

1. Abstract
Anti‐endothelial cell antibodies (AECA) represent a heterogeneous family of autoantibodies directed against structural endothelial proteins, as well as antigens adhering to endothelial cells. Although AECA immunoassays still show a high‐interlaboratory variability, several findings suggest a pathogenic role of these autoantibodies in diseases characterized by endothelial damage. In this chapter, we analyze the knowledge about AECA prevalence, clinical relevance, and their pathogenic role in autoimmune diseases focusing in particular on systemic lupus erythematosus, antiphospholipid syndrome, systemic sclerosis (SSc), and systemic vasculitis.
2. Introduction
An increasing body of evidences in the last two decades have been reported on the presence of autoantibodies directed to a heterogeneous family of structural endothelial proteins, as well as antigens adhering to endothelial cells (ECs) in patients with diseases characterized by endothelial damage (Fig. 1 ). Vascular ECs play an important role in many processes such as blood pressure regulation, coagulation, fibrinolysis, angiogenesis, and blood cell activation during physiological and pathological processes [1]. So far, anti‐endothelial cell antibodies (AECA) have been correlated with both disease activity and vascular involvement in several diseases. Much effort has been made to determine whether AECA mediated‐mechanisms underlie the development of diseases or merely represent markers of vascular damage. Intriguingly, endothelial antigens are readily accessible to circulating antibodies and a pathogenic role of AECA has been suggested by several authors. In particular, recent findings suggest that AECA may activate ECs by an upregulation of the cell adhesion molecule (CAM), or by inducing the secretion of proinflammatory cytokines. It has been suggested that AECA might also activate ECs through an interaction with toll‐like receptor 4 (TLR4) because of the molecular mimicry between beta2‐glycoprotein I (β2‐GPI) and microbial structures that represent the natural ligand of TLR4. Finally, controversial results have been reported regarding the induction of ECs apoptosis by AECA, although it is well known that these autoantibodies bind to apoptotic ECs, thus increasing phagocytosis by macrophages and clearance of apoptotic cells [[2], [3], [4]].
Fig. 1.
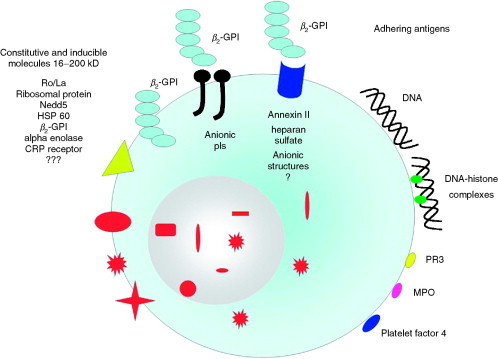
Target antigens of AECA. AECA represent a heterogeneous group of antibodies direct to structural endothelial proteins (see left side of the figure), as well as to antigens adhering to endothelial cells directly or through endothelial receptors (see right side of the figure). Nedd5, intracytoplasmatic protein of the septin family; β2‐GPI, β2‐glycoprotein‐I; HSP 60, heat shock protein 60; CRP, C‐reactive protein; PR3, proteinase 3; MPO, myeloperoxidase.
In this chapter, we analyze the knowledge about AECA in autoimmune diseases focusing in particular on their pathogenic role in systemic lupus erythematosus (SLE), antiphospholipid syndrome (APS), systemic sclerosis (SSc), and vasculitis.
3. Methods of AECA Detection
Since the discovery of AECA, several studies have focused on their role as diagnostic tools for diseases characterized by an inflammation of blood vessels. Despite the growing interest in this field, AECA immunoassays still show a high‐interlaboratory variability, most probably due to the lack of international standardization. Many methods have been developed to detect AECA, including immunofluorescence, immunoassays (ELISA and RIA) using fixed cells, Western blot (WB), and fluorescence‐activated cell sorter analysis (FACS). Nevertheless, only a small number of comparative studies have been done and data concerning sensitivity and specificity of methods to detect AECA are frequently variable and inconclusive (reviewed in Ref. [5]). The major part of our knowledge on AECA reactivity comes from in vitro experiments with human umbilical vein ECs (HUVECs). Nevertheless, the isolation of primary HUVECs is laborious and the AECA reactivity obtained with different donors cannot easily be compared to each other because of their different origin. Moreover, HUVECs present a short life span and can be kept in culture only for short periods. Another point of debate is the fixation procedure of HUVECs, which may influence the AECA reactivity against intracytoplasmic or membrane antigens. Interference from heterophilic antibodies, which recognize fetal calf serum (FCS) proteins, may be avoided by adding FCS to the dilution buffer [6]. Finally, different ECs from macro‐ and microvasculature have been used for AECA detection with different results. ECs from diverse tissues are also heterogeneous not only in respect to small vs large vessels but also in respect to their surface phenotype and protein expression. For instance, Renaudineau et al. [7] showed the highest prevalence of AECA reactivity in a large series of patients with SSc using microvascular bone marrow ECs in comparison to HUVECs. The need for reproducible results has increased the demand for immortalized EC lines, such as EA.hy926, generated by fusion of HUVECs with the human lung carcinoma cell line A549 [8], or the immortalized human microvascular EC line (HMEC‐1) [9]. ECs derived from human umbilical and iliac veins and arteries transfected with a plasmid containing the Simian Virus 40 large T‐antigen, may also be useful for the detection of AECA [10]. Immortalized human glomerular endothelial cell (HGEC) lines were also generated [11]. These cell lines maintain the morphologic and functional characteristics of HGECs even after several weeks in culture, thus providing a standardized substrate for AECA detection. Nevertheless, AECA detection on immortalized ECs may be subject to great variability and scientists interested in autoimmunity against ECs would prefer primary EC lines as substrate because these cells probably present more primary endothelial traits. Although the original cyto‐ELISA remains the most widely used method for the detection of AECA, it is advisable to confirm a positive result using other methods, such as flow cytometry, immunoprecipitation, or WB, which appear to be difficult to use on a routine basis [12]. It is important to point out that heating sera should be avoided because this treatment increases AECA binding by a nonspecific mechanism in sera from both patients and healthy individuals [13]. ANA or RF in serum samples have been shown not to interfere with AECA detection by ELISA with nucleus‐depleted lysates prepared from EA.hy926 [14].
Finally, because it has been reported that different AECA isotypes may be prevalent in different autoimmune disorders, as discussed elsewhere in this chapter, it should be pointed out that the AECA test should include determination of both IgG and IgM in most disorders with the addition of IgA in patients with Henoch‐Schonlein purpura (HSP). In fact, while IgG AECA are predominant in most autoimmune conditions, IgM AECA have been shown to be particularly increased in Kawasaki disease (KD) probably reflecting the acute nature of this disorder. In addition, IgG and IgM AECA may exert diverse pathogenic role due to their differential ability to fix and activate the complement cascade as well as to induce ECs damage via antibody‐dependent cytotoxicity.
4. AECA in Autoimmune Diseases
4.1. AECA in Systemic Lupus Erythematosus and Antiphospholipid Syndrome
SLE is a protean autoimmune disease characterized by the largest number of detectable autoantibodies reactive with antigens localized in the nucleus, cytoplasm, or on the cell membrane (reviewed in Ref. [15]). Anti‐dsDNA antibodies, and complement consumption are established markers of disease activity. A large number of studies have focused on the association of autoantibodies with specific symptoms of SLE: for example, it is well known that anti‐dsDNA antibodies, as well as antibodies to the collagen‐like region of the complement component C1q (anti‐C1q), are associated with renal involvement in SLE [16]. Moreover, anti‐Ro antibodies have been found to be a marker of neonatal lupus and subacute cutaneous lupus, and anti‐β2‐glycoprotein I (anti‐β2‐GPI) and anti‐cardiolipin (aCL) antibodies were significantly correlated with clinical thrombosis, abortion, and fetal loss. Despite the fact that most studies have shown a higher prevalence of AECA in SLE, the clinical relevance of these autoantibodies is still a matter of debate.
The prevalence of AECA in SLE largely range from 15% to 80%, depending on the method and patient selection. Nonetheless, the presence of vasculitis in SLE is frequently associated with AECA although these autoantibodies have been correlated with several other clinical manifestations of the disease such as lupus nephritis, pleurisy, thrombocytopenia, and neuropsychiatric symptoms [[17], [18], [19]]. Inflammatory or thrombotic vascular injury represents one of the most frequent lesions in SLE and endothelial perturbation induced by AECA may be involved in the earliest events in the pathophysiology of vascular tissue damage. Vessels of any size may be affected and therefore the clinical scenario of vasculitis in SLE is extremely wide. In 1987, Hashemi and coworkers described an association between AECA and vasculitis [20]. Thereafter, Del Papa et al. [21] showed that sera from SLE patients with vasculitis immunoprecipitated EC surface proteins ranging from 200 to 25 kDa. Song and coworkers described a higher positivity rate of AECA in patients with digital vasculitis as previously reported [[19], [22]]. In this study IgG‐AECA showed a positive correlation with disease activity in SLE patients. Moreover, a significant correlation between the IgG‐AECA titer and SLE disease activity index (SLEDAI) score modification was also observed in SLE patients after treatment [19]. Thus, AECA may be useful in the follow‐up of patients; other soluble markers of EC dysfunction, such as von Willebrand factor, thrombomodulin, and soluble E‐selectin, seem to be surrogate markers (an epiphenomenon) related to EC damage [23]. It is possible that AECA may represent a marker of cell injury or a sign of polyclonal activation of humoral immune responses in other diseases such as in liver transplantation where AECA correlate with cytomegalovirus (CMV) infection [[24], [25]] and are associated with higher frequency of humoral allograft rejection [25]. AECA have been found in patients with SLE and urticarial vasculitis (UV) as well as in hypocomplementemic urticarial vasculitis syndrome (HUVS) [26]. D'Cruz and colleagues described a high prevalence of AECA, associated with reduced serum levels of complement, in SLE patients with UV, but not in primary UV or in SLE patients without UV [26]. The binding of AECA to ECs may be accompanied by the formation of immune complexes and complement fixation resulting in tissue damage. The anti‐C1q antibodies seem to be a specific marker for HUVS [[26], [27]], but they may also be detected occasionally in SLE patients (with or without UV). It has been suggested that anti‐C1q antibodies could contribute to EC damage by cross‐linking C1q bound to the immune complexes on the surface of ECs [28].
The presence of AECA has also been associated with thrombotic vascular damage in patients with SLE as well as in primary or secondary APS [29]. In this regard, Dieude et al. showed that AECA from SLE patients recognized heat‐shock protein 60 (HSP60) on the surface of HUVECs and induced apoptosis. A relationship between a high titer of anti‐HSP60, the presence of lupus anticoagulant (LAC), and thrombosis in SLE patients was also observed [3]. Thus, the authors suggest that, in the pathophysiologic pathway of thrombosis, the induction of apoptosis by anti‐HSP60 may represent the initial event that leads to aPL binding to the anionic phospholipid on the EC surface which may initiate the thrombotic cascade [3].
Anti‐endothelial protein C receptor (EPCR) autoantibodies can be detected in APS patients and represent a novel risk factor for fetal death [30]. A higher percentage of AECA positivity was also observed in Sneddon's syndrome, a disease characterized by livedo reticularis and ischemic cerebral vascular disease [31].
