

Highlights
► A super high speed qRT-PCR (SHRT-PCR) was developed. RT reaction and PCR (40 cycles) can be completed in less than 20 min. ► Despite the high speed, the sensitivity and specificity of the SHRT-PCR are comparable to conventional qRT-PCR system. ► The SHRT-PCR can be used to test clinical samples.
Keywords: Influenza virus, Typing, Super high-speed quantitative real-time PCR
Abstract
The development of a rapid and sensitive system for detecting influenza viruses is a high priority for controlling future epidemics and pandemics. Quantitative real-time PCR is often used for detecting various kinds of viruses; however, it requires more than 2 h per run. Detection assays were performed with super high-speed RT-PCR (SHRT-PCR) developed according to a newly designed heating system. The new method uses a high-speed reaction (18 s/cycle; 40 cycles in less than 20 min) for typing influenza viruses. The detection limit of SHRT-PCR was 1 copy/reaction and 10−1 plaque-forming unit/reaction for viruses in culture supernatants during 20 min. Using SHRT-PCR, 86 strains of influenza viruses isolated by the Tokyo Metropolitan Institute of Public Health were tested; the results showed 100% sensitivity and specificity for each influenza A and B virus, and swine-origin influenza virus. Twenty-seven swabs collected from the pharyngeal mucosa of outpatients were also tested, showing positive signs for influenza virus on an immunochromatographic assay; the results between SHRT-PCR and immunochromatography exhibited 100% agreement for both positive and negative results. The rapid reaction time and high sensitivity of SHRT-PCR makes this technique well suited for monitoring epidemics and pre-pandemic influenza outbreaks.
1. Introduction
Influenza is a highly contagious disease caused by negative-strand RNA viruses of the family Orthomyxoviridae. Seasonal outbreaks of influenza present global health problems involving morbidity, mortality, and economic losses. Since 2003, the highly pathogenic avian influenza H5N1 virus has spread from Asia to Europe and Africa posing a pandemic threat of a highly lethal and contagious disease (Gambotto et al., 2008, Webster and Govorkova, 2006). A pandemic caused by swine-origin influenza virus (S-OIV) infection occurred in 2009 (Dawood et al., 2009, Itoh et al., 2009, Shinde et al., 2009) causing more than 18,849 deaths in more than 214 countries; the World Health Organization (WHO) announced on September 10, 2010, that the pandemic had transitioned into a post-pandemic phase (WHO, 2010a, WHO, 2010b). The occurrence of outbreaks and pandemics indicates that rapid subtyping of influenza viruses, including avian and swine-origin viruses, is a high priority for public health response systems; rapid virus detection is expected to improve the control of the pandemic spread of influenza and patient care.
Quantitative real-time PCR (qRT-PCR) is commonly used for detecting and subtyping viruses, including influenza viruses (van Elden et al., 2001, WHO, 2009a). This method offers high sensitivity and selectivity, but generally requires approximately 2 h per run; therefore, qRT-PCR is not appropriate for rapid virus detection or subtyping in outbreaks of fast-spreading and/or highly pathogenic viruses at public health centers, hospitals, airports, and other public transportation hubs. Super high-speed qRT-PCR (SHRT-PCR) is a recently developed version of qRT-PCR that is characterized by extremely short reaction times (less than 20 min per run for 40 cycles). In this assay, reaction mixtures of qRT-PCR are applied to thin compact disc (CD)-type sample containers, sealed, and rotated on heat blocks at 3 different temperatures (for denaturing, annealing, and extension temperatures).
The unique structural and thermodynamic properties of heat blocks fixed at 3 different temperatures are critical for the super high-speed polymerase reaction because the blocks allow rapid temperature changes within the samples. While SHRT-PCR is a unique variant of qRT-PCR, it can be applied to detect many kinds of microbes. In addition, the super high-speed reaction is well suited to the detection, diagnosis, and control of rapidly spreading pathogens such as those characteristic of seasonal pandemic influenza viruses. However, the clinical application of the disk-type qRT-PCR has not been reported at present.
This study establishes a new method for high-speed (<20 min) typing of influenza viruses using SHRT-PCR. The analysis targeted the nucleotide sequences of the matrix protein segment of influenza virus A (A-MP), influenza virus B (B-MP), and hemagglutinin (HA) of the 2009 pandemic S-OIV and H5N1 avian influenza viruses. Using this system, 86 strains of influenza viruses isolated from hospitals in Tokyo and the Tokyo Metropolitan Institute of Public Health were rapidly analyzed and subtyped. The typing results were correlated with those using a standardized qRT-PCR method. SHRT-PCR is expected to provide new strategies for controlling the transmission of influenza viruses.
