

Abstract
We have developed a novel multisample detection system by employing a technology combining a tag insertion primer and an electrochemical DNA chip. In the first application, Helicobacter species-infected mouse samples were detected. The primers that insert a different tag sequence in each sample were prepared, and loop-mediated isothermal amplification (LAMP) reaction was carried out. Then amplification products in which a part of the sequence was different in each sample could be obtained. The target sample in which these amplification products were mixed was injected into a cassette that included the DNA chip with immobilized probes. After the cassette was set in the DNA detection system, Genelyzer, the processes of hybridization, washing, and detection were performed by the system automatically. The positive and negative concordance rates of the existing nested polymerase chain reaction (PCR) method and this method were 100% (40/40 samples) and 97.3% (117/120 samples), respectively. This is a simple high-throughput method. Moreover, the cost per sample can be drastically lowered. Therefore, it is expected to contribute to the diagnosis of infectious agents in humans and animals.
Keywords: Electrochemical DNA chip, Multisample detection, Tag insertion primer, Loop-mediated isothermal amplification, Helicobacter species
Diagnosis of human infectious diseases, such as human papillomavirus (HPV), human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV), cytomegalovirus (CMV), influenza, tubercle bacillus, clamydia, and gonococcus, is important for determining appropriate diagnosis and providing effective treatment. Diagnosis of farm animals and laboratory animals is useful for early isolation of the infected individuals and prevention of the spread of infection. Immune-based tests have long been the principal means of diagnosis of these infectious diseases. On the other hand, the demand for genetic diagnosis has risen because of its high sensitivity, specificity, and reproducibility [1], [2], [3]. Genetic analysis methods include real-time polymerase chain reaction (PCR), 1 PCR sequence, TaqMan PCR, invader assay, PCR restriction fragment-length polymorphism (RFLP), allele-specific primer (ASP) PCR, single-strand conformation polymorphism (SSCP), and DNA chip-based technology.
A DNA chip is a powerful tool allowing simultaneous detection of numerous nucleic acid targets in a sample [4], [5]. This technology can be used for gene expression monitoring, mutation, polymorphism, and phenotypic analysis and has become an indispensable tool in biology, biotechnology, and drug discovery [6]. However, because general fluorescence-based DNA chip methods require expensive devices and involve complicated operations, their use is largely limited to research. With regard to infectious diseases, the ability to test many samples for several genes would be advantageous. As for a general DNA chip, because one chip corresponds to one sample, it is unsuitable for multisample processing. To analyze multiple samples simultaneously, a DNA chip that physically separates a reactive part has been reported [7], [8]. But there are disadvantages in that large-scale device remodeling is necessary and the chip becomes more expensive owing to its larger size.
We have been developing an electrochemical DNA chip using an electrochemically active intercalator and DNA probe immobilized on a gold electrode [9], [10]. The principle of the electrochemical DNA chip has been described previously [11]. This method is simple and inexpensive because it requires no labeling step and large and expensive equipment is unnecessary. Therefore, it is suitable for genetic diagnostics. Moreover, in exploiting an advantage of this method, namely that it is easy to develop an automatic system, we have developed a miniature system, Genelyzer, that can be used at the bedside and for on-the-spot examination [12].
In this study, a multisample detection system that combines this electrochemical DNA chip and tag insertion primer was developed mainly for diagnosis of infectious diseases. A feature of this method is the use of a tag for sample identification. In the first application, a screening chip for the detection of Helicobacter species that are the most common contaminants in laboratory rodents was developed [11].