It has been suggested that AECA of various isotypes may be associated with lupus nephritis in SLE [[17], [32], [33]]; in particular, the highest AECA levels have been detected in a large series of SLE patients with diffuse proliferative glomerulonephritis at the time of renal biopsy or with proteinuria and nephrotic syndrome [17]. AECA showed the highest titer in active lupus, whereas the mean antibody titer fell significantly as patients entered remission [34]. These findings have been further validated by a prospective study that suggested a role for AECA, in association with other laboratory parameters, as a marker of disease activity in SLE [35]. Autoantibodies reacting in immunoblot analysis with both 27‐ and 29‐kDa endothelial cell antigens were correlated to the presence of glomerular capillary thrombi and heavy proteinuria [36]. In another study, patients with lupus nephritis, vasculitis, and hypocomplementemia showed IgG‐AECA against a 66‐kDa membrane antigen [18]. Antibodies against ribosomal P protein P0 have been identified by a molecular cloning strategy in SLE patients with active disease and nephritis [37]. In one patient, these autoantibodies correlated with total AECA levels as well as with anti‐DNA antibody, disease activity, and renal involvement in a longitudinal follow‐up. Nevertheless, to date, positive findings of both anti‐dsDNA and anti‐C1q antibodies are of higher specificity for active nephritis in SLE patients [16].
aPL, anti‐β2‐GPI, and AECA have been associated with various neurological manifestations in patients with SLE. These in vivo observations were further validated by a mouse model of APS, which developed neurological dysfunction and hyperactive behavior associated with aPL, anti‐β2‐GPI, and AECA after passive immunization with human aCL mAb [38]. We demonstrated an association between the presence of AECA and psychiatric manifestations, such as psychosis and depression in SLE, suggesting a possible organic mechanism underlying the psychiatric symptoms [39]. In our study no significant correlation was found between aCL, anti‐β2‐GPI, anti‐Ro, antiglial fibrillary acidic protein, antiribosomal P protein, anti‐dsDNA, and anti‐nucleosome antibodies and psychiatric involvement in SLE patients. Moreover, no association was found between AECA reactivity and aCL and anti‐β2‐GPI antibodies. Remarkably, by screening a cDNA library from human umbilical artery ECs (HUAECs) with serum from an SLE patient with acute and active psychosis and elevated AECA serum level, we further identified one strongly reactive clone encoding the C‐terminal region (C‐ter) of Nedd5, an intracytoplasmic protein of the septin family [40]. IgGs specific to Nedd5 C‐ter were present in 14 of 51 SLE patients (27.4%) by ELISA and the mean IgG reactivity to this protein was significantly higher in SLE patients with psychosis or mood disorders. On the contrary, no correlation was observed between the presence of anti‐Nedd5 C‐ter antibodies and the other clinical features. The results of these studies suggest a relationship between AECA and anti‐Nedd5 antibodies with neuropsychiatric involvement in SLE, supporting the hypothesis of a biological origin of these disturbances.
4.2. AECA in Systemic Sclerosis
Systemic sclerosis is a multisystemic disorder of the connective tissue characterized by immune abnormalities, microvascular injury, and fibrotic changes involving skin and visceral organs, such as the lungs, heart, gastrointestinal tract, and kidneys. It is well recognized that vascular injury appears frequently in the clinic course of the disease—that is, Raynaud's phenomenon (RP)—and that vascular dysfunction represents one of the primary events in the pathogenesis of the disease. Thus, it is not surprising that a significant amount of research by both clinical and basic scientists has focused on the identification of the clinical parameters and the pathogenic mechanisms of endothelial dysfunction, with AECA displaying great relevance in both fields of investigation.
AECA reactivity in scleroderma is quite common, having been detected with an overall prevalence varying from 19% to 85%, depending on the method used (ELISA, WB, IF), the Ig isotypes investigated, the substrate (tissue sections, source of human ECs) used, as well as the variability in the subtypes of SSc patients included (limited vs diffuse SSc) in the different studies (reviewed in Ref. [41]).
Considerable effort has focused on the identification of associations between AECA positivity and the clinical manifestations of SSc. An early study involving 31 patients with diffuse SSc (dSSc), 36 with limited SSc (lSSc) and 13 with primary RP (PRP) reported an increased prevalence of AECA with increased severity of clinical manifestations related to SSc [42]. If the analysis was limited to AECA IgG, these authors observed an AECA prevalence of 15% in patients with isolated RP, 39% in lSSc, and 77% in dSSc. An increased prevalence of AECA in dSSc compared to lSSc was also reported by Negi et al. [43] in 76 patients with SSc with a prevalence of 40% vs 13.5%. Although these results suggested an important role for AECA in identifying different clinical subsets of SSc, they have not been confirmed in later studies, in which no difference in AECA reactivity was observed between patients with lSSc and dSSc [[44], [45]].
Although the utility of AECA in differentiating the main clinical subsets of SSc is debatable, significant associations between AECA reactivity and some of the clinical manifestations of SSc have been consistently reported [[42], [43], [44], [45]].
AECA were found to closely associate with capillaroscopy abnormalities as well as with severe digital ischemia and digital ulcers [[42], [43], [44]]. Together with vascular damage, the most striking clinical association of AECA observed in SSc relates to lung involvement. This is particularly important since lung involvement, manifested by either interstitial fibrosis and/or pulmonary hypertension, represents the most frequent cause of death in SSc [46].
Although evaluating lung involvement according to different clinical parameters and diagnostic procedures, at least four reports have demonstrated a strict association between AECA and one or more parameters of lung disease in SSc.
Salojin et al. [42] demonstrated lung involvement in 93% of AECA positive patients with SSc but only in 30% of those displaying no serum AECA reactivity. Unfortunately, no further characterization of lung disease (i.e., isolated reduction in diffusing capacity for carbon monoxide (DLco), pulmonary fibrosis, or pulmonary hypertension) and association with AECA was available in this study. Negi et al. [43] found a significant correlation between AECA and pulmonary arterial hypertension; Pignone et al. [44] reported a strong association with both reduced DLco and with pulmonary artery hypertension but not with pulmonary fibrosis. This observation is of particular interest considering that isolated DLco reduction has been indicated as a predictor of subsequent development of pulmonary hypertension [47]. Finally, Ihn et al. [45] observed a significant association between AECA and reduced percent vital capacity (%VC), decreased DLco as well as pulmonary fibrosis, with the strongest association with severe interstitial lung disease.
Although prospective studies have not yet been carried out, all the available data suggest that AECA may have a profound clinical relevance in identifying SSc patients at major risk of developing more advanced vascular damage and severe pulmonary disease manifested either as interstitial fibrosis and/or pulmonary hypertension. Longitudinal studies on a large number of patients are awaited to confirm the possible clinical utility of AECA as disease markers in SSc.
Putative antigens responsible for AECA reactivity in sera of SSc patients have been identified by several groups as protein bands of various molecular weights obtained by WB analysis of sera reacting against cytoplasmic or membrane proteins extracted from HUVECs or ECs from other sources. However, in the absence of a molecular biology approach to identify the nature of the antigens observed, the exact endothelial antigenic targets recognized by AECA in SSc remain elusive. A first study reported the identification of several membrane endothelial cell specific antigens (more than 20 different bands at WB analysis) as the target of AECA in SSc patients [48]. In particular, reactivity against a membrane antigen of ∼19 kDa was observed in more than 50% of the patients with SSc and in 100% of SSc patients with CREST syndrome and was shown to display anticentromere activity in IFI analysis. Although interesting, this observation has not been confirmed by subsequent reports. Ihn et al. [45] using HUVEC protein extracts observed reactivity of AECA positive sera, as previously assessed by cyto‐ELISA on HUVECs, toward four major antigens recognizable as WB bands of 60, 90, 110, and 140 kDa. The most common response was observed against the 90‐kDa antigen and purified anti‐90 kDa antibodies displayed a cytoplasmic pattern at IFI identical to that observed in AECA positive sera. No such reactivity was observed using the same sera toward protein extracted from dermal fibroblasts, suggesting specificity of the immune response against endothelial antigens. Wusirika et al. [49] reported reactivity of SSc sera with pulmonary involvement against multiple bands between 38 and 110 kDa obtained by WB of protein extracts from HUVECs and pulmonary microvascular ECs. Although no further characterization of AECA antigenic target in SSc is provided in these reports, it is clear that, as observed in other autoimmune conditions, AECA reactivity in SSc is rather heterogeneous and only the identification of the exact antigens responsible for AECA activity of SSc sera may provide important clues in understanding the pathogenic mechanisms underlying endothelial dysfunction and aggression in this disorder.
4.3. AECA in Systemic Vasculitis
Detection of AECA in sera of patients affected by systemic vasculitis is a relatively common finding, with prevalence varying greatly among the different forms of vasculitis.
Early reports demonstrated the ability of sera from children with acute KD to lyse HUVECs when pretreated in vitro with either IFNγ or monokines [[50], [51]]. This effect was observed only in patients with acute febrile KD but not in the convalescent phase of the disease and was dependent on the presence of circulating IgM with AECA activity. However, later studies provided conflicting evidence on the effective role of AECA in KD. Guzman et al. [52] observed AECA in just 17% of 22 patients with KS and AECA were not able to differentiate KD from other febrile disorders of childhood resembling KD. Similar findings were reported by Nash et al. [53] in 58 children with acute KD, which showed no increased AECA reactivity compared to 35 children with febrile infections. Conversely, Kaneko et al. [54] detected IgM AECA in 73% of 22 KD children, half of which displayed complement dependent cytotoxicity in vitro against EC. Finally, Falcini et al. [55] reported AECA reactivity in 26% of KD patients, with increased incidence in the acute compared to the convalescent phase while Fujieda et al. [56] observed IgM AECA in 42% and IgG AECA in 26% of KD children, confirming the increased presence of IgM with AECA reactivity in the acute phase of KD. Although the effective role of AECA in KD is still matter of debate, there is sufficient evidence to support the concept that IgM AECA rise in a subset of patients in the acute febrile phase of the disease and may participate to endothelial inflammation leading to arteritis in KD.
More consistent evidence has been reported on AECA in anti‐neutrophil cytoplasmic antibody (ANCA)‐associated systemic vasculitis. Ferraro et al. observed AECA, mostly of IgG isotype, in 60% of sera of patients with microscopic polyangiitis (MPA) and 40% of those with Wegener's granulomatosis (WG). Importantly, absorption of ANCA did not affect endothelial binding of AECA suggesting that AECA and ANCA represent two different populations of autoantibodies [57]. Frampton et al. described increased AECA levels in ANCA‐associated vasculitis with a significant correlation between the two antibodies. In addition, both AECA and ANCA correlated with disease activity [58]. These earlier results were later confirmed in an extensive evaluation of AECA reactivity in sera from 168 patients with ANCA‐positive MPA or WG. AECA were present in about 60% of patients with ANCA‐associated vasculitis (59% IgG and 68% IgM). Interestingly, and differently from KD, AECA from these patients were not able to induce complement‐dependent cytotoxicity, even after pretreatment of ECs with cytokines, but a minority of sera induced an antibody‐dependent cytotoxicity [59], as described elsewhere in this chapter. Although Del Papa et al. [21] observed in the majority of AECA‐positive WG patients a relatively conserved precipitation pattern by WB analysis of EC surface extracts with five major bands of 25, 68, 125, 155, and 180 kDa, this finding awaits confirmation in larger studies; thus, as in other autoimmune conditions, the nature of the antigen(s) recognized by AECA in ANCA‐associated vasculitis is currently unknown.
Two studies prospectively evaluated AECA (and ANCA) levels in patients with ANCA‐associated vasculitis and assessed their relationship with disease activity [[60], [61]]. Both reports demonstrated that AECA were present in the majority of patients during the observation period and fluctuation of AECA was correlated with disease activity, with increased titer in disease relapses and reduced levels during remission phases. Although encouraging, these results do not provide definitive evidence on the possible role of AECA, alone or in association with ANCAs, as predictive factors of clinical relapses in ANCA‐associated vasculitis.
Less information is available regarding the role of AECA in other forms of systemic vasculitis. In Takayasu arteritis (TA), Eichhorn et al. [62] reported a striking incidence of AECA of 95%, but this finding was not confirmed in a later study reporting an incidence of 33% [63]. Limited evidence is also available for Churg–Strauss syndrome (CSS), with Schmitt et al. [64] reporting AECA in 50% of 16 CSS patients but, in contrast with ANCAs, without any association with disease activity.
