

Abstract
Hereditary angioedema (HAE), a rare but life-threatening condition, manifests as acute attacks of facial, laryngeal, genital, or peripheral swelling or abdominal pain secondary to intra-abdominal edema. Resulting from mutations affecting C1 esterase inhibitor (C1-INH), inhibitor of the first complement system component, attacks are not histamine-mediated and do not respond to antihistamines or corticosteroids. Low awareness and resemblance to other disorders often delay diagnosis; despite availability of C1-INH replacement in some countries, no approved, safe acute attack therapy exists in the United States. The biennial C1 Esterase Inhibitor Deficiency Workshops resulted from a European initiative for better knowledge and treatment of HAE and related diseases. This supplement contains work presented at the third workshop and expanded content toward a definitive picture of angioedema in the absence of allergy. Most notably, it includes cumulative genetic investigations; multinational laboratory diagnosis recommendations; current pathogenesis hypotheses; suggested prophylaxis and acute attack treatment, including home treatment; future treatment options; and analysis of patient subpopulations, including pediatric patients and patients whose angioedema worsened during pregnancy or hormone administration. Causes and management of acquired angioedema and a new type of angioedema with normal C1-INH are also discussed. Collaborative patient and physician efforts, crucial in rare diseases, are emphasized. This supplement seeks to raise awareness and aid diagnosis of HAE, optimize treatment for all patients, and provide a platform for further research in this rare, partially understood disorder.
Key words: AAE, acquired angioedema, angioedema, C1 esterase inhibitor, C1-INH, HAE, HANE, HANO, hereditary angioedema, hereditary angioneurotic edema, angioneurotic edema, chemically induced angioedema, human SERPING1 protein
Abbreviations used: AAE, Acquired angioedema; AAEE, (Italian) Voluntary Association for the Study, Therapy, and Fight Against Hereditary Angioedema; ACE, Angiotensin-converting enzyme; APP, Aminopeptidase P; AT2, Angiotensin II; B19V, Parvovirus B19; BMD, Bone mineral density; BVDV, Bovine viral diarrhea virus; C1, First component of the complement cascade; C1-INH, C1 esterase inhibitor; C1nh, Murine C1 esterase inhibitor gene; C1NH, Human C1 esterase inhibitor gene; C2, Second component of the complement cascade; C3, Third component of the complement cascade; C4, Fourth component of the complement cascade; C5, Fifth component of the complement cascade; CCM, Chemical cleavage of mismatches; CH50, Total hemolytic complement, 50% cell lysis; Cmax, Maximum concentration; CPMP, Committee for Proprietary Medicinal Products; CPV, Canine parvovirus; DHPLC, Denaturing HPLC; FF, (Ovarian) follicular fluid; FFP, Fresh frozen plasma; HAE, Hereditary angioedema; HAE-I, Hereditary angioedema type I; HAE-II, Hereditary angioedema type II; HAEA, US HAE Association; HAV, Hepatitis A virus; HbsAg, Hepatitis B surface antigen; HBV, Hepatitis B virus; HCV, Hepatitis C virus; HK, High molecular weight kininogen; HRT, Hormone replacement therapy; HUVS, Hypocomplementemic urticaria-vasculitis syndrome; LH, Luteinizing hormone; MASP, Mannose-binding protein associated serine protease; MBL, Mannan-binding lectin; MFO, Multifollicular ovary; MGUS, Monoclonal gammopathies of undetermined significance; Mr, Molecular mass; NAT, Nucleic acid amplification technique; NEP, Neutral endopeptidase; OC, Oral contraceptive; OMIM, Online Mendelian Inheritance in Man (database); PCO, Polycystic ovary; PCT, Primary care trust; PREHAEAT, Novel Methods for Predicting, Preventing, and Treating Attacks in Patients with Hereditary Angioedema; PRV, Pseudorabies virus; rhC1-INH, Recombinant human C1 esterase inhibitor; rtPA, Recombinant tissue-type plasminogen activator; SHBG, Sex hormone binding globulin; SSCA, Single-stranded conformational analysis; tPA, Tissue-type plasminogen activator; UK, United Kingdom
Introduction
This supplement, like the 2003 C1 Esterase Inhibitor Deficiency Workshop and the many patient and physician initiatives that inspired it, seeks to further assist clinicians and researchers in the diagnosis, understanding, and management of nonallergic angioedema. It represents the combined scientific effort of nearly 80 scientists, physicians, and patient advocates from around the world, many of whom presented at the Budapest workshop, but all of whom have helped to advance the knowledge of these rare disorders, and it cites the work of hundreds more. May the spirit of scientific solidarity contained herein spark continued efforts toward international unity in improving the knowledge and management of these diseases.
History of angioedema from Quincke and Osler to today
(Angelo Agostoni, MD, Lorenza Zingale, MD,∗ Kayla Williams, BS, MA, MFA, and Marco Cicardi, MD,∗ Milan, Italy, and Cambridge, Mass)
Angioedema in the absence of allergy continues to represent a medical paradox. This uncommon disorder may manifest as facial, laryngeal, genital, or intra-abdominal swelling or swelling of the extremities. Despite its often dramatic presentation, its rarity and its tendency to mimic other, dissimilar disease states often obscure its diagnosis. Even so, the condition has been documented for more than a century. Although a review by Dennehy1 posited that the writer Nathaniel Hawthorne first described familial angioedema in his 1851 novel The House of the Seven Gables,2 the hereditary disorder described therein caused death associated with hemorrhage (“There was an unnatural distortion in the fixedness of Colonel Pyncheon's stare… there was blood on his ruff, and… his hoary beard was saturated with it”) and was thus quite different from angioedema. Indeed, even Dennehy1 wisely attributed the observation to his father-in-law, hinting at a personal understanding of familial—if not heritable—defects.
Nonetheless, true medical descriptions soon appeared. In 1876, John Laws Milton3 described “giant urticaria.” Acute, circumscribed edema of the skin was documented by Heinrich Quincke4 in 1882. By 1888, Sir William Osler5 distinguished an inherited form of angioedema, then known as angio-neurotic edema, and was the first to fully describe its clinical characteristics. A biochemical defect was isolated 75 years later, when Donaldson and Evans6 described similar patients whom they demonstrated were lacking the serum inhibitor directed against the first component of the complement system, C1 esterase inhibitor (C1-INH). At the time of their 1963 publication, the extent of the deficiency was unknown, but immunoelectrophoresis permitted a semiqualitative evaluation indicating that the patients' blood lacked C1-INH.
Since then, further work has been undertaken to better understand the genetics, pathogenesis, and appropriate clinical management of nonallergic angioedema. With a fuller knowledge of its biochemical mechanism has come the gradual dismissal of neurotic from its name. However, today hereditary angioedema (HAE) and its even rarer acquired form, acquired angioedema (AAE), remain little known in clinical practice and thus frequently misdiagnosed and inappropriately treated, often resulting in unnecessary suffering. Similarities to allergic conditions and inappropriate framing as part of the urticaria-angioedema syndrome frequently lead patients with HAE to be considered allergic and treated with antihistamines and corticosteroids, ineffective in this disorder. Abdominal edema may so closely resemble an acute abdomen that some patients with HAE have undergone unnecessary surgical explorations, often more than once. Because untreated edema of the larynx may be fatal, inappropriate management may result in death.
For many, HAE and AAE present an ongoing clinical challenge. Despite the recurrent nature of angioedema attacks, their acute treatment is often suboptimal, sometimes delayed, and often requires lengthy hospital stays. In some countries, including the United States, no safe and effective acute attack therapy is available. Even the prophylactic management of these disorders is inconsistent across centers and nations, and, because of the side effects of antifibrinolytics and steroids currently in use, requires a lifelong, individualized calculation of benefits and risks. These drawbacks are well known to the small community of physicians who deal frequently with these diseases and are a feature of life for those patients who suffer frequent or severe attacks.
Nonallergic angioedema as a model for the treatment of rare diseases
(Kayla Williams, BS, MA, MFA, and Henriette Farkas, MD, PhD,∗ Cambridge, Mass, and Budapest, Hungary)
In recognition of these challenges, several national and international physician and patient initiatives have begun in the past 2 decades. In many ways, the field of nonallergic angioedema, and especially HAE, is becoming an exemplar for the understanding and management of rare diseases. The estimated frequency of HAE is 1:50,000.7 As in many uncommon conditions, HAE's infrequent incidence fosters collaboration, forcing clinicians and researchers to pool their anecdotal experiences and data to attain statistical significance. Nonetheless, HAE is an attractive field because it offers doctors a chance to improve the lives of their patients dramatically through study but also via educated case management. As such, it has brought together a group of motivated and compassionate physicians. The pharmaceutical industry has also been welcomed to the C1 Esterase Inhibitor Deficiency Workshop and other HAE initiatives, fostering free exchanges between academia, industry, and patients.
Indeed, perhaps the most distinctive feature of HAE physician initiatives is their inclusion of patients with HAE, not only in a traditional capacity of raising awareness and research funding but also as ethical advisors and welcomed guests for the presentation of scientific abstracts and talks. The first C1 Esterase Inhibitor Deficiency Workshop, held in Hungary in 1999, was the earliest meeting to follow this model. Since then, the 2 subsequent workshops and other patient-association gatherings in the United States and Canada have followed its inclusive precedent. Such a high level of patient involvement reflects not only the close relationship between knowledgeable physicians and their patients but also regional shortcomings in diagnosis and treatment. Because of the incapacitating and life-threatening aspects of the disease, patients and their families from areas where HAE is largely unknown have been forced to become educated enough to explain the disorder to strangers and, often, emergency department personnel to obtain the proper treatment. Even patients whose cases are managed by a competent local practitioner may have attacks while traveling or when their doctor is unavailable and thus may need to articulate their condition to someone entirely unfamiliar with the disease. By incontrovertible necessity, patients with HAE are one of the best-educated patient populations, and this is especially true in areas where satisfactory therapy for acute attacks is unavailable.
For patients and physicians alike, the Internet facilitates increasingly more communication, both personal and scientific. For patients with HAE, it can help to reduce the isolation of having a rare disease. Many patients first contact their national patient association online and use e-mail to stay in touch with fellow patients. The Internet is also being used by physicians and scientists to support a private patient registry and a public, constantly updated human C1-INH gene (C1NH) mutation database. Through this online contact and regular meetings open to all, information about nonallergic angioedema is shared rapidly among a small, concerned group. Nonetheless, the need to educate more physicians and the general public remains. The rarity of nonallergic angioedema increases the likelihood that clinicians, especially general practitioners or emergency department personnel, may never have seen a case. Patient organizations and other groups have thus worked to create emergency passports for patients with known HAE to carry and educational materials to distribute to emergency departments.
Scientific opportunities and current areas of controversy
(Kayla Williams, BS, MA, MFA, Cambridge, Mass)
Nonallergic angioedema is a puzzle with relatively well-defined borders: many specific C1NH mutations resulting in HAE have been identified, and the symptomatic results are known. However, several central pieces are missing. Despite recognition of functional C1-INH deficiency as the cause of most forms of nonallergic angioedema, the specific mechanism of attack generation has not been definitively described. Likewise, symptoms similar to those of nonallergic angioedema have now been reported in patients with normal amounts of functional C1-INH.8
Multiple pathways have been proposed for the chemical cause of angioedema attacks. The murine HAE model developed by Han et al9 shares similarities with the human form of the disease but diverges from typical HAE in the triggering of angioedema. Despite homozygous C1-INH deficiency, the mice, with few exceptions, have not been observed to have typical angioedema attacks. Attacks, manifesting solely as local increases in vascular permeability, could be provoked by the application of mustard oil. Rather than representing a shortcoming of the mouse model, such a high threshold for attacks might parallel the course of those human heterozygotes, identified via a family member with active HAE, who nonetheless never have an attack (for documentation of such patients, see Agostoni and Cicardi7). The absence of spontaneous attacks despite profound C1-INH deficiency suggests that multiple biological events must transpire for angioedema to manifest.
Equally fascinating is the range of human disorders associated with functional C1-INH deficiency. On the mild end of the spectrum, the American physicians Luong and Nguyen10 have reported a group of apparently unrelated Vietnamese women presenting to their California clinic with lower extremities discomfort of unknown etiology. All of these women were found to have reduced amounts of serum C1-INH, and danazol treatment resolved both the C1-INH deficiency and the discomfort. At the opposite end of the C1-INH deficiency spectrum, some patients with HAE have periods of weekly or near-continuous angioedema attacks. In the most severe cases, laryngeal attacks may extend far enough into the thorax that even tracheostomy cannot maintain airway patency.
It is unclear whether discerning the mechanism of some forms of HAE, AAE, and C1-INH deficiency–associated disorders may elucidate others, but the attraction of a unified theory is obvious. However, among other factors, the inhibitory promiscuity of the C1-INH molecule and its predisposition to mutation may not lend themselves to a simple answer. Nonetheless, given the many proposed pathways for attack generation, information gained toward a full understanding of nonallergic angioedema attacks may lead to a greater knowledge of 1 or more chemical cascades, including the classical complement pathway, kinin generation, and the intrinsic coagulation pathway.
The areas of greatest controversy include which vasoactive peptide is ultimately responsible for the increased vascular permeability that results in angioedema. Bradykinin and second component of the complement cascade (C2)-kinin have been proposed,11., 12. with recent research contributing evidence to the importance of bradykinin.13., 14., 15., 16. Nonetheless, within the current understanding of coagulation, kinin, and complement pathways, neither peptide seems to perfectly explain all of the symptoms of angioedema. Although bradykinin is the only candidate mediator for which there is direct clinical evidence, it is possible that yet another system, intermediary, or molecule may be involved in edema-generating vascular leakage.
Specific triggers for vasoactive peptide release are also unknown. It is proposed that the activation of factor XII is crucial to attack generation,17 and that factor XII activation may be a result of phospholipids released from damaged or apoptotic cells. Recently, endothelial cells have been implicated in the generation, via kallikrein, of bradykinin, both in the presence18 and absence19., 20. of factor XII. These hypotheses explain how illness or localized tissue damage may precipitate attacks but do not account for other triggers, which are themselves not well defined. In large part, triggers seem to vary from patient to patient and, in several attacks, may not be apparent. Of these, the most scientifically documented and explored are hormonal triggers, made all the more interesting by relatively recent reports of patients with normal C1-INH concentrations and HAE-like symptoms provoked or exacerbated by increased levels of estrogen.
The importance of hormones in the regulation of nonallergic angioedema has long been acknowledged via its prophylaxis with androgens. Increasingly, the effects of estrogen, progesterone, and other sex hormones are being explored. In some women, estrogen results in an increased frequency of angioedema attacks,21., 22. but others appear unaffected. Depending on the patient and trimester, pregnancy may reduce or increase the number and severity of attacks.23., 24.
In this supplement, the role of progesterone is debated, with Visy et al finding a positive correlation between serum progesterone values and attack frequency, whereas Bork et al note no increase in attack frequency among patients whose oral contraceptive (OC) contained progesterone and estrogen compared with those receiving estrogen alone. Indeed, Bork et al refer to several published works in which progestins were used, with varying success, to ameliorate HAE symptoms.22a., 23a., 24a. In contrast, danazol, a common prophylaxis, alters multiple biological mechanisms but is known to block progesterone receptors and increase progesterone's metabolic clearance.25 Given these conflicting findings, the influence of progesterone seems a likely area for further study.
About the supplement
After the 2003 Third C1 Esterase Inhibitor Deficiency Workshop in Budapest, participants were invited to further develop the information they presented for publication. The scientific content herein represents the work of participants who responded, some of which, as noted, has now been published elsewhere. In an attempt to survey the field of nonallergic angioedema fairly and completely, the supplement also includes reviews of relevant articles as well as original material covering emergent areas in the field of HAE, AAE, and related disorders. Individual authorship is cited in text where possible and fully attributed in the table of contents.
This supplement would not have been possible without those who organized the Third C1-Esterase Inhibitor Deficiency Workshop: the European C1-INH Deficiency Working Group, the Hungarian Society for Immunology, and the Foundation for the Prevention and Treatment of Fatal Angio-oedematous Diseases. Most especially, I would like to acknowledge Editor Dr. Henriette Farkas for her unfailing compassion, organization, and support, and Professor Dr. Marco Cicardi and Dr. Tony Williams, who first imagined that such a document could be a reality. Dr. Bruce Zuraw's cogent explanation of bradykinin metabolites was greatly appreciated. In addition, Dr. Karen Binkley graciously shared her work on very short notice and Dr. Alvin Davis III provided a valuable review; Dr. Shih-Wen Huang's contribution to the US HAE Association newsletter informed me of the full range of C1-INH deficiencies, Dr. Alvin Schmaier explained the mystery of angiotensin II receptor blocker-associated angioedema, and Dr. Erik Nielsen, both thorough his online Hereditary Angioedema Thesis and quick correspondence, provided information and inspiration. Many thanks go to Dr. Ineke Bos, whose model of the C1 esterase inhibitor molecule graces our cover; Chrystal McDonald who worked tirelessly to secure reprint permissions; Dr. Brunello Wüthrich who provided images of HAE attacks; and Drs. Werner Müller and Georg Dewald for their additions to the text. I would also like to recognize Mr. Anthony Castaldo of the US HAE Association for his review of text pertaining to the patient experience and his indomitable, sustaining sense of humor.
Lastly, I would like to dedicate this supplement to its many contributing authors and all the HAE, AAE, and non-allergic angioedema patients they strive to help.
Kayla Williams
Clinical manifestations and diagnosis
In the first part of this section, Cicardi and Zingale describe the varied ways in which HAE can manifest and discuss other diseases that published case reports and their clinical case series have associated with HAE.
Clinical manifestations of HAE
(Marco Cicardi, MD,∗ and Lorenza Zingale, MD,∗ Milan, Italy)
The symptoms of HAE are caused by the extravasation of plasma into the deeper cutaneous or mucosal layers as a result of 1 or more locally released vasoactive peptides. The edema in HAE is nonwhealing, nonpruritic, and generally unrelieved by antihistamines, suggesting that histamine is not involved in its induction.26 The biological characteristics of the vasoactive peptides released in C1-INH–deficient sera indicate that the peptides belong to the kinin family. However, the discussion is not entirely closed on whether bradykinin, released because of contact system activation, or a peptide originated from C2 on classical complement pathway activation and the generation of plasmin, is the main mediator of symptoms in patients with HAE.11., 12. Nonetheless, recent lines of evidence coming from C1-INH knockout mice, studies in patients' plasma, and analysis of C1-INH mutants from patients with HAE support the bradykinin hypothesis.9., 27., 28. Kinin peptides participate in inflammatory processes and increase vascular permeability, activating intracellular pathways that lead to the release of nitric oxide.29., 30. Vascular leakage can occur without anatomical damage and rapidly revert when the release of mediator molecules ceases. Hence, edema usually resolves within 72 hours. In some cases, it may resolve within 12 hours, but in others, it may persist as long as 5 days. Urticaria, a condition analogous to angioedema but with plasma leakage into the upper cutaneous layers, is typically absent or minimal and short-lasting in patients with HAE.
Typical symptoms
The recurrence of cutaneous angioedema, abdominal pain, and asphyxia caused by laryngeal edema is the full clinical pattern of HAE, present in about 50% of adult patients.7 Attacks usually evolve within a single site, but it is not uncommon for some patients to have simultaneous or closely spaced cutaneous and abdominal involvement. Most patients recognize several hours in advance that an attack is coming. They may have sudden mood changes, anxiety, or complete exhaustion.
Cutaneous symptoms. Skin edema is nonpitting and nonerythematous, with ill-defined margins. It typically affects the face ( Fig 1), extremities, and genitals ( Fig 2). It usually spreads to disfigure the affected site, temporarily depriving it of function. Most often, a single site is affected by an extended edema that grows and then regresses within 2 to 5 days. Alternatively, edema may persist, although reduced in size, and migrate to different cutaneous locations. In contrast with edema of other etiologies, edema associated with HAE does not principally manifest in the perioral region. Edema can localize subcutaneously in any body part, including the trunk.
Fig 1.

Facial edema. Photo: Dermatologische Klinik, Universitätsspital Zurich, Switzerland. Brunello Wüthrich, MD. Reprinted with permission from Swiss Medical Weekly.43a
Fig 2.
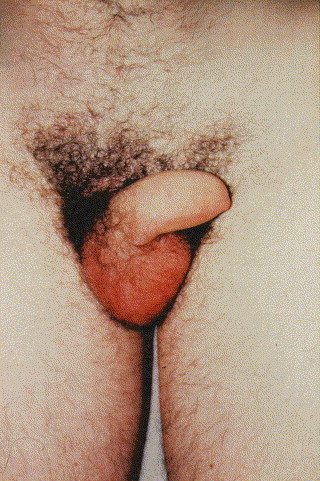
Penile edema. Photo credit: Dr. Martin Ludovic.
Abdominal symptoms. Recurrent abdominal pain, a consequence of gastrointestinal wall edema, is reported by 70% to 80% of patients with HAE.7., 31., 32. This is a distinguishing feature of C1-INH deficiency because abdominal involvement is rarely seen in angioedema of other origins. It presents with symptoms that may vary from mild discomfort to severe, intractable pain accompanied by vomiting and/or diarrhea.33 In this setting, hypovolemia can result from a combination of fluid loss, plasma extravasation, and vasodilation and can progress to hypovolemic shock.34., 35. Ascites resulting from extravasation into the peritoneal cavity, edema of the bowel wall, or changes in splenoportal axis caliber have been described during abdominal attacks as detected by ultrasounds or computed tomography.36., 37., 38., 39., 40., 41. Gastrointestinal endoscopy performed during an abdominal attack revealed gastric involvement. Interestingly, during the healing process after a prominent gastric edema, several small nodules and raised erosions developed over the entire gastric mucosal surface. Within 55 days, the gastric mucosa had returned to normal.42
The similarity between bowel angioedema and surgical emergencies is confirmed by the fact that approximately ⅓ of patients with undiagnosed HAE undergo unnecessary surgery during abdominal attacks.7 However, even after a diagnosis of HAE has been established, differentiating angioedema of the bowel from a surgical emergency remains a critical task for the physician.31 The physical examination can show the presence of an abdominal defense reaction. Moderate or sometimes even marked leukocytosis can be part of an angioedema attack.43 Abdominal ultrasounds and computer-assisted tomography scans demonstrate the presence of free peritoneal fluid and edematous intestinal mucosa.36., 39., 41. However, all of these signs are clearly not specific to angioedema. The authors note that this symptomatic generality should be borne in mind to avoid the situation that occurred with a patient in their case series. Surgery was inappropriately delayed when acute appendicitis was mistaken for intestinal angioedema. The efficacy of C1-INH plasma concentrate in resolving symptoms may help to distinguish angioedema from a true surgical emergency.
Laryngeal symptoms. Laryngeal edema is the most dramatic clinical event for patients with HAE. Half of them have it at least once in their lives, but a history of recurrent episodes of suffocation caused by laryngeal edema is not uncommon, and deaths still occur as a result.44 In the past, 25% to 30% of patients with HAE died from laryngeal edema. This percentage has dramatically dropped for patients who are appropriately diagnosed because of the availability of effective treatments in several countries.45 Nevertheless, because of previous life-threatening experiences, some patients with HAE still carry permanent tracheal cannulae, allowing them to breathe by bypassing the larynx when edema occurs.
As mentioned, angioedema without urticaria is the hallmark of C1-INH deficiency. However, a discrete number of patients, 26% in a survey by Frank et al,31 have erythematous mottling, erythema multiforme, or erythema marginatum, always mild and transient, that inconstantly heralds or attends their angioedema.46 Some patients recognize this symptom as announcing an attack, and when on prophylactic treatment, can still have a rash not followed by swelling. Fig 3 depicts several erythematous rashes experienced by patients before or during attacks of angioedema.
Fig 3.
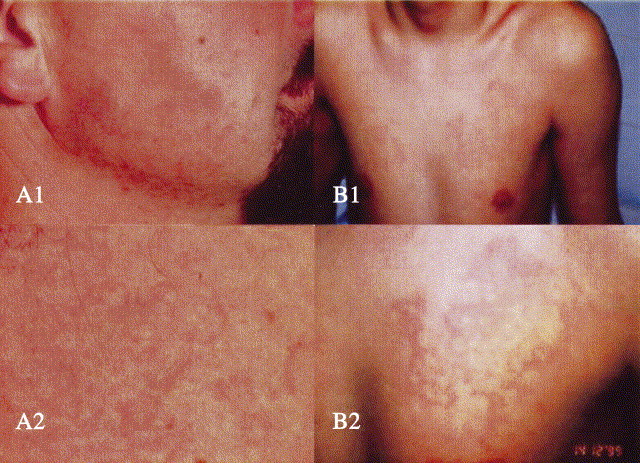
Various erythematous rashes preceding or accompanying angioedema episodes. A1, Facial erythema marginatum; A2, close view. Photo credit: Brunello Wüthrich, MD. Reprinted with permission from Swiss Medical Weekly.43aB1, Mottling on chest; B2, close view. Photo credit: George Harmat, MD. Reprinted with permission from Acta Dermato-Venereologica.46
Unusual symptoms
Reports in the literature suggest that edema caused by C1-INH deficiency could occur in locations other than the characteristic sites of manifestation.47., 48., 49., 50. Frank et al31 reported transient pleuritic symptoms with pleural effusion in 2 patients. Local cerebral edema has been considered responsible for transient seizures and hemiparesis seldom described in patients with HAE.31., 51. This assumption, despite its attraction and its occurrence in other forms of angioedema,49 has not been confirmed so far. Neurologic disorders and the potential manifestation of cerebral edema remain a rarity in patients with HAE.
Although atypical, urinary symptoms mimicking an infection have been described, and in 1 patient, the presence of bladder edema was documented by endoscopy and biopsy.24., 48.
Pulmonary edema as a consequence of C1-INH deficiency has occasionally been suggested but never clearly demonstrated.47 In the authors' experience, such an event was never observed to accompany an angioedema attack. They suggest that the high efficiency of the pulmonary vascular tree in the inactivation of bradykinin accounts for the lungs' protection from its effects.52
Age of onset and frequency of symptoms
C1-INH deficiency is present at birth, and a minority can have perinatal angioedema symptoms. Most commonly, symptoms begin at school age. Half of patients with HAE had symptoms within the first decade of life, and another third had symptoms by the second decade. Asymptomatic adults carrying a C1NH mutation, detected because of the presence of offspring with clinically overt disease, have been described and are estimated to account for 5% of all patients with HAE.7
The frequency at which bouts of angioedema recur is extremely variable among subjects and may vary in the same individual during different stages of life. A survey of the Italian case list showed that slightly less than ⅓ of untreated patients with HAE have more than 1 angioedema attack per month, 40% have 6 to 11 swellings per year, and the remaining 30% are infrequently symptomatic or completely symptom-free. This range of phenotypic expression has no significant correlation with plasma concentrations of C1-INH and is usually inconsistent among family groups. It should therefore be concluded that factors other than C1-INH deficiency intervene to determine a subject's tendency to develop angioedema. These factors might be genetic or environmental. The hypothesis that symptom frequency correlates with specific functional polymorphisms of some of the proteins involved in pathogenesis is attractive but thus far unproven. An initial report suggesting that a polymorphism within the bradykinin receptor could distinguish oligosymptomatic from polysymptomatic patients has not been confirmed.53 Farkas et al54 found that patients with HAE infected with Helicobacter pylori are more susceptible to symptoms than uninfected patients, and that eradication of the infection reduces the frequency and severity of swellings, particularly angioedema of the bowel. If confirmed in a larger group of patients, these findings could support those of several groups suggesting that infections increase susceptibility to angioedema in the general population as well as in patients with HAE.7., 31., 55., 56., 57.
Clinical and laboratory criteria for diagnosis are provided in Table I; a severity scale for the evaluation of nonallergic angioedema is provided in Table II. These tools are based on contributions elaborated from experts from 10 European countries who received a grant from the European Commission for a project called Novel Methods for Predicting, Preventing, and Treating Attacks in Patients with Hereditary Angioedema (PREHAEAT), consisting of a concerted action in the framework of the specific research and a technologic development program, Quality of Life and Management of Living Resources, designed to improve the lives of patients with HAE.
Table I.
Criteria for diagnosis of angioedema caused by C1 inhibitor deficiency
| Clinical criteria |
| Major |
| (1) Self-limiting, noninflammatory subcutaneous angioedema without major urticarial rash, often recurrent and often lasting more than 12 hours |
| (2) Self-remitting abdominal pain without clear organic etiology, often recurrent and often lasting more than 6 hours |
| (3) Recurrent laryngeal edema |
| Minor |
| (4) Family history of recurrent angioedema and/or abdominal pain and/or laryngeal edema |
| Laboratory criteria |
| (1) C1 inhibitor antigenic levels <50% of normal at 2 separate determinations with patient in basal condition and after the first year of age |
| (2) C1 inhibitor functional levels <50% of normal at 2 separate determinations with patient in basal condition and after the first year of age |
| (3) Mutation in C1 inhibitor gene altering protein synthesis and/or function |
| Diagnosis can be established in presence of 1 major (1-3) clinical criterion and 1 laboratory criterion |
Table II.
Criteria for evaluation of disease severity∗
| Attack severity | Score |
|---|---|
| Mild attacks (discomfort noticed, but no disruption of normal daily activity) | 0.5 for each 24 hours |
| Moderate attacks (discomfort sufficient to reduce or affect normal daily activity) | 1 for each 24 hours |
| Severe attacks (inability to work or perform daily activity) | 2 for each 24 hours |
| Need for treatment | |
| Emergency treatment: conservative, substitutive (C1-INH, FFP) | 5 each |
| Emergency treatment: invasive (intubation, tracheotomy) | 25 each |
| Long-term prophylaxis for more than 6 months | 25 |
| Long-term prophylaxis for 3-6 months | 12.5 |
| Score | Class | Degree |
|---|---|---|
| >30 | 1 | Severe |
| 21-30 | 2 | Moderate |
| 11-20 | 3 | Mild |
| 1-10 | 4 | Minimal |
| 0 | 5 | Asymptomatic |
These parameters are determined over the period of 1 year. The sum of the scores defines the severity of the disease for that year.
Diseases associated with C1-INH deficiency
Most often, patients with HAE are substantially healthy apart from problems associated with swelling. However, there are several reports of autoimmune diseases in patients with HAE,58., 59., 60., 61., 62., 63., 64., 65., 66., 67., 68. and systemic lupus, in particular, has been described rather often.62 In a systematic study, 19 of 157 patients with HAE had some kind of autoimmune disorder.60 Moreover, patients with HAE, because of defective control of the classical pathway of complement activation, have a deficiency of the fourth component of the complement cascade (C4) and C2, a condition that increases the risk of autoimmune diseases.69 A large epidemiologic study in 1997 based on 24 major autoimmune diseases estimated the prevalence of autoimmune diseases in Americans to be 1 in 31 (3.2%).70 Given that all autoimmune diseases were not evaluated in this general population study, one cannot definitively conclude that patients with HAE have a higher risk of autoimmune disease, but it appears likely.
The association of HAE with other inherited and noninherited conditions has occasionally been reported, but these observations remain isolated.71., 72., 73., 74.
Last, patients with HAE can be exposed to risk through needed treatments. Several cases of hepatitis C virus (HCV) in the Italian case series were a result of receiving plasma-derived products. These cases occurred before the introduction of viral inactivating procedures for plasma products.75 No cases of HIV were reported, but because of HCV, approximately 5% of their patients now have liver-related problems.
Role of ultrasound investigations in HAE
(George Harmat, MD, PhD, Pál N. Kaposi, MD, PhD, Kálmán Fáy, MD, István Karádi, MD, PhD, DSc, Béla Fekete, MD, PhD, DSc, George Füst, MD, PhD, DSc,∗ Lilian Varga, PhD,∗ and Henriette Farkas, MD, PhD,∗ Budapest, Hungary)
In this section, Harmat et al describe the results of a study of 70 Hungarian patients with HAE in whom ultrasonography was used to evaluate acute abdominal attacks of HAE.
Background and rationale
Ascites can result from diverse causes. The most common etiology, found in approximately 80% of cases, is the decompensated liver (cirrhosis). The remaining 20% result from other pathologies, such as malignancy in the abdomen (10%); various inflammatory diseases and other disorders, such as nephrotic syndrome, exudative enteropathy, chylous ascites, and mesenteric thrombosis; and others. However, HAE is very seldom mentioned as a cause of ascites. This is a real problem, because ascites are a significant diagnostic sign of this uncommon but serious disease.
The most common symptoms of HAE appear in the form of ascites that cause acute abdominal attacks. For diagnosing this state, ultrasonography is the most potent tool.39., 76., 77.
Methods
Ultrasonographic assessment is especially well suited to investigating the cause of abdominal symptoms. This study was performed to evaluate the usefulness of ultrasonographic diagnosis and included 70 patients (26 pediatric) from the Hungarian HAE center database. Of these, 60 had HAE type I and 10 had HAE type II. The male to female ratio was 32:38, and patient age ranged from 2.5 to 66 years. Patient follow-up continued for a decade. In addition to biochemical studies, ultrasound investigations were performed at 6-month intervals.
Patients with typical symptoms of HAE were hospitalized if the presence of other pathologies could be ruled out and if the manifestation was associated with hypovolemia and included recurrent paroxysms of acute colicky pain, nausea and vomiting, or profuse diarrhea, not responding to symptomatic therapy. All hospitalized patients underwent ultrasonography.
During each abdominal attack, ultrasound examinations were performed before treatment and repeated at 24 and 48 hours post-treatment.78., 79. Ultrasonographic investigations were performed by using a Hitachi 451, a Hitachi EUB 40 (Hitachi Medical Systems, Zug, Switzerland), or an Aloka SSD-1700 diagnostic system (Aloka Co, LTD, Tokyo, Japan), with a 3.5-MHz or - MHz convex transducer or a linear 7.5-MHz transducer. Subdiaphragmatic and pelvic regions were scanned with the patient in a supine position. Kidneys were explored and the presence of free peritoneal or retroperitoneal fluid was ascertained with the patient in the supine and lateral positions or, when necessary, standing. Free fluid, when detected, was classified into 1 of 3 categories, as follows:
-
(1)
Small-volume free peritoneal fluid was visible only in the subhepatic or subsplenic space, and in every case, in the Douglas cul-de-sac.
-
(2)
Moderate-volume ascites, in addition to ascites found in these regions, included those identified in the sublienal space and among the intestinal loops. The intestinal walls were also swollen (thickness in excess of 5 mm80).
-
(3)
In large-volume ascites, the intestinal loops floated in peritoneal fluid.
Results
An ultrasound image taken during an acute abdominal attack ( Fig 4) clearly illustrates the abdominal manifestations of HAE. In this medial sagittal section of the pelvic area, a large amount of free peritoneal fluid can be observed in the Douglas cul-de-sac, distal to and well separated from the urinary bladder. A floating intestinal loop can be seen.
Fig 4.
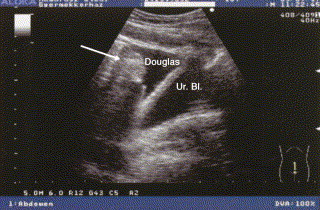
Sagittal sonogram during an abdominal HAE attack. A significant amount of fluid can be seen in the pouch of Douglas, with a swollen intestinal loop visible (arrow) floating in the free fluid.
During the attack, an edematous thickening of the intestinal wall and a thin, echo-free fluid layer around the bowels also could be observed, as illustrated in Fig 5. As shown in Fig 6, a small amount of free fluid may be observed in the triangle among the colon, spleen, and left kidney; here, the intestinal wall is also thickened.
Fig 5.
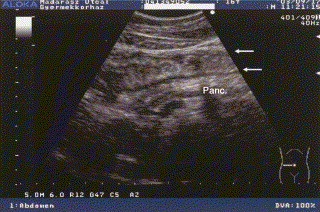
Transverse sonogram during an abdominal HAE attack, showing bowel and pancreas. Longitudinal section of a swollen bowel: the intestinal wall is edematously thickened (arrows); in addition, the reflectivity of the pancreas is increased.
Fig 6.
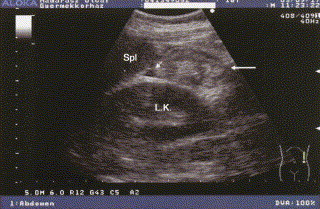
Sonogram during an abdominal attack of HAE, showing kidney and spleen. Section of a thickened intestinal wall (large arrow) and a small amount of fluid (small arrow) between the left kidney and the spleen.
The symptoms of HAE are usually treated by the administration of C1-INH concentrate. Fig 7 compares sonograms taken before and after treatment. At 24 hours posttreatment, the volume of the ascites had decreased significantly; however, clinical symptoms abated within 30 to 60 minutes of the infusion. The free peritoneal fluid and intestinal wall swelling fully disappeared within 48 hours of treatment.
Fig 7.
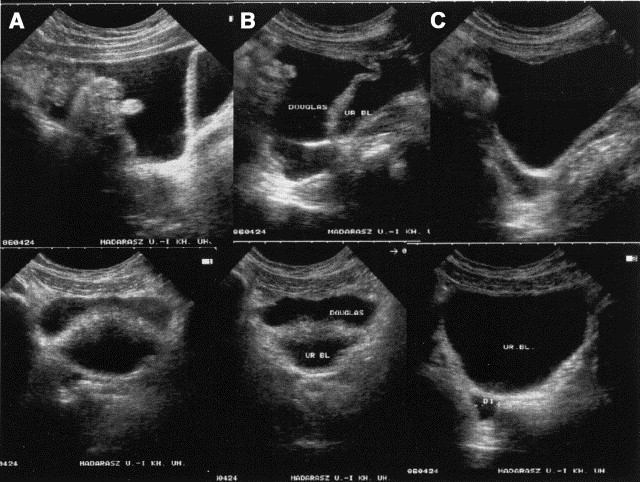
Sagittal and transverse sonograms during an HAE attack before and after treatment. Sagittal sections are shown above, transverse below. A, A large amount of free peritoneal fluid has accumulated in the pouch of Douglas and, in the sagittal section, a floating intestinal loop is visible. The urinary bladder appears below. B, Soon after treatment with C1-INH concentrate, the amount of peritoneal fluid is somewhat decreased. C, Only a minimal amount of fluid is present in the pouch of Douglas 24 hours after C1-INH treatment. Several sonograms have previously been published in slightly different format.78., 81. Transverse panels B and C reprinted with permission from Acta Paediatrica.81 Sagittal panels A-C reprinted with permission from the European Journal of Gastroenterology and Hepatology.78
Edema of the portal veins, biliary ducts, and cholecyst wall, causing gross structural changes in the liver, was also observed. The liver parenchyma generally appeared less echogenic, whereas the walls of the portal vein radicles displayed increased echogenicity, resulting in a so-called starry sky texture that could be observed during the acute phase. Because of local edema, the pancreatic region also displayed an increased echogenicity ( Fig 8). In addition to the hepatic portal vein, the wall of the cholecyst was also echogenic ( Fig 9). After treatment with C1-INH concentrate, the former brightness disappeared, and the echo pattern of the liver returned to normal (sonogram not shown).
Fig 8.
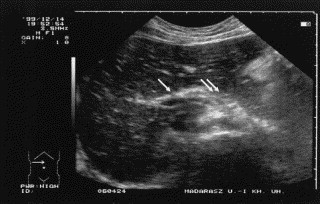
Transverse sonogram during an abdominal HAE attack: liver and pancreas. Increased hepatic reflection (starry sky liver) and thickened, echogenic portal veins (arrow); the pancreatic region is also hyperechoic (double arrows). Reprinted with permission from Acta Paediatrica.81
Fig 9.
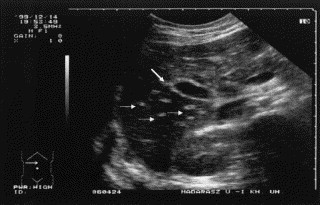
Transverse sonogram during an abdominal HAE attack: liver and cholecyst. Because of edematous swelling, the wall of the cholecyst is hyperechoic (large arrow) and the small portal veins are also more echogenic (small arrows) in contrast with the liver's overall decrease in echogenicity.
Discussion
Early recognition of acute abdominal attacks is of utmost importance because incorrect or delayed diagnosis often leads to unnecessary surgical intervention. In undiagnosed patients, ultrasound examination can be a differential diagnostic means for recognizing HAE in the abdominal organs because of its ability to detect nonspecific but sensitive clues such as thickening of the intestinal wall, free peritoneal fluid, intestinal hypermotility or hypomotility, and echo pattern changes of the liver and pancreas. Ultrasound examination has therefore proven very useful as a complementary, quick, and painless tool for recognizing the early phase symptoms of HAE. Patients presenting with skin symptoms (erythema marginatum) or acute pains, nausea, vomiting, or profuse diarrhea of unknown origins should be immediately hospitalized and investigated with ultrasound. Ultrasound follow-up in known cases of HAE is also capable of proving the efficacy and expeditiousness of acute attack treatment. In rare cases in which patients with known HAE present with abdominal symptoms unresponsive to C1-INH concentrate, ultrasonography may help distinguish between a refractory HAE attack and an unrelated surgical emergency.
Abdominal and pelvic ultrasound examination is a highly reproducible and informative diagnostic tool and thus is indicated during acute abdominal attacks of HAE unresponsive to C1-INH concentrate. Conversely, a search for HAE is warranted when the typical sonographic features are ascertained in a patient with abdominal symptoms.
Classifying HAE and AAE
(Kayla Williams, BS, MA, MFA, and Christoph Bucher, MD,∗ Cambridge, Mass, and Zürich, Switzerland)
Angioedema may be caused by reasons as various as allergies, inherited or acquired deficiencies of C1-INH, or drug reactions.82., 83. For the life of a patient presenting with unexplained airway swelling, the most important etiologic distinction is that between angioedema of allergic, histaminergic origins and the far rarer C1-INH–associated or nonallergic angioedema. When allergic angioedema has been ruled out, nonallergic angioedema is next determined to be hereditary or acquired, and subclassification is pursued.
Allergic angioedema, with histamine as its major mediator, may best be defined by its clinical response to antiallergic drugs such as antihistamines and corticosteroids. In this type of angioedema, reaction of specific IgE antibodies with an allergen induces the release of histamine and other mediators from mast cells. It is often associated with urticaria. In contrast, angioedema caused by C1-INH deficiency is not known to be triggered by an allergic reaction, is not usually associated with hives, and likely has bradykinin as its principal mediator.
Current systems for classifying HAE and AAE describe the disorders in terms of C1-INH deficiency type. Although observed convention supports the classification of major types, some further classifications, such as AAE types, are more fluid. In the case of the more recently described estrogen-sensitive angioedema, a new formal description is suggested here. In the interest of both definition and the elucidation of mechanism that these differences imply, the divisions of HAE and AAE type are presented. For an example of prevalence, Table III presents Agostoni's 573-patient angioedema case series by type.
Table III.
Large nonallergic angioedema case series classified by type
| Classification | Number of patients N = 573 |
|---|---|
| HAE-I | 356 (62.1%) |
| HAE-II | 85 (14.8%) |
| ACE inhibitor–related angioedema | 64 (11.2%) |
| Idiopathic nonhistiminergic angioedema | 43 (7.5%) |
| AAE (with or without antibodies) | 25 (4.4%) |
HAE: Types I and II
(Angelo Agostoni, MD, Konrad Bork, MD,∗ Bettina Fischer, MD, C. Erik Hack, MD, PhD,∗ Christian Drouet, PhD,∗ Alvaro Blanch,∗ Olga Roche,∗ Nicole Monnier, PhD, Christiane Duponchel, Lajos Kalmár, Attila Tordai, MD, PhD,∗ Emanuela Pappalardo, PhD,∗ Roberto Perricone, MD, Margarita Lopez-Trascasa, MD, PhD,∗ Lorenza Zingale, MD,∗ and Marco Cicardi, MD,∗ Milan and Rome, Italy, Mainz, Germany, Amsterdam, The Netherlands, Grenoble and Rouen, France, Madrid, Spain, and Budapest, Hungary)
Hereditary angioedema related to C1 inhibitor deficiency is a well-defined autosomal dominant trait. Its variants include types I (HAE-I) and II (HAE-II), associated with mutations of the C1 inhibitor gene (C1NH or SERPING1),84., 85. and a newly described type not associated with C1-INH deficiency8., 86., 87. further defined and discussed in another section.
The disease results from a large variety of mutations of the C1NH gene, located in the q12-q13.1 subregion of chromosome 11. According to the relative concentrations of antigenic and functional C1-INH, 2 types of HAE have traditionally been described.88 The defective gene produces either no C1-INH (HAE-I) or a dysfunctional C1-INH (HAE-II).6., 84., 85., 88. In either case, it is associated with low functional activity of C1-INH, low levels of C4, and normal levels of the third component of the complement cascade (C3). Concentrations of C1q, other than during angioedema attacks, are normal.
In HAE-I (∼85% of patients with C1-INH–associated HAE), defective expression of 1 allele results in low antigenic and functional concentrations of C1-INH.
In HAE-II (∼15% of patients with C1-INH–associated HAE), concentrations of functional C1-INH are low, but C1-INH antigenic levels are normal or increased, with the presence of a dysfunctional mutant protein.88
For both, C1-INH function is usually 5% to 30% of normal, instead of the 50% expected if the single normal allele were fully expressed. This difference is ascribed to permanent C1 and contact phase activation, with subsequent C1-INH consumption in the periphery.89., 90. Interestingly, the description of low levels of nonfunctional C1-INH mutants in patients with HAE-I has demonstrated that the distinction between HAE-I and HAE-II is not absolute.91 This finding occurred in patients with mutations to exon 8 at the carboxy terminus of the C1NH gene, thought to be responsible for the proper folding necessary for transport outside of the cell and exposure of the reactive site loop. Thus, although these patients with low antigenic concentrations of C1-INH appear to have HAE-I, they are in fact expressing nonfunctional C1-INH that cannot efficiently exit the cell.
Estrogen-dependent and estrogen-associated inherited angioedema (previously HAE type III)
(Karen Binkley, MD, FRCPC, and Alvin E. Davis III, MD, Toronto, Canada, and Boston, Mass)
A type of angioedema, to date manifest only in women, has recently been described.8., 86., 87. Its symptoms closely resemble those associated with functional C1-INH deficiency but occur in the presence of normal C1-INH concentrations; the genetic defect responsible is currently unknown. Although this type of angioedema has been referred to as HAE type III (Online Mendelian Inheritance in Man [OMIM] 300268), others have argued that this designation is both redundant and misleading. The following piece by Binkley and Davis explores their work in a kindred with estrogen-dependent inherited angioedema, more fully describes estrogen-sensitive forms of inherited angioedema, and proposes a rational system of nomenclature.
Overview
The authors investigated a family with symptoms of angioedema restricted to conditions of high estrogen levels. Although this investigation was undertaken to provide better care for the affected family members, it also presented a unique opportunity to better understand the effects of estrogen and androgens on C1-INH.8 However, instead of altered hormonal regulation of C1-INH, this family seemed to possess a completely novel abnormality, as suggested by the absence of identifiable mutations in either the coding or the 5′ regulatory regions of the C1NH gene8 and normal C1-INH function and activity in a pregnant, symptomatic family member.92 The exact mechanisms responsible for angioedema in these patients have yet to be identified.
The importance of kinin degradation pathways and aminopeptidase P (APP) in the control of angioedema generation has been independently recognized in studies of angiotensin-converting enzyme (ACE) inhibitor–related angioedema. Bradykinin and its active metabolite, des-Arg-bradykinin, are metabolized largely by 2 enzymes, ACE and APP.93., 94., 95. With ACE inhibitor administration, APP becomes the primary enzyme responsible for inactivating bradykinin and des-Arg-bradykinin. In fact, individuals with low plasma concentrations of APP appear to be predisposed to developing angioedema during ACE inhibitor treatment, when neither ACE nor APP is available to inactivate these kinins.96
Kinin inactivation pathways might also modulate clinical symptoms in classic HAE. For example, decreasing kinin inactivation in patients with HAE with the use of ACE inhibitors can result in exacerbation of angioedema.97., 98., 99., 100. Given the important contribution of kinin inactivation pathways to the control of angioedema, this may be an avenue for further investigation.
Case histories and investigation of the index family
The index family presented with histories of episodic, HAE-like angioedema.8 These episodes occurred only during pregnancy, OC use, or estrogen replacement therapy. Symptoms began 14 to 21 days after conception, or within 7 to 14 days of starting endogenous hormones. No episodes occurred in the postpartum period. One patient's description was particularly compelling: “My period was just a day or two late, but when one side of my face swelled up, I knew I must be pregnant, because this is just like what happened to my mother and sisters every time they were pregnant.” In affected individuals, symptoms occurred in all pregnancies and with each course of estrogen therapy. Unaffected individuals had no symptoms at any time. There were 8 affected women in 3 generations and 1 obligate male carrier. Transmission was consistent with an autosomal dominant inheritance. Complement values, C1-INH, C1-INH function, prekallikrein, factor XII, and high molecular weight kininogen were normal in 3 patients during asymptomatic periods.
Genetic investigations were undertaken for the following reasons: (1) the patients were asymptomatic at the time of presentation, (2) baseline biochemical investigations were unremarkable, and (3) exposing patients to estrogens for the purpose of detecting resultant biochemical abnormalities was unethical in light of the risk of laryngeal edema.
The striking clinical similarity to classic HAE focused initial investigations on the C1NH gene. However, no abnormalities in the coding sequences of the C1NH gene or in the 5′ regulatory region were detected.8
When patient III-24 became pregnant and developed recurrent angioedema, biochemical investigations were undertaken.92 C1-INH antigen and function were both normal. The mechanism by which increased estrogens precipitate symptoms thus remains under investigation.
Related phenotypes: HAE with normal C1 inhibitor activity in women
Most of the 36 women with angioedema in 10 families reported by Bork et al86 appeared to have a phenotype different than that of estrogen-dependent angioedema, because only 1 of 36 patients had attacks exclusively during pregnancy. In 10 of 36 patients, attacks occurred more frequently during OC use but were not limited to these periods. By extrapolation, 15 of these 36 patients had angioedema apparently unrelated to use of OCs or pregnancy. Age of onset of symptoms in the patients of Bork et al86 was variable and was not reported as directly correlating with onset of exogenous estrogen use or pregnancy. Symptoms in at least 1 patient started as early as 1 year of age, before significant hormonal effects were likely as the authors note. These features are in sharp contrast with those of patients with estrogen-dependent inherited angioedema, in whom episodes of angioedema occurred exclusively during pregnancy or exogenous estrogen therapy, and suggest that a different underlying defect might be responsible for the different phenotypes.
In the women described by Bork et al,86 C1-INH and C4 levels were normal in the affected individuals without symptoms. Normal measurements of C4 and C1-INH during symptomatic periods were also obtained in some individuals.86 Other pedigrees have also been reported.87
Nomenclature
Until further biochemical and molecular genetic studies elucidate the underlying defects in these pedigrees, it remains unclear whether the different pedigrees represent subtle abnormalities in the same underlying pathway or distinct biochemical and clinical entities. Therefore, affected patients can currently be classified only on the basis of phenotype, without reference to the underlying defect.
This has implications for the nomenclature applied to these conditions. The term HAE type III may be misleading because it implies that these patients have a defect similar to HAE-I (inadequate C1-INH concentration) and HAE-II (inadequate C1-INH function). This is clearly not the case, because C1-INH concentration and function are normal in several pedigrees.92., 101. Further confusion arises because the term HAE type III had been previously suggested to apply to a form of angioedema resulting from inadequate C1-INH function caused by a mutation resulting in inappropriate binding to albumin.102., 103. Although HAE type IV was suggested for the patients of Bork et al86 to address this latter concern,103 the term still erroneously implies a defect in C1-INH function. The authors thus suggest that patients should be categorized on the basis of their phenotype and recommend the terms estrogen-dependent inherited angioedema and estrogen-associated inherited angioedema 92., 101. until molecular studies suggest an alternate, rational nomenclature.
Clinical implications
Further studies are required to identify the factors that contribute to angioedema in patients with estrogen-dependent angioedema. Unaffected family members might then be identified through biochemical or genetic assays so that they might use OCs or plan pregnancies freely. Identification of affected family members would allow these individuals to avoid OCs, bypassing a trial of therapy and the attendant risk of laryngeal edema. Should effective treatment became available, affected individuals wishing to use OCs or become pregnant could begin treatment prophylactically or, at least, ensure its availability beforehand.
If a particular factor is conclusively shown to be reduced in these patients, symptomatic individuals might be treated by replacing the missing factor. Other possible treatments include novel strategies to reduce kinin formation or enhance kinin inactivation.
Prenatal diagnosis of fetal status (affected or unaffected) might also be relevant to the management of pregnancy in these individuals. The reported kindred showed significant variation in symptom severity during pregnancy, with some individuals experiencing relatively mild symptoms. In at least 1 affected individual, it is likely that symptoms during a pregnancy with an affected fetus (identified as such only later in life) were accurately recalled as being particularly severe (Binkley, Unpublished data, March 2000). It is interesting to speculate that an affected fetus would not provide the missing factor to the affected pregnant mother, and this might explain the severity of the symptoms. Conversely, an unaffected fetus might act as a source of the otherwise missing factor during pregnancy and might mitigate symptoms in an affected pregnant mother. If pregnancies could be identified early as being at high or low risk for severe angioedema on the basis of fetal status, follow-up and management could be guided accordingly.
At least 1 direction for further study of the mechanisms responsible for symptoms in patients with estrogen-associated angioedema is suggested by the reduced kinin inactivation in ACE inhibitor–associated forms of angioedema. Elucidation of the defect responsible for this phenotype would allow better diagnosis and possibly specific treatment. General strategies to reduce kinin formation and/or enhance inactivation might also be helpful for the amelioration of symptoms.
Concerning HAE-I and HAE-II, just as variations in serum concentrations of APP appear to determine which individuals in a normal population develop angioedema with a second perturbation of kinin metabolism, such as the use of ACE inhibitors,96 it could be speculated that variations in either kinin activation or inactivation pathways might contribute to the differences in severity of angioedema in individuals with a pre-existing perturbation in kinin metabolism, such as a mutation in C1-INH (as occurs in HAE). Thus, it is possible that some of the variation in symptom severity seen between different members of the same family, carrying the same C1-INH mutation, comes from variation in other kinin pathways. Identification of the defects in estrogen-dependent and estrogen-associated angioedema might illuminate potential candidate factors.
Knowledge of kinin production and inactivation pathways and how they are influenced by sex hormones may also offer insight into some perplexing issues regarding the effects of sex hormones on C1-INH values and angioedema symptoms in HAE. Androgens are effective in reducing episodes of angioedema and are used clinically for this purpose in HAE.104., 105. Although androgens increase plasma concentrations of C1-INH,105 the amount of C1-INH increase does not correlate well with symptom diminution.106 It is tempting to speculate that androgens may also increase kinin inactivation pathways, and this, perhaps in combination with slightly higher amounts of C1-INH, contributes to the observed reduction in angioedema with androgen therapy. Further studies will be necessary to explore this possibility as well.
Use of estrogen therapies typically results in some lowering of plasma C1-INH concentration in normal individuals,107 and use of estrogen therapy tends to exacerbate angioedema in patients with HAE.31 However, during pregnancy, estrogen concentrations are high, C1-INH concentrations decrease,108., 109., 110. and paradoxically, episodes of angioedema may decrease, especially in late pregnancy.31 These puzzling observations have long suggested that a second mechanism is important in controlling angioedema. Kinin inactivation pathways may be one such mechanism. Speculation about possible mechanisms of symptom reduction in pregnancy suggests potential fruitful areas for further study. For example, are there hormonal factors in pregnancy, not operative during estrogen therapy, that increase kinin inactivation or other factors and reduce angioedema, despite an estrogen-induced lowering of C1-INH? Is the fetus or placenta a source of kinin inactivation factors or other factors that mitigate the effects of estrogen-induced lowering of C1-INH? Does variation in fetal production of kinin-inactivators or other factors underlie any variation in angioedema symptoms between pregnancies in the same individual, or between individuals in the same families, all with the same C1-INH mutation?
