

Abstract
Human parainfluenza viruses (HPIVs) are distributed worldwide and are involved mainly in the pathogenesis of respiratory tract infections. The development and optimization of three quantitative reverse transcription real time polymerase chain reactions (RT Real Time Qt-PCRs) and an indirect immunofluorescence (IFA) for the detection and quantitation of HPIV-1, -2 and -3 in clinical samples are described. Efficiency, sensitivity, specificity, inter- and intra-assay variability and turnaround time of the two methods were compared. These assays have been validated on 131 bronchoalveolar lavage specimens. Based on the results obtained, the molecular methods represent a valid and rapid tool for clinical management and should be included in diagnostic panels aimed to evaluate suspected respiratory tract infections.
Keywords: Viral diagnosis, Human parainfluenza viruses, RT Real Time PCR, Indirect immunofluorescence
Human parainfluenza viruses (HPIVs) are non-segmented RNA viruses which belong to Paramyxoviridae family (Pringle, 1997) and are distributed widely with a seroprevalence of 50–90% in children and young adults. HPIV-1, -2, -3 are an important cause of upper and lower respiratory tract diseases in both adults and children (Hall, 2001, Henrickson, 2003). HPIVs cause severe clinical manifestations in immunocompromised hosts, such as HPIV-2-related giant cells pneumonia and HPIV-3-associated interstitial pneumonia (Chanock et al., 2001, Sable and Hayden, 1995). The availability of HPIV-specific diagnostic assays allows differentiation from other respiratory pathogens since clinical patterns are overlapping. HPIV infection can be confirmed by viral isolation (time required, 7–10 days), direct/indirect rapid detection of viral antigens in cell culture (LLC-MK2, Hep-2, Vero, HEF) (time required, 2–5 days), and molecular tests (Niesters, 2002, Sable and Hayden, 1995, Vernet, 2004). Currently, IFA is the “gold standard” for virological diagnosis, however molecular methods result in more sensitive, specific, and rapid detection of respiratory viruses (van Elden et al., 2002).
In this study, three quantitative RT Real Time PCR assays (RT Real Time Qt-PCRs) and an indirect immunofluorescence assay (IFA) for HPIV-1, -2, -3 detection were developed and validated on bronchoalveolar lavage specimens. The two diagnostic approaches were compared in terms of sensitivity, specificity, turnaround time, and applicability.
Prototype virus strains of HPIV-1 (strain C-35), HPIV-2 (strain Greer), HPIV-3 (strain C-243) were obtained from American Type Culture Collection (ATCC, Manassas, VA) (VR-94, VR-92, VR-93, respectively). One-hundred thirty-one bronchoalveolar lavage specimens, thawed and liquefied with 1:1 N-acetylcisteine, obtained from 118 patients (male/female, 73/45; median age 51.63 ± 16.12) from different clinical settings were examined. Informed consent was obtained from all the patients or the nearest relatives and the study was approved by the Ethics Committee of the University Hospital San Giovanni Battista of Turin. Human laryngeal epidermoid carcinoma (Hep-2) cells and African green monkey kidney epithelial (Vero) cells were used for primary viral isolation and propagation. Cell lines were obtained from the Zooprofilattic Institute of Lombardy and Emilia Romagna (BS TCL 23 and BS CL 86, respectively). Hep-2 and Vero cells were maintained at 37 °C and 5% CO2 with supplemented MEM (PAA Laboratories GmbH, Pasching, Austria) containing 1% filtered glutamine, 0.15% antibiotic agent (PenStrep, Sigma–Aldrich, Saint Louis, MO), 0.2% antimycotic agent (Fungizone, Bristol-Myers Squibb, Sermoneta, Italy), and 10% fetal calf serum (FCS; PAA Laboratories GmbH). Infection was carried out at 50–60% confluence. Each infected flake (T25, Falcon, Becton Dickinson, Milano, Italy) was inoculated with a solution containing 20 μl of each viral stock and 1.2 ml of MEM with 2% FCS. The medium for HPIV-1 infection contained no FCS and was supplemented with 1 μg/ml of trypsin (GIBCO, Invitrogen, Carlsbad, CA). The flakes which were not inoculated with virus were used as controls. Cells were maintained for 1 h at 37 °C and 5% CO2 to assist viral adsorption. Subsequently, the inoculum was removed and MEM with 2% FCS was added. The cell monolayers were observed daily for cytopathic effect (CPE). For virus recovering, the cellular media were centrifuged for 7 min at 210 × g and the supernatants were kept on ice. The cell monolayers were scraped and a thermal treatment was performed to extract viral particles from cells consisting in rapid freezing on liquid nitrogen followed by defrosting for 5 m at 37 °C for three times. The recovered viral particles were stored at −80 °C. Ninety six-well plates at 50–60% confluence Hep-2 and Vero were inoculated with 100 μl of 10-fold diluted virus or medium (i.e. negative control) for TCID50 assay. The plates were observed daily for CPE.
