

Abstract
Viruses are associated with up to 15% of human cancer. MicroRNAs (miRNAs) encoded by numerous oncogenic viruses including Kaposi’s sarcoma-associated herpesvirus (KSHV) play significant roles in regulating the proliferation and survival of virus-induced cancer cells, hence representing attractive therapeutic targets. Here, we report that specific inhibition of viral miRNAs by carbon dots (Cdots)-mediated delivery of locked nucleic acid (LNA)-based suppressors inhibit the proliferation of KSHV-associated primary effusion lymphoma (PEL) cells. Specifically, a combination of Cdots-LNAs to knock down the levels of KSHV miR-K12-1, miR-K12-4, and miR-K12-11 induces apoptosis and inhibits proliferation of PEL cells. Significantly, these Cdots-LNAs effectively inhibit the initiation of PEL and regress established PEL in a xenograft mouse model. These results demonstrate the feasibility of using Cdots to deliver miRNA suppressors for targeting viral cancers. Our study with viral miRNAs as targets may provide the scientific basis for using antisense drugs for human cancers associated with oncogenic viruses.
Keywords: cancer therapy, viral microRNA, antisense drugs, human oncogenic virus, KSHV, delivery system
Graphical Abstract

Infections by viruses are associated with up to 15% of human cancer.1 The special sequences of viral genomes and the distinct mechanisms of oncogenesis of viruses offer attractive targets for therapy. Numerous oncogenic viruses including Kaposi’s sarcoma-associated herpesvirus (KSHV) and Epstein–Barr virus (EBV) encode microRNAs (miRNAs), some of which are critical for viral infection and the development of cancers induced by these viruses.2,3 Therefore, targeting viral miRNAs represents a promising therapeutic approach.
Numerous methods have been developed to inhibit the functions of miRNAs including peptide nucleic acids,4 cholesterol-conjugated “antagomirs”,5,6 and locked nucleic acid (LNA) oligonucleotides.7,8 Most of these approaches are based on antisense technology, which could potentially be used in therapies of viral cancers.
Antisense gene therapy has evolved to become an attractive therapeutic approach for various diseases including cancer, renal diseases, and cardiovascular diseases.9,10 Antisense drugs can alter the levels of their target RNAs, leading to reduction, restoration, or modification of protein expression. By directly targeting the cause of the pathogenesis, antisense gene therapy has a higher chance of success than therapies targeting downstream pathways.9,11 Over the last two decades, numerous antisense drugs have entered clinical trials for treating a variety of diseases, and five have been approved by the U.S. Food and Drug Administration, including Fomivirsen (1998), Mipomersen (2013), Eteplirsen (2016), Nusinersen (2016), and most recently Tegsedi (2018).12 The features of antisense drugs, including high specificity, ability to directly act on targets that are inaccessible by traditional therapies, and reduced toxicity, render them ideal candidates for therapeutic application.9 However, a key challenge for antisense-based gene therapy and drug development is the delivery of an antisense oligonucleotide to the desired disease site.
Although nucleic acid delivery systems such as viral vectors,13 lipoplex formulations,14–18 cationic transfection agents,19 and inorganic nanoparticles including gold nanoparticles20–23 have been developed, numerous drawbacks exist, including toxicity, instability, and immunogenicity. Besides, these systems usually require chemically modified nucleic acids or conjugation of the nucleic acids on the surface of nanoparticles and further purification processes, posing limitations, which includes multiple processes, a high cost, and time- and labor-consuming nature. Therefore, the development of biocompatible, efficient, easy-to-prepare delivery systems is still an urgent issue for practical and clinical applications of antisense therapy. As a new class of carbon-based nanomaterials, carbon dots (Cdots) have high biological compatibility, well-defined structures, and tunable surface functionalities.24–26 Recently, Cdots have been attracting enormous attention due to the simple and cost-effective synthetic method, and these excellent properties make it a promising system for delivery of oligonucleotides.27,28
Primary effusion lymphoma (PEL) is one of the four malignancies caused by infection of KSHV.29 PEL is an aggressive non-Hodgkin B-cell lymphoma accounting for 4% of AIDS-related lymphomas.30 PEL patients have a poor prognosis with a median survival time of 6.2 months. There is currently no efficient and specific therapy for PEL.
Herein, we report that LNA-based oligonucleotides can be used to cause specific knockdown of KSHV-encoded miR-K12-1, miR-K12-4, and miR-K12-11 (shortened as miR-K1, miR-K4, and miR-K11), leading to induction of apoptosis and suppression of proliferation of PEL cells without cytotoxicity to KSHV-negative B cells (Figure 1). Using Cdots-mediated delivery, these miRNA suppressors effectively inhibit the initiation of PEL and regress established lymphomas in a mouse model of PEL. Compared to the standard R-CHOP therapy (cyclophosphamide, hydroxydaunorubicin, oncovin, and prednisone in combination with chimeric anti-CD20 monoclonal antibody rituximab),31 these miRNA suppressors have higher specificity and efficiency. Our results demonstrate that Cdots-mediated delivery of miRNA suppressors represents an attractive approach for cost-effective therapy of viral cancers.
Figure 1.

Schematic illustration of the strategy for targeting KSHV-encoded miRNAs by Cdots-mediated delivery of locked nucleic acid (LNA) for treating KSHV-induced cancers. LNA-based suppressors of miR-K1, -K4, and -K11 are loaded onto Cdots and delivered into KSHV-infected cells. The released LNAs specifically target their respective KSHV miRNAs by RNase H-mediated degradation. The suppression of KSHV miRNAs induces apoptosis and inhibits cell proliferation.
RESULTS AND DISCUSSION
Efficient Formation of Cdots/LNA Complexes.
The locked nucleic acids were designed complementary to the KSHV miRNAs, which can form highly specific and stable duplexes.32–36 One of the barriers for nucleic acid-based therapy is that the naked nucleic acid can hardly cross the phospholipid membranes of cells. To resolve this problem, we employed Cdots to deliver LNA. As a new class of carbon-based nanomaterials, Cdots have received significant attention due to their low cytotoxicity, high chemical stability, and high drug loading capacity.37,38 Cdots were successfully synthesized by a microwave-assisted pyrolysis method and characterized by transmission electron microscope and X-ray photoelectron spectroscopy (Figure S1). To determine the LNA loading capacity of Cdots, various concentrations of Cdots and a fixed amount of LNA were mixed (Figure 2A), or various concentrations of LNA and a fixed amount of Cdots were mixed (Figure S2), and the formation of Cdots/LNA complexes was examined by agarose gel electrophoresis. We found that the minimal weight of Cdots for efficient loading of 100 pmol of LNA was at least 0.3 μg (Figure 2A). The formation of Cdots/LNA complexes was further confirmed by the increased sizes of Cdots following loading of LNA (Figure 2B and Figure S1, mean 3.7 nm vs 35.0 nm). Importantly, the Cdots/LNA complexes remained stable and relatively uniform in the phosphate-buffered saline (PBS) containing 10% fetal bovine serum (FBS) over time (Figure S3). To understand how the complexes formed between Cdots and LNA, the zeta-potential measuring the surface charge of the complexes was performed (Figure 2C). LNA had negative charges, while Cdots had positive charges. Upon loading of LNA, the Cdots/LNA complexes showed a decrease of surface charge compared to Cdots, indicating that the complexes were formed by electrostatic interaction between the positive charges on the surface of Cdots and the negative charges on the phosphate backbone of LNA. Taken together, LNA can be loaded onto Cdots through electrostatic interaction, forming Cdots/LNA complexes.
Figure 2.