Acquired angioedema is typically caused by ACE inhibitor treatment, and less commonly is caused by autoantibodies directed at C1-INH. General strategies to reduce kinin formation or/and increase kinin inactivation, identified through characterization of the elements of these pathways as well as their regulation, may be applicable to these patients as well.
Moving ahead
The discoveries of estrogen-dependent and estrogen-associated inherited angioedema are likely to focus attention on mechanisms other than C1-INH that control angioedema. Pathways involving kinin production and inactivation may be fruitful areas of further study in these conditions, a better understanding of which might provide new therapeutic opportunities potentially relevant to all types of angioedema.
AAE: Types I and II and subcategories
Angioedema may be acquired, mainly in association with lymphoproliferative disorders or occasionally with autoimmune, neoplastic, or infectious diseases.7 AAE also includes various other types of secondary C1-INH deficiency, angioedema caused by certain antihypertensive medications, urticaria-associated angioedema, and idiopathic angioedema.82., 83. In the laboratory, AAE is characterized by low functional C1-INH, low amounts of C4, and normal amounts of C3. Concentrations of C1q are often very low.
Angioedema caused by acquired C1-INH deficiency: Type I and type II distinguished.
(Marco Cicardi, MD∗, Andrea Zanichelli, Laurence Bouillet, MD, CCA,∗ and Emel Aygören-Pürsün, MD, Milan, Italy, Grenoble, France, and Frankfurt, Germany)
In this section, Cicardi et al review the current classifications of AAE and discuss the possible pathogenic mechanisms on which these distinctions are ostensibly based.
Angioedema caused by acquired deficiency of the inhibitor of the first component of human complement (C1-INH), usually referred to as acquired angioedema, is a rare, life-threatening disease first described by Caldwell et al.111 Characteristic of acquired C1-INH deficiency are the increased consumption of C1-INH and the hyperactivation of the classical pathway of human complement.112 As a consequence, these patients have almost undetectable serum levels and/or activity of C1-INH, C4, C2, and C1q, r, and s. Usually, these abnormalities are constantly present, but temporary normalization of 1 or more of these parameters has been reported.113
The clinical manifestations of the disease mimic those of the inherited defect of C1-INH and include subcutaneous, nonpruritic swelling without accompanying urticaria; involvement of the upper respiratory tract manifested as dysphagia, voice change, or respiratory stridor; and partial obstruction of the gastrointestinal tract presenting as colicky abdominal pain.7 Angioedema caused by acquired C1-INH deficiency differs from HAE by the absence of a family history of angioedema and a late onset of symptoms (in the fourth decade of life or later). Response to treatment varies compared with HAE caused by the C1-INH hypercatabolism characteristic of acquired C1-INH deficiency.114
Acquired C1-INH deficiency is frequently reported in association with B lymphoproliferative diseases. Different forms of B lymphoproliferation can occur, ranging from benign monoclonal gammopathies of undetermined significance (MGUS) to true malignancies.115 Neoplastic lymphatic tissues have been shown to consume C1-INH116 and/or classical pathway complement components,117 suggesting that they were directly involved in the pathogenesis of acquired C1-INH deficiency. Scattered reports describe acquired C1-INH deficiency associated with nonhematologic neoplasm, infections, or autoimmune diseases, whereas 14% of patients with acquired C1-INH deficiency have no other disease.105., 118., 119., 120., 121., 122., 123., 124., 125., 126. Bouillet et al in Grenoble recently observed an acquired C1-INH deficiency state via liver transplantation (Bouillet et al, Personal Communication, May 2003). The liver donor did not have a history of angioedema but was of unknown C1-INH status. It is speculated that a C1-INH deficiency might have been present.
In 1986, autoantibodies inactivating C1-INH were first detected in patients with acquired C1-INH deficiency.127 Initially, autoantibodies inactivating C1-INH were identified in otherwise healthy patients. On the basis of this observation, it was proposed that 2 separate forms of acquired C1-INH deficiency existed: type I, paraneoplastic, mainly associated with lymphatic malignancies; and type II, autoimmune, caused by autoantibodies to C1-INH. The latter form appeared to be characterized further by elevated serum levels of cleaved C1-INH.128., 129. Because cleaved C1-INH was not invariably found to be present in the serum of patients with so-called autoimmune acquired C1-INH deficiency,115 this division has been questioned.130., 131. Furthermore, autoantibodies to C1-INH were later described in patients with associated diseases. These autoantibodies were found to be common in patients with MGUS and frequently exhibit the same isotype of the M component.115., 132., 133. Autoantibodies to C1-INH impair C1-INH function. Although the exact mechanism for such impairment remains controversial,7., 122. the majority of these autoantibodies appear to enhance C1-INH cleavage by target proteases, preventing their inactivation. A recent article on 23 patients with acquired C1-INH deficiency followed for as long as 24 years (median, 8 years) demonstrated that half of the patients with malignancies also had autoantibodies to C1-INH, either at the time of onset of angioedema or later in the course of disease, indicating that autoimmune acquired C1-INH deficiency is not distinct from the acquired C1-INH deficiency that occurs in the setting of malignancies or other diseases. Detection of autoantibodies to C1-INH in a patient with acquired C1-INH deficiency should not decrease the importance of considering the possibility of an associated pathologic condition. Compared with the general population, patients with acquired C1-INH deficiency presented higher risk for B-cell malignancies. In patients with acquired C1-INH deficiency, the risk for progression of MGUS to malignancy was not higher than in other patients with MGUS.134
AAE: Further distinctions
(Angelo Agostoni, MD, Milan, Italy)
New causes of AAE, especially drug-related AAE, are still being discovered, posing the question whether all types of AAE share a common biomechanism if not a common etiology. In the descriptive sections that follow, Agostoni surveys several classes of AAE by cause.
Idiopathic nonhistaminergic angioedema. Cicardi et al135 describe a subset of angioedema patients having normal complement values, no history of provoking drug treatment, and who are unresponsive to antihistamines. This condition, with a clinical presentation similar to that of C1-INH deficiency, is deemed idiopathic nonhistaminergic angioedema. It is possible that this classification might overlap, at least in part, with that of estrogen-sensitive angioedema.
ACE inhibitor-related angioedema. Angioedema may be a consequence of an adverse drug reaction not induced by an allergic or parallergic mechanism.136 ACE inhibitor–related angioedema occurs in 0.1% to 0.5% of patients taking the drug. Decreased bradykinin degradation is implicated because ACE, also known as kinase II, activates both angiotensin I and bradykinin. ACE inhibitor–related angioedema may be an underestimated side effect because it can appear after years of ACE inhibitor use, thus obscuring its relationship with the drug.
Unlike patients with C1-INH deficiency, patients who develop ACE inhibitor–related angioedema show no evidence of the cleavage products of high molecular weight kininogen (HK) in their plasma, despite high plasma concentrations of bradykinin. Because the cleavage of HK generates bradykinin, the pathogenic mechanism of ACE inhibitor–related angioedema probably resides in the catabolic side of bradykinin metabolism instead.16
When ACE is inhibited, APP plays a major role in plasma bradykinin catabolism. To identify patients at risk of developing angioedema during ACE inhibitor treatment, Adam et al96 evaluated blood concentrations of APP. Their results indicated lower plasma concentrations of APP in patients who had previously had ACE inhibitor–associated angioedema, suggesting an inverse relationship between APP concentration and the tendency to develop angioedema.
It is evident that ACE inhibitor use should be avoided in patients with hereditary or acquired C1-INH deficiency.
Angioedema related to other drugs. Rare instances of angioedema have been reported with angiotensin II (AT2) receptor antagonsists,137 although this adverse effect seems to occur less frequently with AT2 receptor antagonists than with ACE inhibitors.136 It is unknown whether the 2 adverse drug reactions share the same mechanism.
Scattered reports have suggested the possibility of angioedema associated with the use of estrogens, fibrinolytic agents, psychotropic agents, and antihypertensives other than ACE inhibitors.
HAE: genetic investigations
(Christian Drouet, PhD,∗ Alvaro Blanch,∗ Olga Roche,∗ Nicole Monnier, PhD, Christiane Duponchel, Lajos Kalmár, Attila Tordai, MD, PhD,∗ Emanuela Pappalardo, PhD,∗ Roberto Perricone, MD, Lorenza Zingale, MD,∗ and Margarita Lopez-Trascasa, MD, PhD,∗ Grenoble and Rouen, France, Madrid, Spain, Budapest, Hungary, and Milan and Rome, Italy)
The molecular diagnosis of angioedema is primarily based on evidence of the decrease or lack of C1-INH function, which is routinely stated by its control capacity toward the target protease C1s in spectrophotometric assays.138 Molecular sizing of circulating C1-INH can subsequently be assessed by SDS-PAGE and immunoblot.
As discussed in greater detail in the Pathogenesis and Pathobiology of HAE and AAE section, C1-INH controls several proteases, including C1r and C1s, the mannose-binding protein associated serine protease (MASP) system, kallikrein, coagulation factors XIIa and XIa, plasmin, and tissue plasminogen activator.139., 140., 141., 142., 143., 144. Therefore, C1-INH plays a key role in regulating the early steps of complement and the contact system of kinin formation.89 This broad inhibitory ability ensues from a property unique to the serpin class: highly efficient complex formation with target proteases.145 Thus, mutations of the C1NH gene typically affect many pathways. To add further complexity, many different C1NH mutations resulting in HAE have been discovered. Through the study of these mutations, it is hoped that more complete knowledge of the many functions of C1-INH can be gained, ultimately contributing to a better biochemical knowledge of HAE.
Mutation analysis of the C1NH gene
(Christian Drouet, PhD,∗ Alvaro Blanch,∗ Olga Roche,∗ Nicole Monnier, PhD, Christiane Duponchel, Lajos Kalmár, Attila Tordai, MD, PhD,∗ Emanuela Pappalardo, PhD,∗ Lorenza Zingale, MD,∗ Roberto Perricone, MD, and Margarita Lopez-Trascasa, MD, PhD,∗ Grenoble and Rouen, France, Madrid, Spain, Budapest, Hungary, and Milan and Rome, Italy)
In this section, Drouet et al review the methods currently available for detecting C1NH mutations and describe the powerful online mutation database that has grown out of such efforts.
C1NH gene
The C1NH gene maps to chromosome 11. Theriault et al,146 by using in situ hybridization in 1990, localized it to 11q11-q13.1; a year later, Janson et al147 mapped it to 11q12-q13.1. It consists of 8 exons distributed over a DNA stretch of 17 kb, with introns containing 17 repetitive Alu sequences148 ( Fig 10). The structural abnormalities in the C1NH gene in patients with HAE have been found to be very heterogeneous. Illustrative examples explain the generation of C1NH gene defects: large deletions or, less frequently, partial duplications involving Alu repeats distributed along the C1NH gene149., 150.; deletions resulting from a peculiar consensus sequence or an alternative secondary structure151; and mutations based on Cytosine-phosphate-guanine (CpG) methylation and subsequent cytosine deamination to thymine.91., 152. As shown in Table IV, more than 150 mutations have been reported in unrelated patients, with pathogenic amino acid substitutions distributed over the entire length of the coding sequence.85., 153. In addition, the frequent de novo mutations in the C1NH gene underline the presence of multiple hot spots, including those containing a CpG dinucleotide.154 All of these lead to an apparent failure to synthesize or secrete functional C1-INH protein.
Fig 10.
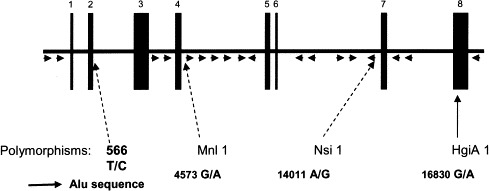
Structure of C1NH gene.85., 153.Arrowheads represent Alu elements.150 The positions of the 4 polymorphic sites are indicated by elongated arrows. Nucleotide numbers refer to the C1NH gene, with number 1 denoting the first nucleotide of exon 1. Accession number X54486.
Table IV.
Mutations in the C1NH gene that lead to HAE∗
| Region | Mutation (genomic nucleotide change) | Phenotype (predicted change) | HAE type | Reference |
|---|---|---|---|---|
| Promoter | −103C>T | ? | I | Verpy et al,172 1996 |
| −40C>G | ? | I | Verpy et al,172 1996 | |
| Exon 1 | None | |||
| Intron 1 | 564G>A | Splicing defect (−1) | I | Verpy et al,172 1996 |
| Exon 2 | 566T>C | ? | I | Verpy et al,172 1996; Cumming et al,183 2003 |
| 589_596dup 8 bp | Frameshift>stop eighth residue of the signal peptide sequence | I | Verpy et al,172 1996 | |
| 602_603insGA | Frameshift>stop ninth residue of the signal peptide sequence | I | Zuraw and Herschbach,84 2000 | |
| Intron 2 | 638G>A | Splicing defect (+1) | I | Kalmar et al,185 2003 |
| 642G>A | Splicing defect (+5) | I | Verpy et al,172 1996 | |
| 2194delA | Splicing defect (−2) | I | Pappalardo et al,154 2000 (neomutation) | |
| Exon 3 | 2238C>T nonsense | Q10X | I | Kalmar et al,185 2003 |
| 2250_2251delAG | Frameshift>stop 34 | I | Verpy et al,172 1996 | |
| 2264_2265delAG | Frameshift>stop 34 | I | Freiberger et al,175 2002 | |
| 2304delC | Frameshift>stop 56 | I | Freiberger et al,175 2002 | |
| 2353C>G nonsense | S48X | I | Freiberger et al,175 2002 | |
| 2394_2558del 165 bp | D62_T116del (423-residue protein lacking N-terminal region¶) | I-II | Bos et al,206 2003 | |
| 2458_2461delCAAC | Frameshift>stop 124 | I | Cumming et al,183 2003 | |
| 2467insA | Frameshift>stop 110 | I | Bowen et al,85 2001 | |
| 2490C>T nonsense | Q94X | I | Pappalardo et al,154 2000 (neomutation) | |
| 2533G>A missense | C108Y | I | Kalmar et al,185 2003 | |
| 2534_2535delCT | Frameshift>Stop 109 | I | Kalmar et al,185 2003 | |
| 2579_2620del 42 bp | L124_A137del | I | Kalmar et al,185 2003 | |
| 2589T>G missense | F127V | I | Bissler et al,177 1997 | |
| 2608A>G missense | H133R | I | Bissler et al,177 1997 | |
| 2650T>C missense | F147S | I | Verpy et al,172 1996 | |
| 2652T>C missense | S148P | I | Pappalardo et al,154 2000 (neomutation) | |
| 2656C>T missense | P149L | I | Verpy et al,172 1996 | |
| 2674T>C missense | L155P | I | Bissler et al,177 1997 | |
| 2679_2680insTT | Frameshift>stop 189 | I | Verpy et al,172 1996 | |
| 2694G>A | Splicing defect (+1) or G162R | I | Verpy et al,172 1996; Pappalardo et al,154 2000; Zuraw and Herschbach,84 2000 (including neomutation) | |
| Intron 3 | 2695G>A | Splicing defect (+2) | I | Kalmar et al,185 2003 |
| 2696_2697insT | Splicing defect | I | Kalmar et al,185 2003 | |
| Exon 4 | 4351G>A missense | G162E | I | Zuraw and Herschbach,84 2000 |
| 4371delA | Frameshift>stop 188 | I | Zuraw and Herschbach,84 2000 | |
| 4371A>G missense | T169P | I | Bissler et al,177 1997 | |
| 4372_4373insA | Frameshift>stop 234 | I | Verpy et al,172 1996 | |
| 4395delT | Frameshift>stop 188 | I | Bissler et al,177 1997 | |
| 4400delC | Frameshift>stop 188 | I | Verpy et al,172 1996 | |
| 4414G>A missense | C183Y, destroys disulfide bridge | I | Zuraw and Herschbach,84 2000 | |
| 4428delC | Frameshift>stop 188 | I | Zuraw and Herschbach,84 2000 | |
| 4453T>A missense | V196D | I | Zuraw and Herschbach,84 2000 | |
| 4460_4461insA | Frameshift>stop 234 | I | Zuraw and Herschbach,84 2000 | |
| 4467C>T nonsense | Q201X | I | Kalmar et al,185 2003 | |
| Exon 5 | 8346T>C missense | F214S | I | Verpy et al,172 1996 |
| 8370AC>T | Frameshift>stop 229 | I | Pappalardo et al,154 2000 (neomutation) | |
| 8377C>A missense | S224R | I | Bissler et al,177 1997 | |
| 8383delC | Frameshift>stop 229 | I | Verpy et al,172 1996 | |
| 8390delC | Frameshift>stop 229 | I | Verpy et al,172 1996 | |
| 8448_8449insAACAC | Frameshift>stop 258 | I | Bissler et al,434 1994 | |
| 8455_8457delCAA | N250del | I | Bissler et al,434 1994; Verpy et al,172 1996 | |
| 8456_8458delAAG | K251del,‡ creates an N-glycosylation site | II | Parad et al,435 1990; Zuraw and Herschbach,84 2000 | |
| 8457_8459delAGA | K251del,‡ creates an N-glycosylation site | II | Bissler et al,434 1994 | |
| 8459A>G missense | I252V | I | Zuraw and Herschbach,84 2000 | |
| 8493_8494delCC | Frameshift>stop 281 | I | Freiberger et al,175 2002 | |
| 8517T>C missense | I271T | I | Pappalardo et al,154 2000 (neomutation) | |
| Intron 5 | 8721A>G | Splicing defect (+2) | I | Bowen et al,85 2001 |
| Exon 6 | 8735C>A missense | T279K | I | Bissler et al,177 1997 |
| 8735C>T missense | T279I | I | Bissler et al,177 1997 | |
| 8737_8739delACA | T280del | I | Pappalardo et al,154 2000 (neomutation) | |
| 8741T>G missense | F281C, introduces a fifth Cys residue | I | Verpy et al,172 1996 | |
| 8755A>G missense | T286A | I | Cumming et al,183 2003 | |
| 8770T>C missense | F291L | I | Siddique et al,436 1995 | |
| 8779_8784del 6 bp | K294_N295del | I | Bissler et al,177 1997 | |
| 8807T>C missense | M303T | I | Verpy et al,172 1996; Pappalardo et al,154 2000 (neomutation) | |
| 8814_8815delCA | Frameshift>stop 314 | I | Verpy et al,172 1996 | |
| 8823C>G nonsense | Y308X | I | Verpy et al,172 1996 | |
| Intron 6 | 8863G>T | Splicing defect (+1) | I | Siddique et al,437 1991 |
| 14019_14047del 29 bp | Splicing defect | I | Verpy et al,172 1996 | |
| 14029_14032AGGT>GCA | Splicing defect (−2) | I | Verpy et al,172 1996 | |
| 14030G>A | Splicing defect (−1) | I | Cumming et al,183 2003 | |
| 14030G>C | Splicing defect (−1) | I | Verpy et al,172 1996; Cumming et al,183 2003 | |
| Exon 7 | 14037C>T nonsense | Q324X | I | Verpy et al,172 1996 |
| 14070delA | Frameshift>stop 341 | I | Pappalardo et al,154 2000 (neomutation) | |
| 14082C>T nonsense | Q339X | I | Ono et al,438 1996 | |
| 14103G>T nonsense | E346X | I | Bissler et al,177 1997 | |
| 14107delA | Frameshift>stop 374 | I | Kalmar et al,185 2003 | |
| 14181A>C missense | T372P | I | Verpy et al,172 1996; Kalmar et al,185 2003 | |
| 14196C>T missense | P377S | I | Bissler et al,177 1997 | |
| 14196C>G missense | P377A | I | Bissler et al,177 1997 | |
| 14206A>T missense | K380I | II | Bissler et al,177 1997 | |
| 14224A>T missense | D386V | I | Kalmar et al,185 2003 | |
| 14233C>A nonsense | S389X | I | Pappalardo et al,154 2000 (neomutation) | |
| Intron 7 | 14252T>A | Splicing defect (+2) | I | Kawachi et al,439 1998 |
| Exon 8 | 16655delT | Frameshift>stop 408 | I | Frangi et al,440 1991 |
| 16657delC | Frameshift>stop 408 | I | Zhi et al,441 2003 | |
| 16661_16662insA | Frameshift>stop 404 | I | Frangi et al,440 1991 | |
| 16676_16677delTG | Frameshift>stop 449 | I | Freiberger et al,175 2002 | |
| 16698delC | Frameshift>stop 427 | I | Siddique et al,442 1992 | |
| 16707delT | Frameshift>stop 427 | I | Blanch et al,178 2002 | |
| 16708delC | Frameshift>stop 427 | I | Pappalardo et al,154 2000 (neomutation) | |
| 16720A>G missense | H421R | I | Zuraw and Herschbach,84 2000; Blanch et al,178 2002 (neomutation) | |
| 16735_16737AAC>T | Frameshift>stop 449 | I | Pappalardo et al,154 2000 (neomutation) | |
| 16742_16743insA | Frameshift>stop 450 | I | Blanch et al,178 2002 | |
| 16749_16775dup 27 bp | G431_V439dup | I | Kalmar et al,185 2003 | |
| 16749_16750insTGT | G431V and insW432 | II | Siddique et al,443 1993 | |
| 16752_16785del 34 bp | Frameshift>stop 513 | I | Bissler et al,151 1994 | |
| 16753T>A missense | V432E‖ | II | Davis et al,159 1992 | |
| 16759C>A missense | A434E‖ | II | Skriver et al,444 1991; Siddique et al,445 1991; Blanch et al,178 2002 | |
| 16764G>A missense | A436T¶ | II | Levy et al,446 1990; Davis et al,159 1992; Aulak et al,161 1993 | |
| 16765C>T missense | A436V | II | Frangi et al,447 1992; Siddique et al,448 1992 | |
| 16770T>C missense | S438P | II | Blanch et al,178 2002 | |
| 16771_16790dup 20 bp | Frameshift>stop 531 | I | Bissler et al,151 1994; Ernst et al,189 1996 | |
| 16774C>T missense | A439V | I | Bissler et al,177 1997 | |
| 16775_16776insC | Frameshift>stop 450 | I | Blanch et al,178 2002 | |
| 16786C>T missense | A443V§ | II | Zahedi et al,160 1995; Zahedi et al,28 1997 | |
| 16788C>T missense | R444C,§ introduces a fifth Cys residue | II | Ariga et al,187 1989; Skriver et al,152 1989; Donaldson and Bissler,449 1992; Bissler et al,177 1997; Nielsen et al,450 1998; Blanch et al,178 2002; Pappalardo et al,154 2000; Freiberger et al,175 2002; Kalmar et al,185 2003 (including neomutation) | |
| 16788C>A missense | R444S§ | II | Aulak et al,451 1990; McPhaden et al,181 1991; Eldering et al,191 1992 | |
| 16789G>A missense | R444H§ | II | Siddique et al,448 1992; Eldering et al,191 1992; Bissler et al,177., ∗, † 1997; Zuraw and Herschbach,84 2000; Freiberger et al,175 2002; Blanch et al,178 2002 (including neomutation); Cumming et al,183 2003 | |
| 16789G>T missense | R444L§ | I-II | Frangi et al,447 1992; Bissler et al,177 1997; Pappalardo et al,154 2000; Zuraw and Herschbach,84 2000; Blanch et al,178 2002 (including neomutation) | |
| 16789G>C missense | R444P | II | Blanch et al,178 2002 | |
| 16791A>C missense | T445P | II | Ocejo-Vinyals et al,452 1995 | |
| 16809G>A missense | V451M¶ | I-II | Verpy et al,91 1995; Eldering et al,155 1995; Verpy et al,172 1996 | |
| 16810T>G missense | V451G | I | Blanch et al,178 2002 | |
| 16810T>A missense | V451E | I | Kalmar et al,185 2003 (neomutation) | |
| 16812C>G missense | Q452E | I-II | Bissler et al,177 1997; Verpy et al,172 1996 | |
| 16822T>C missense | F455S¶ | I | Verpy et al,91 1995; Eldering et al,155 1995; Verpy et al,172 1996 | |
| 16827T>C missense | F457L | I | Bowen et al,85 2001 | |
| 16834T>C missense | L459P intracellular retention | I | Verpy et al,91 1995; Verpy et al,172 1996 | |
| 16834T>G missense | L459R intracellular retention | I | Verpy et al,91 1995; Verpy et al,172 1996 | |
| 16838G>A nonsense | W460X | I | Blanch et al,178 2002 | |
| 16842C>T nonsense | Q462X | I | Ariga et al,453 1993; Siddique et al,454 1993; Blanch et al,178 2002 | |
| 16858C>G missense | P467R intracellular retention | I | Verpy et al,91 1995; Verpy et al,172 1996 | |
| 16861T>A missense | V468D | I | Blanch et al,178 2002 | |
| 16867T>A missense | M470K | I | Bowen et al,85 2001 | |
| 16869G>A missense | G471R | I | Sugiyama et al,186 2001 | |
| 16870G>A missense | G471E | I | Blanch et al,178 2002; Kalmar et al,185 2003 | |
| 16872C>T nonsense | R472X intracellular retention | I | Verpy et al,91 1995; Verpy et al,172 1996; Bissler et al,177 1997; Zuraw and Herschbach,84 2000; Blanch et al,178 2002 (neomutation); Kalmar et al,185 2003 | |
| 16884C>T missense | P476S¶ | I-II | Verpy et al,91 1995; Eldering et al,155 1995; Verpy et al,172 1996 | |
| 16885C>G missense | P476R | I | Kalmar et al,185 2003 | |
| 16893T>A missense | X479R | I | Blanch et al,178 2002 | |
| Alu | Exons 1-3 | > 4 kb deleted | I | Stoppa-Lyonnet et al,150 1991 |
| Exons 1-4 | > 9 kb deleted | I | Stoppa-Lyonnet et al,150 1991 | |
| Segment incl exon 4 | 9 kb deleted | I | Kalmar et al,185 2003 | |
| Segment incl exon 4 | 4 kb deleted | I | Kalmar et al,185 2003 | |
| Segment incl exon 4 | 4 kb deleted | I | Kalmar et al,185 2003 | |
| Exon 4 | 1 kb deleted | I | McPhaden et al,181 1991 | |
| Exon 4 | 2 kb deleted | I | Pappalardo et al,154 2000 | |
| Exon 4 | 2.6 kb deleted | I | Stoppa-Lyonnet et al,455 1990; Stoppa-Lyonnet et al,150 1991 | |
| Exon 4 | 2.6 kb deleted | I | Stoppa-Lyonnet et al,150 1991 | |
| Exon 4 | 2.75 kb deleted | I | Stoppa-Lyonnet et al,150 1991 | |
| Exon 4 | 3.2 kb deleted | I | Stoppa-Lyonnet et al,455 1990; Stoppa-Lyonnet et al,150 1991 | |
| Exon 4 | 3.2 kb deleted | I | Stoppa-Lyonnet et al,455 1990; Stoppa-Lyonnet et al,150 1991 | |
| Exon 4 | 3.5 kb deleted | I | Kalmar et al,185 2003 | |
| Exons 4-6 | 8.5 kb deleted | I | Ariga et al,149 1990 | |
| Exons 4-6 | Unknown | I | Cumming et al,183 2003 | |
| Exons 5-6 | 5.5 kb deleted | I | Pappalardo et al,154 2000 | |
| Exons 5-8 | 17 kb deleted | I | Ariga et al,453 1993 (neomutation) | |
| Exon 7 | 1.7 kb deleted | I | Ariga et al,187 1989 | |
| Exon 7 | 1.7 kb deleted | I | McPhaden et al,181 1991 | |
| Exons 7-8 | Unknown | I | Cumming et al,183 2003 | |
| Exon 8 | 3.5 kb deleted | I | Stoppa-Lyonnet et al,150 1991 | |
| All exons | > 15 kb deleted | I | Duponchel et al,182 2001 |
The nucleic acid site and residue change are used to denote mutations. Nucleotide numbering after Carter et al,148 1991, with nucleotide 1 denoting the first nucleotide of exon 1. All of these naturally occurring mutations of the C1NH gene have been introduced in the HAEdb database (http://hae.biomembrane.hu).
Determined solely at the protein level.
Type II protein phenotypes have been characterized with functional outcomes as follows:
Multimerization and conversion to substrate.
Alteration in specificity.
Conversion to substrate or
multimerization only.
In the context of a normal steady-state C1-INH mRNA content in almost all cases of HAE-I and HAE-II, defective expression has been related to impaired protein secretion,91., 155. transinhibition of C1-INH translation,156 or extensive consumption.157 The consequences of several missense mutations have been determined at the functional and intracellular processing level by transfection of an in vitro mutagenized construct into COS cells. Point mutations help map amino acid residues critical for proper molecular folding and processing, with subsequent conversion of the serpin as a substrate,158 determination of target protease specificity,28., 159., 160. or spontaneous multimerization.89., 155., 161. Hence, many different mutations can lead to dysfunctional C1-INH, as recently reviewed.121., 162.
Strategies for mutation analyses of the C1NH gene
Molecular genetic analysis of C1NH gene anomalies in patients serves as a supplementary diagnostic tool for accurate diagnosis at the molecular level. It contributes to the understanding of DNA mutagenesis processes, protein folding and processing, and the structure-function relationships of the C1-INH serpin.85., 153., 154., 163. Such studies may also help the collection of population distribution data, potentially deepening understanding of the relationship between de novo mutation formation and distinct, independent founder effects in different geographical locations.
C1-INH deficiency is heterogeneous at the gene level and is caused by subtle changes affecting 1 or several nucleotides, large deletions, or duplications.84., 153., 164. This heterogeneity prompted the authors to develop suitable methods for the detection not only of mutations previously identified in probands but also of as yet unknown mutations. De novo C1NH mutations, including exon deletions, account for nearly 25% of cases of angioedema.154 This finding has implications relevant to the genetic epidemiology of and genetic counseling for this disease.
The following sections describe the principles, advantages, critical parameters, and drawbacks of some technical strategies, including successful developments in the detection of C1NH gene anomalies.
Scanning methods for point mutations or small deletions or insertions. Methods based on heteroduplex DNA and single-strand conformation analyses are of proven efficiency in the screening of large segments of genomic or complementary DNA.165., 166. Specific PCR amplification is first performed on exons and flanking intronic sequences or cDNA. Any variation detected is then sequenced to characterize the molecular change. In the following text, whenever possible, methods are accompanied by citations for reviews describing their advantages and limits.
Chemical cleavage of mismatches (CCM) technology is based on selective reactions of mismatched thymine and cytosine with OsO4 and hydroxylamine, respectively. The modified mismatched bases are subjected to piperidine cleavage reactions, and the resulting fragments are separated and identified by gel electrophoresis.167 This method is, in principle, well suited to detect mutations independent of the length and sequence composition of the examined region and capable of detecting nearly all single-base mismatches. It has been successfully developed in a large number of studies (reviewed in Ellis et al168), including those of the C1NH gene to detect pathogenic mutations and analyze its polymorphism.169
Even if all mutations could potentially be detected, the efficiency of chemical modification and cleavage of the CCM depends on type of the mispairing and the stability of the adjacent sequences. In consequence, some mismatches are poorly cleaved, with their mutations subsequently undetectable via CCM.170
The need for a method suited to identifying most anomalies in the C1NH gene, including point mutations and short and medium-sized deletions or insertions, prompted some groups to take advantage of CCM and fluorescent probes with the development of fluorescence-assisted mismatch analysis.154., 171., 172.
Denaturing HPLC (DHPLC) uses an alkylated nonporous poly(styrene-divinylbenzene) matrix and an amphiphilic ion gradient enabling separation of homoduplexes and heteroduplexes by means of ion-pair reverse-phase liquid chromatography (reviewed in Xiao and Oefner173). Illustrative examples of its application in C1NH gene studies are given in Fig 11. The most important advantage of the DHPLC method is its easy automation by using a mechanical sampler.
Fig 11.
Mutation analysis of the C1NH gene by DHPLC showing data with insertion, point mutation, and deletion. Red and black profiles correspond to patient and control samples, respectively. A, A 10-nucleotide insertion in exon 3 causes the presence of heteroduplexes before and after normal homoduplex. B, A point mutation (A → G) in exon 4 causes the presence of heteroduplexes in close vicinity to a normal homoduplex. C, A 5-nucleotide deletion in exon 5 causes the presence of heteroduplexes before normal homoduplex. Drouet et al, Unpublished data, August 2003.
Denaturing gradient gel electrophoresis is based on the migration of double-stranded DNA molecules through polyacrylamide gels containing linearly increasing concentrations of a denaturing agent (reviewed in Fodde and Losekoot174). C1NH-specific primers have been designed to amplify genomic DNA segments ranging from 171 to 408 bp.175 The amplified fragments identified as forming heteroduplexes were directly sequenced.
Single-stranded conformational analysis (SSCA) is an electrophoretic method using a nondenaturing gel, in which the mobility of heat-denatured single strands is dependent on their folding according to individual secondary structure formation. Since its introduction in 1989, it has gained popularity for its technical simplicity, low cost, and high sensitivity sufficient to detect most mutations (reviewed in Nataraj et al176). The method has been used in C1NH gene studies84., 177., 178. ( Fig 12).
Fig 12.
Analysis of C1NH gene mutations by SSCA. A, SSCA of fragment 2144-2360 including part of exon 3; lanes 1, 2, 4 HAE kindreds without mutations in this fragment; lane 3, mutation 2264-5delAG [R18fsX33]. B, SSCA of fragment 2482-2761 including part of exon 3; lane 1, healthy control; lanes 3, 4, HAE kindreds without mutations in this fragment; lane 2, mutation in exon 3: T2650C [F147S]. C, SSCA of fragment 8690-8895 including exon 6; lanes 1, 3, 4, HAE kindreds without mutations in exon 6; lane 2, exon 6 mutation: C8823G [Y308Stop]. Lopez-Trascasa et al, Unpublished data, June 2003.
Analysis of DNA rearrangements. Because large genomic rearrangements account for nearly 20% of the total spectrum of the observed changes (reviewed in Tosi153), methods adapted to analyzing DNA rearrangements are required every time the aforementioned strategies are unsuccessful.
Southern blot analysis. Southern blot analysis was first used for C1NH gene analysis by 2 groups who compared DNA from multiple members of families with HAE against DNA from unrelated patients, yielding the first indication that a defective structural gene was responsible for the disease.179., 180. Digestion of genomic DNA with BclI can detect most deletion/insertion boundaries, because the 17-kb–long C1NH gene lies within a 20-kb BclI fragment ( Fig 13). To localize gene alterations more precisely, other enzymes have been used in single or multiple digestions: BamHI, BglII, EcoRI, HindIII, SacI, SalI, PvuI, and PstI.150., 181.
Fig 13.
Southern blot analysis of patients with HAE-I carrying rearrangements in the C1NH gene. The outer lanes (N) contain control DNA of unaffected individuals and flank different heterozygous deletions/duplications. BclI-digested DNA was hybridized with a full-length cDNA probe. The 20-kb BclI fragment contains the 8 exons of the C1NH gene. Data from Stoppa-Lyonnet et al.150
Quantitative exon multiplex PCR. Southern analyses are difficult to set up and time consuming. To ensure complete molecular characterization of C1-INH deficiencies, a fluorescent multiplex assay has been constructed to amplify 5 exons of C1NH and 1 exon of the BRCA1 gene simultaneously.182 In brief, after C1NH exon amplification, the fluorescence intensities of C1NH exons are compared quantitatively with those of a control exon (eg, BRCA1 exon 5) under conditions in which template concentration is rate limiting. The method has been further validated in exploring large deletions and insertions.183 Reliable estimates of relative gene dosage can be obtained by comparing peak levels in the test DNA with those of appropriate controls in deletion/insertion situations182 ( Fig 14).
Fig 14.
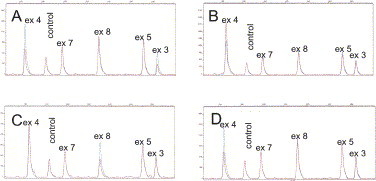
Quantitative fluorescence multiplex analysis: exon-specific multiplex PCR results for exons 3, 4, 5, 7, and 8 from C1NH and a control exon from BRCA1. Red and blue profiles correspond to the patient and control samples, respectively. Deletions of exons 3 and 4 (A), duplication of exon 4 (B), deletion of exon 8 (C), and deletion of exon 4 (D) are clearly visible. ex, Exon. Modified from Duponchel et al.182
C1NH promoter sequence analysis. The promoter of the C1NH gene is unusual because it contains no TATA sequence, but instead contains a TdT-like initiator element at nucleotides −3 to +5 and a polypurine-polypyrimidine tract between nucleotides −17 and −45.184 Only a few mutations have been reported to affect promoter sequence transcriptional activity, and among them, an interesting promoter variant (a C-to-T transition at position −103) was found in an exceptional family with recessive transmission of the disease.172 However, subsequent transcriptional alteration remains to be demonstrated. These anomalies have been successfully detected via mismatch scanning (fluorescence-assisted mismatch analysis) in reactions with external and internal primers specific for the promoter sequence.
Sequencing and expression of sequence data. The most widespread, gold standard technique for mutation detection is direct sequencing. This technique is now commonly available with a high degree of automation and a parallel decrease in labor and cost. The principle is based on dideoxy chain termination reactions using fluorescent dideoxy nucleotides followed by automated gel or capillary electrophoresis. Direct sequencing identifies the complete nucleotide sequence of a selected and PCR-amplified DNA fragment and detects heterozygosity.
The resulting data must be carefully interpreted. Any newly discovered missense mutation has to meet the following criteria: (1) the complete coding sequence and splice sites of the C1NH gene were screened/sequenced; (2) the new mutation is not present in 100 general population chromosomes; and (3) the new mutation segregates with the disease, ie, affected members carry the mutations whereas healthy relatives do not.
Novel interactive, locus-specific mutation database of the C1NH gene
Published C1NH gene mutations were periodically summarized in reviews,85., 153. but such collections became quickly outdated when several new studies described a multitude of novel mutations.154., 175., 178., 185., 186. C1NH gene mutations are also represented in large universal databases (OMIM 606860; Human Gene Mutation Database 119041), but these databases update their contents infrequently, with poor interactions and heavy requests. One of the goals of the European Concerted Action (PREHAEAT) is to perform systematic analyses of C1NH gene mutations in several laboratories for structure-function relationships and consequence on disease expression. To achieve this goal, a mutation database (http://hae.biomembrane.hu) was created with the following purposes: (1) to help the comprehensive collection of information on genetic alterations of the C1NH gene, (2) to create a database in which data can be searched and compared, and (3) to provide additional help in deciding whether a new mutation segregates with the disease.
C1-INH serpin function defect: Contributions from understanding C1NH mutations
The need to ascertain the consequence of C1NH mutation on C1-INH expression and function prompted investigators to develop experimental systems adapted to both transcription and protein expression studies. Patient peripheral blood monocytes and fibroblasts were successfully developed to study C1-INH synthesis in pathological conditions.156., 187., 188., 189. It then became interesting to correlate C1NH gene anomalies with transcription and translation defects, specifically with reference to serpin function, without the need for patient cells. Pathological mutations can now be introduced in C1-INH cDNA expression systems and transfected into cell lines suitable for biosynthesis experiments.91., 155., 190., 191., 192., 193.
Serpin function is routinely assessed in plasma samples (see Varga et al in the following Laboratory Diagnosis section). Some type II mutant C1-INH proteins have been described with respect to their atypical interactions with the target protease.161 A more complete model of nonfunctional C1-INH mutants can yield insights into the C1-INH inhibitory mechanism and aid in the development of a relevant dynamic 3-dimensional model of the C1-INH molecule.162 Knowledge of these type II mutant C1-INH proteins allows every mutation to be associated with its corresponding serpin control failure, as proposed in a 3-category classification scheme162: class I includes mutations that lead to altered exposure of the active site, with consequences on protease specificity (denoted § in Table IV); class II mutations convert C1-INH protein into a substrate, with subsequent inefficient protease trapping (denoted ‡ and ‖ in Table IV); and class III encompasses mutations with a spontaneous insertion of the reactive loop, either into the same molecule or another molecule entirely (denoted ‖ and ¶ in Table IV). Finally, as posited by Cumming et al,183 it is important to correlate mutations with disease expression. However, on the basis of most clinical data, disease expression cannot be attributed to specific mutant proteins. Variable clinical presentation is thought to result from genetic or nongenetic elements distinct from the C1NH gene.
In laboratory practice
Every index case characterized by impaired C1-INH serpin function and ruled not to be of an acquired origin can be submitted to a genetic analysis. To ensure an economical and reliable strategy, an algorithm is proposed in Fig 15. Currently, to be exhaustive, the conjunction of scanning methods and Southern hybridization or exon multiplex technology is required to detect most mispaired or unpaired sequences and rearrangements, respectively. Every nucleotide change detected from the index case has to be analyzed in the family members, keeping in mind that de novo mutations are not uncommon. As stated, and in particular when detected in introns, every new mutation without a known significance has to be distinguished from a polymorphism in the C1NH gene after establishing its absence in the relevant healthy population (from at least 100 chromosomes) and its segregation with the disease.
Fig 15.
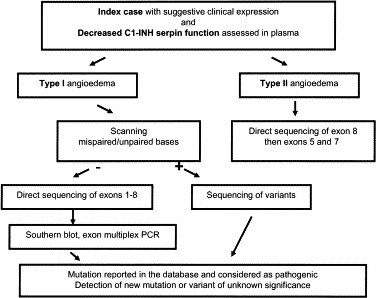
Decision algorithm for mutation analyses of the C1NH gene. This algorithm takes into account the importance of mutations in the coding sequence and in the introns, and the abundance of rearrangements. Depending on the methods used, a preliminary screening of rearrangements may be preferred.
The interpretation of biochemical data on C1-INH serpin function and of C1NH gene anomalies is of great importance to establish a better knowledge of the pathogenic effects of C1NH gene mutations. Moreover, the possibility of C1-INH polymorphisms affecting disease expression should be considered.183 Generating a C1-INH molecular model should be of great help in the understanding and treatment of HAE type II and discerning which C1-INH residues are essential for efficient control of all its target proteases.162
The uniform resource locators for data related to this section are as follows:
-
•
C1 Inhibitor Gene Mutation Database: http://hae.biomembrane.hu/
-
•
eGenome 11 home page: http://genome.chop.edu/
-
•
Ensembl: http://www.ensemble.org/Homo_sapiens (Ensembl Gene ID ENSG00000149131)
-
•
Information about HAE (types I and II) at the OMIM database: http://ncbi.nlm.nih.gov/htbin-post/Omim/ (OMIM ID 106100)
-
•
NCBI LocusLink database: http://www.ncbi.nlm.nih.gov/LocusLink/ (Locus ID 710; OrgSymbol SERPING1)
-
•
Human Gene Mutation Database Cardiff: http://archive.uwcm.ac.uk/uwcm/mg (Human Gene Mutation Database 119041)
This section on mutation analysis was supported by grants from the European Community Quality of Life Program (2001-01359), Programme Hospitalier de Recherche Clinique PHRC 2002, Fondo de Investigaciones Sanitarias FIS (00/0216), Hungarian National Science Research (OTKA) (T034830), Ministry of Education of Hungary (NKFP) (1/024/2001), and Ministry of Health of Hungary (ETT 490/2003). Its authors are indebted to Prs. Marco Cicardi, Mario Tosi, and Joel Lunardi for valuable discussions, and thank Pr. Jean-Yves Cesbron for continuous encouragement. O. Roche and A. Blanch are supported by fellowships from FIS (BEFI 00/9158) and Programa de Formación de Personal Universitario de la Universidad Autonóma de Madrid, respectively.
Pathogenesis and pathobiology of HAE and AAE
From a pathogenetic perspective, the form of angioedema most extensively studied is that resulting from C1-INH deficiency. Nonetheless, the pathogenesis of this clinical condition remains incompletely understood. In the following section, Hack briefly reviews the biochemistry and biology of C1-INH as well as the biochemical changes that occur in persons with C1-INH deficiency during both attacks and symptom-free periods. On the basis of these reviewed data, Hack then discusses the potential pathogenesis of C1-INH deficiency angioedema.
Pathogenesis of angioedema attacks
(C. Erik Hack, MD, PhD,∗ Amsterdam, The Netherlands)
Often, HAE results from a deficiency of C1-INH. An interesting feature, revisited in the previous Clinical Manifestation and Diagnosis section, is the great variance of the disease's clinical course. Some patients are virtually free of attacks even in the absence of treatment, whereas others, despite therapy, have attacks nearly every week. Angioedema attacks result from the extravasation of fluid caused by increased vasopermeability. Unlike angioedema of allergic origins, angioedema caused by HAE typically does not itch. Pain is often not a primary feature of such angioedema, although it may manifest secondary to the localization of the angioedema, eg, severe pain caused by obstruction of the bowel lumen. In spite of decades of research, the pathogenesis of HAE attacks is still unclear.
C1-INH biochemistry and biology
C1-INH belongs to the family of serine protease inhibitors, or serpins, which also includes proteins such as α1-antitrypsin and antithrombin III194., 195., 196., 197. ( Fig 16). Most serpins have only 1 or a few target proteases, but C1-INH is exceptional in this respect. It is the major inhibitor of several proteases, including (1) C1s and C1r—two serine proteases that, together with C1q, constitute the C1 complex of the classical pathway of complement, (2) the mannan-binding lectin (MBL)–associated serine proteases or MASPs, and (3) the contact system proteases factor XIa, factor XIIa, and kallikrein.198 In addition, C1-INH may interact with several proteases such as thrombin,199 plasmin,200 and tissue-type plasminogen activator (tPA).201 Thus, C1-INH regulates the activity of several inflammatory, clotting, and fibrinolytic proteases and is therefore an inhibitor of several pathways of inflammation.198 In comparison with other serpins, C1-INH is a relatively weak inhibitor, approximately 100 times less effective than other serpins. However, the inhibitory activity of C1-INH toward C1r, C1s, and factor XIa can be remarkably potentiated by heparin and other glycosaminoglycans.202., 203. The inhibitory activity of C1-INH to factor XIIa and kallikrein cannot be potentiated. Dextran sulfate is the most effective glycosaminoglycan, enhancing C1-INH function 130-fold in vitro and 60-fold in plasma. However, the in vivo effect of dextran sulfate is transient, probably because of dissociation of the dextran sulfate–C1-INH complex and the subsequent rapid clearance of dextran sulfate.204
Fig 16.

α-1-Antitrypsin tertiary structure conserved among the serpin family α-1-antitrypsin is composed of 3 β-sheets (A in red, B in green, and C in yellow) and 9 α-helices (A-I in gray). The blue strand is the reactive center loop (RCL) with the P1 active amino acid. Reprinted with permission from Silverman et al.197
Mature C1-INH consists of 478 amino acids and is heavily glycosylated (approximately 30% by weight). Although on SDS-PAGE gels it migrates with an apparent molecular weight of 104 kd, its calculated molecular weight is 76 kd. The C1-INH molecule has at least 13 glycosylation sites205 and possibly 20 glycosylation sites, part of them linked to threonine residues. Most carbohydrate groups are located at the N-terminal region; their function is largely unknown. The C1-INH molecule is composed of an N-terminal domain of 113 amino acids and a serpin domain of 365 amino acids. Although the function of the N-terminal domain is unknown, recent evidence suggests that it helps to stabilize the central β-sheet by forming 2 disulfide bridges linking cysteine 101 to cysteine 406, and cysteine 108 to cysteine 183.206 The structure of the serpin domain is homologous to that of other serpins and is essential for the inhibitory capacity of the molecule. Recently, a novel 3-dimensional model of C1-INH was proposed on the basis of the crystal structure of 4 other inhibitory serpins.162
Similar to other serpins, C1-INH inhibits proteases by binding to their active site via its reactive center. This reaction follows the equation
In the first step, a reversible complex is formed between the target protease and C1-INH; C1-INH then undergoes conformational changes caused by insertion of part of its reactive center loop into a 5-stranded β-sheet. This results in the formation of modified C1-INH (C1-INH∗ in the equation) that is tightly bound to the target protease (step 2). Mutations of the residues at the P14, P12, and P10 positions in the reactive center interfere with efficient loop insertion and cause the reaction to proceed to step 3. In that case, C1-INH has become a substrate for its target proteases. Such mutations have been found in some patients with HAE-II (characterized by the presence of dysfunctional C1-INH protein). Complexes between modified C1-INH∗ and proteases are very stable and only very slowly dissociate into an inactive, modified C1-INH (with changed conformation caused by a completed insertion of the cleaved active site loop into the 5-stranded β-sheet) and an active target protease (step 3 in the equation). Most of the stable complexes will be removed from the circulation before dissociation via receptors specific for complexed serpins. This mechanism ensures efficient removal of biologically active proteases.
C1-INH is an acute phase protein, of which plasma concentrations may increase as much as 2-fold during uncomplicated infections. Woo et al207 reported that the synthetic rate of C1-INH may increase as much as 2.5 times the normal rate in patients with rheumatoid arthritis. Cytokines such as interferon-γ stimulate the synthesis of C1-INH.208 In normal volunteers, the fractional catabolic rate of C1-INH is 2.5% of the plasma pool per hour, which results in an apparent plasma half-life time of clearance of about 28 hours.90 The half-life time of clearance of human C1-INH in rabbits is comparable, at 26 hours,209 whereas that in rats is considerably shorter, at about 4.5 hours.210 The apparent half-life time of clearance is markedly longer in patients with HAE, in whom it may exceed 48 hours, as determined by assessing the course of plasma concentrations after the intravenous administration of exogenous C1-INH.211 However, at low plasma concentrations of C1-INH, such as occur in untreated patients with HAE, C1 is autoactivated212 and consumes functional C1-INH. At higher C1-INH concentrations such as occur after the administration of exogenous C1-INH, this autoactivation is inhibited, leading to a decreased consumption of C1-INH. Hence, monitoring of plasma concentrations after a therapeutic dose of C1-INH may lead to an overestimation of the half-life caused by inhibition of consumption of endogenous C1-INH. Removal of sialic acids greatly enhances the clearance of C1-INH from the circulation, yielding an apparent half-life of 3 to 5 minutes,209 presumably by binding to asialoglycoprotein receptors in the liver. Subsequent removal of penultimate galactosyl residues largely restores the clearance rate to a value similar to that of normal C1-INH.209
Activation of cascade systems in HAE
As mentioned, C1-INH regulates the activation of at least 3 different so-called plasma cascade systems, ie, the classical and MBL pathways of complement via its effect on C1r, C1s, and the MASPs; the contact system via its effects on kallikrein and activated factor XII (factor XIIa); and the intrinsic pathway of coagulation via its effect on activated factor XI (factor XIa).213 In addition, it may regulate coagulation to some extent via its effects on thrombin, and it may regulate fibrinolysis through its effect on tPA and plasmin. Except for the MBL pathway, there is evidence for activation of all of these systems in patients with HAE.
The classical pathway of complement consists of C1q, C1r, and C1s, together forming the C1 complex; C4 and C2; as well as the inhibitors C4 binding protein and factor I. Typically, patients with HAE have low C4 and C2 but normal C3 values even during attack-free periods. Low C4 and C2 result from uncontrolled activation of activated C1 because the concentration of activation products of C4, C4b/c, or C4d is elevated.213., 214. Activation of C3 is typically low or absent, probably because of efficient control of activation at the level of C4 by C4 binding protein and factor I. This continuous activation of the classical pathway is attributed to enhanced autoactivation of C1, because this autoactivation process is dependent on functional C1-INH values.212 However, it is now clear that the classic pathway of complement may be activated by apoptotic cells through various mechanisms.215., 216., 217. Hence, one could postulate that the excessive activation of the classic pathway in untreated patients with HAE results from activation of this pathway by cell debris. Indeed, Familian and Hack have observed increased amounts of complement activation products in patients with HAE (Unpublished data, November 2002), reflecting activation via the penetraxin serum amyloid P component, 1 of the proteins that can bind to apoptotic cells.218 Ongoing activation of the classical pathway in untreated patients continuously produces activated C2.
The contact system of coagulation consists of the proteins factor XII (also known as Hageman factor), prekallikrein, and high molecular weight kininogen.219 The system is activated in vitro on contact with negatively charged surfaces, but the nature of potential activators in vivo is currently unknown. Activation of the system yields factor XIIa and kallikrein. C1-INH inhibits both proteases. In addition, kallikrein can also be inhibited to some extent by α2-macroglobulin. Kallikrein can cleave HK, yielding cleaved HK and bradykinin. The latter nonapeptide is a potent inducer of vasodilation and increased vasopermeability. Activation of this system is often assessed by measuring factor XIIa–C1-INH and kallikrein–C1-INH complexes,220 although these parameters are unsuitable to assess activation in HAE. Alternatively, activation can be measured by detecting increased amounts of cleaved HK or decreased antigenic quantities of the contact system proteins. Activation of the contact system in HAE typically occurs during attacks and is hardly detectable during attack-free periods.221 In particular, increased amounts of cleaved HK are found in most, if not all, patients with attacks.17., 222., 223. Remarkably, nearly all HK is often cleaved during attacks, such that native HK is hardly detectable. It is questionable whether all HK cleaved during an attack gives rise to bradykinin, given its extremely potent activity as a vasodilator. Bradykinin is known to cause a significant drop in blood pressure even at low concentrations, yet hypotension is not characteristic for HAE. However, there are no data supporting the cleavage of HK via other degradation pathways during HAE attacks such as would generate minimal or no bradykinin. It should be noted that the measurement of bradykinin in plasma or other body fluids is hampered by its rapid degradation by carboxypeptidases and is only possible using samples collected in special inhibitor cocktails to prevent enzymatic degradation. Few studies have attempted to measure bradykinin according to such protocols in HAE, but these studies have shown increased bradykinin generation during attacks and not during symptom-free periods, particularly when samples are drawn from the site of the edematous swelling.13., 14., 27.
C1-INH is a major inhibitor of factor XIa of the intrinsic pathway of the intrinsic pathway of coagulation143., 224. and also inhibits thrombin,199 although inhibition of this latter enzyme is likely physiologically inconsequential. The role of factor XI in coagulation is not yet resolved. Originally, coagulation was considered to proceed via at least 2 different pathways, the intrinsic and the extrinsic, merging at the level of factor X into a common pathway of thrombin generation. The extrinsic pathway is initiated by exposure of factor VII to tissue factor, whereas the intrinsic pathway is triggered by factor XII activation, eg, upon contact of blood with negatively charged surfaces such as glass. In this view, activation of factor XII led to thrombin generation via activation of factor XI, which then activated factor IX, which together with factor VIIIa activated factor X, which then together with factor Va activated prothrombin. This traditional distinction between intrinsic and extrinsic pathway has been left behind, and the intrinsic pathway is now considered to amplify extrinsic pathway activation in that factor IX amplifies factor VIIa–induced activation of factor X, whereas factor XI, activated by thrombin, activates additional factor IX. According to this revised scheme of coagulation, the activation of factor XI is no longer mediated solely by factor XIIa but also may occur with significant thrombin generation. In the traditional scheme of coagulation, C1-INH is expected to have a major effect on intrinsic pathway activation because it blocks the activity of factor XIIa, kallikrein, and factor XIa, whereas according to the current understanding, C1-INH has only a limited effect on coagulation, with its main effect the inhibition of factor XIa–mediated amplification of factor IX activation. Clinical observations in HAE do not support that C1-INH deficiency is a major risk for thromboembolic disease, although some generation of thrombin may occur during attacks.222 This mild effect of C1-INH is easily understood through the revised scheme of coagulation.