For evaluating IFA sensitivity, 10-fold dilutions (ranging from 102 to 10−2 TCID50/200 μl) of titrated virus were obtained and different variables, such as days of incubation (2–4 days) and primary monoclonal antibody dilutions (MAb; 1:40–1:80–1:160 in albumin supplemented with PBS 1%), were examined. Two hundred microliters of TCID50 dilution were inoculated into shell vials at 50–60% confluence. The inoculum adsorption was enhanced by centrifugation at 210 × g for 45 min. One millilitre of supplemented medium was added and the shell vials were kept in a thermostat. After incubation, the cells were fixed with 1 ml of acetone–methanol (2:1) for 10 min. Subsequently, the slides were incubated with a generic anti-HPIVs MAb (11-040 LOT M1601-Z; Argene, Verniolle, France) for 30 m at 37 °C and then with goat anti-mouse fluorescein-conjugated monoclonal antibody (51-010 LOT N0310; Argene), as secondary antibody, for 30 m at 37 °C. The slides were washed and read immediately using a fluorescence microscope. The presence of bright green fluorescence within intact cells was considered positive. The slides with too few intact cells were considered inadequate for examination.
Optimal IFA conditions were used for reproducibility within a single run (n = 5; intra-assay variability) or different run experiments (n = 5; inter-assay variability) using three dilutions: the dilution of the sensitivity level and the 10-fold above and the 10-fold below ones (Table 1 ).
Table 1.
Test reproducibility. Optimal IFA conditions and correspondent repeatability. IFA sensitivity value is underlined.
| Virus/cells/MAb concentration | Day of observation | TCID50 | No. positive IF assay (%) intra- and inter-test | Reproducibility |
|---|---|---|---|---|
| Test reproducibility | ||||
| HPIV-1/Hep-2/1:80 | 3 | 10−1 | 5/5 (100%) | 100% |
| 10−2 | 5/5 (100%) | 100% | ||
| 10−3 | 0/5 (0%) | 100% | ||
| HPIV-2/Vero/1:40 | 4 | 10−1 | 5/5 (100%) | 100% |
| 10−2 | 5/5 (100%) | 100% | ||
| 10−3 | 3/5 (60%) | 60% | ||
| HPIV-3/Vero/1:40 | 4 | 100 | 5/5 (100%) | 100% |
| 10−1 | 5/5 (100%) | 100% | ||
| 10−2 | 4/5 (80%) | 80% | ||
Nucleic acid extraction was performed using NucliSens EasyMag (bioMeriéux, Marcy l’Etoile, France). Viral cDNA was generated, first by incubation of random primers (50 ng/l) and dNTPs (10 mM) (Invitrogen) with 10 μl of RNA for 5 min at 65 °C. Subsequently, a mix containing 0.1 M DTT, buffer 10× [200 mM Tris–HCl (pH 8.4), 500 mM KCl], SuperScript™ II RT (50 U/l) and RNaseOUT™ (40 U/l) (Invitrogen) was added. The total volume (20 μl) of the reaction mixture was incubated for 10 min at 25 °C, 50 min at 42 °C, 15 min at 70 °C using 9800 Fast Thermal Cycler (Applied Biosystems, Monza, Italy).
