

Highlights
-
•
Activation of RNase L inhibits HBV replication in infected and transfected hepatoma cells.
-
•
Antiviral activity of RNase L depends on its ribonuclease function.
-
•
OAS2 and OAS3 but not OAS1 are involved in RNase L activation in the process.
Keywords: RNase L, Oligoadenylate synthetase, Hepatitis B virus, Interferon, RNA decay
Abstract
RNase L is a cellular endoribonuclease that is activated by 2′,5′-linked oligoadenylates (2–5A), which are unique and specific ligands synthesized by a family of interferon-inducible, dsRNA-activated enzymes named oligoadenylate synthetases. In the typical antiviral pathway, activated RNase L degrades viral and cellular RNAs, thus limiting viral replication and spread. Although the antiviral activity of RNase L has been demonstrated for several RNA viruses, there is little evidence regarding its role against DNA viruses. In the present study, the potential antiviral activity of RNase L against hepatitis B virus (HBV) was explored utilizing the recently reported infection protocol based on human hepatoma HepG2 cells stably complemented with the virus entry factor NTCP. Viral replication and expression in this cell type was markedly inhibited by poly(I:C)- or 2–5A-mediated activation of RNase L; however, the inhibition was significantly reversed by RNase L knockdown. Further analysis in HBV1.2-transfected Huh-7 hepatoma cells indicated that the antiviral activity of RNase L depends on its ribonuclease function. We also provide evidence for the specific roles of OAS family members in this process. These results suggest that HBV replication can be regulated through interferon-mediated RNA decay pathways and that activation of these host antiviral factors may represent a novel therapeutic strategy for HBV infection.
1. Introduction
RNase L is a latent endoribonuclease that is constitutively expressed in almost all mammalian cells. This enzyme is activated upon the binding of a specific ligand, 2′,5′-linked oligoadenylate (2–5A), which is synthesized by a family of enzymes named oligoadenylate synthetases (OASs). OAS expression is induced by type I interferons (IFN-α/β), but the enzymes require dsRNA for the activation of their catalytic activity (Kristiansen et al., 2011). In the typical antiviral pathway, viral dsRNA binds to an OAS, which results in the activation of the OAS and the synthesis of 2–5A. RNase L, activated upon binding to 2–5A, degrades viral and cellular RNAs, thus limiting viral replication and spread (Chakrabarti et al., 2011). In addition to the direct antiviral effect, RNase L can activate intracellular signaling pathways through the small RNA cleavage products that it generates, leading to the induction of type I IFN production (Malathi et al., 2007). The antiviral role of RNase L has been established for a number of RNA viruses. RNase L-deficient mice exhibited increased susceptibility to infection by these viruses (e.g., the picornavirus EMCV and the flavivirus WNV) (Silverman, 2007). Recent findings of viral functions that antagonize OAS or RNase L, most notably the murine MHV coronavirus ns2-encoded phosphodiesterase that can degrade 2–5A, underscore the importance of RNase L pathway in antiviral immunity (Zhao et al., 2012). Compared to its roles against RNA viruses, evidence regarding the antiviral role of RNase L against DNA viruses (e.g., herpesviruses and vaccinia virus) is limited (Rasmussen et al., 2009, Xiang et al., 2002, Zheng et al., 2001), although DNA viruses can produce dsRNA (Weber et al., 2006).
Hepatitis B virus (HBV) is a hepatotropic DNA virus that can cause acute and chronic hepatitis in humans. More than 350 million people worldwide are chronic carriers of the virus and are at risk of progression of the infection to liver cirrhosis and hepatocarcinoma (Lok and McMahon, 2007). IFN-α is currently the approved treatment for chronic hepatitis B (Perrillo, 2009). However, the mechanism by which IFN-α inhibits HBV replication is not completely understood. A recent etiological study that aimed to identify a potential link between naturally occurring RNase L gene variations (e.g., R462Q) and prostate cancer suggested that this gene is correlated with chronic hepatitis B and HIV infections (Arredondo et al., 2012). However, there are few experimental data on the association between HBV and RNase L. In an early study, HBV replication was notably inhibited following poly(I:C) administration to HBV transgenic mice, but the extent of inhibition was not significantly different between groups of mice that were RNase L+/+ or RNase L−/− (Guidotti et al., 2002). While these data indicated that RNase L is not likely to mediate the antiviral activity of IFN against HBV, it was not determined whether the catalytic activity of RNase L was activated in these mice.
In the present study, we adopted human hepatoma cells that permit HBV infection to address whether the OAS/RNase L system plays a role in the inhibition of HBV, although HBV is not known to carry or produce dsRNA. Our data indicate that ligand-mediated activation of these enzymes results in a marked inhibition of HBV replication in both infected and transfected cells. We also provided evidence for differential roles of OAS family members in this process. These results suggest that HBV replication can be regulated through the activation of RNase L-mediated RNA decay pathways.
2. Materials and methods
2.1. Virus and cell cultures
HBV stock was prepared from the culture supernatant of HepG2.2.15 cells (Sells et al., 1987). The cells were maintained in DMEM supplemented with 10% FBS (HyClone), 200 μg/ml G418 (A.G. Scientific) and 50 μg/ml gentamicin (Invitrogen). The cells were split 1:4 every 7 days, and the culture supernatant was concentrated as described (Vincent et al., 2011). To prepare viral DNA the viral stock was treated with 200 μg/ml proteinase K for 1 h at 37 °C in 10 mM Tris (pH 7.5), 10 mM EDTA and 1% SDS, followed by phenol/chloroform/isoamyl alcohol (PCI) treatment. Viral DNA was measured by real-time qPCR with a SYBR qPCR kit (KAPA Biosystems) and a MyiQ system (Bio-Rad Laboratories) using HBV-specific primers (Suppl. Table 1). PCR was conducted by denaturation at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 3 s and annealing/elongation at 60 °C for 20 s. The viral genome copy number was calculated based on a standard curve generated with pGEM-HBV1.2 and was indicated as genome equivalents (Geq).
