

Abstract
Demyelination following infection of mice with the neurotropic coronavirus mouse hepatitis virus strain JHM (MHV) is immune-mediated. It has been demonstrated that MHV-specific CD4 and CD8 T cells are capable of causing demyelination independent of the other T cell subset. Recent work has also demonstrated that activated bystander CD8 T cells mediate significant demyelination. The ability of bystander CD4 T cells to mediate demyelination was investigated using CD4 T cell transgenic mice. The results indicated that bystander CD4 T cells were unable to cause demyelination in MHV-infected mice, despite being recruited into the central nervous system (CNS) and irrespective of activation status. These results suggest that CD4 T cells must recognize antigen in the CNS in order to cause demyelination.
Keywords: T lymphocytes, Cell trafficking, Viral, Demyelination, Transgenic/knockout
1. Introduction
Immunocompetent mice inoculated with MHV-JHM develop acute and chronic demyelinating diseases (Stohlman et al., 1999) that serve as animal models for the human demyelinating disease, multiple sclerosis (MS). By contrast, mice that are immunodeficient, such as those with a genetic disruption of recombination activation gene 1 (RAG1−/−) or mice with severe combined immunodeficiency syndrome (SCID), do not develop demyelination despite harboring large virus titers Houtman and Fleming, 1996a, Wang et al., 1990, Wu and Perlman, 1999. These mice die from viral encephalitis 14–16 days post-inoculation (p.i.) without protection from T cells or antibodies. Adoptive transfer of CD4 or CD8 T cells from immunocompetent mice into MHV-infected RAG1−/− or SCID mice results in inflammation and demyelination within 7 days post-transfer (p.t.). Macrophage/microglia recruitment and concomitant demyelination occur rapidly after adoptive transfer Wu et al., 2000, Wu and Perlman, 1999, in large part because pro-inflammatory cytokines and chemokines are expressed in the central nervous system (CNS) of RAG1−/− mice even in the absence of transferred T cells (Haring et al., 2002).
Although MHV-specific CD4 and CD8 T cells predominate in the CNS after adoptive transfer, recruitment of T cells into the CNS is random with regard to antigen-specificity Bauer et al., 1998, Hickey et al., 1991, Williams and Hickey, 1995. Using MHC class I tetramers and intracellular cytokine staining (ICS), a large fraction of CNS-derived T cells has been determined to be MHV-specific in immunocompetent mice infected with MHV and after adoptive transfer of T cells into infected RAG1−/− mice Haring et al., 2001, Marten et al., 2000a, Marten et al., 2000b, Pewe and Perlman, 1999. Despite these sensitive methods of identifying antigen-specific T cells, populations of T cells with unknown specificities exist in the CNS after MHV infection. Although as yet undetermined viral epitopes may be recognized by some of these T cells, some T cells in the MHV-infected CNS are likely not to be MHV-specific. Some of these T cells could be autoreactive, as detected in the CNS of mice with demyelination induced by Theiler's murine encephalomyelitis virus (TMEV) (Miller et al., 1997), but autoreactive cells have only rarely been detected in the MHV-infected CNS (Watanabe et al., 1983).
Bystander T cells may be defined as T cells that traffic to a site of inflammation, but are not specific for an antigen present in the inciting agent or for a host protein at the site of inflammation. Little is known about the trafficking of bystander T cells into sites of inflammation, if they are important in resolution of infections, or if they participate in immune-mediated pathology. In a recent study, we demonstrated that activated bystander CD8 T cells were efficient mediators of demyelination in MHV-infected mice (Haring et al., 2002). Using two distinct CD8 TCR transgenic mouse strains, both on a RAG2−/− background (P14 and N15), we showed that CD8 T cells with no reactivity to MHV- or CNS-derived antigens could mediate significant demyelination, but only if the T cells were activated either with cognate peptide plus adjuvant, or by infection of the transgenic mice with virus containing the cognate peptide. The amount of demyelination caused by bystander CD8 T cells was approximately one-third to one-half the amount mediated by MHV-specific CD8 T cells at a similar time post infection with MHV (Wu et al., 2000). The presence of demyelination correlated with increased numbers of activated bystander CD8 T cells, as measured by ICS for interferon-γ (IFN-γ), in the CNS at the time of peak disease. These results provide strong evidence that activated bystander CD8 T cells are capable of participating in immunopathology.
Although MHV-specific CD4 and CD8 T cells can independently cause demyelination, the mechanisms used by each cell type in this process are not entirely redundant Pewe et al., 2002, Pewe and Perlman, 2002. It is likely that bystander CD4 and CD8 T cells may also contribute to demyelination autonomously. Herein, we used TCLi/RAG1−/− mice to investigate if bystander CD4 T cells, like bystander CD8 T cells (Haring et al., 2002), participate in the process of immune-mediated demyelination. In addition, we studied the recruitment and activation of bystander CD4 T cells into the CNS after MHV infection.
2. Materials and methods
2.1. Virus
The neuroattenuated J2.2-V-1 (Fleming et al., 1986) variant of MHV-JHM was generously provided by Dr. J. Fleming (University of Wisconsin, Madison, WI). Viruses were propagated and titered as previously described (Perlman et al., 1987).