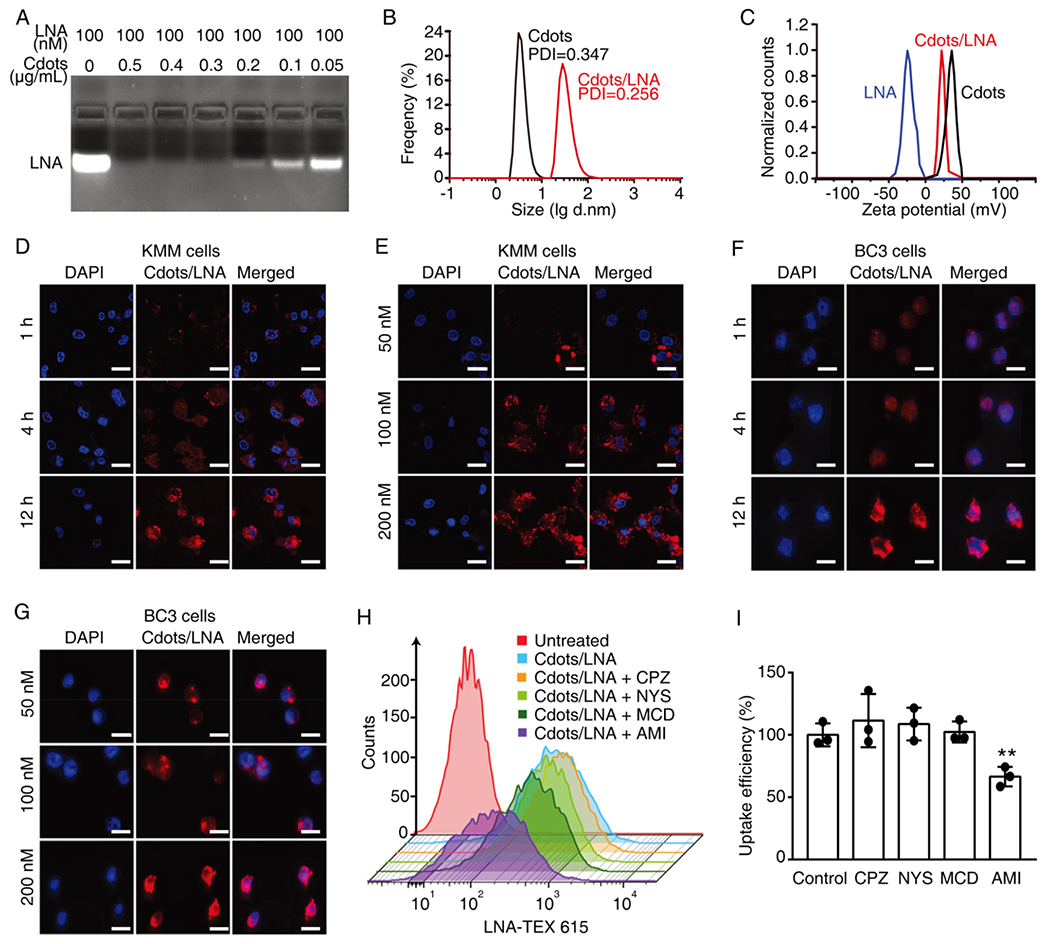
Efficient uptake of Cdots/LNA complexes by both adherent and suspension KSHV-infected cells. (A) Determination of the amount of Cdots required for loading 100 nM LNA analyzed by agarose gel electrophoresis. (B) Determination of the sizes of Cdots and Cdots/LNA complexes containing 1 μg/mL Cdots and 100 nM LNA by dynamic light scattering analysis (DLS). (C) Analysis of zeta-potentials of LNA, Cdots, and Cdots/LNA complexes. (D–G) Uptake of Cdots/LNA by KSHV-infected adherent KMM cells (D and E) and suspension BC3 cells (F and G) treated with Cdots/LNA for different times (D and F) and concentrations (D and G) analyzed by confocal laser scanning microscopy. LNA was conjugated with TEX615 dye, while the nuclei were stained with DAPI. Scale bars represent 20 μm. (H, I) Uptake efficiencies of Cdots/LNA by BC3 cells in the presence of different inhibitors including chlorpromazine hydrochloride (CPZ), nystatin (NYS), methyl-β-cyclodextrin (MCD), and amiloride (AMI) analyzed by flow cytometry (H) and the statistical analyses (I). LNA was conjugated with TEX615. Bars represent mean ± SD (n = 3). Statistical significance was calculated by one-way ANOVA with Tukey’s post hoc test. *P < 0.05; **P < 0.01; ***P < 0.001.
Delivery of Cdots/LNA Complexes into KSHV-Infected Cells.
We next investigated the cellular uptake efficiency of LNA delivered by Cdots in adherent KSHV-transformed primary rat embryonic metanephric mesenchymal cells (KMM).39 LNA conjugated to TEX615 at the 5′ end was employed to track its cellular location. Confocal images showed that Cdots/LNA efficiently entered into KMM cells and remained in the cytoplasm in a time- and dose-dependent fashion (Figure 2D,E and Figure S4). In contrast, LNA itself without the Cdots-mediated delivery was not able to enter into cells (Figure S5). Besides, the fluorescence of Cdots and LNA was not fully colocalized, which indicated that some of the LNA had been released from Cdots/LNA (Figure S6). The efficient cellular uptake of Cdots/LNA complexes by cells could be attributed to the favorable interaction between the negatively charged cell surface and the positively charged Cdots/LNA complexes. Moreover, the cationic polymer on the surface of Cdots might help induce endosomal escape by the proton sponge effect.40
We then examined the cellular uptake of Cdots/LNA complexes by suspension KSHV-infected PEL cells, which are usually refractory to transfection. We observed red fluorescence signal in BC3 cells, following incubation with Cdots/LNA for 1 h, and a time- and dose-dependent uptake of LNA by BC3 cells (Figure 2F,G).
To elucidate the pathway of cellular uptake of Cdots/LNA complexes by cells, we used inhibitors of the endocytic pathways to pretreat the cells and then measured the relative cellular uptake of Cdots/LNA. These included chlorpromazine hydrochloride (CPZ), which inhibits clathrin-dependent endocytosis; nystatin (NYS), which inhibits caveolin-dependent endocytosis; methyl-β-cyclodextrin (MCD), which inhibits cholesterol-dependent membrane fusion; and amiloride (AMI), which inhibits macropinocytosis. Flow cytometry analysis revealed that AMI caused 40% reduction of Cdots/LNA uptake by cells, while other inhibitors, including CPZ, NYS, and MCD, had no effect (Figure 2H,I and Figure S7), indicating that the main cell internalization pathway of Cdots/LNA complexes was likely through macropinocytosis. Additionally, there was no cytotoxicity of Cdots on 293T, KMM, BCBL1, and BJAB cells (Figure S8). Taken together, the above results indicated that LNA could be efficiently delivered by Cdots into both adherent and suspension cells.
Suppression of KSHV MiRNAs by Specific Cdots/LNA.
We then evaluated the knockdown efficiencies of miRNAs following Cdots-mediated delivery of LNA-based miRNA suppressors. We previously showed that a mutant KSHV containing a deletion of a cluster of precursor miRNAs failed to transform primary cells; however, genetic complementation with miR-K1, -K4, or -K11 was sufficient to restore transforming phenotype, indicating that, among the 12 pre-miRNAs encoded by KSHV, miR-K1, -K4, and -K11 are strongly associated with cellular transformation and tumor induction.39 Hence, these viral miRNAs are attractive therapeutic targets of KSHV-induced cancers. We designed three LNAs complementary to the KSHV miR-K1, -K4, and -K11, respectively. Upon binding to their specific miRNAs, LNAs target miRNAs for RNase H-mediated degradation.11 We examined different combinations of LNA and Cdots concentrations for each miRNA suppressor for the knockdown efficiencies and chose a combination of 100 nM LNA and 1.0 μg/mL of Cdots, which gave the optimal knockdown efficiencies for all the suppressors (Figure S9–S11). Under this condition, LNA-K1, -K4, or -K11 delivered by Cdots either alone (designated as Cdots/LNA-K1, -K4, or -K11) or in combination (designated as Cdots/LNA-K1+K4+K11 or Cdots/LNAs) effectively decreased KSHV miR-K1, -K4, or -K11 level by at least 98%, 90%, and 65%, respectively, compared to the untreated and LNA negative control (LNA-NC) in adherent KMM cells and suspension PEL cells BC3, BCP1, and BCBL1 (Figure 3A). Without Cdots-mediated delivery, these LNAs used at the same concentrations had no effect on the levels of KSHV miRNAs (Figure S12). Moreover, the suppressors were specific to their respective miRNAs without interfering with each other (Figure 3A) or other KSHV miRNA (Figure S13). It was worth noting that the individual suppressors had variable efficiencies against their respective targets (Figure 3A). It has been reported that the design, quality, and delivery are the most important elements that could affect the knockdown efficiency of a miRNA suppressor.17 The observed different knockdown efficiencies of these three LNAs in our study were likely due to the design and quality rather than the delivery system. We further confirmed that the suppressors functioned through binding to the corresponding miRNAs. We used the miR-K1, miR-K4, and miR-K11 sensor reporters containing tandem repeats of the respective miRNA binding sites cloned into the pGL3 vector downstream of the luciferase gene.35 Using a dual-luciferase reporter system, we found that LNA-K1, -K4, and -K11 delivered by Cdots efficiently increased the reporter activity, indicating the inhibitory effects of miR-K1, -K4, and -K11 on their sensor reporters were relieved by these suppressors (Figure S14). Examination of known miR-K1 target IκBα,34 miR-K4 target CASTOR1,32 and miR-K11 target BACH141 showed that the suppressors greatly increased the expression of the respective proteins (Figure S15). Together, these results indicated that these miRNA suppressors were fully functional following Cdots-mediated delivery into cells.