Although C1-INH inhibits plasmin and tPA in vitro, and to some extent in vivo,201 this inhibition is weak. Nonetheless, significant plasmin formation, as measured by the formation of plasmin–α2-antiplasmin complexes, occurs during attacks in patients with HAE but hardly at all during symptom-free periods. At present there is no evidence for tPA or urokinase-type plasminogen activator involvement in the formation of plasmin during HAE attacks. As such, this generation most likely results from factor XII activation. Although debated in the literature, convincing in vivo evidence exists to support the activation of plasminogen in human beings by factor XIIa.225., 226.
Angioedema attack pathogenesis: Mediators of angioedema
Angioedema is a frequently experienced side effect for patients receiving ACE inhibitors. This angioedema does not itch, is not associated with urticarial lesions, and does not respond at all to corticosteroids or antihistamines, features also typical of angioedema associated with C1-INH deficiency. ACE inhibitors slow down the processing of C-terminal arginine residues of various vasoactive peptides such as angiotensin and bradykinin, thus prolonging the biological activity of these compounds. The association between ACE inhibitors and angioedema is a strong argument that this condition may result from exposure of the endothelium to increased levels of vasoactive peptides. Considering the similarities between ACE inhibitor–related angioedema and that caused by C1-INH deficiency, the latter form also likely results from enhanced exposure of the endothelium to vasoactive peptides. Studies in a few patients indicate that bradykinin may be the vasoactive peptide involved.13., 14., 27.
Considering that contact activation in HAE mainly, if not exclusively, occurs during attacks, and that activation of this system results in the liberation of bradykinin from HK, most investigators currently believe that C1-INH deficiency angioedema results from the local generation of bradykinin caused by uncontrolled proteolytic activity of factor XIIa and kallikrein ( Fig 17, bottom).15 Kallikrein has 2 important effects in contact activation: it (1) amplifies factor XII activation via so-called reciprocal activation, and (2) releases bradykinin from HK. This mechanism of contact activation and bradykinin generation as the basis for angioedema is supported by observations in the murine model of C1-INH deficiency, because the enhanced vasopermeability in this model can be blocked with bradykinin receptor antagonists.9 According to this mechanism, inhibiting kallikrein or blockading bradykinin receptors (BKR-2) would be therapeutic options; as discussed in the Treatment section of this supplement, both are currently being investigated in clinical trials.
Fig 17.

Different pathways of kinin generation in C1-inhibitor deficiency state. Low C1-inhibitor leads to uncontrolled activation of factor XII, which generates kallikrein and plasmin. Kallikrein liberates bradykinin from HK, whereas plasmin cleaves off C2-kinin from activated C2. Activated C2 is continuously produced during baseline complement activation, which is increased as a result of insufficient control of autoactivation of C1 caused by C1-INH deficiency.
Activation of the contact system holds several attractions as an explanation for angioedema attacks. First, factor XII activation can be triggered by chemical activators and may well occur in bouts. Second, bradykinin is a potent vasoactive peptide that can enhance vasopermeability. Third, ACE inhibitors have been shown to result in the delayed breakdown of bradykinin, and part of their hypotensive effects are actually blocked by bradykinin receptor antagonists.227 However, some strong arguments exist against this mechanism as the sole explanation for angioedema. First, bradykinin is a strong vasodilating agent and among the most hypotensive agents known, whereas HAE attacks are not known to be associated with clear hypotensive reactions. Nonetheless, one could argue that most bradykinin is generated locally, at the site of edema formation, rather than in the circulation. Second, the local injection of bradykinin results in a strong pain sensation,228 whereas primary, localized pain is not typical for HAE attacks. Third, contact activation cannot explain the efficacy of antifibrinolytic agents such as tranexamic acid. It has been suggested that these agents are efficacious by reducing plasmin formation during attacks, which would lead to the reduced consumption of functional C1-INH by plasmin. In spite of this, it is difficult to understand how active plasmin would be capable of mediating C1-INH consumption, because it is rapidly inhibited by a potent plasmin inhibitor, ie, α2-antiplasmin. Moreover, in most patients with HAE, rather than being elevated, the concentration of cleaved C1-INH (free C1-INH∗ in the equation) is fairly low. Finally, although vasopermeability seems to be enhanced and dependent on bradykinin formation in C1-INH–deficient mice, these permeability changes do not appear to occur in bouts and are otherwise atypical for angioedema. For all of these reasons, other mechanisms may be involved in HAE attacks.
Some decades ago, another vasoactive peptide was thought to be involved in angioedema formation. This peptide can be generated by cleavage of the C2b fragment, but not from native C2, by plasmin, and is therefore called C2-kinin.229 This C-terminal fragment of C2b can be generated by C1s or plasmin. Mediation of attacks by this fragment requires activation of multiple plasma cascade systems: (1) complement, to generate activated C2; (2) the contact system, to generate factor XIIa; and (3) the fibrinolytic system, to generate the C2-kinin generating enzyme, plasmin. According to this mechanism, attacks are triggered by factor XII activation that then yields plasmin, which in turn cleaves the C2-kinin sequence from C2b (Fig 17, top). The attractive aspects of this controversial mechanism include its explanation of the efficacy of antifibrinolytic agents. Furthermore, one may postulate that C2-kinin does not induce vasodilation, explaining why HAE attacks are not associated with hypotension and may not induce sensations of pain, and why angioedema in general does not hurt. Nevertheless, it should be noted that the biologic properties of C2-kinin are not well established, and thus, it is not truly known whether this kinin's properties are consistent with the description. Moreover, receptors for this kinin have not been identified, and it is therefore unknown whether their distribution correlates positively with the localization of attacks. In addition, there are no data regarding concentrations of C2-kinin in patients with HAE. Finally, if correct, the C2-kinin mechanism provides no explanation for the profound cleavage of HK and the apparent lack of biological activity of bradykinin.
These mechanisms are not necessarily exclusive and may be simultaneously involved in attacks. Furthermore, the involvement of other, as yet unknown vasoactive peptides in the pathogenesis of attacks cannot be excluded.
Triggers of angioedema attacks
Clinically, HAE attacks are often triggered by trauma or emotional stress. Attacks of HAE are specifically associated with activation of the fibrinolytic and contact systems, whereas complement activation occurs continuously, independent of clinical symptoms. Activation of the fibrinolytic system can occur via 3 plasminogen activators: tPA, urokinase-type plasminogen activator, and factor XII. There is no evidence that the former 2 are involved in plasmin formation during attacks; hence, it likely results from factor XII activation. Thus, attacks are triggered by activation of factor XII. Though factor XII (Hageman factor) was discovered in the 1950s, physiological activators of this contact system protein have not yet been identified. In vitro, factor XII is easily activated by glass and other negatively charged compounds such as dextran sulfate. Obviously, these compounds are not relevant as activators of the contact system in vivo. The author writes that recently, his group has studied the generation of thrombin by phospholipid microparticles released in vivo from activated platelets and damaged cells.230 By using various specific mAbs against tissue factor, factor VII, factor XI, and factor XII, they noticed that a small but significant part of thrombin generation by microparticles purified from human plasma was dependent on factor XII. Indeed, earlier studies have clearly indicated that factor XII can be activated by phospholipids in vitro.231., 232. Apparently, factor XII can be activated by phospholipid microparticles in vivo. Damaged or apoptotic cells generate such particles, and one may therefore postulate that these particles constitute triggers for contact activation. Under normal conditions, ie, at normal concentrations of C1-INH, this weak activation will never yield substantial contact activation. However, in the absence of sufficient amounts of this inhibitor, the system may become profoundly activated by even small amounts of microparticles via the principle of reciprocal activation (factor XII activates prekallikrein into kallikrein, which in turn activates additional factor XII, and so on). Microparticles and damaged cells are likely generated during trauma, which may explain why this is a trigger for angioedema attacks. Emotional stress as a trigger for attacks is more difficult to understand, although one may speculate that it predisposes patients to microtrauma. Support for this hypothesis is that occasional patients have been described who, in spite of acquired C1-INH deficiency, are free of symptoms. Two such patients had detectable antibodies against phospholipids.125 These antibodies might interfere with the binding of factor XII to phospholipid microparticles or damaged cells. Further studies are needed to provide evidence toward this hypothesis of HAE attack generation.
Attack frequency
The angioedema attack rate in persons with C1-INH deficiency may vary widely, ranging from no attacks to an attack every week. The molecular mechanism for the variation in attack rate is unknown. In the previous paragraph, 2 mechanisms to explain the trigger for attacks are discussed. Both mechanisms claim that attacks are initiated by activation of factor XII. Therefore, one may speculate that the frequency of attacks is strongly linked to an individual's tendency toward factor XII activation. If true, attack rates might be influenced by the concentration of factor XII as well as by the amount of factors that compete with factor XII for binding to its activator molecules. As discussed, in the case of phospholipid microparticles, antiphospholipid bodies may prevent factor XII from binding to the particles, conferring protection against the activation of factor XII. Older literature has revealed several proteins capable of competition with factor XII for binding to artificial activators.233., 234. The concentrations of these competing proteins might well determine whether factor XII may become activated on exposure to an activator and therefore influence HAE attack frequency. Future research will test this hypothesis.
In summary
Angioedema attacks caused by C1-INH deficiency are likely mediated by the excessive release of vasoactive peptides such as bradykinin and C2-kinin. This release is initiated by the uncontrolled activation of factor XII, leading to kallikrein and plasmin formation. The trigger for attacks may be phospholipids, released from damaged cells, that activate factor XII. Factor XII's ease of activation may be determined by its concentration as well as the concentration of proteins that compete with it for binding to activators, and may explain the variable frequency of attacks in patients.
Additional discussions of pathogenesis
Others, assuming bradykinin to be the principal mediator of nonallergic angioedema attacks, have begun work toward bridging the gap between C1-INH deficiency and local angioedema generation. Bolstered by the increasing evidence that ACE inhibitors235., 236., 237. can cause AAE in a small percentage of individuals, investigators have sought to determine whether—independent of C1NH gene sequence—an increased liberation of bradykinin, a decrease in its catabolism, or a combination of the 2 may contribute to the angioedema experienced in HAE, AAE, and related disorders.
Murine model of angioedema
(Kayla Williams, BS, MA, MFA, Cambridge, Mass: Review of E. Han, R. C. McFarlane, A. N. Mulligan, J. Scafidi, and A. E. Davis)
In 2002, Han et al9 published their influential mouse model of angioedema. In this model, mice heterozygous and homozygous for disruption to the gene coding for murine C1-INH (C1nh gene) were created by gene trapping. Although human beings homozygous for C1NH mutations have yet to be documented (with 1 exception171., 238.), no obvious phenotypic abnormalities were observed in C1nh heterozygous mice, nor in mice lacking both copies of a functional C1nh gene.
Phenotypic characteristics. Mating of heterozygous-deficient mice produced the expected 1:2:1 Mendelian ratio of C1-INH+/+:C1-INH+/−:C1-INH−/− mice. As observed in human heterozygotes, C1-INH expression was less than 50% of normal in heterozygous mice; homozygous C1-INH knockouts did not produce detectable plasma concentrations of C1-INH. In male but not in female mice, carrying the defective C1-INH gene correlated with a slight but statistically significant decrease in weight. The growth of both heterozygous-deficient and homozygous-deficient mice appeared normal, with no more newborn deaths observed than in wild-type littermates. Limited analysis of serum C4 concentration in heterozygous-deficient and homozygous-deficient mice revealed that homozygotes had 38% to 59% as much C4 as their wild-type littermates, constituting a significant mean difference from wild-type; C4 in heterozygotes was more variable, ranging from 49% to 120% of wild-type. The group also reported that total hemolytic complement activity in the sera of null homozygotes was reduced compared with wild-type. Although the C1-INH+/− and C1-INH−/− mice were not observed to have spontaneous attacks of skin edema, 2 heterozygotes and 3 homozygous knockouts developed spontaneous abdominal distension secondary to intestinal wall edema. Because these mice were euthanized, it is unclear whether these episodes would have spontaneously resolved. No such episodes of abdominal edema were observed in wild-type littermates.
Measuring increased vascular permeability. Evans blue dye was used to visualize differences in vascular permeability between the 3 types of mice. Within 15 to 30 minutes of injection into the tail vein of wild-type mice, the dye caused a faint blue coloration, particularly visible around the nose and eyes and on the feet. After similar injections, C1-INH+/− and C1-INH−/− mice showed a much more saturated blue color in these areas, with homozygous null mice producing the most saturated blue. These differences could be quantitated spectrophotometrically, as shown in Fig 18. The mice were euthanized and the dye extracted from the area in question by using formamide, then read at an absorbance of 600 nm.
Fig 18.
Analysis of vascular permeability. Extravasation of Evans blue dye at 15 to 30 minutes was much more extensive in C1-INH–deficient mice (B) than in wild-type mice (A), particularly after the application of mustard oil to the left ears of the mice. (C) The difference in the amount of extravasation was clearly demonstrated by the rear footpads of mice of each genotype. Administration of human C1-INH resulted in reduced vascular permeability, as did the combination of Bk2R deficiency together with C1-INH deficiency. Reprinted with permission of the American Society for Clinical Investigation.9
Han et al9 noted that unlike in the feet and face, there were no apparent vascular permeability differences in the ears of mice of all 3 genotypes after Evans blue injection. However, when mustard oil, a local irritant, was applied to the ears of injected mice, a pronounced permeability increase correlating with C1nh mutation could be spectrophotometrically demonstrated. This increase could be reversed if the mice were given human C1-INH concentrate. Intestinal vascular permeability was also investigated in a few mice; although of borderline statistical significance because of the sample size, intestinal permeability was also increased in C1-INH−/− versus wild-type mice ( Fig 19).
Fig 19.
Spectrophotometric analysis of vascular permeability in the small intestine. Quantitation of extravasated Evans blue dye in C1INH−/− mice treated with an ACE inhibitor (captopril), a Bk1R antagonist (des-Arg9,[Leu8]-bradykinin; Bk1RA), and a Bk2R antagonist (Hoe140). Reprinted with permission of the American Society for Clinical Investigation.9
Bradykinin type 2 receptor assays. Groups of heterozygous and homozygous C1nh- deficient mice were treated with the plasma kallikrein inhibitor DX-88, the bradykinin receptor 2 antagonist Hoe140 (Icatibant; Jerini, Berlin, Germany), or the ACE inhibitor captopril ( Fig 19, Fig 20). Both interference with plasma kallikrein (and thus contact system–mediated bradykinin generation) and disruption of the bradykinin receptor 2 interaction with its ligand decreased vascular permeability. Treatment with the bradykinin receptor 1 antagonist des-Arg9,[Leu8]-bradykinin had no effect. Treatment with captopril dramatically increased vascular permeability.
Fig 20.
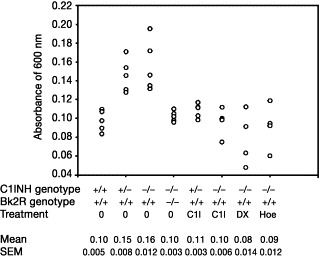
Spectrophotometric analysis of vascular permeability in footpads. Quantitation of Evans blue dye extracted from the paws of mice of the indicated genotypes, either treated or not treated with intravenous C1-INH (C1I), DX-88 (DX), or Hoe140 (Hoe). Reprinted with permission of the American Society for Clinical Investigation.9
When C1nh knockout mice were crossed with bradykinin receptor 2 knockout mice, the resulting offspring were overtly phenotypically normal. As shown in Fig 20, absence of the bradykinin receptor 2 was able to reverse the vascular permeability observed in both C1-INH+/− and C1-INH−/− mice.
In conclusion, although this mouse model does not exclude the possibility of other contributing mechanisms, it strongly supports the hypothesis that bradykinin mediates angioedema.
Bradykinin-mediated angioedema?
(Kayla Williams, BS, MA, MFA, Cambridge, Mass)
In this section, Williams reviews recent information on the role of bradykinin in both drug-related angioedema and HAE and explores the contributing factors that may influence the generation of bradykinin-mediated angioedema.
Drug-related angioedema: A growing problem. As the use of ACE inhibitors, AT2 receptor antagonists (AT2 blockers), and related, next-generation drugs increases, it is projected that the incidence of drug-related angioedema will also increase.235., 239. Although angioedema is a relatively rare adverse effect of such medications, the medications are commonly and increasingly prescribed. The incidence of angioedema during ACE inhibitor treatment has been estimated at between 1 and 7 per 1000 patients.240 These cases of drug-induced angioedema are interesting because, unlike other forms of nonallergic angioedema, the role of bradykinin in ACE inhibitor–related angioedema is fairly well established. Also of note and concern are reported racial differences in the response to this vasoactive peptide.
Bradykinin, race, and ACE. In 1996, Gainer et al published the results of a clinical trial evaluating wheal response to bradykinin among African Americans and white Americans.241 In this randomized, double-blind study, increased bradykinin dose, hypertension, and African American race each correlated with an increased wheal response to bradykinin. Further investigations by the same group compared the effects of short-term ACE inhibition in normotensive and hypertensive individuals of both races. In this study, the hypotensive effects of the ACE inhibitor captopril were shown to be greatly reduced in all subjects by concomitant administration of the bradykinin receptor 2 antagonist Icatibant.227 Icatibant was also shown to significantly alter the change in plasma renin activity in response to ACE inhibition. These results not only confirmed the importance of bradykinin in the short-term effects of ACE inhibitors but also suggested that bradykinin contributes to the effects of ACE inhibition on the renin-angiotensin system. In addition, when considered together, these results suggest that clinicians prescribing ACE inhibitors for patients of Afro-Caribbean descent should be especially aware of the possibility for drug-related angioedema, a warning beginning to be supported by case analysis.242., 243.
Further antihypertensive-related angioedema. Initially, AT2 blockers were thought to be a safer alternative for patients who had angioedema associated with ACE inhibitors. Unlike ACE inhibitors, AT2 blockers have not been shown to increase bradykinin concentrations. Nonetheless, further case reports documented angioedema after therapy with AT2 blockers.137., 244. In a survey of the literature, 32% of these patients also had similar episodes in conjunction with ACE inhibitor treatment.245 Additional documented cases of AT2 blocker–associated angioedema threw bradykinin's role in the pathogenesis of such episodes into doubt246 because increased concentrations of bradykinin are not associated with AT2 blocker use.245 However, very recent work has shown that increased concentrations of AT2, such as would occur during a blockade of AT2 receptors, are associated with an increase in the expression of bradykinin receptors that may be almost 3-fold.247 Thus, increased opportunities for bradykinin binding may explain angioedema in the absence of extra bradykinin. The mediator for this receptor crosstalk effect has been shown to be the AT2 receptor 1.247 (Presumably, this phenomenon could occur in the presence of AT2 receptor 1 blockers only in the absence of a total blockade.)
Meanwhile, the new drug application for the vasopeptidase inhibitor omapatrilat, a dual inhibitor of both ACE and neutral endopeptidase (NEP), was voluntarily withdrawn when the Food and Drug Administration raised questions regarding the comparative incidence of angioedema reported within the new drug application database. In response to these questions, a large (25,000-patient) multinational trial was initiated to compare the efficacy and safety of omapatrilat versus the ACE inhibitor enalapril in patients with heart failure.248 The results of this study have recently been reported.249 Angioedema occurred in 2.17% of subjects receiving omapatrilat compared with 0.68% of subjects receiving an ACE inhibitor alone. Two of the dual inhibitor–treated subjects had angioedema with airway compromise. An editorial250 published after the trial results were released but before their publication noted that the rates of angioedema were much higher in African Americans receiving the dual inhibitor (5.54%) than in those receiving the ACE inhibitor alone (1.62%). Of smokers receiving the dual inhibitor, 3.93% had angioedema, versus 0.81% of smokers receiving the ACE inhibitor alone.
The biologic activity of NEP must account for this several-fold increase in angioedema. Indeed, among its many possible functions, NEP has been shown to metabolize bradykinin to an inactive form.237 A combination of ACE and NEP inhibition could thus be expected to prevent efficiently the degradation of bradykinin to inactive metabolites, thereby increasing the risk of angioedema.251 In certain subgroups with altered responses to bradykinin or altered pathways for its generation or degradation, these effects could be especially pronounced, and such predisposing factors may also be involved in the generation of C1-INH deficiency–related angioedema.
Implications for C1-INH deficiency angioedema. Early investigations have confirmed that bradykinin may be at work in C1-INH deficiency angioedema. The transience of bradykinin has complicated measurements of this nonapeptide; however, several small studies of plasma bradykinin concentrations in patients with angioedema have revealed that high concentrations of this vasoactive peptide occur during edematous attacks.13., 14., 27. This increase in plasma bradykinin was demonstrated both for patients with HAE with C1-INH deficiency and for patients with drug-associated angioedema who had an angioedematous attack in conjunction with ACE inhibitor treatment. Patients receiving ACE inhibitors showed elevated plasma bradykinin concentrations throughout ACE inhibitor therapy, both during attacks and during periods of remission.13 As documented in 1 patient, this increase reverted to normal on discontinuation of ACE inhibitor therapy.14 In contrast, patients with C1-INH deficiency were shown to have high concentrations of bradykinin during their attacks13 but have been documented to have normal or marginally increased amounts of bradykinin during periods of remission.14 At least in C1-INH deficiency, bradykinin increases may be highly localized; in 2 previously studied C1-INH–deficient patients, bradykinin concentrations in blood draining from an angioedematous site were 3 and 8 times as high as those measured in systemic circulation.
Thus, bradykinin appears to be important in various currently identified forms of nonallergic angioedema. Following the example put forth in the large-scale trial of antihypertensive agents that showed (1) African descent and, to a lesser extent, (2) smoking to correlate with increased angioedema risk,250 clinicians and researchers may wish to investigate the interaction of these variables with C1-INH deficiency.
Bradykinin magnified. Even if bradykinin is the major mediator of nonallergic angioedema, many questions regarding the triggering and localization of angioedema attacks remain. Why should patients with ACE inhibitor–induced angioedema have this angioedema in bouts if bradykinin is constantly elevated? What is different about the small percentage of patients taking ACE inhibitors who have angioedema? What regulatory pathways are involved? How, chemically, does one proceed from insufficient functional C1-INH to localized, full-blown angioedema?
The past decade of research indicates that the answers may reside in a surprisingly multifactorial system. Bradykinin-mediated effects can result from increased amounts of bradykinin, the reduced breakdown of bradykinin to inactive metabolites, the increased conversion of bradykinin to active metabolites, and/or the increased uptake of these active metabolites. Each of these mechanisms may be involved in at least 1 form of nonallergic angioedema. Pathways for the breakdown of bradykinin are depicted in Fig 21.
Fig 21.
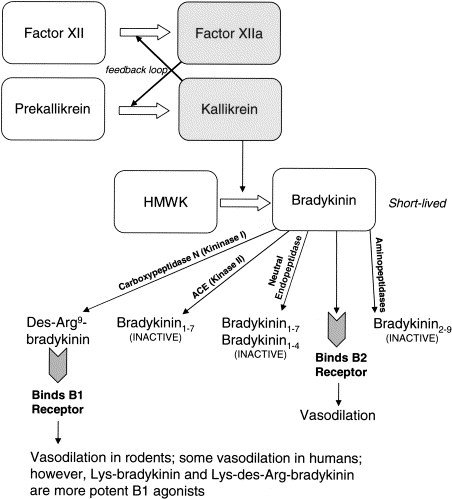
Pathways for bradykinin generation and metabolism. On the endothelium, bradykinin generation likely initiates independent of factor XII; however, once kallikrein is generated, it soon catalyzes factor XII activation, thus amplifying the response.
Decreased or alternative catabolism. Molinaro et al252 have demonstrated that in human plasma, bradykinin is metabolized mainly by 3 metallopeptidases: ACE, APP, and kininase I (carboxypeptidase N). Of these, ACE and APP, respectively, are the first and second major pathways of inactivation. Kininase I represents a minor pathway unless ACE is inhibited, whereupon kininase I transforms bradykinin into its active metabolite des-Arg9-bradykinin, which, in turn, is inactivated by APP and ACE, with APP the major inactivating metallopeptidase.253 NEP, as discussed in the context of vasopeptidase inhibition, likely represents another minor pathway.
Once formed, bradykinin and des-Arg9-bradykinin are short-lived and thus exert their effects locally at the site of their formation, stimulating B2 and B1 receptors, respectively.254., 255. During ACE or vasopeptidase inhibition, patients with low APP activity may be subject to angioedema. In fact, Adam et al96 have found a correlation between low APP activity with a history of angioedema among patients receiving ACE inhibitors. Likewise, in their Estrogen-dependent and Estrogen-associated inherited angioedema piece earlier in this supplement, Binkley and Davis propose reduced APP activity as a contributing element in estrogen-sensitive inherited angioedema. In addition, they note that estrogen may decrease gene expression of ACE.256., 257.
To demonstrate further the delicate balance of enzymes and receptors, it should be noted that Mathews et al258 observed angioedema in a subject with familial serum carboxypeptidase N deficiency. Because the subject's sister had an equally depressed serum carboxypeptidase value but no reported episodes of angioedema,259 it is possible that another defective control mechanism, possibly APP, contributed to the angioedema.
Increased anabolism. As Hack discussed in the Pathogenisis and Pathobiology of HAE and AAE section, C1-INH is known to inhibit not just C1 esterase but also other serine proteases, including factor XII (Hageman factor). In addition, C1-INH inhibits the serine protease kallikrein. Without the controlling effects of C1-INH, it is possible that both of these serine proteases may contribute to an increased generation of bradykinin. This reaction is thought to occur at the endothelial cell surface. The way in which this unchecked bradykinin generation starts has been debated, with recent evidence supporting the secondary role of factor XII,260., 261. and placing the conversion of prekallikrein to kallikrein, likely by an enzyme such as prolylcarboxypeptidase,262., 263. foremost, at least under resting physiologic conditions. It is thought that once kallikrein is present, it catalyzes the activation of factor XII. Only in the presence of activated platelets, neutrophils, or lymphocytes can the high local concentration of free zinc ions necessary for factor XII to bind to the endothelial cell surface—and thus initiate bradykinin release—occur.263 This provides yet another example of how infection or tissue damage might aggravate an existing tendency to angioedema.
In vitro, kallikrein and plasmin together have been shown to exert a synergistic effect on the release of kinins from HK. Of interest, plasmin's digestion of HK in this model could be inhibited by ε-aminocaproic acid.264 Because C1-INH is also known to inhibit plasmin, and antifibrinolytics show at least partial efficacy in the treatment of HAE, this enhancing mechanism might well be operative in C1-INH deficiency angioedema. In addition, because angioedema has been reported in patients treated for acute ischemic stroke who received recombinant tissue-type plasminogen activator (rtPA; often concomitantly with an ACE inhibitor), Molinaro et al252 undertook an in vitro comparison of human plasma incubated with and without rtPA in the presence of an ACE inhibitor. Their results showed that rtPA generates bradykinin that is further metabolized to des-Arg9-bradykinin; this effect could be recreated to a similar extent in the absence of rtPA by the addition of negatively charged glass beads, thus mimicking contact activation.
Joseph et al19., 20. have identified yet another possible mechanism active at the endothelial cell level, showing that the cytosol of endothelial cells contains a factor capable of converting prekallikrein to kallikrein in the absence of factor XII. They identified this factor as heat shock protein 90. Perhaps this mechanism might account for angioedema attacks that occur in the wake of trauma or infection when ruptured cells spill their contents into local circulation. Indeed, increased kallikrein may be a feature of mechanical trauma: an experiment conducted more than 20 years ago demonstrated that mechanically generated blisters in 5 of 5 patients with HAE contained active kallikrein that could be inactivated by C1-INH concentrate or antibodies to plasma kallikrein. In contrast, blister fluids from 6 of 8 healthy subjects did not contain detectable amounts of plasma kallikrein.265
Last, these anabolic effects might be created or magnified by hormonal interactions. As noted by Binkley and Davis,92 estrogens may increase the expression of factor XII107., 266., 267., 268. and prekallikrein.269
Increased uptake. Various factors may increase gene expression of the B1 and B2 receptors. Not normally expressed, the B1 receptor has a high affinity for kinin metabolites.270 Expression of B1 receptors may be induced during inflammation and tissue damage.271., 272. Although B2 receptors are constitutively expressed,273 their expression may be increased by estrogens274 and AT2 receptor antagonists.247
In summary. Despite increasing evidence that bradykinin is indeed the major mediator of nonallergic angioedema, this knowledge does not represent a simple answer given the many variables governing bradykinin and its metabolites. However, an understanding of this web of chemical interactions may yield more successful management algorithms for the various types of nonallergic angioedema. This recent knowledge also suggests a place for bradykinin receptor antagonists and kallikrein inhibitors for the control of nonallergic angioedema, especially when it occurs in the presence of functional C1-INH.
Angioedema attacks: triggers and hormonal influences
The current scientific understanding of the triggering of angioedema attacks ranges from the anecdotal to that supported by relatively large and rigorous studies. Patients with HAE, AAE, or related forms of angioedema, especially patients who have angioedema attacks in the absence of a formal diagnosis, pay particular attention to the events that precede their angioedema attacks; however, even in the same patient, these events may vary among attacks. Various patients have implicated exposure to cold; mechanical trauma sustained during routine activities such as gardening; prolonged sitting or standing; eating certain foods, such as eggs; exposure to pesticides or chemicals in new fabrics and clothing; infection or illness; or excitement or stress in triggering their attacks (K. Williams, Unpublished data, August 2003). However, as with other, better studied triggering events, these conditions or activities did not always predictably provoke attacks.
Ethical concerns and the small patient populations available to would-be investigators prevent the systematic investigation of most patient-reported angioedema attack triggers. Those triggers more readily quantifiable via laboratory assays without necessitating patients' increased risk of developing attacks have received the most scientific attention, largely restricting the study of attack triggers to the possible role of hormones.
Role of sex hormones in HAE
(Laurence Bouillet, MD, CCA,∗ Grenoble, France)
In the first part of this section, Bouillet reviews his group's clinical experience and other reports linking sex hormones and HAE. On the basis of the literature, he proposes possible pathogenic mechanisms by which these influences might manifest. These relationships are discussed with a final view toward patient care.
Current literature
It is currently known that HAE is influenced by the fluctuation of female hormones, but the effects sometimes appear to vary greatly among women. Some patients seem to be estrogen-dependent: puberty, pregnancies, or estroprogestative contraception can precipitate attacks, whereas menopause impairs the disease. However, the course of some women's disease seems to be unaffected by estrogen.
Reports have highlighted the close relationship between female hormones and angioedema. Yip and Cunliffe22 described cases of HAE occurring in a mother and her daughter whose symptoms appeared to be estrogen-influenced. Their first attack happened around puberty; angioedema worsened premenstrually and when they took OCs. Likewise, a woman with HAE-II associated with Turner syndrome was reported as experiencing an increase in both the severity and the frequency of her angioedema attacks on beginning estrogen physiological replacement at age 34 years.275 McGlinchey and McCluskey276 also described HAE manifesting after the initiation of estrogen replacement therapy. Bork et al,277 as discussed later in this supplement, reported their evaluation of the effect of OC use or hormone replacement therapy on the frequency and the severity of HAE attacks, finding that 63% of women with HAE taking these drugs presented with new or worsened angioedema.
Some kinin-mediated angioedema appears to be related to estrogen therapy. The first cases of angioedema during oral contraception were described in a 1986 report by Warin et al277a in which 2 sisters presented with HAE only after taking oral contraception and during pregnancy: C1-INH concentration and complement components were normal. Bouillet's group has also reported 5 women278 whose angioedema attacks manifested after starting oral contraception. A study of C1-INH biological function revealed a lowered C1-INH activity with marked protein cleavage on the immunoblot. Clinical and biological anomalies ceased after oral contraception was discontinued.
Hypoandrogenism has been associated with insufficient production of C1-INH in women taking cyproterone acetate as well in men with hypogonadism. In 1989, Pichler et al279 reported the cases of 4 women taking the contraceptive drug cyproterone acetate who developed clinical HAE reversed by discontinuation of this contraceptive; also described were the cases of 2 men with hypogonadism who had recurrent angioedema successfully cured by androgen treatment.
As discussed in previous sections, 3 reports described a new estrogen-influenced HAE.8., 86., 87. Only women had angioedema, and their attacks were precipitated when estrogen concentrations were increasing because of pregnancies, oral contraception, or hormone replacement therapy. No C1-INH abnormalities have been reported in these cases.
Hormone-related pathogenesis
For these clinical events to be understood, they must be placed in a biochemical context. As discussed, the complement, contact, and fibrinolytic systems are involved in the genesis of angioedema attacks,17., 222., 223. and all are regulated, at least to some extent, by C1-INH. Bradykinin is thought by many to be the principal candidate mediator of angioedema genesis, and a mouse model demonstrated that angioedema is mediated by bradykinin via B2 receptor.9 In human beings, studies have shown local elevations in bradykinin concentration during HAE attacks.13., 14. It is speculated that factor XIIa, kallikrein, and thrombin may be increased as well. An elevation of plasmin–α2-antiplasmin complexes is also noted.
In ovariectomized rats, studies showed that 17β-estradiol favored the increase of factor XII by stimulating its gene transcription.268., 280., 281., 282. This hormone also increased kininogen and kallikrein concentrations.283 In addition, estrogens regulate B2 receptor gene expression and function: when reduced in ovariectomized rats, the vasodepressor response to bradykinin and B2 receptor mRNA levels could be restored by estrogen substitution.274 Progesterone did not modify factor XII concentration but seemed to increase the amount of kallikrein cDNA.283
In healthy women taking oral contraception, there is an increase of the fibrinolytic proteins: plasmin and factors VII, X, and IX are increased, whereas plasminogen activator inhibitor is decreased.266., 284., 285. These effects appear to be estrogen dose–dependent.285 Plasma samples from these women showed enhanced in vitro fibrinolysis.107 The contact system is also altered: factor XII, prekallikrein, kallikrein, and HK increase.286., 287., 288. Last, C1-INH levels are decreased, correlating with the increase in factor XII.107., 286., 287.
Hormone replacement therapy (HRT), although using a weaker estrogen dose than oral contraception, appears to have the same effects. Fibrinolytic proteins are also increased (plasminogen and tPA), whereas plasminogen activator inhibitor decreases.289., 290., 291. HRT increases factor XII, prekallikrein, C3, and C4 concentrations.266., 290., 291. Moreover, some studies have shown HRT to influence the bradykinin system: ACE activity decreases, whereas bradykinin concentration increases.256., 292., 293.
Toward treatment
This imbalance in these 3 systems appears to trigger angioedema attacks in some estrogen-sensitive women. Each physician should identify these estrogen-sensitive patients to manage specifically their contraception needs, pregnancies, and choices regarding HRT. Further studies to determine predictive biological, genetic, and clinical parameters would be of use.
Role of sex hormones in HAE, part II
(Beáta Visy, MD, George Füst, MD, PhD, DSc,∗ Lilian Varga, PhD,∗ George Szendei, MD, Edit Takács, MD, István Karádi, MD, PhD, DSc, Béla Fekete, MD, PhD, DSc, George Harmat, MD, PhD, and Henriette Farkas, MD, PhD,∗ Budapest, Hungary)
Most reported studies have investigated the influence of exogenous sex hormones on the frequency and severity of HAE attacks. Here, Visy et al very briefly summarize a study they undertook measuring serum concentrations of endogenous sex hormones in women with HAE and correlating these results with the occurrence of HAE attacks. This work has since been published in Clinical Endocrinology.294
Objective
The fluctuations of sex hormone concentrations at the beginning of adolescence, in the perimenopausal period, and during pregnancy or OC use can precipitate HAE attacks. Edematous attacks usually disappear after the onset of menopause. The authors undertook their study to establish any relationship between serum concentrations of sex hormones and the incidence of HAE attacks.
Patients and measurements
Serum concentrations of luteinizing hormone (LH), follicle-stimulating hormone, progesterone, estradiol, testosterone, prolactin, and sex hormone binding globulin (SHBG) were measured in 78 patients (mean age, 30.3 years; range, 4-70 years) with HAE. A questionnaire was used to explore the medical history of adult patients to characterize the evolution and properties of attacks.
Results
Twenty-one patients had been symptom-free before menarche. Symptoms in 13 of 21 patients first occurred during adolescence, whereas the remaining 8 patients had no attacks in the pubertal period. Edema during the perimenstrual period was reported by 42.4%. Pregnancy was associated with a higher incidence of attacks in 36%; edema formation was less common in 56%, and 8% had no change in the frequency of symptoms despite pregnancy.
Of the 11 patients using OCs, 7 reported an increase in the frequency and severity of edematous symptoms compared with the period before hormonal contraception had been initiated.
Serum concentrations of sex hormones were normal in the majority of patients; however, more than half of the subjects had progesterone values above the upper limit of the normal range.
During the 1-year follow-up, the attack rate was 5 times higher in female patients with high progesterone concentrations (above the menopausal threshold of 4 nmol/L) than in women with normal or low serum concentrations. The eminent role of this hormone is also confirmed by the observation that only progesterone was higher in more than 50% of subjects than the serum level considered normal for sex, age, and stage of menstrual cycle. Therefore, serum progesterone concentrations above the postmenopausal threshold are predictive of a higher incidence of edematous attacks in female patients with HAE. In addition, multiple logistic regression analysis demonstrated a significantly lower attack frequency during 1-year follow-up in patients with higher (40 nmol/L) SHBG level (odds ratio, 0.25 confidence interval; 0.07-0.90; P = .034). This difference existed independently of age and danazol dose ( Fig 22).
Fig 22.
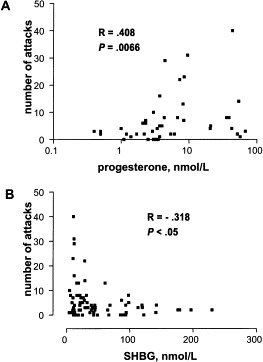
Correlation in female patients with HAE between serum progesterone (A) and SHBG concentrations (B) and the number of edematous attacks in the year after blood sampling. Spearman correlation coefficients and their significance (P values) are indicated.
Looking ahead
In view of these results, the monitoring of progesterone and SHBG concentrations can prove useful in the prediction of HAE attacks. Further investigations might address whether this increased progesterone is a cause or a result of frequent attacks, or whether progesterone itself exerts any direct effects on vascular permeability. Last, progesterone's effects on relevant gene expression might represent another valuable avenue of exploration.
Influence of OCs or HRT on hereditary forms of recurrent angioedema
(Konrad Bork, MD,∗ and Bettina Fischer, MD, Mainz, Germany)
In this section, Bork et al write of a systemic study undertaken after they noticed women in their clinic with new or worsened angioedema in response to oral contraception or HRT. Their study crosses the barriers of C1-INH deficiency and HAE type classifications to investigate the effects of exogenous hormones on HAE. The new type of inherited angioedema recently described by their group has clinical features highly similar to classic HAE-I and HAE-II; however, it is not associated with a C1-INH deficiency. Currently, the genetic defect is unknown. Until now, the disease has been observed exclusively in women8., 86., 87.; however, in 2 families the existence of clinically unaffected male carriers has been deduced.8., 86. In the following report, the authors show that estrogens play a similar role in HAE-I and this new type of inherited angioedema, herein described as HAE with normal C1 inhibitor (HAE type III) in accordance with the original report of this condition by Bork et al.86
Among their case series, the authors observed female patients with HAE who had newly developed recurrent angioedema after receiving OCs or postmenopausal HRT. In addition, other women had a worsening of their pre-existing angioedema after initiating OCs or HRT, suggested that these treatments might trigger HAE. However, this possible relationship has not formally been investigated, nor is angioedema listed as an adverse effect of OCs or HRT.295., 296., 297. Information regarding the relationship between hormone administration and the first occurrence or worsening of HAE symptoms is limited to anecdotal reports.8., 22., 23., 24., 31., 32., 86., 87., 107., 275., 276., 277a., 298., 299.
In this study, a systematic approach was used to obtain information about the relationship between the use of OC or HRT and the clinical manifestation of HAE-I and HAE with normal C1-INH (HAE type III).
Methods
The case series by Bork et al277 included 228 women with recurrent angioedema who had taken OC, HRT, or both.277 Of these, this study investigated 32 women with HAE-I and 39 women with HAE with normal C1-INH (HAE type III). The OCs used by all patients were combinations of estrogen (ethinyl estradiol) and progestin. Medications used for HRT contained only estrogens, as was standard medical practice in Germany until recently.
All patients underwent a thorough clinical examination, and a standardized medical and family history was taken. In particular, patients were questioned about the frequency, intensity, and organ involvement of their angioedema since the very first manifestation of their disease. The start date and duration of administration of sex hormones, with special emphasis on OC and HRT, were recorded. Patients were asked whether their angioedema occurred shortly after beginning this hormonal medication. If yes, the time between starting OC or HRT and the onset of angioedema was recorded. Patients who had recurrent angioedema before the intake of OC or HRT were asked whether their angioedema episodes were more frequent or severe after beginning treatment.
Concentrations of C1-INH, C4, and C1q were assayed by radial immunodiffusion, and C1-INH functional activity was determined by using the chromogenic substrate C2H5CO-Lys(ɛ-Cbo)-Gly-Arg-pNA (Immunochrom C1-INH; Immuno Diagnostics, Vienna, Austria).300., 301.
Results
As shown in Table V, OC, HRT, or both treatments were associated with angioedema attacks in 63% of women with HAE-I and 62% of women with HAE type III. Among the group of patients whose angioedema attacks were newly induced by estrogen-containing medications, there was a high preponderance of HAE type III, whereas in the patient group with an exacerbation of symptoms, there were more patients with HAE-I ( Fig 23).
Table V.
Summary of 71 women with inherited angioedema receiving OCs or HRT: Outcome of medication
| Type | Women N | Angioedema induced or worsened n (%) | Angioedema not influenced n (%) |
|---|---|---|---|
| HAE-I | 32 | 20 (62.5) | 12 (37.5) |
| HAE type III | 39 | 24 (61.5) | 15 (38.5) |
| Total | 71 | 44 | 27 |
Fig 23.

Underlying angioedema disease in 44 women who had induction or exacerbation of recurrent angioedema caused by OCs or HRT.
In 23 women—6 with HAE-I and 17 with HAE type III—occurrence of skin or gastrointestinal angioedema manifested for the first time after the administration of OC. Because of the assumed intolerance, several women tried different OC formulations, and all 33 trials were followed by recurrences of angioedema episodes so severe that all OC had to be discontinued.
The interval between starting OC and episode occurrence ranged from 2 to 90 days (mean, 18 days).
In 9 of the 23 women, symptoms were limited to the period of OC administration and ended after discontinuation. All 9 had HAE type III. In the other 14 patients, angioedema attacks recurred despite OC discontinuation. Episodes occurred less frequently and less severely in 12, whereas symptoms were unchanged in 4 women.
There was 1 occurrence of angioedema for the first time after starting postmenopausal HRT. One woman initially developed recurrent angioedema after starting HRT with estradiol (2 mg/d). The patient continued to take oral estradiol over a period of 4 years. After a consultation in the outpatient clinic, HRT was discontinued. Symptoms persisted but occurred less frequently and were milder. The patient's daughter had recurrent skin angioedema and gastrointestinal pain attacks since age 12 years; therefore, with a normal C1-INH concentration and activity in both mother and daughter, a diagnosis of HAE type III was assumed.
In 18 women, the administration of OC induced a severe exacerbation of their pre-existing angioedematous disease. Because of this exacerbation, 4 women switched to another OC, some trying many different preparations. All of these OC doses were followed by recurrences of severe angioedema such that OC had to be discontinued.
Five women with previous recurrent angioedema of the skin and the gastrointestinal tract developed an exacerbation of their disease after starting HRT. Of these, only 2 were eventually able to tolerate such therapy. In 3 of the 5, their angioedema also had exacerbated when on OC several decades before.
Discussion
The results indicate that in 63% of women with HAE-I and in 62% of women with HAE type III, the use of OC or HRT was associated with either the initial appearance of angioedema attacks or with a severe exacerbation of previously existing angioedema. Correspondingly, although also taking OC or receiving HRT, 37% of women with HAE-I and 38% of women with type III tolerated these medications without any negative influence on their disease; their disease was neither estrogen-dependent nor estrogen-sensitive. The results demonstrate that estrogen sensitivity is not a feature specific to HAE type III because it occurs in a similar percentage of women with HAE-I.
Results from several case reports have suggested that administration of estrogens may exacerbate clinical manifestations of HAE-I. Frank et al31 mentioned 2 young women with an exacerbation of HAE associated with the use of OC. Similar effects of the administration of OC or HRT on the clinical condition of patients with HAE-I have been documented by others.22., 23., 24., 32., 107., 276., 277a., 299. As in the current study, initiation of hormonal therapy was associated with the onset of clinical disease or a marked increase in the frequency and severity of angioedema attacks.
With respect to HAE-II, Fletcher and Weetman275 described a patient with HAE-II and coexisting Turner syndrome who had a severe exacerbation of angioedema attacks after initiation of a HRT with a combination of conjugated estrogen and progesterone.
Another type of HAE, not associated with a C1-INH deficiency, has been recently described.8., 86., 87. This disease has been reported exclusively in women and was termed HAE type III by Bork et al86 and estrogen-dependent inherited angioedema or estrogen-associated inherited angioedema by others.8., 87. Ten of the 36 women described as having HAE type III in a previous study by Bork et al86 took estrogen-containing OC and reported either the first appearance of clinical symptoms or a severe exacerbation of the disease in association with this treatment. In 2 additional families with this new type of HAE,8., 87. of 11 affected women, 4 developed angioedema after starting OC, as did 3 after initiating HRT. Warin et al277a described 2 sisters with estrogen-associated urticaria/angioedema. With the exception of urticaria, the symptoms in these women closely resemble those of patients Bork et al86 have described as having HAE type III.
It is highly probable that the agent responsible for worsening of pre-existing angioedema or provocation of initial symptoms in the women studied was estrogen rather than progestin. Six of the women evaluated in their study had new angioedema attacks or exacerbation of pre-existing disease after receiving estrogens alone, and their symptoms did not differ from those of women who received estrogens plus progestins. Moreover, progestins have even been used for treatment of HAE caused by C1-INH deficiency, and improvement of disease has been observed in at least some of the patients treated.302., 303., 304. Most important in the current context, progestin treatment has not been associated with worsening of angioedema. The causative role of estrogens in disease exacerbation suspected in this and other reports is also supported by the fact that attenuated androgens are effective in preventing edema attacks in HAE caused by C1-INH deficiency. All of these observations are consistent with the view that estrogens and not progestins are responsible for provocation of angioedema attacks or worsening disease in women with inherited angioedema who take OC or receive HRT.
The pathophysiological mechanisms underlying the occurrence of angioedema in association with estrogen-containing medications is not known. As stated, available evidence suggests that bradykinin is involved in the development of symptoms in patients with HAE caused by C1-INH deficiency.9., 14., 30. In addition to its inhibitory effects on C1s and C1r, C1-INH blocks the activity of other plasma proteases including kallikrein and factor XIIa/XIIf, enzymes that participate in bradykinin formation. Bradykinin is generated from HK by kallikrein; the activation of prekallikrein to kallikrein is mediated by activated factor XII. The degradation of bradykinin to des-Arg bradykinin and finally vasoinactive peptides is mainly mediated by carboxypeptidase N (kininase I) and angiotensin I converting enzyme (kininase II).30 Estrogen-containing medications might favor edema formation by influencing both kinin generation and degradation. Gordon et al107 showed that C1-INH levels in the plasma of women taking OC are lowered. Administration of estrogens also leads to increased factor XII levels, possibly via a functional estrogen response element in the promoter of the gene encoding factor XII.30., 107., 280. Plasma concentrations of prekallikrein have also been shown to be increased in women using estrogen-containing OC.269 Estrogens might also influence the degradation of bradykinin via their influence on ACE. It has been demonstrated that HRT in postmenopausal women leads to a decrease in ACE activity.257., 292., 305. Nogawa et al292 also observed an increase in plasma bradykinin levels in such women, a finding consistent with the action of ACE in the breakdown of bradykinin.
The mediator responsible for edema formation in HAE type III is not known. However, many clinical similarities exist between HAE-I and type III, and the percentages of women whose disease is negatively affected by estrogen-containing medications is similar in the 2 conditions. These facts could permit speculation that estrogen-associated effects in women with HAE type III may also be related to the kinin pathway.
However, whether the influence of estrogen-containing OC or HRT on the kinin pathway actually plays a role in favoring angioedema formation in patients with inherited angioedema is far from being clear today. The current results suggest that among patients with underlying HAE, administration of estrogen does not always result in the appearance or worsening of symptoms. For example, other factors that might predispose women with HAE to new or exacerbated symptoms when treated with estrogens could include a functionally or quantitatively relevant genetic polymorphism in the kinin system. Information about such polymorphisms might be important in differentiating women whose angioedema would be aggravated by administration of estrogen-containing medications.
Laboratory diagnosis
Recent guidelines for urticaria and angioedema released by the British Association of Dermatologists306 define HAE as angioedema without urticaria and highlight the need for a specific treatment for this condition. Toward this end, the newly created European PREHAEAT network proposes a list of diagnostic criteria for angioedema caused by functional C1-INH deficiency (Table I). Because of the tendency of nonallergic angioedema to mimic other disorders, these criteria rely on both clinical and laboratory assessments. In addition, in the case of asymptomatic or newborn family members of relatives with known HAE, laboratory measures, and genetic analyses represent the sole means of learning whether they, too, have the disease and might have future attacks. These points underscore the importance of laboratory measures in the definitive diagnosis of HAE and related disorders.
Laboratory findings not only permit more complete diagnosis and classification of angioedema caused by inadequate C1-INH function but also form a key part of the evolving understanding of the genetic and biochemical aspects of these diseases. However, currently, no valuable laboratory criteria are available for the recently detected estrogen-dependent inherited angioedema.8., 86., 87. In the report that follows, Varga et al, in an international collaboration arising out of the Third C1 Esterase Inhibitor Deficiency Workshop, set forth guidelines for the proper laboratory investigation of suspected HAE, AAE, and related disorders.
Diagnostic tools in a routine laboratory: problems and opportunities
(Lilian Varga, PhD,∗ Erik Waage Nielsen, MD, PhD,∗ Lennart Truedsson, MD, PhD,∗ C. Erik Hack, MD, PhD,∗ George Füst, MD, PhD, DSc,∗ and Peter Späth, PhD, Budapest, Hungary, Bodø, Norway, Bern, Switzerland, and Amsterdam, The Netherlands)
For diagnostic purposes, the C1-INH protein phenotypes of the various types of angioedema without an allergic or pseudoallergic background are important ( Table VI). Mutations in the coding region of the C1-INH gene leading to HAE are all heterozygous conditions. (There is 1 known exception, an Italian family with a homozygous defect in the gene control region that might have evolved because of multiple consanguinity. However, even as a homozygous mutation, this defect did not lead to a complete deficiency of C1-INH.171., 238.) HAE-I is defined as the presence of 1 mutant C1-INH gene whose gene product is undetectable in the circulation by routine methods. However, ultrasensitive methods might detect abnormal protein in some patients diagnosed with HAE-I by routine analysis. In these patients, a reduced amount of normal C1-INH is found (Table VI). HAE-I may result from the production of a missing mutant protein, eg, from a mutation resulting in a stop codon, production of a mutant protein that is subsequently degraded intracellularly, production of a mutant protein that is not secreted and that might accumulate intracellularly, or production of a mutant protein that is released but rapidly catabolized or degraded in the circulation.
Table VI.
At-a-glance comparison of bradykinin, complement protein concentrations, and C1-INH function in HAE and AAE vs in HUVS and ACE inhibitor–induced angioedema
| Condition | C1q concentration | C1s concentration | C1-INH concentration | C1-INH function | C4 concentration | C3 concentration | Bradykinin concentration |
|---|---|---|---|---|---|---|---|
| HAE-I | N | N | <30% | <30% | L∗ | N | TR† |
| HAE-II | N | N | N/H | <30% | L | N | TR |
| Estrogen-dependent inherited angioedema | N | N | N | N | N | N | ? |
| Acquired C1-INH deficiency type I | <30% | <30% | <50% | L | <30% | N/L | TR |
| Acquired C1-INH deficiency type II | L/(N)‡ | L | N/L | L | <30% | N/L | TR |
| Angioedema in association with sex hormone balance shifts§ | N | N | N | N | N | N | ? |
| HUVS | <30% | N | N/H | N/L | N/L | ? | |
| ACE inhibitor–induced angioedema | N | N | N | N | N | N | TR |
H, High; L, low; N, normal; TR, transient rise.
A normal C4 concentration was reported in 1 patient by Karim et al.332
A transient rise in bradykinin can be measured during attacks.14
At least 1 patient with normal C1q level was reported.114
Female patients and 1 male patient with recurrent edema caused by androgen deficit.279
Hereditary angioedema type II is defined as 1 C1-INH gene producing a dysfunctional protein that is released and coexists with the normal protein in patients' circulation. The current routine analytic tools for detecting C1-INH antigen are unable to distinguish between normal and abnormal forms of circulating C1-INH. As a consequence, normal or elevated concentrations of the protein are found (Table VI). Notably, the distinction between HAE-I and HAE-II is not absolute because some mutant protein can be detected in selected patients diagnosed with HAE-I by using the most sensitive detection systems rather than routine methods. Thus, the classification or diagnosis of HAE-I or HAE-II might depend on the sensitivity of the analytic system used. Independent of the type of HAE, the affected heterozygous individuals show concentrations of native, functional C1-INH far below 50% of normal.
Estrogen-dependent inherited angioedema patients (formerly HAE type III) show no abnormalities in C1-INH concentration or function (Table VI). Tissue swelling in the presence of normal C1-INH concentration and function in this form of inherited angioedema might be understood through the assumption that attacks are related to transient increases in bradykinin concentration.
For the classification of the acquired forms of angioedema, the presence of a pathogenic autoantibody to C1-INH is relevant: type I shows no such pathogenic autoantibody, whereas in patients with type II, such autoantibodies are detectable. However, the distinction between type I and type II AAE may become obsolete over a disease course, because patients might present with symptoms in the early stage of AAE without detectable autoantibodies, whereas at later stages, autoantibodies can be detected.131 Regarding the distinction between the 2 types of AAE, the diagnostic tools used and their sensitivities or accuracy of assessment might be crucial for classification.132 The presence of anti–C1-INH autoantibodies causes hypercatabolism of C1-INH; increased amounts of a cleaved C1-INH with a reduced molecular weight are found in the circulation in patients with AAE.
Finally, a few patients with recurrent, nonallergic angioedema in association with androgen deficit have been reported.279 The report mentions an affected male patient in addition to affected female patients; disease manifestation was reversible by appropriate correction of androgen levels. Inheritance was not reported. It remains open whether this form fits best to AAE.
According to comprehensive family studies and case reports, the onset of HAE-I can manifest during an affected child's first years of life.307., 308., 309. As early as 1966, a high number of colicky babies were reported in families with HAE.310 Several authors have observed the onset of the disease beginning in children 1.5 years of age.311., 312. Farkas et al79 found clinical manifestations of HAE in children from 2.5 years. In a large HAE-affected family in northern Norway, children with the disease had their first typical symptoms at 3 to 4 years of age. However, in 1 case, a mother noticed different phonation, edema of the mouth and tongue, and enlargement of the uvula in her 6-month-old child, arising during a period of primary teeth eruption.24 Even if HAE often makes it debut later, the statement in a review on the management of HAE in pregnancy in the journal Anesthesia that “HAE is always asymptomatic in infancy”313 is misleading. Early infancy is a likely age for the first manifestations, and physicians dealing with newborns with HAE-affected parents should suspect an underlying C1-INH deficiency as a potential cause of symptoms such as unspecific recurrent abdominal pain, diarrhea, upper respiratory disease, or skin rashes. Therefore, a reliable diagnosis of children born to HAE-affected parents is required as soon as possible. A C1-INH concentration or functional activity <50 % of normal, in conjunction with low C4 concentration, supports a diagnosis of HAE.314
Tools for laboratory diagnosis
Life-threatening laryngeal edema in association with inadequate C1-INH function in untreated or poorly treated patients has been reported to have a lethality of 14% to 30%.7., 31., 315. Because C1-INH functional deficit might remain clinically silent for decades, the high lethality because of acute airway obstruction underscores the importance and significance of awareness of this syndrome and the availability of an expert laboratory to assess C1-INH functional deficiency.