2.2. Mice
C57BL/6.PL (B6.PL) mice were kindly provided by Dr. M. Dailey (University of Iowa, Iowa City, IA). hCLIP transgenic mice (TCLi), possessing a TCR specific for residues 81–104 of the human invariant chain-derived CLIP peptide (hCLIP) presented by I-Ab (Wong et al., 2000), were kindly provided by Dr. A. Rudensky (University of Washington, Seattle, WA). They were crossed onto a RAG1−/− background by breeding with C57BL/6J–RAG1−/− mice maintained at the University of Iowa. Pups were screened for α and β transgenes by PCR as previously described (Wong et al., 2000).
2.3. Adoptive transfer
Adoptive transfers were performed as previously described with minor modifications (Wu et al., 2000). Wild-type MHV-JHM was used to immunize adoptive transfer donors as previously described (Wu and Perlman, 1999), except that B6.PL mice were used instead of C57BL/6 (B6) mice. Briefly, TCLi mice were infected intracranially (i.c.) with 1×103 PFU J2.2-V-1 4 days prior to receiving 5×106 splenocytes from MHV-immune B6.PL donors. TCLi mice in the virus-only experimental group were infected, but did not receive donor cells.
2.4. Peptide treatment
In some experiments, we injected TCLi mice i.p. one time 3 days prior to infection with 100 μg of a peptide encompassing residues 85–104 of hCLIP (hCLIP85) (Morkowski et al., 1995) generously provided by Dr. M. Buchmeier (The Scripps Research Institute, La Jolla, CA) emulsified in Complete Freund's Adjuvant. The sequence of hCLIP (85–104) is: PKPVSKMRMATPLLMQALPM (Morkowski et al., 1995).
2.5. Lymphocyte isolation and FACS analysis
At the time of harvest, mice were given a lethal dose of sodium pentobarbital and perfused with PBS, after which brains and spinal cords were removed. Lymphocytes were isolated from the brains of TCLi mice and intracellular cytokine staining was performed as previously described Pewe et al., 1999, Wu et al., 2000. Antibodies used were: anti-CD16/CD32 (24G.2), FITC-conjugated anti-mouse CD4 (GK1.5), FITC-conjugated anti-mouse CD8 (Lyt-2), biotin-conjugated anti-mouse Thy 1.1 (OX-7), biotin-conjugated anti-mouse Thy 1.2 (53-2.1), PE-conjugated rat Ig, PE-conjugated anti-mouse CD44 (IM7), PE-conjugated anti-mouse CD25 (PC61), PE-conjugated anti-mouse CD69 (H1.2F3), PE-conjugated anti-mouse CD62L (MEL-14), PE-conjugated anti-mouse IFN-γ (XM G1.2). All antibodies were purchased from Pharmingen (San Diego, CA) except for MEL-14, which was generously provided by Dr. M. Dailey. Streptavidin-Tri-Color (Caltag, Burlingame, CA) was used to detect biotin-conjugated antibodies. Analysis was performed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA).
2.6. Quantification of antigen-specific lymphocytes
Numbers of cells responding to epitope hCLIP85, the immunodominant MHV-specific CD4 T cell epitope (residues 133–147 of the transmembrane (M) protein (M133); Haring et al., 2001) or the immunodominant MHV-specific CD8 T cell epitope (residues 510–518 of the surface (S) glycoprotein (gp) (S510); Pewe and Perlman, 1999), were calculated as follows: total number of CNS-derived lymphocytes×the percentage of CD4 or CD8 T cells×the percent Thy 1.1+ or Thy1.2+ when applicable×(the percent of peptide-specific T cells−the percentage of cells that expressed IFN-γ in response to irrelevant peptide or no peptide). Statistical differences indicated in Table 1, Table 2 were determined using an unpaired t-test.
Table 1.
Absolute numbers of bystander and B6.PL lymphocytes isolated from the brains of TCLi mice
| Naive TCLi (n=3) | Experimental group (TCLi mice+MHV) |
|||
|---|---|---|---|---|
| MHV only (n=5) | hCLIP85:CFA (n=4) | Undepleted B6.PLa (n=8) | ||
| Total no.b | 9.3±0.7×104 | 3.9±0.3×105 | 5.5±0.9×105 | 7.8±1.0×105 |
| CD4/Thy 1.1+b | NA | NA | NA | 7.4±1.3×104 |
| CD4/Thy 1.2+b | 5.9±2.7×102 | 2.2±0.4×103 | 2.7±0.5×104c | 9.0±2.4×103c |
| % Demyelinationd | NA | 2.1±0.5% | 1.7±0.5% | 9.4±2.2% |
Donor population.
Absolute number±S.E. per brain.
Significantly more of these cells were isolated from the brains of mice treated with hCLIP85:CFA and recipients of undepleted splenocytes than from mice receiving no treatment or transferred cells (“MHV only”) (P<0.05).
Percentage of the white matter of the spinal cord with demyelination±S.D.
Table 2.