Materials and methods
DNA samples
Standard plasmids inserted into the 16S ribosomal RNA (rRNA) gene sequences of Helicobacter species, Helicobacter hepaticus (Hh), Helicobacter bilis (Hb), Helicobacter typhlonius (Ht), Helicobacter pylori (Hp), and Helicobacter muridarum (Hm), were obtained as custom synthesis products from Invitrogen Japan (Tokyo, Japan). The standard plasmids were used for determination of the detecting condition of this method. Genomic DNAs were extracted from 150 cecal samples of mice. The method for DNA extraction from the cecum by phenol/chloroform and isoamylic alcohol (Nakalai Tesque, Kyoto, Japan) consisted of the addition to each sample of 1 ml of lysis buffer (100 mM Tris–HCl [pH 7.5], 150 mM NaCl, 12.5 mM ethylenediaminetetraacetic acid [EDTA], 65 μg proteinase K/ml, and 1% sodium dodecyl sulfate), followed by incubation in a water bath at 50 °C overnight. Subsequently, the lysate was washed with a mixture of phenol/chloroform and isoamylic alcohol (25:24:1), and the supernatant aqueous phase was recovered by a final washing using chloroform (Nakalai Tesque). Absolute ethanol (Junsei Chemical, Tokyo, Japan) was added to the solution, which was then centrifuged for 5 min at 13,000g. The precipitated DNA was washed with 75% ethanol, and DNA was resuspended in 300 μl of TE buffer (Wako Pure Chemicals, Osaka, Japan). To confirm the accuracy of the DNA chip, these 150 genomic DNAs were also detected by nested PCR analysis.
Nested PCR
The first PCR primer sequences were 5′-CTATGACGGGTATCCGGC-3′ (F1) and 5′-CTCACGACACGAGCTGAC-3′ (R1), and the second PCR primer sequences were 5′-AGGGAATATTGCTCAATGGG-3′ (F2) and 5′-TCGCCTTCGCAATGAGTATT-3′ (R2). The first PCR was carried out in a total volume of 50 μl containing 10× Pyrobest buffer, 0.2 mM each deoxynucleoside triphosphate (dNTP), 0.1 μM each primer (F1 and R1), 1.25 U of Pyrobest (Takara Bio, Shiga, Japan), distilled water, and 1 μl of extracted DNA. The second PCR was subsequently performed using 1 μl of the first PCR product in a total volume of 50 μl containing 10× Pyrobest buffer, 0.2 mM each dNTP, 0.1 μM each primer (F2 and R2), 1.25 U of Pyrobest, and distilled water. Amplification was performed with the same conditions for both the first and second PCRs after preheating at 94 °C for 3 min, followed by 25 cycles of 94 °C for 1 min, 55 °C for 1 min, 72 °C for 1 min, and final extension at 70 °C for 7 min. Secondary PCR products were analyzed by electrophoresis on a 4% agarose gel stained with ethidium bromide and examined under ultraviolet (UV) light.
LAMP reaction
The primers used for amplification are listed in Table 1 . To correspond to the diversity between Helicobacter species, two kinds of FIP primer (FIP-1 and FIP-2) and LPb primer (LPb-1 and LPb-2) were designed and mixed in the same quantity. Loop-mediated isothermal amplification (LAMP) was carried out in 25 μl of reaction mixture containing 3.2 μM each FIP-1, FIP-2, and BIP, 0.4 μM each F3 and B3, 1.6 μM each LPb-1 and LPb-2, 1.4 mM each dNTP, 0.8 M betaine, 20 mM Tris–HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4, 8 mM MgSO4, 0.1% Tween 20, 16 U of Bst DNA polymerase (New England Biolabs, Beverly, MA, USA), and 10, 100, or 1000 copies of standard plasmid or 5 μl of extracted DNA [13]. The mixture was incubated at 63 °C for 60 min and was heated at 95 °C for 5 min to terminate the reaction. The same reaction mixture without a template DNA was used as negative control. The reaction was carried out using a GeneAmp PCR System (model 9700, Applied Biosystems, Foster City, CA, USA). Amplification was confirmed by the presence of the white precipitate of magnesium pyrophosphate in the amplification solution [14].
Table 1.
LAMP primer and amplification results.
