 potential inhibitors identified from the 2.0-ns model were selected for testing according to the same criteria which identified CS11. Interestingly, all active compounds identified using the 2.0-ns model were much less active than the inhibitors identified using the 4.0-ns model; the two most active compounds (CS14 in Fig. 1 and CS21 in Fig. S2 of supplementary information) are structurally analogous to CS11 and showed 25–29% inhibition at a drug concentration of 300 μM. These results demonstrate that the identification of active inhibitors is indeed sensitive to regional structural changes of drug targets.
The identification of CS11 by the use of the computer model suggests that terascale computing can model flexible regions of proteins, addressing a known limitation in crystallography, to yield 3D models useful in virtual screens for drug leads. Serving as a foundation for this study, crystal structures of proteins related to CCP were used in generating the computer model.28 Thus, terascale computing is not in competition with crystallography, but rather serves as a complement to crystallography.
The genome-to-drug-lead approach exemplified herein is not only possible but also essential to improve virtual screening for its speed and for its ability to deal with flexible regions of proteins. In this study, the 3D model of CCP was predicted from the genome in 20 days and an excellent inhibitor lead for CCP was identified by virtual screening in 9 days at the Computer-Aided Molecular Design Laboratory (CAMDL) of the Mayo Clinic. This lead was obtained from a chemical vendor in 30 days and assayed for SARS-CoV inhibition in 3 days at the Southern Research Institute. The 2-month drug-lead identification from the SARS-CoV genome can be shortened to 1 month by the 3.4 teraflops computing resource available at CAMDL in 2005 and to even a shorter process with petascale computing in the near future. All the results presented herein suggest that the genome-to-drug-lead approach can broaden the utilization of genetic information in drug discovery and open a fast track to therapeutics to combat emerging viruses such as SARS-CoV and the highly pathogenic Avian Influenza (HPAI) virus (Bird Flu).
Acknowledgments
Supported by U.S. Defense Advanced Research Projects Agency, U.S. Army Medical Research Acquisition Activity, U.S. Army Research Office, High Performance Computing Modernization Program of the U.S. Department of Defense, University of Minnesota Supercomputing Institute, National Institute of Allergy and Infectious Diseases, the Jay and Rose Phillips Family Foundation, and the Mayo Foundation.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2005.11.018.
Supplementary data
Models of SARS-CoV CCP.
References and notes
- 1.Perola E., Xu K., Kollmeyer T.M., Kaufmann S.H., Prendergast F.G. J. Med. Chem. 2000;43:401. doi: 10.1021/jm990408a. [DOI] [PubMed] [Google Scholar]
- 2.Miller M.A. Nat. Rev. Drug Disc. 2002;1:220. doi: 10.1038/nrd745. [DOI] [PubMed] [Google Scholar]
- 3.Yang H.T., Yang M.J., Ding Y., Liu Y.W., Lou Z.Y. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenwitheesuk E., Samudrala R. Bioorg. Med. Chem. Lett. 2003;13:3989. doi: 10.1016/j.bmcl.2003.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajnarayanan R.V., Dakshanamurthy S., Pattabiraman N. Biochem. Biophys. Res. Commun. 2004;321:370. doi: 10.1016/j.bbrc.2004.06.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du Q., Wang S., Wei D., Sirois S., Chou K. Anal. Biochem. 2005;337:262. doi: 10.1016/j.ab.2004.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du Q., Wang S., Zhu Y., Wei D., Guo H. Peptides. 2004;25:1857. doi: 10.1016/j.peptides.2004.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee V., Wittayanarakul K., Remsungenen T., Parasuk V., Sompornpisut P. Science (Asia) 2003;29:181. [Google Scholar]
- 9.Toney J., Navas-Martin S., Weiss S., Koeller A. J. Med. Chem. 2004;47:1079. doi: 10.1021/jm034137m. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X.W., Yap Y.L. Bioorg. Med. Chem. 2004;12:2517. doi: 10.1016/j.bmc.2004.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R.N. Eng. J. Med. 2003;348:1967. [Google Scholar]
- 12.Marra M.A., Jones S.J.M., Astell C.R., Holt R.A., Brooks-Wilson A. Science. 2003;300:1399. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 13.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R. Science. 2003;300:1394. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 14.Ziebuhr J., Herold J., Siddell S. J. Virol. 1995;69:4331. doi: 10.1128/jvi.69.7.4331-4338.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Biochemistry. 2004;43:4906. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 16.Barnard D.L., Hubbard V.D., Burton J., Smee D.F., Morrey J.D. Antiviral Chem. Chemother. 2004;15:15. doi: 10.1177/095632020401500102. [DOI] [PubMed] [Google Scholar]
- 17.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D. Chem. Biol. 2004;11:1445. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan K., Lai S., Chu C., Tsui E., Tam C. Hong Kong Med. J. 2003;9:399. [PubMed] [Google Scholar]
- 19.Hsu J.T., Kuo C.J., Hsieh H.P., Wang Y.C., Huang K.K. FEBS Lett. 2004;574:116. doi: 10.1016/j.febslet.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kao R.Y., To A.P., Ng L.W., Tsui W.H., Lee T.S. FEBS Lett. 2004;576:325. doi: 10.1016/j.febslet.2004.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keyaerts E., Vijgen L., Maes P., Neyts J., Van Ranst M. Biochem. Biophys. Res. Commun. 2004;323:264. doi: 10.1016/j.bbrc.2004.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morgenstern B., Michaelis M., Baer P., Doerr H., Cinatl J., Jr. Biochem. Biophys. Res. Commun. 2005;326:905. doi: 10.1016/j.bbrc.2004.11.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sainz B., Jr., Mossel E.C., Peters C.J., Garry R.F. Virology. 2004;329:11. doi: 10.1016/j.virol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tan E.L., Ooi E.E., Lin C.Y., Tan H.C., Ling A.E. Emerg. Infect. Dis. 2004;10:581. doi: 10.3201/eid1004.030458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto N., Yang R., Yoshinaka Y., Amari S., Nakano T. Biochem. Biophys. Res. Commun. 2004;318:719. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z., Huang C., Fan K., Wei P., Chen H. J. Chem. Inf. Mod. 2005;45:10. doi: 10.1021/ci049809b. [DOI] [PubMed] [Google Scholar]
- 27.Chen L., Gui C., Luo X., Yang Q., Gunther S. J. Virol. 2005;79:7095. doi: 10.1128/JVI.79.11.7095-7103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pang Y.-P. Proteins. 2004;57:747. doi: 10.1002/prot.20249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pang Y.-P., Perola E., Xu K., Prendergast F.G. J. Comput. Chem. 2001;22:1750. doi: 10.1002/jcc.1129. [DOI] [PubMed] [Google Scholar]
- 30.Pang Y.-P., Xu K., Kollmeyer T.M., Perola E., McGrath W.J. FEBS Lett. 2001;502:93. doi: 10.1016/s0014-5793(01)02672-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Models of SARS-CoV CCP.