

Abstract
The present study describes the development of SYBR Green based real-time polymerase chain reaction (real-time PCR) for detection and quantitation of canine parvovirus type 2 (CPV 2) in faecal samples of dogs. In this assay, the primers were designed and custom-synthesized based on nucleotide sequence of VP2 gene of CPV 2. A standard curve was plotted using 10-fold serial dilution of standard plasmid DNA and Ct value. The standard curve was found to be linear over a 10−7 dilution. The real-time PCR results were expressed as the number of DNA copies of CPV 2 per mg of faecal samples and showed range of 1.0 × 103 to 7.0 × 109 copies of viral DNA per mg of stool samples. The analytical sensitivity of the SYBR Green based real-time PCR was shown to be equivalent to 10 copies. Faecal samples (47) from dogs suspected of CPV 2 infection were analyzed by real-time PCR, haemagglutination (HA) assay and by a conventional PCR and 24, 20 and 22 samples were found positive for CPV 2, respectively. Comparison between the results of three different assays revealed that real-time PCR is more sensitive than HA and conventional PCR and allow the detection of low titers of CPV 2 in infected dogs.
Keywords: Canine parvovirus, Gastroenteritis, Conventional PCR, HA assay, Real-time PCR
Canine parvovirus-2 (CPV 2) causes a highly contagious and often fatal disease in dogs characterized by haemorrhagic diarrhoea and vomiting. CPV 2 emerged in 1978 as the cause of a new disease in dogs throughout the world, when it spread rapidly in domestic dog populations as well as in wild dogs with high morbidity (100%) and frequent mortality up to 10% (Appel et al., 1978). Between 1979 and 1981, the original CPV 2 strain of the virus was replaced around the world by a different genetic and antigenic strain termed as CPV 2a (Parrish et al., 1985). The two viruses differed in 5–6 amino acids, which constitute two different neutralizing antigenic sites on the surface of the capsid. In 1984, a further different antigenic strain was detected, which differed in only a single epitope compared to CPV 2a and designated as CPV type 2b (Parrish et al., 1991). These viruses become distributed globally and replaced original CPV 2 type, indicating that they must have been under strong selection (Decaro et al., 2007). CPV 2 has been isolated for the first time in India by Ramadass and Khadher (1982) and subsequently several outbreaks of the disease have been reported from different parts of the country (Kumar et al., 2003, Phukan et al., 2004, Biswas et al., 2006).
The laboratory diagnosis of infection with CPV 2 relies on the detection of antibody, the virus or both from animals suspected of infection. Antigen detection in faecal samples can be done by agarose gel precipitation test, virus isolation, counter immuno-electrophoresis (CIE) test (Ramadass and Khadher, 1982), virus neutralization test, haemagglutination (HA) test, electron microcopy (EM), fluorescent antibody test (FAT), antigen-capture sandwich ELISA or viral DNA detection by PCR (Mochizuki et al., 1993, Pereira et al., 2000). The HA test is simple, inexpensive and easy to perform but requires continuous supply of red blood cells of porcine origin, and the presence of non-specific agglutinin in faeces makes the HA test less reliable for CPV detection (Mochizuki et al., 1993). Recently, the PCR technique has been used increasingly for the diagnosis of several viral infections. However, conventional PCR methods are time consuming and suffer from carry over contamination. To overcome this problem, real-time PCR employing Taq Man probe (Decaro et al., 2005) and loop-mediated isothermal amplification (LAMP) (Ho-Seong et al., 2006) have been used for the diagnosis of CPV 2 infections in dogs with varying degree of sensitivity and specificity. In this study, real-time PCR employing SYBR Green has been developed for the detection and quantitation of canine parvovirus particles in faecal samples.
The rectal swabs were taken from 47 dogs with diarrhoea suspected for CPV 2 and suspended (in the ratio 1:9) in Hank's balanced salt solution (HBSS) containing streptomycin (100 mg/l) and penicillin (1,00,000 IU/l). It was filtered through a disposable syringe filter (0.45 μm) (Millex, Milipore) and then centrifuged at 14,000 × g at 4 °C for 15 min in a refrigerated centrifuge. The supernatant was pipetted in aliquots of 100 μl, treated with SDS and proteinase K to a final concentration of 1% and 250 μg/ml, respectively and kept at 56 °C for 1 h. The genomic DNA of CPV 2 was extracted by phenol–chloroform method and DNA pellet was resuspended in 10 μl of nuclease free water and kept at −20 °C until used further (Sambrook and Russell, 2001).