| Number | Name | Tag position (distance from 3′ end) | Tag number | Sequence (5′ to 3′) | Amplification resulta |
|
|---|---|---|---|---|---|---|
| Hh | Hp | |||||
| 1 | FIP-1b | – | – | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGCATTTG | ○ | ○ |
| 2 | FIP-2b | – | – | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGCATTTG | ||
| 3 | BIPc | – | – | AAGAGGAATACTCATTGCGA-CTGTTTGCTCCCCACG | – | |
| 4 | F3c | – | – | TACTCGGAATCACTGGGC | – | |
| 5 | B3c | – | – | GGCGTGGACTACCAGGGT | – | |
| 6 | LPb-1c | – | – | GGAACATTACTGACGCTGAT | – | |
| 7 | LPb-2c | – | – | GGAACATTACTGACGCTCAT | – | |
| 8 | FIP-1 3AC | 3 | 2 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGCATACTTG | ○ | ○ |
| 9 | FIP-2 3AC | 3 | 2 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGCATACTTG | ||
| 10 | FIP-1 3TG | 3 | 2 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGCATTGTTG | × | × |
| 11 | FIP-2 3TG | 3 | 2 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGCATTGTTG | ||
| 12 | FIP-1 3CTG | 3 | 3 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGCATCTGTTG | × | × |
| 13 | FIP-2 3CTG | 3 | 3 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGCATCTGTTG | ||
| 14 | FIP-1 3GGA | 3 | 3 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGCATGGATTG | × | × |
| 15 | FIP-2 3GGA | 3 | 3 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGCATGGATTG | ||
| 16 | FIP-1 3ACTG | 3 | 4 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGCATACTGTTG | × | × |
| 17 | FIP-2 3ACTG | 3 | 4 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGCATACTGTTG | ||
| 18 | FIP-1 3TGAC | 3 | 4 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGCATTGACTTG | × | × |
| 19 | FIP-2 3TGAC | 3 | 4 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGCATTGACTTG | ||
| 20 | FIP-1 6AC | 6 | 2 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGACCATTTG | ○ | ○ |
| 21 | FIP-2 6AC | 6 | 2 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGACCATTTG | ||
| 22 | FIP-1 6TG | 6 | 2 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGTGCATTTG | ○ | ○ |
| 23 | FIP-2 6TG | 6 | 2 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGTGCATTTG | ||
| 24 | FIP-1 6CTG | 6 | 3 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGCTGCATTTG | ○ | ○ |
| 25 | FIP-2 6CTG | 6 | 3 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGCTGCATTTG | ||
| 26 | FIP-1 6GGA | 6 | 3 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGGGACATTTG | × | ○ |
| 27 | FIP-2 6GGA | 6 | 3 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGGGACATTTG | ||
| 28 | FIP-1 6ACTG | 6 | 4 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGACTGCATTTG | ○ | × |
| 29 | FIP-2 6ACTG | 6 | 4 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGACTGCATTTG | ||
| 30 | FIP-1 6TGAC | 6 | 4 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGTGACCATTTG | × | ○ |
| 31 | FIP-2 6TGAC | 6 | 4 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGTGACCATTTG | ||
| 32 | FIP-1 9AC | 9 | 2 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAAACCTGCATTTG | ○ | ○ |
| 33 | FIP-2 9AC | 9 | 2 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAAACCTGCATTTG | ||
| 34 | FIP-1 9TG | 9 | 2 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAATGCTGCATTTG | ○ | ○ |
| 35 | FIP-2 9TG | 9 | 2 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAATGCTGCATTTG | ||
| 36 | FIP-1 9CTGd | 9 | 3 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACTGCTGCATTTG | ○ | ○ |
| 37 | FIP-2 9CTGd | 9 | 3 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACTGCTGCATTTG | ||
| 38 | FIP-1 9GGAd | 9 | 3 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAAGGACTGCATTTG | ○ | ○ |
| 39 | FIP-2 9GGAd | 9 | 3 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAAGGACTGCATTTG | ||
| 40 | FIP-1 9ACTG | 9 | 4 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAAACTGCTGCATTTG | ○ | ○ |
| 41 | FIP-2 9ACTG | 9 | 4 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAAACTGCTGCATTTG | ||
| 42 | FIP-1 9TGAC | 9 | 4 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAATGACCTGCATTTG | ○ | ○ |
| 43 | FIP-2 9TGAC | 9 | 4 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAATGACCTGCATTTG | ||
| 44 | FIP-1 9CCTd | 9 | 3 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAACCTCTGCATTTG | ○ | ○ |
| 45 | FIP-2 9CCTd | 9 | 3 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAACCTCTGCATTTG | ||
| 46 | FIP-1 9TCCd | 9 | 3 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAATCCCTGCATTTG | ○ | ○ |
| 47 | FIP-2 9TCCd | 9 | 3 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAATCCCTGCATTTG | ||
| 48 | FIP-1 9ATCd | 9 | 3 | AAGAATTCCACCTACCTCTCCC-TAGCTTAACTACAGAAATCCTGCATTTG | ○ | ○ |
| 49 | FIP-2 9ATCd | 9 | 3 | AAGAATTCCACCTACCTCTCCC-TGGCTTAACCATAGAAATCCTGCATTTG | ||
Note. Tags were inserted in the F2 region in the FIP primer. The underlined bases indicate the tag positions. Amplification results were confirmed by the presence of the white precipitate of magnesium pyrophosphate in the amplification solution.