Laboratory analyses should be performed under at least minimal quality assurance conditions. Test systems using calibration curves with only 3 points must be considered as not meeting the standard; through 3 points, every curve fitting is possible. The test system must also include 2 quality control specimens: 1 normal to high and 1 low. A test is valid only if the quality control measurements are within a range of predefined values. It is advisable to assess the laboratory's own normal range. The number of normal sera/plasma used (in Bern) for such a purpose is >50. A standard specimen is prepared by pooling equal aliquots of each serum/plasma; next, testing individual plasma samples against the pool provides the normal range.
Material for analysis
For complement protein concentration measurements, EDTA-treated plasma is best suited. In the event that citrated plasma is used, normal ranges assessed with citrated plasma must be applied to correct for dilution by the citrate. For assessment of C1-INH function, serum or citrated plasma is convenient; heparinized plasma should not be used.301 A diagnosis of any type of C1-INH deficiency or dysfunction should be confirmed with at least 2 assessments by using samples drawn 1 to 3 months apart with the patient in basal condition.
For the early diagnosis of newborns born to parents with HAE-I, sufficient blood can easily and painlessly be obtained from the placenta or the umbilical cord at birth. Because newborns have a very high hematocrit of 0.6 and a consequently low plasma portion, the amount of protein will be diluted when using citrate as anticoagulant.314 This should be kept in mind when comparing reference values from newborns; citrate dilution of samples must also be considered with samples from adult patients.
C1-INH concentration
The concentration of C1-INH protein can be assessed by several routine methods such as radial immunodiffusion, nephelometry, or ELISA. The mean serum concentration in normal individuals is around 0.2 g/L.
In the sera of newly diagnosed patients with HAE-I or patients with HAE-I patients who do not need medication because of mild symptoms, the mean concentration of C1-INH protein is considerably reduced ( Table VII). In patients with HAE-II, the serum C1-INH concentration is approximately 100% or may be elevated to 250% to 350%. The latter excess is a consequence of an Arg444Cys mutation of the reactive site, resulting in a free sulfhydryl group, enabling the mutated C1-INH protein to bind to other serum proteins (Table VII).
Table VII.
Example C1-INH concentrations in serum samples from patients with angioedema symptoms visiting the Angioedema Center in Hungary∗
| Patients | C1-INH (%) | 25th-75th Percentile | Number of samples |
|---|---|---|---|
| HAE-I | |||
| New† | 22.0 | 11.5-29.5 | 25 |
| Untreated‡ | 19.5 | 7.5-28 | 74 |
| Treated§ | 23.0 | 12.5-30.5 | 45 |
| HAE-II | 287 | 267-327 | 9 |
| Not HAE | 118 | 100-135 | 330 |
C1-INH concentrations are reported as percentage of normal (normal range is 64% to 166%).
New: C1-INH concentration was determined at the first visit of those patients in whom a diagnosis of HAE-I was later proven.
Untreated: Patients had only mild symptoms and required no treatment.
Treated: Patients receiving androgens and/or replacement infusions with C1-INH concentrate.
In the sera of patients with AAE, the concentration of C1-INH can be low or normal. Normal antigenic amounts of C1-INH can be detected if interaction with autoantibodies against the 105-kd C1-INH protein results in a cleaved, nonfunctional 96-kd protein.128
C1-INH function
C1-INH function can be assessed by several methods.316 When analyzing samples from a patient with HAE-I before and 30 minutes after C1-INH concentrate infusion, considerable differences emerge in rise of apparent C1-INH function, depending on which method is applied ( Table VIII). C1-INH function, when assessed by enzyme-based chromogenic assays, results in a linear relationship with the antigenic concentration of the C1-INH protein. The chromogenic substrates used are EtCO-Lys (ɛ-Cbo)-Gly-Arg-pNA (formerly from Immuno, Vienna) or MeCO-Lys (ɛ-Cbo)-Gly-Arg-pNA (Aventis Behring, Marburg, Germany). Some reports favor the assessment of C1-INH function by loss of immunologically reactive C1r. The method uses the natural substrate C1r of C1-INH as the indicator and works exclusively with endogenous complement. By using this inexpensive type of assay system, one can observe a functional threshold value for C1-INH concentration and function.138 Formation of C1-INH–C1r or C1-INH–C1s complexes can be assessed by an ELISA test system that also uses the natural but exogenous substrate of C1-INH as an indicator. By commercial ELISA tests based on the detection of formed C1-INH–C1s complexes, the median functional concentration is less than 50% of normal at the time of diagnosis ( Table IX, new group), although normal functional C1-INH concentrations were observed in 6 of these 25 cases despite a confirmed HAE diagnosis via DNA analysis. Normal functional C1-INH concentrations were detected in 22 of 72 sera (30%) of 36 patients who were not treated with attenuated androgens or C1-INH concentrate (Table IX). This observation is somewhat discordant with numerous observations made in Bern, reported in Table VIII.
Table VIII.
Alteration of C1-INH function measured by 4 different methods and changes in C1-INH concentration vs therapy regimen in 1 patient with HAE∗
| Therapy and parameters | Normal range | Before infusion | 30 min after infusion | P value |
|---|---|---|---|---|
| 1000 U of C1-INH concentrate per week (for 15 doses) | ||||
| Concentration (%) | 55-135 | 18.2 ± 4.9 | 35.2 ± 8.4 | <.001 |
| EtCO-R (%) | 70-130 | 10.6 ± 10.5 | 27.1 ± 22.5 | .055 |
| MeCO-R (%) | 80-125 | 11.9 ± 5.4 | 26.2 ± 10.9 | .001 |
| C1r/C1-INH (%) | 70-135 | 29.8 ± 20.9 | 87.7 ± 40.0 | <.001 |
| C1s/C1-INH (%) | >68 | 19.3 ± 10.5 | 54.1 ± 16.4 | <.001 |
| 500 U of C1-INH concentrate twice per week (for 16 doses) | ||||
| Concentration (%) | 55-135 | 20.5 ± 5.8 | 30.6 ± 6.6 | <.001 |
| EtCO-R (%) | 70-130 | 11.9 ± 11.0 | 23.5 ± 14.7 | .015 |
| MeCO-R (%) | 80-125 | 19.4 ± 9.1 | 26.2 ± 9.7 | .012 |
| C1r/C1-INH (%) | 70-135 | 38.4 ± 9.8 | 73.6 ± 30.0 | <.001 |
| C1s/C1-INH (%) | >68 | 23.8 ± 13.6 | 43.8 ± 16.6 | <.002 |
| 500 U of C1-INH concentrate twice a week (for 31 doses) + danazol 100 mg/d | ||||
| Concentration (%) | 55-135 | 23.2 ± 3.4 | 34.8 ± 4.7 | <.001 |
| EtCO-R (%) | 70-130 | 13.2 ± 16.5 | 25.3 ± 20.3 | .012 |
| MeCO-R (%) | 80-125 | 9.1 ± 6.2 | 20.1 ± 11.9 | <.001 |
| C1r/C1-INH (%) | 70-135 | 47.3 ± 23.6 | 85.6 ± 25.8 | <.001 |
| C1s/C1-INH (%) | >68 | 24.0 ± 11.0 | 42.7 ± 7.2 | .090 |
A previously untreated patient received initial treatment with 1000 U of C1-INH concentrate (Aventis-Behring). Because of the patient's tendency to develop an attack before the next C1-INH concentrate infusion, the weekly dose of C1-INH was divided into two 500 U infusions. Several times, while receiving 1000 U C1-INH concentrate per week, the patient had an attack the day or a day before the next infusion.316 The possible effect of a baseline therapy of danazol in HAE patients is best seen in samples before when comparing therapy with 500 U C1-INH concentrate per week with 500 U C1-INH concentrate per week plus danazol 100 mg/d. R = (ɛ-Cb0)-Gly-Arg-pNA.
Table IX.
C1-INH functional values in samples from patients with angioedematous symptoms visiting the Angioedema Center in Hungary
| Patients | C1-INH function∗ | 25th-75th Percentile | Number of samples |
|---|---|---|---|
| HAE-I | |||
| New | 44.8 | 33.0-64.5 | 25 |
| Untreated | 60.2 | 28.1-71.1 | 74 |
| Treated | 62.6 | 40.4-76.1 | 45 |
| HAE-II | 44.0 | 25.0-59.5 | 9 |
| Not HAE | 92.7 | 82.6-103.7 | 330 |
Functional C1-INH was assessed by using a commercial ELISA kit based on detection of formed C1-INH–C1s complexes. Functional level is reported as percentage of normal, where normal is >69%.
Analysis of other complement components
To exclude or to confirm an acquired form of C1-INH deficiency, functional C1 can be assessed by a hemolytic assay, or 1 of the subcomponents of C1, C1q, C1r, and C1s can be assessed by radial immunodiffusion or by ELISA. In HAE, C1 or its subcomponents are normal, as indicated in Table X, whereas decreased concentration of 1 of these proteins together with deficient C1-INH function or low C1-INH concentration is indicative of acquired C1-INH deficiency. However, low concentrations of C1q are not exclusive to AAE. Levels lower than the normal limit can eventually be found in 10% to 20% of blood samples of newly diagnosed (15%), nontreated (10%), or previously diagnosed patients with HAE. Furthermore, low C1q and C4 occasionally accompanied by consumptional loss of C1-INH is observed in anti-C1q autoantibody associated hypocomplementemic urticaria-vasculitis syndrome (HUVS; Table VI). Diagnosis of AAE can be cumbersome, because normal C1q/C1 values have been observed in this condition, particularly in those patients with autoantibody-mediated C1-INH deficiency.114., 132.
Table X.
C1q, C4, and C3 concentrations in samples from patients with angioedema symptoms visiting the Angioedema Center in Hungary in 2003∗
| Patients | C1q | C4 | C3 | n |
|---|---|---|---|---|
| HAE-I | ||||
| New | 93.9 (64.6-121.0) | 0.06 (0.02-0.12) | 1.03 (0.74-1.18) | 25 |
| Untreated | 97.2 (75.6-132.5) | 0.04 (0-0.08) | 1.04 (0.83-1.19) | 74 |
| Treated | 113.0 (89.4-151.0) | 0.05 (0.01-0.08) | 1.05 (0.75-1.31) | 45 |
| HAE-II | 96.7 (66.2-116.8) | 0.05 (0-0.11) | 1.16 (0.90-1.30) | 9 |
| Not HAE | 130.9 (101.0-172.0) | 0.39 (0.29-0.47) | 1.14 (0.89-1.39) | 330 |
C1q mean concentration is given in mg/L (normal, 60-180 mg/L); C4 concentration is given in g/L (normal, 0.15-0.55); C3 concentration is given in g/L (normal, 0.7-1.8). Values in parentheses indicate the 25th to 75th percentiles.
The concentration of C4 may be assessed by nephelometry or radial immunodiffusion by using polyclonal antibodies. A substantial part of the already diminished C4 actually consists of C4 split products,317 as can be measured with a monoclonal antibody specific for a neoepitope on C4 split-products.318., 319. The ratio of C4 split-products over C4 was 0.39 for a normal subject and 4.34 for the subject's cousin with HAE.320 Functionally active C4 can be determined by hemolytic assay. In AAE with diminished C1, the replacement of C1 must be considered, because the low C1 value may hamper C4 assessment. The antigenic or functional concentration of C4 is very low in almost all newly diagnosed or untreated cases of HAE (Table X) and AAE. In Hungary, a borderline C4 concentration was found in only 1 of 25 newly diagnosed patients with HAE. Thus, for an inexpensive follow-up, assessment of C4 concentration might suffice. Indeed, increased C1-INH concentration and function because of therapy that resulted in a reduction in edematous attacks was associated with a measurable increase of C4 concentration to nearly normal when using polyclonal antibodies to assess C4 ( Fig 24). It should be noted that such a test system is unable to distinguish between natural and cleaved C4.
Fig 24.
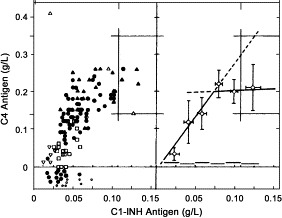
Assessment of C4 antigen concentration by polyclonal anti-C4 antibodies. Assessment of C4 antigen levels by polyclonal anti-C4 antibodies might serve as a simple follow up parameter for therapy. Upper left and right panels depict C1-INH versus C4 antigen concentrations in 111 plasma samples from 21 patients with HAE-I. In the left panel, the individual C1-INH concentrations are plotted against C4 concentrations in plasma samples from patients with various therapeutic managements. Open square, no therapy; inversed open triangle, therapy with tranexamic acid; closed circle, therapy with danazol; closed triangle, therapy with stanozolol. Open squares and triangles represent samples from patients with current or recent bacterial or viral infections and therapy with danazol or stanozolol. Open squares with a crossing line indicate samples from an asymptomatic woman, a member of a family with concomitant heterozygous C1-INH deficiency and factor I protein gene mutation, herself heterozygous for C1-INH only. In the right panel, the means (±1 SD) of concentration ranges are depicted. The lower left panel indicates clinical manifestation of the C1-INH deficiency in 111 patients with HAE-I: closed circles give C1-INH concentration in plasma collected after an attack, and open circles indicate mean C1-INH concentrations of 2 assessments between which an attack occurred.
A diminished antigenic level of C3 is extremely rare in HAE, but in 10% to 20% of cases, even in 5 of 25 in newly diagnosed patients with HAE, a slightly depressed concentration of C3 was measured in Hungary. There is no correlation between the concentration of C1q and C3.
Detection of pathogenic anti–C1-INH autoantibodies
The majority of patients with AAE have autoantibodies to C1-INH in their serum (AAE type II). These antibodies bind epitopes within or close to the reactive site of C1-INH. Anti–C1-INH autoantibodies that bind to C-INH with different and generally low affinities can belong to each class (IgG, IgA, or IgM). Anti–C1-INH autoantibodies are detected by ELISA.
Diagnosis of C1-INH deficiency in newborns at risk
In adults with angioedema caused by C1-INH deficiency, determination of C4 and C1-INH antigenic and functional concentrations nearly always allows a correct diagnosis. However, these complement components show considerable age-specific variations in children. The concentration of C1-INH antigen measured in umbilical cord blood, as a rule, is approximately ⅔ the concentration found in adults.321., 322., 323., 324., 325. A reference range for the common functional C1-INH test in newborn cord blood was published in 1994.320 The percentiles are presented in Table XI. Functional assays even allow the prompt detection of the rare form of HAE, ie, HAE-II, in which the antigenic concentration of C1-INH is normal or even higher than normal, but mostly consists of a dysfunctional protein. Whereas in the 1994 study the mean C1-INH functional activity in healthy newborns was approximately 70% of the mean adult value, Sonntag et al318 found a somewhat higher C1-INH function (∼90%) in their study in citrated cord blood. During the child's first days, C1-INH and C4 values rapidly rise and may reach 1.5 times values found in adults.326 Andrew et al327 found that near-adult values are achieved for C1-INH by 3 months of life. In healthy premature infants, C1-INH concentrations reached the normal adult range by 1 week of age.328 Norman et al329 found large variation in C4, which in their opinion limits the diagnostic usefulness of quantitative C4 concentrations in children.
Table XI.
C1-INH in umbilical cord blood from 80 normal newborns∗
| C1-INH functional assay (%) | C1-INH antigen concentration (g/L) | Birth weight (g) | Minutes from birth to blood collection | |
|---|---|---|---|---|
| Minimum | 45.6 | 0.07 | 2580 | 2 |
| 2.5th percentile | 47.2 | 0.11 | 2825 | 2 |
| 10th percentile | 49.0 | 0.13 | 3030 | 8 |
| Median | 64.5 | 0.17 | 3580 | 14 |
| 90th percentile | 78.0 | 0.21 | 4230 | 23 |
| 97.5th percentile | 85.9 | 0.22 | 4650 | 38 |
| Maximum | 104.3 | 0.25 | 4850 | 59 |
Functional C1-INH was assessed by using a modified chromogenic assay (Aventis Behring).300
Diagnostic problems and strategy
Several consequences result from the aforementioned experiences. To confirm or disprove a suspicion of clinical HAE-I, measuring the concentration of C1-INH protein is satisfactory; however, this is not adequate to detect HAE-II or AAE type II. Assessment of C1-INH function is therefore required. Normal functional C1-INH concentrations can be observed in patients with HAE under therapy when measured by assays based on the formation of C1-INH–C1r/C1s complexes. If the analyzing laboratory is unaware of this fact and receives a sample from a patient not known in the laboratory, a diagnosis of HAE might be missed. In contrast, C1-INH measurements using commercial enzyme-based chromogenic assays yielded a false diagnosis of HAE in 26% of samples.330 The positive predictive value for a diagnosis of C1-INH deficiency was only 36% with the chromogenic assay, whereas it was 92% with the C1-INH–C1s ELISA test kit. The negative predictive value was very high (99%) with the chromogenic assay and was 89% with the C1-INH–C1s ELISA314 (Varga et al, Unpublished data, April 2003). Improper sample handling might account for some of the differences. However, the considerable differences in apparent C1-INH function seen with the different test systems were also seen when assessed in samples collected, frozen in aliquots, and thawed only once (Table VIII).316 Methods that are based on determination of C1-INH–C1r/C1s complexes appear more sensitive to changes at low C1-INH concentrations. Apparent functional levels measured by these methods correlate well with the clinical status and with increase in C4 concentration (Fig 24). Normal functional activity of C1-INH (based on the detection of C1-INH–C1r or C1-INH–C1s complexes) is frequently found not only in patients under successful treatment but also in patients having mild symptoms who therefore do not require treatment. A cautionary note: patients with HAE-II who, because of borderline C1-INH functional concentrations, have only mild symptoms, can escape diagnostic confirmation despite the application of both antigenic and functional determinations of C1-INH.
Measurement of C4 concentration can be used as a screening marker when HAE is suspected. However, low C4 is not sufficient for diagnosis, because this is seen in many conditions with classic pathway activation. If C4 and the antigenic concentration of C1-INH are both normal, C1-INH functional deficiency can be excluded. Low C4 concentration is also a characteristic feature of AAE. Diminished amounts of C1 or its subcomponents, preferably C1q, are indicative of an acquired C1-INH deficiency. However, low C1 (q, r, s) together with low C4 is not a proof for AAE (Table VI). A normal complement profile (normal antigenic C1-INH, C1q, and C4) was reported in 2 patients with AAE.113., 331. In the latter case, a transient improvement in C1-INH function was associated with a transient loss of anti–C1-INH autoantibodies.
On the basis of the listed diagnostic possibilities, the authors suggest an algorithm for the diagnosis of any type of suspected C1-INH deficiency, as shown in Fig 25. The use of such an algorithm is strongly advised because, on the basis of their experiences, only 1% of patients presenting with angioedematous symptoms are proven to have hereditary or, more rarely, acquired C1-INH deficiency.
Fig 25.
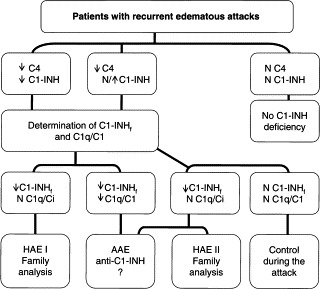
Work-up flow sheet for patients with a clinical history of idiopathic or unknown origin abdominal symptoms or angioedema.
In conclusion, the diagnostic problems of a routine laboratory are caused by the following:
-
(1)
Angioedema within the scope of the Third European C1-INH Deficiency Workshop, ie, not on the basis of allergic or pseudoallergic reactions, can be associated with any type of functional deficiency of C1-INH, but hereditary and nonhereditary forms of angioedema with apparently normal C1-INH function have also been described. An association between hormonal status and the dysregulation of the kinin pathway in these patients is suggested.
-
(2)
One of the predominant kinins, if not the main kinin responsible for angioedema attacks in such patients, is bradykinin. Today, routine assessment of bradykinin is not feasible, nor is the assessment of other kinins that might be involved in edema formation.
-
(3)
In patients with pure functional deficiency of C1-INH, activation of the classical pathway of complement does not progress beyond C2 activation, although mild activation of C3 and the formation of fifth component of the complement cascade (C5) b-9 complexes can be demonstrated during attacks.213., 327. Significant activation of C3 and/or C5 with generation of anaphylatoxins C3a and C5a indicates either a pathophysiologic background other than functional C1-INH deficiency, eg, an immune complex disease, or 2 or more pathological mechanisms coinciding in the patient. Detection of elevated C3 split products such as C3a or C3d should initiate diagnostic measures outside the frame of hereditary or acquired C1-INH deficiency. Detection of C5a in vivo is almost impossible because of rapid binding of this fragment to high-affinity receptors on neutrophilic granulocytes. Hence, elevated C5a levels may be found in neutropenic patients or in patients with fulminate, end-stage disease.
-
(4)
Low levels of functional C1-INH can occur because of in vitro artifacts. A frequent cause is the phenomenon of so-called cold activation, which occurs with some blood samples during storage at 0°C. A normal C4 value points to this phenomenon and rules out a diagnosis of HAE-I or HAE-II, or that of AAE, with 1 reported exception of a patient with HAE-I but normal C4 concentrations.332
Follow-up of patients
According to the literature, C1-INH concentrations in HAE-I patients greater than 35% to 40% of normal are sufficient to provide inhibition of classical pathway activation, a finding that is confirmed by clinical observations. Thus, in such patients, the dose of attenuated androgens should not be increased with the aim to reach antigenic C1-INH levels approximately 50% of normal. Ignorance of this fact might provoke higher and higher doses of, eg, attenuated androgens that bring no added benefit for C1-INH function and might harm the patient's liver. Enzyme-based chromogenic substrate assays are excellent for initial diagnosis of deficient C1-INH function, but seem less well suited for monitoring during therapy (Table VIII). Measurement of C4 concentration is an inexpensive and generally useful parameter, even to follow up therapy, and especially in AAE. Concentrations of C4 close to the lower limit of normal, ie, at the lower 95% limit of confidence, have been found to indicate sufficient function of C1-INH to allow patients to be attack-free (Fig 24).
In summation
For definitive diagnosis of angioedema associated with C1-INH functional deficiency (HAE-I, HAE-II, and AAE), the assessment of C1-INH concentration and/or function and, in some cases, the concentration of at least 1 of the C1 subcomponents are the routine diagnostic tools.
For laboratory diagnosis, measurements should be performed twice, at an interval of at least 1 month, showing C1-INH concentration and/or function <50% of normal. Laboratory diagnosis in newborns is difficult and needs special consideration.
Assessments of other complement parameters, such as C4 concentration, are to be considered surrogate markers, although excellent ones.
No laboratory analyses to reveal estrogen-dependent inherited angioedema are thus far available.
Treatment
In HAE, there are 3 main therapeutic aims: (1) treating acute angioedema attacks, (2) avoiding new attacks via permanent prophylaxis, and (3) avoiding attacks at times of increased risk, eg, after surgery. This section addresses all 3 treatment elements.
Current treatment of HAE and future treatment options
(Konrad Bork, MD,∗ Mainz, Germany)
In this section, Bork describes traditional acute attack treatments as well as what lies ahead. Both short-term and long-term prophylaxis regimens are also discussed.
Current treatment of acute edema episodes
Treatment for acute edema episodes of HAE must be evaluated regarding efficacy, safety, and cost. Laryngeal, pharyngeal, and tongue edema are life-threatening or may progress to a life-threatening state. In these cases, a drug therapy is necessary. During laryngeal edema, according to the state of respiratory distress, further emergency measures may become necessary to keep the upper airways open, such as the use of epinephrine, intubations, or cricothyrotomia. Abdominal attacks caused by intestinal wall edema may be extremely painful and are often associated with vomiting or diarrhea. Severe acute abdominal attacks should be treated; mild abdominal attacks may remain untreated. Skin swelling involving the extremities may also remain untreated.
Drug therapy for acute attacks includes the following:
C1-INH concentrate
C1-INH preparations are purified and pasteurized or vapor-heated concentrates from pooled human plasma and have a half-life of 64 hours. They are effective in preventing and treating acute attacks of HAE caused by C1-INH deficiency and are the treatment of choice where available. In Europe, C1-INH concentrate has been widely used since 1973.333 A randomized, placebo-controlled trial211., 334. was conducted in 11 patients for a total of 55 attacks in the skin, abdomen, and larynx, in which relief was seen after C1-INH concentrate treatment. After C1-INH administration, 69% of the attacks responded after 30 minutes, and 95% responded after 2 hours. Another study focusing on laryngeal edema in 18 patients with HAE showed that, in 192 of 193 laryngeal edema episodes treated with C1-INH concentrate, response occurred within 30 to 60 minutes.45 In the 1 remaining episode, response occurred within 4 hours. In other reports, 8 patients,335., 336. 9 patients,336 and 7 patients337 with attacks of skin, gastrointestinal, or laryngeal edema were successfully treated with C1-INH concentrate.
Because purified C1-INH concentrate is derived from human plasma, virus safety is critical. As mentioned earlier, in 1983 to 1984, 13 patients developed a non-A, non-B hepatitis related to the administration of C1-INH concentrate, probably because of a contaminated batch.7 Since the introduction of viral inactivation steps, transmissions of infectious agents have not been reported.75
Side effects of C1-INH concentrate are rare and include fever and headache. To date, the formation of autoantibodies to C1-INH as a result of C1-INH concentrate administration has not been observed in any patient with HAE.
C1-INH concentrate is expensive because of high production costs, the low number of patients with HAE, and the relatively low dose per patient compared with other blood products, such as those used in hemophilia. C1-INH concentrate is now available in most countries, at least on a named-patient basis, but is not available in the United States.
Fresh frozen plasma
Fresh frozen plasma (FFP) is also effective338 and can be used if C1-INH concentrate cannot be obtained. Because FFP is not virally inactivated, the risk of transmitting infectious agents is relatively high. The administered volume is larger and requires a longer infusion time, which might be critical in emergency situations. The use of FFP is associated with several adverse effects, including urticaria, anaphylactic shock, and hemolysis.
Other
Corticosteroids and antihistamines are usually ineffective. Attenuated androgens require at least a few days to become effective and therefore are usually not administered in acute attacks of HAE. The same is typically true for antifibrinolytics; however, some patients with mild attacks respond to 1g tranexamic acid by mouth every 3 to 4 hours for 12 to 18 hours.339
For abdominal pain during attacks, spasmolytics such as butylscopolamine may be used symptomatically and are of benefit in milder attacks.
Future treatment options for acute edema episodes
Kallikrein inhibitors. The candidate mediator of angioedema caused by C1-INH deficiency is bradykinin, released upon kallikrein activation. More than 25 years ago, aprotinin, a kallikrein inhibitor extracted from bovine lungs, was shown to be effective in treating HAE attacks.340 Because of the risk of fatal anaphylactic reactions,341 its use was discontinued after substitutive therapy with C1-INH concentrate became available.7
A novel kallikrein inhibitor has recently been developed.342 It is a synthetic Kunitz domain protein designed by phage display technology to bind to human kallikrein. It has a high affinity and is highly selective for kallikrein. It is effective in abating acute attacks of HAE but is not yet commercially available.343
Bradykinin antagonists. Because bradykinin is assumed to be the key mediator of HAE, blocking its B2 receptors might be therapeutically useful. Icatibant is a synthetic peptide with a structure similar to bradykinin. However, it contains nonproteinogenic amino acids and is not degraded by the 2 main bradykinin cleaving enzymes, carboxypeptidase and ACE. A potent antagonist, Icatibant has the same affinity for B2 receptors as bradykinin across several tested species, including human beings. It is specific for B2 bradykinin receptors and does not interact with receptors of other peptides. Preliminary studies were performed in human beings with asthma and seasonal allergic rhinitis,344., 345. and Icatibant was also shown effective in reversing increased vascular permeability in the murine HAE model.9 Recently, the results of a phase II single-dose trial for the treatment of acute HAE attacks in 8 patients were reported. In this European trial conducted by Bork et al.345a Icatibant appeared effective and well tolerated.
Recombinant C1-INH. During recent years, a recombinant C1-INH produced in the milk of transgenic rabbits has been developed. However, because of glycosylation differences, it has a shorter half-life than C1-INH derived from human plasma. Initial results in patients with HAE concerning tolerability and safety are encouraging.346 The interim results of a phase II trial conducted by Levi et al346a in acute attacks of HAE appear positive.
Long-term prophylaxis
Long-term prophylactic treatment is indicated for patients with many harmful and disturbing edema episodes, usually patients with more than 1 attack per month. Long-term prophylaxis may be performed with attenuated androgens, antifibrinolytic agents, and C1-INH concentrate. All of these medications are associated with potential adverse effects, limiting their use as standard, life-long prophylactic treatment.
Attenuated androgens. In 1960, Spaulding104 reported the striking efficacy of methyltestosterone in HAE, describing both a decrease in the severity and number of attacks. Later, further androgens, fluoxymestrone in men and oxymetholone in women (with fewer virilizing effects than methyltestosterone), were tested. In 1959, stanozolol was synthesized, having 1 of the largest anabolic/androgenic ratios. In 1963, danazol, a synthetic analogue of ethinyltestosterone and less virilizing than the early synthetic androgens, was developed. In 1976, the attenuated androgen danazol was shown to considerably reduce the number of HAE attacks.347 Since then, attenuated androgens (17-α alkylated androgens), mostly danazol or stanozolol, have been widely used for long-term prophylaxis in HAE.348., 349. Although 17-α alkylated androgens are more efficient than other androgens, they are metabolized by the liver. Thus, hepatotoxicity and the induction of liver cell adenoma or carcinoma are major concerns. Androgens' mechanisms for exerting beneficial effects in patients with HAE are unknown.
Major contraindications for treatment with androgens are pregnancy, lactation, childhood, and prostate cancer. However, as discussed later, there are a few reports on the successful use of androgen treatment in children.
Danazol. A widely used regimen is to start with an induction dose of 400 to 600 mg daily for 4 weeks and then taper down to the lowest maintenance dose that provides symptomatic relief. Most patients' HAE is controlled with 50 to 200 mg daily or every other day. Long-term administration may be associated with several adverse effects, including weight gain, amenorrhea, decreased libido, menstrual irregularities, virilization in women (hirsutism, deepening of the voice, and decreased breast size), acne, muscle cramps, myalgia, fatigue, headaches, hemorrhagic cystitis, arterial hypertension, and hepatic necrosis or cholestasis.348., 349., 350., 351. Recently, hepatocellular adenoma and liver cell carcinoma have been reported in patients with HAE taking danazol for many years.352., 353., 354. Laboratory changes may include decreased sex-hormone binding protein and thyroxine-binding protein, increased aminotransferases and cholesterol, and polyglobulia.
Stanozolol. Stanozolol is used in initial doses of 4 to 6 mg per day for 1 month, then tapered to the minimal effective dose (0.5-2 mg daily). Potential adverse effects may be similar to those of danazol but seem to occur less frequently. However, this needs further confirmation, because fewer patients treated with stanozolol have been reported in the literature compared with patients receiving danazol.
Other androgens. Several patients have been treated with an oxandrolone. A small number of patients do not respond to androgens.
Antifibrinolytic agents. Antifibrinolytic agents may also be used for the continuous prophylactic treatment of HAE.355 However, their efficacy is lower than that of androgens. In about 70% of patients, attacks occur less frequently and are usually milder. A considerable reduction of the number and severity of attacks is reached in only approximately 30% of patients. If long-term prophylaxis is necessary in children, tranexamic acid may be preferable to androgen treatment because androgen treatment should be avoided in childhood. Presumably, antifibrinolytic agents act through the inhibition of plasmin. All antifibrinolytics bear the risk of thromboembolic events.
ε-Aminocaproic acid is effective and reduces the frequency of attacks. The daily dose is usually 8 to 12 g in 4 equally divided doses. It is associated with various side effects such as thrombosis, postural hypotension, muscular pain and weakness associated with an increase in creatine kinase and aldolase, anal pruritus, and myositis.
Because of the many adverse effects associated with ε-aminocaproic acid, tranexamic acid is the antifibrinolytic of choice. The daily tranexamic acid dose is usually 1 to 3 g per day. Side effects are uncommon but include abdominal discomfort, mild transient diarrhea, nausea, headache, and anal pruritus.
C1-INH concentrate. Not only an acute attack therapy, C1-INH concentrate has been found effective in the long-term prophylaxis of HAE. In 1980, the first patients were treated with 500 U C1-INH concentrate 1 or 2 times weekly for 1 year or more.356 A report of 20 patients who received C1-INH concentrate twice weekly was published in 2002.357
Short-term prophylaxis before surgery
Because of the risk of developing angioedema and possibly experiencing life-threatening laryngeal edema after dental surgery, endoscopy, endotracheal intubation, and other surgical procedures, preoperative treatment has been attempted. To date, available information is limited to case reports or small patient series. Furthermore, even in the absence of preoperative prophylaxis, not all patients with HAE develop edema attacks after surgery. Patients who undergo multiple dental surgeries may develop edema episodes after some tooth extractions but not others. It is therefore difficult to assess the efficacy of a recommended preoperative prophylaxis until studies with larger numbers of patients are available.
Such prophylaxis has been performed with FFP, antifibrinolytics, attenuated androgens, and C1-INH concentrate. In several patients, FFP has been used prophylactically before dental surgery; in some patients, mild attacks occurred despite pretreatment with FFP.358., 359., 360. Antifibrinolytics such as ε-aminocaproic acid361 and tranexamic acid362., 363. have also been used for preoperative prophylaxis in some patients. Because antifibrinolytics are less effective in long-term HAE prophylaxis, preoperative treatment focuses on attenuated androgens and C1-INH concentrate. In patients with known HAE caused by C1-INH deficiency, preoperative prophylaxis with attenuated androgens, either danazol or stanozolol, has been shown to be effective in many patients,359., 364., 365., 366. although some patients nonetheless developed swelling.358., 367. For short-term prophylaxis, attenuated androgens may be administered from 5 days before to 3 days after the event, danazol at a dose of 10 mg/kg/d, maximum 600 mg per day, or stanozolol at a dose of 6 mg per day.
Currently, C1-INH concentrate has been used as preoperative prophylaxis in only a few patients; for this purpose, the dose of C1-INH concentrate was 500 or 1000 U.368., 369.
The risk of developing angioedema attacks secondary to dental surgery or other oral manipulations cannot be completely avoided by preoperative prophylaxis. As such, it is important to inform the patient that an angioedema attack might occur, to describe the clinical symptoms of laryngeal edema, and to educate the patient about what to do in case a laryngeal edema occurs.
Treatment of AAE
(Marco Cicardi, MD,∗ Andrea Zanichelli, Laurence Bouillet, MD, CCA, ∗ and Emel Aygören-Pürsün, MD, Milan, Italy, Grenoble, France, and Frankfurt, Germany)
In this section, Cicardi et al describe the typical treatment of AAE and discuss the special considerations unique to this disorder.
From pathogenesis to treatment
Like other acquired forms of protein deficiencies, the course of acquired C1-INH deficiency can be related to the course of the underlying disease. The possibility of reversing the biochemical and/or clinical abnormalities of acquired C1-INH deficiency by curing the associated disease was reported by Cohen et al105 and subsequently confirmed.105., 122., 132., 370., 371., 372. However, the response can be temporary, even without evidence of relapse of the associated disease.105., 373.
Analogous to HAE, patients with acquired C1-INH deficiency have received attenuated androgens for prophylaxis and C1-INH concentrate to treat acute attacks.334., 347. Nevertheless, these patients are frequently resistant to attenuated androgens,114., 371. whereas they tend to benefit from antifibrinolytic agents.114., 133., 374. Antifibrinolytics are thus more effective for long-term prophylaxis in this population and represent the first choice for patients with acquired C1-INH deficiency.134
Replacement therapy with C1-INH plasma concentrate is the treatment of choice for life-threatening laryngeal attacks. However, patients with AAE partially resistant to this treatment have been reported.114., 129., 356., 375. The response to treatment differs from HAE because of the rapid catabolism of C1-INH that characterizes AAE. Higher doses of C1-INH plasma concentrate were required in patients with a slow response to treatment.112 Slow responsiveness is a high-risk condition because it seems to increase with subsequent treatment. A new peptide, DX-88, a synthetic kallikrein inhibitor based on the same functional domain (Kunitz domain) of aprotinin, is under study for treating angioedema in patients with HAE.342., 343. By inhibiting kallikrein activation, this peptide can stop generation of the vasoactive peptide bradykinin from HK,18 the candidate mediator of symptoms in C1-INH deficiency. Bypassing the problem of accelerated C1-INH catabolism, this peptide represents a promising alternative for patients with acquired C1-INH deficiency who are resistant to C1-INH concentrate.
Virus safety of current plasma-derived C1-INH: The Aventis Behring experience
(Mathias Juers, MD, and Albrecht Gröner, PhD, Hattersheim and Marburg, Germany)
For HAE and related conditions, plasma-derived C1-INH concentrate remains the acute attack therapy of choice. In this section, Juers and Gröner detail the steps that are taken to ensure the high margin of virus safety for Berinert P, produced at Aventis Behring GmbH, a ZLB Behring company.
Before the advent of specific therapy, the HAE mortality rate caused by laryngeal edema was as high as 30%; even today, rates as high as 40% mortality associated with HAE have been published.31., 44. The introduction of C1-INH concentrate (Berinert in 1979 in Germany) was followed by the German registration of a pasteurized C1-INH concentrate (Berinert P) in 1985. These drugs significantly changed the treatment procedures for acute attacks in patients with HAE, becoming the treatment standard in the countries where Berinert P is available. In countries where Berinert P or another C1-INH concentrate was not available (for example, the United States), treatment of acute attacks was and sometimes still is usually performed with FFP. However, during the preparation of FFP, no reduction of potentially present viruses is achieved, in contrast with the plasma-derived C1-INH concentrate Berinert P. Similarly, although solvent-detergent treated FFP might seem an alternative, this treatment does not inactivate nonenveloped viruses. The following sections describe the current status of virus safety of Berinert P.
Methods
Berinert P is produced from pooled human plasma, primarily from source plasma, ie, plasma collected by plasmapheresis. In general, plasma-derived products may potentially transmit infectious and/or pathogenic viruses. This risk is minimized by careful selection of donor centers with regard to virus marker rates (epidemiology) and careful selection of donors; each donor, including qualified donors (ie, repeat donors), is evaluated before each donation for vital signs including temperature and blood pressure and subjected to an intensive questionnaire. Details have been described recently.376 Furthermore, every donation is tested with serologic methods for hepatitis B surface antigen (HbsAg) and antibodies against HCV, HIV-1, and HIV-2 as well as elevated alanine aminotransferase levels. In addition, sample pools of donations are tested by using nucleic acid amplification technique (NAT)/PCR for genomic material of hepatitis A virus (HAV), hepatitis B virus (HBV), HCV, HIV-1, and high titers of parvovirus B19 (B19V). Reactive donations are discarded. Furthermore, the plasma pool for fractionation is tested and released for further processing only if the pool is nonreactive (negative) for HbsAg and nonreactive for antibodies against HCV, and HIV-1, and HIV-2 as well as nonreactive for HAV RNA, HBV DNA, HCV RNA, and HIV-1 RNA, and for B19V DNA not exceeding 105 IU/mL. The third main step ensuring virus safety is the elimination and inactivation of possible viral contaminants by the manufacturing procedure, whose capacity for virus reduction is tested in virus validation studies. The virus inactivation/removal steps used in the manufacture of Berinert P use treatment in aqueous stabilized solution at 60°C for 10 hours (pasteurization) and specific chromatography.
Results
In virus validation studies designed to assess the capacity of the manufacturing process to inactivate and/or eliminate intentionally added virus, high overall virus reduction factors were demonstrated ( Table XII). These virus validation studies were performed in accordance with the applicable Committee for Proprietary Medicinal Products (CPMP) Notes for Guidance. Viruses covering a wide range of physicochemical properties were used in these studies (HIV-1 and HAV are relevant viruses, bovine viral diarrhea virus [BVDV] and canine parvovirus [CPV] are specific model viruses for HCV and B19V, respectively, and pseudorabies virus (PRV) is a nonspecific model virus for large enveloped DNA viruses). The results demonstrate that the pasteurization and the manufacturing process of Berinert P effectively inactivate and/or eliminate relevant human pathogenic viruses. To demonstrate that the manufacturing steps explored in the virus validation studies were very robust and that variations in the manufacturing procedure had no effect on the virus reduction capacity, specific studies covering parameters beyond production specification were performed. These studies showed that even under these conditions, a very effective virus inactivation occurred. Regarding analysis of postmarketing surveillance for Berinert P from 1985 until now, no cases of proven virus transmission, according to the assessment of the Global Pharmacovigilance Department at Aventis Behring, were reported, although a total of almost 100 million units of Berinert P were administered.
Table XII.
Mean overall virus reduction factors for HIV, BVDV, PRV, HAV, and CPV for Berinert P
| Virus | Chromatography Virus reduction factor (±SE) [log10] |
Pasteurization Virus reduction factor (±SE) [log10] |
Overall mean virus reduction factor (±SE) [log10] |
||
|---|---|---|---|---|---|
| Run A | Run B | Run A | Run B | Overall | |
| HIV | ≥4.5 ± 0.3 | ≥4.5 ± 0.3 | ≥6.6 ± 0.3 | ≥6.4 ± 0.3 | ≥11.0 ± 0.3 |
| BVDV | ≥5.1 ± 0.3 | ≥5.0 ± 0.2 | ≥9.2 ± 0.2 | ≥9.0 ± 0.2 | ≥14.2 ± 0.3 |
| PRV | ≥6.7 ± 0.2 | ≥6.5 ± 0.2 | 6.7 ± 0.3 | 6.5 ± 0.3 | ≥13.2 ± 0.3 |
| HAV | 3.6 ± 0.1 | ≥3.2 ± 0.2 | ≥6.4 ± 0.3 | ≥6.3 ± 0.2 | ≥9.8 ± 0.2 |
| CPV | ≥6.8 ± 0.2 | 6.6 ± 0.1 | 1.4 ± 0.1 | 1.4 ± 0.1 | 8.1 ± 0.1 |
Discussion
The outbreak of AIDS in the 1980s and the resulting infections caused by blood transfusion and treatment with certain plasma derivatives, mainly factor VIII concentrates, showed that virus safety in plasma proteins is a field of permanent attention, screening, and continuous research. Since the former Behringwerke AG introduced pasteurization in the 1980s (developed by Heimburger primarily for hemophilia products), no HIV transmission via the company's pasteurized products has occurred.377., 378., 379. This pasteurization procedure has been adapted to Aventis Behring's plasma products, including C1-INH concentrate. The pasteurization procedure has been shown to have an excellent track record of virus safety, and the validity of the pasteurization for virus inactivation was confirmed in scientific articles.380., 381., 382. Pasteurization is a very effective and robust virus inactivation method, but there are also limits. Animal parvovirus, for example, is inactivated by pasteurization only to a certain degree. However, because of recent findings, it should be kept in mind that the human B19V is heat-sensitive and can be effectively inactivated by pasteurization; preliminary data from the group's own experiments (data not shown) also confirm this observation for stabilized aqueous solutions.383 In addition, antibodies against B19V in the plasma pool for fractionation and in intermediates derived from that pool will neutralize B19V to a high degree. Furthermore, regarding CPV, the specific chromatographic step was validated for its capacity to remove CPV, and a very effective and robust removal could be demonstrated. In conclusion, on the basis of the data from virus validation studies, 2 independent effective virus reduction steps in the production procedure for Berinert P, effective for a wide range of enveloped and nonenveloped viruses, could be demonstrated.
As outlined in the CPMP Guideline (CPMP/BWP/268/95), the aim of virus validation studies is “(i) to provide evidence that the production process will effectively inactivate/remove viruses which are either known to contaminate the starting materials, or which could conceivably do so, and (ii) provide indirect evidence that the production process might inactivate/remove novel or unpredictable virus contamination.” So far, all new emerging viruses or members of the same virus family, which may be of severe concern in a blood transfusion setting, were shown to be reduced through manufacturing procedures. This was especially demonstrated for West Nile virus and in preliminary studies for the severe acute respiratory syndrome model virus (data not shown).384 The high margin of virus safety of Berinert P and other pasteurized products by Aventis Behring has continuously been confirmed in clinical publications over the years.385., 386., 387., 388., 389. In conclusion, on the basis of virus validation studies and testing the starting material for relevant viruses, the currently established safety and quality standards at Aventis Behring meet all requirements as documented in postmarketing surveillance data regarding the virus safety of Berinert P. Exceeding the requirements established by authorities, Aventis Behring is testing the starting material for Berinert P for HBV, HIV-1, HAV, and B19V via NAT/PCR.390 Therefore, management of donations (eg, as long as 6 months quarantine for donations from first-time donors [applicant donors], and 60 days inventory hold of donations from repeat donors [qualified donors] to discard a donation before entering the fractionation process based on Look Back procedure); NAT/PCR testing for HAV, HBV, HCV (HCV is mandatory in Europe), HIV-1, and B19V; quality assurance reviewing current Good Manufacturing Practice; and the internal release procedures overseen by Aventis Behring's quality control as well as external ones by the competent authority Paul-Ehrlich-Institut all facilitate the virus reduction capacity of the manufacturing procedure, improving the margin of safety beyond the numbers reported for the overall virus reduction factors. Furthermore, 2 independent and effective virus reduction steps are inherent in the manufacturing procedure of Berinert P. In conclusion, since its introduction in 1985, no cases of proven virus transmission have been reported to Aventis Behring Global Pharmacovigilance, although more than almost 100 million units of Berinert P have been sold.
The authors dedicate this section to Prof. Dr. Norbert Heimburger, inventor of the specialized pasteurization step that has protected thousands of patients with hemophilia from HIV transmission during treatment with their pasteurized product.
Access to C1 inhibitor for patients with HAE and AAE in London, UK: an audit
(Hilary J. Longhurst, MA, MRCP, PhD, MRCPath,∗ London, UK)
Although C1-INH concentrate is largely accepted as the current treatment of choice for HAE and AAE among practitioners who regularly treat these diseases, some governments and private healthcare institutions refuse to stock hospitals with this potentially life-saving medication or fund patients to keep a personal dose. In this section, Longhurst reports the results of a local audit investigating the effects of C1-INH concentrate availability on patients' lives.
C1-INH concentrate is the treatment of choice for laryngeal and severe abdominal edema in patients with HAE and AAE and in the United Kingdom (UK) is usually administered in the nearest emergency department.45., 334. However, C1-INH concentrate is not universally available in the UK and cannot be obtained quickly in an emergency. For this reason, the author recommends that patients keep 1 treatment dose (usually 500 IU) of C1-INH concentrate at home to bring to their local hospital for infusion in case of emergency. In addition, she recommends that the local hospital stocks C1-INH concentrate. Patients receive this if their family doctor (general practitioner) or local primary care trust (PCT) agrees to pay for it. Not all UK doctors and PCTs accept the need for patients to hold their own supply of C1-INH concentrate. All of the author's patients carry a letter detailing emergency management of HAE and giving contact numbers at which 24-hour expert medical advice is available. Despite this, several incidents and near misses have resulted from difficulties with access to C1-INH concentrate. The author's group's quality standard was that every patient should have timely access to C1-INH concentrate for treatment of severe or life-threatening attacks.
Methods
Twenty-four patients attending the author's center with confirmed HAE or AAE, under follow-up for 5 to 48 months, were audited. The occurrence of HAE-related events and extent of access to C1-INH concentrate were determined by reviewing patient and pharmacy records.
Results
Of 24 patients, 13 had access to C1-INH concentrate at home: 4 patients had been prescribed C1-INH concentrate before referral to the center, and a further 9 received C1-INH concentrate after a delay of 0 to 6 months. Despite the group's recommendation, 11 patients did not receive C1-INH concentrate. Of these, 3 did not have access to C1-INH at their local hospital.
Patients had medical, psychologic, and economic difficulties attributable to HAE or AAE. Among patients with a home supply of C1-INH concentrate, 3 patients had 4 avoidable adverse effects. One developed severe anxiety and depression requiring outpatient psychiatric care on being diagnosed with AAE. Another was unable to work as a baker on several occasions because of severe hand edema. The third had frequent angioedema and long-standing severe temperomandibular pain, which resolved when she was started on regular C1-INH concentrate. This patient required frequent absences from work.
Of patients who did not have access to C1-INH concentrate at home, 7 patients had 16 avoidable adverse effects. As Fig 26 illustrates, 2 patients had 6 avoidable admissions to hospital, attributable to C1-INH concentrate treatment not being given or being delayed, resulting in suboptimal response to therapy. One man required intubation and a 5-day stay in the intensive care unit when no C1-INH concentrate could be found in the department. The same man was listed for surgery (tracheotomy for laryngeal edema and laparotomy for acute abdominal pain and guarding) on 2 occasions despite carrying a letter giving advice concerning the emergency management of his AAE. On both occasions, after calls to his regular physician, he responded to treatment with C1-INH concentrate, and surgery was canceled. In addition, 3 patients were treated with FFP, a product that is likely to be less safe and possibly less efficacious than C1-INH concentrate.75., 382. One woman received an average of 2 units of FFP each month at her local emergency department, where personnel were unwilling to prescribe C1-INH concentrate.
Fig 26.
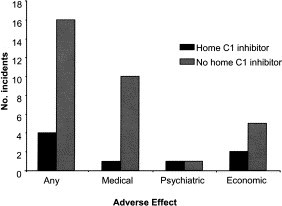
Adverse events attributable to HAE or AAE in patients with and without a supply of C1 inhibitor at home.
One patient had severe anxiety and depression and was unable to work. She improved when started on the home therapy program. However, she had a severe exacerbation of her psychiatric condition when funding difficulties put the future of her home therapy in doubt. Four additional patients had 1 day or more per month of absence from work or school because of symptoms, and 1 patient requested a private prescription of C1-INH concentrate to enable him to travel abroad safely, despite being entitled to C1-INH concentrate funded by the UK National Health Service.
Discussion
Many UK patients have difficulty in accessing appropriate emergency treatment for HAE or AAE, with avoidable medical, psychiatric, and economic consequences. The events recorded here are the most serious. Many patients have ongoing anxiety concerning access to treatment. Some are reluctant to travel; many accept frequent painful attacks and disruption of education or work without complaint (Price; UK HAE network; Personal communication, June 2004). This audit suggests that patients who carried their own supplies of C1-INH concentrate were less likely to have difficulties in accessing appropriate care and were less likely to have ill effects.
Reasons for PCTs' reluctance to fund home C1-INH concentrate included lack of familiarity with the use of C1-INH, reluctance to use a product not licensed in the UK, lack of national management guidelines for HAE and AAE, lack of agreement among UK specialists of the necessity for a home C1-INH supply, expense of the product, and the mistaken belief that the hospital had funds to prescribe C1-INH concentrate.
On the basis of their experience, the author's group recommends that all patients with HAE or AAE should carry their own supply of C1-INH concentrate and that their local hospital should stock additional supplies so that this can be replaced immediately after use.
To improve access in the UK, the author recommends negotiation with UK primary care trusts to fund costs of HAE or AAE treatment for all patients, development of national and international guidelines concerning HAE management and supply of C1-INH concentrate, and improved education for medical staff.
Home therapy for HAE: the Barts experience
(Hilary J. Longhurst, MA, MRCP, PhD, MRCPath,∗ and Caroline O'Grady, RGN, London, UK)
In this companion piece, Longhurst and O'Grady describe a program allowing patients with HAE to self-infuse C1-INH concentrate and detail its outcome.
The benefit and safety of home therapy is well established for many patient groups, including those requiring blood products such as Igs for antibody deficiency and factor VIII for hemophilia A.391., 392.
In the UK, patients with HAE are advised to attend their local emergency department for treatment of severe or potentially life-threatening angioedema attacks. Patients with frequent attacks are also offered prophylaxis with attenuated androgens, such as danazol, or tranexamic acid.83., 339. Despite prophylaxis, a minority of patients with C1-INH deficiency require repeated emergency hospital visits for administration of C1-INH concentrate to treat life-threatening laryngeal swelling and severe abdominal pain. Waiting times in UK emergency departments can be several hours, and personnel may be unfamiliar with managing HAE. As a result, hospital attendance is disruptive to the patient, who may be tempted to wait until the attack is severe, with potentially serious consequences.
Methods
The authors have introduced a home therapy program to improve access to treatment, thereby improving quality of life for people with severe HAE and reducing the use of emergency services.
Patients entering the home therapy program must fulfill the following criteria:
-
(1)
Proven C1-INH deficiency
-
(2)
Prophylactic therapy optimal
-
(3)
Required infusions of C1-INH concentrate at least every 3 months
-
(4)
Infusion partner available
-
(5)
Commitment to program
Of 34 patients considered for the program, 4 patients from the same family were already self-administering (under the care of their general practitioner) and received a refresher course. Six further patients' symptoms were not controlled after optimizing prophylactic therapy. Of these, 4 patients met the criteria for inclusion, 1 patient was refused funding, and 1 patient did not wish to have home therapy. All 4 patients were trained and successfully use C1-INH home therapy.
A minimum of 6 hours training over a period of 3 visits was followed by a formal assessment of patient and infusion partner. Patients kept a record sheet of all attacks and contacted the specialist nurse if they infused. Records were reviewed and infusion technique (using sterile saline) and knowledge were assessed at 3-month intervals.
Case reports
Patient A, a 30-year-old female patient, was initially treated with danazol. This was stopped because of persistently abnormal liver function. Tranexamic acid was only partially effective. She had attacks of severe angioedema affecting the skin, abdomen, and occasionally the larynx, requiring treatment with 1500 IU of C1-INH concentrate at least once a week. Intractable temperomandibular pain developed. She was started on C1-INH concentrate (Berinert P) 1000 IU twice weekly. Her temperomandibular pain improved, and she had fewer angioedema attacks.
Patient B, a 51-year-old female patient, had discontinued danazol because of excessive weight gain and voice abnormalities. She had subsequently received depot methyltestosterone 100 mg subcutaneously every 3 months without side effects for many years, in combination with tranexamic acid 1g twice daily. However, she had cutaneous and severe abdominal angioedema every 3 to 4 days. Her mother had died of HAE despite knowledge of the diagnosis, and the patient developed severe anxiety, necessitating specialist psychiatric treatment. After inclusion on the home therapy program, the patient infused 1000 to 1500 IU C1-INH concentrate approximately once a month, as required for severe symptoms, and reported an improvement in her quality of life. However, the threat of suspension of her home therapy because of funding problems led to an increase in her anxiety and the number of angioedema attacks, a well-recognized consequence of psychologic distress,24 such that 1000 IU of C1-INH concentrate was required once or twice per week. Resolution of the funding problems led to an immediate restoration of well-being and reduction in use of medical services, but she continued to require 4 to 5 infusions of 1000 IU C1-INH concentrate per month.
Patient C, a 33-year-old female patient, had frequent abdominal HAE attacks, requiring an average of 2 days off work per month. She attended her local hospital emergency service approximately once per month and on each occasion received 2 units of FFP. After inclusion in the home therapy program, she reported a greatly improved quality of life, no time off work, and no further exposure to FFP. She infused 500 to 1000 IU of C1-INH concentrate approximately once per month.
