

Abstract
Novel pathogens continue to emerge in human, domestic animal, wildlife and plant populations, yet the population dynamics of this kind of biological invasion remain poorly understood. Here, we consider the epidemiological and evolutionary processes underlying the initial introduction and subsequent spread of a pathogen in a new host population, with special reference to pathogens that originate by jumping from one host species to another. We conclude that, although pathogen emergence is inherently unpredictable, emerging pathogens tend to share some common traits, and that directly transmitted RNA viruses might be the pathogens that are most likely to jump between host species.
Introduction
An emerging pathogen can be defined as the causative agent of an infectious disease whose incidence is increasing following its appearance in a new host population or whose incidence is increasing in an existing host population as a result of long-term changes in its underlying epidemiology [1]. One potential source of an emerging pathogen is a different host species (a ‘reservoir’) in which the pathogen is already established (Table 1 ). Switches from one host species to another (species ‘jumps’) have led to some of the most devastating disease epidemics recorded, including the ongoing HIV/AIDS pandemic in human communities worldwide, the decimation of the European rabbit population by myxomatosis during the mid 20th century, the catastrophic impact of rinderpest on African ruminants during the late 19th century and, more recently, widespread mortality of North Sea seals as a result of distemper 2, 3, 4, 5. It has even been argued that many of the main killer diseases of humans (e.g. measles, TB, influenza and smallpox) emerged through pathogens jumping from domestic animals to humans over the past 10 000 years [6]. Species jumps have also given rise to devastating epidemics of plant pathogens in crop species (e.g. potato late blight in the cultivated potato) and in wild plant species (e.g. the near extinction of American chestnut trees by chestnut blight) 7, 8, 9.
Table 1.
Examples of pathogens considered to have emerged via a species jump
| Pathogen | Original host | New host | Year reported | Refs |
|---|---|---|---|---|
| Transmissible spongiform encephalopathies | ||||
| BSE/vCJD | Cattle | Humans | 1996 | [53] |
| Viruses | ||||
| Rinderpest | Eurasian cattle | African ruminants | Late 1800s | [4] |
| Myxoma virus | Brush rabbit/Brazilian rabbit | European rabbit | 1950s | [3] |
| Ebola virus | Unknown | Humans | 1977 | [54] |
| FPLV/CPV | Cats | Dogs | 1978 | [55] |
| SIV/HIV-1 | Primates | Humans | 1983 | [2] |
| SIV/HIV-2 | Primates | Humans | 1986 | [2] |
| Canine/Phocine distemper virus | Canids | Seals | 1988 | [56] |
| Hendra virus | Bats | Horses and humans | 1994 | [57] |
| Australian bat lyssavirus | Bats | Humans | 1996 | [57] |
| H5N1 influenza A | Chickens | Humans | 1997 | [32] |
| Nipah virus | Bats | Pigs and humans | 1999 | [57] |
| SARS coronavirus | Palm civets | Humans | 2003 | [58] |
| Monkeypox virus | Prairie dogs | Humans | 2003a | [59] |
| Bacteria | ||||
| Escherichia coli O157:H7 | Cattle | Humans | 1982 | [60] |
| Borrelia burgdorferi | Deer | Humans | 1982 | [15] |
| Fungi | ||||
| Phytophthora infestans | Andean potato | Cultivated potato | 1840s | [8] |
| Cryphonectria parasitica | Japanese chestnut | American chesnut | Late 1800s | [9] |
Monkeypox was first reported in humans in 1970, but infections acquired from prairie dogs were not seen until 2003.
Conversely, there are numerous examples of species jumps that have had far less dramatic consequences: for example, BSE/vCJD and Ebola virus in humans which, although undoubtedly serious problems in themselves, show no signs of ‘taking off’ in the way that HIV/AIDS has. Moreover, there are many pathogens that have a long history of routinely jumping between species (e.g. rabies virus into humans from domestic or wild carnivores) without, again, triggering major epidemics in the ‘new’ host population. Understanding the epidemiology and evolutionary biology underlying these differences is crucial for understanding the phenomenon of emerging infectious diseases in human, domestic animal, wildlife and plant populations.