Finally, interesting findings have been reported regarding the presence of IgA AECA in patients with HSP. Fujieda et al. [65] observed IgA AECA in nearly 50% of HSP patients with nephritis but, interestingly, in no HSP patients without nephritis. Similar results were obtained by Yang et al. [66] who detected AECA IgA in 45% and 35% using HUVECs and human dermal microvascular endothelial cell (HDMECs) as substrates, respectively, with HUVEC AECA IgA titers correlating significantly with disease activity.
In summary, detection of AECAs in sera of patients affected by systemic vasculitis is a relatively common finding, with prevalence varying greatly among the different forms of vasculitis. Some of the differences observed may be related to the prevalent use of HUVEC as a substrate to detect AECA, despite the evidence that different form of systemic vasculitis target preferentially vessels of different size. In the future, evaluation of AECA using as substrates ECs from macro‐ as well as microvasculature may be important to assess more precisely AECA reactivity in the various forms of vasculitis. In addition, this approach may also provide information on the preferential localization of antigenic targets in different forms of vasculitis and allow the identification of new antigenic specificities of AECA, which may reveal of pathogenic importance. In this regard, although inconclusive, preliminary attempts to detect AECA by using ECs from different sources in various autoimmune conditions suggest that this may represent a promising experimental approach [[7], [49], [66], [67]].
4.4. AECA in Other Diseases
AECA have been described in a number of systemic autoimmune‐inflammatory diseases other than those already mentioned. For instance, increased AECA production was observed in patients with mixed connective tissue disease (MCTD) [67]. AECA from MCTD patients may activate ECs by the upregulation of E‐selectin expression and may be implicated in the pathogenesis of MCTD. D'Cruz et al. [68] described an interesting association between AECA levels and interstitial lung disease in all forms of idiopathic inflammatory myopathies. In Behcet's disease (BD), the reported prevalence of AECA varied widely [69]. A significant association between AECA and thrombosis and vasculitis as well as disease activity in BD has been reported [[70], [71]]. Dinc et al. [72] described a low prevalence of AECA in a large series of Turkish Behcet's patients without significant differences from healthy controls. A new technique was described by Lee et al. [73] demonstrating for the first time that AECA IgM from sera of patients with active BD reacted strongly with human alpha‐enolase, a protein of the HDMEC.
AECA were frequently detected in inflammatory bowel disease, and although they were associated with both active and extensive colitis, they seem to be less disease specific than other autoantibodies [74]. Nevertheless, at least in Crohn's disease, intestinal vascular injury mediated by AECA may be an important event [75].
Increasing evidence is accumulating on the possibility that AECA may play an important role in the development and progression of atherosclerotic lesions. Farsi et al. [76] described a higher rate of AECA in patients with unstable angina than in those with effort angina. Only 3 out of 12 AECA positive patients were also positive for anti‐β2‐GPI supporting the idea that β2‐GPI is not the major AECA target antigen. Thus, AECA may contribute to the instability of angina and are associated with the high‐restenosis rate after percutaneous transluminal coronary angioplasty. In contrast, patients with or without coronary atherosclerosis, confirmed by angiography, showed similar AECA levels [77]. The target antigen of AECA in acute myocardial infarction may be the endothelial protein C receptor, as reported by Montes et al. [78].
The AECA seem to be involved in the complex pathogenesis of vascular complications in diabetes [79], although others have suggested that they are only associated with coexisting autoimmune disorders [80]. A putative cytotoxic role of AECA vs ECs of the inner ear in immune‐mediated sudden sensorineural deafness has been suggested [[81], [82]].
Understanding the relationship between autoimmune disease and infection has been a topic of interest for several decades. It is noteworthy that autoimmune diseases may be caused or triggered by infections. AECA are a common finding in several infections, such as HCV infection where these autoantibodies are often associated with mixed cryoglobulins [83]. Moreover, AECA associated with aCL, but not anti‐β2‐GPI, were found in HIV infection [84]. AECA and anti‐epithelial human cell antibodies were described in severe acute respiratory syndrome (SARS) caused by infection with the SARS‐associated coronavirus and in patients with leprosy [[85], [86]].
Natural AECA were described in healthy subjects and Ronda et al. [87] showed that these antibodies interact with living ECs, affecting their function after a ligand–receptor‐like mechanism of internalization. Natural AECA showed a more restricted pattern of reactivity compared to AECA from SLE patients [88]. A significantly higher level of IgM‐AECA was found during normal pregnancy compared with that in healthy nonpregnant controls and pregnant patients with SLE [89]. On the contrary, IgG‐AECA levels were significantly higher in the serum of normal pregnant women and pregnant SLE patients than in the serum of healthy nonpregnant controls. Taken together, these findings support a physiological role of AECA in fetal tolerance of normal pregnant women, as well as a pathogenic role in impaired reproductive function frequently found in pregnant SLE.
5. Pathogenic Mechanisms of AECA
5.1. Cytotoxicity and Apoptotic Effects of AECA
The pathogenic ability of AECAs to induce an endothelial perturbation is suggested by the demonstration of complement‐dependent cytotoxicity on HUVECs from sera of patients with systemic vasculitis [[50], [51], [54], [57], [90]] and SLE [91]. In particular, early reports in the 1980s demonstrated that purified IgM and, to a much lesser extent, IgG from patients with acute but not convalescent KD were able to lyse HUVECs, assessed by 111In release, in the presence of fresh (but not heat inactivated) rabbit serum. Conversely, the simple incubation of KD IgM or IgG failed to induce any direct cytotoxicity; in addition, the lytic effect was observed only when HUVECs were prestimulated with either IFNγ [50] or monokines [51] suggesting that exposure of neo‐antigen on endothelial cells was necessary for the binding of cytotoxic antibodies. The different preincubation time required for IFNγ or monokines inducing binding of AECA and conferring cytotoxicity in vitro and absorption experiments with IFNγ‐ or monokine‐treated HUVECs suggested that different epitopes were recognized by AECA from KD following different HUVECs stimulation. Similar results were reported in SLE by Moscato et al. [91] who showed that SLE sera and purified IgGs were able to fix complement in vitro leading to deposition of the complement component C3 and disruption of the endothelial monolayer. Noteworthy, differently from early reports on KD sera, this effect was observed in unstimulated HUVECs under basal conditions and suggests that cytotoxic SLE AECA may recognise different epitopes on ECs, which do not require endothelial activation by inflammatory cytokines.
While complement‐dependent cytotoxicity may be an important mechanism by which AECA induce endothelial perturbation and cell damage in SLE and systemic vasculitis, the ability of AECA to fix complement and induce complement‐dependent cytotoxicity have not been always confirmed in other autoimmune diseases. In particular, reports in the mid‐ and late 1980s failed to demonstrate the ability of sera from patients with SSc to induce complement‐dependent cytotoxicity in vitro. However, it has been shown that other mechanisms, namely antibody‐dependent cellular cytotoxicity (ADCC) and induction of ECs apoptosis, may account for the pathogenic potential of AECA from SSc and other autoimmune diseases, including SLE and vasculitis.
The ability of AECA to induce ECs damage via an ADCC‐like mechanism was originally demonstrated in the 1980s by in vitro evaluation of the cytotoxic effect of SSc sera on HUVEC monolayers [[92], [93], [94], [95], [96]] (Fig. 2A ). The cytotoxic activity on ECs of either venous or arterial origin was assessed by morphological changes, decreased DNA [3H]thymidine incorporation, reduced fibronectin production, or increased 51Cr release and was present only in a minority of sera from SSc patients, ranging from 19% to 41%. In some cases, the same sera were also able to induce a cytotoxic effect on dermal fibroblasts [92], an observation which may have relevance in understanding the mechanisms leading to skin fibroblast dysfunction and which can be partially explained by possible cross‐reactivity between endothelial and fibroblast antigens as suggested by the evidence that absorption of purified IgG from SSc sera on dermal fibroblasts greatly reduced reactivity toward ECs [97]. Although some differences in the various reports exist, it is generally convincing that whole sera or purified IgG from SSc patients were not able to induce a cytotoxic effect on ECs per se or in the simple presence of complement, but that the cytotoxic mechanism was dependent on the presence of effector cells in the culture medium [[96], [97]]. In vitro coculture experiments using ECs, SSc sera, and peripheral blood mononuclear cells (PBMCs) demonstrated that the cytotoxicity was dependent on IgG with AECA reactivity and the presence of Fc receptors on the effector cells with a mechanism highly reminiscent of ADCC [[93], [94], [96]]. The pathogenic relevance of these observations in determining endothelial dysfunction in vivo in patients with SSc is still not known; as discussed previously, only a minority of SSc sera display cytotoxic activity in vitro.
Fig. 2.
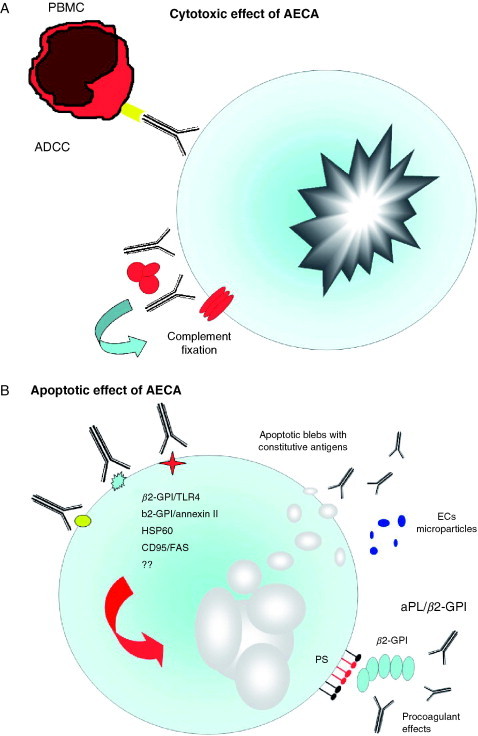
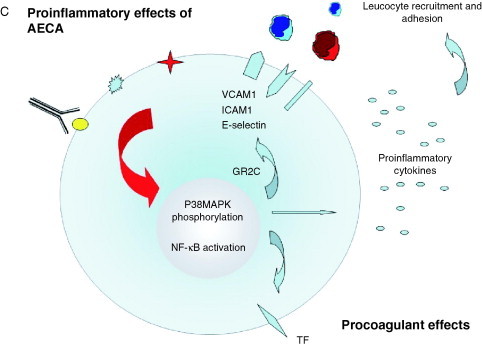
Pathogenic mechanisms of AECA. The binding of AECA to ECs might be able to induce several effects: (A) cytotoxicity through a mechanism resembling ADCC or mediated by complement fixation; (B) apoptosis through interaction with endothelial adhering or inducible antigens (i.e., β2‐GPI and HSP 60) indirectly mediated by Toll 4 and annexin II receptor. Moreover, EC apoptosis may be induced directly through interaction of AECA with CD 95/FAS. As a consequence of EC apoptosis both constitutive antigens in apoptotic blebs and β2‐GPI–anionic phospholipid complex are exposed to the immune system on the endothelial surface. Moreover, endothelial microparticles may be released from ECs; and (C) proinflammatory and procoagulant effects with overexpression or induction of several adhesion molecules, cytokines, and tissue factor, respectively. β2‐GPI, β2‐glycoprotein‐I; TLR4, toll‐like receptor 4; aPL, antiphospholipid antibodies; PS, phosphatidylserine; ADCC, antibody‐dependent cellular cytotoxicity; PBMC, peripheral blood mononuclear cells; TF, tissue factor.