The alignment of virus-specific HN gene, based on available sequences in public databases, was performed with the CLC Free Workbench 3.2 software (CLC bio A/S). Consensus primers and probes were designed within the HN region of HPIV-1–3 using the Primer Express 3.0 software (Applied Biosystems) and OligoPerfect™ Designer software (Invitrogen) (Table 2 ). Primer and probe design was made according to the standard requirements: prevention of primer–dimer formation and melting temperatures (Tm) of the primers up to 60 °C. In order to confirm RNA–DNA extraction and to prevent false negative results, the housekeeping gene glycerin-aldehyde-3-phosphate-dehydrogenase (GAPDH) was co-amplified and used as internal control (VIC® probes, Applied Biosystems) (Table 2).
Table 2.
Probes and primers for plasmid construction and for RT Real Time Qt-PCR.
| Virus | Primer and probe sequences | Region | NCBI sequenze |
|---|---|---|---|
| Parainfluenza virus 1 | Plasmid construction and RT Real Time Qt-PCRa | 7794 nt -8007 nt NC_003461 (Washington/1964) | |
| PIV1FQ (0.9 mM) | 5′ AARGGAAARACCAAATCTCMWCG 3′ | Gene HN | 213 bp |
| PIV1RQ (0.9 mM) | 5′GAGCATCATTGCARACAMTYTG 3′ | Gene HN | |
| PIV 1 probe (0.25 mM) | 5′ FAM-TAACAACTCCGCTCCAAG-MGB 3′ | Gene HN | |
| Parainfluenza virus 2 | Plasmid constructionb | 7379–7780 nt AF533012 (GREER) | |
| PIV2 F CLON | 5′ TTGGAGATTGCCTCGATTTC 3′ | Gene HN | 401 bp |
| PIV2 R CLON | 5′ GGAAGGAGTCCCCTTTATGAGA 3′ | Gene HN | |
| RT Real Time Qt-PCRa | 7447–7722 nt AF533012 (GREER) | ||
| PIV2F-RTD (0.9 mM) | 5′ GCAGCATTTCCAATCTTCAGG 3′ | Gene HN | 275 bp |
| PIV2R-RTD (0.9 mM) | 5′ TAGATCCCGCTTCCAACTGC 3′ | Gene HN | |
| PIV2 probe (0.25 mM) | 5′ FAM-CAAAAGCTGTTCAGTCACTGCTATAC-TAMRA 3′ | Gene HN | |
| Parainfluenza virus 3 | Plasmid construction and RT Real Time Qt-PCRa | 7602–7798 nt U51116 (cp-45) | |
| PIV3FQ (1 mM) | 5′ATCAACTGTGTTCRACTCCHAARG 3′ | Gene HN | 196 bp |
| PIV3RQ (0.9 mM) | 5′TTTGCCTTTRTARTATATCCCTGGT 3′ | Gene HN | |
| PIV 3 probe (0.25 mM) | 5′FAM- TGAYGAAAGATCAGATTATG-MGB 3′ | Gene HN | |
| GAPDH (internal control) | RT Real Time Qt-PCRa | ||
| GAPDHF (0.06 mM) | 5′-GCCAAAAGGGTCATCATCTC-3′ | Exon 6 | 512 bp |
| GAPDHR (0.06 mM) | 5′-GGGGCCATCCACAGTCTTCT-3′ | Exon 8 | |
| GAPDH probe (0.09 mM) | 5′ VIC- TGGTATCGTGGAAGGA-MGB 3′ | Exon 6 | |
Sequences from Primer Express 3.0 software (Applied Biosystem).
Sequences from OligoPerfect™ Designer (Invitrogen).
For the production of plasmid standard, cDNA fragment of virus-specific HN sequences was cloned into the TA vector and propagated in competent Escherichia coli TOP10 cells. Plasmids were created using the Topo TA PCR cloning kit (Invitrogen), according to the manufacturer's specifications. The concentration of the plasmid DNA was quantified by using a high-resolution spectrophotometer.
For optimizing the three RT Real Time Qt-PCRs, two different concentrations of target primers and probe were evaluated: 0.9 mM/0.25 mM and 0.2 mM/0.1 mM for HPIV-1 and HPIV-2; 1 mM/0.25 mM and 0.2 mM/0.1 mM for HPIV-3. Two microliters of cDNA were added to 18 μl of the reaction mix, giving a final reaction volume of 20 μl. Uracil–DNA glycosylase was used to eliminate PCR ‘carry over’ contamination from previous PCRs (Quint et al., 1995, Tetzner et al., 2007). The amplification profile was optimized on the 7300 Real Time PCR System (Applied Biosystems) as follows: one cycle of decontamination at 50 °C for 2 min, one cycle of denaturation at 95 °C for 10 min followed by 45 cycles of amplification at 95 °C for 15 s and 60 °C for 60 s. For each run a standard curve was generated in a 4-log range by 10-fold serial dilutions of the plasmid standard.