2. Materials and methods
2.1. Virus strains, clinical samples, and viral RNA isolation
Regular laboratory strains, including A/WSN/33(H1N1), A/PR8/34(H1N1), A/Aichi/2/68(H3N2), and B/Mass/3/66 were obtained from the American Type Culture Collection (http://www.atcc.org). A/Duck/Hokkaido/Vac-3/07 (H5N1), a low pathogenic H5N1 subtype vaccine strain, was generated by genetic reassortment between 2 low-pathogenic avian influenza viruses, A/Duck/Hokkaido/101/04 (H5N3) and A/Duck/Hokkaido/262/04 (H6N1) (Soda et al., 2008). A/Tokyo and B/Tokyo strains were isolated at Tokyo Metropolitan Institute of Public Health between 2006 and 2009 seasons by the use of Madin–Darby canine kidney cells (MDCK cells) (see Table 1 ). The institutional review boards of the Tokyo Metropolitan Institute of Medical Science and the Tokyo Metropolitan Cancer and Infectious Diseases Center, Komagome Hospital, approved the procedures for use of clinical samples. Samples were collected from the pharyngeal mucosa of patients who had a diagnosis of influenza-like respiratory disease on the basis of signs and symptoms such as fever. Viruses were detected using ESPLINE® (Fujirebio Diagnostics incorporated., Chuo-ku, Tokyo, Japan; http://www.fujirebio.co.jp), immunochromatographic assays, and/or qRT-PCR (CFX96™ Real-Time PCR Detection System; Promega, Madison, WI, USA; http://www.promega.com) using the protocols of the Centers for Disease Control and Prevention (CDC) (WHO, 2009a).
Table 1.
List of influenza viruses isolated by the Tokyo Metropolitan Institute of Public Health (2006–2009 season).
| A H1N1 (seasonal) | A H3N2 (seasonal) | S-OIV | B (Seasonal) |
|---|---|---|---|
| A/Tokyo/13546/06 | A/Tokyo/12546/06 | A/Tokyo/2134/09 | B/Tokyo/13709/06 |
| A/Tokyo/13598/06 | A/Tokyo/12547/06 | A/Tokyo/2214/09 | B/Tokyo/16071/06 |
| A/Tokyo/13599/06 | A/Tokyo/13131/06 | A/Tokyo/2619/09 | |
| A/Tokyo/13711/06 | A/Tokyo/13228/06 | A/Tokyo/3105/09 | B/Tokyo/186/07 |
| A/Tokyo/13230/06 | A/Tokyo/3109/09 | B/Tokyo/641/07 | |
| A/Tokyo/373/07 | A/Tokyo/13232/06 | A/Tokyo/11875/09 | |
| A/Tokyo/10429/07 | A/Tokyo/13235/06 | A/Tokyo/12045/09 | B/Tokyo/15087/08 |
| A/Tokyo/10513/07 | A/Tokyo/13278/06 | A/Tokyo/12571/09 | B/Tokyo/15480/08 |
| A/Tokyo/10514/07 | A/Tokyo/13543/06 | A/Tokyo/12730/09 | B/Tokyo/15972/08 |
| A/Tokyo/10880/07 | A/Tokyo/13545/06 | A/Tokyo/12731/09 | |
| A/Tokyo/10883/07 | A/Tokyo/15167/06 | A/Tokyo/12732/09 | B/Tokyo/S08-2742/09 |
| A/Tokyo/11335/07 | A/Tokyo/15395/06 | A/Tokyo/12801/09 | B/Tokyo/S08-14228/09 |
| A/Tokyo/11630/07 | A/Tokyo/12802/09 | ||
| A/Tokyo/12015/07 | A/Tokyo/136/07 | A/Tokyo/12803/09 | |
| A/Tokyo/12063/07 | A/Tokyo/185/07 | A/Tokyo/13036/09 | |
| A/Tokyo/12064/07 | A/Tokyo/375/07 | A/Tokyo/13081/09 | |
| A/Tokyo/12192/07 | A/Tokyo/573/07 | A/Tokyo/13290/09 | |
| A/Tokyo/15085/07 | A/Tokyo/13292/09 | ||
| A/Tokyo/12371/08 | A/Tokyo/13293/09 | ||
| A/Tokyo/12886/08 | A/Tokyo/15480/08 | A/Tokyo/13296/09 | |
| A/Tokyo/13434/08 | A/Tokyo/15726/08 | A/Tokyo/13470/09 | |
| A/Tokyo/13532/08 | A/Tokyo/13719/09 | ||
| A/Tokyo/14218/08 | A/Tokyo/S08-2668/09 | A/Tokyo/13789/09 | |
| A/Tokyo/14220/08 | A/Tokyo/S08-2679/09 | A/Tokyo/13790/09 | |
| A/Tokyo/14226/08 | A/Tokyo/S08-2703/09 | ||
| A/Tokyo/14703/08 | A/Tokyo/S08-2731/09 | A/Tokyo/S09-1671/10 | |
| A/Tokyo/15139/08 | A/Tokyo/S08-2738/09 | A/Tokyo/S09-1673/10 | |
| A/Tokyo/S08-2740/09 | A/Tokyo/S09-1674/10 | ||
| A/Tokyo/S09-2086/10 | |||
| A/Tokyo/S09-2157/10 |
Total viral RNA was isolated from 140 μL virus-containing cell culture medium or 140 μL phosphate-buffered saline containing resuspended pharyngeal mucosal swabs using a QIAamp® viral RNA Mini Kit (Qiagen, Hilden, Germany; http://www.qiagen.com/). Viral RNA standard was isolated from a 104 plaque-forming unit (PFU) virus-containing cell culture medium as 104 PFU standard RNA. Lower-standard RNA (103 to 10−2 PFU) was prepared from the 104 PFU standard RNA by serial dilution.