Absolute numbers of activated bystander and donor lymphocytes isolated from the brains of TCLi mice
| Naive TCLi (n=3) | Experimental group (TCLi mice+MHV) |
|||
|---|---|---|---|---|
| MHV only (n=5) | hCLIP85:CFA (n=4) | Undepleted B6.PLa (n=8) | ||
| CD4+/IFN-γ+/Thy 1.1+b | NA | NA | NA | 1.0±0.2×104 |
| CD4+/IFN-γ+/Thy 1.2+b | 0.5±0.5×102 | 2.5±1.6×102 | 5.9±1.4×103c | 2.7±1.5×102 |
Donor population.
Absolute number of activated T cells calculated as described in Section 2 ±S.E. per brain.
The number of activated CD4/Thy 1.2+ cells isolated from the brains of TCLi/RAG1−/− mice treated with hCLIP85:CFA was significantly greater (P<0.05) than the number of these cells isolated from the brains of mice infected with MHV only or mice that received adoptive transfer.
2.7. Histology and analysis of demyelination
Spinal cords were harvested and fixed in zinc formalin prior to processing and embedding in paraffin. Following hydration, 8-μm sections of the spinal cord were stained with luxol fast blue (LFB). Images of stained spinal cord sections were digitalized using an Optronics camera attached to a Leitz diaplan light microscope. Quantification of demyelination was performed as previously described (Xue et al., 1999) using Vtrace software (Image Analysis Facility, University of Iowa).
3. Results
3.1. Antigen recognition in the CNS is necessary for CD4 T cells to mediate demyelination
Adoptive transfer of CD4 or CD8 T cells from MHV-immune, immunocompetent mice to infected RAG1−/− recipients results in significant levels of demyelination (Wu et al., 2000). Previously, we also demonstrated that activated bystander CD8 T cells are capable of mediating demyelination after infection with MHV (Haring et al., 2002). In the current study, we assessed the ability of bystander CD4 T cells to participate in immune-mediated demyelination using TCLi/RAG1−/− mice, which contain only CD4 T cells specific for a peptide derived from the human CLIP fragment and are thus not specific for MHV-derived epitopes. We analyzed spinal cords for myelin damage after infection of TCLi/RAG1−/− mice with MHV with or without activation of TCLi CD4 T cells with cognate peptide (hCLIP85). Analysis of spinal cords from MHV-infected TCLi/RAG1−/− mice 14 days p.i. revealed the presence of low levels of demyelination, equivalent to those observed in MHV-infected non-transgenic RAG1−/− mice at this same time p.i. (Fig. 1 , Table 1). To determine if highly activated bystander CD4 T cells were capable of causing demyelination, spinal cords were harvested from TCLi/RAG1−/− mice treated with 100 μg hCLIP85:CFA 3 days prior to infection with MHV. The clinical diseases in infected TCLi/RAG1−/− mice that were untreated or treated with peptide/CFA were indistinguishable, with all mice showing signs of encephalitis (hunching, lethargy and ruffled fur). Analysis of spinal cords from peptide-treated, infected TCLi mice day 14 p.i. revealed no demyelination above background levels (Fig. 1, Table 1). These results indicate that, unlike activated bystander CD8 T cells, bystander CD4 T cells were unable to mediate demyelination after infection of the CNS with MHV.
Fig. 1.
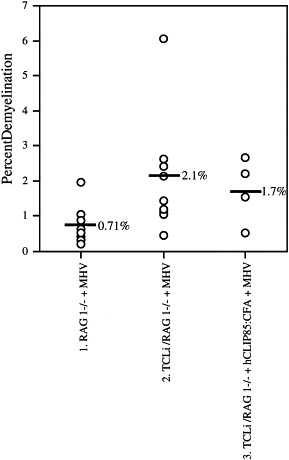
Bystander CD4 T cells were unable to cause demyelination. Little demyelination was detected in MHV-infected RAG1−/− mice (column 1), in MHV-infected TCLi mice (column 2), or in hCLIP85:CFA-treated, MHV-infected TCLi mice (column 3). Black horizontal lines in each column represent the mean percent demyelination for each group. Demyelination±S.D. was 0.7±0.2%, 2.1±0.5% and 1.7±0.5% for Groups 1, 2 and 3, respectively. Demyelination values in columns 2 and 3 are not statistically different from control values in column 1.
3.2. Bystander CD4 T cell recruitment into the CNS is increased after specific activation and in the presence of other T cells
We previously demonstrated that the ability of activated bystander CD8 T cells to mediate demyelination correlated with an increased number of these cells in the CNS.
Given the inability of activated CD4 T cells to mediate demyelination, we investigated if and to what extent bystander CD4 T cells trafficked into the CNS after MHV infection alone or after treatment with cognate peptide followed by infection with MHV. Minimal numbers of T cells were isolated from the brains of uninfected TCLi/RAG1−/− mice (Table 1). To determine if CNS infection with MHV resulted in recruitment and activation of bystander CD4 T cells, we analyzed leukocytes isolated from TCLi/RAG1−/− mice infected with MHV. We detected no sequence homology between peptide hCLIP85 and MHV by computer analysis, excluding an obvious explanation for potential observed recruitment of TCLi cells into the CNS. MHV-infected TCLi/RAG1−/− mice succumbed to encephalitis 13–14 days p.i., a disease course indistinguishable from that in non-transgenic RAG1−/− mice infected with MHV (Wu and Perlman, 1999). As shown in Fig. 2A and Table 1, only a small number of TCLi T cells were recruited into the CNS after MHV infection alone. CD4+ T cells constituted 1% of the leukocytes isolated from the brains of infected TCLi/RAG1−/− mice, equivalent to approximately 2000 TCLi cells per MHV-infected brain (Table 1). These results suggest that the necessary signals to recruit large numbers of bystander CD4 T cells were not produced in the infected TCLi/RAG1−/− CNS.