The real-time PCR conditions such as annealing temperature and primer concentration were optimized by a conventional PCR using primer set pCPV-2RT. The primers pCPV-2RT {F 5′-CAT TGG GCT TAC CAC CAT TT-3′ (20mer) and R 5′-CCA ACC TCA GCT GGT CTC AT-3′ (20mer)} based on the VP2 gene (160 bp) of the CPV 2 genome corresponding to nucleotides 3136–3155 to 3276–3295 (GenBank Accession No. EU009205) were designed and custom-synthesized using Primer 3 software. The designed primers had similar predicted annealing temperatures and little potential for dimerization. The annealing temperatures tested in gradient PCR indicated that the optimal annealing temperature for pCPV-2RT (F&R) was 55 °C. The conventional PCR mixture constituted of 10 μl of target DNA, 0.5 μl each of forward and reverse primers (10 pmol/μl), 2.5 μl of 10× Taq Buffer, 1.5 μl of MgCl2 (25 mM), 1 μl of dNTP mix (10 pmol), 1 μl of Taq DNA polymerase (1 U/μl) and nuclease free water up to 25 μl. The cyclic condition comprised of initial denaturation at 94 °C for 5 min, followed by 30 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s and final extension at 72 °C for 5 min. The PCR product was checked subsequently by running on a 1% agarose gel with ethidium bromide and 100 bp DNA ladder.
The PCR using the primer set pCPV-2bN was also carried out to amplify a sequence of 990 bp amplicon from 2785–2817 to 3742–3774 of VP2 gene of CPV 2 (Nandi et al., 2007). The 990 bp amplicons were gel eluted and cloned in pGEM-T Easy vector. The plasmid DNA was extracted using the QIAquick gel extraction kit (Qiagen, Germany) and dissolved in TE buffer (10 mM Tris–HCl, pH 7.5, 1 mM EDTA). Serial 10-fold dilutions of plasmid DNA containing 108 to 10 copies per 10 μl were made in TE buffer in order to generate a standard curve. The standard curve was plotted with known copy number of target DNA having corresponding Ct values (Fig. 1 ). The assay shows linearity over the dilution range of 107 with an R 2 value (the square of the correlation coefficient) of 0.998 and a reaction efficiency of 96.4% (Fig. 1). The slope value of −3.412 indicates the reproducibility and the detection limit of the assay.
Fig. 1.
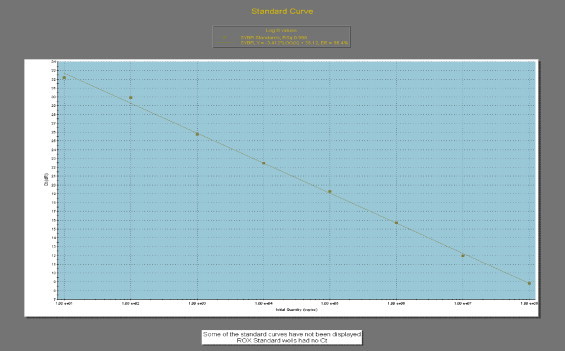
Standard curve (plot of the Ct values against the different plasmid DNA concentrations) indicating the linearity and efficiency of the SYBR® Green reactions for VP2 gene of CPV 2. The assays were linear over a 107 dilution range with R2 values (square of the correlation coefficient) of 0.998, the slope value of −3.412 and reaction efficiency of 96.4%.
Quantitative real-time PCR was undertaken using the Platinum SYBR® Green QPCR Master Mix (Invitrogen, USA) and the Mx3000P spectro-fluorometric thermal cycler operated by the MxPro™ PCR software. Each real-time PCR was placed in a total volume of 25 μl, which contained 10 μl of template DNA, 5 pmol of each primers, 12.5 μl of QPCR master mix, and nuclease-free water (qsp). The PCR conditions consisted of initial denaturation at 95 °C for 10 min, 40 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s and melting curve analysis. The amplification and denaturation data was acquired for further calculations. No template control (NTC) was used as real-time PCR negative control which indicates no reagent contamination by the target DNA.