For the infection assay, HepG2 cells (ATCC HB-8065) were stably transfected with the NTCP gene (a generous gift from Wenhui Li of NIBS, China) and maintained in DMEM with 2 μg/ml blasticidin (Invivogen). The NTCP-supplemented cells were incubated for 7 days before infection in primary hepatocyte maintenance media (PMM). PMM consists of Williams E medium supplemented with 10% FBS, 10 ng/ml EGF (Invitrogen), 5 μg/ml transferrin, 3 μg/ml insulin, 2 mM l-glutamine, 18 μg/ml hydrocortisone, 40 ng/ml dexamethasone, 5 ng/ml sodium selenite, 2% DMSO (all from Sigma) and 50 μg/ml gentamicin. The cells were seeded in 24-well plates (∼105 cells/well) and inoculated the following day with HBV at ∼2000 Geq/cell in PMM (without DMSO) containing 4% PEG8000. The viral inoculum was removed 16 h later, and the cells were further incubated (up to 9 days in most experiments) in PMM; the media was changed every 3 days.
For the transfection assay, Huh-7 cells grown in DMEM with 5% FBS were transfected with 1 μg/well of pGEM-HBV1.2, a replicon containing the 1.2-mer HBV genome. Viral polymerase gene expression was nullified in the HBV1.2(P-) construct due to a T to C mutation in the first ATG codon and a deletion of T from the second ATG codon; these changes did not alter the expression of the core gene that overlaps in the same region of the genome (Ryu et al., 2008). In HBV1.2(X-), the expression of the viral X gene was blocked with two stop codons introduced adjacent to the first and second ATG codons of the X gene by substitution of T for C at nt positions 22 and 259 (Cha et al., 2009). HBV1.2(C-42) expresses an assembly-defective core protein due to deletion of the 42nd codon (Leu) of the capsid gene (Koschel et al., 2000).
2.2. Viral DNA and RNA analyses
Capsid-associated viral DNA was extracted from infected cells that were lysed for 20 min on ice in buffer [50 mM Tris–HCl (pH 7.5), 1 mM EDTA, 0.2% NP-40 and 150 mM NaCl]. The lysate was clarified by centrifugation at 15,000g, and the supernatant was transferred to a new tube and gently mixed for 12 h with an anti-HBc antibody (Dako) and Protein A/G PLUS-Agarose (Santa Cruz Biotechnology). The beads were collected by centrifugation at 1000g and washed 3 times in the lysis buffer. Viral DNA was extracted with 200 μg/ml of proteinase K for 1 h at 37 °C in 10 mM Tris (pH 7.5), 10 mM EDTA and 1% SDS; it was then extracted by PCI and measured by southern blot and real-time PCR as described above.
Viral cccDNA was extracted by PCI from infected cells that were lysed for 4 h at 65 °C in lysis buffer [50 mM Tris–HCl (pH 8.0), 50 mM EDTA, 100 mM NaCl and 1% SDS] supplemented with proteinase K (200 μg/ml). The extracted DNA (∼500 ng) was treated with 10 U of plasmid-safe DNase (Epicentre Technologies) for 8 h at 37 °C followed by DNase inactivation at 70 °C for 30 min. An aliquot (∼20 ng) of extracted DNA was denatured at 95 °C for 5 min and amplified by qPCR using cccDNA-specific primers (Suppl. Table 1). The amplification protocol included 45 cycles of denaturation at 95 °C for 30 s, annealing at 62 °C for 25 s and elongation at 72 °C for 45 s. The HBV cccDNA copy number was calculated based on a standard curve generated with pGEM-HBV1.2.
Viral pgRNA was measured by real-time qPCR. Total RNA was extracted from infected cells with RNAiso plus (Takara), treated with RQ1 DNase and reverse transcribed with the ImProm-II RT system (Promega). cDNA derived from 50 ng of total RNA was amplified by qPCR as described for the capsid DNA assay. Transcripts of RNase L, p53, OAS1, OAS2 and OAS3 were measured by real-time PCR from 1 μg of total cellular RNA under the same conditions described above. GAPDH mRNA was used as a normalization control. Viral RNA and capsid DNA were measured by northern and southern blotting as previously described (Park et al., 2011).
2.3. Immunostaining and immunoblotting
Infected cells were fixed with 3.7% paraformaldehyde, permeabilized with 0.5% Triton X-100 and immunostained with anti-HBcAg (Dako) followed by Alexa 568-conjugated goat anti-rabbit IgG (Invitrogen). RNase L was immunoblotted and detected with anti-RNase L (Invitrogen) or anti-Flag antibody (Sigma). Tubulin was probed with an antibody obtained from Applied Biological Materials.
2.4. Other reagents
The RNase L expression plasmid p3xFlag-RNase L and a nuclease-defective construct, R667A, have been previously described (Kwon et al., 2013). Poly(I:C) was obtained from Sigma. Trimeric 2′,5′ adenylates, either in 5′-monophosphorylated (p2–5A) or unphosphorylated (2–5A) forms were provided by ChemGenes Corp. PCR primers and siRNAs were purchased from Integrated DNA Technologies and GenePharma, respectively, and their nucleotide sequences are shown in Suppl. Table 1. JetPEI (Polyplus) was used for DNA transfection. Lipofectamine 2000 (Invitrogen) was used for transfection of poly(I:C), 2–5As and siRNAs. IFN-α was obtained from R&D Systems, Inc.
3. Results
3.1. Inhibition of HBV replication by poly(I:C)
Studies on HBV have been hampered by the lack of a robust cell culture system that permits the full life cycle of viral growth. A recent study identified a bile acid transporter protein named sodium taurocholate cotransporting polypeptide (NTCP) that binds the N-terminal pre-S1 domain of HBV envelope protein L and mediates the entry of HBV and its surrogate virus HDV (Yan et al., 2012). It was shown that supplementation of human hepatoma cells such as Huh-7 and HepG2 with NTCP supports HBV infection. To address the role of RNase L in this process, we prepared a pool of HepG2 cells stably transfected with the NTCP gene. While the NTCP-expressing cells were routinely maintained in DMEM, for experiments involving HBV infection, the cells were incubated in PMM for several days before infection, which appeared to render the cells more susceptible to viral infection. Infected cells were further incubated (up to 9 days in most experiments) in PMM, and the media was changed every 3 days. Immunostaining indicated that the number of cells positive for HBcAg, the viral capsid protein, increased with time, although it was less than 5% of the total cells at 9 days post-infection (Fig. 1 A). To follow viral replication, we measured the viral pregenomic RNA (pgRNA) by RT-qPCR. At 9 days post-infection, pgRNA reached up to ∼2 × 106 copies per μg of total cellular RNA (Fig. 1B). In contrast to these results, expression of HBcAg and pgRNA was markedly reduced when infected cells were transfected with poly(I:C). Viral DNA in the capsids, measured by real-time PCR of intracellular viral capsids immunoprecipitated with anti-HBc antibody, was similarly decreased. Intracellular viral cccDNA was also significantly reduced at 9 days post-infection, while no inhibition was observed at 2 days post-infection (Suppl. Fig. S1). These results demonstrate the profound antiviral effect of poly(I:C) on HBV expression and replication in this cell-based infection system.