Fig. 2.
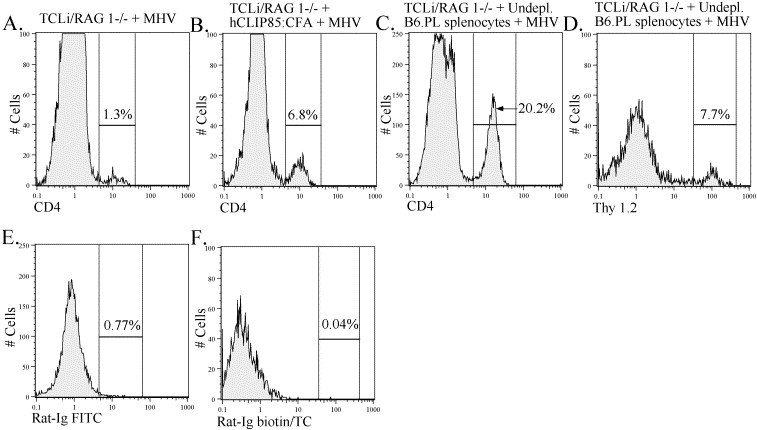
Bystander CD4 T cell recruitment into the CNS was increased in the presence of other T cells and after in vivo activation with cognate peptide. Lymphocytes were isolated from the brains of TCLi mice that were infected with MHV (A), infected TCLi mice that were previously treated with hCLIP85:CFA (B, E) and infected TCLi mice that received 5×106 undepleted splenocytes from an MHV-immunized B6.PL mouse (C, D, F). Lymphocytes were stained for CD4 and Thy 1.2 (transgenic CD4 T cell marker) as described in Section 2. Lymphocytes from individual animals were analyzed. Histograms shown are representative of results obtained from five TCLi-infected mice, four hCLIP85:CFA-treated, infected TCLi mice and eight infected TCLi mice receiving adoptively transferred splenocytes. Percentages represent the percent of lymphocytes that fall within the indicated gate. Panels D and F were gated on CD4+ cells.
To investigate if peripherally activated TCLi T cells trafficked to the CNS, we treated transgenic mice with cognate antigen prior to infection. As shown in Fig. 2B, this resulted in a dramatic increase in the number of bystander CD4 T cells trafficking to the CNS 14 days after infection. Almost 27,000 TCLi cells were isolated per infected brain after treatment with peptide hCLIP85:CFA (Table 1), indicating that entry was much more efficient if T cells were in an activated state, in agreement with previous reports Brabb et al., 2000, Hickey et al., 1991, Krakowski and Owens, 2000. Thus, as in CD8 T cell transgenic mice, peripheral activation resulted in recruitment of a large number of activated transgenic T cells to the CNS in MHV-infected TCLi/RAG1−/− mice; however, unlike our results with the CD8 T cell transgenic mice, an increase in demyelination in TCLi/RAG1−/− mice was not observed.
These results showed that peripheral activation with peptide hCLIP85:CFA resulted in increased trafficking to the CNS, but did not indicate whether bystander cells could be recruited or were activated under more physiological conditions. T cells specific for MHV-derived epitopes are efficiently recruited into the CNS both in immunocompetent mice and in adoptive transfer recipients Haring et al., 2001, Pewe and Perlman, 1999, Pewe and Perlman, 2002, Wu et al., 2000. Next, we investigated the effect of virus-specific T cells on trafficking of bystander CD4 T cells into the CNS. C57BL/6.PL (B6.PL) mice were used as donors for adoptive transfer of undepleted splenocytes to TCLi/RAG1−/− mice to facilitate differentiation between Thy 1.1+ B6.PL CD4 T cells and Thy 1.2+ TCLi CD4 T cells. Recruitment of bystander CD4 T cells into the CNS of infected TCLi mice was greater after adoptive transfer of undepleted splenocytes when compared to infected TCLi/RAG1−/− mice that did not receive transferred cells. These mice were analyzed on days 7–9 p.i. or days 10–12 p.t. of immune splenocytes when they were moribund. The combined CD4 T cell frequency after adoptive transfer was approximately 20% of CNS-derived lymphocytes (Fig. 2C), of which 7–8% were TCLi cells (Fig. 2D), equivalent to approximately 9000 cells (Table 1).
To determine if the recruitment of bystander CD4 T cells into the inflamed CNS was preferentially induced by CD4 or CD8 T cells, splenocyte populations singly depleted of either T cell subset were adoptively transferred into infected TCLi mice. Recruitment of TCLi transgenic T cells into the CNS was unchanged when CD8 T cells were depleted from the splenocyte preparation prior to transfer; however, when CD4 T cells were depleted, there was a significant drop in the numbers of recruited bystander CD4 T cells (data not shown). Together, these results suggest that MHV-specific T cells in the CNS produce factors, as yet unidentified, that are necessary for the recruitment of bystander CD4 T cells and that are not present at sufficient levels without the transfer of additional T cells. Highly activated CD4 T cells appear capable of trafficking into the inflamed CNS without the aid of these putative factors.