Pathogen population dynamics
Epidemiological theory has a well developed conceptual framework for evaluating the spread of infection through a host population (Box 1). The expected size of an outbreak depends upon the number of introductions, so-called ‘primary’ cases of infection, and the potential for transmission of the pathogen from one new host to another (Box 1, Figure Ia). This transmission potential can be expressed in terms of the basic reproduction number, R 0, and pathogens that enter a new host population via a species jump can be placed in two categories depending on its value. If R 0 is <1 in the new host population then, even if the new host repeatedly acquires the pathogen, there will be only limited spread of infection within that population. This category of emerging pathogen is unlikely to constitute the greatest disease threat; examples in humans include the Ebola, monkeypox and avian influenza viruses and the vCJD agent. Conversely, if R 0 is >1 in the new host population, then there is a finite chance (Box 1, Figure Ib) that a major epidemic will occur. This category is likely to constitute the greatest disease threat; examples in humans include HIV, influenza type A virus and SARS coronavirus.
Box 1. Epidemic thresholds and R0, the basic reproduction number.
The basic reproduction number, R 0, is the average number of secondary cases of infection generated by a single primary case introduced into a large population of previously unexposed hosts [5]. R 0 is related to the transmissibility of the pathogen (new infections per unit time) and the duration of infectiousness. It is sometimes referred to as a measure of transmission potential (with analogies to r max as used in simple ecological theory) because it refers to the situation where there are no density-dependent constraints on the spread of infection. R 0 defines an important threshold: if R 0>1, each primary infection will, on average, generate more than one secondary infection and the pathogen is capable of invading the host population; if R 0<1, each primary case will, on average, fail to replace itself (although short chains of transmission are still possible) and each single introduction will lead to no more than a minor outbreak.
The expected final size of an outbreak of an infectious disease, I final can be related to R 0 and I 0, the number of primary cases of infection, using a modified version of the Kermack–McKendrick equation (Equation I, [26]):
| [Eqn I] |
where N is the size of the susceptible population.
The behaviour of this equation is illustrated in Figure I a. For R 0<1, the size of an outbreak is determined mainly by the number of primary cases, I 0. For R 0>1, the size of an outbreak is determined mainly by the size of the susceptible population, N. For R 0 close to 1, the size of an outbreak is sensitive to the precise value of R 0.
Even for R 0>1, a major epidemic is not inevitable: it is possible that the infection will die out without causing a major epidemic owing to demographic stochasticity. The probability of a major epidemic occurring can be related to R 0 and I 0 using Equation II [17]:
| [Eqn II] |
The behaviour of this equation is illustrated in Figure Ib. For R 0 values not much above 1, there is a high probability that a major epidemic will not occur unless there are many primary cases. Even for larger R 0 values, there is a good chance that an epidemic will not occur if there are only a few primary cases.
The probability that, following a single cross-species transmission event by a pathogen with R 0<1in the new host, the pathogen adapts to its new host during an outbreak is approximated by Equation III [19]:
| [Eqn III] |
where μ is the (small) probability that the required genetic change occurs during a single infection. The behaviour of this equation is illustrated in Figure Ic. If the new R 0 is >1, then the evolved pathogen will give rise to a major epidemic with the probability given by Equation II.
Figure I.
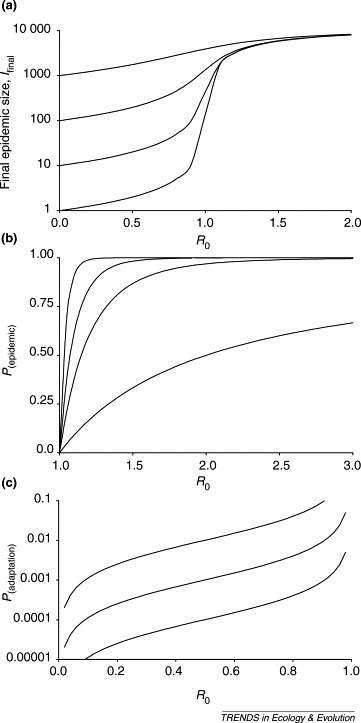
Impact of R0 on the population dynamics of pathogen emergence. (a) Relationship between final epidemic size (log scale) and R0 from Equation I with N =10 000, for I0=1, 10, 100 and 1000. (b) Relationship between the probability of a major epidemic, P(epidemic), and R0 from Equation II, for I0=1, 5, 10 and 25. Redrawn with permission from [17]. (c) Approximate relationship (valid when μ≪1 and R0 is <1 and not too close to 1) between the probability that the pathogen adapts during an outbreak so that R0 becomes >1, P(adaptation), and the original value of R0 from Equation III for μ=0.0001, 0.001 and 0.01. Redrawn with permission from [19].