Figure 3.
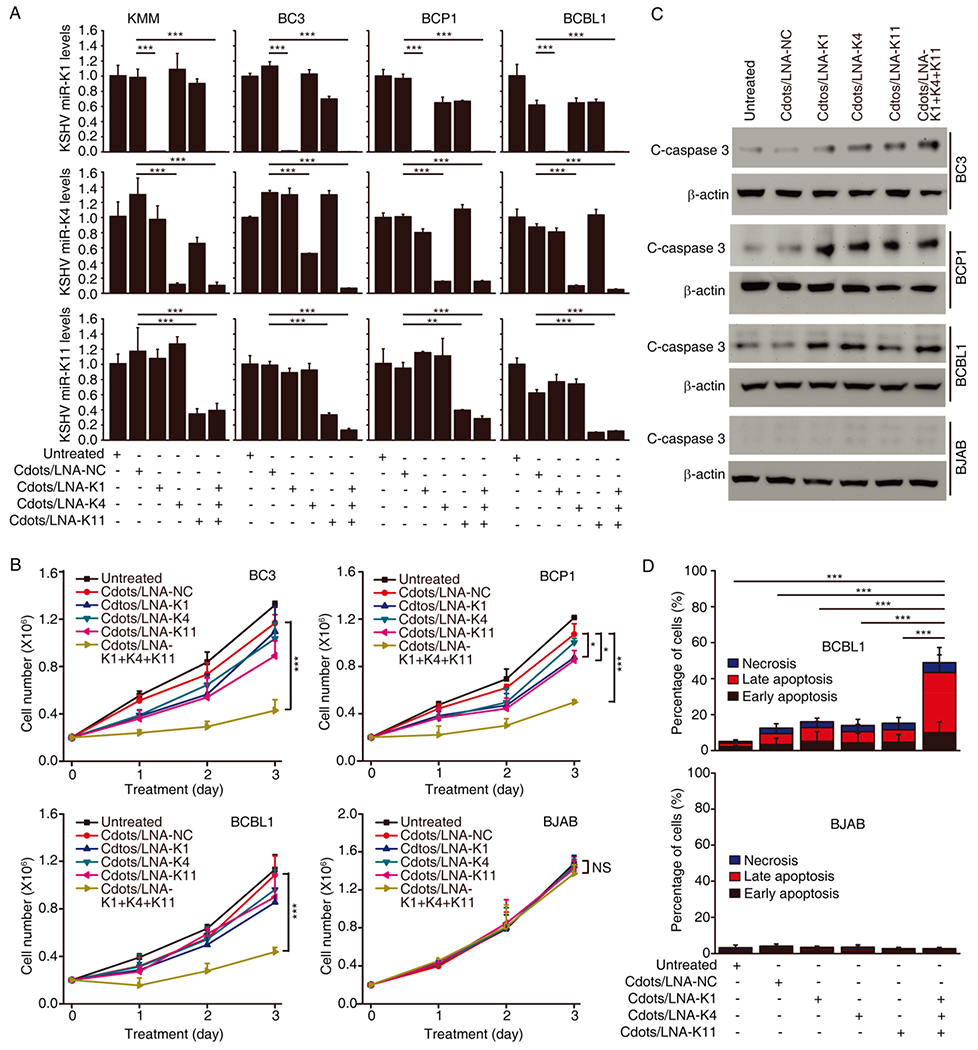
Specific suppression of KSHV miRNAs by Cdots-mediated delivery of LNA-based suppressors inhibits proliferation and induces apoptosis of PEL cells. (A) Relative levels of KSHV miR-K1, -K4, or -K11 measured by quantitative real-time reverse transcription PCR (RT-qPCR) after treatment with 100 nM of Cdots/LNA-K1, -K4, or -K11, the combination of three suppressors, or Cdots/LNA-NC for 3 days in KMM, BC3, BCP1, and BCBL1 cells. (B) Cell proliferation of KSHV-positive BC3, BCP1, and BCBL1 cells and KSHV-negative BJAB cells after treatment with 100 nM of Cdots/LNA-K1, -K4, or -K11, the combination of three suppressors, or Cdots/LNA-NC. (C, D) Cleaved caspase 3 analyzed by Western blotting (C) and apoptosis analyzed by flow cytometry (D) after treatment with 100 nM of Cdots/LNA-K1, -K4, or -K11, the combination of three suppressors, or Cdots/LNA-NC for 3 days. Bars represent mean ± SD (n = 3). Statistical significance was calculated by one-way ANOVA with Tukey’s post hoc test. NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Cdots/LNAs-Mediated Suppression of KSHV miR-K1, -K4, and -K11 Inhibits Cell Proliferation and Induces Apoptosis.
We next investigated the effect of Cdots-mediated suppression of KSHV miR-K1, -K4, and -K11 on the proliferation of KSHV-positive PEL cell lines BC3, BCP1, and BCBL1 and a KSHV-negative Burkitt lymphoma cell line BJAB. Inhibition of individual miRNA only weakly reduced the proliferation of PEL cell lines (Figure 3B), which was likely due to the high functional redundancies of these miRNAs.36 However, simultaneous Cdots-mediated delivery of LNA-K1, -K4, and -K11 significantly decreased the proliferation of all three PEL lines. When BCBL1 cells were treated with Cdots-mediated delivery of individual LNA-K1, LNA-K4, or LNA-K11 as well as their combination at a concentration of 100 or 300 nM, respectively, there was not much difference between the groups treated with individual LNAs at either 100 or 300 nM (Figure S16). However, the combination of the three LNAs at 100 nM showed a significant decrease in cell proliferation compared to the group treated with individual LNAs at 300 nM. The results indicated that the simultaneous delivery of three different LNAs exhibited synergistic suppression effects on tumor cell proliferation. Furthermore, none of the LNAs had any effect on the proliferation of BJAB cells, demonstrating the specificities of the suppressors to KSHV miRNAs. Consistent with these results, individual suppressors and a combination of all three suppressors induced cleaved caspase 3 (C-caspase 3) (Figure 3C) and apoptosis (Figure 3D) in PEL cells, suggesting that the reduced cell proliferation was likely due to the induction of apoptosis. In contrast, no change of cleaved caspase 3 level or apoptosis was observed in BJAB cells, which was in accordance with the results of cell proliferation. These results indicated that simultaneous Cdots-mediated delivery of LNA-K1, -K4, and -K11 inhibited the proliferation of PEL cells by inducing apoptosis but had no effect on KSHV-negative cells.
Cdots-Mediated Delivery of LNA to PEL in Vivo.