3.3. TCLi cells were partly activated after recruitment by MHV-specific T cells
Activation of bystander CD8 T cells was required for demyelination to occur in our previous study (Haring et al., 2002). To determine whether bystander CD4 T cells recruited into the CNS were in a functionally activated state, we measured intracellular IFN-γ production after stimulation with specific peptide Haring et al., 2001, Varga and Welsh, 1998, Varga and Welsh, 2000, Whitmire et al., 1998. Minimal bystander activation of TCLi T cells was observed after MHV infection alone (Fig. 3A , Table 2). In mice treated with hCLIP85:CFA prior to infection, a large fraction (25%, equivalent to approximately 6000 cells/brain; Table 2) of CNS-derived TCLi T cells produced IFN-γ in response to stimulation with cognate peptide (Fig. 3C, Table 2) in the absence of adoptive transfer of MHV-immune cells. After adoptive transfer of undepleted splenocytes from MHV-immune mice, very few TCLi cells responded to stimulation with cognate peptide by making IFN-γ (Fig. 3E) despite the presence of a large number of activated MHV-specific T cells (Fig. 3G,I and Table 2). Thus, we observed no functional activation of bystander CD4 T cells in the absence of cognate antigen.
Fig. 3.
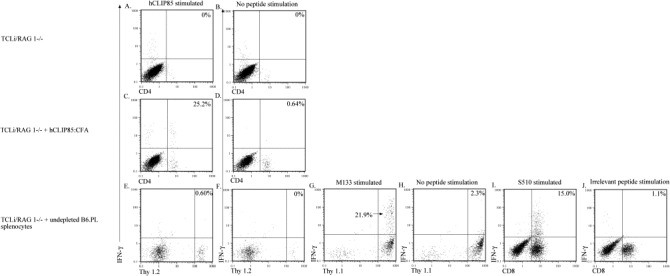
Bystander activation of TCLi CD4 T cells as measured by intracellular cytokine staining was minimal. Lymphocytes were isolated from the brains of mice in each of the experimental groups listed at the left of each row. Cells were stained for intracellular IFN-γ as described in Section 2. The peptide used to stimulate the cells in each panel is indicated at the top of each column. Panels A, C and E show staining of TCLi transgenic T cells after in vitro stimulation with hCLIP85. Panels B, D and F show staining of TCLi cells with no peptide stimulation and serve as controls for panels A, C and E. Panel G shows staining of transferred B6.PL CD4 T cells after stimulation with MHV-specific CD4 T cell peptide M133. Panel H shows staining of B6.PL CD4 T cells with no peptide stimulation and serves a control for G. Panels E–H are gated on CD4+ T cells. Panel I shows staining of B6.PL CD8 T cells after stimulation with MHV-specific CD8 T cell peptide S510. Panel J shows staining of B6.PL CD8 T cells after stimulation with an irrelevant peptide (Ova257) and serves as a control for panel I. Percentages are the percent of CD4 or CD8 T cells that fall in the indicated quadrant.
Partial activation of bystander T cells did occur in the presence of MHV-specific T cells. Insufficient numbers of T cells for extensive phenotypic analysis were isolated from the brains of uninfected TCLi/RAG1−/− mice. Phenotypic analysis of spleen-derived TCLi cells from naive mice revealed that they were 4.2±1.5% CD69+ and 95.3±1.0% CD62L+ (n=3); however, 54.7±4.6% of CNS-derived TCLi cells were CD69+ and 48.9±8.1% were CD62L+ after adoptive transfer of undepleted splenocytes (n=3). T cells were important for this partial activation because minimal bystander activation of TCLi CD4 T cells was detected after transfer of CD4 or CD8 T cell depleted splenocytes as measured by ICS and staining for surface activation markers (data not shown). These results suggest that even in the presence of MHV-specific T cells, bystander cells did not contribute to demyelination since, although recruited to the CNS, they were only partly activated.
4. Discussion
The results of the current study showed that unlike bystander CD8 T cells, bystander CD4 T cells were not able to mediate demyelination, even if specifically activated with cognate peptide, despite being recruited into the CNS. Although no clear mechanism for this inability has been elucidated, it is most likely a result of the difference in the mechanisms by which CD4 and CD8 T cells mediate demyelination. MHV-infected RAG1−/− mice that received adoptive transfer of CD8 T cell-enriched splenocytes from an MHV-immune donor underwent a protracted disease course. These mice survived >11 days p.t. and developed overwhelming demyelination, approaching 40% of the spinal cord. In contrast, recipients of CD4 T cell-enriched splenocytes became moribund within 6–7 days p.t. and exhibited significantly less demyelination (approximately 10% of the spinal cord) (Wu et al., 2000). These data indicate that MHV-specific CD4 and CD8 T cells cause demyelination by mechanisms that are not completely overlapping. The putative effector function(s) missing from bystander CD4 T cells, which resulted in the inability to cause demyelination, has not been identified. Using a panel of probes for chemokines and cytokines, we have been unable to identify any factors that are critical for demyelination Haring et al., 2002, Pewe et al., 2002, Pewe and Perlman, 2002. Our results suggest that macrophage/microglia recruitment or activation is crucial for demyelination to occur. Future studies will be directed at understanding these processes after adoptive transfer of MHV-specific and non MHV-specific T cells.