2.2. Plasmid and RNA in vitro generation
Viral RNA was isolated from each virus subtype using a QIAamp viral RNA Mini Kit (Qiagen, Hilden, Germany; http://www.qiagen.com/). Extracted RNA was transcribed into cDNA using a ReverTra Ace Kit (Toyobo, Osaka, Japan http://www.toyobo.co.jp/e/) with the Uni12 primer (AGC AAA AGC AGG) and cloned into the pCR2-TOPO vector (Invitrogen, Carlsbad, CA, USA; http://www.invitrogen.com/) containing T7 promoter. Standard RNA was transcribed using the T7 RiboMax Express Large Scale RNA Production System (Promega, Madison, WI, USA; http://www.promega.com/). RNA concentrations were measured by qRT-PCR using CDC protocols and the CFX96™ Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA; http://www.bio-rad.com/).
2.3. SHRT-PCR
SHRT-PCR was conducted using a unique qRT-PCR testing unit: UR-104MK IV (Trust Medical, Hyogo, Japan; http://www.trustmedical.jp/). Methodological information is presented in Fig. 1 . Two reaction mixture recipes were used for the SHRT-PCR. The original reaction mixture includes RUSH™ DNA polymerase (Trust Medical, Hyogo, Japan; http://www.trustmedical.jp/) and ReverTra Ace (Toyobo, Osaka, Japan; http://www.toyobo.co.jp/e/). The commercial reaction mixture was from the qRT-PCR kit, RNA-Direct™ SYBR® Green Real-time PCR Master Mix (Toyobo, Osaka, Japan; http://www.toyobo.co.jp/e/). The DNA polymerase (derived from the thermophilic bacteria Thermus thermophiles) of the kit has reverse transcriptase activity. Therefore, the enzyme enables both the reverse transcription and PCR steps. The details of these recipes are described in Table 2 .
Fig. 1.
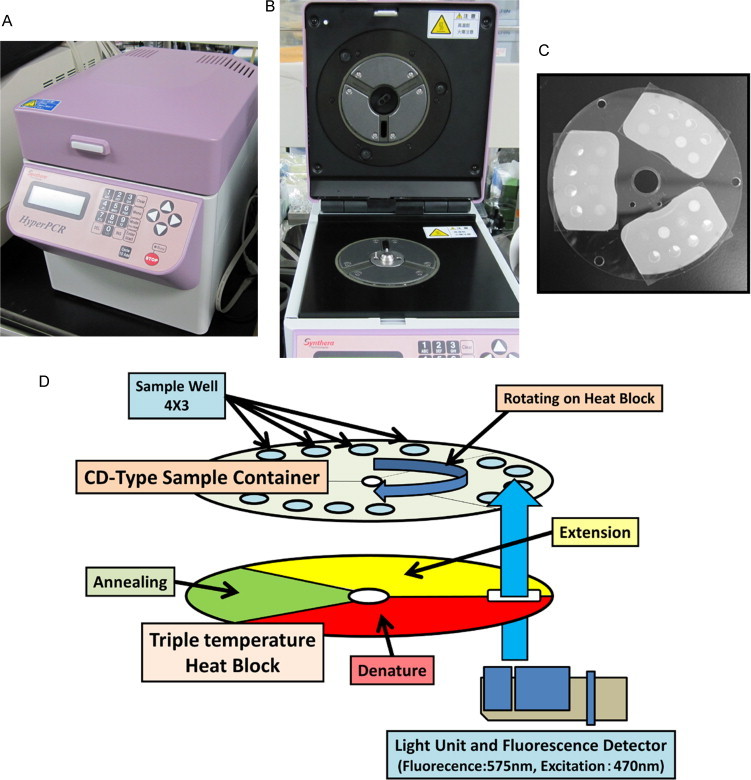
SHRT-PCR equipment and principles: SHRT-PCR unit, UK-104MK IV (A), triple heat blocks (B), compact disc-type sample container (C), and schematic diagram of SHRT-PCR technique (D). The disc-type thin sample container is rotated over heat blocks at 3 different temperatures (for denaturing, annealing, and extension), resulting in rapid temperature changes in the samples.