The linearity range was evaluated using 10-fold dilutions (from 1010 to 100 copies/reaction) of each plasmid. The intra- and inter-assay coefficients of variability (CV) were evaluated using different concentrations of plasmid standard (ranging from 105 to 102 copies/reaction) within a single run (n = 10) or different run experiments (n = 10).
Different concentrations of TCID50 (ranged from 102 to 10−5/reaction) were amplified by the RT Real Time Qt-PCRs in order to compare sensitivity and specificity of the two diagnostic approaches.
The generic anti-HPIVs MAb was tested for potential cross-reactivity with unrelated viruses and bacteria (influenza virus A H1N1, ATCC VR-98; A H3N2, ATCC VR-544; influenza virus B, ATCC VR-296; adenovirus, ATCC VR-5; RSV, ATCC VR-1580; CMV, ATCC VR-538; HSV 1-2, ATCC VR-2021; human rhinoviruses, ATCC VR-283/VR-330/VR-486; human coxsackievirus type B1, ATCC VR-28; echovirus types 1, ATCC VR-31; 6, ATCC VR-36; enterovirus type 68, ATCC VR-561; human coronavirus types 229E, ATCC VR-740 and OC43, ATCC VR-1558; Streptococcus pneumoniae, ATCC 6301; Legionella pneumophila, ATCC 33152; Mycoplasma pneumoniae, ATCC 15377; Chlamydia pneumoniae, ATCC 53592). Similarly, primers and probes were tested using the above strains and human sequences based on the data available at the BLAST alignment software (http://blast.ncbi.nlm.nih.gov/Blast.cgi). No significant homology to any other sequences was found.
Viral CPE consisted of cell rounding and focal destruction of the monolayer with syncytial formation. Vero and Hep-2 cells showed a different susceptibility to HPIVs infection: for HPIV-1, TCID50/ml 6 × 103 on Hep-2 (Vero cells were unable to support growth in the presence of trypsin) at day 8; for HPIV-2, 106 and 3 × 106 on Vero and Hep-2, respectively, on day 5; for HPIV-3, 3 × 105 and 2.1 × 1010 on Vero and Hep-2, respectively, at day 5. HPIV-1-IFA sensitivity was 10−2 TCID50 at day 3 (Fig. 1A). No significant difference was observed on day 4. Single positive cells showing a granular cytoplasmic fluorescence were seen at lower TCID50 (e.g. 10−1 to 10−2), while at higher TCID50 (e.g. 102 to 101) rare syncytial formations were appreciable. There was no difference between 1:40 and 1:80 MAb dilutions in terms of fluorescence intensity, while a decrement using 1:160 dilution was evident (data not shown). On day 4, HPIV-2-IFA sensitivity on Vero and Hep-2 cells was 10−2 TCID50 and 10−1 TCID50, respectively (Fig. 1B). A granular cytoplasmic fluorescence in infected cellular foci with a “spread” infection pattern at lower TCID50 (e.g. 10−1 to 10−2) was observed; while at higher TCID50 (e.g. 102 to 101), the fluorescence involved the nucleus. With Vero cells, the stain intensity resulted dependent on MAb dilution and reduced constantly with the increase of MAb dilution from 1:40 to 1:160; while no significant difference was observed on Hep-2 cells. On day 4, HPIV-3-IFA sensitivity on Vero and Hep-2 cells was 10−1 TCID50 and 101 TCID50, respectively (Fig. 1C). A lower intensity of fluorescence was seen with high MAb dilutions, such as 1:80 and 1:160, in comparison to 1:40. Since Hep-2 cells had a very low sensitivity on day 4 post-infection, cell culture was prolonged until day 7; however, this resulted in only 1-log increment of sensitivity. A widespread and homogeneous cytoplasmic fluorescence pattern in both cellular models at lower TCID50 (e.g. 10−1 to 10−2) was observed; while at higher TCID50 (e.g. 102 to 101), syncytial formation was seen. Interestingly, HPIV-3 infection pattern was “all or nothing”, as the first dilution higher than that of sensitivity threshold (spread staining) appeared completely negative. In Table 1 the reproducibility and sensitivity (i.e. the lowest TCID50 concentration detectable at a frequency of 100%) of IFA are reported.