Fig. 1.
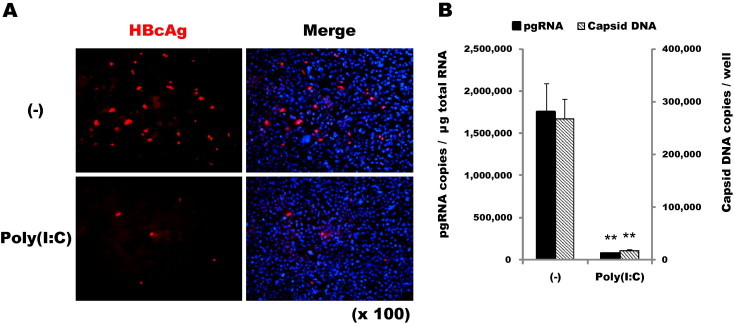
Inhibition of HBV replication by poly(I:C). (A) HepG2-NTCP cells (∼105 cells/well) were inoculated with HBV (at ∼2000 Geq/cell) followed 16 h later by either mock-transfection (–) or transfection with 100 ng/well of poly(I:C). The cells were incubated in PMM for up to 9 days; the medium was changed every 3 days. Cells immunostained for HBcAg at 9 days post-infection are shown in red. The image (magnified ×100) is overlapped on the right with nuclear (DAPI) staining. (B) Intracellular viral pgRNA and capsid DNA at 9 days post-infection were measured by RT-qPCR and real-time qPCR, respectively. The average copy numbers of triplicate measurements are shown as bars with standard errors indicated at the top (∗∗P < 0.01).
3.2. Antiviral activity and expression of RNase L
The antiviral effect of poly(I:C) observed above was likely due to the type I IFN-inducing response mediated by dsRNA-binding cellular receptor proteins such as TLR3 and RIG-I. Indeed, we observed strong induction of IFN-β and OAS2 mRNAs in HepG2-NTCP cells approximately 12 h after poly(I:C) transfection (Suppl. Fig. S2). Among the IFN-related cellular factors, we focused on RNase L because while the induction of OAS depends on the IFN response, the activation of its endonuclease function requires dsRNA. To more specifically address the antiviral activity, if any, of RNase L against HBV, we transfected the infected cells with synthetic 5′-monophosphorylated trimeric 2′,5′-adenylates (p2–5A), a specific activator of RNase L. Viral pgRNA expression was inhibited by up to ∼70% in a p2–5A dose-dependent manner, whereas no inhibition was observed in the cells either mock-transfected or transfected with unphosphorylated 2–5A (Fig. 2 A). Because p2–5A is a unique and specific activator for RNase L, these results strongly indicated that the observed inhibition of HBV was the result of RNase L activation. However, unlike the situation in liver tissue, where abundant expression of RNase L was observed, Huh-7 and HepG2 cells express only low levels of RNase L (Kwon et al., 2013, Malathi et al., 2007, Zhou et al., 2005).
Fig. 2.
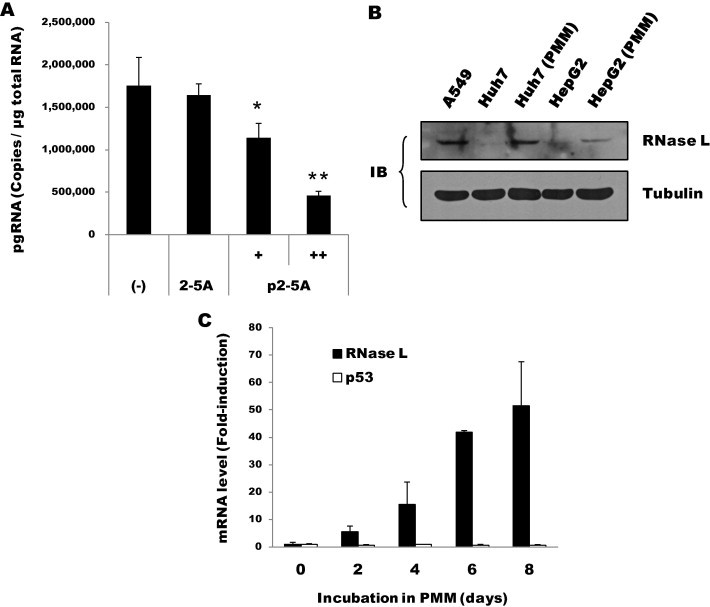
Antiviral activity and expression of RNase L. (A) Antiviral activity mediated by p2–5A. At 16 h after infection, HepG2-NTCP cells were mock-transfected (–) or transfected with 2–5A (100 μM) or p2–5A (30 and 100 μM). Intracellular pgRNA at 9 days post-infection was measured by RT-qPCR (∗P < 0.05, ∗∗P < 0.01). (B) Aliquots of Huh-7 or HepG2 cells either grown in DMEM or further incubated in PMM for 5 days were immunoblotted for RNase L (∼82 kDa). Human lung carcinoma (A549) cells grown in DMEM were also probed. Tubulin served as a loading control. (C) RNase L mRNA levels in HepG2 cells incubated in PMM for the indicated periods were measured by RT-qPCR and are shown as fold-induction relative to the initial level. For comparative purposes, p53 mRNA is also shown.