Symbols: ○, white precipitate was confirmed; ×, white precipitate was not confirmed.
Primers without tag insertion.
Primers used for every amplification.
Tag insertion primers finally selected. These primers were also confirmed to amplify in Hb, Ht, and Hm plasmids.
Preparation of DNA chip
The DNA chip substrates used in this study were prepared as described previously [10]. Oligonucleotide probes with a thiol group at the 3′ end were obtained as custom synthesis products from Invitrogen Japan. The sequences of the probes are listed in Table 2 . Five negative control probes whose sequences are irrelevant to Helicobacter 16S rRNA sequences were also prepared. Each working electrode was spotted with 0.1 μl of the probe solution containing the 3 μM oligonucleotide probe, 100 mM sodium chloride, 100 μM 6-mercapto-1-hexanol (Sigma–Aldrich, St. Louis, MO, USA), and 20 mM Pipes buffer by using a spotter. The negative control probes were mixed evenly so that the final concentration became 3 μM. Then the chip was washed with distilled water and stored at −20 °C. For each probe, four electrodes were assigned.
Table 2.
Sequence of probe DNA.
| Number | Name | Sequence (5′ to 3′) |
|---|---|---|
| 1 | Probe for 9CTG | AGTTTCAAATGCAGCAGTTCT |
| 2 | Probe for 9GGA | AGTTTCAAATGCAGTCCTTCT |
| 3 | Probe for 9CCT | AGTTTCAAATGCAGAGGTTCT |
| 4 | Probe for 9TCC | AGTTTCAAATGCAGGGATTCT |
| 5 | Probe for 9ATC | AGTTTCAAATGCAGGATTTCT |
| 6 | Negative control | TGCTTCTACACAGTCTCCTGTACCTGGGCA |
| 7 | TGGTCCTGGCACTGATAATAGGGAATGTAT | |
| 8 | CAAGGTCATAATAATGGTATTTGTTGGGGC | |
| 9 | AGGTCATCCGGGACAGCCTCGCCAAGTTTT | |
| 10 | AGTAGTTATGTATATGCCCCCTCGCCTAGT |
Note. The underlined bases indicate the tag positions.
Hybridization reaction, washing, and electrochemical detection by Genelyzer
DNA chip sample (50 μl) containing the LAMP products from each of five suspected samples and 2× SSC (saline–sodium citrate) was injected into a cassette that included the DNA chip with immobilized probes. After setting the cassette in the Genelyzer system, hybridization reaction, washing, and electrochemical detection are performed automatically. The procedure from hybridization to electrochemical detection has been described previously [15]. The hybridization reaction was carried out at 64 °C for 10 min, and washing was carried out at 44 °C for 3 min. Anodic peak current (I pa) values were measured from the voltammogram of Hoechst 33258 (Wako Pure Chemicals), and increased current (ΔI pa) values show the values in the case that the I pa values on the probes for Helicobacter detection were subtracted from those on the negative control probe. The ΔI pa values exactly indicate the signals corresponding to the hybridization. An average of ΔI pa from four electrodes () was adopted for analysis [15].
Results and discussion
Examination of tag insertion position and tag number
The principle of the multisample detection DNA chip is shown in Fig. 1 . This figure assumes the case where the target nucleic acid, such as bacteria and the virus to be detected, exists in sample 1 and 3 but does not exist in sample 2. First, the primers that insert a different tag sequence in each sample are prepared. Second, the amplification reactions are performed independently in each sample using these primers. In samples 1 and 3, when the tag insertion primer binds to the target nucleic acid, the tag part causes the loop out and the amplification reaction can proceed. Then amplification products in which a part of the sequence is different in each sample can be obtained. The F2 region, between the F2 and F1 regions (F2–F1), the B2 region, and between the B2 and B1 regions (B2–B1) of the LAMP products are partially single-stranded loop [15]. Therefore, when the tag part is inserted into the F2 or B2 region of the LAMP primer, the tag part is introduced into the single-stranded loop portion of the amplification product. Finally, the target sample in which the amplification products from each sample are mixed is detected by the DNA chip. The target sequence that hybridized with the probe is designed to include both the tag part and the F2–F1 or B2–B1 region.