We further examined the therapeutic effects of Cdots/LNAs on PEL in vivo using an intraperitoneal (i.p.) xenograft model of PEL.42,43 BCBL1 cells were engineered to stably express a cassette of luciferase designated as BCBL1-Luc.43 We detected the bioluminescence signal in BCBLl-Luc cells in vitro with the number of cells linearly correlated with the bioluminescence signal (Figure S17, R2 = 0.9930), hence allowing the monitoring of PEL growth in vivo by bioluminescence imaging. We confirmed that Cdots-mediated delivery of LNA-K1, -K4, and -K11 could effectively cause knockdown of KSHV miR-K1, -K4, and -K11, respectively, in BCBLl-Luc cells (Figure S18). TEX615-conjugated LNA fluorescence signal was detected in vitro, and the concentration of LNA was linearly correlated with fluorescence signal (Figure S19, R2 = 0.9983), allowing in vivo monitoring of the delivery efficiency and biodistribution of the miRNA suppressors. Upon i.p. injection of the Cdots/LNAs into mice, the area and intensity of the fluorescence reached a maximum level at 4 h postinjection and then decreased over a period of 24 h (Figure 4A). We then injected Cdots/LNA into mice with established PEL and observed strong colocalization of fluorescence signals with bioluminescence signals in the abdominal cavity, suggesting efficient uptake of Cdots/LNAs by tumor cells (Figure 4B). In contrast, no fluorescence signal was detected in PEL in mice without treating with Cdots/LNA (Figure 4B). Furthermore, we did not observe any acute toxicity or bodyweight loss in the animals after two months of Cdots/LNA delivery. These results indicated that Cdots/LNA could be efficiently delivered to PEL in vivo.
Figure 4.

In vivo biodistribution of LNAs delivered by Cdots in mice with and without PEL. (A) Biodistribution of Cdots/LNAs in Nod/Scid mice at different time points following injection monitored by fluorescence imaging of the TEX615 dye. (B) Biodistribution of Cdots/LNAs 4 h after the injection in Nod/Scid mice intraperitoneally engrafted with BCBL1-Luc cells.
Inhibition of the Initiation and Growth of PEL Tumor by Cdots/LNAs-Mediated Suppression of KSHV MiRNAs.
To determine whether suppression of KSHV miR-K1, -K4, and -K11 could inhibit the initiation and growth of PEL, we daily injected Cdots/LNAs into mice 3 days after inoculation of BCBL1-Luc cells and performed bioluminescence imaging and measurement of abdomen size at day 21 postengraftment (Figure 5A). We detected bioluminescence signal in 4 of 9 mice treated with Cdots/LNAs compared to 10 of 10 mice treated with PBS and 8 of 10 mice treated with Cdots/LNA-NC, respectively (Figure 5B, Cdots/LNAs vs PBS, 44.4% vs 100%, P = 0.003; and Cdots/LNAs vs Cdots/LNA-LC, 44.4% vs 80%, P = 0.05), indicating effective inhibition of PEL initiation by miRNA suppressors. Additionally, mice treated with Cdots/LNAs had significantly lower levels of luminescence compared to those treated with PBS or Cdots/LNA-NC (Figure 5C). Furthermore, mice treated with PBS or Cdots/LNA-NC had higher body weights as a result of PEL development than those treated with Cdots/LNAs starting at day 12 postinoculation (Figure 5D), which was in accordance with the results of bioluminescence signals. Finally, mice treated with PBS or Cdots/LNA-NC had more severe abdominal distention caused by developed lymphomatous ascites than those treated with Cdots/LNAs (Figure 5E). Collectively, these results indicated that Cdots-mediated combined delivery of LNA-K1, -K4, and -K11 effectively inhibited the initiation and growth of PEL.
Figure 5.

Cdots/LNAs-mediated suppression of KSHV miRNAs inhibits the initiation and progression of PEL. (A) Time line of the experiment. (B, C) In vivo luminescence images of mice with the indicated treatments at day 21 postinoculation of PEL cells (B) and the quantified luminescent signals (C). (D) Weights of different groups of mice recorded twice a week for 3 weeks. (E) Photographs of different groups of mice taken at day 21. Bars represent mean ± SD (n = 10). Statistical significance was calculated by one-way ANOVA with Tukey’s post hoc test. *P < 0.05; **P < 0.01; ***P < 0.001.
Regression of Established PEL Tumors by Cdots/LNAs-Mediated Suppression of KSHV MiRNAs.
We further determined whether Cdots/LNAs-mediated suppression of KSHV miRNAs could regress established PEL tumors. We first inoculated mice with BCBL1-Luc cells for 3 weeks, at which time the mice had well-established PEL as confirmed by bioluminescence imaging. We then randomized the mice into three groups, followed by daily i.p. injection of PBS, Cdots/LNA-NC, and Cdots/LNAs, respectively, for 3 weeks. Bioluminescence imaging was then performed to monitor the PEL tumors (Figure 6A). Compared with mice treated with PBS or Cdots/LNA-NC, mice treated with Cdots/LNAs had a dramatically reduced luminescence signal (Figure 6B,C). In fact, in every animal treated with Cdots/LNAs, the luminescence signal was weaker after than before the treatment, indicating effective regression of the established lymphomas by the miRNA suppressors. In contrast, in every animal treated with PBS or Cdots/LNA-NC, the luminescence signal was stronger after than before the treatment. Consistent with these results, mice treated with Cdots/LNAs had significant loss of ascites and body weights, confirming the regression of the lymphomas (Figure 6D,E). Furthermore, mice treated with Cdots/LNAs had a significant extended average life span compared to those treated with PBS or Cdots/LNA-NC (Figure 6F). To confirm that the PEL was derived from the inoculated cells, we collected the ascites after the treatment and examined the expression of KSHV latent genes. We detected the expression of LANA, vFLIP, and vCyclin genes in ascites collected from all the mice, thus confirming that the pleural effusion mostly contained PEL cells rather than the mouse cells (Figure S20). Examination of levels of KSHV miR-K1, -K4, and -K11 showed that all three miRNAs were significantly decreased in ascites of mice treated with Cdots/LNAs compared with those from mice treated with PBS or Cdots/LNA-NC, indicating that the miRNA suppressors were indeed delivered to the tumor cells and were fully functional (Figure 6G–I). Of note, we observed solid tumors in numerous organs including liver, kidney, and gastric area in mice treated with PBS or Cdots/LNA-NC (Figure S21) but not in those treated with Cdots/LNAs, suggesting that the miRNAs suppressors also effectively inhibited the invasion of tumor cells and formation of solid tumors. Taken together, Cdots-mediated suppression of KSHV miR-K1, -K4, and -K11 was highly efficient and specific in both preventing the initiation and growth of PEL and regressing established PEL in vivo.
Figure 6.

Cdots/LNAs-mediated suppression of KSHV miRNAs regresses established PEL. (A) Time line of the experiment. (B, C) In vivo luminescence images of mice before and after the indicated treatments for 7 days (B) and quantification of luminescent signals (C). (D) Total volumes of ascites extracted and recorded. (E) Average weights over time in the indicated treatment groups. (F) Survival curves of mice with the indicated treatment. (G–I) Levels of KSHV miR-K1 (G), -K4 (H), and -K11 (I) quantified by RT-qPCR in cells extracted from the ascites. Bars represent mean ± SD (n = 4). Statistical significance was calculated by one-way ANOVA with Tukey’s post hoc test. *P < 0.05; **P < 0.01; ***P < 0.001.
As an aggressive subset of diffuse large B-cell lymphoma (DLBCL), PEL often occurs as a liquid effusion in pleural, pericardial, and peritoneal cavities with some developing solid tissue masses.30,44 Current treatment for DLBCL is chemotherapy with R-CHOP.30,31 Unfortunately, this regimen often causes drug resistance in PEL with a median survival time of 6.2 months. Hematopoietic stem cell transplant (SCT), though, is an option for chemoresistant patients and is a highly specialized, costly, and resource-intensive procedure.45 Since R-CHOP was approved more than 15 years ago, there has been no front-line therapy or meaningful improvement in the survival rate.30,31
One important characteristic of PEL is the association with KSHV and dependence on KSHV for cell survival. The seven human cancer viruses are responsible for close to 15% of human cancers.1 While these cancer viruses have different molecular characteristics, they share a similar mechanism for inducing tumorigenesis, which is subversion of the host machinery, resulting in dysregulation of cell differentiation and proliferation, apoptosis, genome stability, and recognition by the immune system.1 Considering the distinct genetic compositions, the viral genomes in whole or in part or their products have been regarded as ideal therapeutic targets for combating viral cancers.