Interestingly, we identified RNase L proteins in an amount that was detectable by immunoblotting of hepatoma cells that were incubated for 5 days in PMM, whereas it was not detected in the same type of cells grown in DMEM (Fig. 2B). Our analysis indicated a steady increase of RNase L mRNA in the two hepatoma cell lines during incubation in PMM (up to ∼50-fold in 8 days), whereas the level of p53 mRNA remained stable (Fig. 2C). Although we could not explain this phenomenon, the elevated expression of RNase L provided an opportunity to address the role of RNase L through siRNA-mediated suppression. Inhibition of pgRNA by p2–5A was almost fully reversed in the RNase L knockdown cells, demonstrating a specific role for RNase L in the inhibition of HBV (Fig. 3 ). Compared with this result, poly(I:C)-mediated inhibition was rescued by ∼50%, suggesting that other effectors in addition to RNase L also contributed to the inhibition.
Fig. 3.
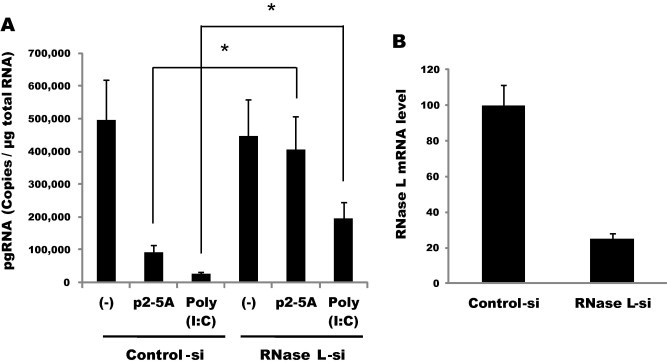
Viral expression was rescued in RNase L knockdown cells. (A) HepG2-NTCP cells transfected in suspension with siRNAs (50 nM) were plated and infected on the following day with HBV. The cells were further transfected 16 h later with p2–5A (100 μM) or poly(I:C) (100 ng/well). Viral pgRNA at 9 days post-infection was quantified by RT-qPCR (∗P < 0.05). (B) The knockdown efficiency of RNase L was determined by real-time PCR.
3.3. Ribonuclease-dependent antiviral activity of RNase L
The antiviral activity of RNase L was further characterized in another hepatoma cell line, Huh-7, following transient transfection of the viral genome construct HBV1.2. Including the 3.5-kb pgRNA, all viral mRNAs (2.4, 2.1 and 0.9 kb in length) were markedly reduced when the replicon cells were further transfected with RNase L (Fig. 4 A). As in the infection experiments, poly(I:C) was required for the antiviral activity of RNase L, although a mild inhibitory effect was observed even without poly(I:C) (Fig. 4B). This mild inhibitory effect of RNase L (shown in the lane 2 vs. lane 1 of Fig. 4A) suggested that some OAS activation occurred under these conditions, most likely due to non-specific dsRNAs (e.g., read-through transcripts) produced off the plasmid DNA templates. In contrast, no inhibition was observed with R667A, which harbors a missense mutation in the ribonuclease domain of RNase L (Dong et al., 2001). The strong antiviral effect and concomitant occurrence of rRNA cleavage (shown in the lane 5) confirmed the poly(I:C)-mediated activation of ectopically expressed RNase L. Poly(I:C) alone (without ectopic RNase L expression) had no effect (lanes 1 vs. 4; lanes 3 vs. 6). This result is consistent with the low level of endogenous RNase L expression, as we reported previously (Kwon et al., 2013), and no induction of IFN-β or ISG expression was observed in this type of hepatoma cell upon poly(I:C) transfection (described below) as reported (Li et al., 2005). Therefore, the observed antiviral activity of transfected poly(I:C) was primarily contributed by the activated ribonuclease function of RNase L. The activation of RNase L was also confirmed with p2–5A transfected Huh-7 cells (Suppl. Fig. S3).
Fig. 4.
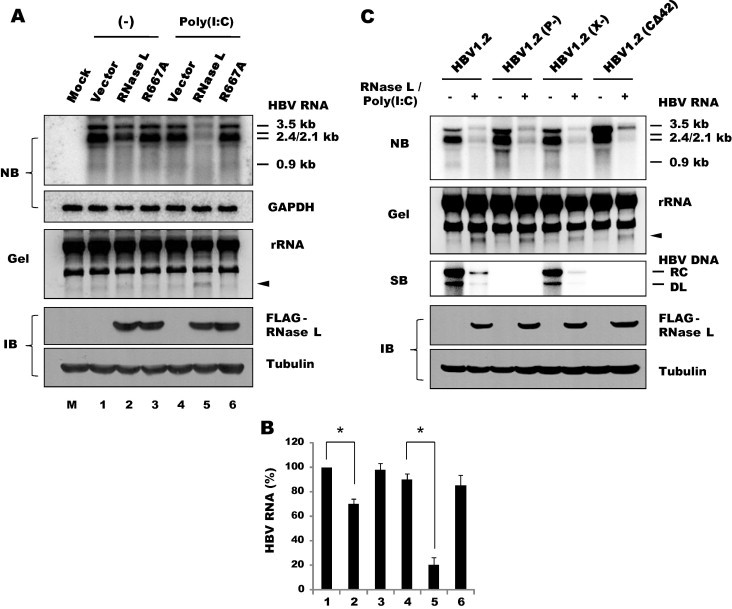
Ribonuclease-dependent antiviral activity of RNase L. (A) Huh-7 cells were transfected with 1 μg/well each of pGEM-HBV1.2 and either a wild-type p3×Flag-RNase L or a nuclease-defective R667A construct. After 24 h, the cells were either mock-transfected (–) or transfected with 1 μg/well of poly(I:C). RNA was extracted after 24 h and separated on a 1.5% agarose gel containing formaldehyde. Viral transcripts (3.5, 2.4, 2.1 and 0.9 kb in length) were analyzed by northern blotting. GAPDH mRNA was probed as a control. An agarose gel with rRNA (28S and 18S) and RNase L-specific cleavage products (indicated with arrowheads) is shown in a reverse phase image. An aliquot of transfected cells was immunoblotted for RNase L with an anti-Flag antibody. Tubulin was probed as a loading control. (B) The intensity of HBV RNA shown in Fig. 4A was quantified using Multi Gauge V3.0 (Fuji film). An averages of duplicate experiments are shown as bars with standard errors indicated at the top (∗P < 0.05). (C) Huh-7 cells were co-transfected with RNase L and HBV genomes that carry mutations in the pol, HBx or core genes. The cells were transfected 24 h later with poly(I:C). Viral capsid DNA was analyzed in 24 h by southern blotting. Relaxed circular (RC) and double-stranded linear (DL) forms of viral DNA are indicated on the right. RNA and proteins were analyzed as described above.