Fig.1.
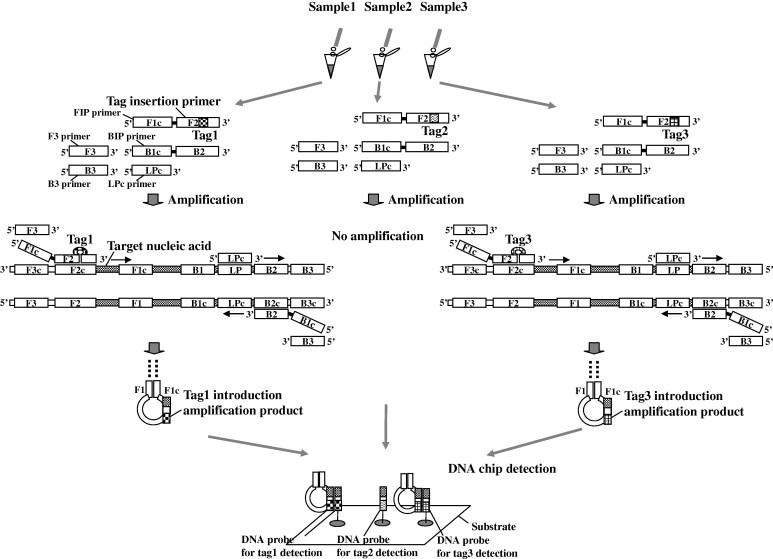
Principle of multisample detection DNA chip. In the LAMP method, six primer regions are set and four primers are used for amplification. The F3, F2, and F1 regions are placed in this order from the 5′ terminal side of a forward strand of a target nucleic acid, and the B3c, B2c, and B1c regions are placed in this order from the 3′ terminal side of the forward strand of a target nucleic acid. The complementary regions of F3, F2, F1, B3c, B2c, and B1c in the reverse strand are called F3c, F2c, F1c, B3, B2, and B1 regions, respectively. Primers constituting four basic primers are FIP (F1c + F2), BIP (B1c + B2), F3, and B3. In addition, the amplification period can be shortened by optionally using a primer called a loop primer, LPc. FIP primer hybridizes to F2c in the target nucleic acid and initiates complementary strand synthesis, and then F3 primer hybridizes to F3c in the target nucleic acid and initiates strand displacement DNA synthesis, releasing an FIP-linked complementary strand. When F1c and F1 that exist in the FIP-linked complementary strand bind, the F2 region and F2–F1 region become a single-stranded loop. Similarly, the B2 region and B2–B1 region become a single-stranded loop. In this multisample detection DNA chip, first, the primers that insert a different tag sequence in each sample are prepared. Second, the amplification reactions are performed independently in each sample using these primers. In samples 1 and 3 where the target nucleic acid exists, when the tag insertion primer binds to the target nucleic acid, the tag part causes the loop out and the amplification reaction can proceed. Then amplification products in which a part of the sequence was different in each sample can be obtained. Finally, the target sample in which the amplification products from each sample are mixed is detected by the DNA chip.