The key difference between the two categories lies in the origin of infections within the new host population: if R 0 is <1, then a large proportion of infections will be acquired directly from the original source host population; if R 0 is >1 (and the outbreak takes off), then most infections will be acquired from within the new host population, the resulting positive feedback potentially fuelling a major epidemic. There is a transition between these behaviours in the region R 0≈1, where the size of an epidemic is highly sensitive to small changes in the transmission potential (Box 1, Figure Ia). This is especially relevant to pathogen emergence because it implies that relatively small changes in R 0 can have large impacts on the incidence of infection. As has been widely discussed, there are many reasons why R 0 might change. These include:
• Changes in host ecology and environment. For example, urbanization has been cited as a key factor increasing the transmission potential of many human viral and bacterial infections 10, 11, as has the extremely high densities of European wheat and barley crops for diseases such as yellow rust and powdery mildew, respectively 12, 13. Another example is the ongoing concern that climate change might be associated with changing distributions of vector-borne diseases, such as tick-borne encephalitis and Lyme disease 14, 15, 16;
• Changes in host behaviour and movements. For example, patterns of sexual behaviour directly affect the potential spread of sexually transmitted diseases 10, 17, and global travel exacerbated the spread of SARS [18];
• Changes in host phenotype. For example, immunosuppression during hospital treatments or due to the effects of HIV/AIDS has been cited as contributing to the spread of numerous infections (e.g. the fungal pathogen Pneumocystis carinii) 10, 11, 17. The loss of cross-immunity (where acquired immune responses induced by exposure to one microorganism are at least partially protective against infection with another; a quite general concept that, for example, underlies the efficacy of BCG vaccination) might also increase the potential for invasions by new pathogens, as has been suggested for several pairings: yaws and syphilis; leprosy and TB; yellow fever and dengue fever; smallpox and monkeypox; and vivax malaria and falciparum malaria 17, 19, 20;
• Changes in host genetics. For example, the loss of major histocompatibility complex haplotypes or other genetic diversity in inbred livestock or small populations might increase susceptibility to infection [21]. Among plants, the use of single cultivars increases the vulnerability of many crop species to widespread epidemics of pathogens spilling over from closely related wild host species: for example, commercial banana plantations composed of a single clone are at risk from various races of Panama disease, Fusarium oxysporum [22];
• Changes in pathogen genetics (discussed in detail below).
The biology of jumps
The discussion above considers the fate of primary cases of infection introduced into a new host population. But to understand species jumps, we need to consider in greater detail the biological processes occurring around the jump itself.
The first step is exposure of the new host species to the pathogen (Figure 1 ). The rate of exposure will be a function of the ecologies and behaviour of the two host species and of the transmission biology of the pathogen itself (including the biology of any vectors involved). Indeed ‘ecological’ change, in its broadest sense, is associated with most instances of disease emergence 11, 23, 24; for example, those of phocine distemper, Lyme disease, BSE and hepatitis C. For vector-borne diseases, exposure of new host species might be facilitated by the pathogen jumping between vector species or populations, as has been suggested for Venezuelan equine encephalitis virus (VEEV) [25].
Figure 1.

Preventing cross-species transmission. As part of a campaign to reduce the risk to public health in Matongo, Tanzania, local people in the region are encouraged to have their domestic dogs vaccinated against rabies. Reproduced with permission from T. Lembo.
The second step is for the pathogen to be able to infect the new host; that is, for pathogen and host to be ‘compatible’. Pathogens have highly variable host ranges: some only naturally infect a single species (e.g. the mumps virus or Plasmodium falciparum in humans), whereas others can infect hosts from different taxonomic orders or even classes (e.g. rabies virus or the protozoan Blastocystis hominis) [26]. The reasons for this variation are poorly understood, although certain factors, such as an indirect route of transmission, are known to be associated with a broad host range [26]. For viruses, one such factor is the use of cell receptors that are phylogenetically conserved [23]. Crucial to the ability of a cell-free virus to infect hosts is the presence of appropriate cell receptors on host cells. When receptors are conserved across a range of potential host species, the hosts are likely to be predisposed to infection by the viruses using these receptors. For example, use of conserved receptors might explain the wide host ranges of foot-and-mouth diseases virus (FMDV), which uses the integrin vitronectin, and rabies virus, which uses the nicotinic acetylcholine receptor [27].