Because the first step in the HBV replication cycle is the synthesis of viral mRNA and pgRNA off the viral genome, downregulation of viral transcripts by RNase L would subsequently affect viral DNA replication as well. As expected, the capsid-associated viral DNA was barely detectable in RNase L-activated cells, indicating that the downregulation of viral transcripts resulted in the severe inhibition of viral DNA synthesis, including the new synthesis of viral relaxed-circular (RC) and duplex-linear (DL) DNAs (Fig. 4C, 2nd lane of the southern blot). To further substantiate this result, we examined HBV with mutations that have critical effects on viral DNA replication. HBV1.2(P-) is incapable of DNA replication because viral Pol gene expression is nullified due to a T to C mutation in the first ATG codon and a T deletion in the second ATG codon; however, these mutations do not alter the expression of the core gene that overlaps the same region (Ryu et al., 2008). HBV1.2(C-42) is also replication-defective because the core gene lacks the 42nd codon (Leu) and thus cannot form assembled capsids (Koschel et al., 2000). Our analyses indicated that the transcripts of these mutants, which were not affected despite their replication defects, were degraded by poly(I:C) and RNase L similar to those of the wild-type genome. A similar result was obtained for the third mutant, HBV1.2(X-), in which the viral X gene was nullified due to stop codons introduced adjacent to the first and second ATG codons (Cha et al., 2009). Despite nullification of this critical protein, viral transcription and DNA replication were not affected as reported previously for this type of hepatoma cell (Melegari et al., 1998). Likewise, all viral transcripts of this mutant were degraded by poly(I:C) and RNase L similar to those of the wild-type genome. Thus, RNase L-dependent downregulation of viral RNAs (and DNA) as observed for wild-type HBV was observed for all three mutants regardless of their replication capability.
3.4. Differential expression and antiviral roles of OAS
RNase L activation requires 2–5A, which is synthesized by OAS. In humans, three closely related and linked genes, OAS1, OAS2 and OAS3, code for OAS proteins. OAS expression is induced by type I IFNs, but dsRNA is required for the activation of their catalytic activity. Our RT-PCR analysis showed that OAS1 and OAS3, but not OAS2, were constitutively expressed in Huh-7 and HepG2 cells before all three genes were further induced by IFN-α treatment (Fig. 5 ). As mentioned above, poly(I:C) had no effect on the expression pattern of the three OAS genes in Huh-7 cells, yet it induced all three OAS genes in HepG2 cells. Therefore, it is likely that in Huh-7 cells, the constitutively expressed endogenous OAS1 and/or OAS3 proteins were catalytically activated by poly(I:C) and contributed to RNase L activation as observed above. To address this hypothesis, we attempted to knock down OAS expression using a specific siRNA. Despite the notable suppression of OAS1 (by up to 60%), inhibition mediated by RNase L and poly(I:C) was not affected (Fig. 6 ), suggesting that OAS1 does not play a major role in this process. Interestingly, a moderate rescue of viral RNA was observed in the OAS1 knockdown cells even prior to the transfection of RNase L and poly(I:C). Most likely, OAS1 was activated by non-specific dsRNAs produced off the plasmid DNA templates, resulting in the activation of endogenous RNase L that was present at a low level.
Fig. 5.
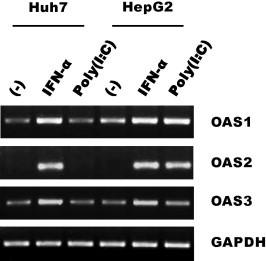
Expression of OAS in hepatoma cells. OAS1, OAS2 and OAS3 mRNAs were amplified by RT-PCR from 1 μg of RNA extracted from Huh-7 or HepG2 cells at 24 h following treatment with IFN-α (1000 U/ml) or transfection with poly(I:C). GAPDH mRNA was amplified as a control. An EtBr-stained agarose gel is shown.
Fig. 6.
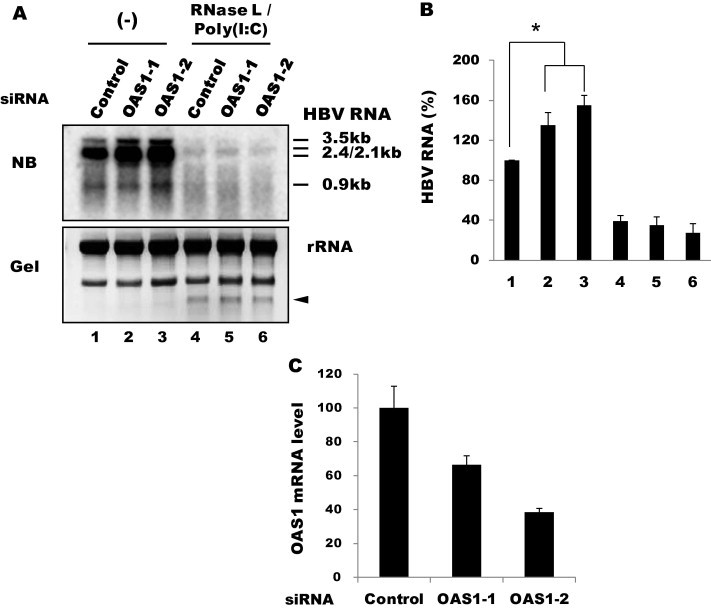
Effect of OAS1 knockdown. (A) Huh-7 cells were transfected with siRNAs (100 nM) against OAS1, followed by transfection 10 h later with HBV1.2 and RNase L. The cells were further transfected 24 h later with poly(I:C) (1 μg/well in a 6-well plate). RNA was extracted after 24 h. Viral transcripts were analyzed by northern blotting. (B) HBV RNA shown in Fig. 6A was measured using Multi Gauge V3.0 (Fuji film). Averages of triplicate experiments are shown as bars with standard errors indicated at the top (∗P < 0.05). (C) The knockdown efficiency of the siRNAs was determined by RT-qPCR.