First, the tag insertion position and the tag number in which the amplification reaction was not obstructed were examined. As shown in Fig. 2 , the tag was inserted 3, 6, and 9 bases from the 3′ end of the F2 region, and the tag numbers 2, 3, and 4 were examined. Two kinds of tag sequence were prepared for each design, and 1000 copies/reaction of the Hh and Hp plasmids were used. As shown in Table 1, amplification reaction could be conducted for all primers inserted into the 9 bases from the 3′ end of the F2 region. For primers inserted into 6 bases from the 3′ end, tag numbers 3 and 4 showed unstable amplification, although tag number 2 showed amplification. For primers inserted into 3 bases from the 3′ end, all primers showed unstable amplification. Second, the sensitivity of the tag insertion primers was evaluated. The primers that inserted five tags (1.CTG, 2.GGA, 3.CCT, 4.TCC, and 5.ATC) into 9 bases from the 3′ end of the F2 region and the primer without tag insertion were prepared, and 10, 100, and 1000 copies/reaction of Hh, Hb, Ht, Hp, and Hm plasmids were used. As a result, amplification was confirmed for all primers and all 1000 copies/reaction plasmids. For 10 and 100 copies/reaction plasmids, all primers showed unstable amplification. The sensitivity of the five tag primers was almost the same as that of the primer without tag insertion. In addition, primers that inserted tag numbers 5, 6, 7, 8, and 9 into 9 bases from the 3′ end were examined using 1000 copies/reaction of Hh and Hp plasmids, and tag numbers 5, 6, 7, and 8 (all except 9) could be amplified. Primers that inserted tag number 3 into 7 and 8 bases from the 3′ end were also examined using 1000 copies/reaction of Hh and Hp plasmids, and both primers could be amplified. These results indicated that the tag introduction products can be obtained by optimizing the tag insertion position and tag number.
Fig.2.
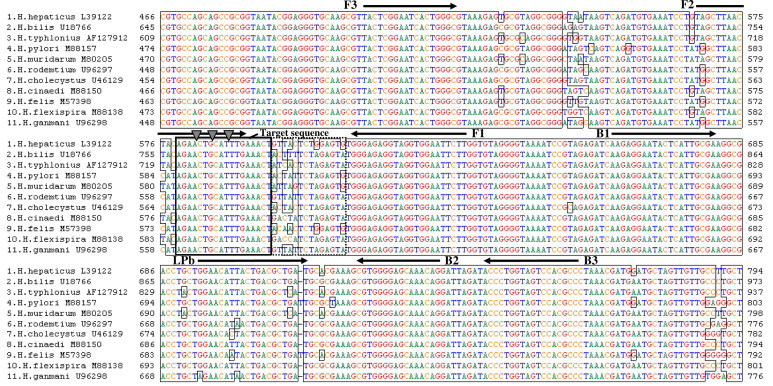
Position of LAMP primer and target sequence. The sequence alignment of the 16S rRNA gene of Helicobacter species is shown. The numbers after species names are GeneBank accession numbers. The arrows, bold-line squares, and triangles indicate the LAMP primer positions, target sequence positions, and tag insertion positions, respectively. Tag sequences that inserted LAMP primers are listed in Table 1. The dotted-line squares indicate the specific sequence regions between Helicobacter species.
Detection of the tag introduction products by DNA chip
The above-mentioned five kinds of tag introduction products were detected by the DNA chip. As shown in Fig. 3 , the ΔI pa that originated from each tag introduction product was confirmed. Next, the data of 148 samples for which equipment, operator, day of operation, and number of positive product are different were collected for the reliability evaluation and the threshold setting of the DNA chip. The means ± 3SD (standard deviation) values of the of the positive and negative samples were calculated as described previously [16]. As shown in Table 3 , the means ± 3SD values of positive and negative samples had clearly divided in all of the five tags. Based on these statistical results, the thresholds of the positive and negative samples were set as 25 nA or more and 10 nA or less, respectively.
Fig.3.
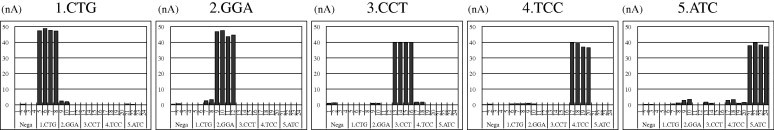
Detection results of five tag introduction products by Genelyzer. 1.CTG is the detection result of a target sample containing a positive product from CTG tag insertion primer and negative products from GGA, CCT, TCC, and ATC tag insertion primers. Similarly, 2.GGA, 3.CCT, 4.TCC, and 5.ATC are the detection results of target samples containing one positive product and four negative products. Here 5 μl of each product was used for detection.
Table 3.