However, even if capable of infecting a different host species, pathogens are usually, although not always, significantly less infectious to it. This is referred to as the ‘species barrier’, and it can be substantial, implying that much higher doses are required to infect the new host: for example, the dose of rabies virus from foxes required to infect dogs and cats has been shown experimentally to be up to a million times greater than that required to infect other foxes [28].
The third and final step in a successful species jump is for the pathogen to be sufficiently transmissible between individuals within the new host population. As discussed above, this relates to the value of R 0 and, therefore, whether the pathogen can successfully invade the new host population.
Evolution and host adaptation
If R 0 is >1, then there is a sense in which there is ‘an epidemic waiting to happen’; numerous recent examples include the introductions of West Nile virus into North American birds, phocine distemper virus into North Sea seals, and Dutch elm disease into elms in the UK and USA. Indeed, emerging pathogens are often a special concern because the absence of shared evolutionary history with the new host implies an absence of evolved constraints on susceptibility and pathogenicity, which might, at least in some instances, enable disease outbreaks of large magnitude and unusual severity 21, 29.
Conversely, if R 0 is <1, then the arguments presented earlier imply that each primary case will result in a chain of transmission in the new host population that will stutter to extinction. This, however, might be overly optimistic because of the possibility that the pathogen evolves so that R 0 becomes >1 and, as a result, could go on to generate a major epidemic [19].
This evolution, or ‘adaptation’, of the pathogen can involve genetic changes ranging from a few nucleotide substitutions (e.g. canine parvovirus, CPV [30]), through gene capture from other organisms (e.g. Salmonella enterica and Escherichia coli [31]), to recombination or reassortment (e.g. H5N1 influenza [32] and Ophiostoma novo-ulmi, the agent of Dutch elm disease [33]) and hybridization (e.g. Phytophthora alni in alder trees in north-west Europe appears to be an allopolyploid recombinant between a newly introduced pathogen of hard woods, P. cambivora, and a related specialist pathogen of raspberries and strawberries [34]). Adaptation might be so rapid that pathogen lineages adapt to different host tissues or vector cells versus host cells 25, 35.
The probability of successful adaptation occurring depends on several factors such as: (i) the number of primary infections, I 0; (ii) the initial R 0 of the infection in the new host population; (iii) the number of mutations or other genetic changes required; and (iv) the likelihood of these changes occurring and how R 0 changes at each step. It is relatively simple to see that the probability of emergence increases linearly with I 0, but is much more sensitive to the evolution of R 0, particularly when this is close to 1 [19]. This is because the probability of each (rare) evolutionary step is proportional to the expected size of the initial outbreak and, hence, the number of opportunities for the required genetic change(s) to occur (Box 1, Figure Ic). The expected size of an outbreak is related non-linearly to R 0 (Box 1, Figure Ia) and, because conditions are likely to differ from outbreak to outbreak, outbreak sizes in practice tend to be highly overdispersed, with occasional larger outbreaks providing more opportunities for adaptation (Figure 2 ).
Figure 2.
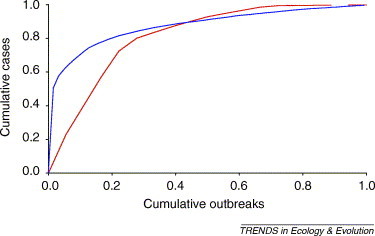
Examples of overdispersed outbreak sizes. Plots show the cumulative fraction of all cases against the cumulative fraction of outbreaks ordered from the largest to the smallest for outbreaks of human infection with Ebola virus in sub-Saharan Africa from 1976 to 2004 [red line; 18 outbreaks, 428 cases; data from ProMed (http://www.promedmail.org), the World Health Organization (http://www.who.int) and the Centers for Disease Control and Prevention (http://www.cdc.gov)] and Escherichia coli O157 in Scotland from 1996 to 2003 (blue line; 63 outbreaks, 1008 cases; data from Health Protection Scotland (http://www.hps.scot.nhs.uk)). For both examples, most outbreaks are small (<10 cases) and only a few outbreaks are large (hundreds of cases), as indicated by the strongly convex shape of the plots. This is consistent with a general trend for disease outbreak size distributions to follow a power law with exponent >2, indicative of severe overdispersion [26].