Fig. 1.
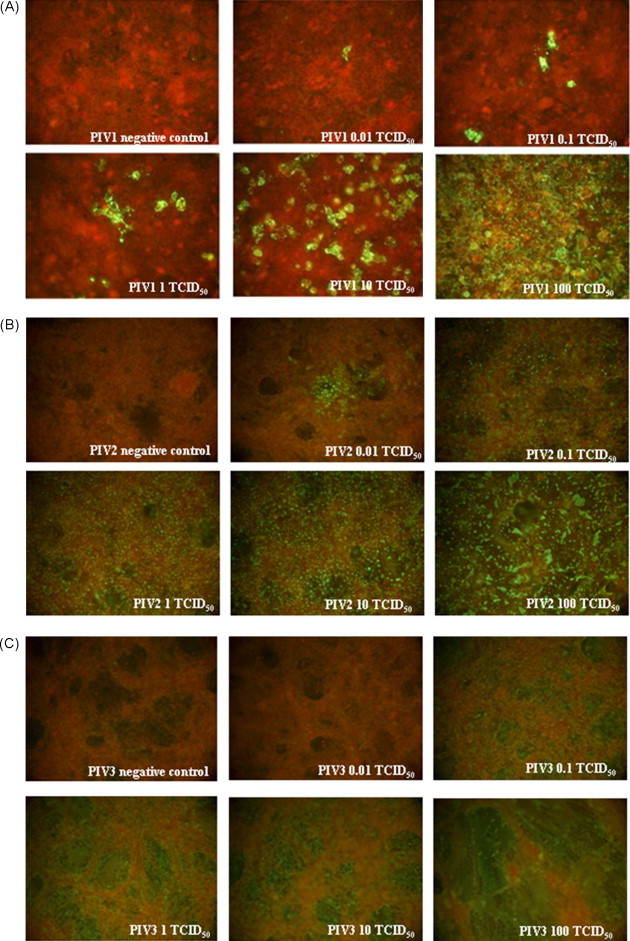
Sensitivity of HPIV-1, -2 and -3 on optimal cell model. (A) HPIV-1 on Hep-2 cells at day 3 post-infection (1:80 MAb dilution). (B) HPIV-2 on Vero cells at day 4 post-infection (1:40 MAb dilution). (C) HPIV-3 on Vero cells at day 4 post-infection (1:40 MAb dilution).
For the RT Real Time Qt-PCRs, the optimal parameters in obtaining the lowest detection limit with a high specificity resulted in the following concentrations: both primers 0.9 mM and probe 0.25 mM for HPIV-1 and HPIV-2; forward primer 1 mM and reverse 0.9 mM, and probe 0.25 mM for HPIV-3 amplification.
The dynamic range of the three RT Real Time Qt-PCR assays was assessed by carrying out serial dilutions of the plasmid standard (from 1010 to 100 copies/reaction) and was as follows: 108 to 100 copies/reaction (Fig. 2A) for HPIV-1; 107 to 101 (Fig. 2B) for HPIV-2; 107 to 101 (Fig. 2C) for HPIV-3. The sensitivity of the RT Real Time Qt-PCRs (defined as the lowest concentration of target quantified at a frequency of 100%) was found to be 1 copy/reaction for HPIV-1 and 100 copies/reaction for HPIV-2 and HPIV-3. The RT Real Time Qt-PCR sensitivity was found to be 10−4 TCID50/reaction for HPIV-1 and HPIV-2 and 10−3 for HPIV-3 (data not shown). The inclusion of an internal control, GAPDH target, did not induce loss of primary target sensitivity during amplification (data not shown). The results of intra- and inter-assay reproducibility are summarized in Table 3 .
Fig. 2.