Statistical results of 148 samples and threshold setting.
| Number | Tag | Match/Mismatch | Number of sample | Mean of (nA) | SD of (nA) | Mean of − 3SD (nA) | Mean of + 3SD (nA) | Threshold setting (nA) |
|---|---|---|---|---|---|---|---|---|
| 1 | CTG | Match | 70 | 47.5 | 2.9 | 38.8 | – | 25 |
| Mismatch | 78 | −1.6 | 0.8 | – | 0.8 | 10 | ||
| 2 | GGA | Match | 68 | 44.2 | 3.9 | 32.5 | – | 25 |
| Mismatch | 80 | 0.8 | 1.6 | – | 5.6 | 10 | ||
| 3 | CCT | Match | 68 | 40.9 | 3.4 | 30.7 | – | 25 |
| Mismatch | 80 | −1.5 | 0.9 | – | 1.2 | 10 | ||
| 4 | TCC | Match | 70 | 38.8 | 3.6 | 28.0 | – | 25 |
| Mismatch | 78 | −1.5 | 1.6 | – | 3.3 | 10 | ||
| 5 | ATC | Match | 70 | 37.0 | 3.6 | 26.2 | – | 25 |
| Mismatch | 78 | 0.2 | 2.2 | – | 6.8 | 10 |
Practical verification using field samples
To evaluate the practicality of the DNA chip, the comparison between the existing nested PCR method and DNA chip was performed by using 150 field samples. All 40 positive samples by nested PCR were also positive by DNA chip. For the 110 negative samples by nested PCR, 107 samples were negative and 3 samples were positive by DNA chip (Table 4 ). For 3 samples whose results were divided between the two methods, the samples diluted by 10 times in TE buffer showed unstable amplification by DNA chip. When the sensitivity of nested PCR and DNA chip had been compared by using the same sample, DNA chip showed sensitivity superior to that of the nested PCR method by approximately an order of magnitude. It is speculated that the bacterial concentrations of these 3 samples were so low that it was impossible to detect them by nested PCR. These results demonstrated the practicality and accuracy of the DNA chip.
Table 4.
Results of practical examination.
| Positive | Negative | |
|---|---|---|
| Nested PCR | 40 | 110 |
| DNA chip | 43 | 107 |
In this study, where tag insertion primers were used, it was shown that five samples were easily detected by DNA chip. Simultaneous detection of more samples will become possible by increasing the kinds of tag sequence and optimization of tag insertion position and tag number. The advantages of this method are as follows. First, the cost per sample can be drastically lowered through multisample detection by one DNA chip. Second, this method does not require any configuration change for the equipment and the DNA chip. Third, there is no false-positive detection by the nonspecific reaction of primers that sometimes occurs in LAMP reaction. Fourth, genotyping and species identification can be analyzed according to the application. The target sequence selected for this study was a common region between Helicobacter species for screening use. There is a relatively specific region between each species in the vicinity of this target sequence (Fig. 2). So, if this specific region is set as a target sequence, nearly every species can be identified. Moreover, with the expansion of this method, two or more target nucleic acids can be detected simultaneously by multiplex amplification. We are now developing the DNA chip for multisample detection that identifies several infectious canine diseases such as parvovirus, distemper virus, and enteric coronavirus. In addition, with a view to eventually offering a lineup of automatic systems, we have developed a roll-type system that detects 24 cassettes by automatic operation and a fully automatic system that includes the nucleic acid extraction and amplification. The roll-type system was developed for examination centers and has given satisfactory results with this developed Helicobacter detection DNA chip. The combined technology of the electrochemical DNA chip for multisample detection and the automatic systems is expected to contribute to infectious disease diagnosis in the near future.
Conclusions
We have developed a novel multisample detection system using a tag insertion primer and an electrochemical DNA chip. In the first application, a screening chip that detected Helicobacter species was developed. By optimizing the tag design, the prospect of amplification without decreasing the detection sensitivity was indicated. Furthermore, practicality and accuracy of the DNA chip were confirmed by the results of the practical verification test using the field samples. This method is simple, and the cost per sample can be drastically lowered. We plan to expand the application field of this multisample detection technology to include genotyping, species identification, simultaneous detection of plural target nucleic acids, and so forth and expect it to contribute to the diagnosis of infectious disease in humans and animals.
Footnotes
Abbreviations used: PCR, polymerase chain reaction; rRNA, ribosomal RNA; Hh, Helicobacter hepaticus; Hb, Helicobacter bilis; Ht, Helicobacter typhlonius; Hp, Helicobacter pylori; Hm, Helicobacter muridarum; LAMP, loop-mediated isothermal amplification; SD, standard deviation.