We are beginning to understand the biology underlying host adaptation in just a few instances. For example, virus receptor use can be labile [27] and just a few point mutations on the viral capsid can enable the use of new receptors: for example, FMDV will switch to using additional receptors after accumulating just a few amino acid substitutions in cell culture [36]; feline panleukopenia virus (FPLV) evolved into CPV by acquiring the ability to use the canine transferrin receptor as a result of a few changes in the capsid amino acid sequence [30]; and the adaptation of VEEV to equines is associated with changes in the envelope glycoprotein [25].
But, even though it is sometimes possible to point to genetic differences between pathogens in the original and new host, it is often difficult to ascribe these changes to: (i) events in the original host population before the jump (i.e. predisposition of novel genotypes to jump species); (ii) events during the ‘adaptation’ phase where there was a shift from R 0<1 to R 0>1 in the new host; or (iii) subsequent divergence once the pathogen is established in the new host.
Host jumping is likely to have been an important driver of pathogen diversity through evolutionary time. Evidence of historically deeper host jumps is provided by incongruencies in the phylogenetic topologies of host species and their respective pathogens [37]. For example, in a recent study of RNA viruses, hantaviruses, spumaviruses and avian sarcoma leucosis viruses showed significant levels of congruence with their host species, whereas arenaviruses and lyssaviruses showed no congruence [38].
Future work
There are several additional issues that could also be analyzed using the above framework for assessing the likelihood of a successful invasion (many of which apply to biological invasions in general [39]). For example, fluctuating transmission rates can increase persistence times for pathogens with R 0<1 [40]. It is also possible that, rather than single introduction(s) into a new host population, there are periods when the pathogen is spreading in a mixture of host species (e.g. as has been proposed for human sleeping sickness [41]); this could increase persistence times in the new host but, simultaneously, reduce selection pressure on the pathogen to adapt to the new host. And the new host population itself is unlikely to be homogeneous: some individuals might be more susceptible to novel pathogens (e.g. owing to immunosuppression) and/or more exposed to them (e.g. owing to their behaviour or spatial location [42]) and/or more likely to transmit infection (so-called ‘supershedders’). Such heterogeneities can increase outbreak sizes [5]. The structure of the new host population might also be important: contrast Ebola, which usually affects remote communities, with SARS, which arose in a region with high human population density, large numbers of movements and extensive travel. Similarly, the HIV/AIDS epidemic apparently took off once it had escaped remote communities and entered urban populations [17]. The general issue here is the relationship between ‘samplers’ (individuals with high risk of acquiring novel infections) and ‘spreaders’ (individuals with high potential for transmitting a novel infection onwards within the new host population): the closer the epidemiological linkage between these groups the greater the potential for successful invasions by new pathogens. The mechanism of genetic change of the pathogen is also likely to be important: if it can evolve not only by mutation, but also by recombination (e.g. influenza viruses and SARS coronavirus 32, 43) or by gene capture (e.g. pathogenicity islands or antimicrobial resistance genes in bacteria 44, 45) or by hybridization (e.g. P. alni) then this might influence the epidemiology of species jumps by requiring the same host to be co-infected with two different pathogens, perhaps from different sources.
Combating emerging pathogens
The emergence of a new pathogen following a species jump represents the successful colonization of a new habitat (reflecting this, emerging pathogens have been compared to weeds [46]). Although it is extremely hard to predict which pathogens are most likely to jump between host species, there are some hints that some progress can be made. For example, perhaps the most striking feature of the list of examples of species jumps given in Table 1 is that the pathogens involved are mostly, and disproportionately, single-stranded RNA viruses. Although Table 1 should not be regarded as an exhaustive survey, this might well be a genuine effect reflecting the typically broader host ranges and much higher mutation rates of RNA viruses [26], facilitating both the initial infection of a new host and subsequence adaptation to that host. Further refining this observation, none of the RNA viruses listed are transmitted by arthropod vectors, perhaps reflecting the constraints imposed on a small genome by having to be compatible with a vector as well as a definitive host. In support of this, both emerging and long-recognised zoonotic RNA arboviruses tend to be poorly transmissible between humans 24, 25, although experimental evidence for such constraints is inconclusive [25].
