

Abstract
Pan-viral DNA array (PVDA) and high-throughput sequencing (HTS) are useful tools to identify novel viruses of emerging diseases. However, both techniques have difficulties to identify viruses in clinical samples because of the host genomic nucleic acid content (hg/cont). Both propidium monoazide (PMA) and ethidium bromide monoazide (EMA) have the capacity to bind free DNA/RNA, but are cell membrane-impermeable. Thus, both are unable to bind protected nucleic acid such as viral genomes within intact virions. However, EMA/PMA modified genetic material cannot be amplified by enzymes. In order to assess the potential of EMA/PMA to lower the presence of amplifiable hg/cont in samples and improve virus detection, serum and lung tissue homogenates were spiked with porcine reproductive and respiratory virus (PRRSV) and were processed with EMA/PMA. In addition, PRRSV RT-qPCR positive clinical samples were also tested. EMA/PMA treatments significantly decreased amplifiable hg/cont and significantly increased the number of PVDA positive probes and their signal intensity compared to untreated spiked lung samples. EMA/PMA treatments also increased the sensitivity of HTS by increasing the number of specific PRRSV reads and the PRRSV percentage of coverage. Interestingly, EMA/PMA treatments significantly increased the sensitivity of PVDA and HTS in two out of three clinical tissue samples. Thus, EMA/PMA treatments offer a new approach to lower the amplifiable hg/cont in clinical samples and increase the success of PVDA and HTS to identify viruses.
Keywords: DNA array, High-throughput sequencing, Virus identification, Porcine reproductive and respiratory syndrome virus, PRRSV, Propidium monoazide, PMA, Ethidium bromide monoazide, EMA
1. Introduction
The emergence of new viral diseases represents a constant threat to human and animal health. Fortunately, in the past decade, accesses to novel technologies have improved the detection and identification of unknown viruses in clinical samples. Most of these novel virus identification processes are based on viral genome detection using new technologies like pan-viral DNA microarrays (PVDA) and high-throughput sequencing (HTS).
The first PVDA, which contained 1600 oligonucleotides probes targeting highly conserved DNA sequences of 140 distinct selected viral genomes, was reported in 2002 (Wang et al., 2002). Since then, the PVDA has been further developed and includes, in its latest version, 36,000 oligonucleotides probes targeting approximately 1500 distinct viral genomes (Chen et al., 2011). This technology has been used to rapidly identify viruses involved in human illness, like severe acute respiratory syndrome (SARS) (Wang et al., 2003), and in animal diseases (Chen et al., 2011). Use of this technology is of interest as the results can be generally obtained within a day and does not require other advanced technologies for results interpretation (Chen et al., 2011). However, PVDA is dependent on the selected probes it contains and their tolerance to nucleotide mismatch during the DNA hybridization process required for the detection and identification of viruses in a clinical sample (Delwart, 2012).
Decreasing costs have made HTS technology more accessible and consequently, its use in identifying novel or unknown viruses affecting humans, animals or plants has increased (Massart et al., 2014, Lipkin and Firth, 2013, Quinones-Mateu et al., 2014). It has even led to the discovery of unforeseen viruses in clinical samples (Bellehumeur et al., 2013). Metagenomic sequencing has the potential to determine the entirety of the nucleic acid sequences within a sample, including viral nucleic sequences of interest (Hall et al., 2014). The metagenomic DNA sequences obtained with HTS are then compared to a genomic database in order to identify the nucleic acid sequences associated with known viruses (Firth and Lipkin, 2013). One major benefit to metagenomic sequencing of clinical samples is the potential to detect and assemble the genome of novel viruses (Barzon et al., 2013).
Although both PVDA and HTS have led to the discovery of new viruses in the last years, especially from isolated viruses, both techniques are negatively impacted by the presence of nucleic acid found in clinical samples, mainly host genomic DNA/RNA (Firth and Lipkin, 2013, Kang et al., 2011). The high host to viral DNA ratio in extracted clinical samples greatly decreases PVDA sensitivity since most of the amplified labeled DNA corresponds to host DNA (Kang et al., 2011). For sequencing, depending on the method of tissue preparation and viral particle concentration, again the high host to viral DNA ratio decreases the sensitivity of the technology (Yang et al., 2011). As more reads must be obtained in order to detect the presence of a virus in a clinical sample, this can increase sequencing costs and lower throughput while creating a potential bioinformatics bottleneck. Thus, in order to improve viral detection in clinical samples with HTS and PVDA, the levels of host genomic DNA must be lowered. This is generally done by treating samples with a combination of ultracentrifugation, filtration and/or nuclease treatment (typically DNase and/or RNase treatment) (Hall et al., 2014, Clem et al., 2007). As these methods can introduce bias in viral identification (9) the development of alternative methods to lower host genomic material in clinical samples is of interest.
Ethidium bromide monoazide (EMA) and its analog propidium monoazide (PMA), when combined with PCR, allow the quantification of living cells such as bacteria (Nocker et al., 2007, Nocker et al., 2009, Gatteschi et al., 2012, Codony et al., 2012a). Both are azide-bearing, DNA/RNA-intercalating dyes that only cross damaged lipid membrane barriers. Both dyes can bind and covalently crosslink DNA/RNA when the azide group is converted to a highly reactive nitrene radical upon exposure to bright visible light. Thereafter, they are easily inactivated and the unbound inactivated EMA/PMA remains free in solution. EMA/PMA-generated DNA/RNA cross-linking strongly inhibits reverse-transcription and PCR amplification of the EMA/PMA modified genomes while unmodified genomes from presumptively living bacteria (which possesses intact membranes) can be amplified (Codony et al., 2012a). Interestingly, EMA/PMA treatments have been used to distinguish infectious from non-infectious viruses such as Hepatitis A virus, coxsakievirus, echovirus, norovirus and poliovirus, suggesting that intact virus particles have the potential to protect their genetic material from EMA/PMA chemicals (Elizaquivel et al., 2014).