References
- 1.Zaravinos A., Mammas I.N., Sourvinos G., Spandidos D.A. Molecular detection methods of human papillomavirus (HPV) Intl. J. Biol. Markers. 2009;24:215–222. doi: 10.1177/172460080902400401. [DOI] [PubMed] [Google Scholar]
- 2.Scott J.D., Gretch D.R. Molecular diagnostics of hepatitis C virus infection: a systematic review. JAMA. 2007;297:724–732. doi: 10.1001/jama.297.7.724. [DOI] [PubMed] [Google Scholar]
- 3.Ratcliff R.M., Chang G., Kok T., Sloots T.P. Molecular diagnosis of medical viruses. Curr. Issues Mol. Biol. 2007;9:87–102. [PubMed] [Google Scholar]
- 4.Chee M., Yang R., Hubbell E., Berno A., Huang X.C., Stern D., Winkler J., Lockhart D.J., Morris M.S., Fodor S.P. Accessing genetic information with high-density DNA arrays. Science. 1996;274:610–614. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- 5.DeRisi J.L., Iyer V.R., Brown P.O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 6.Ramsay G. DNA chips: state-of-the-art. Nat. Biotechnol. 1998;16:40–44. doi: 10.1038/nbt0198-40. [DOI] [PubMed] [Google Scholar]
- 7.Shen R., Fan J.B., Campbell D., Chang W., Chen J., Doucet D., Yeakley J., Bibikova M., Wickham G.E., McBride C., Steemers F., Garcia F., Kermani B.G., Gunderson K., Oliphant A. High-throughput SNP genotyping on universal bead arrays. Mutat. Res. 2005;573:70–82. doi: 10.1016/j.mrfmmm.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 8.Chang Y.J., Hu C.Y., Yin L.T., Chang C.H., Su H.J. Dividable membrane with multi-reaction wells for microarray biochips. J. Biosci. Bioeng. 2008;106:59–64. doi: 10.1263/jbb.106.59. [DOI] [PubMed] [Google Scholar]
- 9.Hashimoto K., Ito K., Ishimori Y. Sequence-specific gene detection with a gold electrode modified with DNA probes and an electrochemically active dye. Anal. Chem. 1994;66:3830–3833. doi: 10.1021/ac00093a045. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto K., Ishimori Y. Preliminary evaluation of electrochemical PNA array for detection of single base mismatch mutations. Lab Chip. 2001;1:61–63. doi: 10.1039/b103851f. [DOI] [PubMed] [Google Scholar]
- 11.Goto K., Horiuchi H., Shinohara H., Motegi K., Hashimoto K., Hongo S., Gemma N., Hayashimoto N., Itoh T., Takakura A. Specific and quantitative detection of PCR products from Clostridium piliforme, Helicobacter bilis, H. hepaticus, and mouse hepatitis virus infected mouse samples using a newly developed electrochemical DNA chip. J. Microbiol. Methods. 2007;69:93–99. doi: 10.1016/j.mimet.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Hongo S., Okada J., Hashimoto K., Tuji K., Nikaido M., Gemma N. Development of an automated DNA detection system using an electrochemical DNA chip technology. SICE J. Control Meas. Syst. Integr. 2008;1:265–270. [Google Scholar]
- 13.Notomi T., Okayama H., Masubuchi H., Yonekawa T., Watanabe K., Amino N., Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:e63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mori Y., Nagamine K., Tomita N., Notomi T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 2001;23:150–154. doi: 10.1006/bbrc.2001.5921. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura N., Ito K., Takahashi M., Hashimoto K., Kawamoto M., Yamanaka M., Taniguchi A., Kamatani N., Gemma N. Detection of six single-nucleotide polymorphisms associated with rheumatoid arthritis by a loop-mediated isothermal amplification method and an electrochemical DNA chip. Anal. Chem. 2007;79:9484–9493. doi: 10.1021/ac0715468. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura N., Ito K., Takahashi M., Hongo S., Hashimoto K., Kawamoto M., Taniguchi A., Kamatani N., Gemma N. Clinical verification of a combination technology of a loop-mediated isothermal amplification method and an electrochemical DNA chip for personalized medicine. Clin. Biochem. 2009;42:1158–1161. doi: 10.1016/j.clinbiochem.2009.03.016. [DOI] [PubMed] [Google Scholar]