A second notable feature of Table 1 is that there is no obvious indication of close taxonomic relatedness between the original and the new host species. Consistent with this, a systematic survey of emerging zoonotic pathogens of humans [47] found that the most probable reservoirs were (in rank order): (i) ungulates; (ii) carnivores; (iii) rodents; (iv) primates; (v) birds and other non-mammalian hosts; (vi) bats; and (vii) marine mammals. It has even been suggested that nanoviruses jumped from plants to vertebrates [48]. A broad host range seems to be more important to the potential for a pathogen to jump between species than is the relatedness of the hosts involved 23, 24, 47.
The unpredictability of pathogen emergence means that the first line of defence has to be effective surveillance, requiring identification and monitoring of high-risk populations or individuals or locations, and even the setting up of ‘sentinel’ systems [11]. Recent work using agent-based simulation models points the way to the efficient design of surveillance systems based on an understanding of the contact network within the host population(s) [49]. An effective public health and/or veterinary response then requires prompt, coordinated action by multi-disciplinary teams, as exemplified by the recent global effort led by the World Health Organization (http://www.who.int) to combat SARS [50]. The importance of rapid identification, assessment and action cannot be overstated: often, the single biggest factor affecting the scale of an epidemic is the speed with which effective interventions are put in place 51, 52.
Acknowledgements
We thank Louise Matthews and Eddie Holmes for valuable discussions and Robert Webster and Kate Snedeker for assistance with data collation. M.E.J.W. and D.T.H. gratefully acknowledge the support of the Wellcome Trust.
References
- 1.Woolhouse M.E.J., Dye C. Population biology of emerging and re-emerging pathogens – preface. Philos. Trans. R. Soc. Lond. Ser. B. 2001;356:981–982. [Google Scholar]
- 2.Hahn B.H. AIDS as a zoonosis: scientific and public health implications. Science. 2000;287:607–614. doi: 10.1126/science.287.5453.607. [DOI] [PubMed] [Google Scholar]
- 3.Fenner F., Fantini B. CABI; 1999. Biological Control of Vertebrate Pests: The History of Myxomatosis – an Experiment in Evolution. [Google Scholar]
- 4.Sinclair A.R.E., Norton-Griffiths M. University of Chicago Press; 1995. Serengeti: Dynamics of an Ecosystem. [Google Scholar]
- 5.Hudson P.J., editor. The Ecology of Wildlife Diseases. Oxford University Press; 2002. [Google Scholar]
- 6.Diamond J. Evolution, consequences and future of plant and animal domestication. Nature. 2002;418:700–707. doi: 10.1038/nature01019. [DOI] [PubMed] [Google Scholar]
- 7.Anderson P.K. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol. Evol. 2004;19:535–544. doi: 10.1016/j.tree.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 8.Goodwin S.B. Panglobal distribution of a single clonal lineage of the Irish potato famine fungus. Proc. Natl. Acad. Sci. U. S. A. 1994;91:11591–11595. doi: 10.1073/pnas.91.24.11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milgroom M.G. Intercontinental population structure of the chestnut blight fungus, Cryphonectria parasitica. Mycologia. 1996;88:179–190. [Google Scholar]
- 10.McMichael A.J. Environmental and social influences on emerging infectious diseases: past, present and future. Philos. Trans. R. Soc. Lond. Ser. B. 2004;359:1049–1058. doi: 10.1098/rstb.2004.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smolinski M.S., editor. Microbial Threats to Health: Emergence, Detection and Response. National Academies Press; 2003. [PubMed] [Google Scholar]
- 12.Hovmoller M.S. Clonality and long-distance migration of Puccinia striiformis f.sp tritici in north-west Europe. Plant Pathol. 2002;51:24–32. [Google Scholar]
- 13.Wolfe M.S. Barley mildew in Europe – population biology and host-resistance. Euphytica. 1992;63:125–139. [Google Scholar]