The analytical specificity of CPV 2 DNA detection by real-time or conventional PCR was evaluated using DNA preparations of CPV 2 strains including CPV 2 (from vaccine), CPV 2a, CPV 2b and CPV 2c maintained at the Virus Laboratory, Centre for Animal Disease Research and Diagnosis (CADRAD) (Nandi et al., 2010) and produced amplification signal. The analytical specificity was also evaluated with other canine pathogens using a vaccine strain of canine adenovirus type 1 (CAdV-1), canine distemper virus and canine coronavirus and without non-specific amplification other than amplicon specific for CPV was evident. The analytical sensitivity of the real-time PCR was examined using 10-fold dilutions of the standard plasmid DNA ranging from 108 to 10 copies/10 μl and the detection limit was found to be 10 copies/10 μl. The analytical sensitivity was also estimated with CPV 2 having titre of 104.5 TCID50/ml (Biocan, Bioveta, Czech Republic). The overall detection limit was shown to be equivalent to 0.05 TCID50 per reaction.
A standard curve was plotted over a range of target DNA concentrations (1.0 × 108 to 1.0 × 10 copies of plasmid DNA per 25 μl reaction). The linear portion of the standard curve was found to span from 1.0 × 108 to 1.0 × 10, therefore, a lower detection limit (or cutoff) of 10 copies per 25 μl reaction was established. There was a cut-off value corresponding to a threshold cycle (Ct) of 32.18 and was applied to test samples. The samples with a Ct >32.18 were taken as not quantifiable (i.e. below the detection limit of the test) for CPV 2 DNA and 23 samples were considered negative. Faecal samples negative for CPV 2 also showed non-specific melting temperature (T m), and in positive faecal samples specific T m was detected (Fig. 2 ). Extracted samples with a copy number exceeding 1.0 × 108 per 25 μl reactions, or a Ct < 8.77 were diluted 1:10 or more using nuclease free water and re-tested. The dilution factor was taken into consideration when calculating the copies of CPV 2 in the sample. The results were expressed as the number of copies of CPV 2 genomes per mg of faecal samples (Table 1 ). The 24 samples found positive in this study had 1.0 × 103 to 7.0 × 109 copies of viral DNA per mg of stool samples in real- time PCR. The results were in accordance with the findings of Decaro et al. (2005), who showed virus titre ranging from 1.0 × 103 to 7.43 × 1011 copies/mg of faeces by real-time PCR.
Fig. 2.
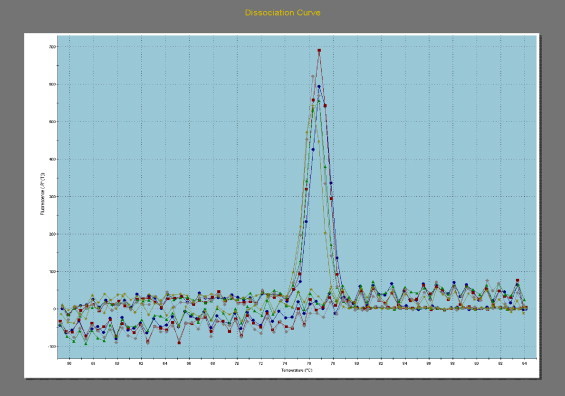
Dissociation curve of five faecal samples indicating its melting temperature.
Table 1.
Results of 24 faecal samples found positive in real-time PCR.
| Sr. No. | Samples | HA assay | Conventional PCR | Real time titer (per mg faecal sample) |
|---|---|---|---|---|
| 1 | F 6 | − | + | 1.6 × 104 |
| 2 | F 7 | + | + | 6.5 × 107 |
| 3 | F 9 | − | − | 6.3 × 103 |
| 4 | F 10 | − | + | 6.4 × 104 |
| 5 | F 11 | − | − | 3.1 × 103 |
| 6 | F 12 | + | + | 1.5 × 109 |
| 7 | F 14 | + | + | 4.2 × 108 |
| 8 | F 15 | + | + | 2.1 × 109 |
| 9 | F 16 | + | + | 5.8 × 107 |
| 10 | F17 | + | + | 4.3 × 108 |
| 11 | R 1 | + | + | 1.1 × 108 |
| 12 | R 2 | + | + | 1.8 × 107 |
| 13 | R 3 | + | + | 6.1 × 107 |
| 14 | R 4 | + | + | 4.5 × 109 |
| 15 | R 5 | + | + | 5.8 × 109 |
| 16 | R 6 | + | + | 1.1 × 109 |
| 17 | R 7 | + | + | 1.6 × 109 |
| 18 | R 8 | + | + | 7.8 × 106 |
| 19 | R 9 | + | + | 1.3 × 109 |
| 20 | R 10 | + | + | 5.3 × 108 |
| 21 | R 11 | + | + | 7.3 × 105 |
| 22 | R 12 | + | + | 3.2 × 105 |
| 23 | R 13 | + | + | 6.1 × 107 |
| 24 | R 14 | + | + | 1.2 × 108 |
The canine parvovirus haemagglutinates pig red blood cell and rhesus monkey erythrocyte at 4 °C. The pig RBCs were used for the HA test because of its easy availability and sensitivity. Pig blood was collected in Alsever's solution, washed three times in PBS and suspended to 1% (v/v) in ice cold PBS containing 0.1% bovine serum albumin (BSA). Haemagglutination test was carried out following the method described by Kumar et al. (2003). Out of 47 faecal samples, 20 samples were found positive with a HA titre of 64 or greater. Both the negative control well and the positive control well exhibited the button formation and mat formation respectively (Kumar et al., 2003).