Viral miRNAs regulate critical pathways of host cell proliferation and survival.2,3 Antisense oligonucleotides are the most direct way to target these miRNAs. The recent developments of modifications of oligonucleotides and elucidation of the mechanism of action have greatly advanced the antisense-based approaches for therapies.18,19 The approvals of the antisense oligonucleotides for the treatment of spinal muscular atrophy and Duchenne muscular dystrophy represent landmarks in this regard. Therefore, exploiting the delivery of specific miRNA inhibitors targeting critical viral miRNAs could be a promising therapeutic approach.
In this study, we have explored antisense oligonucleotides to specifically suppress KSHV miRNAs as a therapeutic strategy for PEL. Previous works have shown that KSHV miRNAs play important roles in KSHV-transformed cells.31 In particular, KSHV miR-K1, -K4, and -K11 regulate the host proliferation and survival pathways. miR-K1 activates the NF-κB pathway and promotes cell cycle progression by targeting IκBα and p21, respectively.34,46 miR-K11 is a functional ortholog of miR-155, which is a human oncogenic miRNA directly targeting SH2-containing inositol phosphatase, a negative regulator of cell proliferation and survival.41,47 More recently, miR-K4 has been shown to target arginine sensor CASTOR1 to activate the mTORC1 pathway.32 Due to its high affinity, high specificity, and high nuclease stability, we chose LNA oligonucleotides to inhibit the functions of these KSHV miRNAs. Our results have shown that combined targeting of KSHV miR-K1, -K4, and -K11 is sufficient to induce apoptosis and inhibit the proliferation of PEL cells.
A key challenge of antisense therapy is the effective delivery of antisense oligonucleotides to the desired disease site. Although several delivery systems including viral vectors, lipoplex formulations, cationic transfection agents, and nano-particles-based delivery have been developed, drawbacks have been identified, including toxicity, immunogenicity, and instability as well as the high cost and the time- and labor-consuming nature.13–23 As a new class of carbon-based nanomaterials, carbon dots have low cytotoxicity, high chemical stability, and special optical properties. In this study, we synthesized polyethylenimine-coated Cdots as the carriers for LNA delivery. We showed the high capacity and the fast and simple way of loading of Cdots for LNA through electrostatic interactions. The Cdots/LNA complexes had a positively charged surface and could be efficiently taken up by both adherent and suspension cells (Figure 2). Furthermore, Cdots-mediated delivery of LNA-K1, -K4, and -K11 could effectively and specifically suppress the functions of their respective miRNAs and inhibit the proliferation of PEL cells (Figure 3). Importantly, we found these suppressors had no side effects on KSHV-negative cells and no effect on other KSHV miRNAs. Finally, we showed combined delivery of LNA-K1, -K4, and -K11 by Cdots effectively inhibited the initiation and progression of PEL and regressed established PEL in a xenograft mouse model (Figures 4–6). Since cancer viruses reside inside the virus-induced cancer cells but not the normal cells, targeting viral miRNAs is an ideal therapy for combating viral cancers.
CONCLUSIONS
In conclusion, we have reported that LNA oligonucleotides delivered by Cdots can be employed for specific knockdown of KSHV miR-K1, miR-K4, and miR-K11 to induce apoptosis and suppress the cell proliferation of KSHV-positive lymphoma cells through activation of cleaved caspase 3. This antisense drug can effectively inhibit the initiation of primary effusion lymphoma in a xenograft Nod/Scid mouse model. Moreover, this antisense drug can also induce tumor regression in a fully established PEL mice model and greatly increase the animal survival rate. This study employed miRNAs of an oncogenic virus as the therapeutic targets with nanoparticles-based antisense oligonucleotides, which have demonstrated that the use of antisense drugs is an attractive therapeutic approach for human cancers associated with oncogenic viruses..
MATERIALS AND METHODS
Chemicals and Reagents.
Citric acid (Cat. No. A104-500, Fisher Scientific) and branched polyethylenimine (PEI, Cat. No. 215515, Beantown Chemical) were used without further purification. Chlorpromazine hydrochloride (Cat. No. C8138), nystatin (Cat. No. N4014), methyl-β-cyclodextrin (Cat. No. 332615), and amiloride (Cat. No. 129876) were purchased from Sigma-Aldrich. Ultrapure water (Millipore Milli-Q) with a resistivity of 18.2 MΩ was used for all the solutions.
Cell Lines and Cell Culture.
All the cell lines were maintained at 37 °C with 5% CO2. KSHV-transformed rat primary embryonic mesenchymal precursor cells (KMM) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Cat. No. 25-500, Genesee Scientific) supplemented with 10% fetal bovine serum (FBS) and 150 μg/mL hygromycin. PEL cell lines BC3, BCP1, and BCBL1 and Burkitt’s lymphoma line BJAB were cultured in RPMI-1640 (GE Healthcare Life Science, Cat. No. SH30027) supplemented with 10% FBS (Cat. No. F2442, Sigma-Aldrich) and penicillin/streptomycin. BCBL1 cells expressing hPGK-promoter-driven firefly luciferase (BCBL1-Luc) were established by infection with Lenti-hPGK-Luc and cultured the same way as PEL cell lines.
Locked Nucleic Acids.
Locked nucleic acids with designed sequences were ordered from EXIQON. The LNA sequences are as follows:
LNA-NC:
5′-+A*+C*g*t*+C*t*a*+T*a*+C*g*+C*c*+C*+A*-3′
LNA-K1:
5′-+C*+C*c*+A*g*+T*t*+T*+C*c*+T*g*+T*a*+A*-3′
LNA-K4:
5′-+T*a*c*+T*g*+C*g*g*+T*t*+T+*A*g*+C*-3′
LNA-K11:
5′-+C*+A*g*g*+C*t*+A*+A*g*c*+A*t*t*+A*-3′
Whereas DNA bases are written in small letters, LNA bases are written as +N, and phosphorothioate linkages are indicated by an asterisk.
Synthesis of Cdots.
Cdots were synthesized by a microwave-assisted pyrolysis method. Briefly, 25 mg of PEI (2.5 mmol) was first dissolved in 2.5 mL of water in a baker. Then, 500 mg of citric acid (2.6 mmol) in 2.5 mL of water was added. The solution was stirred for 30 min at room temperature to form a homogeneous solution. Then the beaker containing a transparent solution was placed at the center of the rotation plate of a microwave oven (1150 W) and heated for 3 min. After the mixture cooled to room temperature, 5 mL of water was added, and the solution was sonicated for 15 min. The Cdots were isolated from the suspension through centrifugation at 13 000 rpm for 10 min. The supernatants were further dialyzed against deionized water using a molecular weight cutoff membrane (14 kDa) for 2 days to eliminate the excess citric acid and free PEI. The obtained Cdots solution was stored at 4 °C for further use.
Preparation of Cdots/LNA Complexes and Agarose Gel Electrophoresis.
The Cdots/LNA complexes were formed through electrostatic interaction between the positively charged Cdots and the negatively charged LNA. Different ratios of Cdots and LNA were mixed in PBS (pH 7.4) for 10 min at room temperature. The complexes were examined by 1% agarose gel in 1× TBE buffer with a constant voltage of 80 V for 20 min. The gel was stained with ethidium bromide, and the image was acquired using a ChemiDoc MP imaging system (Bio-Rad). The size distribution and zeta potential were measured by dynamic light scattering (DLS) through a Malvern Zeta Nanosizer.
Cellular Uptake of Cdots/LNA.
KMM cells and B cells were seeded in a 24-well plate at a density of 2 × 104 cells/well. On the following day, Cdots (1 μg) and LNA (100 pmol) were mixed in opt-MEM reduced serum medium for 10 min. The mixture was added into the respective wells with three different final LNA concentrations (50, 100, and 200 nM). After incubation at 37 °C with 5% CO2 for the indicated duration (1, 4, and 12 h), the cells were washed with PBS three times. An Olympus IX 81 confocal laser scanning microscope was used for visualizing the fluorescence. DAPI or Cdots were observed using an excitation wavelength of 405 nm and an emission wavelength of 425–475 nm. An excitation wavelength of 555 nm and an emission wavelength of 615–645 nm was employed to track TEX 615 dye-modified LNA.