Patient D, a 29-year-old female patient who had infrequent attacks without prophylaxis, developed severe abdominal attacks, occurring once or twice a week from the 13th week of her first pregnancy. She was treated with 1000 IU of C1-INH concentrate twice weekly from week 16 until 5 weeks after delivery of a healthy boy. She had only 1 mild abdominal attack during her pregnancy and continued full time work until the 36th week. After delivery, her attacks were infrequent, despite discontinuation of C1-INH concentrate. She chose to withdraw from the home therapy program but plans to retrain if necessary during any future pregnancies.
Family E, a 59-year-old mother, her 2 sons (30 and 20 years), and her daughter (31 years), had been trained in the use of C1-INH concentrate by their family doctor, and had been using this for treatment of acute attacks for 20 years without significant side effects. This family was the inspiration for the establishment of home therapy service. All 4 members of this family have been able to complete tertiary education, pursue demanding professional careers, and travel extensively, including to regions where medical care is not readily available, despite, in 1 case, severe HAE.
Results
In all cases, home therapy resulted in improved quality of life as a result of improved access to treatment and reduced use of emergency services. No patient used C1-INH concentrate inappropriately. No adverse incidents occurred.
Discussion
Despite the existence of successful home therapy programs for Ig and factor VIII,391., 392. C1-INH concentrate home therapy is not universally accepted in the UK. No literature exists for its use elsewhere, although anecdotal reports suggest that it is being used successfully in several European countries (Bork, Personal communication, April 2004; Levi, Personal commnuication, May 2003]. UK practitioners are concerned about safety of home infusion, maintenance of patient skills, possible inappropriate use leading to increased C1-INH concentrate costs, and the fact that C1-INH concentrate is unlicensed in the UK. The authors' patients have reported no side effects from C1-INH concentrate other than occasional difficulties with venous access. Despite concerns that patients on home therapy might delay going to hospital if venous access could not be established or C1-INH concentrate was ineffective, this has not occurred. Patients also retain the option of hospital treatment if conditions for home therapy are not optimal. These results show that use of C1-INH concentrate is appropriate and that, in general, C1-INH concentrate usage is not increased by home therapy. Use of emergency and routine medical services and time off work is reduced, and all patients report great improvements in their quality of life.
Recombinant C1 inhibitor
(Jan H. Nuijens, MD, PhD, and C. Erik Hack, MD, PhD,∗ Leiden and Amsterdam, The Netherlands)
As discussed, recombinant C1-INH is among the therapies currently being investigated for the future treatment of HAE. In this section, Nuijens and Hack describe the potential advantages of such a therapy and the steps being taken toward realizing this means of treatment.
Functional deficiency of the plasma protein C1-INH in patients with HAE and AAE accounts for the recurrent attacks of localized submucosal and/or subcutaneous swelling that can cause serious discomfort and are sometimes life-threatening.7 Intravenous supplementation with human plasma–derived C1-INH is the preferred treatment of severe attacks of angioedema, and this treatment appears very effective and well tolerated.335., 393., 394., 395. However, plasma-derived products are of limited availability for reasons related to their human blood origin, supply, safety concerns, and/or development costs. Three different plasma-derived C1-INH preparations are licensed by regulatory authorities in 1 to 3 European countries each. Even where licensed and/or available, treatment with plasma-derived C1-INH often is restricted to laryngeal attacks with risk for suffocation, severe abdominal attacks, and preoperative prophylactic use. These restrictions have grown out of viral safety concerns, although the risks of transmission of human pathogenic viruses may nowadays be considered minimal.75 Long-term prophylactic treatment with plasma-derived C1-INH, although apparently safe and efficacious, occurs only on a very small scale.356 Plasma-derived C1-INH has also been tried in several serious pathological conditions such as sepsis, vascular leakage syndromes, and acute myocardial infarction, ie, in conditions associated with excessive activation of the complement and contact systems and/or absolute or relative deficiency of functional C1-INH.198., 396. Although its application in these conditions appeared safe, its efficacy was not yet demonstrated unequivocally.
Rationale for production of recombinant human C1-INH
Until very recently, all animal and clinical studies with C1-INH used human blood plasma-derived C1-INH. Although the advantage of this C1-INH preparation is that it has normal clearance kinetics, normal specificity, and no antigenicity, the apparent disadvantages are that the source material is limited and potentially contaminated with blood-borne pathogens. In particular, the limited source is an obstacle to develop C1-INH for indications other than HAE because these indications require more C1-INH than for the treatment of HAE (eg, approximately 15-25 U/kg body weight for an HAE attack vs 200 U/kg for the treatment of myocardial infarction). Hence, availability of recombinant C1-INH is warranted. Major advantages of production of human C1-INH via recombinant DNA technology are that the recombinant material is not derived from a human source, that the production can more easily be controlled, and that the production can be scaled up to meet the requirements for the investigational treatment of HAE as well as other potential indications.198 On cloning of C1-INH by Bock et al,205 recombinant functional C1-INH was expressed in COS-1 cells by Eldering et al190 in the late 1980s. The C1-INH expressed in this way was fully active, but expression levels were low (approximately 1 mg/L), thus allowing only structure-function relationship studies.190 Expression in Escherichia coli was also attempted. Though much larger quantities of protein were expressed, only a fraction (a few percent) was active, whereas the majority was denatured (Hack et al, Unpublished data, January 1990). Similar observations have been made by Lamark et al,397 although these investigators could obtain more functional C1-INH preparations by using different vectors and different strains of E coli. Hence, other expression systems are needed to produce recombinant C1-INH for clinical applications. Two such systems, expression in transgenic animals and a yeast system, are discussed here.
Generation of recombinant human C1-INH transgenic rabbits and pharmaceutical development of transgenic recombinant human C1-INH
Expression of C1INH via the mammary gland of transgenic animals was first attempted in rabbits, among others, because of rabbits' short generation time. In addition, a lactating rabbit may produce 10 L milk per year; because of this, transgenic rabbits may yield sufficient quantities of recombinant human C1-INH (rhC1-INH) for the HAE indication. A mammary gland–specific expression vector containing a bovine milk-specific promoter sequence (αS1 casein) functionally linked to the gene encoding human C1-INH was introduced into the rabbit germ line by microinjection of fertilized oocytes isolated from superovulated female donor animals. A production line was selected with a relatively low transgene copy number and that expressed rhC1-INH in milk at levels exceeding 10 g/L. To ensure continuous availability of a herd of transgenic animals from the selected founder line and to restrict potential rearrangements of the transgene through subsequent generations, a Master Transgenic Bank and a Manufacturing Working Transgenic Bank have been established. Production herds are housed in specially designed and validated facilities subject to stringent environmental monitoring. Animal health is continuously monitored, and the rabbits are regularly tested for the presence of a wide range of rabbit and human pathogens. Rabbits showing any abnormality are excluded from being milked. This health monitoring system ensures that the production herd is a specified pathogen-free rabbit population. Milk is collected by using a specially designed rabbit-milking device. The raw milk is analyzed for compliance with internal criteria before release for downstream processing. The purification of rhC1-INH from raw milk is composed of skimming the milk, cation-exchange chromatography, viral inactivation by solvent-detergent incubation, anion-exchange chromatography, zinc-chelating chromatography, virus removal by nanofiltration, ultrafiltration, final filtration, filling, and lyophilization. Stability studies of vialed rhC1-INH have indicated a shelf life of at least 2 years. The downstream process of rhC1-INH has been the subject of a virus removal or inactivation validation study using a panel of viruses representing different viral classes and sizes (enveloped and nonenveloped, single-stranded and double-stranded RNA and DNA viruses).
Characterization of transgenic rhC1-INH protein
Nonreduced and reduced SDS-PAGE of rhC1-INH showed rather broad (single-chain) protein bands with an estimated relative molecular mass (Mr) of approximately 90,000. The observed heterogeneity in Mr on SDS-PAGE of rhC1-INH as well as the difference in Mr (approximately 15,000 and 10,000 on nonreduced and reduced SDS-PAGE) of rhC1-INH and plasma-derived C1-INH most likely results from differential glycosylation. This follows from the observation that both the N-terminus and C-terminus of the protein are intact as determined by N-terminal and C-terminal amino acid sequence analyses. Moreover, liquid chromatography–mass spectrometry peptide mapping data revealed approximately 70% recovery of the amino acid sequence of rhC1-INH and conformity with the expected amino acid sequence. The theoretical molecular mass of the polypeptide backbone of C1-INH is approximately 53 kd. The use of matrix-assisted laser desorption ionization–time of flight mass spectroscopy revealed a molecular mass of approximately 67 kd and approximately 73 kd for rhC1-INH and plasma-derived C1-INH, respectively, and suggests that approximately 21% and approximately 28% of the molecular mass of rhC1-INH and plasma-derived C1-INH can be ascribed to carbohydrate structures. Monosaccharide analysis of the major monosaccharides in glycoproteins (Fuc, GalNac, GlcNac, Gal, Man, and Neu5Ac) confirmed that rhC1-INH consists of approximately 21% carbohydrates. N-glycolyl neuraminic acid, a sialic acid that is not found in normal human tissue, as well as α-galactosyl were not detected on monosaccharide analysis of rhC1-INH. Mass spectrometric and nuclear magnetic resonance analyses of the glycans released from rhC1-INH indeed showed the presence of at least 4 different O-linked and at least 7 different N-linked carbohydrate structures, including oligomannose and hybrid and complex-type carbohydrates.398 N-linked glycan profiling indicated that most of the N-linked complex structures of rhC1-INH appeared to be monosialylated. The inhibitory potency of rhC1-INH toward the target proteases C1s, kallikrein, factor XIa, and factor XIIa was found to be comparable with that of plasma-derived C1-INH. In addition, no difference in the inhibition of plasmin and thrombin was observed. Physicochemical characterization to demonstrate identity, purity, potency, and consistency between different batches of rhC1-INH revealed that it was consistently being produced as an active, intact molecule, with purity exceeding 99.98 %, containing no detectable amounts of cleaved rhC1-INH, endogenous rabbit C1-INH (less than 1 ppm), and rabbit milk proteins (less than 20 ppm).
Summary of experiments in animals with transgenic rhC1-INH
The purpose of the preclinical safety program was to assess the initial safe dose in human beings and to identify safety parameters for clinical monitoring. The package contained safety pharmacology in dogs (625 U/kg), acute single-dose toxicity in rats and dogs (25-1250 U/kg), 2-week subchronic toxicity in rats (25-625 U/kg), repeated dose toxicity in dogs (625 U/kg/day for 5 days), and local tolerance in rabbits (intravenous, perivenous, or intra-arterial microinfusions). Overall, no adverse clinical signs were observed and no or negligible effects were reported on clinical laboratory parameters. On the macroscopic level, some minor findings were reported, but histopathological analyses revealed no treatment-related findings. rhC1-INH was cleared more rapidly from the circulation of the rat and the dog in comparison with plasma-derived C1-INH. In rat serum samples from animals in the highest dosage group (single-dose, 1250 U/kg; subchronic, 625 U/kg), a mild specific immune response was noted. There was no evidence for the generation of neutralizing antibodies. On the basis of these studies, it was concluded that rhC1-INH may be considered safe.
Phase I study of rhC1-INH in asymptomatic subjects with HAE
An open-label phase I study of the safety, tolerability, pharmacokinetics, and pharmacodynamics of ascending intravenous doses of rhC1-INH (6.25-100 U/kg) in 12 asymptomatic subjects with HAE was conducted at the Centre for Human Drug Research (Leiden, the Netherlands; Cohen et al). The study subjects were invited to participate by Levi (Department of Internal Medicine, Academic Medical Centre, Amsterdam, The Netherlands) in close communication with the Dutch Association of Patients with HAE and Quincke's Edema. All subjects were without angioedema symptoms at the time of the trial and received rhC1-INH on 2 occasions with an interval of at least 5 weeks ( Table XIII). The subjects were assessed clinically, and blood samples were taken to evaluate laboratory safety parameters and immunogenicity as well as the clearance and biological effects of rhC1-INH. Assays of pharmacokinetics and pharmacodynamics parameters were performed at Sanquin Research (Hack, Amsterdam, the Netherlands).
Table XIII.
Dosing scheme for phase I trial of rhC1-INH
| Study period 1 | Washout | Study period 2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Group A | 6.25 | 25 | |||||||
| Group B | 12.5 | 50 | |||||||
| Group C | 25 | 100 | |||||||
| Group D | 50 | 100 | |||||||
Safety results were as follows. RhC1-INH appeared safe and well tolerated: no probable product-related adverse events (eg, allergic reactions) or changes in electrocardiogram, vital signs, or routine laboratory parameters were observed. Neither clinically significant increases in anti–C1-INH or antirabbit milk protein nor evidence of C1-INH neutralizing antibodies was noted.
Pharmacokinetics results were as follows. Infusion of rhC1-INH resulted in dose-dependent increases of functional C1-INH response parameters maximum concentration (above baseline), area under effect curve above baseline, dose-normalized area under effect curve, and of time above 0.4 U/mL, whereas dose-normalized maximum concentration appeared constant (approximately 0.02 U/mL/U/kg). The profiles of functional C1-INH showed a full initial recovery and a dose-dependent clearance of rhC1-INH that indicated a saturable mechanism of elimination. After the infusion of rhC1-INH at 100 U/kg, a clearance of approximately 13 mL/min, a half-life of about 3 hours, a volume of distribution of about 3 L, and an endogenous infusion rate of about 2 U/min were observed by using a standard 1-compartment model of analysis. Application of a 1-compartment model with Michaelis-Menten elimination provided dose-independent estimates of Vmax, Km, volume of distribution, and endogenous infusion rate of approximately 45 U/min, approximately 0.6 U/mL, approximately 3 L, and approximately 12 U/min, respectively. Thus, the half-life of rhC1-INH in the subjects with HAE appeared reduced compared with historical data of plasma-derived C1-INH.211., 334., 335. This likely relates to the differential glycosylation and lower degree of sialylation in rhC1-INH. The increased clearance of rhC1-INH from the bloodstream likely occurs mainly through endocytic asialoglycoprotein and mannose receptors on hepatocytes.209 After dosing with rhC1-INH at 100 U/kg, functional C1-INH was at least 2-fold the normal level for about 2 hours and remained above 0.4 U/mL for about 9 hours. Activation of the cascade systems releasing inflammatory mediators responsible for the generation of edema is thought to be sufficiently controlled with functional C1-INH levels exceeding 0.4 U/mL.138
Pharmacodynamics results were as follows. rhC1-INH displayed biological activity in the subjects as evidenced by plasma C4 increases and inhibition of C4 cleavage. The dose-dependent increase in mean normalized C4 was highly variable within dosage groups and was approximately 2-fold increased after the infusion of rhC1-INH at 100 U/kg. C4 peak levels occurred at approximately 12 hours postinfusion, similar to C4 responses after plasma-derived C1-INH, and thereafter gradually declined to baseline.211., 335. An immediate dose-dependent effect of rhC1-INH on plasma C4b/c was observed. The magnitude and the duration of the decrease in C4b/c appeared dependent on the dose of rhC1-INH. Cleavage of C4 appeared to occur once functional C1-INH dropped below a level of approximately 70% of normal.
Taken together, the phase I results suggest that an adequate dosage of rhC1-INH will be able to correct C1-INH activity in blood for a sufficiently long period to halt the progression of a swelling episode and to allow the resolution of edema. That is, the results warrant clinical studies of the safety and efficacy of rhC1-INH in symptomatic patients with HAE.
Phase II and phase II/III studies of transgenic rhC1-INH for the treatment of acute attacks in patients with HAE
A phase II exploratory open-label study of the safety, tolerability, and efficacy as well as pharmacokinetics and pharmacodynamics of rhC1-INH at 100 U/kg in symptomatic patients with HAE with severe attacks of angioedema in the abdomen, the facial-oro-pharyngeal region, and/or the genito-urinary region is currently being performed in the Netherlands (Levi). On the basis of the initial encouraging results of the phase II study, a multicenter phase II/III study of rhC1-INH at sites in several European countries is being initiated.
Expression of wild-type C1-INH in the yeast Pichia pastoris
The yeast Pichia pastoris is increasingly used as an expression system to produce recombinant proteins on a large scale.399 Hence, recombinant C1-INH has been expressed in this system as well. However, development of yeast-expressed rhC1-INH for clinical use is still at an early stage. Initial expression of C1-INH was achieved by using the vectors pPicZαA and pPic9.400 Without optimization, expression of as much as 2 g of recombinant protein per liter was observed. Though some of the C1-INH preparations produced in P pastoris were fully active, compared with plasma C1-INH, other preparations were inactive because of proteolytic inactivation of the recombinant molecule by contaminating proteases. Inactivation in particular was observed when the yeast was cultured for more than 3 days. The recombinant protein was secreted in the culture supernatant and constituted at least 20%, in most cases >50%, of the total amount of protein, which may make this system attractive for commercial production applications. Recombinant C1-INH was purified to >95% homogeneity on SDS-PAGE on laboratory scale by using HPLC cation-exchange chromatography.
Plasma C1-INH is a heavily glycosylated molecule, owing approximately 28% of its molecular mass to carbohydrate groups. In the original publication by Bock et al205 describing the nucleotide sequence of cDNA coding for human C1-INH, 13 glycosylation sites were identified. However, the number of potential glycosylation sites (N-linked and O-linked) may be as high as 28. The function of these carbohydrate groups is not completely clear, although there is conclusive evidence that incomplete glycosylation and/or undersialylation of the carbohydrate groups is involved in the clearance of the molecule in vivo.209 On the basis of migration patterns of yeast-expressed C1-INH on SDS-PAGE as well as current knowledge of glycosylation by yeast (mainly mannose), it is expected that carbohydrate groups on yeast-expressed C1-INH will need to be modified to achieve normal clearance in vivo. Indeed, in a rat model, accelerated clearance of Pichia-expressed C1-INH was observed (half-life, 20 minutes for Pichia–C1-INH vs 3.5 hours for plasma derived C1-INH; Bos et al).400., 401.
Expression of C1-INH mutants
C1-INH mutants have been made for different purposes, ie, to study the structure and function, to study glycosylation, and to modify function toward applicability in inflammatory conditions. Regarding application in inflammatory conditions, C1-INH, like many other serpins, can be catalytically inactivated by neutrophil elastase, which may limit its use in inflammatory diseases. To improve the potential performance of C1-INH at sites of inflammation, it may be beneficial to enhance the resistance of the molecule to catalytic inactivation by elastase, because this would reduce the amount of inhibitor needed. C1-INH mutants with amino acid replacements at the so-called P3 and P5 positions of the reactive center (this is the domain interacting with target and nontarget proteases) have been developed.402 These mutants have a nearly normal function but are much more resistant to inactivation by elastase than wild-type C1-INH. Until now, these mutants have never been tested in animal models to prove the principle that they indeed perform better in inflammatory conditions.
As indicated, Pichia-expressed C1-INH is glycosylated differently (mainly mannose) than plasma-derived C1-INH (mainly complex-type carbohydrate groups). Most glycosylation sites are located in the N-terminal part of the molecule. The function of this part is currently unknown, although according to the latest data, the most C-terminal amino acids of this N-terminal part serve to stabilize the so-called central β-sheet of C1-INH, which is essential for serpin function.206 To evaluate the role of the N-terminal portion in the functional activity of C1-INH, deletion mutants lacking 76 or 98 amino acids of the N-terminus have been expressed in Pichia. The functional activity of these mutants has yet to be studied in detail but at first glance looks normal. In contrast, a mutant lacking the complete N-terminal part was inactive. The mutant lacking 98 amino acids is interesting in that it seems to have normal functional activity despite having only 3 carbohydrate groups left. Experiments with endoglycosidase H and concanavalin A reveal that these groups are N-linked (Asn 216; Asn 231 and Asn 330).
For the future
Recombinant protein technology not only offers the possibility of producing large amounts of protein at acceptable costs but also allows for mutation of the protein to adapt its function. Expression of wild-type human C1-INH has been successfully achieved in transgenic rabbits, and this recombinant inhibitor is now in phase II clinical development for the treatment of acute attacks in patients with HAE. Pichia-expressed C1-INH may constitute another alternative to plasma-derived C1-INH but is still at a very early stage of development.
DX-88: efficacy and safety of a kallikrein inhibitor in HAE and AAE
(Kayla Williams, BS, MA, MFA, Cambridge, Mass: Review of T. Gonzales-Quevado, T. Caballero, M. Cicardi, K. Bork, and A. Williams; and A. Williams and L. G. Baird)
This section reviews the work of Gonzales-Quevado et al343 and Williams et al342 in which they briefly describe the apparent success of a kallikrein inhibitor in relieving attacks of HAE and AAE.
Kallikrein inhibition and nonallergic angioedema
The kallikrein inhibitor DX-88 was generated via phage display, a technique for rapidly identifying target-specific protein binders. The phage display process involves generating many possible binders specific for each desired target, in this case human plasma kallikrein, and then selecting the binder with the highest affinity. As such, this process resulted in a molecule with an extremely high (44-pmol/L) binding affinity for human plasma kallikrein. Kallikrein is known to catalyze the conversion of kininogen to bradykinin.254 Assuming bradykinin is the major mediator of nonallergic angioedema,28 plasma kallikrein inhibition should be beneficial during angioedema attacks. Kallikrein has also been shown to influence the renin-angiotensin system by promoting the conversion of prorenin to renin262; however, as discussed in the pathogenesis section of this supplement, the renin-angiotensin system likely plays a much lesser role in angioedema generation.
Phase I normal volunteer study
In a study conducted in Scotland, DX-88 was administered to 12 healthy male or female volunteers; pharmacokinetic and safety parameters were assessed. Two patients each received 10 or 20 mg DX-88, and 4 patients each received 40 or 80 mg DX-88. Half-life measurements ranged from 0.55 to 1.71 hours.
Phase II European study
An open-label study of DX-88 for the treatment of nonallergic angioedema attacks was conducted in 4 centers in Europe. Of the 9 treated patients, 7 patients had HAE, and 2 had AAE. Patients presented with facial/lip, hand, genital, or abdominal attacks; 1 patient was treated for simultaneous hand and abdominal edema. In this dose-ranging study, 3 patients each received 10 mg, 40 mg, or 80 mg intravenous DX-88. One patient (40 mg DX-88; genital edema) had a drug-related anaphylactoid reaction. Patient-reported times to the start of attack resolution ranged from 25 to 240 minutes (mean, 92 minutes); patient-reported times to complete resolution ranged from 2 to 72 hours (mean, 38.6 hours). The authors note that DX-88 could potentially advance the treatment of HAE by reducing patients' reliance on anabolic steroids or plasma-derived products.
Treatment-associated conditions
Attenuated anabolic steroids are the most common prophylaxis for HAE and associated disorders. Although these agents are effective in many patients, they carry a wide range of side effects, many of which are undesirable. Their mechanism of action in these diseases, and indeed the full extent of their systemic effects, is not entirely known. As illustrated by the following investigations, attenuated androgens such as danazol may have other, at least transiently positive effects in addition to their better-documented risks.
Investigation of bone turnover in patients with HAE
(Tímea Kollár, MD, Béla Fekete, MD, PhD, DSc, Peter Lakatos, MD, PhD, DSc, Beáta Visy, MD, Éva Németh, and Henriette Farkas, MD, PhD,∗ Budapest, Hungary)
In this section, Kollár et al summarize their study of the effects of danazol on bone turnover in patients with HAE receiving danazol prophylaxis.
Attenuated androgens effectively prevent swelling attacks in HAE.351 The attenuated anabolic steroid danazol is mainly used in the therapy of many estrogen-dependent diseases such as endometriosis, cystic fibrotic mastopathy, primary menorrhagia, primary pubertas praecox, and gynecomastia.403., 404. However, its other, less known clinical indication is the prophylaxis of HAE.351
The high androgen and low estrogen levels caused by this drug25., 405. are responsible for some side effects. Among these, seborrhea, acne, hirsutism, weight gain, hair loss, voice changes, clitoral hypertrophy, vaginitis, irregular menstruation or amenorrhea, and reduced breast mass are the most common.349
Methods
This study investigated the effects of danazol on bone metabolism. Low estrogen concentrations have previously been shown to play a role in the development and progression of osteoporosis. The authors' current investigation sought to discover how reduced estrogen production caused by danazol therapy influences bone turnover. According to available data, danazol's effects on bone metabolism have been investigated only in patients with endometriosis.406., 407., 408., 409. These investigations gave contradictory results. In addition, the authors note that the danazol doses given to treat endometriosis in these studies far exceed the doses administered in HAE. Rock et al410 have reported a temporary increase in bone mineral density (BMD) lasting only for the 24 weeks of treatment, which then decreased such that BMD values measured at the 72nd week were lower than the initial values. According to Dodin et al,411 the lumbar bone mass slightly increased by the end of the third and sixth months.366, Dmowski et al412 found no significant change in BMD during the danazol treatment.
The current investigation measured the degree to which the minimum effective dose of danazol for HAE control influenced bone turnover. All patients cared for at the Hungarian HAE Center were involved in this study. The control group contained 50 patients not treated with danazol. Concentrations of alkaline phosphatase, calcium, phosphate, parathyroid hormone, dehydroepiandrosterone sulfate, β-cross laps, and osteocalcin were measured in blood serum. Dual-energy X-ray absorptiometry was also performed in patients older than 20 years.
Results
Significant correlations have been found by using multiple logistic regression analysis between the variable of β-cross laps, osteocalcin values, z score values detected by the femoral neck, and danazol treatment. β-Cross laps and osteocalcin concentration (markers of bone resorption) have shown a negative correlation, whereas z score values (markers of bone formation) have shown a positive correlation with danazol treatment. In the authors' study, it was evident that β-cross lap concentration was lower in the danazol-treated group than in the control group. Changes in serum osteocalcin concentrations, like those in β-cross lap values, showed a negative correlation. Osteocalcin concentrations in patients treated with danazol were considerably decreased. However, the authors note that both osteocalcin and β-cross lap concentrations oscillated in the normal range. z Scores measured by the femoral neck showed that positive results were much more frequently detected in the danazol-treated group, whereas values in the untreated control group were always between 0 and −0.5. Thus, bone mineral density was significantly higher in the danazol-treated group.
The authors propose that the increased androgen level may have compensated for the deleterious effect of decreased estrogens, citing that the anabolic effect of androgens on bone metabolism have been well documented in the literature.
Discussion
Comparing the results of this study with those reported in the literature on the effects of high-dose danazol treatment in endometriosis, the authors suggest that the minimum effective dose treatment in HAE does not cause osteoporosis and, indeed, may protect the bone from mineral loss.
Ovarian function and follicular fluid in HAE: effects of danazol
(Roberto Perricone, MD, Caterina De Carolis, MD, Carlo Perricone, Roberto Giacomelli, MD, and Luigi Fontana, MD, Rome and L'Aquila, Italy)
Here, Perricone et al describe the full arc of their past research in HAE and ovulation, culminating in their investigating the influence of HAE management doses of danazol on ovarian function.
Ovulation is the result of complex, interrelated mechanisms initiated by the surge of LH and characterized by resumption of meiosis and germinal vesicle breakdown, initiation of luteinization of the granulosa cells, rupture of the follicle wall, and release of a fertilizable ovum.413 Hormonal control of ovulation and biochemical and morphologic changes of the preovulatory follicle, including collagenolysis and vascular changes, have been extensively studied.
Ovarian follicular fluid (FF) is an enzyme reservoir controlling the permeability of the capillaries of the antrum and proteolytically degrading collagen, thus leading to the rupture of the follicle wall. The plasminogen-activator-plasminogen hypothesis is the most likely explanation of the mechanism initiating the cascade that leads to follicular rupture. The preovulatory follicle, stimulated by gonadotropins, secretes urokinase-type plasminogen activator, which can convert plasminogen to plasmin. A latent collagenase activated by plasmin attacks collagen, and the resulting telopeptide-free collagen can be attacked and degraded by nonspecific proteases. The decrease of the tensile strength of the follicle wall then allows the rupture of the follicle by the action of the existing intrafollicular pressure.413
In addition to several multifactorial systems (eg, kinins and fibrinolysis) that have been demonstrated and characterized in human FF, the presence of active complement has also been demonstrated in amounts similar to those present in normal human serum.414 Plasmin's activation of complement in FF is very important for the function of the enzymatic multifactorial mechanism of ovulation. After complement activation in FF, biologically active peptides such as C3a, C4a, and C5a are released via the complement cascade. Complement activation is also important for the assembly of the membrane attack complex on the follicle wall.414
Complement activation and ovulation: consequences in HAE
It has been clearly demonstrated that women of reproductive age with HAE who were not under danazol treatment and off any therapy, including OCs, frequently had cystic ovaries with an ultrasound pattern of polycystic ovaries (PCOs; approximately 35%) or of multifollicular ovaries (MFOs; approximately 55%).415 Although PCOs in women not affected with HAE are associated with increased LH and testosterone concentrations, and MFOs with almost normal values of these hormones, in patients with HAE, normal concentrations of LH and testosterone (and of prolactin, cortisol, and ACTH) are almost always demonstrable independently from the ultrasound pattern (PCO, MFO, normal) but in the presence of significantly reduced follicle stimulating hormone and slightly increased LH:follicle stimulating hormone ratio. Menstrual irregularities and oligomenorrhea are frequent features of the patients studied, and hirsutism is sometimes present in patients with HAE both with PCO and MFO.
Neuroendocrine connection?
In HAE, the role of C1-INH deficiency in the development of clinical symptoms and in the management of the disease has been explored.31 Nevertheless, some interesting questions remain. The proposed relationship between physical and/or psychologic stress and the onset of acute episodes has yet to be proven fully. Likewise, the effects of repeated stressful situations, such as the attacks themselves, on patients' neuroendocrine systems have yet to be completely explained.
However, some possible links between the neuroendocrine system and HAE have been studied and reported. Perricone et al416 have previously found high plasma β-endorphin concentrations without simultaneous elevation of ACTH during HAE attacks as well as during symptom-free periods. This provides evidence of a possible massive release of β-endorphin from the readily disposable pool present in the pituitary and/or for an increase in the turnover of the peptide as evaluated by β-endorphin:β-lipotropin ratios and, in turn, of a pituitary abnormality in the patients.
Also in the group of patients with HAE affected with cystic ovaries (studied off therapy and in symptom-free periods), significantly high plasma β-endorphin concentrations and normal ACTH have been detected.415 The very high prevalence of PCO and MFO in HAE again emphasizes the possible presence of hypothalamic-pituitary abnormality in these patients. In fact, both PCO and MFO are linked to hypothalamic-pituitary dysfunction,417 and in HAE, 1 of the features of such a dysfunction, ie, high plasma β-endorphin concentrations and normal ACTH, is present in many patients, including men, postmenopausal women, and girls who are not of reproductive age.416
Influences on ovulation
The presence of both complement and ovarian abnormalities in patients with HAE and the role of FF complement in ovulation induced the authors to study complement function in the FF of women of reproductive age affected with HAE. They found that impaired complement function is present in FF obtained from female patients of reproductive age affected with HAE. In fact, contrary to what had been observed both in previously studied women not affected with HAE415 and in controls, HAE patients' FF showed a dramatically decreased classic pathway activity as well as undetectable functional and antigenic C1-INH, and very low C4. In addition, the authors observed a slightly reduced or within normal serum range values for alternative complement pathway activity, including C3 and factor B. This complement profile was also detected415 in the patients' sera.418
It is well known that the multifactorial mechanism of ovulation involves 2 relevant steps modulated by C1-INH: (1) the formation of plasmin, and (2) the plasmin-dependent complement classical pathway activation.414 Activation of FF complement is important for the correct continuation of the ovulatory process, and the lack of C1-INH, which facilitates the formation of plasmin and plasmin-dependent complement activation, can, in theory, be beneficial. On the contrary, when the activation of complement is massive, as occurs during HAE attacks, a significant release of biologically active complement products can induce consistent inflammation and, in turn, pain. Abdominal pain is frequently encountered in female patients with HAE during attacks and during ovulation as well. However, because complement is chronically consumed during remissions and dramatically consumed during HAE attacks, the potential of complement activation in HAE FF is indeed the result of complex interrelated phenomena.
If significant consumption of complement components occurs as a result of HAE attacks shortly before the periovulatory period, FF complement will represent a defective reservoir of components unable to support further activation during ovulation. Such a situation occurred in 1 patient: a few days before ovulation, an attack characterized by edema of the extremities and abdominal pain occurred; thereafter, the patient manifested neither abdominal pain nor detectable FF complement activities and C3 cleavage fragments during the periovulatory period. In another patient, the periovulatory period (as well as the time of the study) was far away from attacks (30 days); at this time, abdominal pain (without other symptoms of the disease) in the presence of reduced but detectable FF complement activities and of C3 cleavage fragments occurred.
Notwithstanding the presence of some important mechanisms of proven efficacy that can compensate for the complement deficiency (eg, the activation of a latent collagenase and the action of lysosomal enzymes),413 it is possible that, in the presence of impaired complement function, the rupture of the follicle wall is more difficult. However, once significant time has elapsed since the last HAE attack, complement function can ameliorate; if ovulation coincides with such a condition, it will meet an only mildly reduced complement that will be capable of participating in the rupture of the follicle wall. These considerations can explain the apparently undiminished reproductive capacity of female patients with HAE. On the other hand, HAE patients' FF complement supports a certain degree of activation by seminal plasma,419 possibly caused by the relative integrity of the alternative pathway supporting its participation in the phenomenon. It is nonetheless possible that the more difficult rupture of some follicles in HAE and the consequent altered intraovaric regulation by atresic follicles can result in the PCO and MFO frequently observed in the disease.415 It is known that in cystic ovaries, the rupture of the cysts is very painful; thus, the authors cannot exclude that in patients with HAE, the rupture of ovarian cysts might contribute to the genesis of the dramatic abdominal pain during attacks.
Danazol and ovulation: conclusions
One of the mainstays in the management of HAE is long-term therapy with danazol, a mild androgen capable of correcting low C1-INH concentrations and of greatly reducing the typical symptoms of the disease.350 The authors therefore studied the effects of danazol on ovarian FF C1-INH and on ovarian ultrasound pattern. Six patients with HAE affected with cystic ovaries (3 with PCO and 3 with MFO) who had been off any therapy for at least 1 year were treated with danazol (200 mg daily). As a result of danazol treatment, HAE clinical symptoms consistently improved, and C1-INH serum concentrations increased. After 6 months of therapy, these patients were monitored by pelvic ultrasound scanning, and their ovaries were found normal. Serum C1-INH and FF C1-INH also increased. Furthermore, when the ultrasound pattern of the ovaries was studied in another group of patients, women with HAE of reproductive age who were usually under danazol treatment, cystic ovaries were found in only 1 of 9 patients (11%).
Taken together, these data suggest a further link between the neuroendocrine and immune systems, describing a pathology involving both hypothalamic-pituitary dysregulation and an immunologic disorder.
Pediatric HAE
Recent research has contributed to a less anecdotal and more systematic appreciation of the differences between adult and pediatric HAE and the special age-related concerns that attend HAE from the first months of life through puberty. Here, 2 prospective follow-ups of pediatric case series are reported.
Clinical manifestation and therapy in children with HAE: a prospective follow-up
(Inmaculada Martinez-Saguer, MD, Eva Rusicke, Emel Aygören-Pürsün, MD, and Wolfhart Kreuz, MD, PhD, Frankfurt/Main and Frankfurt, Germany)
In this section, Martinez-Saguer et al detail the results of a prospective follow-up of their pediatric case series in Germany and summarize their treatment recommendations on the basis of these data.
Most patients have the first manifestation of HAE during childhood. Unfortunately, treatment of acute attacks that manifest with circumscribed swelling of the skin and the subcutaneous tissues can be performed successfully only by administering C1-INH concentrate.420 Other medications that are usually helpful in edema of other origins (eg, antihistamines or corticoids) are not effective here.421 The authors report that ⅔ of their patients, when anamnestically well evaluated, had edema of the mucous membranes of the respiratory tract at least once; remarkably high lethality rates (as high as 30%) have been associated with this disease in the past.31., 44., 421. Even recently, a mortality rate as high as 12.5% was published in an article summarizing experiences with Austrian, Swiss, and German patients with HAE.422 As previously discussed, bradykinin seems to be the major mediator of edema, but discussions have not yet been finalized.9 Because this disease still bears unknown details thus far identified, the authors chose to examine the baseline parameters of patients with HAE to broaden the general database available and to enhance the understanding of the medical and scientific community.
Methods
Data from 245 patients with HAE have been collected in the database at the Department for Pediatric Hematology, Oncology and Hemostaseology of the Johann Wolfgang Goethe University, Frankfurt/Main, Germany. As of this March 2003 analysis, this database contained a total of 56 pediatric patients (age <18 years) with diagnosed HAE. These patients had been followed up for a period of as long as 12.9 years. The baseline characteristics of a total of 45 patients from 35 different families with HAE were analyzed.
The authors further monitored C1-INH plasma activity and C1-INH plasma antigen and supplemented these values with analyses of C3 and C4 and measurement of the total hemolytic activity (CH50) in the patients' plasma. Finally, the frequency and localization of attacks, the history of each patient, and the efficacy and safety of replacement therapy with C1-INH concentrate (Berinert P) were investigated.
Antigenic C1-INH levels were determined by radial immunodiffusion with NOR-Partigen plates (Dade Behring, Deerfield, Ill). Functional C1-INH concentration was determined by chromogenic assay with Berichrom C1-Inhibitor (Dade Behring).
Results and discussion
All patients showed low C1-INH activity, with a median activity of 22% of normal (normal value, 64% to 146%; range, 6.4% to 47%). Decreased C4 (45/45 patients), C3 (9/45 patients), and CH50 (24/45 patients) were found at baseline. Median age at HAE diagnosis was 4.8 years (range, 0.1-12.3 years). Of 46 pediatric patients, 26 were symptomatic as of March 2003. These 26 patients presented with subcutaneous edema of the extremities (17 patients), face (13 patients), or larynx (3 patients), or gastrointestinal manifestations (20 patients). The initial manifestation of HAE in children occurred at a median age of 4.8 years (range, 0.3-9.7 years) and was associated with low C1-INH activity and decreased plasma concentrations of C4. The authors found that C1-INH concentration did not correlate with the number and severity of attacks, with each patient seeming to have an individual threshold. Four patients presented symptoms in the first year of life (1 patient, face; 3 patients, extremities). Decreased amounts of CH50 were present in 24 of 45 patients (58%).
All acute HAE attacks were successfully treated with 10 to 30 units C1-INH concentrate per kilogram body weight. In these pediatric HAE patients, C1-INH concentrate was effective and well tolerated. No side effects or seroconversions for HAV, HBV, HCV, hepatitis G virus, HIV-1, or HIV-2 were observed. The gold standard of treatment for acute attacks in pediatric patients with HAE is the administration of a C1-INH concentrate, preferably 1 that is specifically virus-inactivated. The authors always used a pasteurized C1-INH concentrate (Berinert P) and did not observe any problems regarding safety or efficacy.
In 1998, the transmission of hepatitis G virus in patients with angioedema treated with a steam-heated concentrate of C1-INH was reported.423 The authors have not observed a seroconversion with regard to hepatitis G virus in their patients treated with pasteurized C1-INH concentrate.386., 423. As treatment of pediatric patients with HAE with C1-INH concentrate is not registered all over the world, some pediatric patients have to be treated with danazol therapy.312 Introduced in 1976 by Gelfand et al,347 this treatment seems to have a rationale in long-term prophylaxis of patients with HAE.347 However, because this treatment sometimes loses efficacy over time despite increased doses of danazol424 and is often associated with severe side effects,352 especially in children, the authors recommend the treatment of pediatric HAE exclusively with C1-INH concentrate if possible on a named-patient basis through personal importation programs. In their experience, this approach should also be feasible for women of childbearing age and all other patients with HAE who cannot tolerate the side effects associated with danazol treatment.
Clinical management of HAE in children
(Henriette Farkas, MD, PhD,∗ George Harmat, MD, PhD, George Füst, MD, PhD, DSc,∗ Lilian Varga, PhD,∗and Beáta Visy, MD, Budapest, Hungary)
Although excellent reviews on HAE have been published,7., 31. searching the literature yields only a few publications on the treatment and follow-up care of pediatric patients. This paucity of relevant information prompted Farkas et al to review their decade-plus experience treating children with HAE.
Methods
Between 1987 and 2000, ninety patients were registered in the Hungarian HAE Center database. What follows is a review of the clinical data of 26 pediatric patients from 19 families. The male to female ratio was 11:15, and the age at time of diagnosis ranged from 2.5 to 15 years (mean, 7.3 years).
CH50 total complement levels were measured by the standard hemolytic titration method, using sensitized sheep erythrocytes.425 The C4 complement fraction was determined by single radial immunodiffusion using anti-C4 antibodies (ATAB, Stillwater, Minn), and results were expressed as the percentage of standard, normal serum values.426 The C1-INH concentration was also measured by radial immunodiffusion; C1-INH activity was measured by kinetic assay using a commercially available diagnostic kit (Behringwerke AG, Marburg, Germany). The activity of the C1-INH in the serum was determined by photometry and expressed as the percentage of standard, normal values.
In the follow-up protocol, the first occurrence, frequency, and localization of clinical manifestations, duration of the disease, as well as potential precipitating factors of edematous attacks were ascertained.
-
•
A pedigree analysis was performed in all cases.
-
•
Long-term prophylaxis was elected if edematous attacks recurred frequently (≥1 attack per month) or a life-threatening episode could be identified in the history.
-
•The following tests were performed in patients receiving long-term danazol prophylaxis:
-
-Anthropometric assessment of growth (comparison of body height and weight development to age-specific normal values).
-
-Bone age determination (hand X-rays).
-
-Development of secondary sexual characteristics and time of puberty compared with control population.
-
-Mental development (performance at school).
-
-
-
•
Short-term prophylaxis was administered before surgical or diagnostic procedures performed on the head and neck.
-
•
Changes in serum complement fractions and clinical manifestations were monitored during therapy. Potentially drug-related adverse effects of treatment were also recorded.
-
•
In patients undergoing long-term prophylaxis, laboratory tests (complete blood count, liver function tests, creatine kinase activity, and urinalysis) were repeated at 3-month to 4-month intervals, and abdominal ultrasound was performed semiannually during the first 2 years of treatment. Subsequently, asymptomatic individuals or patients with mild symptoms were checked every 6 months to detect potential liver damage.
-
•
Patients with an acute attack of submucosal (laryngeal or severe abdominal) edema were always hospitalized and abdominal ultrasound performed if the attack involved the gastrointestinal tract. Hospitalization for an acute abdominal attack was considered justified when the presence of other acute abdominal pathologies could not be ruled out with certainty and/or manifestations were severe—that is, the attack was associated with obvious signs of hypovolemia (paleness of skin, prostration, dehydration, tachyarrhythmia) and included recurrent paroxysms of acute colicky pain unresponsive to symptomatic therapy, nausea and vomiting, or profuse diarrhea.
-
•
The C1-INH concentrate used in this study was screened for hepatitis B and C by the manufacturer. Nevertheless, in observance of the safety precautions pertinent to blood products, HbsAg and HCV specific antibody were tested by using commercially available immunoassays in all patients who had received C1-INH concentrate.
Acute edematous attacks were treated by administering C1-INH concentrate (Berinert P injection; Centeon, Vienna, Austria), and ε-aminocaproic acid (Acepramin; Pannon Pharma, Pecsvarad, Hungary), tranexamic acid (Exacyl; Sanofi Synthe Labo-Chinoin, Budapest, Hungary), and danazol (Danoval; KRKA, Novo Mesto, Slovenia) were used for prophylaxis.
Results
The initial manifestations of HAE were observed within an age range of 2.5 to 12 years (median, 5.9 years). Six children were asymptomatic at the time of diagnosis, which was established only after HAE was ascertained in their symptomatic parents. Pedigree analysis revealed familial occurrence of the disease in 24 cases (from 19 families) but failed to identify HAE in first-degree relatives of 2 children; therefore, in these patients, C1-INH deficiency must have resulted from a new C1NH mutation ( Table XIV). The localization of clinical signs was variable. In the majority of patients (20 children; 76%), edema formation involved subcutaneous tissues, whereas gastrointestinal manifestations occurred in 14 patients (53%) and laryngeal edema developed in 7 patients (26%). Subcutaneous edema of the extremities or genitals usually persisted for 2 to 3 days, then resolved spontaneously. Edematous swelling of the skin was not accompanied by itching but caused an unpleasant sensation of distension within the involved region. In 8 children, generalized, nonpruritic skin rash (erythema marginatum) was observed before and during edematous attacks of HAE. The exploration of potential precipitating causes invariably identified mechanical trauma; however, upper airway infection was another important factor ( Table XV). According to the results of serum complement studies, 21 children had HAE type I, whereas 5 had HAE type II ( Table XVI).
Table XIV.
Demographic properties of 26 pediatric patients
| Number | Sex | Age at diagnosis (y) | Age at onset of clinical manifestations (y) | Length of follow-up (y) | Pedigree analysis |
|---|---|---|---|---|---|
| 1 | Female | 10 | 9 | 1 | P |
| 2 | Female | 10 | 5 | 2 | Ø |
| 3 | Male | 5 | Ø | 1 | M |
| 4 | Male | 7 | 7 | 11 | M, sibling |
| 5 | Male | 13 | 12 | 11 | M, sibling |
| 6 | Male | 11 | 11 | 1 | M, sibling |
| 7 | Male | 5 | 4 | 6 | M |
| 8 | Male | 7 | 5 | 5 | P |
| 9 | Male | 8 | 5 | 6 | P |
| 10 | Male | 7 | 7 | 6 | P |
| 11 | Male | 11 | 3 | 2 | Ø |
| 12 | Female | 7 | 3 | 1 | M, sibling |
| 13 | Female | 3 | Ø | 1 | P |
| 14 | Female | 5 | 2.5 | 1 | M |
| 15 | Female | 3 | 6 | 9 | M, sibling |
| 16 | Female | 7 | 5 | 9 | M, sibling |
| 17 | Female | 6 | 6 | 7 | M |
| 18 | Female | 14 | 9 | 6 | P |
| 19 | Female | 3 | 3 | 19 | M |
| 20 | Female | 15 | Ø | 1 | P |
| 21 | Female | 8 | 6 | 11 | P |
| 22 | Male | 8 | 4 | 2 | P |
| 23 | Male | 12 | Ø | 1 | P, sibling |
| 24 | Female | 11 | Ø | 1 | M, sibling |
| 25 | Female | 1 | 6 | 1 | M, sibling |
| 26 | Female | 9 | Ø | 1 | M, sibling |
P, Father; M, mother.
Table XV.
Clinical characteristics of 26 pediatric patients
| N | Involved site |
Precipitating factors |
|||||
|---|---|---|---|---|---|---|---|
| Subcutaneous | Laryngeal | Abdominal | Trauma | Stress | Infection | Menstruation | |
| 1 | + | − | + | + | − | + | − |
| 2 | + | − | + | + | − | − | − |
| 3 | None | ||||||
| 4 | + | + | + | + | − | + | − |
| 5 | + | − | − | + | − | + | − |
| 6 | + | − | − | + | − | − | − |
| 7 | + | + | + | + | − | + | − |
| 8 | + | + | + | + | + | + | − |
| 9 | + | − | − | + | − | − | − |
| 10 | + | − | − | + | − | − | − |
| 11 | + | − | + | + | − | + | − |
| 12 | + | + | + | + | − | + | − |
| 13 | None | ||||||
| 14 | + | − | − | + | − | − | − |
| 15 | + | − | + | − | − | − | − |
| 16 | + | − | + | + | + | + | + |
| 17 | + | − | + | + | + | − | − |
| 18 | + | − | + | + | + | + | + |
| 19 | + | + | + | + | + | + | + |
| 20 | None | ||||||
| 21 | + | + | + | + | + | + | + |
| 22 | + | + | − | + | + | + | − |
| 23 | None | ||||||
| 24 | None | ||||||
| 25 | + | − | + | + | − | + | − |
| 26 | None | ||||||
Table XVI.
Complement values at the time of diagnosis
| N | CH50 (48-103 CH50/mL) | C4 (36% to 144%) | C1-INH conc. (64% to 166%) | C1-INH activity (>70%) | Type of HAE |
|---|---|---|---|---|---|
| 1 | 15.1 | 30 | 19 | 1.4 | I |
| 2 | 31 | 15 | 233 | 22.5 | II |
| 3 | 31 | 24 | 43 | 47.5 | I |
| 4 | 41 | 9.2 | 214 | 0 | II |
| 5 | 23 | 21 | 297 | 0 | II |
| 6 | 5 | 5 | 5 | 1.5 | I |
| 7 | 41 | 5 | 24.1 | 0 | I |
| 8 | 17.1 | 2 | 19.4 | 14.5 | I |
| 9 | 48.6 | 23 | 17 | 36.1 | I |
| 10 | 28.8 | 17.2 | 30.8 | 17.5 | I |
| 11 | 27 | 6.4 | 23 | 9.7 | I |
| 12 | 5 | 38 | 5.8 | 7.2 | I |
| 13 | 50 | 20.8 | 36.4 | 10.2 | I |
| 14 | 51 | 6.5 | 21 | 13 | I |
| 15 | 41 | 17 | 250 | 14.3 | II |
| 16 | 22 | 15 | 311 | 6.8 | II |
| 17 | 58.8 | 12.4 | 21.1 | 12 | I |
| 18 | 39.6 | 6.2 | 15.1 | 15.5 | I |
| 19 | 24 | 15 | 18 | 0 | I |
| 20 | 26 | 18 | 48 | 23.5 | I |
| 21 | 38.4 | 15 | 12.6 | 35.5 | I |
| 22 | 5 | 12 | 15 | 21 | I |
| 23 | 29 | 8.1 | 19 | 21.2 | I |
| 24 | 47 | 25 | 30 | 59.8 | I |
| 25 | 87 | 14 | 36 | 63.7 | I |
| 26 | 69 | 5 | 23 | 49.1 | I |
All children with an acute edematous attack manifesting as laryngeal edema or severe acute abdominal complaints were hospitalized. Emergency treatment was implemented by the parenteral administration of 500 U C1-INH concentrate exclusively ( Table XVII). The injection accomplished substantial symptomatic relief within 30 minutes to 1 hour. This was accompanied by the regression of edematous swelling; however, complete resolution of clinical signs and symptoms took 24 to 48 hours. Administration of an additional 500 U dose (ie, a 1000 U cumulative dose) was necessary in 2 patients (1 with laryngeal and diffuse facial edema, 1 with acute abdominal attack) to achieve a satisfactory rate of improvement. All patients hospitalized with an acute abdominal attack underwent abdominal ultrasound examination. Ascites was invariably ascertained in all cases, whereas edema of the intestinal wall was detected in 80% of patients (Fig 7, A). As evidenced by follow-up ultrasound, appropriate treatment (C1-INH concentrate) achieved complete disappearance of free peritoneal fluid and edematous swelling of the intestinal wall within 48 hours.
Table XVII.
Frequency of symptoms and type of treatment in 21 pediatric patients
| Symptoms |
Therapy∗ |
||||||
|---|---|---|---|---|---|---|---|
| Number | N | R | F | Tranexamic acid | Danazol | Duration of treatment | C1-INH concentrate cumulative dose/attack |
| 1 | • | 2 g/d | Long-term (1 y) | 2 × 500 U | |||
| 2 | • | 2 g/d | For symptoms only | ||||
| 3 | • | ||||||
| 4 | • | 200 mg/d | Long-term (11 y) | 3 × 500 U | |||
| 5 | • | 100 mg/d | Long-term (11 y) | ||||
| 6 | • | ||||||
| 7 | • | 100 mg/2 d | Long-term (6 y) | 3 × 500 U + 1 × 1000 U | |||
| 8 | • | 100 mg/3 d | Long-term (4 y) | 4 × 500 U | |||
| 9 | • | ||||||
| 10 | • | ||||||
| 11 | • | 100 mg/2 d | Long-term (2 y) | 2 × 500 U | |||
| 12 | • | 1 g/d | Long-term (1 y) | 3 × 500 U | |||
| 13 | • | ||||||
| 14 | • | ||||||
| 15 | • | ||||||
| 16 | • | 2 g/d | For symptoms only | ||||
| 17 | • | 2 g/d | For symptoms only | ||||
| 18 | • | 100 mg/2 d | Long-term (5 y) | 3 × 500 U | |||
| 19 | • | 200 mg/d | Long-term (11 y) | 2 × 500 U + 1 × 1000 U | |||
| 20 | • | ||||||
| 21 | • | 2 g/d | Long-term (1 y) | 1 × 500 U | |||
| 22 | • | 100 mg/d | Long-term (8 y) | ||||
| 23 | • | ||||||
| 24 | • | ||||||
| 25 | • | 2 g/d | For symptoms only | ||||
| 26 | • | ||||||
N, None; R, rare (<1 attack/mo); F, frequent (>1 attack/mo).
ε-Aminocaproic acid is not shown because it had no effect or caused intolerance, and therefore, it was replaced by tranexamic acid.
Long-term drug prophylaxis with antifibrinolytic agents (tranexamic acid or ε-aminocaproic acid) was initiated in 11 cases because of frequent (weekly or monthly) recurrence of edematous attacks or the presence of life-threatening attacks in the history. Complete remission was achieved in 3 girls maintained on 1 to 2 g/d tranexamic acid; however, antifibrinolytic agents were ineffective in 8 cases. Transferring these patients to treatment with 100 to 200 mg danazol/d completely eliminated serious edematous attacks (Table XVII). To taper the cumulative dose, the dosage interval of danazol (100 mg) was increased to 2 to 3 days after 6 months of clinical remission. Switching to this intermittent regimen (ie, doses repeated every other day or at 3-day intervals) from continuous dosage was successful in 4 cases. In patients maintained on long-term prophylaxis, doubling the daily dose for several days aborted prodromal symptoms or prevented the progression of mild clinical manifestations to full-blown attacks. A similar protective effect was observed in cases exposed to a precipitating factor (eg, upper airway infection or mechanical trauma).
No adverse effects potentially related to long-term prophylaxis necessitating the withdrawal of treatment were observed. The bone age, skeletal growth, and weight development were not substantially different from the age-specific averages of the Hungarian pediatric population. The age at appearance of secondary sexual characteristics and the onset of puberty were also similar to the national average in 7 children, but in 1 girl, menarche was delayed (to the age of 19 years), and her menstruation was irregular. Hirsutism was not observed in patients receiving long-term danazol prophylaxis. Monitoring of laboratory parameters revealed normal values in all 11 children tested, and abdominal ultrasound showed no abnormalities. The efficacy of danazol treatment was demonstrated by the significant increases of C4 and C1-INH concentrations in the serum ( Fig 27).
Fig 27.
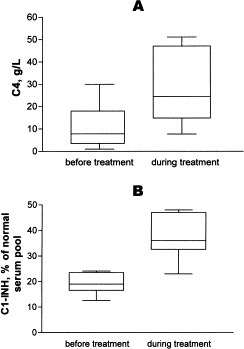
Effect of long-term danazol treatment on serum concentrations of C4 (A) and C1-INH (B) in children with HAE. Baseline values are compared with those measured in the first serum sample obtained after treatment had been initiated. C4 levels were determined in 8 children. Patients with high concentrations of nonfunctional C1-INH (233, 214, and 297%) were excluded from the comparison. Danazol treatment significantly increased both C4 and C1-INH levels (P = .002 and P = .0156, respectively; Wilcoxon signed-rank test).
Nine children had only mild, subcutaneous symptoms, and even these resolved spontaneously within 2 to 3 days. Accordingly, long-term prophylaxis was withheld, and treatment was administered only when prodromal symptoms and signs occurred. In 4 patients, tranexamic acid (2 g/d) given for 2 to 3 days reduced the severity and duration of subcutaneous manifestations. Follow-up visits were scheduled at 6-month intervals for these children.
Short-term prophylaxis was necessary in 2 boys (age 11 and 14 years) undergoing dental extraction, because both had facial and laryngeal edema precipitated by similar procedures in their history. Danazol (300 mg/d) initiated 4 days before and continued 4 days after dental treatment successfully prevented edema formation.