Compared with OAS1, viral transcripts were partially but significantly rescued by OAS3 siRNAs, although smearing of the signals might be still indicative of RNA degradation, consistent with incomplete suppression of OAS3 (Fig. 7 ). This result indicated that OAS3 plays an important role in activating RNase L in the presence of poly(I:C). However, unlike OAS1, OAS3 knockdown had no effect without poly(I:C). Because both OAS1 and OAS3 are constitutively expressed in these cells prior to poly(I:C) treatment, as shown in Fig. 5, these results suggest that the two enzymes differ in their requirement for dsRNA; specifically, OAS3 might be activated more efficiently with poly(I:C). In vitro data indicated that OAS3 was more sensitive to dsRNA (∼100 times less dsRNA is required for activation) (Marie et al., 1997). To examine the role of OAS2, we stimulated its expression by IFN-α treatment following transfection with OAS2 siRNA. Viral RNA was partially rescued with one of the two siRNAs, and this rescue was correlated with the extent of OAS2 mRNA suppression (Fig. 8 ). IFN-α treatment alone did not show apparent inhibition of HBV transcripts, as we previously reported for this type of hepatoma cell (Park et al., 2011). Our results indicate that OAS2 and OAS3, but not OAS1, are dependent on poly(I:C) under these conditions. Thus, OAS family members differ in their requirement for dsRNA and contribute differentially to RNase L activation under different conditions.
Fig. 7.
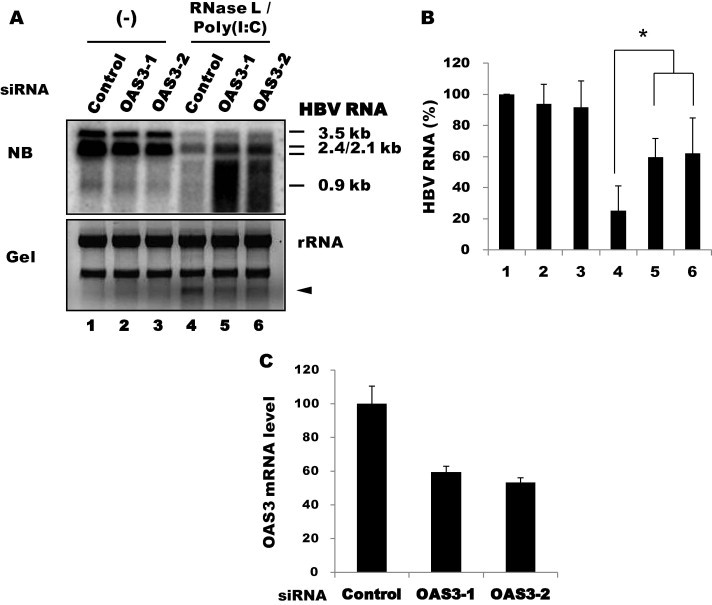
Effect of OAS3 knockdown. (A) Huh-7 cells were transfected with siRNAs (100 nM) against OAS3, followed by transfection 10 h later with HBV1.2 and RNase L. The cells were further transfected 24 h later with poly(I:C) (1 μg/well in a 6-well plate). RNA was extracted in 12 h. Viral transcripts were analyzed by northern blotting. (B) HBV RNA shown in Fig. 7A was quantified as above. Averages of triplicate experiments are shown as bars with standard errors indicated at the top (∗P < 0.05). (C) The knockdown efficiency of the siRNAs was determined by RT-qPCR.
Fig. 8.
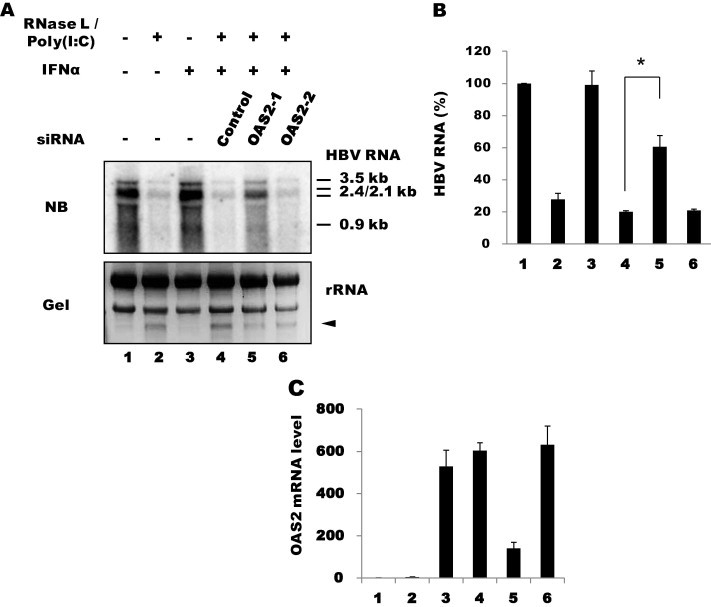
Effect of OAS2 knock down. (A) Huh-7 cells were transfected with poly(I:C) and OAS2 siRNAs, followed by transfection 12 h later with HBV1.2 and RNase L. The cells were then treated 12 h later with IFN-α (1000 U/ml). RNA was extracted in 24 h and analyzed by northern blotting. (B) HBV RNA shown in Fig. 8A was quantified as above. Averages of duplicate experiments are shown as bars with standard errors indicated at the top (∗P < 0.05). (C) The knockdown efficiency of the siRNAs was determined by RT-qPCR.