Similarly, Del Papa et al. [98] showed that IgG AECA from patients with WG, can be cytotoxic for ECs in the presence of human normal PBMC but not in the presence of polymorphonuclear leukocytes or adherent mononuclear cells. Nevertheless, it is well known that AECA represent an extremely heterogeneous family of autoantibodies, not only because of the variety of their target antigens, but also for the heterogeneity of their effects [99].
Together with cytotoxicity, apoptosis of ECs mediated by binding of AECA has emerged as a novel mechanism by which AECA may exert a pathogenic role in autoimmune conditions (Fig. 2B). In particular, studies have focused on the apoptotic effect of AECA from SSc patients, since Sgonc et al. [100] provided evidence that ECs in the deeper dermis of patients with SSc undergo apoptosis at very early stages in the course of the disease process, and only in areas characterized by early or prefibrotic skin lesions. These results were also confirmed in an avian model of SSc, University of California at Davis (UCD) lines 200/206 chicken, which spontaneously developed a scleroderma‐like disease characterized by perivascular lymphocytic infiltration of the dermis with fibrosis of skin and internal organs and the presence of serum autoantibodies such as antinuclear antibodies, aCL antibodies, and AECA [[100], [101], [102], [103]]. In addition, in most cases, endothelial cell apoptosis was associated with positive staining of microvessels for Ig, supporting the hypothesis that AECA could mediate vascular damage. Sgonc et al. [104] later demonstrated that AECA from a subset of patients with SSc were able to induce apoptosis of HDMECs, but not HUVECs, through ADCC via CD95/Fas, as demonstrated by in vitro inhibition of NK‐mediated EC apoptosis with the use of blocking anti‐FasL antibodies.
5.2. Proinflammatory Effects of AECA
There is convincing evidence to support the notion that AECA are able to induce overexpression of endothelial adhesion molecules, such as E‐selectin, ICAM‐1, VCAM‐1, as well as secretion of proinflammatory cytokines by ECs leading to an upregulation of leukocyte adhesion to ECs (Fig. 2C). This suggests an important role for AECA in contributing to vascular damage in autoimmune conditions such as SLE as well as in other form of vasculitis [[105], [106], [107], [108]]. In particular, AECA from patients with WG induced the secretion of IL‐1β, IL‐6, IL‐8, and MCP‐1 by ECs [109]. Several of these effects may be related to an autocrine or paracrine action of IL‐1 generated by ECs in response to AECA [110]. Moreover, IgM AECA may stimulate endothelin‐1 release from HUVECs, thus increasing vascular damage [111] as suggested in preeclampsia [112]. Expression of potential antigens on ECs has been suggested because the treatment of these cells with tumor necrosis factor alpha increased IgG binding from sera of some patients with active SLE [113]. The heterogeneity of the AECA effects was observed by Bordron et al. [99] who clearly showed that AECA from different patients induced activation of ECs, while AECA from others induced apoptosis. The activation of ECs was further validated by Yazici et al. [114], who described proinflammatory effects on live ECs due to a monoclonal IgG AECA generated by hybridoma formation with human SLE B cells. In particular, this mAb bound to a 42‐kDa EC cell membrane protein and induced an overexpression of E‐selectin and ICAM‐1 through NF‐κB activation. Similar findings were observed with mAb AECA from a patient with TA, which bound and activated macrovascular ECs but not microvascular ECs [115] and with mAb from a patient with WG [116]. The proinflammatory phenotype of ECs due to binding of AECA may also be dependent on the origin of ECs. Affinity‐purified AECA F(ab)2 from patients with thrombotic thrombocytopenic purpura (TTP) bound to and activated only microvascular ECs but not large vessel ECs [117]. On the contrary, in uremic patients undergoing hemodialysis AECA reactivity with HUVECs but not with microvascular or EaHy 929 EC lines was observed [118].
Carvalho et al. [110] were the first to report that AECA from SSc patients are able to functionally affect in vitro interactions between ECs and leukocytes by increasing adhesion of the myelomonocytic cell line U937 to cultured HUVECs. This effect was reproducibly mediated by both sera as well as purified IgG from AECA positive SSc patients binding to ECs and related to de novo increased expression of selectins and adhesion molecules (i.e., E‐selectin, ICAM‐1, and VCAM‐1) by HUVECs. In this in vitro system, leukocyte–endothelial cell interactions were not dependent on Fc engagement on U937 cells since Fab fragments also induced leukocyte adhesion with a similar efficacy compared to whole IgG. However, as suggested by the kinetics of leukocyte adhesion and adhesion molecule expression, the effect of AECA was dependent on the direct stimulation of soluble mediator(s) from HUVECs since the ability of inducing leukocyte adhesion to ECs was maintained using IgG‐depleted AECA‐conditioned HUVEC medium. Inhibition experiments using a cocktail of blocking antibodies suggested that the effect AECA was, at least in part, mediated by stimulation of cytokine production, and in particular IL‐1, from HUVECs in vitro. Although an exhaustive proof of the ability of AECA from SSc patients to directly induce cytokine production in HUVECs was missing in this study, AECA from other rheumatic conditions (i.e., WG as reported previously) have been shown to be to upregulate cytokine expression from ECs in vitro [109].
5.3. AECA and Thrombotic Vascular Involvement
The role of aPL and AECA in the pathogenesis of APS is now well investigated. Since the first description of AECA, it was evident that antibodies directed against negatively charged phospholipids can explain part of the AECA reactivity [119]. In fact, absorption with cardiolipin liposomes partially inhibited AECA reactivity [[119], [120]]. Thus, it is likely that AECA directed against aPL may be involved in thrombotic diathesis via more than one mechanism. First, these antibodies may activate ECs through apoptotic or nonapoptotic mechanisms [121] (Fig. 2B and C); second, these autoantibodies may induce significant increases in tissue factor (TF) transcription and expression by ECs [122], as well as by monocytes [[123], [124]].
Incubation of EC with IgG, from patients positive for aPL, increased the expression of VCAM‐1, E‐selectin, and MCP‐1 as previously reported for AECA [125]. Studies have shown that upregulation of adhesion molecules on ECs by aPL correlates with an increased adhesion of leukocytes to ECs in mouse microcirculation and with enhanced thrombosis in vivo [126]. Pretreatment of ECs with fluvastatin further increased the expression of EC activation as well as TF expression [125], and pretreatment with aspirin decreased VCAM‐1 at the cell surface of ECs [127]. On the contrary, a previous study showed that fluvastatin was able to inhibit the expression of TF expression on ECs induced by aPL from patients with APS as well as by affinity‐purified polyclonal IgG‐aPL and IgM human monoclonal anti‐β2‐GPI antibodies [[128], [129]]. Moreover, fluvastatin in a mouse model significantly diminishes aPL‐mediated thrombosis and EC activation in vivo [130].
A novel pathway for EC activation induced by aPL/anti‐β2‐GPI antibodies may be due to the cross‐linking or clustering of annexin A2 on the endothelial surface [121].
Dieude et al. [3] showed that AECA from SLE patients with APS bind to the surface of ECs and share reactivity against a 60‐kDa EC surface polypeptide that was identified as human HSP60. Moreover, the incubation of ECs with purified anti‐HSP60 antibodies induced apoptosis as demonstrated by the exposure of the phosphatidylserine on the plasma membrane [131]. Although AECA with this reactivity were not exclusive to SLE patients an intriguing association was observed between these antibodies and LAC. This may represent the first pathogenic hit for induction of AECA and for endothelial perturbations followed by a second hit represented by aPL antibodies binding to ECs. In line with these results anti‐HSP60 purified from patients with coronary‐artery disease may cause apoptosis of nonstressed ECs; this can be considered a primary event in the pathogenesis of atherosclerosis [132]. According to this hypothesis, Chen et al. [133] showed that aPL binding to ECs occurs only after activation of these cells. Worda et al. [134] reported evidence of in vivo binding of AECA to the microvascular endothelium associated with a significant increase in EC apoptosis. In line with these evidences, AECA might induce an endothelial perturbation with the release of endothelial microparticles that were described in sera from patients with APS [135]. Taken together, these findings support the idea that AECA may be pathogenic and may be able to induce aPL production. The effects of aPL on ECs may be mediated by an overexpression of TF. In line with this hypothesis, Vega‐Ostertag et al. [122] showed that treatment with IgG from patients with aPL induced significant increases in TF transcription, expression, and activity in HUVECs and was mediated by the phosphorylation of p38 MAPK and activation of NF‐κB.
AECA represent a heterogeneous family of autoantibodies directed to several antigens, including partially overlapping specificities. Several findings suggest β2‐GPI as the most relevant antigen of APS also showing AECA reactivity [136]. β2‐GPI is a plasma protein able to bind to anionic phospholipids; this apolipoprotein represents a cofactor required for the generation of the antigenic epitopes recognized by some aPL. Pure anti‐β2‐GPI antibodies have been also detected [[137], [138], [139]]. The binding of β2‐GPI to several cells, such as apoptotic thymocytes, trophoblast cells, macrophages, platelets, and ECs, has been previously reported [140]. In particular, the immunoreactivity of β2‐GPI has been largely investigated in ECs but whether this reactivity is due to plasma adhesion or to a direct intracellular synthesis of the glycoprotein by ECs is still debated. The disappearance of β2‐GPI immunoreactivity has been described following extensive washing of HUVECs or culture in serum‐free medium [141]. Interestingly, such a phenomenon was not found using primary human EC culture from skin or brain microcirculation suggesting the capability of these cell types to synthesize β2‐GPI [141]. In this regard, our group clearly demonstrated β2‐GPI mRNA expression in ECs cultured both in the absence and presence of β2‐GPI in the culture medium [140]. It is conceivable that ECs constitutively express β2‐GPI mRNA, since culture in the absence of β2‐GPI for several days does not influence β2‐GPI mRNA synthesis in ECs. This finding is also supported by the observation that in ECs β2‐GPI is located and accumulates in late endosome [[142], [143]]. The same results were observed for astrocytes, neurons, and human monocytes [124]. There is evidence that β2‐GPI is also expressed in vivo on trophoblast vessels in term placentas as well as in circulating monocytes [124]. The immunoreactivity of β2‐GPI expressed by several cells involved in the photogenic scenario of APS may be due, in part, to a direct β2‐GPI synthesis. On the other hand, it is possible to speculate that the β2‐GPI synthesis may be increased in vitro by stress conditions, such as serum depletion, from the culture medium. In fact, it is important to point out that plasma protein, such as β2‐GPI may also be absorbed on the cell membrane directly or through an endothelial receptor(s). In line with such a hypothesis, it has been suggested that β2‐GPI might interact with anionic EC membrane structures such as heparan sulfate (HS), annexin A2, and apolipoprotein E receptor 2 (reviewed in Ref. [144]). Although a previous study showed a minimal influence of β2‐GPI for AECA reactivity [120], to date it is well accepted that β2‐GPI on ECs may be a target for AECA. Raschi et al. [145] clearly demonstrated that both human monoclonal IgM antibodies anti‐β2‐GPI as well as affinity‐purified polyclonal anti‐β2‐GPI induce phosphorylation of the IL‐1 receptor‐activated kinase (IRAK) and suggest that β2‐GPI might interact with the TLR4 because of his homology with microbial structures.
6. Conclusions
AECA represent a heterogeneous family of autoantibodies directed to several antigens, including partially overlapping specificities. Despite the fact that most studies have shown a higher prevalence of AECA in autoimmune diseases, the clinical relevance of these autoantibodies is still a matter of debate and need to be confirmed by longitudinal studies on a large number of patients. Nevertheless, there are many evidences about the pathogenic role of AECA in SLE, APS, SSc, and vasculitis. Moreover, in the future, evaluation of AECA using as substrates ECs from macro‐ and microvasculature may be important to assess more precisely AECA reactivity in the various forms of autoimmune diseases. For these reasons, we believe that a more precise definition of the AECA role in autoimmune diseases has to be included in the research agenda.