For studying the mechanism of the Cdots/LNA cellular internalization, inhibitors including chlorpromazine hydrochloride (inhibiting clathrin-dependent endocytosis), nystatin (inhibiting caveolin-dependent endocytosis), methyl-β-cyclodextrin (inhibiting cholesterol-dependent membrane fusion), and amiloride (inhibiting macropinocytosis) were used. B cells were seeded in a 24-well plate with a density of 2 × 105 cells/well. Cells were then washed with PBS three times and treated with inhibitors in serum-free medium at 37 °C for 1 h (0.5 μg/mL of chlorpromazine hydrochloride, 50 μg/mL of methyl-β-cyclodextrin, 25 μg/mL of nystatin, or 20 μg/mL of amiloride). Then the medium was removed and fresh medium containing the inhibitors and Cdots/LNA was added for another 2 h. Cells were washed three times with PBS, and intracellular TEX615 fluorescence were measured by flow cytometry (BD LSRFortessa).
Cell Proliferation Assay.
KMM, BC3, BCP1, BCBL1, and BJAB cells were plated at a density of 2 × 105 cells/well in 24-well plate. The cells were treated with different reagents, including Cdots/LNA-K1, Cdots/LNA-K4, Cdots/LNA-K11, a combination of all three suppressors, or Cdots/LNA-NC. The total concentration of Cdots and LNA for each well was set as 1 μg/mL and 100 nM, respectively. The number of cells was counted daily for 3 days after the treatment. Briefly, the cell suspension was collected, and then an aliquot of cells was diluted in Trypan Blue (Sigma, Cat. No. T8154) with a dilution factor of 2 by gently pipetting. Then 10 μL of the above mixture was transferred under the coverslip on a hemocytometer. The unstained cells were counted and calculated.
Cell Apoptosis Assay.
BCBL1 and BJAB cells were plated at a density of 2 × 105 cells/well in a 24-well plate. The cells were treated with different reagents, including Cdots/LNA-K1, Cdots/LNA-K4, Cdots/LNA-K11, a combination of all three suppressors, or Cdots/LNA-NC. The total concentration of Cdots and LNA for each well was set as 1 μg/mL and 100 nM, respectively. Three days after the treatment, cells were centrifuged and washed twice with PBS. Apoptotic cells were detected by staining with DAPI and phycoerythrin-cyanine 7-conjugated antiannexin V antibody (25-8103-74; EBioscience) for 15 min. Flow cytometry was carried out in a BD LSRFortessa system, and analysis was performed with FlowJo V10.
Western-Blotting Analysis.
Cells were suspended in 1× sample buffer containing 62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 50 mM DTT, and 0.01% bromophenol blue and boiled at 100 °C for 10 min. Total cell lysates were separated in SDS polyacrylamide gels and then transferred to nitrocellulose membranes. The membranes were incubated with 5% milk and primary and secondary antibodies, respectively. The signal was detected with Luminiata Crescendo Western HRP substrate (EMD Millipore). Primary antibodies included β-actin (#sc-47778, Santa Cruz), cleaved caspase-3 (#9664, Cell Signaling Technology), IκBα (#sc-371, Santa Cruz), and BACH1 (#sc-271211, Santa Cruz). An antibody for CASTOR1 was prepared in rabbits by using purified peptide YTLMVDEEGF-KEL-C (Chemipeptide). The signal was visualized with a ChemiDoc MP imaging system (Bio-Rad).
RNA Extraction and Quantification.
Cells with the indicated treatments were lysed by TRIzol Reagent (Cat. No. T9424, Sigma). Total RNA was isolated by chloroform extraction and isopropanol precipitation. RT-qPCR of KSHV latent genes was carried out as previously described. miRNAs were quantified by stem-loop qRT-PCR. Briefly, a reverse transcriptase reaction was first carried out by incubating RNA sample with a stem-loop RT primer for 30 min at 16 °C, 30 min at 42 °C, 5 min at 85 °C, and held at 4 °C in the T100 thermal cycler (Bio-Rad). qPCR was performed in the CFX Connect Real-Time System (Bio-Rad). The reaction was performed in a 96-well plate at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and then 60 °C for 1 min. The relative miRNA levels were calculated after calibration with β-actin and converted to fold changes using one of the untreated samples as a standard. The primers used are 5′-ATC ATT GCT CCT CCT GAG CG-3′ (F) and 5′-CGG ACT CGT CAT ACT CCT GC-3′ (R) for β-actin; 5′-GCA GAC ACT GAA ACG CTG AA-3′ (F) and 5′-AGG TGA GCC ACC AGG ACT TA-3′ (R) for LANA; 5′-GGA TGC CCT AAT GTC AAT GC-3′ (F) and 5′-GGC GAT AGT GTT GGG AGT GT-3′ (R) for vFLIP; and 5′-GCT GAT AAT AGA GGC GGG CAA TGA G-3′ (F) and 5′-GTT GGC GTG GCG AAC AGA GAG GCA GTC-3′ (R) for vCyclin. The miRNA RT primers are 5′-GTC GTA TCC AGT GCA GGG TCC GAG GTA TTC GCA CTG GAT ACG ACG CTT AC-3′ for KSHV miR-K1; 5′-GTC GTA TCC AGT GCA GGG TCC GAG GTA TTC GCA CTG GAT ACG ACC AGA TC-3′ for KSHV miR-K2; 5′-GTC GTA TCC AGT GCA GGG TCC GAG GTA TTC GCA CTG GAT ACG ACC CTA GA-3′ for KSHV miR-K4; 5′-GTC GTA TCC AGT GCA GGG TCC GAG GTA TTC GCA CTG GAT ACG ACT CGG AC-3′ for KSHV miR-K11. Primers used for RT-qPCR of miRNAs are 5′-CGC GCA TTA CAG GAA ACT GGG-3′ (F) and 5′-GTG CAG GGT CCG AGG T-3′ (R) for KSHV miR-K1; 5′-CGT GCA ACT GTA GTC CGG GTC-3′ (F) and 5′-GTG CAG GGT CCG AGG T-3′ (R) for KSHV miR-K2; 5′-CGC GCA GCT AAA CCG CAG TAC-3′ (F) and 5′-GTG CAG GGT CCG AGG T-3′ (R) for KSHV miR-K4; and 5′-CGC GCT TAA TGC TTA GCC TGT-3′ (F) and 5′-GTG CAG GGT CCG AGG T-3′ (R) for KSHV miR-K11.
Luciferase Reporter Assay.
The dual-luciferase reporter assay was used to examine the activity of sensor reporters of KSHV miR-K1, -K4, and -K11, kind gifts from Bryan Cullen at Duke University Medical Center. The assay consisted of two reporters: a renilla luciferase expression construct pRL-TK used as an internal control and a firefly luciferase expression construct of a miRNA sensor reporter containing tandem repeats of the miRNA binding site cloned into the pGL3 vector. The luciferase reporter assay was carried out in 48-well plates. For each well, cells were treated with Cdots/LNA-NC or Cdots/LNA targeting a respective miRNA at a final LNA concentration of 100 nM. After 12 h, 20 ng of the miRNA sensor reporter and pRL-TK was cotransfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Cells were collected 36 h after transfection and analyzed using a dual-luciferase reporter assay system (Promega). Firefly luciferase activity of each sample was normalized to renilla luciferase activity.
Animal Experiments.
All animal-related experiments were performed in full compliance with institutional guidelines and approved by the Animal Use and Care Administration Advisory Committee of the University of Pittsburgh. Female Nod/Scid mice and female BALB/c mice, all at 4–5 weeks old, were purchased from Envigo. All animals were housed under pathogen-free conditions. To establish the PEL model, BCBL1-Luc cells were intraperitoneally administrated into the Nod/Scid mice at 107 cells/mouse.
For in vivo biodistribution of Cdots/LNA, Cdots/LNA was intraperitoneally injected into the mice at 50 μg Cdots and 5 nmol LNA/mouse. TEX615 fluorescence signal conjugated to LNA was monitored at the indicated time points using a Xenogen IVIS-200 imaging system with excitation wavelength at 500–550 nm and emission wavelength at 575–650 nm. For live bioluminescence imaging, mice received an intraperitoneal injection of d-luciferin at 50 mg/kg mice body weight. At 12 min postinjection, images of the mice were taken using the Xenogen IVIS-200 imaging system. Data were analyzed by Living Images 4.5 software.