4. Discussion
Among IFN-related cellular factors, RNase L has been shown to inhibit a number of RNA viruses and few DNA viruses. In the typical antiviral pathway, viral dsRNA activates OAS, which in turn activates RNase L through the synthesis of 2–5A. It is thought that RNase L-mediated viral RNA decay leads to the inhibition of these viruses. In this study, we attempted to address whether the OAS/RNase L system plays a role in cellular restriction or IFN-mediated inhibition of HBV, although HBV is not known to carry or produce dsRNA. With the newly established HepG2-NTCP cell culture system, which permits HBV infection, we showed that viral gene expression and replication can be profoundly reduced by 2–5A- or poly(I:C)-mediated activation of RNase L. In HBV1.2-transfected Huh-7 cells, it was further confirmed that this type of inhibition occurred mostly through viral RNA degradation via the ribonuclease function of RNase L. In contrast to the effect of the specific ligand 2–5A, the poly(I:C)-mediated antiviral effect was not fully rescued by RNase L knock down. While the latter result implies that other effectors also contribute to the inhibition observed, it might be worth considering the ways by which poly(I:C) might have functioned in our study and in previous studies performed with HBV-transgenic animals. Intravenous administration of poly(I:C) resulted in a dramatic reduction of viral replication; however, no significant difference was observed between groups of RNase L+/+ or RNase L−/− animals with respect to the extent of inhibition (Guidotti et al., 2002). A subsequent study showed that poly(I:C) inhibited HBV through induction of type I IFN responses, as evidenced by intrahepatic ISG expression and requirement for the cytokine receptors in this process (Isogawa et al., 2005). While these data indicate that RNase L is not likely to mediate the antiviral activity of IFN against HBV, it was not determined whether the catalytic activity of RNase L was activated in these mice. Most likely, poly(I:C) that was injected intravenously into the animals triggered IFN induction via TLR signaling. However, the lack of apparent difference in viral replication between RNase L+/+or RNase L−/− animals (despite intrahepatic induction of OAS) suggested that RNase L was most likely not activated. Consistent with this notion, the steady state level of viral RNA was not affected in these animals, whereas HBV DNA was profoundly inhibited. In contrast, all forms of HBV RNAs were profoundly reduced in our cell-based assays, indicating the efficient activation of RNase L in poly(I:C)-transfected cells. This direct RNase L-activating effect of poly(I:C) was more evident in the Huh-7 cell, in which TLR3-mediated signaling is known to be inefficient (Li et al., 2005).
Our RNase L knockdown experiment performed in HepG2 cells, showed ∼50% rescue of poly(I:C)-mediated viral RNA reduction, suggesting that other factors in addition to RNase L contributed to the inhibition. Indeed, we observed strong induction of IFN-α and OAS2 mRNAs in HepG2 cells within 12 h of poly(I:C) transfection. Studies exploring how the IFN-mediated antiviral responses (those triggered by the secreted IFNs or by the direct activation of PRR with PAMP) inhibit HBV indicated that various steps of viral replication are affected (reviewed in Chang et al., 2012). Although many of these studies highlight posttranscriptional inhibition mechanisms, cellular effectors responsible for this type of regulation have not been identified. Recently, it was reported that zinc finger antiviral protein (ZAP), a cellular protein known initially as a retroviral restriction factor, inhibits HBV through the viral RNA decay process (Mao et al., 2013). While ZAP itself retains no ribonuclease activity, it is known to bind viral (and cellular) RNAs through its N-terminal domain, which contains four zinc finger motifs and stimulates RNA degradation by recruiting various host RNA processing machineries, including exosomes. According to these authors, expression of at least one isoform of ZAP was induced by IFN signaling, and ectopic expression of ZAP markedly reduced viral RNA. While RNase L differs from ZAP in that it requires specific ligands to activate its ribonuclease function, it is also possible that RNase L acts in combination with other cellular factors or as part of the cellular machinery during viral RNA decay.
Regarding the suggested relationship between naturally occurring RNase L gene variation (e.g., R462Q) and chronic hepatitis B (Arredondo et al., 2012), we did not find any defect in HBV inhibition with this variant in our transfection assay (data not shown). This result was consistent with the reduced 2–5A-mediated oligomerization activity but intact ribonuclease function of this allele (Xiang et al., 2003). We showed that OAS1 and OAS3 were constitutively expressed in Huh-7 cells before their expression was further induced, along with that of OAS2, by IFN-α treatment. Elevated OAS activity has been found in the sera of hepatitis C patients following IFN therapy, and the level of OAS activity was found to be correlated with their virological responses to this therapy (Kim et al., 2006). Among chronic hepatitis B patients, the frequency of a nonsense mutation in the R567 codon of OAS3 was reported to be higher among non-responders to IFN therapy compared with responders (Ren et al., 2011). Although we did not test this allele of OAS3, our knockdown data support the role of OAS3, along with that of OAS2, in the activation of RNase L.
In summary, our data indicated that HBV replication can be regulated through the activation of RNase L-mediated RNA decay pathways with exogenously provided ligands. Specifically, small molecules that mimic natural ligands (such as 2–5A) would represent a novel therapeutic strategy.
Acknowledgments
We are grateful to Wenhui Li for providing us with the NTCP-expressing plasmid. This work was supported by the Basic Science Research Program of the National Research Foundation (#2012-008126) of the Republic of Korea. Some preliminary studies were supported by a grant from Korea University in 2011–2012.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.antiviral.2014.01.021.
Appendix A. Supplementary data
This document contains supplementary information.