Table 2.
Reaction mixtures for SHRT-PCR.
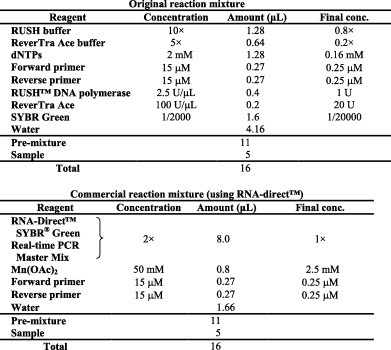
The reaction mixture (11 μL) and extracted RNA (5 μL) were added to a CD-type sample container with 12 sample wells. Then, the container was sealed with a sheet of film and placed in the UK-104MK IV. The reaction conditions and primers are described in Table 3, Table 4 .
Table 3.
Reaction conditions for SHRT-PCR.
| Step | Temperature | Duration | Cycles |
|---|---|---|---|
| Reaction conditions using original reaction mixture | |||
| cDNA synthesis | 55 °C | 3 min | Hold |
| Inactivation of ReverTra Ace | 95 °C | 1 min | Hold |
| Denaturing | 95 °C | 6 s each (total 18 s) | 40 cycles |
| Annealing | 58 °C | ||
| Extension | 75 °C | ||
| Reaction conditions using RNA-Direct™ pre-mixture | |||
| Inactivation of antibody | 90 °C | 30 s | Hold |
| cDNA synthesis | 55 °C | 5 min | Hold |
| Pre-denature | 90 °C | 30 s | Hold |
| Denaturing | 90 °C | Each 6 s (total 18 s) | 40 cycles |
| Annealing | 52 °C | ||
| Extension | 70 °C | ||
Table 4.
Primers for SHRT-PCR.
| Target subtype and gene | Primer name | Sequence (5′ to 3′) |
|---|---|---|
| Influenza virus A matrix gene | A-MP-F | CTT CTA ACC GAG GTC GAA ACG TA |
| A-MP-R | TTG GAC AAA GCG TCT ACG CTG C | |
| Influenza virus B matrix gene | B-MP-F | CAG GGC TCA TAG CAG AGC |
| B-MP-R | AAG AGA TCT CAG CAC TCC AAT GTT GC | |
| S-OIV HA gene | SO-HA-F | GAG CTA AGA GAG CAA TTG A |
| SO-HA-R | TAG CAC GAG GAC TTC TTT CC | |
| H5 HA gene | H5-F | ACA TGC CCA AGA CAT ACT GGA AAA GAC ACA CAA CGG |
| H5-R | ATG TAA GAC CAT TCC GGC ACA TTG ATG A |
2.4. Primers
Between 2006 and 2009, consensus viral gene sequences and conserved bases were identified from at least 200 viral gene sequences according to the Influenza Virus Database of the National Center for Biotechnology Information. Typing primers were designed to correspond to the conserved regions of the matrix gene segment of influenza A and B viruses. Primers for the detection of S-OIV and H5 subtype viruses were designed to correspond to the conserved region of the HA gene segment for the S-OIV and H5 subtypes. In addition, subtyping primers were checked for mismatching consensus sequences of the HA gene segment of other subtypes (<50% matching). The fragment sizes of SHRT-PCR were 100–200 nucleotides. Multiple sets of primers were tested on SHRT-PCR, and the primers which have the best sensitivity for each gene were selected (Table 4).
3. Results
3.1. Analytical sensitivity of SHRT-PCR for the detection of influenza viruses
The goal of this study was to establish a highly sensitive, high-speed detection system for influenza viruses. SHRT-PCR was used for detecting RNA segments of influenza viruses. SHRT-PCR is a unique qRT-PCR technique characterized by extremely short reaction times. Information about the methodology of SHRT-PCR is presented in Fig. 1.
SHRT-PCR was performed on duplicates to generate and measure control RNA by in vitro transcription. Two types of RT-PCR enzyme mixture were used in the method: original mixture and RNA-Direct™ SYBR® Green Real-time PCR Master Mix. The original mixture containing RUSH™ polymerase, whose reaction speed is slightly high, was optimized for SHRT-PCR; meanwhile, the RNA-Direct™ SYBR® Green Real-time PCR Master Mix is a commercial qRT-PCR kit that was taken into consideration because of the possibility of other enzymes that can be used for SHRT-PCR. Under this condition using RNA-Direct™ SYBR® Green Real-time PCR Master Mix, the limits of detection (LOD) for the influenza A virus (A-MP), influenza B virus (B-MP), and S-OIV (HA gene) were 1 copy/reaction (Fig. 2 A, C, E); the LOD for the H5 subtype (HA gene) was 10 copies/reaction (Fig. 2G). LODs using the original mixture were similar to these results (data not shown). All analyses were performed within 15–20 min. There was no marked difference in SHRT-PCR results between the 2 mixtures. RNA-Direct™ SYBR® Green Real-time PCR Master Mix was prepared as a premixed solution, meaning it has improved usability in clinical practice compared with the original mixture. Thus, RNA-Direct™ SYBR® Green Real-time PCR Master Mix was selected for use in the following experiment.