The elimination of known precipitating factors also substantially reduced the incidence of edematous attacks.
Discussion
In 80% of HAE cases, a known family trait of the disease facilitates diagnosis. In the remaining 20%, in which no afflicted relatives can be identified, HAE results from a new gene mutation.393 In the Hungarian pediatric population analyzed by the authors, the proportion of such cases was 93% and 7%, respectively. According to the literature, 85% of patients with HAE have type I and 15% have type II disease.31 The percentage distribution of type I and type II cases was 81% and 19% in the Hungarian series.
In general, the clinical manifestations of HAE first develop before 6 years of age7; the median age of the Hungarian study population was 5.9 years. However, no occurrences have been observed in newborns, and only 1 infant (19-month-old) case has been reported in the literature.320 According to the experience of the authors, adolescence is often associated with substantial changes in the activity of the disease, particularly in girls, warranting closer follow-up during this period. In particular, clustering of edematous attacks during the menstrual period was observed in 4 girls. Nowadays, contraception is initiated in early adolescence. However, OCs can precipitate acute attacks, and therefore, these agents are not recommended for girls with HAE.7
The incidence and severity of characteristic manifestations show substantial interindividual variation. Less than 5% of patients with HAE are asymptomatic, and 25% had symptoms only sporadically. Manifestations of the disease are common in 70% of patients and severe in 30% of this subpopulation.7 Eleven of the 26 children followed up in this study had frequent and severe attacks. Usually, mechanical trauma is reported as the most common precipitating factor. Through their experience, the authors confirm this and identify upper airway infection as another potential triggering factor.
Considering the importance of physical activity and sports on growth and development, restrictions should be determined carefully and individually. In children, as in adults, edema of the extremities is the dominant manifestation; furthermore, abdominal attacks are more common than laryngeal edema.7., 31. Nevertheless, even rare occurrences of laryngeal edema should be considered real emergencies. Because of the small diameter of upper airways in children, relatively mild swelling of the mucosal lining causes substantial obstruction, and suffocation can therefore rapidly ensue. In view of these risks, referral to intensive care is justified to monitor the clinical course and ascertain the efficacy of treatment. In some patients, repeated administration of C1-INH concentrate may be necessary to achieve satisfactory control of edematous manifestations. This fact and the ineffectiveness of conventional agents (eg, antihistamines, corticosteroids, and epinephrine) account for the invariably high mortality of the disease and emphasize the importance of accurate diagnosis.
The striking similarity between the clinical manifestations of abdominal HAE attacks and surgical emergencies is an apparent diagnostic pitfall. Failure to evade this trap often leads to unnecessary interventions on patients already prostrated by the consequences of the acute edematous attack.36., 427. The authors reviewed the medical history of their patients before diagnosis, revealing that 2 children had undergone exploration or laparoscopy during an acute abdominal attack, 1 of them twice. These interventions were clearly unwarranted, because no abnormality other than the edematous swelling of intestinal wall was ascertained. To avoid this diagnostic pitfall, the authors recommend close monitoring of the patient, volume replacement, parenteral medication, and assessment of therapeutic efficacy, all achievable by observing the patient for 24 to 48 hours. Abdominal ultrasound is a noninvasive and readily accessible diagnostic test that yields reproducible results, features particularly advantageous in pediatric practice.39 The authors identify free peritoneal fluid and edema of the intestinal wall as invaluable sonographic clues for distinguishing abdominal HAE attacks from other pathologies commonly associated with ascites formation (such as intra-abdominal inflammation, autoimmune disease, tumors, and so forth). They recommend that, when all other acute abdominal disorders had been ruled out, C1-INH concentrate should be administered to afebrile patients without laboratory abnormalities, particularly to those with known HAE. Prompt symptomatic relief (or lack thereof) after C1-INH concentrate administration can then confirm or refute the tentative diagnosis.
During life-threatening HAE attacks, appropriate measures can include the administration of FFP. This preparation contains C1-INH; however, its use may be associated with alloimmunization or the transmission of infections.7 The most appropriate therapy is the intravenous administration of C1-INH concentrate. Its dosage is the same for children and adults (ie, 500-1000 U).320 In this study, acute HAE attacks were treated with C1-INH concentrate exclusively. This approach was highly effective, resolving clinical symptoms within an hour, and was not associated with adverse effects. C1-INH concentrate has been suggested as an ideal agent for permanent substitution, particularly for pediatric patients. Unfortunately, the short (72-hour) half-life and high cost ($310 per vial) of the concentrate preclude its use for continuous administration.428 Furthermore, C1-INH concentrate is a blood product, and the theoretical risk of blood-borne infections cannot be completely excluded. Nevertheless, viral (HIV, HBV, HCV) safety of the concentrate is promoted by heat treatment as well as the establishment and continuous validation of safe plasma pools.75., 334. In the Hungarian case series, transmission of viral infections could not be demonstrated after the administration of C1-INH concentrate.
In pediatric patients, antifibrinolytic agents are the first choices for long-term prophylaxis because their safety profile is more favorable than that of attenuated androgens.7., 31., 36., 429. Nevertheless, antifibrinolytic agents can cause adverse effects such as muscle weakness, myalgia, elevation of creatine kinase activity, vascular thrombosis, postural hypotension, and myonecrosis.31 The experience of the authors showed tranexamic acid to be better tolerated than ε-aminocaproic acid, which often caused gastrointestinal discomfort. However, when these agents fail to achieve satisfactory improvement, treatment with attenuated androgens (eg, danazol, stanozolol) is necessary.312 Numerous studies performed on adult patients have demonstrated the efficacy of 17α-ethyltestosterone (danazol) manifested by the substantial relief of symptoms as well as the elevation of serum C4 and C1-INH concentrations.347 Their mechanism of action has not yet been fully elucidated, and limited experience with their pediatric use has been obtained only in patients treated for idiopathic thrombocytopenic purpura.430 Potential adverse effects of danazol include weight gain, myalgia, headache, tremor, libido changes, elevation of serum transaminase levels, microhematuria, menstruation irregularities, and hirsutism.349 In addition, 2 reports have been published on hepatocellular adenoma formation observed in adults undergoing long-term, high-dose danazol therapy.353 Nevertheless, using the lowest effective maintenance dose and intermittent dosage regimens can prevent adverse effects that would require the withdrawal of treatment. In the study population, the longest duration of danazol prophylaxis was 11 years. No drug-related adverse reactions or impairment of growth was recorded in these pediatric patients, with the exception of 1 case of delayed menarche with subsequent irregular menstruation probably caused by long-term treatment with 200 mg/d danazol. Compared with age-specific percentiles of the Hungarian population, somatomental development was normal in all cases. Treatment prevented edematous attacks and elevated C4 and C1-INH concentrations significantly. Effective therapy eliminated the need for frequent hospitalizations for acute symptoms—along with their unfavorable psychologic sequelae—and thereby improved the patients' quality of life.
The authors emphasize the essential importance of close follow-up, with appropriate tests performed at regular intervals. The diagnosis of HAE can be made as early as at birth by assessing C1-INH antigenicity and function.320 Prenatal screening is not yet feasible. Importantly, baseline complement concentrations are of no use for predicting the severity and frequency of future attacks; for example, some patients are asymptomatic despite extremely low C4 concentrations and low amounts of antigenic and functional C1-INH. Finally, when administered properly, long-term prophylaxis usually provides reliable protection against acute attacks.
During a patient's first 2 years of long-term prophylaxis, laboratory tests should be performed every 3 to 4 months and abdominal ultrasounds scheduled at 6-month intervals. The clinical course of HAE and reactions to treatment should be monitored to recognize potential adverse effects and ascertain the rate of somatomental development as well as the need for therapy modification. In view of their favorable experience, the authors suggest that danazol administered in the lowest effective dose seems to be well tolerated by pediatric patients and is probably appropriate for long-term therapy. Nevertheless, adverse reactions can occur sporadically. In particular, 1 case of delayed menarche and irregular menstruation was observed in this series; however, the abnormality was readily reversible on discontinuing danazol treatment.
Short-term prophylaxis is indicated for patients undergoing surgical or diagnostic interventions of the head and neck region. Such procedures most commonly include dental procedures and tonsillectomy, but the potential consequences of endotracheal intubation for general anesthesia must also be emphasized.365., 431. Although short-term prophylaxis is required less frequently for children than for adults, antifibrinolytic agents or attenuated androgens provide adequate protection if administered in higher than usual doses. Nevertheless, prophylaxis with C1-INH concentrate is the method of choice in patients with a history of severe attacks precipitated by similar procedures.432
In addition, follow-up should also include the education of patients and their parents, as well as advice on the most suitable means of lifestyle modification. The activity of patients' self-help groups can lend vigor to such efforts. Early health education and meticulous follow-up initiated in childhood can successfully prevent social stigmatization and guarantee an improved the quality of life for adulthood. It is prudent to provide patients with HAE with a medical information card summarizing essential knowledge and methods of emergency help in several languages. When justified by their history, patients should be supplied with C1-INH concentrate to be kept at hand, ie, in the refrigerator at home, for emergencies. The data contained on the medical information card can prove an extremely useful aid for medical professionals relatively unfamiliar with HAE and help to ensure patients' safety.
This article first appeared in a slightly different form as Farkas H, Harmat G, Fust G, Varga L, Visy B. Clinical management of hereditary angio-oedema in children. Pediatr Allergy Immunol 2002;13:153-61. It is reprinted with permission.
Patient initiatives
Although all patient and physician groups share the common goals of increased knowledge of HAE and related disorders and optimized therapy, patient and physician experiences differ among countries. The following accounts demonstrate the range of perspectives, treatment options, and concerns found in Europe and the United States, concluding with a short history of past C1-INH Deficiency Workshops in which representatives from these and other countries have met to further their common aims.
An international approach to patient registries
(Marco Cicardi, MD,∗ and John Jakenfelds, MD, Milan, Italy, and Chalfont St. Giles, UK)
The concept of a European patient registry grew out of the first C1-INH Deficiency Workshop. In this section, Cicardi and Jakenfelds describe the process of creating the European HAE Register and how the register will work.
In 1999, the European group of patients, clinicians, and scientists interested in HAE met for the first C1-INH Workshop in Visegrád, a Hungarian town near Budapest. Workshop members identified the creation of a register containing the clinical and laboratory data of patients with HAE as an obvious first step.
There is a real need for a better insight into the full presentation of HAE, the different ways it is managed, and the various outcomes (including clinical, functional, and economical variables) associated with different disease management strategies. Currently, there are significant differences in the way patients with HAE are treated in various HAE expert centers. There is no consensus on the state-of-the-art treatment of HAE, and there is a lack of data to develop uniform, evidence-based treatment guidelines. As a result, an enormous gap exists between resources already or potentially available and their exploitation.
Furthermore, the low awareness of this disease causes only a minority of patients to reach correct diagnosis and treatment. Small and dispersed case lists and the lack of epidemiologic data lessen the value of clinical studies and discourage public and private investment in research aimed at developing new therapeutic strategies or tailoring existing ones to HAE. Given this situation, an international patient register could play a significant role in the development of a better understanding of HAE and its management.
First steps
Two pharmaceutical companies, Pharming and Baxter, encouraged the establishment of an HAE register in 2001. Baxter already had a product for the treatment of the disease (plasma-derived C1-INH) in some European countries, and Pharming was developing a recombinant version (rhC1-INH). Pharming became the major contributor until 2002, when a group of HAE experts from 10 different European countries received a grant from the European Commission for a project called PREHAEAT, consisting of a concerted action in the framework of the specific research and technologic development program, Quality of Life and Management of Living Resources. Establishing a European Register for HAE was the first work package of the project and was started accordingly by using the Pharming-funded register as a starting point for further refinement.
In this new register, physicians will enter completely anonymized patient data into a Web-based data capture tool. Princeton Healthcare will be responsible for the central server and software, accessible by all sites and individuals wishing to enter data ( Fig 28). Princeton is registered under the UK Data Protection Act to store personal information as part of its ongoing work with clinical systems, which have been in use for several years.
Fig 28.
Flow of data in the European HAE Register. PC, Personal computer.
The register will work by using the Internet. By connecting to the Web and entering the site's name, the password-protected entry screen will be viewable ( Fig 29). On filling out details, the user will be taken to a menu of options, including the entry of new data or the viewing of reports covering anonymized patient records (either individual or for the group under treatment at the clinic). Only the treating physician, by knowing a code, will be able to link an individual report to an individual patient. Access initially will be restricted to the physicians participating in the concerted action and eventually will be open to any HAE-treating physician on request. Efforts will be taken to publicize maximally the existence of the register.
Fig 29.

Window for accessing the European HAE Register.
Access to data: Ownership and management
Each patient owns his or her own data. In order for the data to be entered into the register, patients must give consent to share their anonymized data. All clinicians contributing to the register will have access to their own data as individual or group reports. The board, consisting of the participants to the concerted action and of 2 patient association representatives, will manage the anonymized database and may access aggregated reports but not individual data sets. No single member of the board can use or publish the data without the consent of the board and the contributors as a whole.
The goal of the register is to develop a better understanding of the current benchmarks of care for HAE and the outcomes that result to improve the ways in which patients are treated.
For each patient, there will be an initial entry form and 1 or more follow-up visit forms ( Fig 30). The initial entry form is structured to capture the natural course of HAE in a patient. It will contain the past history before diagnosis of HAE was recognized and/or specific treatment established. It will contain anagraphic data, information on characteristics and severity of symptoms, presence of associated diseases, and laboratory data at the time of diagnosis.
Fig 30.
European HAE Register visit form.
The follow-up visit forms can be replicated indefinitely. They will collect the same data, updated according to the changes occurring in the course of the disease, and detailed data on the different therapies. Specific care has been taken to record effectiveness and side effects of HAE treatments.
Expected achievements and perspectives
This register is expected to become a reference database in HAE. Efforts have already begun to promote the harmonization of similar data collection systems on this topic around the world such that compatible formats will allow pooled data analysis.
Information describing the natural history of HAE and the effect of treatment should be obtained. This database should also be used to provide policy makers and the pharmaceutical industry with data showing exactly how the disease affects the lives of patients who have it.
HAE therapy in the United States
(Anthony J. Castaldo, Washington, DC)
Castaldo, the author of this section, is a Founding Board Member of the United States HAE Association.
The absence of an approved HAE acute attack therapy in the United States of America represents a catastrophic unmet medical need. Patients are suffering needlessly and in some cases are dying. Indeed, the United States HAE Association (HAEA) is aware of 4 HAE-related deaths over the period of the past 40 months. The most pressing near-term goal of the HAEA is to work with industry, the research community, and regulators to encourage US-based clinical trials that will result in licensure of an acute attack therapy. Clearly, the lack of a therapy to treat an HAE attack once it has begun poses a tragic and unnecessary risk to every US HAE patient's life.
Fortunately, a phase II clinical trial for an acute HAE attack therapy sponsored by Dyax Corp is now up and running in the United States. The patient community is excited about the prospects for Dyax's therapy on the basis of some promising safety and efficacy data developed during phase I and II trials conducted in Europe. The HAEA is working closely with Dyax to recruit patients and complete this vital clinical trial. If the US phase II results are as promising as results seen in phase I/II European trials, the researchers hope that Dyax will request an expedited licensing review by the Food and Drug Administration. The HAEA is committed to ensuring that patients, industry, and the Food and Drug Administration work together and strike an intelligent balance that permits an expeditious path to meeting a catastrophic unmet medical need while simultaneously protecting the public health and safety.
Meetings such as the one in Budapest have provided the opportunity for patients with HAE to interact and share information. This networking has spawned a movement that should soon result in the creation of a legally recognized international HAE patient organization. Clearly, an international HAE organization can provide the world's HAE groups with information, analysis, and guidance on key issues regarding HAE management, diagnosis, and the direction of future research. Indeed, researchers active in establishing an international HAE group have already begun the process of examining the state-of-the-art for HAE treatment in different parts of the world. This process has uncovered some strongly held views among the world's patients who are not satisfied with the current treatment options and standards. This drumbeat of concern is prompting international leaders to re-examine the HAE treatment paradigm that almost always features androgens for prophylaxis, with acute attack therapy (C1-INH concentrate) administered only for gastrointestinal and/or life-threatening attacks. There is some very exciting and innovative work going on in Germany that quantifies patient dissatisfaction with the current treatment norm. Specifically, the German data show that when given an option, many patients discontinue androgens and opt for on-demand (usually home-based) treatment with an acute attack therapy that they infuse during the attack prodrome.
In sum, the treatment of HAE in the United States is best described as medieval. Without access to an acute attack therapy, patients must rely exclusively on prophylaxis with androgens. These drugs are highly toxic and have harsh side effects, particularly in women. The HAEA is working hard to assist companies interested in licensing an acute attack therapy in the United States. There is much hope that once formally established, an international HAE organization will help to create and rationalize a partnership between the world's HAE patients, researchers, regulators, and industry to ensure new therapies can be expeditiously tested, approved, and marketed.
The Hungarian experience: assisting the few to live a better life
(István Nagy, Arianna Kitzinger, and Henriette Farkas, MD, PhD,∗ Székesfehérvár, Sopron, and Budapest, Hungary)
In this section, Hungarian physician Farkas and patient-researchers Nagy and Kitzinger describe the importance of the Hungarian HAE Center and its infrastructure, as well as their case series and approach to HAE.
Creating an infrastructure
The Hungarian Complement Laboratory in Budapest was founded by Prof. George Füst in 1970. The first article detailing 3 HAE patients' cases was published in 1972.433 These achievements were followed by several milestones leading to the establishment of the Hungarian HAE Center:
-
•
Accumulating relevant knowledge from the international literature, adapting foreign experiences to domestic conditions, and developing Hungarian protocols for clinical work-up and treatment
-
•
Undertaking retrospective screening for patients by using archived laboratory test results (complement values)
-
•
Communicating pertinent knowledge to medical professionals, graduate and postgraduate education programs, and others via publications
-
•
Establishing a results-based referral system: if laboratory complement measurements are suggestive of HAE, accessibility information for the Hungarian HAE Center is printed on the laboratory test slip
The development of research ran in parallel with the increasing number of patients diagnosed with HAE. Fig 31 illustrates the rate of case accrual. Currently, there are 96 patients in Hungary (total population: 10 million people), of whom 28 live in Budapest and its suburbs.
Fig 31.
Hungarian HAE Center patient accrual.
Hungarian HAE Center
The headquarters of the Hungarian HAE Center are located on the premises of Semmelweis University in Budapest. Each facility has its assignments, as shown in Fig 32.
Fig 32.
Hungarian HAE Center: flow of responsibilities.
Assignments of the Allergy and Angioedema Outpatients' Clinic at Semmelweis University include the following:
-
•
Differential diagnosis, diagnostic work-up, and treatment of patients with angioedematous symptoms from all over the country
-
•
Development of national guidelines for the recognition, treatment, and follow-up of patients with C1-INH deficiency
-
•
Maintenance of the Hungarian HAE Registry (which currently contains the data of 92 patients), as well as the serum, plasma and DNA bank
-
•
Patient education and counseling
-
•
Supply of all patients with a multilingual infocard and C1-INH concentrate for emergencies (the Hungarian Ministry of Health grants full social security reimbursement for the price of the injection, relieving patients of copayment; patients can store the preparation at hand, readily available whenever and wherever emergency occurs)
-
•
Management of patient follow-up (laboratory check-up at 3-month to 6-month intervals, abdominal ultrasound every 6 months, particularly if the patient takes danazol)
-
•
Maintenance of a telephone hotline service for patients and doctors from other specialties
During an acute, life-threatening attack, adult patients are hospitalized and treated at the Third Department of Internal Medicine of Semmelweis University, Budapest. Pediatric patients are admitted to the Madarász Street Children's Hospital, Budapest. Adults' and children's health care institutions provide emergency services for patients with HAE around the clock.
The Complement Laboratory functions as a unit of the HAE Center and is properly equipped to perform total complement profiling (CH50, C4, C1q, C1-INH concentration and functional activity, anti–C1-INH antibodies, anti-C1q antibodies). The Molecular Biology Laboratory and the Research Laboratory of the Third Department of Internal Medicine conduct genetic testing and mutation analysis on all patients with HAE. Furthermore, the center participates in undergraduate and postgraduate education and also pursues scientific activities.
Case management standards
The Hungarian algorithm for HAE treatment, in the light of its symptoms, therapy, and management is as follows:
-
•
Mild or infrequent subcutaneous edema does not require special treatment. Self-management of symptoms will suffice, and medical monitoring can consist of yearly complement measurements.
-
•
Severe or frequent subcutaneous and submucosal edematous symptoms are treated by either long-term prophylaxis (danazol, ɛ-aminocaproic-acid, tranexamic-acid) or short-term prophylaxis (danazol, ɛ-aminocaproic-acid, tranexamic-acid, C1-INH concentrate). Such patients require regular medical and self-control. Medical control involves liver function monitoring and blood cell and complement measurements twice yearly.
-
•
In case of acute abdominal or laryngeal edematous attacks, inpatient therapy with C1-INH concentrate is needed. Patients experiencing these symptoms also require regular medical and self-control. Postattack follow-up is necessary.
Hungarian HAE patients' association
Established in 1996, the Hungarian HAE Patients' Association organizes annual meetings at which patients can meet doctors and exchange views with each other. A foundation has been established to support patients with HAE, and a Hungarian HAE Web site is now being created.
The Hungarian HAE Patients' Association recently collected 26 patient diaries, drawing attention to topics important among the patient population. The association is now beginning to make a systematic survey of social background and its effect on the disease, which ideally will lead to a deeper understanding of HAE and an improvement in patients' quality of life.
The patients' association maintains fruitful relationships with foreign patients' self-help groups. As with all uncommon diseases, the elucidation of its epidemiologic features and pathomechanism, the establishment of current diagnostic algorithms, and the development of effective and safe treatment protocols all require international effort. This recognition led to the establishment of the European C1-INH Deficiency Working Group in 1998. During its first conference held in Rhodes, Greece, this professional organization identified accomplishing these goals as its ultimate objective. Since that time, the Hungarian HAE Work Group has been the main organizer of the European C1-INH Deficiency Workshops, the basis for this supplement. The next workshop is planned for 2005 in Budapest.
The Hungarian HAE Work Group values its international connections and is
-
•
An original member of the European C1-INH Working Group
-
•
A member of the European HAE Board
-
•
A participating center of the ATHERNET European Union (EU) Project
-
•
A subcenter of PREHAEAT EU Project
Much progress has been made since the first steps taken in the 1970s. In addition to the results described, the association is glad to welcome several humanitarian changes. Patients with the diagnosis of HAE are now under regular medical control, in contrast with perpetual uncertainty of the past. As a result of patient education and counseling (ie, sufficient familiarity with the disease), patients are understood and helped more effectively by those in their environment. Instead of isolation, patients feel integration, trust, and confidence: a major change and an impressive outcome in the case of this life-threatening disease. As shown in Table XVIII, all of this progress has helped patients to find an appropriate lifestyle.
Table XVIII.
What has changed through the establishment of the Hungarian HAE Center?
| Relationship | Then | Now |
|---|---|---|
| Patient-doctor | Uncontrolled situation | Under control |
| Patient | Uncertainty | Certainty, access to medicine |
| Patient-environment | Sympathy but lack of comprehension | Becoming understood and helped effectively |
| Patient-patient | Isolation | Integration |
However, without doctors' professional knowledge and its perpetual updating, these developments in Hungarian HAE research could not have been achieved. Many doctors regard their job as their hobby and also feel a commitment to help at both the domestic and international levels. Their compassionate care for patients is both noticed and appreciated by those in need.
Portions of this article appeared in Farkas H, Varga L. The Hungarian HAE experience. Transfus Apheresis Sci 2003;29:229-33. They are reprinted with permission.
The German experience: HAE and self-medication with Berinert P
(Ursula Rauch, Aldenhoven/Siersdorf, Germany)
In this section, Rauch, a member of the German HAE patient association, HAE Vereinigung e. V., writes of her experiences as she presented them at the 2003 C1 Esterase Inhibitor Deficiency Workshop in Budapest. The German HAE patient association's motto is Nicht Mehr Allein, “no longer alone.”
To introduce myself, I would like to tell you a little bit about my career as an HAE patient. I think all the HAE-affected here have had the same or similar experiences.
So, here is the short version of my story. I am 44 years old and have HAE-I. I had my first attack at the age of 1½, diagnosed as allergy; it was followed by tests and experiments with antiallergic remedies and corticoids, which were, of course, useless. There was an increase in my attacks during puberty, and I received the correct diagnosis at age 16. There was no remedy at that time, but it was a relief to know it was HAE and to stop the antiallergy experiments.
Sometimes I went for 2 months without an attack; sometimes I would spend 2 weeks with a permanent attack; but to a certain degree I managed to arrange my life around my disease.
But after my second pregnancy, the attacks greatly increased. I often needed help to care for my children. I became depressed and could not hold down a job because I was ill too often. Not 1 week went by without an attack, and I often had nonstop attacks that lasted 8 to 10 days.
In 2000, I finally found our support group, got a lot of information, and was told about the HAE Center in Frankfurt. I made an appointment at the university hospital in Frankfurt, where I received very good counseling, and 4 weeks later I went to my first HAE group meeting.
With the help of the physicians of the university hospital in Frankfurt and my practitioner, I learned to infuse myself in an emergency. So I was much more independent and felt much safer than before.
After training with normal saline, when I tried my first injection, I went to my practitioner in order to have a supervisor—and it did work!
And today?
No endless pain, vomiting, and breakdowns of my circulation.
No waiting for emergency medical personnel, especially on Saturday or Sunday, or at night.
No more useless claims that corticoids always help, even though my emergency passport says differently.
No hurrying to a hospital, breathless and in panic, because of a laryngeal edema.
No waiting there for a tired physician who already has 30 hours of work behind him.
No enduring questions about whether my husband could have beaten me because my face looks just like a boxer's.
I was able to participate in further professional education. I look after my children myself. I can again go on vacations, which always seemed too dangerous before. And I am active in the patient group as much as I can be. I learned how helpful and important it is to work together and fight for a better and more effective therapy.
To me, it is absolutely incredible that in most countries, families do not have even 1 package of C1 inhibitor at home for emergency cases, and that there are doctors who give androgens to children, even though they are still growing and the doctors should know how dangerous these androgens can be.
And it is horrible that a lot of affected people are still misdiagnosed and therefore constantly living in danger.
We must create the opportunity for every HAE-affected person to get the right diagnosis and the same help, so that children can grow up as normally as possible, without missing school very often, and so that we all can go to work and enjoy life—just like the nonaffected, without horrible pains and horrible fears about the risk of suffocating to death or dying of pain.
We are a minority—in every country—but together we are strong enough not to be stopped.
The Italian experience
(Vincenzo Penna and Marco Cicardi, MD,∗ Milan and Peschiera Borromeo, Italy)
In this section, Penna and Cicardi of the Voluntary Association for the Study, Therapy, and Fight against Hereditary Angioedema (AAEE)—ONLUS, describe the goals and accomplishments of their organization.
In recent years, patient associations have had a fundamental role in the fight against rare diseases. Many organizations now exist in various parts of the world, even for HAE, to perform the important task of sharing information and maintaining a connection between patients and doctors. Such associations perform indispensable functions, useful to both doctors and researchers.
Aims
AAEE was founded as a nonprofit voluntary association in Milan on March 29, 1980, and has the following aims:
-
•
To spread knowledge of the disease and allow correct diagnosis
-
•
To make high-quality medical care and adequate treatment available to all patients
-
•
To issue an officially recognized health identification document describing the disease and its emergency therapy and containing the contact information of doctors who can be called for further information
-
•
To promote social and scientific meetings and congresses at a national and international level
-
•
To encourage meetings and exchanges among patients, between patients and doctors, and among doctors
Services
AAEE represents approximately 400 members and, in more than 20 years of activity, has created a series of services to benefit patients:
-
•
In collaboration with the doctors of the Diagnosis and Cure Center in Milan, a 24-hour emergency telephone number was activated.
-
•
All patients have an emergency identification document indicating their diagnosis and its suggested treatment in more than 1 language.
-
•
A direct link has been established with the pharmaceutical company that produces C1-INH concentrate to locate this life-saving medicine at any time.
-
•
The Minister of Health and Welfare has added HAE to the list of rare diseases for which patients are exempted from paying for medicine.
-
•
A multilingual Internet site (http://www.angioedemaereditario.org) offers medical and scientific information, useful addresses, and contact information for all HAE associations worldwide.
-
•
Regional representatives permit a more direct connection between patients and the association.
-
•
Gynecologic and dental consultations are available.
-
•
To spread medical and scientific information as well as the association's activities, radio and television broadcasts have been organized.
-
•
Every year, a national meeting is held in addition to regional meetings to exchange information between doctors and patients and between patients and the association.
-
•
Some patients who have social roles in the community offer their experience and assistance to the association to deal correctly with various bureaucratic problems.
-
•
There are now 3 other centers in addition to the 1 in Milan available to patients who need treatment, in Rome, Palermo, and Civitanova Marche.
Recent initiatives
Recently, the association prepared a very simple form to collect data on the frequency of HAE attacks in Italian patients. Information will be collected over the period of the next 6 months as a brief trial. This will help AAEE evaluate replies received from patients to make all necessary modifications and adapt the survey as much as possible to each involved country's needs and requirements. Data will be collected in a proposed specific database.
To make better use of the information, the AAEE intends to put their Internet site at everyone's disposal. In the last few weeks, the international pages have been modified, transforming them into a very simple but functional place where HAE information and links throughout the world can easily be found.
With the representatives of other associations, the members of AAEE are working to create an international umbrella organization. After last year's short meeting on HAE in Palermo, where the first ideas were elaborated and after the Budapest workshop, an international statute proposal now exists. During the last meeting, which took place in Milan on October 4 and 5, 2003, an international committee and a medical panel of doctors and researchers were formed.
Conclusion
(Kayla Williams, BS, MA, MFA, Cambridge, Mass)
Clinical manifestations
Hereditary angioedema and related disorders are characterized by acute episodes of unexplained swelling that are not responsive to antihistamines or corticosteroids. Attacks may manifest in 1 or more locations, most frequently the upper airways, face, hands, feet, genitalia, or intra-abdominal contents. Untreated laryngeal swelling can result in death; angioedema of the small intestine can result in obstruction and mimic an acute abdomen. Ultrasound may be useful to distinguish between abdominal pain attributable to HAE and a true surgical emergency. Occasionally, erythema marginatum or a similar, nonwhealing, nonurticarial rash may precede angioedema attacks; at other times, there may be no warning. Episodes of HAE may present as early as late infancy and may vary, even within the same individual, in frequency, severity, and location. Attacks typically resolve within 72 hours but may persist as long as 5 days. AAE, although much rarer than HAE, often follows lymphoproliferative disorders or occasionally autoimmune, neoplastic, or infectious diseases. Drug-induced angioedema may closely follow initiation of a drug regimen (often an antihypertensive agent) or appear many months later, whereas estrogen-dependent angioedema seems to follow initiation of pregnancy, OCs, or HRT more immediately.
Gene mutations and pathogenesis
Hereditary angioedema types I and II are associated with various mutations to the C1NH gene, located on chromosome 11. A frequently updated online database of these mutations is available to the public at http://hae.biomembrane.hu; at the time of writing, 150 different C1NH mutations were known. The progression from low functional C1-INH concentrations to attack genesis is still under debate; however, bradykinin has been identified as a mediator of angioedema attacks, and, unlike other proposed mediators, has been found to correlate with attack localization in early studies. Nonetheless, this nonapeptide is unstable and cannot be measured via routine laboratory analyses. It is hypothesized that the local generation of bradykinin results in regional increases in vascular permeability. The presence of other mechanisms, intermediaries, and moderating factors has not been discounted.
Triggers and hormonal influences
Case reports have shown that in some women, the occurrence or frequency of HAE attacks appears to be influenced by estrogen. In these patients, puberty, pregnancy, or taking estrogen-containing OCs or HRT may initiate attacks. Patients have also been described with normal concentrations of functional C1-INH who nonetheless have angioedema attacks in response to increases in estrogen. Bork et al found that approximately 62% of women with HAE-I or HAE type III have a worsening of their attacks because of pregnancy or exogenous estrogen.
Laboratory diagnosis
C1-INH antigenic and functional assays are sufficient to diagnose HAE-I, HAE-II, and AAE. Low C4 concentration is often a reliable confirmation but is not exclusive to C1-INH deficiencies. Currently, no routine laboratory methods can detect estrogen-dependent angioedema (formerly HAE type III).
Prophylaxis
Ideal prophylactic regimens are highly individualized and should consider the severity and frequency of the patient's attacks. 17-α Alkylated androgens, antifibrinolytics, or C1-INH concentrate have been used successfully for HAE maintenance prophylaxis and/or short-term prophylaxis surrounding surgical or dental procedures. Bork et al describe C1-INH as an effective, if less frequently used, prophylactic agent. In some patients, antifibrinolytics may be sufficient to prevent attacks; however, attenuated androgens more often reduce the frequency and severity of attacks. Patients receiving long-term androgens, especially pediatric patients, should be monitored for undesirable hepatic effects and other consequences such as virilization or delayed menarche; Farkas et al suggest laboratory work-ups at intervals of 3 to 4 months. Androgen overdosing is to be avoided; Varga and other members of the Workshop laboratory diagnosis panel note that, in HAE-I, antigenic C1-INH concentrations approaching 35% to 40% of normal are often sufficient to prevent attacks, and therefore, aiming steroid prophylaxis to achieve a 50% antigenic concentration subjects patients to an unnecessary risk.
For the prophylaxis of AAE, antifibrinolytic agents are recommended because patients with this condition are frequently resistant to attenuated androgens.
Associated effects of prophylactic regimens
Danazol is a common prophylaxis for patients with HAE who have frequent, severe angioedema attacks. Despite known associated adverse effects such as virilization, weight gain, and decreases in hepatic function, the administration of danazol may result in an improvement of some conditions. Contrary to findings in patients receiving high-dose danazol treatment for endometriosis, Kollár et al reported that patients with HAE receiving the minimum effective dose of danazol necessary to control their angioedema attacks did not have osteoporosis. Indeed, their data suggest that such a danazol regimen may have a protective effect against bone loss. Perricone et al noted a higher prevalence of polycystic or multifollicular ovaries in women with HAE but found that danazol administered in HAE-management doses, at least in a small sample of patients, improved ovarian condition.
Acute attack treatment
Treatment varies depending on the location and severity of angioedema. Peripheral edema often requires no treatment. For attacks involving the airways or severe abdominal manifestations, C1-INH concentrate is the current acute attack therapy of choice for patients with HAE. In countries where this is not available, personal importation of C1-INH concentrate may be an option; alternatively, the administration of FFP or an antifibrinolytic, although less effective, may be of some benefit in emergencies. Supportive measures such as rehydration, antiemetics, and pain control may be necessary depending on the site and severity of the attack.
Future treatment options
New uses for existing therapy, such as prophylactic or home administration of C1-INH concentrate, have been studied on a small scale and proven successful. New therapies, including 2 versions of recombinant C1-INH, a kallikrein inhibitor, and a bradykinin receptor-2 antagonist, are in development.
Afterword
(Marco Cicardi, MD,∗ Milan, Italy)
Since the first comprehensive clinical description of HAE in 1888, knowledge of the disease has been expanding. As have many other genetic defects, HAE has benefited in the last 20 years from the spread of molecular biology, which brought a tremendous advance in the understanding of the molecular basis of the disease. However, what appears remarkably unique to HAE is that within the last 4 years, this bulk of information has been directly transferred into therapeutic approaches. The dream of research from the bench to the bedside is becoming real in HAE, and the rule that there is no interest in developing drugs for treating a small number of patients seems to have vanished. Revolutionary new techniques for identifying and producing drugs, such as peptide selection from phage display libraries and transgenic animals as source of recombinant proteins, have been implemented to obtain agents active in HAE. Currently, 3 completely new compounds, a kallikrein inhibitor, a bradykinin receptor antagonist, and a recombinant C1-INH, are under clinical evaluation for the treatment of HAE.
This disease is facing the new pharmacology. Chemicals antagonizing biologic systems are being substituted with disease-tailored agents, and in HAE, this does not necessarily mean gene therapy. This close relationship between a pharmacologic agent and a disease results in the need for a close relationship between patients and industry. In 1999, the first C1 Esterase Inhibitor Deficiency Workshop was held in Hungary with the idea of having patients, scientists, physicians, and industries contributing with equal authority. Since that time, a real HAE community has been created, and patient support groups are actively involved in designing and carrying clinical studies and are supporting initiatives, such as the HAE register, aimed to improve the understanding of the disease and its treatment.
Aside from the new scientific knowledge or the new therapeutic compounds, recent years' frank and open discussion between different parties is a major achievement for the advancement of the HAE cause.
Many sections of this supplement have repeatedly highlighted how disabling and risky it has been for most patients living with HAE, but patients can now have great hopes for the future. The major concern remains the difficulty that many patients still have in reaching correct diagnosis and treatment. Deaths caused by HAE and similar angioedematous disorders still occur in developed countries because the majority of physicians, ignoring the existence of HAE, still label these patients as allergic. We hope that this supplement will help bring more HAE, AAE, and nonallergic edema patients to the correct diagnosis.
Pr Marco Cicardi, MD
Università degli Studi di Milano
Dipartimento di Medicina Interna
Ospedale S. Giuseppe
Milan, Italy
Acknowledgments
Disclosure of potential conflict of interest: A. Agostoni is an employee of the University of Milan. K. Binkley has received financial support from Dyax Corp. to cover the costs associated with preparation of this manuscript. C. Bucher's work in the field of hereditary angioedema is supported by the Swiss Federal Office for Education and Science; the European project is supported by Pharming; he is a consultant with the Swiss branch of Aventis Behring (now ZLB Behring) and has close contacts/collaboration with representatives of companies developing new medications for patients with hereditary angioedema. M. Cicardi has consultant arrangements with the following companies: Dyax Corp. for development of Dx88, Pharming for development of recombinant C1-INH, Jerini for development of Icatibant, and Aventis for trial on C1-INH concentrate. A. E. Davis III has consultant arrangements with Dyax Corp., Jerini, Lev Pharmaceuticals, and Pharming, and he receives grants/research support from Dyax Corp., Lev Pharmaceuticals, and Pharming. G. Füst is a collaborator of ZLB Behring. A. Gröner is an employee of Aventis Behring. C. E. Hack is an employee of Sanquin Research, a not-for-profit Blood Transfusion Institute in The Netherlands, and has consulting agreements with Genmab and Lev Pharmaceuticals. M. Juers is an employee of Aventis Behring GmbH, a ZLB Behring company, and has Aventis stock options. H. Longhurst has received support/funding from Dyax Corp., Pharming, Aventis Behring, and Jerini. J. Nuijens is employed by Pharming Group NV, a biotechnology company developing recombinant C1NH. C. O'Grady has received support/funding from Dyax Corp., Pharming, Aventis Behring, Jerini, and Baxter. P. Späth is a collaborator of ZLB Behring. K. Williams is an independent writing contractor paid for this and other projects by Dyax Corp.; her husband is a senior-level employee of Dyax Corp. and owns shares of Dyax Corp. stock. All other authors—none.
Milan, Rome, L'Aquila, and Peschiera Borromeo, Italy, Frankfurt, Mainz, Bonn, Marburg, Hattersheim, Frankfut/Main, and Aldenhoven/Siersdorf, Germany, Toronto, Canada, Madrid, Spain, Grenoble and Rouen, France, Zurich and Bern, Switzerland, Washington, DC, Boston and Cambridge, Mass, Budapest, Sopron, and Székesféhérvar, Hungary, Amsterdam and Leiden, The Netherlands, Chalfont St. Giles and London, England, Bodø, Norway, and Lund, Sweden
Footnotes
Disclosure of potential conflict of interest: See page S121.
Supported by Kallikrein, LLC, a joint venture of Dyax Corp., and Genzyme Corporation, Cambridge, Mass.