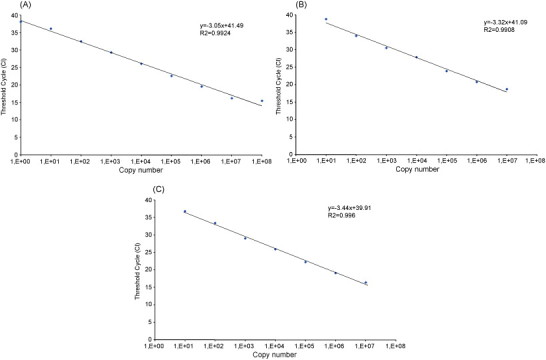
Dynamic range of HPIV-1, -2 and -3 genome quantification with the three RT Real Time PCR assays. Number of cycle threshold (Ct) is plotted versus copy number. (A) HPIV-1 RT Real Time PCR assay, from 100 to 108 copies. (B–C) HPIV-2 and 3 RT Real Time PCR from 101 to 107 copies, respectively.
Table 3.
Intra-assay and inter-assay reproducibility of parainfluenza viruses standard curve by RT Real Time Qt-PCRs.
| Standard dilution (plasmid copies/reaction) | Intra-test (%) |
Inter-test (%) |
||||
|---|---|---|---|---|---|---|
| HPIV-1 | HPIV-2 | HPIV-3 | HPIV-1 | HPIV-2 | HPIV-3 | |
| Coefficient of variability | ||||||
| 102 | 0.29 | 2.90 | 2.32 | 1.00 | 1.15 | 3.88 |
| 103 | 0.96 | 0.11 | 1.71 | 0.42 | 0.44 | 4.04 |
| 104 | 0.79 | 0.55 | 2.46 | 0.39 | 1.25 | 0.27 |
| 105 | 0.77 | 1.33 | 1.89 | 1.11 | 0.59 | 1.56 |
| y = 34.9 − 2.6 log(x) R2 = 0.992 | y = 37.9 − 2.6 log(x) R2 = 0.990 | y = 38.4 − 2.9 log(x) R2 = 0.969 | y = 40.3 − 3.5 log(x) R2 = 0.990 | y = 42.3 − 3.2 log(x) R2 = 0.989 | y = 43.1 − 4.0 log(x) R2 = 0.965 | |
Equations of linear regression curves are indicated.
For RT Real Time Qt-PCR quantitation, the following formula was used: lower limit of virus-specific dynamic range × 220 (correction factor derived from analytic procedure). The inferior limit was 220 GEq/ml of bronchoalveolar lavage for HPIV-1 and 2200 GEq/ml for HPIV-2 and HPIV-3.
In the case of bronchoalveolar lavage specimens, 3/131 (2.3%) were positive by IFA versus 18/131 (13.7%) by RT Real Time Qt-PCRs; 2 (1.5%) for HPIV-1, 6 (4.6%) for HPIV-2, and 13 (9.9%) for HPIV-3 (three specimens with co-infections: one HPIV-1 + 2 and two HPIV-2 + 3). Viral loads were as follows: both specimens <220 for HPIV-1, ranging from <2200 to 54,560 GEq/ml for HPIV-2 (mean 21,413; median 7480), and from <2200 to 293,700 (mean 30,180; median 2200) for HPIV-3.
The development and standardisation of three “in-house” RT Real Time Qt-PCRs and an IFA for the detection of HPIV-1, -2 and -3 in clinical samples are described. Different IFA conditions were evaluated, such as cellular models, days post-infection, and MAb concentrations. According to the features of the replication cycle of HPIV (Vainionpaa and Hyypia, 1994), a cytoplasmic staining of infected cells was observed, although it became nuclear (HPIV-2 infection) or syncytial (HPIV-3 infection) on day 3–4 post-infection. The two cellular models showed different susceptibility to HPIVs infection. In the case of HPIV-1, Hep-2 had a sensitivity of 10−2 TCID50 on day 3 post-infection using a MAb concentration of 1:80 (similar to that obtained by others (Aguilar et al., 2000) on NCI-H292 cells), while TCID50 calculation on Vero cells was not attained because of their inefficient growth in the presence of trypsin, necessary for attachment of HPIV-1. Vero cells were more suitable for detection of HPIV-2 and -3. HPIV-2 sensitivity was 10−2 TCID50 on day 4 post-infection using 1:40 MAb dilution versus 2 × 10−2 found by Aguilar et al. (2000). The typical “spread” staining reflects HPIV-2 infection strategy, in which contiguous cells are infected subsequently. HPIV-3 had a sensitivity of 10−1 TCID50 on Vero cells on day 4 post-infection using 1:40 MAb dilution versus ∼3 × 101 found by Aguilar et al. (2000). The pattern of HPIV-3 infection (i.e. “all or nothing”) is likely to reflect the simultaneous infection of both nuclear and cytoplasmic structures. It should be note that, although the primary MAb is able to recognize simultaneously HPIV-1, -2 and -3 without differentiating the virus, but not HPIV-4a–b, as indicated by the manufacturer, the fluorescence pattern observed was specific for each virus, thus permitting discrimination by IFA in the absence of co-infections. Also the intensity differed in relation to the MAb dilution and cellular model employed: staining on Vero cells was MAb dilution-dependent, with high dilutions (1:80–1:160) resulting in unfocused and blurred fluorescence; whereas on Hep-2 cells, 1:40 or 1:80 showed a similar intensity, with a reduction at 1:160 dilution.