References
- 1.Risau W, Flamme I. Vasculogenesis. Ann Rev Cell Dev Biol. 1995;11:73–91. doi: 10.1146/annurev.cb.11.110195.000445. [DOI] [PubMed] [Google Scholar]
- 2.Williams JM, Colman R, Brookes CJ, Savage CO, Harper L. Anti‐endothelial cell antibodies from lupus patients bind to apoptotic endothelial cells promoting macrophage phagocytosis but do not induce apoptosis. Rheumatology. 2005;44:879–884. doi: 10.1093/rheumatology/keh633. [DOI] [PubMed] [Google Scholar]
- 3.Dieude M, Senecal JL, Raymond Y. Induction of endothelial cell apoptosis by heat‐shock protein 60‐reactive antibodies from anti‐endothelial cell autoantibody‐positive systemic lupus erythematosus patients. Arthritis Rheum. 2004;50:3221–3231. doi: 10.1002/art.20564. [DOI] [PubMed] [Google Scholar]
- 4.Pittoni V, Valesini G. The clearance of apoptotic cells: Implications for autoimmunity. Autoimmun Rev. 2002;1:154–161. doi: 10.1016/s1568-9972(02)00032-0. [DOI] [PubMed] [Google Scholar]
- 5.Revelen R, D'Arbonneau F, Guillevin L, Bordron A, Youinou P, Dueymes M. Comparison of cell‐ELISA, flow cytometry and Western blotting for the detection of antiendothelial cell antibodies. Clin Exp Rheumatol. 2002;20:19–26. [PubMed] [Google Scholar]
- 6.Revelen R, Bordron A, Dueymes M, Youinou P, Arvieux J. False positivity in a cyto‐ELISA for anti‐endothelial cell antibodies caused by heterophile antibodies to bovine serum proteins. Clin Chem. 2000;46:273–278. [PubMed] [Google Scholar]
- 7.Renaudineau Y, Grunebaum E, Krause I, Praprotnik S, Revelen R, Youinou P. Anti‐endothelial cell antibodies (AECA) in systemic sclerosis—increased sensitivity using different endothelial cell substrates and association with other autoantibodies. Autoimmunity. 2001;33:171–179. doi: 10.3109/08916930109008045. [DOI] [PubMed] [Google Scholar]
- 8.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII‐related antigen established by hybridization. Proc Natl Acad Sci USA. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC. HMEC‐1: Establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 10.van Leeuwen EB, Wisman GB, Tervaert JW, Palmans LL, van Wijk RT, Veenstra R. An SV40 large T‐antigen immortalized human umbilical vein endothelial cell line for anti‐endothelial cell antibody detection. Clin Exp Rheumatol. 2001;19:283–290. [PubMed] [Google Scholar]
- 11.Harada T, Batsford S, Morioka T, Yao J, Arakawa M, Gejyo F. Establishment of immortalized human glomerular endothelial cell lines and their application. Nephron Exp Nephrol. 2005;99:e38–e45. doi: 10.1159/000083096. [DOI] [PubMed] [Google Scholar]
- 12.Renaudineau Y, Dugue C, Dueymes M, Youinou P. Antiendothelial cell antibodies in systemic lupus erythematosus. Autoimmun Rev. 2002;1:365–372. doi: 10.1016/s1568-9972(02)00063-0. [DOI] [PubMed] [Google Scholar]
- 13.D'Cruz DP, Keser G, Direskeneli H, Khamashta MA, Hughes GR. Anti‐endothelial cell antibodies in systemic vasculitis and systemic lupus erythematosus (SLE): Effects of heat inactivation on binding and specificity. Clin Exp Immunol. 1999;115:567–570. doi: 10.1046/j.1365-2249.1999.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nissou M.F, Ponard D, Arvieux J, Arvieux J, Dumestre‐Perard C, Gaudin P. Detection of antiendothelial cell antibodies by an enzyme‐linked immunosorbent assay using antigens from cell lysate: Minimal interference with antinuclear antibodies and rheumatoid factors. Clin Diagn Lab Immunol. 2003;10:934–939. doi: 10.1128/CDLI.10.5.934-939.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: More than 100 different antibodies found in SLE patients. Semin Arthritis Rheum. 2004;34:501–537. doi: 10.1016/j.semarthrit.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Oelzner P, Deliyska B, Funfstuck R, Hein G, Herrmann D, Stein G. Anti‐C1q antibodies and antiendothelial cell antibodies in systemic lupus erythematosus: Relationship with disease activity and renal involvement. Clin Rheumatol. 2003;22:271–278. doi: 10.1007/s10067-003-0724-3. [DOI] [PubMed] [Google Scholar]
- 17.D'Cruz DP, Houssiau FA, Ramirez G, Baguley E, McCutcheon J, Vianna J. Antibodies to endothelial cells in systemic lupus erythematosus: A potential marker for nephritis and vasculitis. Clin Exp Immunol. 1991;85:254–261. doi: 10.1111/j.1365-2249.1991.tb05714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li JS, Liu MF, Lei HY. Characterization of anti‐endothelial cell antibodies in the patients with systemic lupus erythematosus: A potential marker for disease activity. Clin Immunol Immunopathol. 1996;79:211–216. doi: 10.1006/clin.1996.0070. [DOI] [PubMed] [Google Scholar]
- 19.Song J, Park YB, Lee WK, Lee KH, Lee SK. Clinical associations of anti‐endothelial cell antibodies in patients with systemic lupus erythematosus. Rheumatol Int. 2000;20:1–7. doi: 10.1007/s002960000060. [DOI] [PubMed] [Google Scholar]
- 20.Hashemi S, Smith CD, Izaguirre CA. Anti‐endothelial cell antibodies: Detection and characterization using a cellular enzyme‐linked immunosorbent assay. J Lab Clin Med. 1987;109:434–440. [PubMed] [Google Scholar]
- 21.Del Papa N, Conforti G, Gambini D, La Rosa L, Tincani A, D'Cruz D. Characterization of the endothelial surface proteins recognized by anti‐endothelial antibodies in primary and secondary autoimmune vasculitis. Clin Immunol Immunopathol. 1994;70:211–216. doi: 10.1006/clin.1994.1031. [DOI] [PubMed] [Google Scholar]
- 22.Yoshio T, Masuyama J, Sumiya M, Minota S, Kano S. Antiendothelial cell antibodies and their relation to pulmonary hypertension in systemic lupus erythematosus. J Rheumatol. 1994;21:2058–2063. [PubMed] [Google Scholar]
- 23.Constans J, Dupuy R, Blann AD, Resplandy F, Seigneur M, Renard M. Anti‐endothelial cell autoantibodies and soluble markers of endothelial cell dysfunction in systemic lupus erythematosus. J Rheumatol. 2003;30:1963–1966. [PubMed] [Google Scholar]
- 24.Varani S, Muratori L, De Ruvo N, Vivarelli M, Lazzarotto T, Gabrielli L. Autoantibody appearance in cytomegalovirus‐infected liver transplant recipients: Correlation with antigenemia. J Med Virol. 2002;66:56–62. doi: 10.1002/jmv.2111. [DOI] [PubMed] [Google Scholar]
- 25.Toyoda M, Petrosian A, Jordan SC. Immunological characterization of anti‐endothelial cell antibodies induced by cytomegalovirus infection. Transplantation. 1999;68:1311–1318. doi: 10.1097/00007890-199911150-00016. [DOI] [PubMed] [Google Scholar]
- 26.D'Cruz DP, Wisnieski JJ, Asherson RA, Khamashta MA, Hughes GR. Autoantibodies in systemic lupus erythematosus and urticarial vasculitis. J Rheumatol. 1995;22:1669–1673. [PubMed] [Google Scholar]
- 27.D'Cruz D. Vasculitis in systemic lupus erythematosus. Lupus. 1998;7:270–274. doi: 10.1191/096120398678920082. [DOI] [PubMed] [Google Scholar]
- 28.Wener MH, Uwatoko S, Mannik M. Antibodies to the collagen‐like region of C1q in sera of patients with autoimmune rheumatic diseases. Arthritis Rheum. 1989;32:544–551. doi: 10.1002/anr.1780320506. [DOI] [PubMed] [Google Scholar]
- 29.Cervera R, Khamashta MA, Font J, Ramirez G, D'Cruz D, Montalban J. Antiendothelial cell antibodies in patients with the antiphospholipid syndrome. Autoimmunity. 1991;11:1–6. doi: 10.3109/08916939108994701. [DOI] [PubMed] [Google Scholar]
- 30.Hurtado V, Montes R, Gris JC, Bertolaccini ML, Alonso A, Martinez‐Gonzalez MA. Autoantibodies against EPCR are found in antiphospholipid syndrome and are a risk factor for fetal death. Blood. 2004;104:1369–1374. doi: 10.1182/blood-2004-03-0793. [DOI] [PubMed] [Google Scholar]
- 31.Frances C, Le Tonqueze M, Salohzin KV, Kalashnikova LA, Piette JC, Godeau P. Prevalence of anti‐endothelial cell antibodies in patients with Sneddon's syndrome. J Am Acad Dermatol. 1995;33:64–68. doi: 10.1016/0190-9622(95)90012-8. [DOI] [PubMed] [Google Scholar]
- 32.Wang MX, Walker RG, Kincaid‐Smith P. Endothelial cell antigens recognized by IgA autoantibodies in patients with IgA nephropathy: Partial characterization. Nephrol Dial Transplant. 1992;7:805–810. [PubMed] [Google Scholar]
- 33.Wang MX, Walker RG, Kincaid‐Smith P. Clinicopathologic associations of anti‐endothelial cell antibodies in immunoglobulin A nephropathy and lupus nephritis. Am J Kidney Dis. 1993;22:378–386. doi: 10.1016/s0272-6386(12)70139-7. [DOI] [PubMed] [Google Scholar]
- 34.Perry GJ, Elston T, Khouri NA, Chan TM, Cameron JS, Frampton G. Antiendothelial cell antibodies in lupus: Correlations with renal injury and circulating markers of endothelial damage. Q J Med. 1993;86:727–734. [PubMed] [Google Scholar]
- 35.Chan TM, Cheng IK. A prospective study on anti‐endothelial cell antibodies in patients with systemic lupus erythematosus. Clin Immunol Immunopathol. 1996;78:41–46. doi: 10.1006/clin.1996.0006. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Geng H, Zhao M, Zou W, EJ, Zheng X, Wang H. The significance of anti‐endothelial cell antibodies in patients with lupus nephritis and immunoblotting analysis of the target components. Chin Med J. 1999;112:597–602. [PubMed] [Google Scholar]
- 37.Frampton G, Moriya S, Pearson JD, Isenberg DA, Ward FJ, Smith TA. Identification of candidate endothelial cell autoantigens in systemic lupus erythematosus using a molecular cloning strategy: A role for ribosomal P protein P0 as an endothelial cell autoantigen. Rheumatology. 2000;39:1114–1120. doi: 10.1093/rheumatology/39.10.1114. [DOI] [PubMed] [Google Scholar]