The conventional PCR was standardized using primer pCPV 2 FP {5′-GAA GAG TGG TTG TAA ATA ATA-3′ (21mer)} and pCPV 2 RP {5′-CCT ATA TCA CCA AAG TTA GTA G-3′ (22mer)} in order to amplify 681 bp product targeting VP2 gene of CPV 2 (3025–3706 nucleotide) (Pereira et al., 2000). It was carried out in 25 μl volume containing 2.5 μl of Taq DNA polymerase buffer, 1.5 μl of MgCl2 (25 mM), 1 μl of each dNTP (10 mM), 10 pmol each of the forward and reverse primer, 1 μl of Taq DNA polymerase (1 U/μl) and 10 μl of template DNA. Amplification was performed in a thermocycler (Applied Biosystems) and initial denaturation was done at 95 °C for 5 min. The cyclic conditions consisted of 30 cycles of 95 °C for 30 s, 55 °C for 2 min and 72 °C for 2 min followed by final extension at 72 °C for 5 min (Nandi et al., 2009). A total of 47 faecal samples were screened and 22 samples were found positive. In the positive cases and in positive control, a DNA band of 681 bp in length was visualized in 1% agarose gel under UV transilluminator.
On comparing the results of the real-time PCR with the HA assay and the conventional PCR, it was evident that samples found positive by the conventional PCR and the HA assay were also positive by the real-time PCR. Only two samples found negative by the conventional PCR were positive for CPV 2 by the real-time PCR assay with a titre in range of 103 copies/mg of faeces. The result was in agreement with the finding of Decaro et al. (2005). Four samples showing negative results in HA assay was found to be CPV 2 positive by real-time PCR.
Canine parvovirus causes a severe gastroenteritis of dogs characterized by depression, anorexia, vomiting, haemorrhagic diarrhoea and leukopaenia. In this study, the development of a simple and rapid SYBR Green based real-time PCR for the detection and quantitation of CPV 2 DNA in the faeces of dogs. The real-time quantitative PCR is based on continuous optical monitoring of a fluorogenic PCR reaction (Heid et al., 1996). The CPV 2 real-time PCR assay was demonstrated to be more sensitive than both the HA and conventional PCR, being able to detect as few as 10 copies of CPV 2 DNA. This real-time PCR was highly reproducible and standard curve was linear over a range of seven orders of magnitude, from 10 to 108 copies, allowing a precise calculation of CPV 2 DNA in samples containing a wide range from 10 to 109 copies of viral DNA. Although there are various qualitative methods available for the diagnosis of CPV 2, the quantitation of number of virus particles in faecal samples can be done by quantitative real-time PCR. The standard procedure was followed for absolute quantitation of CPV particles by plotting the standard curve with a known number of targets DNA and corresponding Ct values were obtained.
The comparison of the real-time PCR with the HA assay and conventional PCR proved to be useful in assessing the sensitivity of the method. The SYBR Green based real-time PCR demonstrated greater sensitivity over HA assay and conventional PCR as it detected the samples with low copy number of virus found negative in other two assays. Real-time PCR has several advantages over conventional PCR, allowing a large increase in throughput and enabling simultaneous processing of several samples. The CPV 2 real-time PCR assay will also help in gaining new insights into the pathogenesis of CPV 2 infection, with particular regard to the amounts of CPV shedding in infected dogs or in animals challenged during vaccine trials. The possibility of determining precisely the duration and the amounts of viral shedding in the faeces is of fundamental importance to evaluate the efficacy of vaccines. Therefore, the real-time PCR can be a valuable diagnostic test with high sensitivity and specificity and can detect virus shedding during the initial stages of infection in dogs as well as in carrier animals and accordingly, the appropriate prevention and control strategy can be implemented.
Acknowledgement
We thank the Director, Indian Veterinary Research Institute, Izatnagar, for providing facilities to carry out the work.