Considering the three RT Real Time Qt-PCR assays, the identical thermal profile was used, thus permitting to amplify the targets in the same work session. The assays have been optimized by identifying an improved primer/probe concentration in order to obtain the highest amplification efficiency and by evaluating the dynamic range, sensitivity, and reproducibility of each virus.
The sensitivity of the two diagnostic approaches were compared in terms of amplification of fixed TCID50 concentrations and was as follows: 10−4 for HPIV-1 and -2 and 10−3 for HPIV-3 by RT Real Time Qt-PCR versus 10−2 for HPIV-1/2 and 10−1 for HPIV-3 by IFA. The sensitivity data obtained by the RT Real Time Qt-PCRs were superior to those obtained by other investigators that employed multiplex RT-PCR or RT Real Time PCR protocols, including HPIVs (Osiowy, 1998, Aguilar et al., 2000, Templeton et al., 2004, Hamano-Hasegawa et al., 2008, Cordey et al., 2009). As regards multiplexing, these differences could be due to the decrease of sensitivity attributed to the simultaneous amplification of different targets.
As shown by the evaluation of clinical specimens, the sensitivity of the RT Real Time Qt-PCRs, expressed in TCID50, was higher than IFA (data not shown); this is likely to be due to the fact that the detection limit of IFA was higher in comparison to that of the molecular methods.
Another factor to be considered in large volume laboratories and for a prompt clinical decision is the turnaround time. In this study, IFA required from 3 (for HPIV-1) to 4 days (for HPIV-2/-3), while the results of the RT Real Time Qt-PCRs were obtained within 5 h, according to previous studies (Kuypers et al., 2006). These molecular methods should be included in the diagnostic workup able to detect a wide range of respiratory viruses for the clinical management of patients with suspected airway infections (Tivjelung-Lindell et al., 2009). Another relevant advantage is the automation of molecular protocols (from nucleic acid extraction to quantification of fluorescence signal), thus limiting the drawbacks derived from a labour intensive, time-consuming, and operator-dependent method, such as IFA.
In conclusion, molecular methods resulted in a significant improvement over IFA for HPIV-1–3 in terms of sensitivity, applicability, and turnaround time and represent a valid tool for clinical management of patients with suspected respiratory tract infections.
Acknowledgments
This study was supported by “Compagnia di San Paolo”. We thank Paolo Solidoro and Daniela Libertucci, Division of Pneumology, Azienda Ospedaliero-Universitaria S. Giovanni Battista of Turin for biological samples.