- 14.Kovats R.S. Early effects of climate change: do they include changes in vector-borne disease? Philos. Trans. R. Soc. Lond. Ser. B. 2001;356:1057–1068. doi: 10.1098/rstb2001.0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Randolph S.E. The shifting landscape of tick-borne zoonoses: tick-borne encephalitis and Lyme borreliosis in Europe. Philos. Trans. R. Soc. Lond. Ser. B. 2001;356:1045–1056. doi: 10.1098/rstb.2001.0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers D.J., Randolph S.E. The global spread of malaria in a future, warmer world. Science. 2000;289:1763–1766. doi: 10.1126/science.289.5485.1763. [DOI] [PubMed] [Google Scholar]
- 17.May R.M. Infectious disease dynamics: what characterizes a successful invader? Philos. Trans. R. Soc. Lond. Ser. B. 2001;356:901–910. doi: 10.1098/rstb.2001.0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peiris J.S.M., Guan Y. Confronting SARS: a view from Hong Kong. Philos. Trans. R. Soc. Lond. Ser. B. 2004;359:1075–1079. doi: 10.1098/rstb.2004.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antia R. The role of evolution in the emergence of infectious diseases. Nature. 2003;426:658–661. doi: 10.1038/nature02104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maitland K. Plasmodium vivax and P. falciparum: biological interactions and the possibility of cross-species immunity. Parasitol. Today. 1997;13:227–231. doi: 10.1016/s0169-4758(97)01061-2. [DOI] [PubMed] [Google Scholar]
- 21.Woolhouse M.E.J. Biological and biomedical implications of the coevolution of pathogens and their hosts. Nat. Genet. 2002;32:569–577. doi: 10.1038/ng1202-569. [DOI] [PubMed] [Google Scholar]
- 22.Ploetz R.C. Panama disease – return of the first banana menace. Int. J. Pest Manage. 1994;40:326–336. [Google Scholar]
- 23.Woolhouse M.E.J. Population biology of emerging and re-emerging pathogens. Trends Microbiol. 2002;10:S3–S7. doi: 10.1016/s0966-842x(02)02428-9. [DOI] [PubMed] [Google Scholar]
- 24.Taylor L.H. Risk factors for human disease emergence. Philos. Trans. R. Soc. Lond. Ser. B. 2001;356:983–989. doi: 10.1098/rstb.2001.0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weaver S.C., Barrett A.D.T. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2004;2:789–801. doi: 10.1038/nrmicro1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woolhouse M.E.J. Population biology of multi-host pathogens. Science. 2001;292:1109–1112. doi: 10.1126/science.1059026. [DOI] [PubMed] [Google Scholar]
- 27.Baranowski E. Evolution of cell recognition by viruses. Science. 2001;292:1102–1105. doi: 10.1126/science.1058613. [DOI] [PubMed] [Google Scholar]
- 28.Blancou J., Aubert M.F.A. Transmission du virus de la rage: importance del la barrière d'espèce. Bull. Acad. Natl. Méd. 1997;181:301–312. [PubMed] [Google Scholar]
- 29.Stearns S.C., editor. Evolution in Health & Disease. Oxford University Press; 1999. [Google Scholar]
- 30.Hueffer K. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J. Virol. 2003;77:1718–1726. doi: 10.1128/JVI.77.3.1718-1726.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wain J. Acquisition of virulence-associated factors by the entric pathogens Escherichia coli and Salmonella enterica. Philos. Trans. R. Soc. Lond. Ser. B. 2001;356:1027–1034. doi: 10.1098/rstb.2001.0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li K.S. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature. 2004;430:209–213. doi: 10.1038/nature02746. [DOI] [PubMed] [Google Scholar]
- 33.Brasier C.M. Rapid evolution of introduced plant pathogens via interspecific hybridization. Bioscience. 2001;51:123–133. [Google Scholar]
- 34.Brasier C.M. Origin of a new Phytophthora pathogen through interspecific hybridization. Proc. Natl. Acad. Sci. U. S. A. 1999;96:5878–5883. doi: 10.1073/pnas.96.10.5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weaver S.C. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J. Virol. 1999;73:4316–4326. doi: 10.1128/jvi.73.5.4316-4326.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baranowski E. Cell recognition by foot-and-mouth disease virus that lacks the RGD integrin-binding motif: flexibility in aphthovirus receptor usage. J. Virol. 2000;74:1641–1647. doi: 10.1128/jvi.74.4.1641-1647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Page R.D.M., editor. Tangled Trees – Phylogeny, Cospeciation, and Coevolution. University of Chicago Press; 2003. [Google Scholar]