Previous work demonstrated that the ability of MHV-specific CD8 T cells to mediate demyelination after adoptive transfer was not dependent upon perforin or TNF production by donor cells, but was largely dependent on IFN-γ production by these cells (Pewe and Perlman, 2002). IFN-γ production was not necessary, however, for CD4 T cell-mediated demyelination after adoptive transfer (Pewe et al., 2002). The ability of myelin-specific CD8 T cells to induce demyelination was also dependent on IFN-γ production by these cells in another animal model of MS, experimental autoimmune encephalomyelitis (Huseby et al., 2001). The mechanisms downstream of IFN-γ that ultimately lead to the development of demyelination in either model are unknown. Several possible mechanisms exist, including the increased production by glia of chemokines involved in the recruitment of additional inflammatory cells into the CNS, or the upregulation of antigen presenting molecules on resident cells of the CNS, which may increase the ability of MHV-infected cells to be recognized by incoming T cells. The mechanisms by which CD4 T cells mediate demyelination after MHV infection are less understood.
In the presence of virus only, low numbers of transgenic T cells were recruited to the CNS, probably due to decreased integrity of the blood brain barrier (BBB) and increased expression of adhesion molecules on the vascular endothelium Houtman and Fleming, 1996b, Williams and Hickey, 1995. Infection of RAG1−/− mice with MHV resulted in the expression of several chemokines including MIP-2, MCP-3, MIP-1β, MCP-1, IP-10 and RANTES (Haring et al., 2002), which were likely to contribute to the inflammatory process. After adoptive transfer of MHV-specific T cells, recruitment of bystander CD4 T cells was increased. This increase may reflect enhanced production of chemokines, such as RANTES and IP-10 in the CNS after adoptive transfer of CD4 T cell-enriched splenocytes (Pewe et al., 2002). IP-10, Mig and RANTES are all required for maximal mononuclear cell infiltration into the CNS of MHV-infected immunocompetent mice Lane et al., 2000, Liu et al., 2000, Liu et al., 2001a. Continuous production of IP-10 is also required for maximal inflammation during chronic infection since neutralization of this chemokine resulted in significantly decreased T cell recruitment into the MHV-infected CNS (Liu et al., 2001b).
Our results, showing that bystander activation did not occur to an appreciable extent, are in general agreement with the work of others. One study, using mice transgenic for an LCMV-specific TCR infected with vaccinia virus (VV), suggested that bystander activation of naive CD8 T cells occurred at low levels. However, Welsh and co-workers, in subsequent studies, showed that this was due to the activation of T cells with dual reactivity to LCMV and a second virus (VV or Pichinde (PV)). Only T cells responding to a subset of LCMV-specific epitopes were elicited by VV or PV. Notably, the anti-LCMV CD8 T cell response elicited by VV was protective against subsequent challenge with LCMV Brehm et al., 2002, Selin et al., 1994, Selin et al., 1998. Also consistent with the specificity of the cross-reactive response, mice doubly transgenic for pancreatic expression of the LCMV gp and for an LCMV gp-specific T cell receptor did not develop diabetes after infection with VV. However, infection with LCMV did result in diabetes, indicating that a response with the correct specificity was able to cause disease (Ehl et al., 1997).
Naive CD4 T cell bystander activation, although documented infrequently, has been demonstrated most convincingly in a murine model of herpes stromal keratitis (HSK). In this model, mice transgenic for an ovalbumen-specific CD4 T cell TCR were infected by scarification of the cornea with herpes simplex virus strain RE. Transgenic CD4 T cells trafficked to the eye, were activated, based on surface molecule and cytokine expression, and caused HSK. However, HSK lesions occurred with greater frequency and severity if the bystander CD4 T cells were activated prior to infection Deshpande et al., 2001, Gangappa et al., 1999. These results were similar to those obtained in our previous study of demyelination mediated by bystander CD8 T cells, although we were unable to detect bystander activation in that study (Haring et al., 2002). Together, these results suggest that bystander activation is uncommon. If activated by cognate antigen, however, bystander CD4 and CD8 T cells are capable of mediating immunopathological changes.
In conclusion, we demonstrated that recruitment, but not activation of bystander CD4 T cells, occurred in the intense inflammatory milieu induced by MHV. Our results also indicated that peripheral activation of bystander CD4 T cells did not enhance their ability to mediate demyelination after infection with MHV, suggesting that CD4 T cell-mediated demyelination, unlike that mediated by CD8 T cells, required antigen recognition within the CNS.