- 38.Ziporen L, Shoenfeld Y, Levy Y, Korczyn AD. Neurological dysfunction and hyperactive behavior associated with antiphospholipid antibodies. A mouse model. J Clin Invest. 1997;100:613–619. doi: 10.1172/JCI119572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conti F, Alessandri C, Bompane D, Bombardieri M, Spinelli FR, Rusconi AC. Autoantibody profile in systemic lupus erythematosus with psychiatric manifestations: A role for anti‐endothelial‐cell antibodies. Arthritis Res Ther. 2004;6:R366–R372. doi: 10.1186/ar1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Margutti P, Sorice M, Conti F, Delunardo F, Racaniello M, Alessandri C. Screening of an endothelial cDNA library identifies the C‐terminal region of Nedd5 as a novel autoantigen in systemic lupus erythematosus with psychiatric manifestations. Arthritis Res Ther. 2005;7:R896–R903. doi: 10.1186/ar1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Renaudineau Y, Revelen R, Levy Y, Salojin K, Gilburg B, Shoenfeld Y. Anti‐endothelial cell antibodies in sclerosis. Clin Diagn Lab Immunol. 1999;6:156–160. doi: 10.1128/cdli.6.2.156-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salojin KV, Le Tonqueze M, Saraux A, Nassonov EL, Dueymes M, Piette JC. Antiendothelial cell antibodies: Useful markers of systemic sclerosis. Am J Med. 1997;102:178–185. doi: 10.1016/s0002-9343(96)00404-4. [DOI] [PubMed] [Google Scholar]
- 43.Negi VS, Tripathy NK, Misra R, Nityanand S. Antiendothelial cell antibodies in scleroderma correlate with severe digital ischemia and pulmonary arterial hypertension. J Rheumatol. 1998;25:462–466. [PubMed] [Google Scholar]
- 44.Pignone A, Scaletti C, Matucci‐Cerinic M, Vazquez‐Abad D, Meroni PL, Del Papa N. Anti‐endothelial cell antibodies in systemic sclerosis: Significant association with vascular involvement and alveolo‐capillary impairment. Clin Exp Rheumatol. 1998;16:527–532. [PubMed] [Google Scholar]
- 45.Ihn H, Sato S, Fujimoto M, Igarashi A, Yazawa N, Kubo M. Characterization of autoantibodies to endothelial cells in systemic sclerosis (SSc): Association with pulmonary fibrosis. Clin Exp Immunol. 2000;119:203–209. doi: 10.1046/j.1365-2249.2000.01115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steen VD, Conte C, Owens GR, Medsger TA., Jr Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994;37:1283–1289. doi: 10.1002/art.1780370903. [DOI] [PubMed] [Google Scholar]
- 47.Stupi AM, Steen VD, Owens GR, Barnes EL, Rodnan GP, Medsger TA., Jr Pulmonary hypertension in the CREST syndrome variant of systemic sclerosis. Arthritis Rheum. 1986;29:515–524. doi: 10.1002/art.1780290409. [DOI] [PubMed] [Google Scholar]
- 48.Hill MB, Phipps JL, Cartwright RJ, Milford Ward A, Greaves M, Hughes P. Antibodies to membranes of endothelial cells and fibroblasts in scleroderma. Clin Exp Immunol. 1996;106:491–497. doi: 10.1046/j.1365-2249.1996.d01-867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wusirika R, Ferri C, Marin M, Knight DA, Waldman WJ, Ross P., Jr. The assessment of anti‐endothelial cell antibodies in scleroderma‐associated pulmonary fibrosis. A study of indirect immunofluorescent and western blot analysis in 49 patients with scleroderma. Am J Clin Pathol. 2003;120:596–606. doi: 10.1309/8HVC-MJMY-NPUQ-PBD2. [DOI] [PubMed] [Google Scholar]
- 50.Leung DY, Collins T, Lapierre LA, Geha RS, Pober JS. Immunoglobulin M antibodies present in the acute phase of Kawasaki syndrome lyse cultured vascular endothelial cells stimulated by gamma interferon. J Clin Invest. 1986;77:1428–1435. doi: 10.1172/JCI112454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leung DY, Geha RS, Newburger JW, Burns JC, Fiers W, Lapierre LA. Two monokines, interleukin 1 and tumor necrosis factor, render cultured vascular endothelial cells susceptible to lysis by antibodies circulating during Kawasaki syndrome. J Exp Med. 1986;164:1958–1972. doi: 10.1084/jem.164.6.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guzman J, Fung M, Petty RE. Diagnostic value of anti‐neutrophil cytoplasmic and anti‐endothelial cell antibodies in early Kawasaki disease. J Pediatr. 1994;124:917–920. doi: 10.1016/s0022-3476(05)83180-4. [DOI] [PubMed] [Google Scholar]
- 53.Nash MC, Shah V, Reader JA, Dillon MJ. Anti‐neutrophil cytoplasmic antibodies and anti‐endothelial cell antibodies are not increased in Kawasaki disease. Br J Rheumatol. 1995;34:882–887. doi: 10.1093/rheumatology/34.9.882. [DOI] [PubMed] [Google Scholar]
- 54.Kaneko K, Savage CO, Pottinger BE, Shah V, Pearson JD, Dillon MJ. Antiendothelial cell antibodies can be cytotoxic to endothelial cells without cytokine pre‐stimulation and correlate with ELISA antibody measurement in Kawasaki disease. Clin Exp Immunol. 1994;98:264–269. doi: 10.1111/j.1365-2249.1994.tb06136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Falcini F, Trapani S, Turchini S, Farsi A, Ermini M, Keser G. Immunological findings in Kawasaki disease: An evaluation in a cohort of Italian children. Clin Exp Rheumatol. 1997;15:685–689. [PubMed] [Google Scholar]
- 56.Fujieda M, Oishi N, Kurashige T. Antibodies to endothelial cells in Kawasaki disease lyse endothelial cells without cytokine pretreatment. Clin Exp Immunol. 1997;107:120–126. doi: 10.1046/j.1365-2249.1997.d01-894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferraro G, Meroni PL, Tincani A, Sinico A, Barcellini W, Radice A. Anti‐endothelial cell antibodies in patients with Wegener's granulomatosis and micropolyarteritis. Clin Exp Immunol. 1990;79:47–53. doi: 10.1111/j.1365-2249.1990.tb05125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frampton G, Jayne DR, Perry GJ, Lockwood CM, Cameron JS. Autoantibodies to endothelial cells and neutrophil cytoplasmic antigens in systemic vasculitis. Clin Exp Immunol. 1990;82:227–232. doi: 10.1111/j.1365-2249.1990.tb05431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savage CO, Pottinger BE, Gaskin G, Lockwood CM, Pusey CD, Pearson JD. Vascular damage in Wegener's granulomatosis and microscopic polyarteritis: Presence of anti‐endothelial cell antibodies and their relation to anti‐neutrophil cytoplasm antibodies. Clin Exp Immunol. 1991;85:14–19. doi: 10.1111/j.1365-2249.1991.tb05675.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan TM, Frampton G, Jayne DR, Perry GJ, Lockwood CM, Cameron JS. Clinical significance of anti‐endothelial cell antibodies in systemic vasculitis: A longitudinal study comparing anti‐endothelial cell antibodies and anti‐neutrophil cytoplasm antibodies. Am J Kidney Dis. 1993;22:387–392. doi: 10.1016/s0272-6386(12)70140-3. [DOI] [PubMed] [Google Scholar]
- 61.Gobel U, Eichhorn J, Kettritz R, Briedigkeit L, Sima D, Lindschau C. Disease activity and autoantibodies to endothelial cells in patients with Wegener's granulomatosis. Am J Kidney Dis. 1996;28:186–194. doi: 10.1016/s0272-6386(96)90300-5. [DOI] [PubMed] [Google Scholar]
- 62.Eichhorn J, Sima D, Thiele B, Lindschau C, Turowski A, Schmidt H. Anti‐endothelial cell antibodies in Takayasu arteritis. Circulation. 1996;94:2396–2401. doi: 10.1161/01.cir.94.10.2396. [DOI] [PubMed] [Google Scholar]
- 63.Navarro M, Cervera R, Font J, Reverter JC, Monteagudo J, Escolar G. Anti‐endothelial cell antibodies in systemic autoimmune diseases: Prevalence and clinical significance. Lupus. 1997;6:521–526. doi: 10.1177/096120339700600608. [DOI] [PubMed] [Google Scholar]
- 64.Schmitt WH, Csernok E, Kobayashi S, Klinkenborg A, Reinhold‐Keller E, Gross WL. Churg‐Strauss syndrome: Serum markers of lymphocyte activation and endothelial damage. Arthritis Rheum. 1998;41:445–452. doi: 10.1002/1529-0131(199803)41:3<445::AID-ART10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 65.Fujieda M, Oishi N, Naruse K, Hashizume M, Nishiya K, Kurashige T. Soluble thrombomodulin and antibodies to bovine glomerular endothelial cells in patients with Henoch‐Schonlein purpura. Arch Dis Child. 1998;78:240–244. doi: 10.1136/adc.78.3.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang YH, Wang SJ, Chuang YH, Lin YT, Chiang BL. The level of IgA antibodies to human umbilical vein endothelial cells can be enhanced by TNF‐alpha treatment in children with Henoch‐Schonlein purpura. Clin Exp Immunol. 2002;130:352–357. doi: 10.1046/j.1365-2249.2002.01964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bodolay E, Csipo I, Gal I, Sipka S, Gyimesi E, Szekanecz Z. Anti‐endothelial cell antibodies in mixed connective tissue disease: Frequency and association with clinical symptoms. Clin Exp Rheumatol. 2004;22:409–415. [PubMed] [Google Scholar]
- 68.D'Cruz D, Keser G, Khamashta MA, Direskeneli H, Targoff IN, Miller F. Antiendothelial cell antibodies in inflammatory myopathies: Distribution among clinical and serologic groups and association with interstitial lung disease. J Rheumatol. 2000;27:161–164. [PubMed] [Google Scholar]
- 69.Pivetti‐Pezzi P, Priori R, Catarinelli G, Meroni PL, Federici AB, Abdulaziz M. Markers of vascular injury in Behcet's disease associated with retinal vasculitis. Ann Ophthalmol. 1992;24:411–414. [PubMed] [Google Scholar]
- 70.Aydintug AO, Tokgoz G, D'Cruz DP, Gurler A, Cervera R, Duzgun N. Antibodies to endothelial cells in patients with Behcet's disease. Clin Immunol Immunopathol. 1993;67:157–162. doi: 10.1006/clin.1993.1059. [DOI] [PubMed] [Google Scholar]
- 71.Direskeneli H, Keser G, D'Cruz D, Khamashta MA, Akoglu T, Yazici H. Anti‐endothelial cell antibodies, endothelial proliferation and von Willebrand factor antigen in Behcet's disease. Clin Rheumatol. 1995;14:55–61. doi: 10.1007/BF02208085. [DOI] [PubMed] [Google Scholar]
- 72.Dinc A, Takafuta T, Jiang D, Melikoglu M, Saruhan‐Direskeneli G, Shapiro SS. Anti‐endothelial cell antibodies in Behcet's disease. Clin Exp Rheumatol. 2003;21:S27–S30. [PubMed] [Google Scholar]
- 73.Lee KH, Chung HS, Kim HS, Oh SH, Ha MK, Baik JH. Human alpha‐enolase from endothelial cells as a target antigen of anti‐endothelial cell antibody in Behcet's disease. Arthritis Rheum. 2003;48:2025–2035. doi: 10.1002/art.11074. [DOI] [PubMed] [Google Scholar]
- 74.Romas E, Paspaliaris B, d'Apice AJ, Elliott PR. Autoantibodies to neutrophil cytoplasmic (ANCA) and endothelial cell surface antigens (AECA) in chronic inflammatory bowel disease. Aust N Z J Med. 1992;22:652–659. doi: 10.1111/j.1445-5994.1992.tb04865.x. [DOI] [PubMed] [Google Scholar]
- 75.Aldebert D, Notteghem B, Reumaux D, Lassalle P, Lion G, Desreumaux P. Anti‐endothelial cell antibodies in sera from patients with inflammatory bowel disease. Gastroenterol Clin Biol. 1995;19:867–870. [PubMed] [Google Scholar]