- 38.Jackson A.P., Charleston M.A. A cophylogenetic perspective of RNA-virus evolution. Mol. Biol. Evol. 2004;21:45–57. doi: 10.1093/molbev/msg232. [DOI] [PubMed] [Google Scholar]
- 39.Sakai A.K. The population biology of invasive species. Annu. Rev. Ecol. Syst. 2001;32:305–332. [Google Scholar]
- 40.Lewontin R.C., Cohen D. On population growth in a randomly varying environment. Proc. Natl. Acad. Sci. U. S. A. 1969;62:1056–1060. doi: 10.1073/pnas.62.4.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hide G. The origins, dynamics and generation of Trypanosoma brucei rhodesiense epidemics in East Africa. Parasitol. Today. 1996;12:50–55. doi: 10.1016/0169-4758(96)80654-5. [DOI] [PubMed] [Google Scholar]
- 42.Wolfe N.D. Naturally acquired simian retrovirus infections in central African hunters. Lancet. 2004;363:932–937. doi: 10.1016/S0140-6736(04)15787-5. [DOI] [PubMed] [Google Scholar]
- 43.Stavrinides J., Guttman D.S. Mosaic evolution of the severe acute respiratory syndrome coronavirus. J. Virol. 2004;78:76–82. doi: 10.1128/JVI.78.1.76-82.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ochman H. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 45.Groisman E.A., Ochman H. How Salmonella became a pathogen. Trends Microbiol. 1997;5:343–349. doi: 10.1016/S0966-842X(97)01099-8. [DOI] [PubMed] [Google Scholar]
- 46.Dobson A., Foufopoulos J. Emerging infectious pathogens of wildlife. Philos. Trans. R. Soc. Lond. Ser. B. 2001;356:1001–1012. doi: 10.1098/rstb.2001.0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cleaveland S. Diseases of humans and their domestic mammals: pathogen characteristics, host range and the risk of emergence. Philos. Trans. R. Soc. Lond. Ser. B. 2001;356:991–999. doi: 10.1098/rstb.2001.0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gibbs M.J., Weller G.F. Evidence that a plant virus switched hosts to a vertebrate and then recombined with a vertebrate-infecting virus. Proc. Natl. Acad. Sci. U. S. A. 1999;96:8022–8027. doi: 10.1073/pnas.96.14.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eubank S. Modelling disease outbreaks in realistic urban social networks. Nature. 2004;429:180–184. doi: 10.1038/nature02541. [DOI] [PubMed] [Google Scholar]
- 50.Stohr K. A multicentre collaboration to investigate the cause of severe acute respiratory syndrome. Lancet. 2003;361:1730–1733. doi: 10.1016/S0140-6736(03)13376-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilesmith J.W. An epidemiologist's view of bovine spongiform encephalopathy. Philos. Trans. R. Soc. Lond. Ser. B. 1994;343:357–361. doi: 10.1098/rstb.1994.0029. [DOI] [PubMed] [Google Scholar]
- 52.Haydon D.T. The construction and analysis of epidemic trees with reference to the 2001 UK foot-and-mouth outbreak. Proc. R. Soc. Lond. Ser. B. 2003;270:121–127. doi: 10.1098/rspb.2002.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bruce M.E. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 54.Morvan J.A. Ebola virus and forest ecosystem. Bull. Soc. Path. Exot. 2000;93:172–175. [PubMed] [Google Scholar]
- 55.Parrish C.R. Canine parvovirus 2: a probable example of interspecies transfer. In: Morse S., editor. Emerging Viruses. Oxford University Press; 1993. pp. 194–202. [Google Scholar]
- 56.Mahy B.W.J. Seal plague virus. In: Morse S., editor. Emerging Viruses. Oxford University Press; 1993. pp. 184–193. [Google Scholar]
- 57.Mackenzie J.S., Field H.E. Emerging encephalitogenic viruses: lyssaviruses and henipaviruses transmitted by frugivorous bats. Arch. Virol. 2004;18(Suppl.):97–111. doi: 10.1007/978-3-7091-0572-6_8. [DOI] [PubMed] [Google Scholar]
- 58.He J.F. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666–1669. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- 59.Guarner J. Monkeypox transmission and pathogenesis in prairie dogs. Emerg. Inf. Dis. 2004;10:426–431. doi: 10.3201/eid1003.030878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaper, J.B. and O'Brien, A.D., eds (1998) Escherichia coli O157:H7 and Other Shiga Toxin-Producing E. coli strains, American Society for Microbiology