References
- Bauer J., Bradl M., Hickey W., Forss-peter S., Breitscopf H., Linington C., Wekerle H., Lassmann H. T-cell apoptosis in inflammatory brain lesions: destruction of T cells does not depend on antigen recognition. Am. J. Pathol. 1998;153:715–724. doi: 10.1016/s0002-9440(10)65615-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabb T., von Dassow P., Ordonez N., Schnabel B., Duke B., Goverman J. In situ tolerance within the central nervous system as a mechanism for preventing autoimmunity. J. Exp. Med. 2000;192:871–880. doi: 10.1084/jem.192.6.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm M.A., Pinto A.K., Daniels K.A., Schneck J.P., Welsh R.M., Selin L.K. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat. Immunol. 2002;3:627–634. doi: 10.1038/ni806. [DOI] [PubMed] [Google Scholar]
- Deshpande S., Zheng M., Lee S., Banerjee K., Gangappa S., Kumaraguru U., Rouse B. Bystander activation involving T lymphocytes in herpetic stromal keratitis. J. Immunol. 2001;167:2902–2910. doi: 10.4049/jimmunol.167.5.2902. [DOI] [PubMed] [Google Scholar]
- Ehl S., Hombach J., Aichele P., Hengartner H., Zinkernagel R.M. Bystander activation of cytotoxic T Cells: studies on the mechanism and evaluation of in vivo significance in a transgenic mouse model. J. Exp. Med. 1997;185:1241–1251. doi: 10.1084/jem.185.7.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming J.O., Trousdale M.D., El-Zaatari F., Stohlman S.A., Weiner L.P. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J. Virol. 1986;58:869–875. doi: 10.1128/jvi.58.3.869-875.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangappa S., Deshpande S., Rouse B. Bystander activations of CD4+ T cells can represent an exclusive means of immunopathology in a virus infection. Eur. J. Immunol. 1999;29:3674–3682. doi: 10.1002/(SICI)1521-4141(199911)29:11<3674::AID-IMMU3674>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Haring J.S., Pewe L.L., Perlman S. High magnitude, virus-specific CD4 T-cell response in the central nervous system of coronavirus-infected mice. J. Virol. 2001;75:3043–3047. doi: 10.1128/JVI.75.6.3043-3047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring J., Pewe L., Perlman S. Bystander CD8 T cell-mediated demyelination after viral infection of the central nervous system. J. Immunol. 2002;169:1550–1555. doi: 10.4049/jimmunol.169.3.1550. [DOI] [PubMed] [Google Scholar]
- Hickey W.F., Hsu B.L., Kimura H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- Houtman J.J., Fleming J.O. Dissociation of demyelination and viral clearance in congenitally immunodeficient mice infected with murine coronavirus JHM. J. Neurovirol. 1996;2:101–110. doi: 10.3109/13550289609146543. [DOI] [PubMed] [Google Scholar]
- Houtman J.J., Fleming J.O. Pathogenesis of mouse hepatitis virus-induced demyelination. J. Neurovirol. 1996;2:361–376. doi: 10.3109/13550289609146902. [DOI] [PubMed] [Google Scholar]
- Huseby E., Liggitt D., Brabb T., Schnabel B., Ohlen C., Governman J. A pathogenic role for myelin-specific CD8+ T cells in a model for multiple sclerosis. J. Exp. Med. 2001;194:669–676. doi: 10.1084/jem.194.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakowski M., Owens T. Naive T lymphocytes traffic to inflamed central nervous system, but require antigen recognition for activation. Eur. J. Immunol. 2000;30:1002–1009. doi: 10.1002/(SICI)1521-4141(200004)30:4<1002::AID-IMMU1002>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Lane T.E., Liu M.T., Chen B.P., Asensio V.C., Samawi R.M., Paoletti A.D., Campbell I.L., Kunkel S.L., Fox H.S., Buchmeier M.J. A central role for CD4+ T-cells and RANTES in virus-induced central nervous system inflammation and demyelination. J. Virol. 2000;74:1415–1424. doi: 10.1128/jvi.74.3.1415-1424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.T., Chen B.P., Oertel P., Buchmeier M.J., Armstrong D., Hamilton T.A., Lane T.E. The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J. Immunol. 2000;165:2327–2330. doi: 10.4049/jimmunol.165.5.2327. [DOI] [PubMed] [Google Scholar]
- Liu M.T., Armstrong D., Hamilton T.A., Lane T.E. Expression of Mig (monokine induced by interferon-gamma) is important in T lymphocyte recruitment and host defense following viral infection of the central nervous system. J. Immunol. 2001;166:1790–1795. doi: 10.4049/jimmunol.166.3.1790. [DOI] [PubMed] [Google Scholar]
- Liu M.T., Keirstead H.S., Lane T.E. Neutralization of the chemokine CXCL10 reduces inflammatory cell invasion and demyelination and improves neurological function in a viral model of multiple sclerosis. J. Immunol. 2001;167:4091–4097. doi: 10.4049/jimmunol.167.7.4091. [DOI] [PubMed] [Google Scholar]
- Marten N.W., Stohlman S.A., Atkinson R.D., Hinton D.R., Fleming J.O., Bergmann C.C. Contributions of CD8+ T cells and viral spread to demyelinating disease. J. Immunol. 2000;164:4080–4088. doi: 10.4049/jimmunol.164.8.4080. [DOI] [PubMed] [Google Scholar]