References
- Arredondo M., de Bethencourt F., Trevino A., Collado A., Torres P., Barbolla L., Soriano V., de Mendoza C. Short communication: RNASEL alleles and susceptibility to infection by human retroviruses and hepatitis viruses. AIDS Res. Hum. Retroviruses. 2012;28:1259–1261. doi: 10.1089/aid.2012.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha M.Y., Ryu D.K., Jung H.S., Chang H.E., Ryu W.S. Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J. Gen. Virol. 2009;90:978–986. doi: 10.1099/vir.0.009928-0. [DOI] [PubMed] [Google Scholar]
- Chakrabarti A., Jha B.K., Silverman R.H. New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 2011;31:49–57. doi: 10.1089/jir.2010.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Block T.M., Guo J.-T. The innate immune response to hepatitis B virus infection: implication for pathogenesis and therapy. Antiviral Res. 2012;96:405–413. doi: 10.1016/j.antiviral.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Dong B., Niwa M., Walter P., Silverman R.H. Basis for regulated RNA cleavage by functional analysis of RNase L and Ire1p. RNA. 2001;7:361–373. doi: 10.1017/s1355838201002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti L.G., Morris A., Mendez H., Koch R., Silverman R.H., Williams B.R., Chisari F.V. Interferon-regulated pathways that control hepatitis B virus replication in transgenic mice. J. Virol. 2002;76:2617–2621. doi: 10.1128/JVI.76.6.2617-2621.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogawa M., Robek M.D., Furuichi Y., Chisari F.V. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 2005;79:7269–7272. doi: 10.1128/JVI.79.11.7269-7272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.I., Kim S.R., Sasase N., Taniguchi M., Harada S., Kinoshita K., Kim S.H., Akimoto Y., Shikata M., Kimura N., Izawa S., Ohtani A., Nakao K., Motojima M., Kinoshita M., Hirai M., Ohzu M., Hirooka T., Nabeshima S., Ishii F., Tanaka K., Hotta H. 2′-,5′-Oligoadenylate synthetase response ratio predicting virological response to PEG-interferon-alpha2b plus ribavirin therapy in patients with chronic hepatitis C. J. Clin. Pharm. Ther. 2006;31:441–446. doi: 10.1111/j.1365-2710.2006.00761.x. [DOI] [PubMed] [Google Scholar]
- Koschel M., Oed D., Gerelsaikhan T., Thomssen R., Bruss V. Hepatitis B virus core gene mutations which block nucleocapsid envelopment. J. Virol. 2000;74:1–7. doi: 10.1128/jvi.74.1.1-7.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen H., Gad H.H., Eskildsen-Larsen S., Despres P., Hartmann R. The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J. Interferon Cytokine Res. 2011;31:41–47. doi: 10.1089/jir.2010.0107. [DOI] [PubMed] [Google Scholar]
- Kwon Y.C., Kang J.I., Hwang S.B., Ahn B.Y. The ribonuclease L-dependent antiviral roles of human 2′,5′-oligoadenylate synthetase family members against hepatitis C virus. FEBS Lett. 2013;587:156–164. doi: 10.1016/j.febslet.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Li K., Chen Z., Kato N., Gale M., Jr., Lemon S.M. Distinct poly(I–C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 2005;280:16739–16747. doi: 10.1074/jbc.M414139200. [DOI] [PubMed] [Google Scholar]
- Lok A.S., McMahon B.J. Chronic hepatitis B. Hepatology. 2007;45:507–539. doi: 10.1002/hep.21513. [DOI] [PubMed] [Google Scholar]
- Malathi K., Dong B., Gale M., Jr., Silverman R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao R., Nie H., Cai D., Zhang J., Liu H., Yan R., Cuconati A., Block T.M., Guo J.-T., Guo H. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathogens. 2013;9 doi: 10.1371/journal.ppat.1003494. e1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie I., Blanco J., Rebouillat D., Hovanessian A.G. 69-kDa and 100-kDa isoforms of interferon-induced (2′–5′)oligoadenylate synthetase exhibit differential catalytic parameters. Eur. J. Biochem. 1997;248:558–566. doi: 10.1111/j.1432-1033.1997.t01-1-00558.x. [DOI] [PubMed] [Google Scholar]
- Melegari M., Scaglioni P.P., Wands J.R. Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J. Virol. 1998;72:1737–1743. doi: 10.1128/jvi.72.3.1737-1743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I.H., Baek K.W., Cho E.Y., Ahn B.Y. PKR-dependent mechanisms of interferon-alpha for inhibiting hepatitis B virus replication. Mol. Cells. 2011;32:167–172. doi: 10.1007/s10059-011-1059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrillo R. Benefits and risks of interferon therapy for hepatitis B. Hepatology. 2009;49:S103–S111. doi: 10.1002/hep.22956. [DOI] [PubMed] [Google Scholar]
- Rasmussen S.B., Jensen S.B., Nielsen C., Quartin E., Kato H., Chen Z.J., Silverman R.H., Akira S., Paludan S.R. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene-like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 2009;90:74–78. doi: 10.1099/vir.0.005389-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S., Yu H., Zhang H., Liu Y., Huang Y., Ma L., Wei L., Wu H., Chen X. Polymorphisms of interferon-inducible genes OAS associated with interferon-alpha treatment response in chronic HBV infection. Antiviral Res. 2011;89:232–237. doi: 10.1016/j.antiviral.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Ryu D.K., Kim S., Ryu W.S. Hepatitis B virus polymerase suppresses translation of pregenomic RNA via a mechanism involving its interaction with 5′ stem-loop structure. Virology. 2008;373:112–123. doi: 10.1016/j.virol.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Sells M.A., Chen M.L., Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA. 1987;84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman R.H. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007;81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I.E., Zannetti C., Lucifora J., Norder H., Protzer U., Hainaut P., Zoulim F., Tommasino M., Trepo C., Hasan U., Chemin I. Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS One. 2011;6:e26315. doi: 10.1371/journal.pone.0026315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F., Wagner V., Rasmussen S.B., Hartmann R., Paludan S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Condit R.C., Vijaysri S., Jacobs B., Williams B.R., Silverman R.H. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 2002;76:5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Wang Z., Murakami J., Plummer S., Klein E.A., Carpten J.D., Trent J.M., Isaacs W.B., Casey G., Silverman R.H. Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′,5′-oligoadenylates. Cancer Res. 2003;63:6795–6801. [PubMed] [Google Scholar]
- Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H., Fu L., Song M., Chen P., Gao W., Ren B., Sun Y., Cai T., Feng X., Sui J., Li W. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;1 doi: 10.7554/eLife.00049. e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Jha B.K., Wu A., Elliot R., Ziebuhr J., Gorbalenya A.E., Silverman R.H., Weiss S.R. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe. 2012;11:607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Silverman R.H., Zhou A., Goto T., Kwon B.S., Kaufman H.E., Hill J.M. Increased severity of HSV-1 keratitis and mortality in mice lacking the 2–5A-dependent RNase L gene. Invest. Ophthalmol. Vis. Sci. 2001;42:120–126. [PubMed] [Google Scholar]
- Zhou A., Molinaro R.J., Malathi K., Silverman R.H. Mapping of the human RNASEL promoter and expression in cancer and normal cells. J. Interferon Cytokine Res. 2005;25:595–603. doi: 10.1089/jir.2005.25.595. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This document contains supplementary information.