References
- Appel M.J.G., Cooper B.J., Greisen H., Carmichael L.E. Status report: canine viral enteritis. J. Am. Vet. Med. Assoc. 1978;173:1516–1518. [Google Scholar]
- Biswas S., Das P.J., Ghosh S.K., Pradhan N.R. Detection of canine parvovirus (CPV) DNA by polymerase chain reaction and its prevalence in dogs in and around Kolkata, West Bengal. Ind. J. Anim. Sci. 2006;76(4):324–325. [Google Scholar]
- Decaro N., Elia G., Martella V., Desario C., Campolo M., Trani L.D., Tarsitanoa E., Tempestaa M., Buonavoglia C.V. A real-time PCR assay for rapid detection and quantitation of canine parvovirus 2 in the faeces of dogs. Vet. Microbiol. 2005;105:19–28. doi: 10.1016/j.vetmic.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Decaro N., Desario C., Addie D.D., Martella V., Vieira M.J., Elia G., Davis C., Thompson G., Truyen U., Buonavoglia C. The study of molecular epidemiology of canine parvovirus, Europe. Emerg. Infect. Dis. 2007;13:1222–1224. doi: 10.3201/eid1308.070505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid C.A., Stevens J., Livak K.J., Williams P.M. Real time quantitative PCR. Gen. Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Ho-Seong C., Jong K., Nam-Yong P. Detection of canine parvovirus in faecal samples using loop-mediated isothermal amplification. J. Vet. Diagn. Invest. 2006;18:81–84. doi: 10.1177/104063870601800111. [DOI] [PubMed] [Google Scholar]
- Kumar P., Garg S.K., Bandyopadhyay S.K., Singh R., Shrivastava S. Hemagglutinating activity of canine parvovirus. Ind. J. Anim. Sci. 2003;73(2):123–125. [Google Scholar]
- Mochizuki M., SanGabriel M.C., Nakatani H., Yoshida M. Comparison of polymerase chain reaction with virus isolation and haemagglutination assays for the detection of canine parvoviruses in faecal specimens. Res. Vet. Sci. 1993;55:60–63. doi: 10.1016/0034-5288(93)90035-e. [DOI] [PubMed] [Google Scholar]
- Nandi S., Kumar M., Anbazhagan R., Chidri S., Chauhan R.S. A sensitive method to detect canine parvoviral DNA in the stool samples by polymerase chain reaction. Ind. J. Comp. Microbiol. Immunol. Infect. Dis. 2007;27(1 & 2):56–57. [Google Scholar]
- Nandi S., Chidri S., Kumar M. Molecular characterization and phylogenetic analysis of a canine parvovirus isolate in India. Vet. Med. Czech. 2009;54:483–490. [Google Scholar]
- Nandi S., Chidri S., Kumar M., Chauhan R.S. Occurrence of canine parvovirus type 2c in the dogs with haemorrhagic enteritis in India. Res. Vet. Sci. 2010;88:169–171. doi: 10.1016/j.rvsc.2009.05.018. [DOI] [PubMed] [Google Scholar]
- Parrish C.R., O’Connell P.H., Evermann J.F., Carrmichael L.E. Natural variation of canine parvovirus. Science. 1985;230:1046–1048. doi: 10.1126/science.4059921. [DOI] [PubMed] [Google Scholar]
- Parrish C.R., Aquadro C.F., Strassheim M.L., Evermann J.F., Sgro J.Y., Mohammed H.O. Rapid antigenic type replacement and DNA sequence evolution of canine parvovirus. J. Virol. 1991;65:6544–6552. doi: 10.1128/jvi.65.12.6544-6552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira C.A., Monezi T.A., Mehnert D.U., D’Angelo M., Durigon E.L. Molecular characterization of canine parvovirus in Brazil by polymerase chain reaction assay. Vet. Microbiol. 2000;75:127–133. doi: 10.1016/s0378-1135(00)00214-5. [DOI] [PubMed] [Google Scholar]
- Phukan A., Deka D., Boro P.K. Occurrence of canine parvovirus infection in and around Guwahati. Ind. J. Anim. Sci. 2004;74(4):930–931. [Google Scholar]
- Ramadass P., Khadher T.G.A. Diagnosis of canine parvovirus infection by agar gel precipitation test and fluorescent antibody technique. Cherion. 1982;11:323–326. [Google Scholar]
- Sambrook J., Russell D.W. Molecular Cloning; A Laboratory Manual. 3rd edition. Cold spring Harbor laboratory Press; New York: 2001. Preparation and analysis of genomic DNA. pp. 545–547. [Google Scholar]