- Marten N.W., Stohlman S.A., Bergmann C.C. Role of viral persistence in retaining CD8(+) T cells within the central nervous system. J. Virol. 2000;74:7903–7910. doi: 10.1128/jvi.74.17.7903-7910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S.D., Vanderlugt C., Begolka W., Pao W., Yauch R., Neville K., Katz-Levy Y., Carrizosa A., Kim B. Persistent infection with Theiler's virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997;3:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- Morkowski S., Goldrath A.W., Eastman S., Ramachandra L., Freed D.C., Whiteley P., Rudensky A. T cell recognition of major histocompatibility complex class II complexes with invariant chain processing intermediates. J. Exp. Med. 1995;182:1403–1413. doi: 10.1084/jem.182.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S., Schelper R., Bolger E., Ries D. Late onset, symptomatic, demyelinating encephalomyelitis in mice infected with MHV-JHM in the presence of maternal antibody. Microb. Pathog. 1987;2:185–194. doi: 10.1016/0882-4010(87)90020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pewe L., Perlman S. Immune response to the immunodominant epitope of mouse hepatitis virus is polyclonal, but functionally monospecific in C57Bl/6 mice. Virology. 1999;255:106–116. doi: 10.1006/viro.1998.9576. [DOI] [PubMed] [Google Scholar]
- Pewe L., Perlman S. Cutting edge: CD8 T cell-mediated demyelination is IFN-g dependent in mice infected with a neurotropic coronavirus. J. Immunol. 2002;168:1547–1551. doi: 10.4049/jimmunol.168.4.1547. [DOI] [PubMed] [Google Scholar]
- Pewe L., Heard S.B., Bergmann C.C., Dailey M.O., Perlman S. Selection of CTL escape mutants in mice infected with a neurotropic coronavirus: quantitative estimate of TCR diversity in the infected CNS. J. Immunol. 1999;163:6106–6113. [PubMed] [Google Scholar]
- Pewe L., Haring J., Perlman S. CD4 T-cell-mediated demyelination is increased in the absence of gamma interferon in mice infected with mouse hepatitis virus. J. Virol. 2002;76:7329–7333. doi: 10.1128/JVI.76.14.7329-7333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selin L., Nahill S., Welsh R. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J. Exp. Med. 1994;179:1933–1943. doi: 10.1084/jem.179.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selin L., Varga S., Wong I., Welsh R. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J. Exp. Med. 1998;188:1705–1715. doi: 10.1084/jem.188.9.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohlman S.A., Bergmann C.C., Perlman S. Mouse hepatitis virus. In: Ahmed R., Chen I., editors. Persistent Viral Infections. Wiley; New York: 1999. pp. 537–557. [Google Scholar]
- Varga S., Welsh R. Detection of a high frequency of virus-specific CD4+ T cells during acute infection with lymphocytic choriomeningitis virus. J. Immunol. 1998;161:3215–3218. [PubMed] [Google Scholar]
- Varga S.M., Welsh R. High frequency of virus-specific interleukin-2-producing CD4+ T cells and Th1 predominance during lymphocytic choriomeningitis virus infection. J. Virol. 2000;74:4429–4432. doi: 10.1128/jvi.74.9.4429-4432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Stohlman S.A., Fleming J.O. Demyelination induced by murine hepatitis virus JHM strain (MHV-4) is immunologically mediated. J. Neuroimmunol. 1990;30:31–41. doi: 10.1016/0165-5728(90)90050-W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R., Wege H., ter Meulen V. Adoptive transfer of EAE-like lesions from rats with coronavirus-induced demyelinating encephalomyelitis. Nature. 1983;305:150–153. doi: 10.1038/305150a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmire J.K., Asano M.S., Murali-Krishna K., Suresh M., Ahmed R. Long-term CD4 Th1 and Th2 memory following acute lymphocytic choriomeningitis virus infection. J. Virol. 1998;72:8281–8288. doi: 10.1128/jvi.72.10.8281-8288.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K.C., Hickey W.F. Traffic of hematogenous cells through the central nervous system. Curr. Top. Microbiol. Immunol. 1995;202:221–245. doi: 10.1007/978-3-642-79657-9_15. [DOI] [PubMed] [Google Scholar]
- Wong P., Goldrath A., Rudensky A. Competition for specific intrathymic ligands limits positive selection in a TCR transgenic model of CD4+ T cell development. J. Immunol. 2000;164:6252–6259. doi: 10.4049/jimmunol.164.12.6252. [DOI] [PubMed] [Google Scholar]
- Wu G.F., Perlman S. Macrophage infiltration, but not apoptosis, is correlated with immune-mediated demyelination following murine infection with a neurotropic coronavirus. J. Virol. 1999;73:8771–8780. doi: 10.1128/jvi.73.10.8771-8780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.F., Dandekar A.A., Pewe L., Perlman S. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J. Immunol. 2000;165:2278–2286. doi: 10.4049/jimmunol.165.4.2278. [DOI] [PubMed] [Google Scholar]
- Xue S., Sun N., van Rooijen N., Perlman S. Depletion of blood-borne macrophages does not reduce demyelination in mice infected with a neurotropic coronavirus. J. Virol. 1999;73:6327–6334. doi: 10.1128/jvi.73.8.6327-6334.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]