Reprint requests: Dr. Tony Williams, Dyax Corp., 300 Technology Square, Cambridge, MA 01239; e-mail: williams@dyax.com
Cover
Carbon backbone of a 3-dimensional model of C1-inhibitor, with amino acids involved in HAE. This model is made by homology modeling and is based on the crystal structure of 4 other serpins. The α-helices are depicted in red, the β-strands in light blue. Amino acids mutated in HAE patients are depicted in yellow ball-and-stick. The reactive site, P1-Arg, is depicted in magenta ball-and-stick. Ineke Bos. Adapted with permission from Bos et al.456
References
- 1.Dennehy J.J. Hereditary angioneurotic edema: report of a large kindred with defect in C'1 esterase inhibitor and review of the literature. Ann Intern Med. 1970;73:55–59. doi: 10.7326/0003-4819-73-1-55. [DOI] [PubMed] [Google Scholar]
- 2.Hawthorne N. Random House; New York: 2001. The house of the seven gables. [Google Scholar]
- 3.Milton I.L. On giant urticaria. Edinburgh Med J. 1876;22:513–514. [PMC free article] [PubMed] [Google Scholar]
- 4.Quincke H.I. Über akutes umschriebenes Hautödem [About an acute described skin edema] Monatshe Prakt Dermatol. 1882;1:129–131. [Google Scholar]
- 5.Osler W. Hereditary angio-neurotic oedema. Am J Med Sci. 1888;95:362–367. doi: 10.1097/MAJ.0b013e3181b2803f. [DOI] [PubMed] [Google Scholar]
- 6.Donaldson V.H., Evans R.R. A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C' 1-esterase. Am J Med. 1963;35:37–44. doi: 10.1016/0002-9343(63)90162-1. [DOI] [PubMed] [Google Scholar]
- 7.Agostoni A., Cicardi M. Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Baltimore) 1992;71:206–215. doi: 10.1097/00005792-199207000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Binkley K.E., Davis A., 3rd Clinical, biochemical, and genetic characterization of a novel estrogen-dependent inherited form of angioedema. J Allergy Clin Immunol. 2000;106:546–550. doi: 10.1067/mai.2000.108106. [DOI] [PubMed] [Google Scholar]
- 9.Han E.D., MacFarlane R.C., Mulligan A.N., Scafidi J., Davis A.E., 3rd Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. 2002;109:1057–1063. doi: 10.1172/JCI14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luong K.V., Nguyen L.T. Complement abnormalities with lower extremities discomfort in Vietnamese immigrants. Allergy Asthma Proc. 1997;18:239–244. doi: 10.2500/108854197778594098. [DOI] [PubMed] [Google Scholar]
- 11.Donaldson V.H., Rosen F.S., Bing D.H. Role of the second component of complement (C2) and plasmin in kinin release in hereditary angioneurotic edema (H.A.N.E.) plasma. Trans Assoc Am Physicians. 1977;90:174–183. [PubMed] [Google Scholar]
- 12.Fields T., Ghebrehiwet B., Kaplan A.P. Kinin formation in hereditary angioedema plasma: evidence against kinin derivation from C2 and in support of “spontaneous” formation of bradykinin. J Allergy Clin Immunol. 1983;72:54–60. doi: 10.1016/0091-6749(83)90052-0. [DOI] [PubMed] [Google Scholar]
- 13.Nussberger J., Cugno M., Cicardi M. Bradykinin-mediated angioedema. N Engl J Med. 2002;347:621–622. doi: 10.1056/NEJM200208223470820. [DOI] [PubMed] [Google Scholar]
- 14.Nussberger J., Cugno M., Amstutz C., Cicardi M., Pellacani A., Agostoni A. Plasma bradykinin in angio-oedema. Lancet. 1998;351:1693–1697. doi: 10.1016/S0140-6736(97)09137-X. [DOI] [PubMed] [Google Scholar]
- 15.Davis A.E. The pathogenesis of hereditary angioedema. Transfus Apheresis Sci. 2003;29:195–203. doi: 10.1016/j.transci.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Cugno M., Nussberger J., Cicardi M., Agostoni A. Bradykinin and the pathophysiology of angioedema. Int Immunopharmacol. 2003;3:311–317. doi: 10.1016/S1567-5769(02)00162-5. [DOI] [PubMed] [Google Scholar]
- 17.Cugno M., Cicardi M., Coppola R., Agostoni A. Activation of factor XII and cleavage of high molecular weight kininogen during acute attacks in hereditary and acquired C1-inhibitor deficiencies. Immunopharmacology. 1996;33:361–364. doi: 10.1016/0162-3109(96)00086-0. [DOI] [PubMed] [Google Scholar]
- 18.Bossi F., Fischetti F., Durigutto P., Bulla R., Zingale L.C., Pellis V. Endothelial cells contribute to vascular leakage in patients with C1 inhibitor deficiency and angioedema. Mol Immunol. 2004;41:212. [abstract] [Google Scholar]
- 19.Joseph K., Tholanikunnel B.G., Kaplan A.P. Heat shock protein 90 catalyzes activation of the prekallikrein-kininogen complex in the absence of factor XII. Proc Natl Acad Sci U S A. 2002;99:896–900. doi: 10.1073/pnas.022626899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joseph K., Tholanikunnel B.G., Kaplan A.P. Activation of the bradykinin-forming cascade on endothelial cells: a role for heat shock protein 90. Int Immunopharmacol. 2002;2:1851–1859. doi: 10.1016/s1567-5769(02)00186-8. [DOI] [PubMed] [Google Scholar]
- 21.Andre F., Veysseyre-Balter C., Rousset H., Descos L., Andre C. Exogenous oestrogen as an alternative to food allergy in the aetiology of angioneurotic oedema. Toxicology. 2003;185:155–160. doi: 10.1016/s0300-483x(02)00584-x. [DOI] [PubMed] [Google Scholar]
- 22.Yip J., Cunliffe W.J. Hormonally exacerbated hereditary angioedema. Australas J Dermatol. 1992;33:35–38. doi: 10.1111/j.1440-0960.1992.tb00050.x. [DOI] [PubMed] [Google Scholar]
- Frank M.M. Effect of sex hormones on the complement-related clinical disorder of hereditary angioedema. Arthritis Rheum. 1979;22:1295–1299. doi: 10.1002/art.1780221118. [DOI] [PubMed] [Google Scholar]
- 23.Boeckers M., Bork K. [Contraception and pregnancy in hereditary angioedema] Dtsch Med Wochenschr. 1987;112:507–509. doi: 10.1055/s-2008-1068085. [DOI] [PubMed] [Google Scholar]
- Wautier J.L., Caen J.P. [Norgestrienone, a possible therapeutic agent in hereditary angioneurotic edema] Presse Med. 1986;15:2023. [PubMed] [Google Scholar]
- 24.Nielsen E.W., Gran J.T., Straume B., Mellbye O.J., Johansen H.T., Mollnes T.E. Hereditary angio-oedema: new clinical observations and autoimmune screening, complement and kallikrein-kinin analyses. J Intern Med. 1996;239:119–130. doi: 10.1046/j.1365-2796.1996.418764000.x. [DOI] [PubMed] [Google Scholar]
- Laurent J., Jamin C., Lagrue G. [Hereditary angioneurotic edema: norgestrienone is not effective in every case] Presse Med. 1987;16:2132. [PubMed] [Google Scholar]
- 25.Barbieri R.L., Ryan K.J. Danazol: endocrine pharmacology and therapeutic applications. Am J Obstet Gynecol. 1981;141:453–463. doi: 10.1016/0002-9378(81)90611-6. [DOI] [PubMed] [Google Scholar]
- 26.Hadjiyannaki K., Lachmann P.J. Hereditary angio-oedema: a review with particular reference to pathogenesis and treatment. Clin Allergy. 1971;1:221–233. doi: 10.1111/j.1365-2222.1971.tb03021.x. [DOI] [PubMed] [Google Scholar]
- 27.Nussberger J., Cugno M., Cicardi M., Agostoni A. Local bradykinin generation in hereditary angioedema. J Allergy Clin Immunol. 1999;104:1321–1322. doi: 10.1016/s0091-6749(99)70030-8. [DOI] [PubMed] [Google Scholar]
- 28.Zahedi R., Wisnieski J., Davis A.E., 3rd Role of the P2 residue of complement 1 inhibitor (Ala443) in determination of target protease specificity: inhibition of complement and contact system proteases. J Immunol. 1997;159:983–988. [PubMed] [Google Scholar]
- 29.Venema R.C. Post-translational mechanisms of endothelial nitric oxide synthase regulation by bradykinin. Int Immunopharmacol. 2002;2:1755–1762. doi: 10.1016/s1567-5769(02)00185-6. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan A.P., Joseph K., Silverberg M. Pathways for bradykinin formation and inflammatory disease. J Allergy Clin Immunol. 2002;109:195–209. doi: 10.1067/mai.2002.121316. [DOI] [PubMed] [Google Scholar]
- 31.Frank M.M., Gelfand J.A., Atkinson J.P. Hereditary angioedema: the clinical syndrome and its management. Ann Intern Med. 1976;84:580–593. doi: 10.7326/0003-4819-84-5-580. [DOI] [PubMed] [Google Scholar]
- 32.Winnewisser J., Rossi M., Spath P., Burgi H. Type I hereditary angio-oedema: variability of clinical presentation and course within two large kindreds. J Intern Med. 1997;241:39–46. doi: 10.1046/j.1365-2796.1997.76893000.x. [DOI] [PubMed] [Google Scholar]
- 33.Cicardi M., Bergamaschini L., Cugno M., Beretta A., Zingale L.C., Colombo M. Pathogenetic and clinical aspects of C1 inhibitor deficiency. Immunobiology. 1998;199:366–376. doi: 10.1016/S0171-2985(98)80041-7. [DOI] [PubMed] [Google Scholar]
- 34.Bork K. [Hypovolemic shock caused by ascites in hereditary angioedema] Med Klin (Munich) 1998;93:554. doi: 10.1007/BF03042666. [DOI] [PubMed] [Google Scholar]
- 35.Cohen N., Sharon A., Golik A., Zaidenstein R., Modai D. Hereditary angioneurotic edema with severe hypovolemic shock. J Clin Gastroenterol. 1993;16:237–239. doi: 10.1097/00004836-199304000-00016. [DOI] [PubMed] [Google Scholar]
- 36.Talavera A., Larraona J.L., Ramos J.L., Lopez T., Maraver A., Arias J. Hereditary angioedema: an infrequent cause of abdominal pain with ascites. Am J Gastroenterol. 1995;90:471–474. [PubMed] [Google Scholar]
- 37.Dinkel H.P., Maroske J., Schrod L. Sonographic appearances of the abdominal manifestations of hereditary angioedema. Pediatr Radiol. 2001;31:296–298. doi: 10.1007/s002470000409. [DOI] [PubMed] [Google Scholar]
- 38.Campanile E., Scuderi R., Ierna D., Neri S. Changes in splenoportal axis calibre and flow in a patient affected by hereditary angioedema. Panminerva Med. 2001;43:307–310. [PubMed] [Google Scholar]
- 39.Sofia S., Casali A., Bolondi L. Sonographic findings in abdominal hereditary angioedema. J Clin Ultrasound. 1999;27:537–540. doi: 10.1002/(sici)1097-0096(199911/12)27:9<537::aid-jcu9>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 40.De Backer A.I., De Schepper A.M., Vandevenne J.E., Schoeters P., Michielsen P., Stevens W.J. CT of angioedema of the small bowel. AJR Am J Roentgenol. 2001;176:649–652. doi: 10.2214/ajr.176.3.1760649. [DOI] [PubMed] [Google Scholar]
- 41.Wong R.C., Phan T.G., Adelstein S., Basten A. Image of the month: hereditary angioedema (HAE) Gastroenterology. 1999;116:514. doi: 10.1016/s0016-5085(99)70170-8. 777. [DOI] [PubMed] [Google Scholar]
- 42.Hara T., Shiotani A., Matsunaka H., Yamanishi T., Oka H., Ishiguchi T. Hereditary angioedema with gastrointestinal involvement: endoscopic appearance. Endoscopy. 1999;31:322–324. doi: 10.1055/s-1999-14. [DOI] [PubMed] [Google Scholar]
- 43.Calbo L., Quattrocchi P., Ferlazzo B. Abdominal attack of hereditary angioedema associated with marked leucocytosis: a case report. Ital J Gastroenterol. 1992;24:464–465. [PubMed] [Google Scholar]
- Wüthrich B., Devay J., Späth P. [Hereditary or acquired angioedema caused by functional deficiency of C1 inhibitor–a still unfamiliar disease picture] Schweiz Med Wochenschr. 1999;129:285–291. [PubMed] [Google Scholar]
- 44.Bork K., Siedlecki K., Bosch S., Schopf R.E., Kreuz W. Asphyxiation by laryngeal edema in patients with hereditary angioedema. Mayo Clin Proc. 2000;75:349–354. doi: 10.4065/75.4.349. [DOI] [PubMed] [Google Scholar]
- 45.Bork K., Barnstedt S.E. Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med. 2001;161:714–718. doi: 10.1001/archinte.161.5.714. [DOI] [PubMed] [Google Scholar]
- 46.Farkas H., Harmat G., Fay A., Fekete B., Karadi I., Visy B. Erythema marginatum preceding an acute oedematous attack of hereditary angioneurotic oedema. Acta Derm Venereol. 2001;81:376–377. doi: 10.1080/000155501317140188. [DOI] [PubMed] [Google Scholar]
- 47.Legendre M., Chiche J.F., Molina C., Grouffal C., Betail G. [Respiratory manifestations in hereditary angioneurotic edema] Rev Pneumol Clin. 1985;41:251–258. [PubMed] [Google Scholar]
- 48.Van Dellen R.C., Myers R.P. Bladder involvement in hereditary angioedema. Mayo Clin Proc. 1980;55:277–278. [PubMed] [Google Scholar]
- 49.Sunder T.R., Balsam M.J., Vengrow M.I. Neurological manifestations of angioedema: report of two cases and review of the literature. JAMA. 1982;247:2005–2007. doi: 10.1001/jama.247.14.2005. [DOI] [PubMed] [Google Scholar]
- 50.Krause K.H., Rentrop U., Mehregan U. [Cerebral manifestations in angioneurotic edema (author's translation)] J Neurol Sci. 1979;42:429–435. doi: 10.1016/0022-510x(79)90175-8. [DOI] [PubMed] [Google Scholar]
- 51.Landerman N.S. Hereditary angioneurotic edema, I: case reports and review of the literature. J Allergy. 1962;33:316–329. doi: 10.1016/0021-8707(62)90031-x. [DOI] [PubMed] [Google Scholar]
- 52.Bonner G., Preis S., Schunk U., Toussaint C., Kaufmann W. Hemodynamic effects of bradykinin on systemic and pulmonary circulation in healthy and hypertensive humans. J Cardiovasc Pharmacol. 1990;15(suppl 6):S46–S56. [PubMed] [Google Scholar]
- 53.Lung C.C., Chan E.K., Zuraw B.L. Analysis of an exon 1 polymorphism of the B2 bradykinin receptor gene and its transcript in normal subjects and patients with C1 inhibitor deficiency. J Allergy Clin Immunol. 1997;99:134–146. doi: 10.1016/s0091-6749(97)70310-5. [DOI] [PubMed] [Google Scholar]
- 54.Farkas H., Fust G., Fekete B., Karadi I., Varga L. Eradication of Helicobacter pylori and improvement of hereditary angioneurotic oedema. Lancet. 2001;358:1695–1696. doi: 10.1016/S0140-6736(01)06720-4. [DOI] [PubMed] [Google Scholar]
- 55.Iamandescu I.B. Contributions to the study of the favouring role of chronic urinary infections in inducing and starting drug-allergic-type reactions. Med Interne. 1990;28:53–60. [PubMed] [Google Scholar]
- 56.Watson R.D., Gershwin M.E. Acquired angioedema associated with sinusitis. J Investig Allergol Clin Immunol. 2000;10:129–134. [PubMed] [Google Scholar]
- 57.Wedi B., Kapp A. Helicobacter pylori infection and skin diseases. J Physiol Pharmacol. 1999;50:753–776. [PubMed] [Google Scholar]
- 58.Gudat W., Bork K. Hereditary angioedema associated with subacute cutaneous lupus erythematosus. Dermatologica. 1989;179:211–213. doi: 10.1159/000248363. [DOI] [PubMed] [Google Scholar]
- 59.Duncan I.J., Tymms K.E., Carney G. Rheumatoid arthritis and hereditary angioedema. J Rheumatol. 1988;15:700–702. [PubMed] [Google Scholar]
- 60.Brickman C.M., Tsokos G.C., Balow J.E., Lawley T.J., Santaella M., Hammer C.H. Immunoregulatory disorders associated with hereditary angioedema, I: clinical manifestations of autoimmune disease. J Allergy Clin Immunol. 1986;77:749–757. doi: 10.1016/0091-6749(86)90424-0. [DOI] [PubMed] [Google Scholar]
- 61.Brickman C.M., Tsokos G.C., Chused T.M., Balow J.E., Lawley T.J., Santaella M. Immunoregulatory disorders associated with hereditary angioedema, II: serologic and cellular abnormalities. J Allergy Clin Immunol. 1986;77:758–767. doi: 10.1016/0091-6749(86)90425-2. [DOI] [PubMed] [Google Scholar]
- 62.Koide M., Shirahama S., Tokura Y., Takigawa M., Hayakawa M., Furukawa F. Lupus erythematosus associated with C1 inhibitor deficiency. J Dermatol. 2002;29:503–507. doi: 10.1111/j.1346-8138.2002.tb00316.x. [DOI] [PubMed] [Google Scholar]
- 63.Sturfelt G., Rydgren L., Truedsson L., Alm P., Sjoholm A.G. C1 inhibitor deficiency in a patient with rheumatoid arthritis—increased risk of adverse effects of penicillamine? J Rheumatol. 1996;23:378–381. [PubMed] [Google Scholar]
- 64.Hory B., Haultier J.J. Glomerulonephritis and hereditary angioedema: report of 2 cases. Clin Nephrol. 1989;31:259–263. [PubMed] [Google Scholar]
- 65.Ley S.J., Williams R.C., Jr. A family with hereditary angioedema and multiple immunologic disorders. Am J Med. 1987;82:1046–1051. doi: 10.1016/0002-9343(87)90173-2. [DOI] [PubMed] [Google Scholar]
- 66.Agnello V. Association of systemic lupus erythematosus and SLE-like syndromes with hereditary and acquired complement deficiency states. Arthritis Rheum. 1978;21:S146–S152. [PubMed] [Google Scholar]
- 67.Pan C.G., Strife C.F., Ward M.K., Spitzer R.E., McAdams A.J. Long-term follow-up of non-systemic lupus erythematosus glomerulonephritis in patients with hereditary angioedema: report of four cases. Am J Kidney Dis. 1992;19:526–531. doi: 10.1016/s0272-6386(12)80830-4. [DOI] [PubMed] [Google Scholar]
- 68.Suzuki Y., Nihei H., Mimura N., Hara M. A case of hereditary angioneurotic edema associated with systemic lupus erythematosus. Jpn J Med. 1986;25:281–287. doi: 10.2169/internalmedicine1962.25.281. [DOI] [PubMed] [Google Scholar]
- 69.Alper C.A. Inherited deficiencies of complement components in man. Immunol Lett. 1987;14:175–181. doi: 10.1016/0165-2478(87)90098-8. [DOI] [PubMed] [Google Scholar]
- 70.Jacobson D.L., Gange S.J., Rose N.R., Graham N.M. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223–243. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- 71.Fernandez P.G., Day J.H., Simpson N.E., Zachariah P.K. Hereditary angioneurotic edema and Charcot-Marie-Tooth disease in the same family. Can Med Assoc J. 1978;119:455–458. [PMC free article] [PubMed] [Google Scholar]
- 72.Christie D.R., Kirk J.A., Clarke C.L., Boyages J. Association of hereditary angioedema and hereditary breast cancer. Cancer Genet Cytogenet. 1997;95:159–162. doi: 10.1016/s0165-4608(96)00218-x. [DOI] [PubMed] [Google Scholar]
- 73.Farkas H., Visy B., Fekete B., Karadi I., Kovacs J.B., Kovacs I.B. Association of celiac disease and hereditary angioneurotic edema. Am J Gastroenterol. 2002;97:2682–2683. doi: 10.1111/j.1572-0241.2002.06059.x. [DOI] [PubMed] [Google Scholar]
- 74.Farkas H., Gyeney L., Nemesanszky E., Kaldi G., Kukan F., Masszi I. Coincidence of hereditary angioedema (HAE) with Crohn's disease. Immunol Invest. 1999;28:43–53. doi: 10.3109/08820139909022722. [DOI] [PubMed] [Google Scholar]
- 75.Cicardi M., Mannucci P.M., Castelli R., Rumi M.G., Agostoni A. Reduction in transmission of hepatitis C after the introduction of a heat-treatment step in the production of C1-inhibitor concentrate. Transfusion. 1995;35:209–212. doi: 10.1046/j.1537-2995.1995.35395184276.x. [DOI] [PubMed] [Google Scholar]
- 76.Laurent J., Toulet R., Lagrue G. Ultrasonography in the diagnosis of hereditary angioneurotic oedema. Lancet. 1988;1:761. doi: 10.1016/s0140-6736(88)91558-9. [DOI] [PubMed] [Google Scholar]
- 77.Burghardt W., Wernze H. Ultrasonography for early diagnosis of hereditary angioneurotic oedema. Lancet. 1987;2:165. doi: 10.1016/s0140-6736(87)92371-3. [DOI] [PubMed] [Google Scholar]
- 78.Farkas H., Harmat G., Kaposi P.N., Karadi I., Fekete B., Fust G. Ultrasonography in the diagnosis and monitoring of ascites in acute abdominal attacks of hereditary angioneurotic oedema. Eur J Gastroenterol Hepatol. 2001;13:1225–1230. doi: 10.1097/00042737-200110000-00016. [DOI] [PubMed] [Google Scholar]
- 79.Farkas H., Harmat G., Fust G., Varga L., Visy B. Clinical management of hereditary angio-oedema in children. Pediatr Allergy Immunol. 2002;13:153–161. doi: 10.1034/j.1399-3038.2002.01014.x. [DOI] [PubMed] [Google Scholar]
- 80.Mittelstadt C.A., editor. General ultrasound. Churchill-Livingstone; Edinburgh: 1992. [Google Scholar]
- 81.Farkas H., Harmat G., Fekete B., Karadi I., Visy B., Varga L. Acute abdominal attack of hereditary angioneurotic oedema associated with ultrasound abnormalities suggestive of acute hepatitis. Acta Paediatr. 2002;91:971–974. doi: 10.1080/080352502760148720. [DOI] [PubMed] [Google Scholar]
- 82.Greaves M., Lawlor F. Angioedema: manifestations and management. J Am Acad Dermatol. 1991;25:155–161. doi: 10.1016/0190-9622(91)70183-3. discussion 61-5. [DOI] [PubMed] [Google Scholar]
- 83.Nzeako U.C., Frigas E., Tremaine W.J. Hereditary angioedema: a broad review for clinicians. Arch Intern Med. 2001;161:2417–2429. doi: 10.1001/archinte.161.20.2417. [DOI] [PubMed] [Google Scholar]
- 84.Zuraw B.L., Herschbach J. Detection of C1 inhibitor mutations in patients with hereditary angioedema. J Allergy Clin Immunol. 2000;105:541–546. doi: 10.1067/mai.2000.104780. [DOI] [PubMed] [Google Scholar]
- 85.Bowen B., Hawk J.J., Sibunka S., Hovick S., Weiler J.M. A review of the reported defects in the human C1 esterase inhibitor gene producing hereditary angioedema including four new mutations. Clin Immunol. 2001;98:157–163. doi: 10.1006/clim.2000.4947. [DOI] [PubMed] [Google Scholar]
- 86.Bork K., Barnstedt S.E., Koch P., Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 2000;356:213–217. doi: 10.1016/S0140-6736(00)02483-1. [DOI] [PubMed] [Google Scholar]
- 87.Martin L., Degenne D., Toutain A., Ponard D., Watier H. Hereditary angioedema type III: an additional French pedigree with autosomal dominant transmission. J Allergy Clin Immunol. 2001;107:747–748. doi: 10.1067/mai.2001.114242. [DOI] [PubMed] [Google Scholar]
- 88.Rosen F.S., Pensky J., Donaldson V., Charache P. Hereditary angioneurotic edema: two genetic variants. Science. 1965;148:957–958. doi: 10.1126/science.148.3672.957. [DOI] [PubMed] [Google Scholar]
- 89.Davis A.E., 3rd C1 inhibitor and hereditary angioneurotic edema. Annu Rev Immunol. 1988;6:595–628. doi: 10.1146/annurev.iy.06.040188.003115. [DOI] [PubMed] [Google Scholar]
- 90.Quastel M., Harrison R., Cicardi M., Alper C.A., Rosen F.S. Behavior in vivo of normal and dysfunctional C1 inhibitor in normal subjects and patients with hereditary angioneurotic edema. J Clin Invest. 1983;71:1041–1046. doi: 10.1172/JCI110831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Verpy E., Couture-Tosi E., Eldering E., Lopez-Trascasa M., Spath P., Meo T. Crucial residues in the carboxy-terminal end of C1 inhibitor revealed by pathogenic mutants impaired in secretion or function. J Clin Invest. 1995;95:350–359. doi: 10.1172/JCI117663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Binkley K.E., Davis A.E., 3rd Estrogen-dependent inherited angioedema. Transfus Apheresis Sci. 2003;29:215–219. doi: 10.1016/j.transci.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 93.Decarie A., Raymond P., Gervais N., Couture R., Adam A. Serum interspecies differences in metabolic pathways of bradykinin and [des-Arg9]BK: influence of enalaprilat. Am J Physiol. 1996;271:H1340–H1347. doi: 10.1152/ajpheart.1996.271.4.H1340. [DOI] [PubMed] [Google Scholar]
- 94.Erdos E.G., Skigel R.A. Metabolism of bradykinin in health and disease. In: Farmer S.G., editor. Handbook of immunopharmacology: the kinin system. Academic Press; London: 1997. pp. 111–141. [Google Scholar]
- 95.Blais C., Jr., Marc-Aurele J., Simmons W.H., Loute G., Thibault P., Skidgel R.A. Des-Arg9-bradykinin metabolism in patients who presented hypersensitivity reactions during hemodialysis: role of serum ACE and aminopeptidase P. Peptides. 1999;20:421–430. doi: 10.1016/s0196-9781(99)00020-0. [DOI] [PubMed] [Google Scholar]
- 96.Adam A., Cugno M., Molinaro G., Perez M., Lepage Y., Agostoni A. Aminopeptidase P in individuals with a history of angio-oedema on ACE inhibitors. Lancet. 2002;359:2088–2089. doi: 10.1016/S0140-6736(02)08914-6. [DOI] [PubMed] [Google Scholar]
- 97.Shepherd G.M. Possible contraindication of angiotensin converting enzyme inhibitors in patients with hereditary angioedema. Am J Med. 1990;88:446. doi: 10.1016/0002-9343(90)90514-e. [DOI] [PubMed] [Google Scholar]
- 98.Binkley K.E., Leznoff A. Exacerbation of angioedema by an angiotensin converting enzyme inhibitor in a patient with variant form hereditary angioedema. Can J Allergy Clin Immunol. 1996;1:134–135. [Google Scholar]
- 99.Agostoni A., Cicardi M., Cugno M., Zingale L.C., Gioffre D., Nussberger J. Angioedema due to angiotensin-converting enzyme inhibitors. Immunopharmacology. 1999;44:21–25. doi: 10.1016/s0162-3109(99)00107-1. [DOI] [PubMed] [Google Scholar]
- 100.Berkun Y., Shalit M. Hereditary angioedema first apparent in the ninth decade during treatment with ACE inhibitor. Ann Allergy Asthma Immunol. 2001;87:138–139. doi: 10.1016/S1081-1206(10)62208-9. [DOI] [PubMed] [Google Scholar]
- 101.Binkley K.E., Davis A.E. Reply (Hereditary angioedema type III: an additional French pedigree with autosomal dominant transmission) J Allergy Clin Immunol. 2001;107:747–748. doi: 10.1067/mai.2001.114242. [DOI] [PubMed] [Google Scholar]
- 102.Day N.K., Good R.A. Inherited and acquired deficiencies of C1 esterase inhibitor in man. In: Rother K., Till G., editors. The complement system. Springer-Verlag; Heidelberg: 1988. [Google Scholar]
- 103.Kranke B., Salmhofer W., Aberer W. Hereditary angioedema and normal C1-inhibitor activity in women. Lancet. 2000;356:1440–1441. doi: 10.1016/S0140-6736(05)74078-2. [DOI] [PubMed] [Google Scholar]
- 104.Spaulding W.B. Methyltestosterone therapy for hereditary episodic edema (hereditary angioneurotic edema) Ann Intern Med. 1960;53:739–745. [Google Scholar]
- 105.Gelfand J.A., Boss G.R., Conley C.L., Reinhart R., Frank M.M. Acquired C1 esterase inhibitor deficiency and angioedema: a review. Medicine (Baltimore) 1979;58:321–328. doi: 10.1097/00005792-197907000-00004. [DOI] [PubMed] [Google Scholar]
- 106.Sheffer A.L., Fearon D.T., Austen K.F. Hereditary angioedema: a decade of management with stanozolol. J Allergy Clin Immunol. 1987;80:855–860. doi: 10.1016/s0091-6749(87)80277-4. [DOI] [PubMed] [Google Scholar]
- 107.Gordon E.M., Ratnoff O.D., Saito H., Donaldson V.H., Pensky J., Jones P.K. Rapid fibrinolysis, augmented Hageman factor (factor XII) titers, and decreased C1 esterase inhibitor titers in women taking oral contraceptives. J Lab Clin Med. 1980;96:762–769. [PubMed] [Google Scholar]
- 108.Ogston D., Walker J., Campbell D.M. C1 inactivator level in pregnancy. Thromb Res. 1981;23:453–455. doi: 10.1016/0049-3848(81)90206-1. [DOI] [PubMed] [Google Scholar]
- 109.Halbmayer W.M., Hopmeier P., Mannhalter C., Heuss F., Leodolter S., Rubi K. C1-esterase inhibitor in uncomplicated pregnancy and mild and moderate preeclampsia. Thromb Haemost. 1991;65:134–138. [PubMed] [Google Scholar]
- 110.Cohen A.J., Laskin C., Tarlo S. C1 esterase inhibitor in pregnancy. J Allergy Clin Immunol. 1992;90:412–413. doi: 10.1016/s0091-6749(05)80025-9. [DOI] [PubMed] [Google Scholar]
- 111.Caldwell J.R., Ruddy S., Schur P.H., Austen K.F. Acquired C1 inhibitor deficiency in lymphosarcoma. Clin Immunol Immunopathol. 1972;1:39–52. [Google Scholar]
- 112.Melamed J., Alper C.A., Cicardi M., Rosen F.S. The metabolism of C1 inhibitor and C1q in patients with acquired C1-inhibitor deficiency. J Allergy Clin Immunol. 1986;77:322–326. doi: 10.1016/s0091-6749(86)80111-7. [DOI] [PubMed] [Google Scholar]
- 113.Spath P.J., Wuthrich B., Matter L., Loos M., Alsenz J. Acquired angioedema and anti-C1-inhibitor autoantibody. Arch Intern Med. 1989;149:1213. doi: 10.1001/archinte.1989.00390050159036. 1216. [DOI] [PubMed] [Google Scholar]
- 114.Cicardi M., Bisiani G., Cugno M., Spath P., Agostoni A. Autoimmune C1 inhibitor deficiency: report of eight patients. Am J Med. 1993;95:169–175. doi: 10.1016/0002-9343(93)90257-p. [DOI] [PubMed] [Google Scholar]
- 115.Cicardi M., Beretta A., Colombo M., Gioffre D., Cugno M., Agostoni A. Relevance of lymphoproliferative disorders and of anti-C1 inhibitor autoantibodies in acquired angio-oedema. Clin Exp Immunol. 1996;106:475–480. doi: 10.1046/j.1365-2249.1996.d01-866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schreiber A.D., Zweiman B., Atkins P., Goldwein F., Pietra G., Atkinson B. Acquired angioedema with lymphoproliferative disorder: association of C1 inhibitor deficiency with cellular abnormality. Blood. 1976;48:567–580. [PubMed] [Google Scholar]
- 117.Hauptmann G., Petitjean F., Lang J.M., Oberling F. Acquired C1 inhibitor deficiency in a case of lymphosarcoma of the spleen: reversal of complement abnormalities after splenectomy. Clin Exp Immunol. 1979;37:523–531. [PMC free article] [PubMed] [Google Scholar]
- 118.Cicardi M., Frangi D., Bergamaschini L., Gardinali M., Sacchi G., Agostoni A. Acquired C1 inhibitor deficiency with angioedema symptoms in a patient infected with Echinococcus granulosus. Complement. 1985;2:133–139. doi: 10.1159/000467853. [DOI] [PubMed] [Google Scholar]
- 119.Farkas H., Csepregi A., Nemesanszky E., Par A., Gyeney L., Varga L. Acquired angioedema associated with chronic hepatitis C. J Allergy Clin Immunol. 1999;103:711–712. doi: 10.1016/s0091-6749(99)70248-4. [DOI] [PubMed] [Google Scholar]
- 120.Farkas H., Gyeney L., Majthenyi P., Fust G., Varga L. Angioedema due to acquired C1-esterase inhibitor deficiency in a patient with Helicobacter pylori infection. Z Gastroenterol. 1999;37:513–518. [PubMed] [Google Scholar]
- 121.Farkas H., Szongoth M., Bely M., Varga L., Fekete B., Karadi I. Angiooedema due to acquired deficiency of C1-esterase inhibitor associated with leucocytoclastic vasculitis. Acta Derm Venereol. 2001;81:298–300. doi: 10.1080/00015550152572985. [DOI] [PubMed] [Google Scholar]
- 122.Sheffer A.L., Austen K.F., Rosen F.S., Fearon D.T. Acquired deficiency of the inhibitor of the first component of complement: report of five additional cases with commentary on the syndrome. J Allergy Clin Immunol. 1985;75:640–646. doi: 10.1016/0091-6749(85)90087-9. [DOI] [PubMed] [Google Scholar]
- 123.Pascual M., Widmann J.J., Schifferli J.A. Recurrent febrile panniculitis and hepatitis in two patients with acquired complement deficiency and paraproteinemia. Am J Med. 1987;83:959–962. doi: 10.1016/0002-9343(87)90659-0. [DOI] [PubMed] [Google Scholar]
- 124.Nakamura S., Yoshinari M., Saku Y., Hirakawa K., Miishima C., Murai K. Acquired C1 inhibitor deficiency associated with systemic lupus erythematosus affecting the central nervous system. Ann Rheum Dis. 1991;50:713–716. doi: 10.1136/ard.50.10.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barilla-LaBarca M.L., Gioffre D., Zanichelli A., Cicardi M., Atkinson J.P. Acquired C1 esterase inhibitor deficiency in two patients presenting with a lupus-like syndrome and anticardiolipin antibodies. Arthritis Rheum. 2002;47:223–226. doi: 10.1002/art.10342. [DOI] [PubMed] [Google Scholar]
- 126.Frigas E. Angioedema with acquired deficiency of the C1 inhibitor: a constellation of syndromes. Mayo Clin Proc. 1989;64:1269–1275. doi: 10.1016/s0025-6196(12)61290-7. [DOI] [PubMed] [Google Scholar]
- 127.Jackson J., Sim R.B., Whelan A., Feighery C. An IgG autoantibody which inactivates C1-inhibitor. Nature. 1986;323:722–724. doi: 10.1038/323722a0. [DOI] [PubMed] [Google Scholar]
- 128.Jackson J., Sim R.B., Whaley K., Feighery C. Autoantibody facilitated cleavage of C1-inhibitor in autoimmune angioedema. J Clin Invest. 1989;83:698–707. doi: 10.1172/JCI113934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Alsenz J., Lambris J.D., Bork K., Loos M. Acquired C1 inhibitor (C1-INH) deficiency type II: replacement therapy with C1-INH and analysis of patients' C1-INH and anti-C1-INH autoantibodies. J Clin Invest. 1989;83:1794–1799. doi: 10.1172/JCI114084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Whaley K., Sim R.B., He S. Autoimmune C1-inhibitor deficiency. Clin Exp Immunol. 1996;106:423–426. doi: 10.1046/j.1365-2249.1996.d01-879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.D'Incan M., Tridon A., Ponard D., Dumestre-Perard C., Ferrier-Le Bouedec M., Betail G. Acquired angioedema with C1 inhibitor deficiency: is the distinction between type I and type II still relevant? Dermatology. 1999;199:227–230. doi: 10.1159/000018252. [DOI] [PubMed] [Google Scholar]
- 132.Fremeaux-Bacchi V., Guinnepain M.T., Cacoub P., Dragon-Durey M.A., Mouthon L., Blouin J. Prevalence of monoclonal gammopathy in patients presenting with acquired angioedema type 2. Am J Med. 2002;113:194–199. doi: 10.1016/s0002-9343(02)01124-5. [DOI] [PubMed] [Google Scholar]
- 133.Bouillet-Claveyrolas L., Ponard D., Drouet C., Massot C. Clinical and biological distinctions between type I and type II acquired angioedema. Am J Med. 2003;115:420–421. doi: 10.1016/s0002-9343(03)00396-6. [DOI] [PubMed] [Google Scholar]
- 134.Cicardi M., Zingale L.C., Pappalardo E., Folcioni A., Agostoni A. Autoantibodies and lymphoproliferative diseases in acquired C1-inhibitor deficiencies. Medicine (Baltimore) 2003;82:274–281. doi: 10.1097/01.md.0000085055.63483.09. [DOI] [PubMed] [Google Scholar]
- 135.Cicardi M., Bergamaschini L., Zingale L.C., Gioffre D., Agostoni A. Idiopathic nonhistaminergic angioedema. Am J Med. 1999;106:650–654. doi: 10.1016/s0002-9343(99)00123-0. [DOI] [PubMed] [Google Scholar]
- 136.Agostoni A., Cicardi M. Drug-induced angioedema without urticaria. Drug Saf. 2001;24:599–606. doi: 10.2165/00002018-200124080-00004. [DOI] [PubMed] [Google Scholar]
- 137.Irons B.K., Kumar A. Valsartan-induced angioedema. Ann Pharmacother. 2003;37:1024–1027. doi: 10.1345/aph.1C520. [DOI] [PubMed] [Google Scholar]
- 138.Spath P.J., Wuthrich B., Butler R. Quantification of C1-inhibitor functional activities by immunodiffusion assay in plasma of patients with hereditary angioedema—evidence of a functionally critical level of C1-inhibitor concentration. Complement. 1984;1:147–159. doi: 10.1159/000467830. [DOI] [PubMed] [Google Scholar]
- 139.Sim R.B., Reboul A., Arlaud G.J., Villiers C.L., Colomb M.G. Interaction of 125I-labelled complement subcomponents C-1r and C-1s with protease inhibitors in plasma. FEBS Lett. 1979;97:111–115. doi: 10.1016/0014-5793(79)80063-0. [DOI] [PubMed] [Google Scholar]
- 140.de Agostini A., Lijnen H.R., Pixley R.A., Colman R.W., Schapira M. Inactivation of factor XII active fragment in normal plasma: predominant role of C-1-inhibitor. J Clin Invest. 1984;73:1542–1549. doi: 10.1172/JCI111360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Harpel P.C., Lewin M.F., Kaplan A.P. Distribution of plasma kallikrein between C-1 inactivator and alpha 2-macroglobulin in plasma utilizing a new assay for alpha 2-macroglobulin-kallikrein complexes. J Biol Chem. 1985;260:4257–4263. [PubMed] [Google Scholar]
- 142.Huisman L.G., van Griensven J.M., Kluft C. On the role of C1-inhibitor as inhibitor of tissue-type plasminogen activator in human plasma. Thromb Haemost. 1995;73:466–471. [PubMed] [Google Scholar]
- 143.Wuillemin W.A., Minnema M., Meijers J.C., Roem D., Eerenberg A.J., Nuijens J.H. Inactivation of factor XIa in human plasma assessed by measuring factor XIa-protease inhibitor complexes: major role for C1-inhibitor. Blood. 1995;85:1517–1526. [PubMed] [Google Scholar]
- 144.Matsushita M., Thiel S., Jensenius J.C., Terai I., Fujita T. Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J Immunol. 2000;165:2637–2642. doi: 10.4049/jimmunol.165.5.2637. [DOI] [PubMed] [Google Scholar]
- 145.Potempa J., Korzus E., Travis J. The serpin superfamily of proteinase inhibitors: structure, function, and regulation. J Biol Chem. 1994;269:15957–15960. [PubMed] [Google Scholar]
- 146.Theriault A., Whaley K., McPhaden A.R., Boyd E., Connor J.M. Regional assignment of the human C1-inhibitor gene to 11q11-q13.1. Hum Genet. 1990;84:477–479. doi: 10.1007/BF00195824. [DOI] [PubMed] [Google Scholar]
- 147.Janson M., Larsson C., Werelius B., Jones C., Glaser T., Nakamura Y. Detailed physical map of human chromosomal region 11q12-13 shows high meiotic recombination rate around the MEN1 locus. Proc Natl Acad Sci U S A. 1991;88:10609–10613. doi: 10.1073/pnas.88.23.10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carter P.E., Duponchel C., Tosi M., Fothergill J.E. Complete nucleotide sequence of the gene for human C1 inhibitor with an unusually high density of Alu elements. Eur J Biochem. 1991;197:301–308. doi: 10.1111/j.1432-1033.1991.tb15911.x. [DOI] [PubMed] [Google Scholar]
- 149.Ariga T., Carter P.E., Davis A.E., 3rd Recombinations between Alu repeat sequences that result in partial deletions within the C1 inhibitor gene. Genomics. 1990;8:607–613. doi: 10.1016/0888-7543(90)90246-q. [DOI] [PubMed] [Google Scholar]
- 150.Stoppa-Lyonnet D., Duponchel C., Meo T., Laurent J., Carter P.E., Arala-Chaves M. Recombinational biases in the rearranged C1-inhibitor genes of hereditary angioedema patients. Am J Hum Genet. 1991;49:1055–1062. [PMC free article] [PubMed] [Google Scholar]
- 151.Bissler J.J., Donaldson V.H., Davis A.E., 3rd Contiguous deletion and duplication mutations resulting in type 1 hereditary angioneurotic edema. Hum Genet. 1994;93:265–269. doi: 10.1007/BF00212020. [DOI] [PubMed] [Google Scholar]
- 152.Skriver K., Radziejewska E., Silbermann J.A., Donaldson V.H., Bock S.C. CpG mutations in the reactive site of human C1 inhibitor. J Biol Chem. 1989;264:3066–3071. [PubMed] [Google Scholar]
- 153.Tosi M. Molecular genetics of C1 inhibitor. Immunobiology. 1998;199:358–365. doi: 10.1016/S0171-2985(98)80040-5. [DOI] [PubMed] [Google Scholar]
- 154.Pappalardo E., Cicardi M., Duponchel C., Carugati A., Choquet S., Agostoni A. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol. 2000;106:1147–1154. doi: 10.1067/mai.2000.110471. [DOI] [PubMed] [Google Scholar]
- 155.Eldering E., Verpy E., Roem D., Meo T., Tosi M. COOH-terminal substitutions in the serpin C1 inhibitor that cause loop overinsertion and subsequent multimerization. J Biol Chem. 1995;270:2579–2587. doi: 10.1074/jbc.270.6.2579. [DOI] [PubMed] [Google Scholar]
- 156.Kramer J., Rosen F.S., Colten H.R., Rajczy K., Strunk R.C. Transinhibition of C1 inhibitor synthesis in type I hereditary angioneurotic edema. J Clin Invest. 1993;91:1258–1262. doi: 10.1172/JCI116290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lachmann P.J., Rosen F.S. The catabolism of C1(-)-inhibitor and the pathogenesis of hereditary angio-edema. Acta Pathol Microbiol Immunol Scand Suppl. 1984;284:35–39. [PubMed] [Google Scholar]
- 158.Davis A.E., 3rd C1 inhibitor. Functional analysis of naturally-occurring mutant proteins. Adv Exp Med Biol. 1997;425:185–194. [PubMed] [Google Scholar]
- 159.Davis A.E., 3rd, Aulak K., Parad R.B., Stecklein H.P., Eldering E., Hack C.E. C1 inhibitor hinge region mutations produce dysfunction by different mechanisms. Nat Genet. 1992;1:354–358. doi: 10.1038/ng0892-354. [DOI] [PubMed] [Google Scholar]
- 160.Zahedi R., Bissler J.J., Davis A.E., 3rd, Andreadis C., Wisnieski J.J. Unique C1 inhibitor dysfunction in a kindred without angioedema, II: identification of an Ala443–Val substitution and functional analysis of the recombinant mutant protein. J Clin Invest. 1995;95:1299–1305. doi: 10.1172/JCI117780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Aulak K.S., Eldering E., Hack C.E., Lubbers Y.P., Harrison R.A., Mast A. A hinge region mutation in C1-inhibitor (Ala436–Thr) results in nonsubstrate-like behavior and in polymerization of the molecule. J Biol Chem. 1993;268:18088–18094. [PubMed] [Google Scholar]
- 162.Bos I.G., Hack C.E., Abrahams J.P. Structural and functional aspects of C1-inhibitor. Immunobiology. 2002;205:518–533. doi: 10.1078/0171-2985-00151. [DOI] [PubMed] [Google Scholar]
- 163.Bissler J.J., Meng Q.S., Emery T. C1 inhibitor gene sequence facilitates frameshift mutations. Mol Med. 1998;4:795–806. [PMC free article] [PubMed] [Google Scholar]
- 164.Davis A.E., 3rd . C1 inhibitor gene and hereditary angioedema. In: Volanakis J., Frank M., editors. The human complement system in health and disease. Marcel Dekker; New York: 1998. pp. 455–480. [Google Scholar]
- 165.Grompe M. The rapid detection of unknown mutations in nucleic acids. Nat Genet. 1993;5:111–117. doi: 10.1038/ng1093-111. [DOI] [PubMed] [Google Scholar]
- 166.Taylor G.R., Deeble J. Enzymatic methods for mutation scanning. Genet Anal. 1999;14:181–186. doi: 10.1016/s1050-3862(98)00029-1. [DOI] [PubMed] [Google Scholar]
- 167.Cotton R.G., Rodrigues N.R., Campbell R.D. Reactivity of cytosine and thymine in single-base-pair mismatches with hydroxylamine and osmium tetroxide and its application to the study of mutations. Proc Natl Acad Sci U S A. 1988;85:4397–4401. doi: 10.1073/pnas.85.12.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ellis T.P., Humphrey K.E., Smith M.J., Cotton R.G. Chemical cleavage of mismatch: a new look at an established method. Hum Mutat. 1998;11:345–353. doi: 10.1002/(SICI)1098-1004(1998)11:5<345::AID-HUMU1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 169.Verpy E., Couture-Tosi E., Tosi M. C1 inhibitor mutations which affect intracellular transport and secretion in type I hereditary angioedema. Behring Inst Mitt. 1993:120–124. [PubMed] [Google Scholar]
- 170.Cotton R.G. Current methods of mutation detection. Mutat Res. 1993;285:125–144. doi: 10.1016/0027-5107(93)90060-s. [DOI] [PubMed] [Google Scholar]
- 171.Verpy E., Biasotto M., Meo T., Tosi M. Efficient detection of point mutations on color-coded strands of target DNA. Proc Natl Acad Sci U S A. 1994;91:1873–1877. doi: 10.1073/pnas.91.5.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Verpy E., Biasotto M., Brai M., Misiano G., Meo T., Tosi M. Exhaustive mutation scanning by fluorescence-assisted mismatch analysis discloses new genotype-phenotype correlations in angiodema. Am J Hum Genet. 1996;59:308–319. [PMC free article] [PubMed] [Google Scholar]
- 173.Xiao W., Oefner P.J. Denaturing high-performance liquid chromatography: a review. Hum Mutat. 2001;17:439–474. doi: 10.1002/humu.1130. [DOI] [PubMed] [Google Scholar]
- 174.Fodde R., Losekoot M. Mutation detection by denaturing gradient gel electrophoresis (DGGE) Hum Mutat. 1994;3:83–94. doi: 10.1002/humu.1380030202. [DOI] [PubMed] [Google Scholar]
- 175.Freiberger T., Kolarova L., Mejstrik P., Vyskocilova M., Kuklinek P., Litzman J. Five novel mutations in the C1 inhibitor gene (C1NH) leading to a premature stop codon in patients with type I hereditary angioedema. Hum Mutat. 2002;19:461. doi: 10.1002/humu.9029. [DOI] [PubMed] [Google Scholar]
- 176.Nataraj A.J., Olivos-Glander I., Kusukawa N., Highsmith W.E., Jr. Single-strand conformation polymorphism and heteroduplex analysis for gel-based mutation detection. Electrophoresis. 1999;20:1177–1185. doi: 10.1002/(SICI)1522-2683(19990101)20:6<1177::AID-ELPS1177>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 177.Bissler J.J., Aulak K.S., Donaldson V.H., Rosen F.S., Cicardi M., Harrison R.A. Molecular defects in hereditary angioneurotic edema. Proc Assoc Am Physicians. 1997;109:164–173. [PubMed] [Google Scholar]
- 178.Blanch A., Roche O., Lopez-Granados E., Fontan G., Lopez-Trascasa M. Detection of C1 inhibitor (SERPING1/C1NH) mutations in exon 8 in patients with hereditary angioedema: evidence for 10 novel mutations. Hum Mutat. 2002;20:405–406. doi: 10.1002/humu.9073. [DOI] [PubMed] [Google Scholar]
- 179.Cicardi M., Igarashi T., Rosen F.S., Davis A.E., 3rd Molecular basis for the deficiency of complement 1 inhibitor in type I hereditary angioneurotic edema. J Clin Invest. 1987;79:698–702. doi: 10.1172/JCI112873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Stoppa-Lyonnet D., Tosi M., Laurent J., Sobel A., Lagrue G., Meo T. Altered C1 inhibitor genes in type I hereditary angioedema. N Engl J Med. 1987;317:1–6. doi: 10.1056/NEJM198707023170101. [DOI] [PubMed] [Google Scholar]
- 181.McPhaden A.R., Birnie G.D., Whaley K. Restriction fragment length polymorphism analysis of the C1-inhibitor gene in hereditary C1-inhibitor deficiency. Clin Genet. 1991;39:161–171. doi: 10.1111/j.1399-0004.1991.tb03006.x. [DOI] [PubMed] [Google Scholar]
- 182.Duponchel C., Di Rocco C., Cicardi M., Tosi M. Rapid detection by fluorescent multiplex PCR of exon deletions and duplications in the C1 inhibitor gene of hereditary angioedema patients. Hum Mutat. 2001;17:61–70. doi: 10.1002/1098-1004(2001)17:1<61::AID-HUMU7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 183.Cumming S.A., Halsall D.J., Ewan P.W., Lomas D.A. The effect of sequence variations within the coding region of the C1 inhibitor gene on disease expression and protein function in families with hereditary angio-oedema. J Med Genet. 2003;40:e114. doi: 10.1136/jmg.40.10.e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zahedi K., Bissler J.J., Prada A.E., Prada J.A., Davis A.E., 3rd The promoter of the C1 inhibitor gene contains a polypurine-polypyrimidine segment that enhances transcriptional activity. J Immunol. 1999;162:7249–7255. [PubMed] [Google Scholar]
- 185.Kalmar L., Bors A., Farkas H., Vas S., Fandl B., Varga L. Mutation screening of the C1 inhibitor gene among Hungarian patients with hereditary angioedema. Hum Mutat. 2003;22:498. doi: 10.1002/humu.9202. [DOI] [PubMed] [Google Scholar]
- 186.Sugiyama E., Ozawa T., Taki H., Maruyama M., Yamashita N., Ohta M. Hereditary angioedema with a de novo mutation of exon 8 in the C1 inhibitor gene showing recurrent edema of the hands around the peripheral joints: importance for the differential diagnosis of joint swelling. Arthritis Rheum. 2001;44:974–977. doi: 10.1002/1529-0131(200104)44:4<974::AID-ANR155>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 187.Ariga T., Igarashi T., Ramesh N., Parad R., Cicardi M., Davis A.E., 3rd Type I C1 inhibitor deficiency with a small messenger RNA resulting from deletion of one exon. J Clin Invest. 1989;83:1888–1893. doi: 10.1172/JCI114095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kramer J., Katz Y., Rosen F.S., Davis A.E., 3rd, Strunk R.C. Synthesis of C1 inhibitor in fibroblasts from patients with type I and type II hereditary angioneurotic edema. J Clin Invest. 1991;87:1614–1620. doi: 10.1172/JCI115175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ernst S.C., Circolo A., Davis A.E., 3rd, Gheesling-Mullis K., Fliesler M., Strunk R.C. Impaired production of both normal and mutant C1 inhibitor proteins in type I hereditary angioedema with a duplication in exon 8. J Immunol. 1996;157:405–410. [PubMed] [Google Scholar]
- 190.Eldering E., Nuijens J.H., Hack C.E. Expression of functional human C1 inhibitor in COS cells. J Biol Chem. 1988;263:11776–11779. [PubMed] [Google Scholar]
- 191.Eldering E., Huijbregts C.C., Lubbers Y.T., Longstaff C., Hack C.E. Characterization of recombinant C1 inhibitor P1 variants. J Biol Chem. 1992;267:7013–7020. [PubMed] [Google Scholar]
- 192.Zahedi R., Aulak K.S., Eldering E., Davis A.E., 3rd Characterization of C1 inhibitor-Ta: a dysfunctional C1INH with deletion of lysine 251. J Biol Chem. 1996;271:24307–24312. doi: 10.1074/jbc.271.39.24307. [DOI] [PubMed] [Google Scholar]
- 193.Zahedi R., MacFarlane R.C., Wisnieski J.J., Davis A.E., 3rd C1 inhibitor: analysis of the role of amino acid residues within the reactive center loop in target protease recognition. J Immunol. 2001;167:1500–1506. doi: 10.4049/jimmunol.167.3.1500. [DOI] [PubMed] [Google Scholar]
- 194.Hunt L.T., Dayhoff M.O. A surprising new protein superfamily containing ovalbumin, antithrombin-III, and alpha 1-proteinase inhibitor. Biochem Biophys Res Commun. 1980;95:864–871. doi: 10.1016/0006-291x(80)90867-0. [DOI] [PubMed] [Google Scholar]
- 195.Salvesen G.S., Catanese J.J., Kress L.F., Travis J. Primary structure of the reactive site of human C1-inhibitor. J Biol Chem. 1985;260:2432–2436. [PubMed] [Google Scholar]
- 196.Tosi M., Duponchel C., Bourgarel P., Colomb M., Meo T. Molecular cloning of human C1 inhibitor: sequence homologies with alpha 1-antitrypsin and other members of the serpins superfamily. Gene. 1986;42:265–272. doi: 10.1016/0378-1119(86)90230-1. [DOI] [PubMed] [Google Scholar]
- 197.Silverman G.A., Bird P.I., Carrell R.W., Church F.C., Coughlin P.B., Gettins P.G. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins: evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem. 2001;276:33293–33296. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- 198.Caliezi C., Wuillemin W.A., Zeerleder S., Redondo M., Eisele B., Hack C.E. C1-Esterase inhibitor: an anti-inflammatory agent and its potential use in the treatment of diseases other than hereditary angioedema. Pharmacol Rev. 2000;52:91–112. [PubMed] [Google Scholar]
- 199.Cugno M., Bos I., Lubbers Y., Hack C.E., Agostoni A. In vitro interaction of C1-inhibitor with thrombin. Blood Coagul Fibrinolysis. 2001;12:253–260. doi: 10.1097/00001721-200106000-00005. [DOI] [PubMed] [Google Scholar]
- 200.Harpel P.C. C1 inactivator inhibition by plasmin. J Clin Invest. 1970;49:568–575. doi: 10.1172/JCI106267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Booth N.A., Walker E., Maughan R., Bennett B. Plasminogen activator in normal subjects after exercise and venous occlusion: t-PA circulates as complexes with C1-inhibitor and PAI-1. Blood. 1987;69:1600–1604. [PubMed] [Google Scholar]
- 202.Wuillemin W.A., te Velthuis H., Lubbers Y.T., de Ruig C.P., Eldering E., Hack C.E. Potentiation of C1 inhibitor by glycosaminoglycans: dextran sulfate species are effective inhibitors of in vitro complement activation in plasma. J Immunol. 1997;159:1953–1960. [PubMed] [Google Scholar]
- 203.Wuillemin W.A., Eldering E., Citarella F., de Ruig C.P., ten Cate H., Hack C.E. Modulation of contact system proteases by glycosaminoglycans: selective enhancement of the inhibition of factor XIa. J Biol Chem. 1996;271:12913–12918. doi: 10.1074/jbc.271.22.12913. [DOI] [PubMed] [Google Scholar]
- 204.Bos I.G., van Mierlo G.J., Bleeker W.K., Rigter G.M., te Velthuis H., Dickneite G. The potentiation of human C1-inhibitor by dextran sulphate is transient in vivo: studies in a rat model. Int Immunopharmacol. 2001;1:1583–1595. doi: 10.1016/s1567-5769(01)00073-x. [DOI] [PubMed] [Google Scholar]
- 205.Bock S.C., Skriver K., Nielsen E., Thogersen H.C., Wiman B., Donaldson V.H. Human C1 inhibitor: primary structure, cDNA cloning, and chromosomal localization. Biochemistry. 1986;25:4292–4301. doi: 10.1021/bi00363a018. [DOI] [PubMed] [Google Scholar]
- 206.Bos I.G., Lubbers Y.T., Roem D., Abrahams J.P., Hack C.E., Eldering E. The functional integrity of the serpin domain of C1-inhibitor depends on the unique N-terminal domain, as revealed by a pathological mutant. J Biol Chem. 2003;278:29463–29470. doi: 10.1074/jbc.M302977200. [DOI] [PubMed] [Google Scholar]
- 207.Woo P., Lachmann P.J., Harrison R.A., Amos N., Cooper C., Rosen F.S. Simultaneous turnover of normal and dysfunctional C1 inhibitor as a probe of in vivo activation of C1 and contact activatable proteases. Clin Exp Immunol. 1985;61:1–8. [PMC free article] [PubMed] [Google Scholar]
- 208.Prada A.E., Zahedi K., Davis A.E., 3rd Regulation of C1 inhibitor synthesis. Immunobiology. 1998;199:377–388. doi: 10.1016/S0171-2985(98)80042-9. [DOI] [PubMed] [Google Scholar]
- 209.Minta J.O. The role of sialic acid in the functional activity and the hepatic clearance of C1-INH. J Immunol. 1981;126:245–249. [PubMed] [Google Scholar]
- 210.de Smet B.J., de Boer J.P., Agterberg J., Rigter G., Bleeker W.K., Hack C.E. Clearance of human native, proteinase-complexed, and proteolytically inactivated C1-inhibitor in rats. Blood. 1993;81:56–61. [PubMed] [Google Scholar]
- 211.Kunschak M., Engl W., Maritsch F., Rosen F.S., Eder G., Zerlauth G. A randomized, controlled trial to study the efficacy and safety of C1 inhibitor concentrate in treating hereditary angioedema. Transfusion. 1998;38:540–549. doi: 10.1046/j.1537-2995.1998.38698326333.x. [DOI] [PubMed] [Google Scholar]
- 212.Ziccardi R.J. A new role for C-1-inhibitor in homeostasis: control of activation of the first component of human complement. J Immunol. 1982;128:2505–2508. [PubMed] [Google Scholar]
- 213.Nielsen E.W., Johansen H.T., Hogasen K., Wuillemin W., Hack C.E., Mollnes T.E. Activation of the complement, coagulation, fibrinolytic and kallikrein-kinin systems during attacks of hereditary angioedema. Scand J Immunol. 1996;44:185–192. doi: 10.1046/j.1365-3083.1996.d01-298.x. [DOI] [PubMed] [Google Scholar]
- 214.Milgrom H., Curd J.G., Kaplan R.A., Muller-Eberhard H.J., Vaughan J.H. Activation of the fourth component of complement (C4): assessment by rocket immunoelectrophoresis and correlation with the metabolism of C4. J Immunol. 1980;124:2780–2785. [PubMed] [Google Scholar]
- 215.Kim S.J., Gershov D., Ma X., Brot N., Elkon K.B. I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med. 2002;196:655–665. doi: 10.1084/jem.20020542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gershov D., Kim S., Brot N., Elkon K.B. C-reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med. 2000;192:1353–1364. doi: 10.1084/jem.192.9.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Korb L.C., Ahearn J.M. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol. 1997;158:4525–4528. [PubMed] [Google Scholar]
- 218.Familian A., Zwart B., Huisman H.G., Rensink I., Roem D., Hordijk P.L. Chromatin-independent binding of serum amyloid P component to apoptotic cells. J Immunol. 2001;167:647–654. doi: 10.4049/jimmunol.167.2.647. [DOI] [PubMed] [Google Scholar]
- 219.Colman R.W. Surface-mediated defense reactions: the plasma contact activation system. J Clin Invest. 1984;73:1249–1253. doi: 10.1172/JCI111326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nuijens J.H., Huijbregts C.C., Eerenberg-Belmer A.J., Abbink J.J., Strack van Schijndel R.J., Felt-Bersma R.J. Quantification of plasma factor XIIa-C1-inhibitor and kallikrein-C1-inhibitor complexes in sepsis. Blood. 1988;72:1841–1848. [PubMed] [Google Scholar]
- 221.Schapira M., Silver L.D., Scott C.F., Schmaier A.H., Prograis L.J., Jr., Curd J.G. Prekallikrein activation and high-molecular-weight kininogen consumption in hereditary angioedema. N Engl J Med. 1983;308:1050–1053. doi: 10.1056/NEJM198305053081802. [DOI] [PubMed] [Google Scholar]
- 222.Cugno M., Cicardi M., Bottasso B., Coppola R., Paonessa R., Mannucci P.M. Activation of the coagulation cascade in C1-inhibitor deficiencies. Blood. 1997;89:3213–3218. [PubMed] [Google Scholar]
- 223.Cugno M., Hack C.E., de Boer J.P., Eerenberg A.J., Agostoni A., Cicardi M. Generation of plasmin during acute attacks of hereditary angioedema. J Lab Clin Med. 1993;121:38–43. [PubMed] [Google Scholar]
- 224.Wuillemin W.A., Hack C.E., Bleeker W.K., Biemond B.J., Levi M., ten Cate H. Inactivation of factor XIa in vivo: studies in chimpanzees and in humans. Thromb Haemost. 1996;76:549–555. [PubMed] [Google Scholar]
- 225.Levi M., Hack C.E., de Boer J.P., Brandjes D.P., Buller H.R., ten Cate J.W. Reduction of contact activation related fibrinolytic activity in factor XII deficient patients: further evidence for the role of the contact system in fibrinolysis in vivo. J Clin Invest. 1991;88:1155–1160. doi: 10.1172/JCI115416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hack C.E. The role of factor XII in contact system activation. Blood. 1998;92:703–704. [PubMed] [Google Scholar]
- 227.Gainer J.V., Morrow J.D., Loveland A., King D.J., Brown N.J. Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N Engl J Med. 1998;339:1285–1292. doi: 10.1056/NEJM199810293391804. [DOI] [PubMed] [Google Scholar]