Fig. 2.
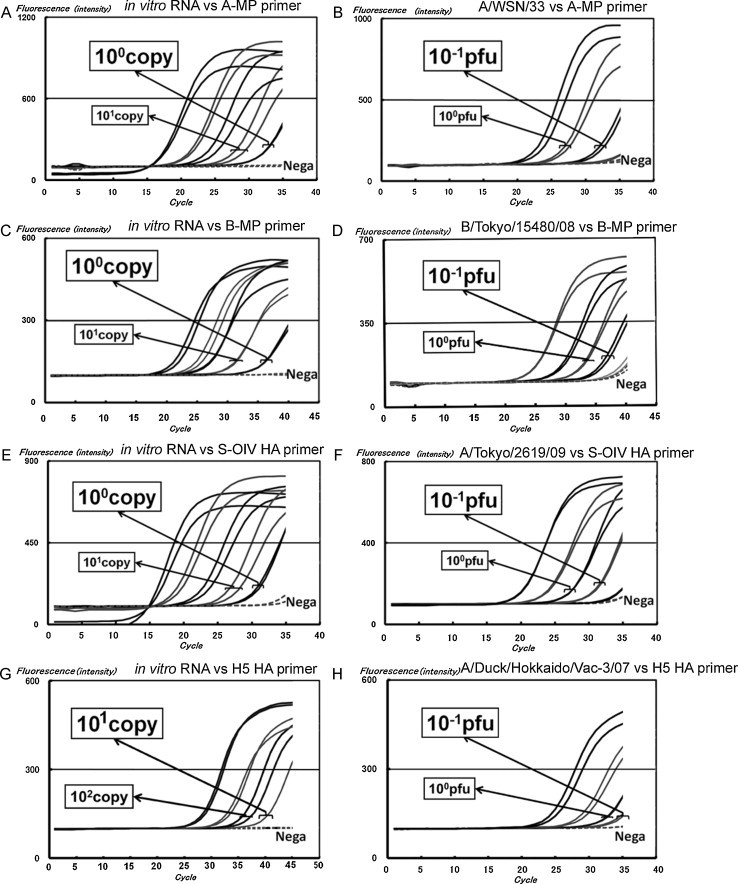
SHRT-PCR results for in vitro-generated viral RNA and influenza viruses. The RNA transcripts and viral titers were 1–104 copies/reaction and 10−1 to 102 PFU/reaction, respectively. Legends for (A)–(H) are shown within the figure.
Laboratory strains A/WSN/33 (H1N1), A/PR/8/34 (H1N1), A/Aichi/2/68 (H3N2), and B/Mass/3/66 as well as 86 other influenza viruses isolated at the Tokyo Metropolitan Institute of Public Health were tested by the system. All viruses could be detected by the appropriate primer sets. Typical results using RNA-Direct™ SYBR® Green Real-time PCR Master Mix are illustrated for A/WSN/33 by A-MP (Fig. 2B), B/Tokyo/15480/08 by B-MP (Fig. 2D), and A/Tokyo/2619/09 (H1N1 pdm) by the S-OIV HA gene (Fig. 2F) as well as A/Duck/Hokkaido/Vac-3/07 (H5N1) by the H5 HA gene (Fig. 2H); the LOD for all samples was 10−1 PFU. The number of PFUs was constantly 1–2 orders of magnitude less than the number of generated RNAs. Influenza A virus from a total of 86 influenza viruses (25 strains of seasonal H1N1, 23 strains of seasonal H3N2, 29 strains of S-OIV, and 9 strains of virus B) isolated by the use of MDCK cells in the 2006–2009 seasons at the Tokyo Metropolitan Institute of Public Health (Table 5 ). The SHRT-PCR system successfully identified 100% (77 of 77) of influenza A virus (i.e., seasonal H1N1, seasonal H3N2, and S-OIV), 100% (29 of 29) of S-OIV, and 100% (9 of 9) of influenza B virus samples. No cross reactions with the incorrect type or subtype of influenza virus were identified.
Table 5.