- 76.Farsi A, Domeneghetti MP, Brunelli T, Gori AM, Fedi S, Gensini GF. Activation of the immune system and coronary artery disease: The role of anti‐endothelial cell antibodies. Atherosclerosis. 2001;154:429–436. doi: 10.1016/s0021-9150(00)00482-2. [DOI] [PubMed] [Google Scholar]
- 77.George J, Meroni PL, Gilburd B, Raschi E, Harats D, Shoenfeld Y. Anti‐endothelial cell antibodies in patients with coronary atherosclerosis. Immunol Lett. 2000;73:23–27. doi: 10.1016/s0165-2478(00)00192-9. [DOI] [PubMed] [Google Scholar]
- 78.Montes R, Hurtado V, Alonso A, Foco L, Zonzin P, Mannucci PM. Autoantibodies against the endothelial receptor of protein C are associated with acute myocardial infarction in young women. J Thromb Haemost. 2005;3:1454–1458. doi: 10.1111/j.1538-7836.2005.01297.x. [DOI] [PubMed] [Google Scholar]
- 79.Kluz J, Adamiec R. The role of anti‐endothelial cell antibodies in the pathogenesis of atherosclerosis and diabetic angiopathy. Przegl Lek. 2003;60:751–754. [PubMed] [Google Scholar]
- 80.Wangel AG, Kontiainen S, Scheinin T, Schlenzka A, Wangel D, Maenpaa J. Anti‐endothelial cell antibodies in insulin‐dependent diabetes mellitus. Clin Exp Immunol. 1992;88:410–413. doi: 10.1111/j.1365-2249.1992.tb06463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ottaviani F, Cadoni G, Marinelli L, Fetoni AR, De Santis A, Romito A. Anti‐endothelial autoantibodies in patients with sudden hearing loss. Laryngoscope. 1999;109:1084–1087. doi: 10.1097/00005537-199907000-00014. [DOI] [PubMed] [Google Scholar]
- 82.Cadoni G, Agostino S, Manna R, De Santis A, Fetoni AR, Vulpiani P. Clinical associations of serum antiendothelial cell antibodies in patients with sudden sensorineural hearing loss. Laryngoscope. 2003;113:797–801. doi: 10.1097/00005537-200305000-00006. [DOI] [PubMed] [Google Scholar]
- 83.Cacoub P, Ghillani P, Revelen R, Thibault V, Calvez V, Charlotte F. Anti‐endothelial cell auto‐antibodies in hepatitis C virus mixed cryoglobulinemia. J Hepatol. 1999;31:598–603. doi: 10.1016/s0168-8278(99)80337-7. [DOI] [PubMed] [Google Scholar]
- 84.Weiss L, You JF, Giral P, Alhenc‐Gelas M, Senger D, Kazatchkine MD. Anti‐cardiolipin antibodies are associated with anti‐endothelial cell antibodies but not with anti‐beta 2 glycoprotein I antibodies in HIV infection. Clin Immunol Immunopathol. 1995;77:69–74. doi: 10.1016/0090-1229(95)90138-8. [DOI] [PubMed] [Google Scholar]
- 85.Yang YH, Huang YH, Chuang YH, Peng CM, Wang LC, Lin YT. Autoantibodies against human epithelial cells and endothelial cells after severe acute respiratory syndrome (SARS)‐associated coronavirus infection. J Med Virol. 2005;77:1–7. doi: 10.1002/jmv.20407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dugue C, Perraut R, Youinou P, Renaudineau Y. Effects of anti‐endothelial cell antibodies in leprosy and malaria. Infect Immun. 2004;72:301–309. doi: 10.1128/IAI.72.1.301-309.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ronda N, Leonardi S, Orlandini G, Gatti R, Bellosta S, Bernini F. Natural anti‐endothelial cell antibodies (AECA) J Autoimmun. 1999;13:121–127. doi: 10.1016/s0896-8411(99)99999-7. [DOI] [PubMed] [Google Scholar]
- 88.Ronda N, Haury M, Nobrega A, Kaveri SV, Coutinho A, Kazatchkine MD. Analysis of natural and disease‐associated autoantibody repertoires: Anti‐endothelial cell IgG autoantibody activity in the serum of healthy individuals and patients with systemic lupus erythematosus. Int Immunol. 1994;6:1651–1660. doi: 10.1093/intimm/6.11.1651. [DOI] [PubMed] [Google Scholar]
- 89.Mendonca LL, Khamashta MA, Cuadrado MJ, Bertolaccini ML, Hughes GR. Natural immune response involving anti‐endothelial cell antibodies in normal and lupus pregnancy. Arthritis Rheum. 2000;43:1511–1515. doi: 10.1002/1529-0131(200007)43:7<1511::AID-ANR14>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 90.Tripathy NK, Upadhyaya S, Sinha N, Nityanand S. Complement and cell mediated cytotoxicity by antiendothelial cell antibodies in Takayasu's arteritis. J Rheumatol. 2001;28:805–808. [PubMed] [Google Scholar]
- 91.Moscato S, Pratesi F, Bongiorni F, Scavuzzo MC, Chimenti D, Bombardieri S. Endothelial cell binding by systemic lupus antibodies: Functional properties and relationship with anti‐DNA activity. J Autoimmun. 2002;18:231–238. doi: 10.1006/jaut.2002.0583. [DOI] [PubMed] [Google Scholar]
- 92.Cohen S, Johnson AR, Hurd E. Cytotoxicity of sera from patients with scleroderma. Effects on human endothelial cells and fibroblasts in culture. Arthritis Rheum. 1983;26:170–178. doi: 10.1002/art.1780260208. [DOI] [PubMed] [Google Scholar]
- 93.Marks RM, Czerniecki M, Andrews BS, Penny R. The effects of scleroderma serum on human microvascular endothelial cells. Induction of antibody‐dependent cellular cytotoxicity. Arthritis Rheum. 1988;31:1524–1534. doi: 10.1002/art.1780311209. [DOI] [PubMed] [Google Scholar]
- 94.Holt CM, Lindsey N, Moult J, Malia RG, Greaves M, Hume A. Antibody‐dependent cellular cytotoxicity of vascular endothelium: Characterization and pathogenic associations in systemic sclerosis. Clin Exp Immunol. 1989;78:359–365. [PMC free article] [PubMed] [Google Scholar]
- 95.Penning CA, Wright JK, Ashby JC, Cunningham J, Rowell NR, Hughes P. Serum‐induced enhancement of peripheral blood mononuclear cell‐mediated cytotoxicity towards human target cells in systemic sclerosis. J Clin Lab Immunol. 1983;12:77–81. [PubMed] [Google Scholar]
- 96.Penning CA, Cunningham J, French MA, Harrison G, Rowell NR, Hughes P. Antibody‐dependent cellular cytotoxicity of human vascular endothelium in systemic sclerosis. Clin Exp Immunol. 1984;57:548–556. [PMC free article] [PubMed] [Google Scholar]
- 97.Rosenbaum J, Pottinger BE, Woo P, Black CM, Loizou S, Byron MA. Measurement and characterisation of circulating anti‐endothelial cell IgG in connective tissue diseases. Clin Exp Immunol. 1988;72:450–456. [PMC free article] [PubMed] [Google Scholar]
- 98.del Papa N, Meroni PL, Barcellini W, Sinico A, Radice A, Tincani A. Antibodies to endothelial cells in primary vasculitides mediate in vitro endothelial cytotoxicity in the presence of normal peripheral blood mononuclear cells. Clin Immunol Immunopathol. 1992;63:267–274. doi: 10.1016/0090-1229(92)90232-d. [DOI] [PubMed] [Google Scholar]
- 99.Bordron A, Revelen R, D'Arbonneau F, Dueymes M, Renaudineau Y, Jamin C. Functional heterogeneity of anti‐endothelial cell antibodies. Clin Exp Immunol. 2001;124:492–501. doi: 10.1046/j.1365-2249.2001.01528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sgonc R, Gruschwitz MS, Dietrich H, Recheis H, Gershwin ME, Wick G. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest. 1996;98:785–792. doi: 10.1172/JCI118851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Haynes DC, Gershwin ME. Diversity of autoantibodies in avian scleroderma. An inherited fibrotic disease of White Leghorn chickens. J Clin Invest. 1984;73:1557–1568. doi: 10.1172/JCI111362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Van de WJ, Gershwin ME, Abplanalp H, Wick G, von der MK. Serial observations and definition of mononuclear cell infiltrates in avian scleroderma, an inherited fibrotic disease of chickens. Arthritis Rheum. 1984;27:807–815. doi: 10.1002/art.1780270712. [DOI] [PubMed] [Google Scholar]
- 103.Van de WJ, Gershwin ME. Animal model of human disease. Avian scleroderma. An inherited fibrotic disease of White Leghorn chickens resembling progressive systemic sclerosis. Am J Pathol. 1985;120:478–482. [PMC free article] [PubMed] [Google Scholar]
- 104.Sgonc R, Gruschwitz MS, Boeck G, Sepp N, Gruber J, Wick G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody‐dependent cell‐mediated cytotoxicity via CD95. Arthritis Rheum. 2000;43:2550–2562. doi: 10.1002/1529-0131(200011)43:11<2550::AID-ANR24>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 105.Papa ND, Raschi E, Moroni G, Panzeri P, Borghi MO, Ponticelli C. Anti‐endothelial cell IgG fractions from systemic lupus erythematosus patients bind to human endothelial cells and induce a pro‐adhesive and a pro‐inflammatory phenotype in vitro. Lupus. 1999;8:423–429. doi: 10.1177/096120339900800603. [DOI] [PubMed] [Google Scholar]
- 106.Carvalho D, Savage CO, Isenberg D, Pearson JD. IgG anti‐endothelial cell autoantibodies from patients with systemic lupus erythematosus or systemic vasculitis stimulate the release of two endothelial cell‐derived mediators, which enhance adhesion molecule expression and leukocyte adhesion in an autocrine manner. Arthritis Rheum. 1999;42:631–640. doi: 10.1002/1529-0131(199904)42:4<631::AID-ANR5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 107.Triolo G, Accardo‐Palumbo A, Triolo G, Carbone MC, Ferrante A, Giardina E. Enhancement of endothelial cell E‐selectin expression by sera from patients with active Behcet's disease: Moderate correlation with anti‐endothelial cell antibodies and serum myeloperoxidase levels. Clin Immunol. 1999;91:330–337. doi: 10.1006/clim.1999.4687. [DOI] [PubMed] [Google Scholar]
- 108.Muller Kobold AC, van Wijk RT, Franssen CF, Molema G, Kallenberg CG, Tervaert JW. In vitro upregulation of E‐selectin and induction of interleukin‐6 in endothelial cells by autoantibodies in Wegener's granulomatosis and microscopic polyangiitis. Clin Exp Rheumatol. 1999;17:433–440. [PubMed] [Google Scholar]
- 109.Del Papa N, Guidali L, Sironi M, Shoenfeld Y, Mantovani A, Tincani A. Anti‐endothelial cell IgG antibodies from patients with Wegener's granulomatosis bind to human endothelial cells in vitro and induce adhesion molecule expression and cytokine secretion. Arthritis Rheum. 1996;39:758–766. doi: 10.1002/art.1780390507. [DOI] [PubMed] [Google Scholar]