- 228.Ueno A., Oh-ishi S. Roles for the kallikrein-kinin system in inflammatory exudation and pain: lessons from studies on kininogen-deficient rats. J Pharmacol Sci. 2003;93:1–20. doi: 10.1254/jphs.93.1. [DOI] [PubMed] [Google Scholar]
- 229.Strang C.J., Cholin S., Spragg J., Davis A.E., 3rd, Schneeberger E.E., Donaldson V.H. Angioedema induced by a peptide derived from complement component C2. J Exp Med. 1988;168:1685–1698. doi: 10.1084/jem.168.5.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Joop K., Berckmans R.J., Nieuwland R., Berkhout J., Romijn F.P., Hack C.E. Microparticles from patients with multiple organ dysfunction syndrome and sepsis support coagulation through multiple mechanisms. Thromb Haemost. 2001;85:810–820. [PubMed] [Google Scholar]
- 231.Griep M.A., Fujikawa K., Nelsestuen G.L. Binding and activation properties of human factor XII, prekallikrein, and derived peptides with acidic lipid vesicles. Biochemistry. 1985;24:4124–4130. doi: 10.1021/bi00336a047. [DOI] [PubMed] [Google Scholar]
- 232.Schousboe I., Rasmussen M.S., Lintner R. Dextran sulphate inhibits phospholipid and sulphatide mediated autoactivation of factor XII. Blood Coagul Fibrinolysis. 1994;5:503–509. [PubMed] [Google Scholar]
- 233.Dumenco L.L., Everson B., Culp L.A., Ratnoff O.D. Inhibition of the activation of Hageman factor (factor XII) by platelet factor 4. J Lab Clin Med. 1988;112:394–400. [PubMed] [Google Scholar]
- 234.Henry M.L., Everson B., Ratnoff O.D. Inhibition of the activation of Hageman factor (factor XII) by beta 2-glycoprotein I. J Lab Clin Med. 1988;111:519–523. [PubMed] [Google Scholar]
- 235.Zanoletti E., Bertino G., Malvezzi L., Benazzo M., Mira E. Angioneurotic edema of the upper airways and antihypertensive therapy. Acta Otolaryngol. 2003;123:960–964. doi: 10.1080/00016480310005129. [DOI] [PubMed] [Google Scholar]
- 236.Agostoni A. [Vasopeptidases and their inhibitors] Recenti Prog Med. 2002;93:519–522. [PubMed] [Google Scholar]
- 237.Campbell D.J. Vasopeptidase inhibition: a double-edged sword? Hypertension. 2003;41:383–389. doi: 10.1161/01.HYP.0000054215.71691.16. [DOI] [PubMed] [Google Scholar]
- 238.La Rocca E., Amoroso S., Brai M., Di Leonardo S., Arico M. [Hereditary angioedema: genealogic studies and clinical considerations on 2 genetic forms in a case study of 10 patients] G Ital Dermatol Venereol. 1986;121:203–208. [PubMed] [Google Scholar]
- 239.Pavletic A. Late angioedema caused by ACE inhibitors underestimated. Am Fam Physician. 2002;66:956. 958. [PubMed] [Google Scholar]
- 240.Angioedema due to ACE inhibitors: common and inadequately diagnosed. Prescrire Int. 1998;7:92–93. [PubMed] [Google Scholar]
- 241.Gainer J.V., Nadeau J.H., Ryder D., Brown N.J. Increased sensitivity to bradykinin among African Americans. J Allergy Clin Immunol. 1996;98:283–287. doi: 10.1016/s0091-6749(96)70151-3. [DOI] [PubMed] [Google Scholar]
- 242.Ajayi A.A., Adigun A.Q. Angioedema and cough in Nigerian patients receiving ACE inhibitors. Br J Clin Pharmacol. 2000;50:81–82. doi: 10.1046/j.1365-2125.2000.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Gibbs C.R., Lip G.Y., Beevers D.G. Angioedema due to ACE inhibitors: increased risk in patients of African origin. Br J Clin Pharmacol. 1999;48:861–865. doi: 10.1046/j.1365-2125.1999.00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Abdi R., Dong V.M., Lee C.J., Ntoso K.A. Angiotensin II receptor blocker-associated angioedema: on the heels of ACE inhibitor angioedema. Pharmacotherapy. 2002;22:1173–1175. doi: 10.1592/phco.22.13.1173.33517. [DOI] [PubMed] [Google Scholar]
- 245.Warner K.K., Visconti J.A., Tschampel M.M. Angiotensin II receptor blockers in patients with ACE inhibitor-induced angioedema. Ann Pharmacother. 2000;34:526–528. doi: 10.1345/aph.19294. [DOI] [PubMed] [Google Scholar]
- 246.Chiu A.G., Krowiak E.J., Deeb Z.E. Angioedema associated with angiotensin II receptor antagonists: challenging our knowledge of angioedema and its etiology. Laryngoscope. 2001;111:1729–1731. doi: 10.1097/00005537-200110000-00011. [DOI] [PubMed] [Google Scholar]
- 247.Tan Y., Hutchison F.N., Jaffa A.A. Mechanisms of angiotensin II-induced expression of B2 kinin receptors. Am J Physiol Heart Circ Physiol. 2004;286:H926–H932. doi: 10.1152/ajpheart.00757.2003. [DOI] [PubMed] [Google Scholar]
- 248.Tabrizchi R. Omapatrilat: Bristol-Myers Squibb. Curr Opin Investig Drugs. 2001;2:1414–1422. [PubMed] [Google Scholar]
- 249.Kostis J.B., Packer M., Black H.R., Schmieder R., Henry D., Levy E. Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am J Hypertens. 2004;17:103–111. doi: 10.1016/j.amjhyper.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 250.Coats A.J. Omapatrilat—the story of Overture and Octave. Int J Cardiol. 2002;86:1–4. doi: 10.1016/s0167-5273(02)00389-3. [DOI] [PubMed] [Google Scholar]
- 251.Ruschitzka F., Corti R., Quaschning T., Hermann M., Luscher T.F. Vasopeptidase inhibitors—concepts and evidence. Nephrol Dial Transplant. 2001;16:1532–1535. doi: 10.1093/ndt/16.8.1532. [DOI] [PubMed] [Google Scholar]
- 252.Molinaro G., Gervais N., Adam A. Biochemical basis of angioedema associated with recombinant tissue plasminogen activator treatment: an in vitro experimental approach. Stroke. 2002;33:1712–1716. doi: 10.1161/01.str.0000017284.77838.87. [DOI] [PubMed] [Google Scholar]
- 253.Cyr M., Lepage Y., Blais C., Jr., Gervais N., Cugno M., Rouleau J.L. Bradykinin and des-Arg(9)-bradykinin metabolic pathways and kinetics of activation of human plasma. Am J Physiol Heart Circ Physiol. 2001;281:H275–H283. doi: 10.1152/ajpheart.2001.281.1.H275. [DOI] [PubMed] [Google Scholar]
- 254.Bhoola K.D., Figueroa C.D., Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- 255.Hall J.M. Bradykinin receptors. Gen Pharmacol. 1997;28:1–6. doi: 10.1016/s0306-3623(96)00174-7. [DOI] [PubMed] [Google Scholar]
- 256.Sumino H., Ichikawa S., Ohyama Y., Nakamura T., Kanda T., Sakamoto H. Effects of hormone replacement therapy on serum angiotensin-converting enzyme activity and plasma bradykinin in postmenopausal women according to angiotensin-converting enzyme-genotype. Hypertens Res. 2003;26:53–58. doi: 10.1291/hypres.26.53. [DOI] [PubMed] [Google Scholar]
- 257.Proudler A.J., Ahmed A.I., Crook D., Fogelman I., Rymer J.M., Stevenson J.C. Hormone replacement therapy and serum angiotensin-converting-enzyme activity in postmenopausal women. Lancet. 1995;346:89–90. doi: 10.1016/s0140-6736(95)92114-1. [DOI] [PubMed] [Google Scholar]
- 258.Mathews K.P., Curd J.G., Hugli T.E. Decreased synthesis of serum carboxypeptidase N (SCPN) in familial SCPN deficiency. J Clin Immunol. 1986;6:87–91. doi: 10.1007/BF00915368. [DOI] [PubMed] [Google Scholar]
- 259.Mathews K.P., Pan P.M., Gardner N.J., Hugli T.E. Familial carboxypeptidase N deficiency. Ann Intern Med. 1980;93:443–445. doi: 10.7326/0003-4819-93-3-443. [DOI] [PubMed] [Google Scholar]
- 260.Rojkjaer R., Hasan A.A., Motta G., Schousboe I., Schmaier A.H. Factor XII does not initiate prekallikrein activation on endothelial cells. Thromb Haemost. 1998;80:74–81. [PubMed] [Google Scholar]
- 261.Rojkjaer R., Schmaier A.H. Activation of the plasma kallikrein/kinin system on endothelial cell membranes. Immunopharmacology. 1999;43:109–114. doi: 10.1016/s0162-3109(99)00069-7. [DOI] [PubMed] [Google Scholar]
- 262.Schmaier A.H. The plasma kallikrein-kinin system counterbalances the renin-angiotensin system. J Clin Invest. 2002;109:1007–1009. doi: 10.1172/JCI15490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Schmaier A.H. The physiologic basis of assembly and activation of the plasma kallikrein/kinin system. Thromb Haemost. 2004;91:1–3. [PubMed] [Google Scholar]
- 264.Kleniewski J., Blankenship D.T., Cardin A.D., Donaldson V. Mechanism of enhanced kinin release from high molecular weight kininogen by plasma kallikrein after its exposure to plasmin. J Lab Clin Med. 1992;120:129–139. [PubMed] [Google Scholar]
- 265.Curd J.G., Prograis L.J., Jr., Cochrane C.G. Detection of active kallikrein in induced blister fluids of hereditary angioedema patients. J Exp Med. 1980;152:742–747. doi: 10.1084/jem.152.3.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Gordon E.M., Williams S.R., Frenchek B., Mazur C.A., Speroff L. Dose-dependent effects of postmenopausal estrogen and progestin on antithrombin III and factor XII. J Lab Clin Med. 1988;111:52–56. [PubMed] [Google Scholar]
- 267.Jespersen J., Kluft C. Increased euglobulin fibrinolytic potential in women on oral contraceptives low in oestrogen—levels of extrinsic and intrinsic plasminogen activators, prekallikrein, factor XII, and C1-inactivator. Thromb Haemost. 1985;54:454–459. [PubMed] [Google Scholar]
- 268.Farsetti A., Misiti S., Citarella F., Felici A., Andreoli M., Fantoni A. Molecular basis of estrogen regulation of Hageman factor XII gene expression. Endocrinology. 1995;136:5076–5083. doi: 10.1210/endo.136.11.7588244. [DOI] [PubMed] [Google Scholar]
- 269.Fossum S., Hoem N.O., Johannesen S., Korpberget M., Nylund E., Sandem S. Contact factors in plasma from women on oral contraception—significance of factor XI for the measured activity of factor XII. Thromb Res. 1994;74:477–485. doi: 10.1016/0049-3848(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 270.Farmer S.G., Burch R.M. Biochemical and molecular pharmacology of kinin receptors. Annu Rev Pharmacol Toxicol. 1992;32:511–536. doi: 10.1146/annurev.pa.32.040192.002455. [DOI] [PubMed] [Google Scholar]
- 271.Ni A., Chao L., Chao J. Transcription factor nuclear factor kappaB regulates the inducible expression of the human B1 receptor gene in inflammation. J Biol Chem. 1998;273:2784–2791. doi: 10.1074/jbc.273.5.2784. [DOI] [PubMed] [Google Scholar]
- 272.Regoli D., Pheng L.H., Allogho S.N., Nguyen-Le X.K., Gobeil F. Receptors for kinins: from classical pharmacology to molecular biology. Immunopharmacology. 1996;33:24–31. doi: 10.1016/0162-3109(96)00077-x. [DOI] [PubMed] [Google Scholar]
- 273.Prado G.N., Taylor L., Zhou X., Ricupero D., Mierke D.F., Polgar P. Mechanisms regulating the expression, self-maintenance, and signaling-function of the bradykinin B2 and B1 receptors. J Cell Physiol. 2002;193:275–286. doi: 10.1002/jcp.10175. [DOI] [PubMed] [Google Scholar]
- 274.Madeddu P., Emanueli C., Song Q., Varoni M.V., Demontis M.P., Anania V. Regulation of bradykinin B2-receptor expression by oestrogen. Br J Pharmacol. 1997;121:1763–1769. doi: 10.1038/sj.bjp.0701255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Fletcher A., Weetman A.P. Coexistence of hereditary angioedema and Turner's syndrome. Postgrad Med J. 1998;74:41–42. doi: 10.1136/pgmj.74.867.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.McGlinchey P.G., McCluskey D.R. Hereditary angioedema precipitated by estrogen replacement therapy in a menopausal woman. Am J Med Sci. 2000;320:212–213. doi: 10.1097/00000441-200009000-00014. [DOI] [PubMed] [Google Scholar]
- 277.Bork K., Fischer B., Dewald G. Recurrent episodes of skin angioedema and severe attacks of abdominal pain induced by oral contraceptives or hormone replacement therapy. Am J Med. 2003;114:294–298. doi: 10.1016/s0002-9343(02)01526-7. [DOI] [PubMed] [Google Scholar]
- Warin R.P., Cunliffe W.J., Greaves M.W., Wallington T.B. Recurrent angioedema: familial and oestrogen-induced. Br J Dermatol. 1986;115:731–734. doi: 10.1111/j.1365-2133.1986.tb06656.x. [DOI] [PubMed] [Google Scholar]
- 278.Bouillet L., Ponard D., Drouet C., Jullien D., Massot C. Angioedema and oral contraception. Dermatology. 2003;206:106–109. doi: 10.1159/000068456. [DOI] [PubMed] [Google Scholar]
- 279.Pichler W.J., Lehner R., Spath P.J. Recurrent angioedema associated with hypogonadism or anti-androgen therapy. Ann Allergy. 1989;63:301–305. [PubMed] [Google Scholar]
- 280.Citarella F., Misiti S., Felici A., Farsetti A., Pontecorvi A., Fantoni A. Estrogen induction and contact phase activation of human factor XII. Steroids. 1996;61:270–276. doi: 10.1016/0039-128x(96)00037-2. [DOI] [PubMed] [Google Scholar]
- 281.Gordon E.M., Johnson T.R., Ramos L.P., Schmeidler-Sapiro K.T. Enhanced expression of factor XII (Hageman factor) in isolated livers of estrogen- and prolactin-treated rats. J Lab Clin Med. 1991;117:353–358. [PubMed] [Google Scholar]
- 282.Gordon E.M., Douglas J.G., Ratnoff O.D., Arafah B.M. The influence of estrogen and prolactin on Hageman factor (factor XII) titer in ovariectomized and hypophysectomized rats. Blood. 1985;66:602–605. [PubMed] [Google Scholar]
- 283.Chen L.M., Chung P., Chao S., Chao L., Chao J. Differential regulation of kininogen gene expression by estrogen and progesterone in vivo. Biochim Biophys Acta. 1992;1131:145–151. doi: 10.1016/0167-4781(92)90069-c. [DOI] [PubMed] [Google Scholar]
- 284.Norris L.A., Bonnar J. The effect of oestrogen dose and progestogen type on haemostatic changes in women taking low dose oral contraceptives. Br J Obstet Gynaecol. 1996;103:261–267. doi: 10.1111/j.1471-0528.1996.tb09716.x. [DOI] [PubMed] [Google Scholar]
- 285.Thiery M., Vermeulen A., Baele G., Deslypere J.P. Effects of a very low-estrogen oral contraceptive on clotting factors, carbohydrate metabolism and plasma lipids and lipoproteins. Med Sci Res. 1987;15:1231–1232. [PubMed] [Google Scholar]
- 286.Hoem N.O., Johannesen S., Hauge G., Rud A.C., Sandem S., Briseid K. Contact activation factors in plasma from women using oral contraceptives—increased levels of factor XII, kinin-free high molecular weight kininogen and acetone-activated kallikrein. Thromb Res. 1991;64:427–434. doi: 10.1016/0049-3848(91)90343-u. [DOI] [PubMed] [Google Scholar]
- 287.Wessler S. Estrogen-associated thromboembolism. Ann Epidemiol. 1992;2:439–443. doi: 10.1016/1047-2797(92)90093-6. [DOI] [PubMed] [Google Scholar]
- 288.Campbell S.J., Mackie I.J., Robinson G.E., Machin S.J. Contact factor mediated fibrinolysis is increased by the combined oral contraceptive pill. Br J Obstet Gynaecol. 1993;100:79–84. doi: 10.1111/j.1471-0528.1993.tb12956.x. [DOI] [PubMed] [Google Scholar]
- 289.Luyer M.D., Khosla S., Owen W.G., Miller V.M. Prospective randomized study of effects of unopposed estrogen replacement therapy on markers of coagulation and inflammation in postmenopausal women. J Clin Endocrinol Metab. 2001;86:3629–3634. doi: 10.1210/jcem.86.8.7768. [DOI] [PubMed] [Google Scholar]
- 290.Madsen J.S., Kristensen S.R., Gram J., Bladbjerg E.M., Henriksen F.L., Christensen K. Positive impact of hormone replacement therapy on the fibrinolytic system: a long-term randomized controlled study in healthy postmenopausal women. J Thromb Haemost. 2003;1:1984–1991. doi: 10.1046/j.1538-7836.2003.00362.x. [DOI] [PubMed] [Google Scholar]
- 291.Teede H.J., McGrath B.P., Smolich J.J., Malan E., Kotsopoulos D., Liang Y.L. Postmenopausal hormone replacement therapy increases coagulation activity and fibrinolysis. Arterioscler Thromb Vasc Biol. 2000;20:1404–1409. doi: 10.1161/01.atv.20.5.1404. [DOI] [PubMed] [Google Scholar]
- 292.Nogawa N., Sumino H., Ichikawa S., Kumakura H., Takayama Y., Nakamura T. Effect of long-term hormone replacement therapy on angiotensin-converting enzyme activity and bradykinin in postmenopausal women with essential hypertension and normotensive postmenopausal women. Menopause. 2001;8:210–215. doi: 10.1097/00042192-200105000-00011. [DOI] [PubMed] [Google Scholar]
- 293.Gallagher P.E., Li P., Lenhart J.R., Chappell M.C., Brosnihan K.B. Estrogen regulation of angiotensin-converting enzyme mRNA. Hypertension. 1999;33:323–328. doi: 10.1161/01.hyp.33.1.323. [DOI] [PubMed] [Google Scholar]
- 294.Visy B., Fust G., Varga L., Szendei G., Takacs E., Karadi I. Sex hormones in hereditary angioneurotic oedema. Clin Endocrinol (Oxf) 2004;60:508–515. doi: 10.1111/j.1365-2265.2004.02009.x. [DOI] [PubMed] [Google Scholar]
- 295.Parfitt K., editor. The complete drug reference. 32nd ed. Pharmaceutical Press; London: 1999. [Google Scholar]
- 296.McEvoy G.K., editor. AHFS drug information 2000. American Society of Health-System Pharmacists; Bethesda: 2000. [Google Scholar]
- 297.Klasco R.K., editor. DRUGDEX® system. Thomson MICROMEDEX; Greenwood Village Colo: 2001. [Google Scholar]
- 298.Reference deleted in proof
- 299.Borradori L., Marie O., Rybojad M., Vexiau P., Morel P., Spath P. Hereditary angioedema and oral contraception. Dermatologica. 1990;181:78–79. doi: 10.1159/000247874. [DOI] [PubMed] [Google Scholar]
- 300.Munkvad S., Jespersen J., Gram J., Overgaard K., Ranby M. Effects of methylamine and heparin on a rapid chromogenic assay of C1-esterase inhibitor in plasma. Clin Chem. 1990;36:737–741. [PubMed] [Google Scholar]
- 301.Nielsen E.W., Johansen H.T., Straume B., Mollnes T.E. Effect of time, temperature and additives on a functional assay of C1 inhibitor. J Immunol Methods. 1994;173:245–251. doi: 10.1016/0022-1759(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 302.Reference deleted in proof
- 303.Reference deleted in proof
- 304.Reference deleted in proof
- 305.Schunkert H., Danser A.H., Hense H.W., Derkx F.H., Kurzinger S., Riegger G.A. Effects of estrogen replacement therapy on the renin-angiotensin system in postmenopausal women. Circulation. 1997;95:39–45. doi: 10.1161/01.cir.95.1.39. [DOI] [PubMed] [Google Scholar]
- 306.Grattan C., Powell S., Humphreys F. Management and diagnostic guidelines for urticaria and angio-oedema. Br J Dermatol. 2001;144:708–714. doi: 10.1046/j.1365-2133.2001.04175.x. [DOI] [PubMed] [Google Scholar]
- 307.Blohme G., Ysander L., Korsan-Bengtsen K., Laurell A.B. Hereditary angioneurotic oedema in three families: symptomatic heterogeneity, complement analysis and therapeutic trials. Acta Med Scand. 1972;191:209–219. [PubMed] [Google Scholar]
- 308.Ohela K. Hereditary angioneurotic oedema in Finland: clinical, immunological and genealogical studies. Acta Med Scand. 1977;201:415–427. [PubMed] [Google Scholar]
- 309.Zeana C. Hereditary angioedema due to deficit of C1 esterase inhibitor. Med Interne. 1989;27:143–147. [PubMed] [Google Scholar]
- 310.Donaldson V.H., Rosen F.S. Hereditary angioneurotic edema: a clinical survey. Pediatrics. 1966;37:1017–1027. [PubMed] [Google Scholar]
- 311.Madalinski K., Sabbouh K., Chorazykiewicz M., Gregorek H. C1-INH defect as an example of deficiency disease. Immunol Invest. 1991;20:133–141. doi: 10.3109/08820139109050781. [DOI] [PubMed] [Google Scholar]
- 312.Barakat A., Castaldo A.J. Hereditary angioedema: danazol therapy in a 5-year-old child. Am J Dis Child. 1993;147:931–932. doi: 10.1001/archpedi.1993.02160330021005. [DOI] [PubMed] [Google Scholar]
- 313.Cox M., Holdcroft A. Hereditary angioneurotic oedema: current management in pregnancy. Anaesthesia. 1995;50:547–549. doi: 10.1111/j.1365-2044.1995.tb06050.x. [DOI] [PubMed] [Google Scholar]
- 314.Gompels M.M., Lock R.J., Morgan J.E., Osborne J., Brown A., Virgo P.F. A multicentre evaluation of the diagnostic efficiency of serological investigations for C1 inhibitor deficiency. J Clin Pathol. 2002;55:145–147. doi: 10.1136/jcp.55.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Matter L., Schopfer K., Voegelin H.P., Fueter R. [Hereditary angioedema: description of a sibship and review] Schweiz Med Wochenschr. 1981;111:1238–1245. [PubMed] [Google Scholar]
- 316.Späth P., Wüthrich B. Inherited and acquired deficiencies of C1 esterase inhibitor in humans. In: Rother K., Till G., Hänsch G., editors. The complement system. Springer-Velag; Berlin Heidelberg: 1998. pp. 353–410. [Google Scholar]
- 317.Nielsen E.W., Johansen H.T., Gaudesen O., Osterud B., Olsen J.O., Hogasen K. C3 is activated in hereditary angioedema, and C1/C1-inhibitor complexes rise during physical stress in untreated patients. Scand J Immunol. 1995;42:679–685. doi: 10.1111/j.1365-3083.1995.tb03711.x. [DOI] [PubMed] [Google Scholar]
- 318.Sonntag J., Bethge K., Strauss E., Dudenhausen J.W., Obladen M. Contact system in healthy term newborns: reference values in cord blood. Thromb Res. 1997;87:339–342. doi: 10.1016/s0049-3848(97)00136-9. [DOI] [PubMed] [Google Scholar]
- 319.Wolbink G.J., Bollen J., Baars J.W., ten Berge R.J., Swaak A.J., Paardekooper J. Application of a monoclonal antibody against a neoepitope on activated C4 in an ELISA for the quantification of complement activation via the classical pathway. J Immunol Methods. 1993;163:67–76. doi: 10.1016/0022-1759(93)90240-8. [DOI] [PubMed] [Google Scholar]
- 320.Nielsen E.W., Johansen H.T., Holt J., Mollnes T.E. C1 inhibitor and diagnosis of hereditary angioedema in newborns. Pediatr Res. 1994;35:184–187. doi: 10.1203/00006450-199402000-00012. [DOI] [PubMed] [Google Scholar]
- 321.Ferlazzo B., Barrile A., Crisafi A., Quattrocchi P., Bonanno D., Giacobbe G. [Determination of complement proteins in the umbilical cord in the early diagnosis of hereditary angioneurotic edema] Recenti Prog Med. 1991;82:29–30. [PubMed] [Google Scholar]
- 322.Johnson U., Truedsson L., Gustavii B. Complement components in 100 newborns and their mothers determined by electroimmunoassay. Acta Pathol Microbiol Immunol Scand [C] 1983;91:147–150. [PubMed] [Google Scholar]
- 323.Spath P.J., Wuthrich B. Angioedema: a review on the acquired, allergic or non-allergic, and the hereditary forms. Recenti Prog Med. 1990;81:513–531. [PubMed] [Google Scholar]
- 324.Davis C.A., Vallota E.H., Forristal J. Serum complement levels in infancy: age related changes. Pediatr Res. 1979;13:1043–1046. doi: 10.1203/00006450-197909000-00019. [DOI] [PubMed] [Google Scholar]
- 325.Intrator L., Laurent J., Branellec A., Drogoul F., Lucas M., Proust A. [Complement fractions in cord blood: value for the neonatal detection of hereditary angioneurotic edema] Presse Med. 1985;14:1668–1669. [PubMed] [Google Scholar]
- 326.Roach B., Kim Y., Jerome E., Michael A.F. Influence of age and sex on serum complement components in children. Am J Dis Child. 1981;135:918–920. doi: 10.1001/archpedi.1981.02130340030011. [DOI] [PubMed] [Google Scholar]
- 327.Andrew M., Paes B., Milner R., Johnston M., Mitchell L., Tollefsen D.M. Development of the human coagulation system in the full-term infant. Blood. 1987;70:165–172. [PubMed] [Google Scholar]
- 328.Andrew M., Massicotte-Nolan P.M., Karpatkin M. Plasma protease inhibitors in premature infants: influence of gestational age, postnatal age, and health status. Proc Soc Exp Biol Med. 1983;173:495–500. doi: 10.3181/00379727-173-41676. [DOI] [PubMed] [Google Scholar]
- 329.Norman M.E., Gall E.P., Taylor A., Laster L., Nilsson U.R. Serum complement profiles in infants and children. J Pediatr. 1975;87:912–916. doi: 10.1016/s0022-3476(75)80904-8. [DOI] [PubMed] [Google Scholar]
- 330.Gompels M.M., Lock R.J., Unsworth D.J., Johnston S.L., Archer C.B., Davies S.V. Misdiagnosis of hereditary angio-oedema type 1 and type 2. Br J Dermatol. 2003;148:719–723. doi: 10.1046/j.1365-2133.2003.05231.x. [DOI] [PubMed] [Google Scholar]
- 331.Zuraw B.L., Altman L.C. Acute consumption of C1 inhibitor in a patient with acquired C1-inhibitor deficiency syndrome. J Allergy Clin Immunol. 1991;88:908–918. doi: 10.1016/0091-6749(91)90248-m. [DOI] [PubMed] [Google Scholar]
- 332.Karim Y., Griffiths H., Deacock S. Normal complement C4 values do not exclude hereditary angioedema. J Clin Pathol. 2004;57:213–214. doi: 10.1136/jcp.2003.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Brackertz D., Kueppers F. Hereditary angioneurotic oedema. Lancet. 1973;2:680. doi: 10.1016/s0140-6736(73)92519-1. [DOI] [PubMed] [Google Scholar]
- 334.Waytes A.T., Rosen F.S., Frank M.M. Treatment of hereditary angioedema with a vapor-heated C1 inhibitor concentrate. N Engl J Med. 1996;334:1630–1634. doi: 10.1056/NEJM199606203342503. [DOI] [PubMed] [Google Scholar]
- 335.Gadek J.E., Hosea S.W., Gelfand J.A., Santaella M., Wickerhauser M., Triantaphyllopoulos D.C. Replacement therapy in hereditary angioedema: successful treatment of acute episodes of angioedema with partly purified C1 inhibitor. N Engl J Med. 1980;302:542–546. doi: 10.1056/NEJM198003063021002. [DOI] [PubMed] [Google Scholar]
- 336.Bergamaschini L., Cicardi M., Tucci A., Gardinali M., Frangi D., Valle C. C1 INH concentrate in the therapy of hereditary angioedema. Allergy. 1983;38:81–84. doi: 10.1111/j.1398-9995.1983.tb01590.x. [DOI] [PubMed] [Google Scholar]
- 337.Visentin D.E., Yang W.H., Karsh J. C1-esterase inhibitor transfusions in patients with hereditary angioedema. Ann Allergy Asthma Immunol. 1998;80:457–461. doi: 10.1016/S1081-1206(10)63067-0. [DOI] [PubMed] [Google Scholar]
- 338.Pickering R.J., Good R.A., Kelly J.R., Gewurz H. Replacement therapy in hereditary angioedema: successful treatment of two patients with fresh frozen plasma. Lancet. 1969;1:326–330. doi: 10.1016/s0140-6736(69)91295-1. [DOI] [PubMed] [Google Scholar]
- 339.Carugati A., Pappalardo E., Zingale L.C., Cicardi M. C1-inhibitor deficiency and angioedema. Mol Immunol. 2001;38:161–173. doi: 10.1016/s0161-5890(01)00040-2. [DOI] [PubMed] [Google Scholar]
- 340.Marasini B., Cicardi M., Martignoni G.C., Agostoni A. Treatment of hereditary angioedema. Klin Wochenschr. 1978;56:819–823. doi: 10.1007/BF01489716. [DOI] [PubMed] [Google Scholar]
- 341.Proud G., Chamberlain J. Letter: anaphylactic reaction to aprotinin. Lancet. 1976;2:48–49. doi: 10.1016/s0140-6736(76)93012-9. [DOI] [PubMed] [Google Scholar]
- 342.Williams A., Baird L.G. DX-88 and HAE: a developmental perspective. Transfus Apheresis Sci. 2003;29:255–258. doi: 10.1016/S1473-0502(03)00170-8. [DOI] [PubMed] [Google Scholar]
- 343.Gonzales-Quevado T., Caballero T., Cicardi M., Bork K., Williams A. The synthetic Kunitz domain protein DX-88 to treat angioedema in patients with hereditary angioedema. Int Immunopharmacol. 2002;2:1318. [abstract] [Google Scholar]
- 344.Turner P., Dear J., Scadding G., Foreman J.C. Role of kinins in seasonal allergic rhinitis: icatibant, a bradykinin B2 receptor antagonist, abolishes the hyperresponsiveness and nasal eosinophilia induced by antigen. J Allergy Clin Immunol. 2001;107:105–113. doi: 10.1067/mai.2001.111145. [DOI] [PubMed] [Google Scholar]
- 345.Akbary A.M., Wirth K.J., Scholkens B.A. Efficacy and tolerability of Icatibant (Hoe 140) in patients with moderately severe chronic bronchial asthma. Immunopharmacology. 1996;33:238–242. doi: 10.1016/0162-3109(96)00065-3. [DOI] [PubMed] [Google Scholar]
- Jerini announces positive phase II study results with Icatibant for the treatment of Hereditary Angioedema [press release]. Berlin: Jerini Pharmaceuticals; January 13, 2004
- 346.Nuijens J., Van Doorn M., Van Dam T., Burggraaf K., Levi M., Hack E. A phase I study of recombinant human C1-INH in asymptomatic patients with hereditary angioedema-HAE. Int Immunopharmacol. 2002;2:1315–1316. doi: 10.1016/j.jaci.2005.05.019. [abstract] [DOI] [PubMed] [Google Scholar]
- Pharming features clinical results of C1 inhibitor at investigator meeting [press release]. Leiden: Pharming; January 19, 2004
- 347.Gelfand J.A., Sherins R.J., Alling D.W., Frank M.M. Treatment of hereditary angioedema with danazol: reversal of clinical and biochemical abnormalities. N Engl J Med. 1976;295:1444–1448. doi: 10.1056/NEJM197612232952602. [DOI] [PubMed] [Google Scholar]
- 348.Cicardi M., Bergamaschini L., Tucci A., Agostoni A., Tornaghi G., Coggi G. Morphologic evaluation of the liver in hereditary angioedema patients on long-term treatment with androgen derivatives. J Allergy Clin Immunol. 1983;72:294–298. doi: 10.1016/0091-6749(83)90034-9. [DOI] [PubMed] [Google Scholar]
- 349.Cicardi M., Castelli R., Zingale L.C., Agostoni A. Side effects of long-term prophylaxis with attenuated androgens in hereditary angioedema: comparison of treated and untreated patients. J Allergy Clin Immunol. 1997;99:194–196. doi: 10.1016/s0091-6749(97)70095-2. [DOI] [PubMed] [Google Scholar]
- 350.Zurlo J.J., Frank M.M. The long-term safety of danazol in women with hereditary angioedema. Fertil Steril. 1990;54:64–72. doi: 10.1016/s0015-0282(16)53638-3. [DOI] [PubMed] [Google Scholar]
- 351.Cicardi M., Bergamaschini L., Cugno M., Hack E., Agostoni G., Agostoni A. Long-term treatment of hereditary angioedema with attenuated androgens: a survey of a 13-year experience. J Allergy Clin Immunol. 1991;87:768–773. doi: 10.1016/0091-6749(91)90120-d. [DOI] [PubMed] [Google Scholar]
- 352.Crampon D., Barnoud R., Durand M., Ponard D., Jacquot C., Sotto J.J. Danazol therapy: an unusual aetiology of hepatocellular carcinoma. J Hepatol. 1998;29:1035–1036. doi: 10.1016/s0168-8278(98)80140-2. [DOI] [PubMed] [Google Scholar]
- 353.Bork K., Pitton M., Harten P., Koch P. Hepatocellular adenomas in patients taking danazol for hereditary angio-oedema. Lancet. 1999;353:1066–1067. doi: 10.1016/S0140-6736(99)00110-5. [DOI] [PubMed] [Google Scholar]
- 354.Bork K., Schneiders V. Danazol-induced hepatocellular adenoma in patients with hereditary angio-oedema. J Hepatol. 2002;36:707–709. doi: 10.1016/s0168-8278(02)00035-1. [DOI] [PubMed] [Google Scholar]
- 355.Lundh B., Laurell A.B., Wetterqvist H., White T., Granerus G. A case of hereditary angioneurotic oedema, successfully treated with epsilon-aminocaproic acid: studies on C'1 esterase inhibitor, C'1 activation, plasminogen level and histamine metabolism. Clin Exp Immunol. 1968;3:733–745. [PMC free article] [PubMed] [Google Scholar]
- 356.Bork K., Witzke G. Long-term prophylaxis with C1-inhibitor (C1 INH) concentrate in patients with recurrent angioedema caused by hereditary and acquired C1-inhibitor deficiency. J Allergy Clin Immunol. 1989;83:677–682. doi: 10.1016/0091-6749(89)90082-1. [DOI] [PubMed] [Google Scholar]
- 357.Martinez-Saguer I., Müller W., Aygören Pürsün E., Rusicke E., Stoll H., Klarmann D. Pharmacokinetic parameters of C1-inhibitor concentrate in 40 patients with hereditary angio-oedema (HAE)—a prospective study. Haemophilia. 2002;8:574. [Google Scholar]
- 358.Degroote D.F., Smith G.L., Huttula G.S. Acute airway obstruction following tooth extraction in hereditary angioedema. J Oral Maxillofac Surg. 1985;43:52–54. doi: 10.1016/s0278-2391(85)80014-8. [DOI] [PubMed] [Google Scholar]
- 359.Atkinson J.C., Frank M.M. Oral manifestations and dental management of patients with hereditary angioedema. J Oral Pathol Med. 1991;20:139–142. doi: 10.1111/j.1600-0714.1991.tb00908.x. [DOI] [PubMed] [Google Scholar]
- 360.Phillips K.M., Glick M., Cohen S.G. Hereditary angioedema: report of case. Spec Care Dentist. 1989;9:23–26. doi: 10.1111/j.1754-4505.1989.tb01016.x. [DOI] [PubMed] [Google Scholar]
- 361.Pence H.L., Evans R., Guernsey L.H., Gerhard R.C. Prophylactic use of epsilon aminocaproic acid for oral surgery in a patient with hereditary angioneurotic edema. J Allergy Clin Immunol. 1974;53:298–302. doi: 10.1016/0091-6749(74)90109-2. [DOI] [PubMed] [Google Scholar]
- 362.Sheffer A.L., Fearon D.T., Austen K.F., Rosen F.S. Tranexamic acid: preoperative prophylactic therapy for patients with hereditary angioneurotic edema. J Allergy Clin Immunol. 1977;60:38–40. doi: 10.1016/0091-6749(77)90080-x. [DOI] [PubMed] [Google Scholar]
- 363.Crosher R. Intravenous tranexamic acid in the management of hereditary angio-oedema. Br J Oral Maxillofac Surg. 1987;25:500–506. doi: 10.1016/0266-4356(87)90143-4. [DOI] [PubMed] [Google Scholar]
- 364.Hardie J., Ringland T., Yang W.H., Wagner V. Potentially fatal hereditary angioedema: a review and case report. J Can Dent Assoc. 1990;56:1096–1099. [PubMed] [Google Scholar]
- 365.Farkas H., Gyeney L., Gidofalvy E., Fust G., Varga L. The efficacy of short-term danazol prophylaxis in hereditary angioedema patients undergoing maxillofacial and dental procedures. J Oral Maxillofac Surg. 1999;57:404–408. doi: 10.1016/s0278-2391(99)90280-x. [DOI] [PubMed] [Google Scholar]
- 366.Lehmann A., Lang J., Boldt J., Saggau W. Successful off-pump coronary artery bypass graft surgery in a patient with hereditary angioedema. J Cardiothorac Vasc Anesth. 2002;16:473–476. doi: 10.1053/jcan.2002.125132. [DOI] [PubMed] [Google Scholar]
- 367.Peled M., Ardekian L., Schnarch A., Laufer D. Preoperative prophylaxis for C1 esterase-inhibitor deficiency in patients undergoing oral surgery: a report of three cases. Quintessence Int. 1997;28:169–171. [PubMed] [Google Scholar]
- 368.Leimgruber A., Jaques W.A., Spaeth P.J. Hereditary angioedema: uncomplicated maxillofacial surgery using short-term C1 inhibitor replacement therapy. Int Arch Allergy Immunol. 1993;101:107–112. doi: 10.1159/000236506. [DOI] [PubMed] [Google Scholar]
- 369.Langton D., Weiner J., Fary W. C1-esterase inhibitor concentrate prevents upper airway obstruction in hereditary angio-oedema. Med J Aust. 1994;160:383–384. [PubMed] [Google Scholar]
- 370.Bain B.J., Catovsky D., Ewan P.W. Acquired angioedema as the presenting feature of lymphoproliferative disorders of mature B-lymphocytes. Cancer. 1993;72:3318–3322. doi: 10.1002/1097-0142(19931201)72:11<3318::aid-cncr2820721130>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 371.Bouillet L., Ponard D., Drouet C., Dumestre C., Pernollet M., Bonerandi J.J. [Acquired angioneurotic edema: clinical and biological characteristics in 9 patients] Presse Med. 2000;29:640–644. [PubMed] [Google Scholar]
- 372.Phanish M.K., Owen A., Parry D.H. Spontaneous regression of acquired C1 esterase inhibitor deficiency associated with splenic marginal zone lymphoma presenting with recurrent angio-oedema. J Clin Pathol. 2002;55:789–790. doi: 10.1136/jcp.55.10.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Cohen S.H., Koethe S.M., Kozin F., Rodey G., Arkins J.A., Fink J.N. Acquired angioedema associated with rectal carcinoma and its response to danazol therapy: acquired angioedema treated with danazol. J Allergy Clin Immunol. 1978;62:217–221. doi: 10.1016/0091-6749(78)90210-5. [DOI] [PubMed] [Google Scholar]
- 374.Cugno M., Cicardi M., Agostoni A. Activation of the contact system and fibrinolysis in autoimmune acquired angioedema: a rationale for prophylactic use of tranexamic acid. J Allergy Clin Immunol. 1994;93:870–876. doi: 10.1016/0091-6749(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 375.Donaldson V.H., Bernstein D.I., Wagner C.J., Mitchell B.H., Scinto J., Bernstein I.L. Angioneurotic edema with acquired C1-inhibitor deficiency and autoantibody to C1-inhibitor: response to plasmapheresis and cytotoxic therapy. J Lab Clin Med. 1992;119:397–406. [PubMed] [Google Scholar]
- 376.Gröner A., Nowak T., Römisch J. Purity, activity, and virus safety of a pasteurized antithrombin concentrate. Semin Thromb Hemost. 2002;28(suppl 1):79–86. doi: 10.1055/s-2002-30200. [DOI] [PubMed] [Google Scholar]
- 377.Heimburger N., Schwinn H., Gratz P., Kumpe G., Herchenhan B. A factor VIII concentrate, highly purified and heated in solution. Haemostasis. 1981;10(suppl 1):204. [PubMed] [Google Scholar]
- 378.Hilfenhaus J. Virus-safe plasma proteins: elimination of viruses of risk by the manufacturing procedure. Transf Sci. 1990;11:35S–41S. [Google Scholar]
- 379.Hilfenhaus J., Nowak T. Inactivation of hepatitis A virus by pasteurization and elimination of picornaviruses during manufacture of factor VIII concentrate. Vox Sang. 1994;67(suppl 1):62–66. [PubMed] [Google Scholar]
- 380.Schimpf K., Brackmann H.H., Kreuz W., Kraus B., Haschke F., Schramm W. Absence of anti-human immunodeficiency virus types 1 and 2 seroconversion after the treatment of hemophilia A or von Willebrand's disease with pasteurized factor VIII concentrate. N Engl J Med. 1989;321:1148–1152. doi: 10.1056/NEJM198910263211702. [DOI] [PubMed] [Google Scholar]
- 381.Schimpf K., Mannucci P.M., Kreuz W., Brackmann H.H., Auerswald G., Ciavarella N. Absence of hepatitis after treatment with a pasteurized factor VIII concentrate in patients with hemophilia and no previous transfusions. N Engl J Med. 1987;316:918–922. doi: 10.1056/NEJM198704093161505. [DOI] [PubMed] [Google Scholar]
- 382.Chandra S., Groener A., Feldman F. Effectiveness of alternative treatments for reducing potential viral contaminants from plasma-derived products. Thromb Res. 2002;105:391–400. doi: 10.1016/s0049-3848(02)00044-0. [DOI] [PubMed] [Google Scholar]
- 383.Blümel J., Schmidt I., Willkommen H., Löwer J. Inactivation of parvovirus B19 during pasteurization of human serum albumin. Transfusion. 2002;42:1011–1018. doi: 10.1046/j.1537-2995.2002.00158.x. [DOI] [PubMed] [Google Scholar]
- 384.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 385.Kreuz W., Auerswald G., Brueckmann C., Linde R., Sutor A., Schramm W. Virus safety of pasteurized clotting factor concentrates: an eleven year follow up. Thromb Hemost. 1991;65:824. [Google Scholar]
- 386.Klarmann D., Kreuz W., Joseph-Steiner J., Ehrenforth S. Hepatitis C and pasteurized C1-inhibitor concentrate. Transfusion. 1996;36:84–85. doi: 10.1046/j.1537-2995.1996.36196190522.x. [DOI] [PubMed] [Google Scholar]
- 387.Bork K. Prospective virus safety follow-up after therapy with a pasteurized C1-inhibitor concentrate. Biomed Prog. 1992;5:59–61. [Google Scholar]
- 388.Martinez-Saguer I., Heller C., Fischer D., Ettinghausen C.E., Kreuz W. Prophylactic treatment with pasteurised C1 inhibitor in herditary angioedema (HAE)—a prospective 32 months follow up. Blood. 1999;94:2339. [Google Scholar]
- 389.Rusicke E., Martinez-Saguer I., Aygören-Pürsuen E., Becker S., Funk M., Heller C. Prospective follow up of clinical manifestation and therapy in children with hereditary angioedema (HAE) Ann Hematol. 2003;82(suppl 1):S73. [Google Scholar]
- 390.Weimer T., Streichert S., Watson C., Gröner A. High-titer screening PCR: a successful strategy for reducing the parvovirus B19 load in plasma pools for fractionation. Transfusion. 2001;41:1500–1504. doi: 10.1046/j.1537-2995.2001.41121500.x. [DOI] [PubMed] [Google Scholar]
- 391.Brennan V.M., Salome-Bentley N.J., Chapel H.M. Prospective audit of adverse reactions occurring in 459 primary antibody-deficient patients receiving intravenous immunoglobulin. Clin Exp Immunol. 2003;133:247–251. doi: 10.1046/j.1365-2249.2003.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Santagostino E., Gringeri A., Mannucci P.M. State of care for hemophilia in pediatric patients. Paediatr Drugs. 2002;4:149–157. doi: 10.2165/00128072-200204030-00002. [DOI] [PubMed] [Google Scholar]
- 393.Cicardi M., Agostoni A. Hereditary angioedema. N Engl J Med. 1996;334:1666–1667. doi: 10.1056/NEJM199606203342510. [DOI] [PubMed] [Google Scholar]
- 394.Agostoni A., Bergamaschini L., Martignoni G., Cicardi M., Marasini B. Treatment of acute attacks of hereditary angioedema with C1-inhibitor concentrate. Ann Allergy. 1980;44:299–301. [PubMed] [Google Scholar]
- 395.Bork K., Hardt J., Schicketanz K.H., Ressel N. Clinical studies of sudden upper airway obstruction in patients with hereditary angioedema due to C1 esterase inhibitor deficiency. Arch Intern Med. 2003;163:1229–1235. doi: 10.1001/archinte.163.10.1229. [DOI] [PubMed] [Google Scholar]
- 396.Nuijens J.H., Eerenberg-Belmer A.J., Huijbregts C.C., Schreuder W.O., Felt-Bersma R.J., Abbink J.J. Proteolytic inactivation of plasma C1- inhibitor in sepsis. J Clin Invest. 1989;84:443–450. doi: 10.1172/JCI114185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Lamark T., Ingebrigtsen M., Bjornstad C., Melkko T., Mollnes T.E., Nielsen E.W. Expression of active human C1 inhibitor serpin domain in Escherichia coli. Protein Expr Purif. 2001;22:349–358. doi: 10.1006/prep.2001.1445. [DOI] [PubMed] [Google Scholar]
- 398.Koles K., Van Berkel P.H., Pieper F.R., Nuijens J.H., Mannesse M.L., Vliegenthart J.F. N- and O-glycans of recombinant human C1 Inhibitor expressed in the milk of transgenic rabbits. Glycobiology. 2004;14:51–64. doi: 10.1093/glycob/cwh010. [DOI] [PubMed] [Google Scholar]
- 399.Hamilton S.R., Bobrowicz P., Bobrowicz B., Davidson R.C., Li H., Mitchell T. Production of complex human glycoproteins in yeast. Science. 2003;301:1244–1246. doi: 10.1126/science.1088166. [DOI] [PubMed] [Google Scholar]
- 400.Bos I.G., de Bruin E.C., Karuntu Y.A., Modderman P.W., Eldering E., Hack C.E. Recombinant human C1-inhibitor produced in Pichia pastoris has the same inhibitory capacity as plasma C1-inhibitor. Biochim Biophys Acta. 2003;1648:75–83. doi: 10.1016/s1570-9639(03)00107-9. [DOI] [PubMed] [Google Scholar]
- 401.Hack C.E., Bos I.G.A., de Bruin E.C., Karuntu Y.A., Modderman P.W., Eldering E. Recombinant human C1-inhibitor produced in Pichia pastoris has the same inhibitory capacity as plasma C1-inhibitor. Abstracts of the International C1-Esterase Deficiency Workshop. 2003;3:34–35. doi: 10.1016/s1570-9639(03)00107-9. [DOI] [PubMed] [Google Scholar]
- 402.Eldering E., Huijbregts C.C., Nuijens J.H., Verhoeven A.J., Hack C.E. Recombinant C1 inhibitor P5/P3 variants display resistance to catalytic inactivation by stimulated neutrophils. J Clin Invest. 1993;91:1035–1043. doi: 10.1172/JCI116260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Lauersen N.H., Wilson K.H. The effect of danazol in the treatment of chronic cystic mastitis. Obstet Gynecol. 1976;48:93–98. [PubMed] [Google Scholar]
- 404.Dmowski W.P. Danazol-induced pseudomenopause in the management of endometriosis. Clin Obstet Gynecol. 1988;31:829–839. doi: 10.1097/00003081-198812000-00009. [DOI] [PubMed] [Google Scholar]
- 405.Dmowski W.P., Headley S., Radwanska E. Effects of danazol on pulsatile gonadotropin patterns and on serum estradiol levels in normally cycling women. Fertil Steril. 1983;39:49–55. doi: 10.1016/s0015-0282(16)46757-9. [DOI] [PubMed] [Google Scholar]
- 406.Dawood M.Y., Lewis V., Ramos J. Cortical and trabecular bone mineral content in women with endometriosis: effect of gonadotropin-releasing hormone agonist and danazol. Fertil Steril. 1989;52:21–26. doi: 10.1016/s0015-0282(16)60782-3. [DOI] [PubMed] [Google Scholar]
- 407.Stevenson J.C., Lees B., Gardner R., Shaw R.W. A comparison of the skeletal effects of goserelin and danazol in premenopausal women with endometriosis. Horm Res. 1989;32(suppl 1):161–163. doi: 10.1159/000181335. discussion 164. [DOI] [PubMed] [Google Scholar]
- 408.Ylikorkala O., Nilsson C.G., Hirvonen E., Viinikka L. Evidence of similar increases in bone turnover during nafarelin and danazol use in women with endometriosis. Gynecol Endocrinol. 1990;4:251–260. doi: 10.3109/09513599009024979. [DOI] [PubMed] [Google Scholar]
- 409.Whitehouse R.W., Adams J.E., Bancroft K., Vaughan-Williams C.A., Elstein M. The effects of nafarelin and danazol on vertebral trabecular bone mass in patients with endometriosis. Clin Endocrinol (Oxf) 1990;33:365–373. doi: 10.1111/j.1365-2265.1990.tb00501.x. [DOI] [PubMed] [Google Scholar]
- 410.Rock J.A., Truglia J.A., Caplan R.J. Zoladex (goserelin acetate implant) in the treatment of endometriosis: a randomized comparison with danazol. The Zoladex Endometriosis Study Group. Obstet Gynecol. 1993;82:198–205. [PubMed] [Google Scholar]
- 411.Dodin S., Lemay A., Maheux R., Dumont M., Turcot-Lemay L. Bone mass in endometriosis patients treated with GnRH agonist implant or danazol. Obstet Gynecol. 1991;77:410–415. [PubMed] [Google Scholar]
- 412.Dmowski W.P., Rana N., Pepping P., Cain D.F., Clay T.H. Excretion of urinary N-telopeptides reflects changes in bone turnover during ovarian suppression and indicates individually variable estradiol threshold for bone loss. Fertil Steril. 1996;66:929–936. doi: 10.1016/s0015-0282(16)58685-3. [DOI] [PubMed] [Google Scholar]
- 413.Lipner H. Mechanism of mammalian ovulation. In: Knobil E., Neill J., editors. The physiology of reproduction. Raven Press; New York: 1988. pp. 447–486. [Google Scholar]
- 414.Perricone R., de Carolis C., Moretti C., Santuari E., de Sanctis G., Fontana L. Complement, complement activation and anaphylatoxins in human ovarian follicular fluid. Clin Exp Immunol. 1990;82:359–362. doi: 10.1111/j.1365-2249.1990.tb05453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Perricone R., Pasetto N., De Carolis C., Vaquero E., Noccioli G., Panerai A.E. Cystic ovaries in women affected with hereditary angioedema. Clin Exp Immunol. 1992;90:401–404. doi: 10.1111/j.1365-2249.1992.tb05858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Perricone R., Moretti C., De Carolis C., De Sanctis G., Gnessi L., Fabbri A. Increased plasma beta-endorphin levels in hereditary angioedema. Immunopharmacology. 1989;18:89–96. doi: 10.1016/0162-3109(89)90061-1. [DOI] [PubMed] [Google Scholar]
- 417.Wortsman J., Wehrenberg W.B., Gavin J.R., 3rd, Allen J.P. Elevated levels of plasma beta-endorphin and gamma 3-melanocyte stimulating hormone in the polycystic ovary syndrome. Obstet Gynecol. 1984;63:630–634. [PubMed] [Google Scholar]
- 418.Perricone R., De Carolis C., Giacomello F., Giacomelli R., De Sanctis G., Fontana L. Impaired human ovarian follicular fluid complement function in hereditary angioedema. Scand J Immunol. 2000;51:104–108. doi: 10.1046/j.1365-3083.2000.00652.x. [DOI] [PubMed] [Google Scholar]
- 419.Perricone R., Pasetto N., De Carolis C., Vaquero E., Piccione E., Baschieri L. Functionally active complement is present in human ovarian follicular fluid and can be activated by seminal plasma. Clin Exp Immunol. 1992;89:154–157. doi: 10.1111/j.1365-2249.1992.tb06895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Bork K., Kreuz W., Witzke G. [Hereditary angioneurotic edema. Clinical picture, diagnosis, patient management and drug therapy] Dtsch Med Wochenschr. 1984;109:1331–1335. doi: 10.1055/s-2008-1069374. [DOI] [PubMed] [Google Scholar]
- 421.Spaeth P.J. Inherited and acquired deficiency of C1 esterase inhibitor in humans. In: Rother K., Till G., Haensch G., editors. The complement system. 2nd rev ed. Springer Verlag; Berlin, Heidelberg: 1998. p. 366. [Google Scholar]
- 422.Goring H.D., Bork K., Spath P.J., Bauer R., Ziemer A., Hintner H. [Hereditary angioedema in the German-speaking region] Hautarzt. 1998;49:114–122. doi: 10.1007/s001050050710. [DOI] [PubMed] [Google Scholar]
- 423.De Filippi F., Castelli R., Cicardi M., Soffredini R., Rumi M.G., Silini E. Transmission of hepatitis G virus in patients with angioedema treated with steam-heated plasma concentrates of C1 inhibitor. Transfusion. 1998;38:307–311. doi: 10.1046/j.1537-2995.1998.38398222877.x. [DOI] [PubMed] [Google Scholar]
- 424.Purtell A., Curd J.G. Loss of danazol responsiveness in angioedema with C1 inhibitor deficiency. J Allergy Clin Immunol. 1983;71:127. [Google Scholar]
- 425.Mayer M.M. Complement and complement fixation. In: Kabat E.A., Mayer M.M., editors. Experimental immunochemistry. 2nd ed. Thomas; Springfield, Ill: 1962. p. 213. [Google Scholar]
- 426.Kohler P.F., Muller-Eberhard H.J. Immunochemical quantitation of the third, fourth and fifth components of human complement: concentrations in the serum of healthy adults. J Immunol. 1967;99:1211–1216. [PubMed] [Google Scholar]
- 427.Weinstock L.B., Kothari T., Sharma R.N., Rosenfeld S.I. Recurrent abdominal pain as the sole manifestation of hereditary angioedema in multiple family members. Gastroenterology. 1987;93:1116–1118. doi: 10.1016/0016-5085(87)90576-2. [DOI] [PubMed] [Google Scholar]
- 428.Farkas H., Harmat G., Gyeney L., Fust G., Varga L. Danazol therapy for hereditary angio-oedema in children. Lancet. 1999;354:1031–1032. doi: 10.1016/s0140-6736(05)76637-x. [DOI] [PubMed] [Google Scholar]
- 429.Frank M.M., Sergent J.S., Kane M.A., Alling D.W. Epsilon aminocaproic acid therapy of hereditary angioneurotic edema: a double-blind study. N Engl J Med. 1972;286:808–812. doi: 10.1056/NEJM197204132861503. [DOI] [PubMed] [Google Scholar]
- 430.Weinblatt M.E., Kochen J., Ortega J. Danazol for children with immune thrombocytopenic purpura. Am J Dis Child. 1988;142:1317–1319. doi: 10.1001/archpedi.1988.02150120071043. [DOI] [PubMed] [Google Scholar]
- 431.Lakshminrusimha S., Kumar S.R. Hereditary angio-oedema in children. Pediatr Asthma Allergy Immunol. 1996;10:155–159. [Google Scholar]
- 432.Maves K.K., Weiler J.M. Tonsillectomy in a patient with hereditary angioedema after prophylaxis with C1 inhibitor concentrate. Ann Allergy. 1994;73:435–438. [PubMed] [Google Scholar]
- 433.Raffai I., Fust G., Koo E., Hajos M. [Hereditary angioneurotic edema, analysis of 3 cases] Orv Hetil. 1972;113:2471–2474. [PubMed] [Google Scholar]
- 434.Bissler J.J., Cicardi M., Donaldson V.H., Gatenby P.A., Rosen F.S., Sheffer A.L. A cluster of mutations within a short triplet repeat in the C1 inhibitor gene. Proc Natl Acad Sci U S A. 1994;91:9622–9625. doi: 10.1073/pnas.91.20.9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Parad R.B., Kramer J., Strunk R.C., Rosen F.S., Davis A.E., 3rd Dysfunctional C1 inhibitor Ta: deletion of Lys-251 results in acquisition of an N-glycosylation site. Proc Natl Acad Sci U S A. 1990;87:6786–6790. doi: 10.1073/pnas.87.17.6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Siddique Z., McPhaden A.R., Whaley K. Characterisation of nucleotide sequence variants and disease-specific mutations involving the 3′ end of the C1-inhibitor gene in hereditary angio-oedema. Hum Hered. 1995;45:98–102. doi: 10.1159/000154267. [DOI] [PubMed] [Google Scholar]
- 437.Siddique Z., McPhaden A.R., Lappin D.F., Whaley K. An RNA splice site mutation in the C1-inhibitor gene causes type I hereditary angio-oedema. Hum Genet. 1991;88:231–232. doi: 10.1007/BF00206079. [DOI] [PubMed] [Google Scholar]
- 438.Ono H., Kawaguchi H., Ishii N., Nakajima H. A point mutation in exon 7 of the C1-inhibitor gene causing type I hereditary angioedema. Hum Genet. 1996;98:452–453. doi: 10.1007/s004390050238. [DOI] [PubMed] [Google Scholar]
- 439.Kawachi Y., Hibi T., Yamazaki S., Otsuka F. A novel donor splice site mutation in the C1 inhibitor gene of a patient with type I hereditary angioneurotic edema. J Invest Dermatol. 1998;110:837–839. doi: 10.1046/j.1523-1747.1998.00170.x. [DOI] [PubMed] [Google Scholar]
- 440.Frangi D., Cicardi M., Sica A., Colotta F., Agostoni A., Davis A.E., 3rd Nonsense mutations affect C1 inhibitor messenger RNA levels in patients with type I hereditary angioneurotic edema. J Clin Invest. 1991;88:755–759. doi: 10.1172/JCI115373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Zhi Y.X., Zhang H.Y., Huang S.Z. [Identification of a novel mutation of C1 inhibitor gene in a Chinese family with hereditary angioedema] Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2003;25:664–666. [PubMed] [Google Scholar]
- 442.Siddique Z., McPhaden A.R., McCluskey D., Whaley K. A single base deletion from the C1-inhibitor gene causes type I hereditary angio-oedema. Hum Hered. 1992;42:231–234. doi: 10.1159/000154075. [DOI] [PubMed] [Google Scholar]
- 443.Siddique Z., McPhaden A.R., Whaley K. C1-inhibitor gene nucleotide insertion causes type II hereditary angio-oedema. Hum Genet. 1993;92:189–190. doi: 10.1007/BF00219690. [DOI] [PubMed] [Google Scholar]
- 444.Skriver K., Wikoff W.R., Patston P.A., Tausk F., Schapira M., Kaplan A.P. Substrate properties of C1 inhibitor Ma (alanine 434-glutamic acid): genetic and structural evidence suggesting that the P12-region contains critical determinants of serine protease inhibitor/substrate status. J Biol Chem. 1991;266:9216–9221. [PubMed] [Google Scholar]
- 445.Siddique Z., McPhaden A., Whaley K. Identification of type I hereditary angio-oedema (HAE) mutations. Clin Exp Immunol. 1991;85S:11. [abstract] [Google Scholar]
- 446.Levy N.J., Ramesh N., Cicardi M., Harrison R.A., Davis A.E., 3rd Type II hereditary angioneurotic edema that may result from a single nucleotide change in the codon for alanine-436 in the C1 inhibitor gene. Proc Natl Acad Sci U S A. 1990;87:265–268. doi: 10.1073/pnas.87.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Frangi D., Aulak K.S., Cicardi M., Harrison R.A., Davis A.E., 3rd A dysfunctional C1 inhibitor protein with a new reactive center mutation (Arg-444-Leu) FEBS Lett. 1992;301:34–36. doi: 10.1016/0014-5793(92)80204-t. [DOI] [PubMed] [Google Scholar]
- 448.Siddique Z., McPhaden A.R., Whaley K. Type II hereditary angio-oedema associated with two mutations in one allele of the C1-inhibitor gene around the reactive-site coding region. Hum Hered. 1992;42:298–301. doi: 10.1159/000154086. [DOI] [PubMed] [Google Scholar]
- 449.Donaldson V.H., Bissler J.J. C1-inhibitors and their genes: an update. J Lab Clin Med. 1992;119:330–333. [PubMed] [Google Scholar]
- 450.Nielsen E.W., Fure H., Winge P., Mollnes T.E. Identification of a C→T mutation in the reactive-site coding region of the C1-inhibitor gene and its detection by an improved mutation-specific polymerase chain reaction method. Scand J Immunol. 1998;47:273–276. doi: 10.1046/j.1365-3083.1998.00290.x. [DOI] [PubMed] [Google Scholar]
- 451.Aulak K.S., Cicardi M., Harrison R.A. Identification of a new P1 residue mutation (444Arg-Ser) in a dysfunctional C1 inhibitor protein contained in a type II hereditary angioedema plasma. FEBS Lett. 1990;266:13–16. doi: 10.1016/0014-5793(90)81494-9. [DOI] [PubMed] [Google Scholar]
- 452.Ocejo-Vinyals J.G., Leyva-Cobian F., Fernandez-Luna J.L. A mutation unique in serine protease inhibitors (serpins) identified in a family with type II hereditary angioneurotic edema. Mol Med. 1995;1:700–705. [PMC free article] [PubMed] [Google Scholar]
- 453.Ariga T., Hoshioka A., Kohno Y., Sakamaki T., Matsumoto S. A de novo deletion in the C1 inhibitor gene in a case of sporadic hereditary angioneurotic edema. Clin Immunol Immunopathol. 1993;69:103–105. doi: 10.1006/clin.1993.1156. [DOI] [PubMed] [Google Scholar]
- 454.Siddique Z., McPhaden A.R., Fothergill J.E., Whaley K. A point mutation in the C1-inhibitor gene causes type I hereditary angiooedema. Hum Hered. 1993;43:155–158. doi: 10.1159/000154171. [DOI] [PubMed] [Google Scholar]
- 455.Stoppa-Lyonnet D., Carter P.E., Meo T., Tosi M. Clusters of intragenic Alu repeats predispose the human C1 inhibitor locus to deleterious rearrangements. Proc Natl Acad Sci U S A. 1990;87:1551–1555. doi: 10.1073/pnas.87.4.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Bos I.G., Hack C.E., Abrahams J.P. Structural and functional aspects of C1-inhibitor. Immunobiology. 2002;205:518–533. doi: 10.1078/0171-2985-00151. [DOI] [PubMed] [Google Scholar]