Testing of clinically isolated viruses during the 2006–2009 seasons isolated by the Tokyo Metropolitan Institute of Public Health.
| Sample type identified by amino acid sequencing (no. of samples) | Influenza A virus positive | S-OIV positive | Influenza B virus positive |
|---|---|---|---|
| Typing of isolated influenza virus from TMIPH using SHRT-PCR | |||
| H1N1 (S-OIV) (29) | 100% (29 of 29) | 100% (29 of 29) | 0% (0 of 29) |
| Seasonal H1N1 (25) | 100% (25 of 25) | 0% (0 of 8) | 0% (0 of 25) |
| Seasonal H3N2 (23) | 100% (23 of 23) | 0% (0 of 8) | 0% (0 of 23) |
| Influenza B virus (9) | 0% (0 of 9) | 0% (0 of 9) | 100% (9 of 9) |
3.2. Identification of clinical samples
SHRT-PCR is a variant of quantitative real-time PCR. However, the system has only 12 sample wells at present; thus, the maximum number of target samples with quantitative analysis is only 4 (2 for duplicated assay) because 2 negative control wells and at least 6 wells for standard RNAs are necessary. Thus, based on actual clinical diagnoses, qualitative assays were conducted to identify clinical samples. A cutoff value of 10 copies was decided upon to avoid unpredicted nonspecific peaks from being misidentified as a positive signal. This is because the fluorescence peak for 10 copies is completely separate from the nonspecific peaks of clinical samples. This cutoff value is also used in qRT-PCR in the CDC protocol.
Pharyngeal mucosal swabs from 27 patients with suspected respiratory disease were tested by SHRT-PCR, standard qRT-PCR using CDC protocols, and immunochromatography using ESPLINE®. The results are presented in Table 6 . The results of immunochromatography and SHRT-PCR matched exactly (Table 6). Although 1 positive sample detected by immunochromatography and SHRT-PCR was identified as a negative sample by qRT-PCR, there were 5.41 copies of RNA, which is less than the 10-copy cutoff value; the SHRT-PCR reaction curve for this sample was slightly sharper than that for the 10-copy cutoff value. These 2 qRT-PCR measurements indicate a similar case for the detection of very few copies of the sample.
Table 6.
Testing of pharyngeal mucosa of patients at Komagome Hospital.
| Clinical samples identified by ESPLINE™ (case) | Influenza A virus positive CDC protocol | Influenza A virus positive SHRT-PCR | Influenza B virus positive SHRT-PCR |
|---|---|---|---|
| Typing of pharyngeal mucosal swabs from patients using SHRT-PCR | |||
| Type A positive (17) | 94.1% (16 of 17) | 100% (17 of 17) | 0% (0 of 17) |
| Type B positive (0) | ND | ND | ND |
| A and B negative (10) | 0% (0 of 10) | 0% (0 of 10) | 0% (0 of 10) |
4. Discussion
Clinical diagnostic tests for influenza viruses in outpatient departments or clinics are typically based on immunochromatographic detection of influenza virus antigens (Chan et al., 2007). The immunochromatographic assay is easy to use and provides immediate results; however, the LOD (about 102–104 pfu/mL; Bai et al., 2006, Chan et al., 2007, Miyagawa et al., 2011) is insufficient for detecting influenza in preclinical stages, i.e., in the absence of signs and symptoms. Additionally, immunochromatography is based on an antigen–antibody reaction, implying that it is not suitable for detecting emerging or re-emerging influenza viruses that are precursors of epidemics or pandemics.
In contrast to clinical diagnoses, general surveillance of influenza viruses is usually based on genetic analyses such as qRT-PCR (Bose et al., 2009, He et al., 2009, WHO, 2009a). Although qRT-PCR is both highly sensitive (about 1–101 copies/reaction) and specific, it is more time consuming (>2 h) than immunochromatographic techniques. However, genetic analyses are more flexible than antigen–antibody-based approaches because the assays can be tailored to fit emerging and re-emerging influenza viral RNA on the basis of specific primers.
In this study, an innovative qRT-PCR method is described with a greatly improved reaction speed for detecting and typing influenza viruses. SHRT-PCR completes the analysis within an extremely short time (<20 min) compared with conventional qRT-PCR systems. In addition, the LODs and specificities for detecting influenza viruses are equal to those of conventional methods (Table 7 ).
Table 7.
Comparison of SHRT-PCR and currently existing detection methods.
| Target | Time/run | Sensitivity | Format | |
|---|---|---|---|---|
| SHRT-PCR | Viral RNA | 15–20 min | 1–10 copy or 10−1 pfu | 12 well/plate |
| Regular qRT-PCR | Viral RNA | 90–120 min | 1–10 copy | 96 or 384 well/plate |
| Immunochromatography | Viral protein | 15 min | 102–104 pfu | 1 sample/strip |
The SHRT-PCR system detects the highly conserved sequence of the corresponding viral genome, and the newly designed primer sets targeted for typing MP segments do not exhibit any cross reactions among other influenza viruses (Table 5). The LODs range from 1 to 10 copies/reaction (Fig. 2), which was sufficiently sensitive for detecting influenza viruses in 27 of 27 clinical cases (Table 6). The results indicate that SHRT-PCR provides the potential to rapidly diagnose and detect infections. Furthermore, SHRT-PCR is expected to be more advantageous than regular immunochromatography or qRT-PCR, especially in the emerging stages of epidemics or pandemics.