For the tumor initiation experiment, 30 female Nod/Scid mice were intraperitoneally injected with 107 BCBL1-Luc cells. At day 3 after inoculation, mice were treated with PBS, Cdots/LNA-NC, or Cdots/LNAs with a total amount of 50 μg of Cdots and 5 nmol of LNA every day for 3 weeks. The body weights of the mice were measured twice every week. On day 21, mice were injected with d-luciferin at 50 mg/kg and imaged with the Xenogen IVIS-200 imaging system. Photographs of the mice were also taken using a digital camera.
For the tumor regression experiment, 15 Nod/Scid mice engrafted with 107 BCBL1-Luc cells for 3 weeks were randomly split into 3 groups by weight. Then, the mice were treated with PBS, Cdots/LNA-NC, or Cdots/LNAs with a total amount of 50 μg of Cdots and 5 nmol of LNA every day. On day 28, mice were injected with d-luciferin at 50 mg/kg and imaged with the Xenogen IVIS-200 imaging system. The ascites volume was measured either when the mice were euthanized because of the abnormal gait or impaired health or at day 42. The weight of the mice was recorded twice a week as a surrogate of tumor progression.
Statistical Analysis.
All results are expressed as the mean ± SD. Biological replicates were used in all experiments. One-way analysis of variance (ANOVA) was performed and Tukey’s post hoc test was used for multiple comparisons when the result was significant (P < 0.05). Statistical differences in survival were determined by Kaplan–Meier survival analysis with the log-rank test. All statistical analyses were performed with IBM SPSS statistics 20.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge financial support by grants from the NIH (CA096512, CA124332, CA132637, CA213275, DE025465, and CA197153).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b06333.
TEM images of Cdots and Cdots/LNA; X-ray photoelectron spectroscopy spectra of Cdots; agarose gel electrophoresis of Cdots/LNA; DLS data of Cdots/LNA in PBS containing serum; uptake of LNA-TEX615 by KMM cells; cytotoxicity of Cdots; qPCR data for detecting levels of KSHV miR-K1, miR-K4, and miR-K11; dual-luciferase reporter assay; Western-blotting results of proteins targeted by KSHV miRNAs; expression of KSHV latent genes in ascites; images of solid tumors (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsnano.9b06333
The authors declare no competing financial interest.
Contributor Information
Enguo Ju, University of Pittsburgh, Pittsburgh, Pennsylvania.
Tingting Li, University of Pittsburgh, Pittsburgh, Pennsylvania.
Zhen Liu, Beijing University of Chemical Technology, Beijing, People’s Republic of China.
Suzane Ramos da Silva, University of Pittsburgh, Pittsburgh, Pennsylvania.
Shan Wei, University of Pittsburgh, Pittsburgh, Pennsylvania.
Xinquan Zhang, University of Pittsburgh, Pittsburgh, Pennsylvania.
Xian Wang, University of Pittsburgh, Pittsburgh, Pennsylvania.
Shou-Jiang Gao, University of Pittsburgh, Pittsburgh, Pennsylvania.
REFERENCES
- (1).Sarid R; Gao SJ Viruses and Human Cancer: From Detection to Causality. Cancer Lett. 2011, 305, 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Qin J; Li W; Gao S-J; Lu C KSHV MicroRNAs: Tricks of the Devil. Trends Microbiol. 2017, 25, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhu Y; Haecker I; Yang Y; Gao SJ; Renne R Gamma-Herpesvirus-Encoded MiRNAs and Their Roles in Viral Biology and Pathogenesis. Curr. Opin. Virol 2013, 3, 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fabani MM; Abreu-Goodger C; Williams D; Lyons PA; Torres AG; Smith KG; Enright AJ; Gait MJ; Vigorito E Efficient Inhibition of MiR-155 Function In Vivo by Peptide Nucleic Acids. Nucleic Acids Res. 2010, 38, 4466–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Krützfeldt J; Rajewsky N; Braich R; Rajeev KG; Tuschl T; Manoharan M; Stoffel M Silencing of MicroRNAs In Vivo with ‘Antagomirs’. Nature 2005, 438, 685–689. [DOI] [PubMed] [Google Scholar]
- (6).Ma L; Reinhardt F; Pan E; Soutschek J; Bhat B; Marcusson EG; Teruya-Feldstein J; Bell GW; Weinberg RA Therapeutic Silencing of MiR-10b Inhibits Metastasis in a Mouse Mammary Tumor Model. Nat. Biotechnol 2010, 28, 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Elmén J; Lindow M; Schütz S; Lawrence M; Petri A; Obad S; Lindholm M; Hedtjärn M; Hansen HF; Berger U; Gullans S; Kearney P; Sarnow P; Straarup EM; Kauppinen S LNA-Mediated MicroRNA Silencing in Non-Human Primates. Nature 2008, 452, 896–899. [DOI] [PubMed] [Google Scholar]
- (8).Elmén J; Lindow M; Silahtaroglu A; Bak M; Christensen M; Lind-Thomsen A; Hedtjärn M; Hansen JB; Hansen HF; Straarup EM; McCullagh K; Kearney P; Kauppinen S Antagonism of MicroRNA-122 in Mice by Systemically Administered LNA-AntimiR Leads to Up-Regulation of a Large Set of Predicted Target mRNAs in the Liver. Nucleic Acids Res. 2008, 36, 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rinaldi C; Wood MJA Antisense Oligonucleotides: The Next Frontier for Treatment of Neurological Disorders. Nat. Rev. Neurol 2018, 14, 9–21. [DOI] [PubMed] [Google Scholar]
- (10).Rupaimoole R; Slack FJ MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discovery 2017, 16, 203–222. [DOI] [PubMed] [Google Scholar]
- (11).Bennett CF; Baker BF; Pham N; Swayze E; Geary RS Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol 2017, 57, 81–105. [DOI] [PubMed] [Google Scholar]
- (12).Stein CA; Castanotto D FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther 2017, 25, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tripathy SK; Black HB; Goldwasser E; Leiden JM Immune Responses to Transgene-Encoded Proteins Limit the Stability of Gene Expression after Injection of Replication-Defective Adenovirus Vectors. Nat. Med 1996, 2, 545–550. [DOI] [PubMed] [Google Scholar]
- (14).Love KT; Mahon KP; Levins CG; Whitehead KA; Querbes W; Dorkin JR; Qin J; Cantley W; Qin LL; Racie T; Frank-Kamenetsky M; Yip KN; Alvarez R; Sah DWY; de Fougerolles A; Fitzgerald K; Koteliansky V; Akinc A; Langer R; Anderson DG Lipid-Like Materials for Low-Dose, In Vivo Gene Silencing. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nguyen DN; Green JJ; Chan JM; Langer R; Anderson DG Polymeric Materials for Gene Delivery and DNA Vaccination. Adv. Mater 2009, 21 , 847–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rush AM; Nelles DA; Blum AP; Barnhill SA; Tatro ET; Yeo GW; Gianneschi NC Intracellular mRNA Regulation with Self-Assembled Locked Nucleic Acid Polymer Nanoparticles. J. Am. Chem. Soc 2014, 136, 7615–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Babar IA; Cheng CJ; Booth CJ; Liang X; Weidhaas JB; Saltzman WM; Slack FJ Nanoparticle-Based Therapy in an In Vivo MicroRNA-155 (miR-155)-Dependent Mouse Model of Lymphoma. Proc. Natl. Acad. Sci. U. S. A 2012, 109, E1695–E1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Putnam D; Gentry CA; Pack DW; Langer R Polymer-Based Gene Delivery with Low Cytotoxicity by a Unique Balance of Side-Chain Termini. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 1200–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ibrahim AF; Weirauch U; Thomas M; Grünweller A; Hartmann RK; Aigner A MicroRNA Replacement Therapy for MiR-145 and MiR-33a Is Efficacious in a Model of Colon Carcinoma. Cancer Res. 2011, 71, 5214–5224. [DOI] [PubMed] [Google Scholar]