References
- Aguilar J.C., Perez-Brena M.P., Garcia N., Cruz M.L., Erdman D.D., Echevarria J.E. Detection and identification of Human Parainfluenza viruses 1, 2, 3 and 4 in clinical samples of pediatric patients by multiplex reverse transcription-PCR. J. Clin. Microbiol. 2000;38:1191–1195. doi: 10.1128/jcm.38.3.1191-1195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanock R.M., Murphy B.R., Collins P.L. Parainfluenza viruses. In: Fields B.N., Knipe D.M., Howley P.M., Chanock R.M., Monath T.M., Melnick J.L., Roizman B., Straus S.E., editors. Virology. 4th ed. Lippincott Williams Wilkins Publishers; Philadelphia: 2001. pp. 1341–1379. [Google Scholar]
- Cordey S., Thomas Y., Cherpillod P., van Belle S., Tapparel C., Kaiser L. Simultaneous detection of parainfluenza viruses 1 and 3 by real-time reverse transcription-polymerase chain reaction. J. Virol. Methods. 2009;156:166–168. doi: 10.1016/j.jviromet.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C.B. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 2001;344:1917–1928. doi: 10.1056/NEJM200106213442507. [DOI] [PubMed] [Google Scholar]
- Hamano-Hasegawa K., Morozumi M., Nakayama E., Chiba N., Murayama S.Y., Takayanagi R., Iwata S., Sunakawa K., Ubukata K. Comprehensive detection of causative pathogens using real-time PCR to diagnose pediatric community-acquired pneumonia. J. Infect. Chemother. 2008;14:424–432. doi: 10.1007/s10156-008-0648-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrickson K.J. Parainfluenza viruses. Clin. Microbiol. Rev. 2003;16:242–264. doi: 10.1128/CMR.16.2.242-264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuypers J., Wright N., Ferrenberg J., Huang M.L., Cent A., Corey L., Morrow R. Comparison of real-time PCR assays with fluorescent-antibody assays for diagnosis of respiratory virus infections in children. J. Clin. Microbiol. 2006;44:2382–2388. doi: 10.1128/JCM.00216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesters H.G.M. Clinical virology in real time. J. Clin. Virol. 2002;25:S3–S12. doi: 10.1016/s1386-6532(02)00197-x. [DOI] [PubMed] [Google Scholar]
- Osiowy C. Direct detection of respiratory syncytial virus, parainfluenza virus, and adenovirus in clinical respiratory specimens by a multiplex reverse transcription-PCR assay. J. Clin. Microbiol. 1998;36:3149–3154. doi: 10.1128/jcm.36.11.3149-3154.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle C.R. The order mononegavirale-current status. Arch. Virol. 1997;157:556–559. [PubMed] [Google Scholar]
- Quint W.G.V., Heijtink R.A., Schirm J., Gerlich W.H., Niesters H.G.M. Reliability of methods for hepatitis B virus DNA detection. J. Clin. Microbiol. 1995;33:225–228. doi: 10.1128/jcm.33.1.225-228.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sable C.A., Hayden F.G. Orthomyxoviral and paramyxoviral infections in transplant patients. Infect. Dis. Clin. North. Am. 1995;9:987–1003. [PubMed] [Google Scholar]
- Templeton K.E., Scheltinga S.A., Beersma M.F., Kroes A.C., Claas E.C. Rapid and sensitive method using multiplex real-time PCR for diagnosis of infections by influenza A and influenza B viruses, respiratory syncytial virus, and parainfluenza viruses 1, 2, 3, and 4. J. Clin. Microbiol. 2004;42:1564–1569. doi: 10.1128/JCM.42.4.1564-1569.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetzner R., Dietrich D., Distler J. Control of carry-over contamination for PCR-based DNA methylation quantification using bisulfite treated DNA. Nucleic Acids Res. 2007;35:e4. doi: 10.1093/nar/gkl955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivjelung-Lindell A., Rotzén-Ostlund M., Gupta S., Ullstrand R., Grillner L., Zweygberg-Wirgart B., Allander T. Development and implementation of a molecular diagnostic platform for daily rapid detection of 15 respiratory viruses. J. Med. Virol. 2009;81:167–175. doi: 10.1002/jmv.21368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainionpaa R., Hyypia T. Biology of parainfluenza viruses. Clin. Microbiol. Rev. 1994;7:265–275. doi: 10.1128/cmr.7.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Elden L.J., van Kraaij M.G., Nijhuis M., Hendriksen K.A., Dekker A.W., Rozenberg-Arska M., van Loon A.M. Polymerase chain reaction is more sensitive than viral culture and antigen testing for the detection of respiratory viruses in adults with hematological cancer and pneumonia. Clin. Infect. Dis. 2002;34:177–183. doi: 10.1086/338238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernet G. Molecular diagnostics in virology. J. Clin. Virol. 2004;31:239–247. doi: 10.1016/j.jcv.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]