The WHO defines 6 phases of the pandemic stage of a disease (WHO, 2009b). Phase 4 is defined as the verification of a community-level outbreak of an animal or reassortant virus and the implementation of a pandemic containment operation. Because the time for political and clinical preparation for next phase directly depends on the speed of containment of infectious patients, quick detection of the influenza virus is essential for the success of the operation. At phase 4, SHRT-PCR can play a critical role in diagnosing infected patients at public health centers, hospitals, and public transportation hubs (e.g., airports). SHRT-PCR can also be applied to detect other rapidly spreading pathogens such as the SARS coronavirus (Poon et al., 2003, WHO, 2003) and foot-and-mouth disease virus (Oleksiewicz et al., 2001, Reid et al., 2001). Rapid containment is critical for limiting the spread of these viruses, and containment depends on rapid and sensitive detection.
Despite its advantages of rapidity and sensitivity, SHRT-PCT is subject to certain limitations. The SHRT-PCR system used in this study is limited to a sample capacity of 12. It is necessary for a quantitative assay to generate a standard curve with multiple defined amounts of samples; thus, SHRT-PCR may be more useful for performing qualitative rather than quantitative assays. The limitation of the number of samples can be resolved by increasing the number of sample wells on the sample container and increasing the design capacity of the testing unit. For clinical applications for public health surveillance, SHRT-PCR will be more useful than immunochromatography but less useful than qRT-PCR for influenza virus typing. This is because surveillance programs should be able to deal with large numbers of clinical samples. Overall, SHRT-PCR is a sufficiently powerful method to provide a basis for rapid pandemic containment at the WHO phase 4 stage.
Acknowledgements
This work was supported by grants from the New Energy and Industrial Technology Development Organization and the Japan Society for the Promotion of Science. We are grateful to Trust Medical Co. Ltd. and members of the Department of Microbiology, Tokyo Metropolitan Institute of Public Health, and the Department of Infectious Disease, Komagome Hospital, for their technical assistance. We thank Dr. Kohara and his laboratory members for their scientific advice as well as all members of our laboratory for their advice and assistance.
References
- Bai G.R., Sakoda Y., Mweene A.S., Fujii N., Minakawa H., Kida H. Improvement of a rapid diagnosis kit to detect either influenza A or B virus infections. J. Vet. Med. Sci. 2006;68:35–40. doi: 10.1292/jvms.68.35. [DOI] [PubMed] [Google Scholar]
- Bose M.E., Beck E.T., Ledeboer N., Kehl S.C., Jurgens L.A., Patitucci T., Witt L., LaGue E., Darga P., He J., Fan J., Kumar S., Henrickson K.J. Rapid semiautomated subtyping of influenza virus species during the 2009 swine origin influenza A H1N1 virus epidemic in Milwaukee, Wisconsin. J. Clin. Microbiol. 2009;47:2779–2786. doi: 10.1128/JCM.00999-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.H., Lam S.Y., Puthavathana P., Nguyen T.D., Long H.T., Pang C.M., Chan K.M., Cheung C.Y., Seto W.H., Peiris J.S. Comparative analytical sensitivities of six rapid influenza A antigen detection test kits for detection of influenza A subtypes H1N1, H3N2 and H5N1. J. Clin. Virol. 2007;38:169–171. doi: 10.1016/j.jcv.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Dawood F.S., Jain S., Finelli L., Shaw M.W., Lindstrom S., Garten R.J., Gubareva L.V., Xu X., Bridges C.B., Uyeki T.M. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med. 2009;360:2605–2615. doi: 10.1056/NEJMoa0903810. [DOI] [PubMed] [Google Scholar]
- Gambotto A., Barratt-Boyes S.M., de Jong M.D., Neumann G., Kawaoka Y. Human infection with highly pathogenic H5N1 influenza virus. Lancet. 2008;371:1464–1475. doi: 10.1016/S0140-6736(08)60627-3. [DOI] [PubMed] [Google Scholar]