- (20).Zheng D; Giljohann DA; Chen DL; Massich MD; Wang XQ; Iordanov H; Mirkin CA; Paller AS Topical Delivery of siRNA-Based Spherical Nucleic Acid Nanoparticle Conjugates for Gene Regulation. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 11975–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lee J-H; Lee K; Moon SH; Lee Y; Park TG; Cheon J All-In-One Target-Cell-Specific Magnetic Nanoparticles for Simultaneous Molecular Imaging and siRNA Delivery. Angew. Chem., Int. Ed 2009, 48, 4174–4179. [DOI] [PubMed] [Google Scholar]
- (22).Giljohann DA; Seferos DS; Prigodich AE; Patel PC; Mirkin CA Gene Regulation with Polyvalent siRNA–Nanoparticle Conjugates. J. Am. Chem. Soc 2009, 131, 2072–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rosi NL; Giljohann DA; Thaxton CS; Lytton-Jean AKR; Han MS; Mirkin CA Oligonucleotide-Modified Gold Nanoparticles for Intracellular Gene Regulation. Science 2006, 312, 1027–1030. [DOI] [PubMed] [Google Scholar]
- (24).Li H; Kang Z; Liu Y; Lee S-T Carbon Nanodots: Synthesis, Properties and Applications. J. Mater. Chem 2012, 22, 24230–24253. [Google Scholar]
- (25).Miao P; Han K; Tang Y; Wang B; Lin T; Cheng W Recent Advances in Carbon Nanodots: Synthesis, Properties and Biomedical Applications. Nanoscale 2015, 7, 1586–1595. [DOI] [PubMed] [Google Scholar]
- (26).Pei X; Xiong D; Fan J; Li Z; Wang H; Wang J Highly Efficient Fluorescence Switching of Carbon Nanodots by CO2. Carbon 2017, 117, 147–153. [Google Scholar]
- (27).Liu C; Zhang P; Zhai X; Tian F; Li W; Yang J; Liu Y; Wang H; Wang W; Liu W Nano-Carrier for Gene Delivery and Bioimaging Based on Carbon Dots with PEI-Passivation Enhanced Fluorescence. Biomaterials 2012, 33, 3604–3613. [DOI] [PubMed] [Google Scholar]
- (28).Kim S; Choi Y; Park G; Won C; Park Y-J; Lee Y; Kim B-S; Min D-H Highly Efficient Gene Silencing and Bioimaging Based on Fluorescent Carbon Dots In Vitro and In Vivo. Nano Res. 2017, 10, 503–519. [Google Scholar]
- (29).Goncalves PH; Uldrick TS; Yarchoan R HIV-Associated Kaposi Sarcoma and Related Diseases. AIDS 2017, 31, 1903–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cesarman E Gammaherpesvirus and Lymphoproliferative Disorders in Immunocompromised Patients. Cancer Lett. 2011, 305, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Coiffier B; Lepage E; Briere J; Herbrecht R; Tilly H; Bouabdallah R; Morel P; Van Den Neste E; Salles G; Gaulard P; Reyes F; Lederlin P; Gisselbrecht C CHOP Chemotherapy Plus Rituximab Compared with CHOP Alone in Elderly Patients with Diffuse Large-B-Cell Lymphoma. N. Engl J. Med 2002, 346, 235–242. [DOI] [PubMed] [Google Scholar]
- (32).Li T; Ju E; Gao SJ Kaposi Sarcoma-Associated Herpesvirus MiRNAs Suppress CASTOR1-Mediated mTORC1 Inhibition to Promote Tumorigenesis. J. Clin. Invest 2019, 129, 3310–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yoo SM; Zhou FC; Ye FC; Pan HY; Gao SJ Early and Sustained Expression of Latent and Host Modulating Genes in Coordinated Transcriptional Program of KSHV Productive Primary Infection of Human Primary Endothelial Cells. Virology 2005, 343, 47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lei X; Bai Z; Ye F; Xie J; Kim CG; Huang Y; Gao SJ Regulation of NF-KappaB Inhibitor IkappaBalpha and Viral Replication by a KSHV MicroRNA. Nat. Cell Biol 2010, 12, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gottwein E; Cai X; Cullen BR A Novel Assay for Viral MicroRNA Function Identifies a Single Nucleotide Polymorphism that Affects Drosha Processing. J. Virol 2006, 80, 5321–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Moody R; Zhu Y; Huang YF; Cui XD; Jones T; Bedolla R; Lei XF; Bai ZQ; Gao SJ KSHV MicroRNAs Mediate Cellular Transformation and Tumorigenesis by Redundantly Targeting Cell Growth and Survival Pathways. PLoS Pathog. 2013, 9, No. e1003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gong N; Ma X; Ye X; Zhou Q; Chen X; Tan X; Yao S; Huo S; Zhang T; Chen S; Teng X; Hu X; Yu J; Gan Y; Jiang H; Li J; Liang XJ Carbon-Dot-Supported Atomically Dispersed Gold as a Mitochondrial Oxidative Stress Amplifier for Cancer Treatment. Nat. Nanotechnol 2019, 14, 379–387. [DOI] [PubMed] [Google Scholar]
- (38).Du J; Xu N; Fan J; Sun W; Peng X Carbon Dots for In Vivo Bioimaging and Theranostics. Small 2019, 15, 1805087. [DOI] [PubMed] [Google Scholar]
- (39).Jones T; Ye F; Bedolla R; Huang Y; Meng J; Qian L; Pan H; Zhou F; Moody R; Wagner B; Arar M; Gao SJ Direct and Efficient Cellular Transformation of Primary Rat Mesenchymal Precursor Cells by KSHV. J. Clin. Invest 2012, 122, 1076–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Varkouhi AK; Scholte M; Storm G; Haisma HJ Endosomal Escape Pathways for Delivery of Biologicals. J. Controlled Release 2011, 151, 220–228. [DOI] [PubMed] [Google Scholar]
- (41).Gottwein E; Mukherjee N; Sachse C; Frenzel C; Majoros WH; Chi JT; Braich R; Manoharan M; Soutschek J; Ohler U; Cullen BR A Viral MicroRNA Functions as an Orthologue of Cellular MiR-155. Nature 2007, 450, 1096–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gruffaz M; Zhou S; Vasan K; Rushing T; Michael QL; Lu C; Jung JU; Gao SJ Repurposing Cytarabine for Treating Primary Effusion Lymphoma by Targeting Kaposi’s Sarcoma-Associated Herpesvirus Latent and Lytic Replications. mBio 2018, 9, No. e00756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).He M; Tan B; Vasan K; Yuan H; Cheng F; Ramos da Silva S; Lu C; Gao SJ SIRT1 and AMPK Pathways are Essential for the Proliferation and Survival of Primary Effusion Lymphoma Cells. J. Pathol 2017, 242, 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Alizadeh AA; Eisen MB; Davis RE; Ma C; Lossos IS; Rosenwald A; Boldrick JC; Sabet H; Tran T; Yu X; Powell J; Yang L; Marti GE; Moore T; Hudson J Jr; Lu L; Lewis DB; Tibshirani R; Sherlock G; Chan WC; et al. Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profiling. Nature 2000, 403, 503–511. [DOI] [PubMed] [Google Scholar]
- (45).Neelapu SS; Locke FL; Bartlett NL; Lekakis LJ; Miklos DB; Jacobson CA; Braunschweig I; Oluwole OO; Siddiqi T; Lin Y; Timmerman JM; Stiff PJ; Friedberg JW; Flinn IW; Goy A; Hill BT; Smith MR; Deol A; Farooq U; McSweeney P; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med 2017, 377, 2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gottwein E; Cullen BR A Human Herpesvirus MicroRNA Inhibits p21 Expression and Attenuates p21-Mediated Cell Cycle Arrest. J. Virol 2010, 84, 5229–5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Skalsky RL; Samols MA; Plaisance KB; Boss IW; Riva A; Lopez MC; Baker HV; Renne R Kaposi’s Sarcoma-Associated Herpesvirus Encodes an Ortholog of MiR-155. J. Virol 2007, 81, 12836–12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.