- 110.Carvalho D, Savage CO, Black CM, Pearson JD. IgG antiendothelial cell autoantibodies from scleroderma patients induce leukocyte adhesion to human vascular endothelial cells in vitro. Induction of adhesion molecule expression and involvement of endothelium‐derived cytokines. J Clin Invest. 1996;97:111–119. doi: 10.1172/JCI118377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yoshio T, Masuyama J, Mimori A, Takeda A, Minota S, Kano S. Endothelin‐1 release from cultured endothelial cells induced by sera from patients with systemic lupus erythematosus. Ann Rheum Dis. 1995;54:361–365. doi: 10.1136/ard.54.5.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yamamoto T, Takahashi Y, Kuno S, Geshi Y, Sasamori Y, Mori H. Effects of anti‐endothelial cell antibody in pre‐eclampsia on endothelin‐1 release from cultured endothelial cells. Immunol Cell Biol. 1997;75:340–344. doi: 10.1038/icb.1997.52. [DOI] [PubMed] [Google Scholar]
- 113.Quadros NP, Roberts‐Thomson PJ, Gallus AS. Sera from patients with systemic lupus erythematosus demonstrate enhanced IgG binding to endothelial cells pretreated with tumour necrosis factor alpha. Rheumatol Int. 1995;15:99–105. doi: 10.1007/BF00302125. [DOI] [PubMed] [Google Scholar]
- 114.Yazici ZA, Raschi E, Patel A, Testoni C, Borghi MO, Graham AM. Human monoclonal anti‐endothelial cell IgG‐derived from a systemic lupus erythematosus patient binds and activates human endothelium in vitro. Int Immunol. 2001;13:349–357. doi: 10.1093/intimm/13.3.349. [DOI] [PubMed] [Google Scholar]
- 115.Blank M, Krause I, Goldkorn T, Praprotnik S, Livneh A, Langevitz P. Monoclonal anti‐endothelial cell antibodies from a patient with Takayasu arteritis activate endothelial cells from large vessels. Arthritis Rheum. 1999;42:1421–1432. doi: 10.1002/1529-0131(199907)42:7<1421::AID-ANR16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 116.Levy Y, Gilburd B, George J, Del Papa N, Mallone R, Damianovich M. Characterization of murine monoclonal anti‐endothelial cell antibodies (AECA) produced by idiotypic manipulation with human AECA. Int Immunol. 1998;10:861–868. doi: 10.1093/intimm/10.7.861. [DOI] [PubMed] [Google Scholar]
- 117.Praprotnik S, Blank M, Levy Y, Tavor S, Boffa MC, Weksler B. Anti‐endothelial cell antibodies from patients with thrombotic thrombocytopenic purpura specifically activate small vessel endothelial cells. Int Immunol. 2001;13:203–210. doi: 10.1093/intimm/13.2.203. [DOI] [PubMed] [Google Scholar]
- 118.George J, Aron A, Levy Y, Gilburd B, Ben‐David A, Renaudineau Y. Anti‐cardiolipin, anti‐endothelial‐cell and anti‐malondialdehyde‐LDL antibodies in uremic patients undergoing hemodialysis: Relationship with vascular access thrombosis and thromboembolic events. Hum Antibodies. 1999;9:125–131. [PubMed] [Google Scholar]
- 119.Vismara A, Meroni PL, Tincani A, Harris EN, Barcellini W, Brucato A. Relationship between anti‐cardiolipin and anti‐endothelial cell antibodies in systemic lupus erythematosus. Clin Exp Immunol. 1988;74:247–253. [PMC free article] [PubMed] [Google Scholar]
- 120.Matsuda J, Gotoh M, Gohchi K, Kawasugi K, Tsukamoto M, Saitoh N. Anti‐endothelial cell antibodies to the endothelial hybridoma cell line (EAhy926) in systemic lupus erythematosus patients with antiphospholipid antibodies. Br J Haematol. 1997;97:227–232. doi: 10.1046/j.1365-2141.1997.d01-2147.x. [DOI] [PubMed] [Google Scholar]
- 121.Zhang J, McCrae KR. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti‐beta2 glycoprotein I antibodies. Blood. 2005;105:1964–1969. doi: 10.1182/blood-2004-05-1708. [DOI] [PubMed] [Google Scholar]
- 122.Vega‐Ostertag M, Casper K, Swerlick R, Ferrara D, Harris EN, Pierangeli SS. Involvement of p38 MAPK in the up‐regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheum. 2005;52:1545–1554. doi: 10.1002/art.21009. [DOI] [PubMed] [Google Scholar]
- 123.Cuadrado MJ, Lopez‐Pedrera C, Khamashta MA, Camps MT, Tinahones F, Torres A. Thrombosis in primary antiphospholipid syndrome: A pivotal role for monocyte tissue factor expression. Arthritis Rheum. 1997;40:834–841. doi: 10.1002/art.1780400509. [DOI] [PubMed] [Google Scholar]
- 124.Conti F, Sorice M, Circella A, Alessandri C, Pittoni V, Caronti B. Beta‐2‐glycoprotein I expression on monocytes is increased in anti‐phospholipid antibody syndrome and correlates with tissue factor expression. Clin Exp Immunol. 2003;132:509–516. doi: 10.1046/j.1365-2249.2003.02180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dunoyer‐Geindre S, Dimitrova Y, Fish RJ, Satta N, Reber G, Kruithof EK. Fluvastatin increases the expression of adhesion molecules, monocyte chemoattractant protein‐1 and tissue factor in HUVEC stimulated by patient IgG fractions containing antiphospholipid antibodies. Thromb Haemost. 2005;93:339–345. doi: 10.1160/TH04-05-0297. [DOI] [PubMed] [Google Scholar]
- 126.Espinola RG, Liu X, Colden‐Stanfield M, Hall J, Harris EN, Pierangeli SS. E‐selectin mediates pathogenic effects of antiphospholipid antibodies. J Thromb Haemost. 2003;1:843–848. doi: 10.1046/j.1538-7836.2003.00119.x. [DOI] [PubMed] [Google Scholar]
- 127.Dunoyer‐Geindre S, Kruithof EK, Boehlen F, Satta‐Poschung N, Reber G, de Moerloose P. Aspirin inhibits endothelial cell activation induced by antiphospholipid antibodies. J Thromb Haemost. 2004;2:1176–1181. doi: 10.1111/j.1538-7836.2004.00801.x. [DOI] [PubMed] [Google Scholar]
- 128.Ferrara DE, Swerlick R, Casper K, Meroni PL, Vega‐Ostertag ME, Harris EN. Fluvastatin inhibits up‐regulation of tissue factor expression by antiphospholipid antibodies on endothelial cells. J Thromb Haemost. 2004;2:1558–1563. doi: 10.1111/j.1538-7836.2004.00896.x. [DOI] [PubMed] [Google Scholar]
- 129.Meroni PL, Raschi E, Testoni C, Tincani A, Balestrieri G, Molteni R. Statins prevent endothelial cell activation induced by antiphospholipid (anti‐beta2‐glycoprotein I) antibodies: Effect on the proadhesive and proinflammatory phenotype. Arthritis Rheum. 2001;44:2870–2878. doi: 10.1002/1529-0131(200112)44:12<2870::aid-art475>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 130.Ferrara DE, Liu X, Espinola RG, Meroni PL, Abukhalaf I, Harris EN. Inhibition of the thrombogenic and inflammatory properties of antiphospholipid antibodies by fluvastatin in an in vivo animal model. Arthritis Rheum. 2003;48:3272–3279. doi: 10.1002/art.11449. [DOI] [PubMed] [Google Scholar]
- 131.Bordron A, Dueymes M, Levy Y, Jamin C, Ziporen L, Piette JC. Anti‐endothelial cell antibody binding makes negatively charged phospholipids accessible to antiphospholipid antibodies. Arthritis Rheum. 1998;41:1738–1747. doi: 10.1002/1529-0131(199810)41:10<1738::AID-ART6>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 132.Bason C, Corrocher R, Lunardi C, Puccetti P, Olivieri O, Girelli D. Interaction of antibodies against cytomegalovirus with heat‐shock protein 60 in pathogenesis of atherosclerosis. Lancet. 2003;362:1949–1950. doi: 10.1016/S0140-6736(03)15016-7. [DOI] [PubMed] [Google Scholar]
- 133.Chen Q, Stone PR, Woon ST, Ching LM, Hung S, McCowan LM. Antiphospholipid antibodies bind to activated but not resting endothelial cells: Is an independent triggering event required to induce antiphospholipid antibody‐mediated disease? Thromb Res. 2004;114:101–111. doi: 10.1016/j.thromres.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 134.Worda M, Sgonc R, Dietrich H, Niederegger H, Sundick RS, Gershwin ME. In vivo analysis of the apoptosis‐inducing effect of anti‐endothelial cell antibodies in systemic sclerosis by the chorionallantoic membrane assay. Arthritis Rheum. 2003;48:2605–2614. doi: 10.1002/art.11179. [DOI] [PubMed] [Google Scholar]
- 135.Dignat‐George F, Camoin‐Jau L, Sabatier F, Arnoux D, Anfosso F, Bardin N. Endothelial microparticles: A potential contribution to the thrombotic complications of the antiphospholipid syndrome. Thromb Haemost. 2004;91:667–673. doi: 10.1160/TH03-07-0487. [DOI] [PubMed] [Google Scholar]
- 136.Meroni PL, Raschi E, Testoni C, Tincani A, Balestrieri G. Antiphospholipid antibodies and the endothelium. Rheum Dis Clin North Am. 2001;27:587–602. doi: 10.1016/s0889-857x(05)70222-2. [DOI] [PubMed] [Google Scholar]
- 137.Galli M, Comfurius P, Maassen C, Hemker HC, de Baets MH, van Breda‐Vriesman PJ. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet. 1990;335:1544–1547. doi: 10.1016/0140-6736(90)91374-j. [DOI] [PubMed] [Google Scholar]
- 138.Hunt J, Krilis S. The fifth domain of beta 2‐glycoprotein I contains a phospholipid binding site (Cys281‐Cys288) and a region recognized by anticardiolipin antibodies. J Immunol. 1994;152:653–659. [PubMed] [Google Scholar]
- 139.Sorice M, Circella A, Griggi T, Garofalo T, Nicodemo G, Pittoni V. Anticardiolipin and anti‐beta 2‐GPI are two distinct populations of autoantibodies. Thromb Haemost. 1996;75:303–308. [PubMed] [Google Scholar]
- 140.Caronti B, Calderaro C, Alessandri C, Conti F, Tinghino R, Palladini G. Beta2‐glycoprotein I (beta2‐GPI) mRNA is expressed by several cell types involved in anti‐phospholipid syndrome‐related tissue damage. Clin Exp Immunol. 1999;115:214–219. doi: 10.1046/j.1365-2249.1999.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Meroni PL, Tincani A, Sepp N, Raschi E, Testoni C, Corsini E. Endothelium and the brain in CNS lupus. Lupus. 2003;12:919–928. doi: 10.1191/0961203303lu503oa. [DOI] [PubMed] [Google Scholar]
- 142.Dunoyer‐Geindre S, Kruithof EK, Galve‐de Rochemonteix B, Rosnoblet C, Gruenberg J, Reber G. Localization of beta2‐glycoprotein 1 in late endosomes of human endothelial cells. Thromb Haemost. 2001;85:903–907. [PubMed] [Google Scholar]
- 143.Sorice M, Ferro D, Misasi R, Pittoni V, Longo A, Circella A. Evidence for anticoagulant activity and beta2‐GPI accumulation in late endosomes of endothelial cells induced by anti‐LBPA antibodies. Thromb Haemost. 2002;87:735–741. [PubMed] [Google Scholar]
- 144.Meroni PL, Raschi E, Testoni C, Parisio A, Borghi MO. Innate immunity in the antiphospholipid syndrome: Role of toll‐like receptors in endothelial cell activation by antiphospholipid antibodies. Autoimmun Rev. 2004;3:510–515. doi: 10.1016/j.autrev.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 145.Raschi E, Testoni C, Bosisio D, Borghi MO, Koike T, Mantovani A. Role of the MyD88 transduction signaling pathway in endothelial activation by antiphospholipid antibodies. Blood. 2003;101:3495–3500. doi: 10.1182/blood-2002-08-2349. [DOI] [PubMed] [Google Scholar]