- He J., Bose M.E., Beck E.T., Fan J., Tiwari S., Metallo J., Jurgens L.A., Kehl S.C., Ledeboer N., Kumar S., Weisburg W., Henrickson K.J. Rapid multiplex reverse transcription-PCR typing of influenza A and B virus, and subtyping of influenza A virus into H1, 2, 3, 5, 7, 9, N1 (human), N1 (animal), N2, and N7, including typing of novel swine origin influenza A (H1N1) virus, during the 2009 outbreak in Milwaukee, Wisconsin. J. Clin. Microbiol. 2009;47:2772–2778. doi: 10.1128/JCM.00998-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y., Shinya K., Kiso M., Watanabe T., Sakoda Y., Hatta M., Muramoto Y., Tamura D., Sakai-Tagawa Y., Noda T., Sakabe S., Imai M., Hatta Y., Watanabe S., Li C., Yamada S., Fujii K., Murakami S., Imai H., Kakugawa S., Ito M., Takano R., Iwatsuki-Horimoto K., Shimojima M., Horimoto T., Goto H., Takahashi K., Makino A., Ishigaki H., Nakayama M., Okamatsu M., Takahashi K., Warshauer D., Shult P.A., Saito R., Suzuki H., Furuta Y., Yamashita M., Mitamura K., Nakano K., Nakamura M., Brockman-Schneider R., Mitamura H., Yamazaki M., Sugaya N., Suresh M., Ozawa M., Neumann G., Gern J., Kida H., Ogasawara K., Kawaoka Y. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature. 2009;460:1021–1025. doi: 10.1038/nature08260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa E., Kogaki H., Uchida Y., Fujii N., Shirakawa T., Sakoda Y., Kida H. Development of a novel rapid immunochromatographic test specific for the H5 influenza virus. J. Virol. Methods. 2011;173:213–219. doi: 10.1016/j.jviromet.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Oleksiewicz M.B., Donaldson A.I., Alexandersen S. Development of a novel real-time RT-PCR assay for quantitation of foot-and-mouth disease virus in diverse porcine tissues. J. Virol. Methods. 2001;92:23–35. doi: 10.1016/s0166-0934(00)00265-2. [DOI] [PubMed] [Google Scholar]
- Poon L.L., Chan K.H., Wong O.K., Yam W.C., Yuen K.Y., Guan Y., Lo Y.M., Peiris J.S. Early diagnosis of SARS coronavirus infection by real time RT-PCR. J. Clin. Virol. 2003;28:233–238. doi: 10.1016/j.jcv.2003.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid S.M., Ferris N.P., Hutchings G.H., Zhang Z., Belsham G.J., Alexandersen S. Diagnosis of foot-and-mouth disease by real-time fluorogenic PCR assay. Vet. Rec. 2001;149:621–623. doi: 10.1136/vr.149.20.621. [DOI] [PubMed] [Google Scholar]
- Shinde V., Bridges C.B., Uyeki T.M., Shu B., Balish A., Xu X., Lindstrom S., Gubareva L.V., Deyde V., Garten R.J., Harris M., Gerber S., Vagasky S., Smith F., Pascoe N., Martin K., Dufficy D., Ritger K., Conover C., Quinlisk P., Klimov A., Bresee J.S., Finelli L. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005–2009. N. Engl. J. Med. 2009;360:2616–2625. doi: 10.1056/NEJMoa0903812. [DOI] [PubMed] [Google Scholar]
- Soda K., Sakoda Y., Isoda N., Kajihara M., Haraguchi Y., Shibuya H., Yoshida H., Sasaki T., Sakamoto R., Saijo K., Hagiwara J., Kida H. Development of vaccine strains of H5 and H7 influenza viruses. Jpn. J. Vet. Res. 2008;55:93–98. [PubMed] [Google Scholar]
- van Elden L.J., Nijhuis M., Schipper P., Schuurman R., van Loon A.M. Simultaneous detection of influenza viruses A and B using real-time quantitative PCR. J. Clin. Microbiol. 2001;39:196–200. doi: 10.1128/JCM.39.1.196-200.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster R.G., Govorkova E.A. H5N1 influenza–continuing evolution and spread. N. Engl. J. Med. 2006;355:2174–2177. doi: 10.1056/NEJMp068205. [DOI] [PubMed] [Google Scholar]
- WHO, 2003. PCR primers for SARS developed by WHO Network Laboratories. http://www.who.int/csr/sars/primers/en/(Accessed March 8, 2011)
- WHO, 2009a. CDC Protocol of real-time RT-PCR for swine influenza A (H1N1). http://www.who.int/csr/resources/publications/swineflu/CDCRealtimeRTPCR_SwineH1Assay-2009_20090430.pdf (Accessed January 11, 2011)
- WHO, 2009b. WHO pandemic phase descriptions and main actions by phase. http://www.who.int/entity/csr/disease/influenza/pandemic_phase_descriptions_and_actions.pdf (Accessed March 7, 2011)
- WHO, 2010a. Influenza updates. http://www.who.int/csr/don/2010_09_10/en/index.html (Accessed March 9, 2011)
- WHO, 2010b. Pandemic (H1N1) 2009 – update 112. http://www.who.int/csr/don/2010_08_06/en/index.html (Accessed March 9, 2011)