In theory, EMA/PMA could be used to prevent host genomic amplification during the PCR steps that are conducted within PVDA and HTS assays, while the viral genome within intact virions are inaccessible to the dyes during treatment before amplification. The main objective of this study was to determine if EMA or PMA treatments can increase the efficacy of PVDA and HTS to detect viruses in clinical samples.
2. Materials and methods
2.1. Cell and virus strains
MARC-145 cells were maintained as described previously and were used for virus production (Beura et al., 2010). The Porcine reproductive and respiratory syndrome virus (PRRSV) strain used to spike tissue and sera samples was the IAF-Klop reference strain (Gagnon et al., 2003). The PRRSV IAF-Klop strain stock was obtained following three cycles of freeze-thaw of PRRSV MARC-145 infected cells. Afterward, the virus stocks were maintained at −70 °C until needed. The infectious dose of the stocks was calculated from MARC-145 infected cells by the Kärber method as described previously (Gagnon et al., 2008). Virus titers were expressed in tissue culture infectious dose 50% per mL (TCID50/mL).
2.2. PRRSV spiked tissues and positive clinical samples
Lung and blood samples were collected from negative control and PRRSV experimentally infected piglets. Animals care was done according to the guidelines of the Canadian Council of Animal Care and the protocol approved by the Institutional Animal Care Committee (Protocol 12-Rech-1669). The PRRSV strain involved in this infection was PRRSV FMV12-1425619 (GenBank accession number KJ1888950). Sera of non-infected and infected piglets were collected at different time post-infection (pi) and kept at −70 °C until needed. Viral load in samples was determined with a specific PRRSV RT-qPCR assay as previously described (Bellehumeur et al., 2013). Lung samples were collected at necropsy at 28 days pi and stored at −70 °C until needed. Three infected lung samples (PRRSV titers: (1) 4929, (2) 4336 and (3) 9408 TCID50/g) were selected. Two sera samples were selected from PRRSV positive clinical sera samples (PRRSV titers of (1) 1059 and (2) 7413 TCID50/mL) submitted to the Molecular Diagnostic Laboratory (MDL) of the University of Montreal were selected and stored at −70 °C until needed. PRRSV negative swine lung samples or PRRSV negative swine sera samples were spiked with a known quantity of the PRRSV IAF-Klop strain to a final concentration of either 5000 TCID50/mL or 50,000 TCID50/mL. Lung tissue samples (spiked samples or PRRSV positive clinical samples; 100 mg of tissue in 1 mL of PBS with glass beads) were homogenized twice for 5 min in a Mini BeadBeater 96 Homogenizer, centrifuged 1 min at 10,000 rpm in a table top centrifuge and kept at 4 °C until used. Serum samples (spiked samples or PRRSV positive clinical samples) were kept at 4 °C once thawed.
2.3. Samples processing
2.3.1. Ultracentrifugation
Lung tissue homogenate and serum samples (spiked with PRRSV or clinical samples) were ultracentrifuged for 3 h at 25,000 rpm in a Sorvall TH-641 swinging bucket rotor at 4 °C through 1 mL of a 20% sucrose cushion in TNE buffer (20 mM Tris–HCl (pH 8.0), 150 mM NaCl and 2 mM EDTA). The virus pellets were re-suspended in TNE buffer to the initial sample volume prior to ultracentrifugation. Non-ultracentrifugated sample aliquots were kept at 4 °C for the duration of the ultracentrifugation step. Clinical samples (lung tissue and serum) were assessed by ultracentrifugation only.
2.3.2. Ethidium bromide monoazide and propidium monoazide treatments
EMA and PMA (Biotium, Hayward, CA) were reconstituted according to manufacturer's recommendation. Stock solutions were then diluted in RNase-free water to a working concentration of 2 mM. Both stock and working solutions were kept at −20 °C until used. Lung tissue homogenates with and without ultracentrifugation (spiked or clinical samples) and sera samples (spiked or clinical samples) were subsequently treated with EMA (final concentrations of 100 μM), PMA (final concentrations of 100 μM), or with an equivalent volume of water. Treated samples were then incubated in the dark for 5 min at room temperature, 5 min on ice, and then exposed during 10 min to two 500 W halogen light sources (at a distance of 20 cm from the light source). Micro-centrifuge tubes were kept on ice during light exposure to avoid excessive heating.
2.4. Total nucleic acid extraction
Following treatments, total DNA and RNA were extracted using a phenol-chloroform-isoamyl alcohol. Briefly, 200 μL of a phenol solution at pH 7.6–7.8 (UltraPure™ buffer-saturated phenol; Invitrogen, Burlington, ON), 200 μL of molecular grade chloroform (Fisher scientific, Ottawa, ON) and 20 μL of isoamyl alcohol (Fisher scientific) were added to each sample. Samples were then homogenized and centrifuged in a table-top microcentrifuge for 1 min at 13,000 rpm. The supernatant was kept and assessed for a second phenol-chloroform step. Finally, the supernatant was treated twice with 200 μL of chloroform and total DNA and RNA precipitated by adding 500 μL of ethanol and 20 μL of sodium acetate (3M, pH 5.2) and incubation at −70 °C overnight followed by centrifugation for 30 min at 4 °C in a table-top microcentrifuge at 13,000 rpm. The pellet representing total nucleic acid was resuspended in 50 μL of RNase-free water and stored in a freezer at −70 °C until used.
2.5. PRRSV and host genome quantification by qPCR and RT-qPCR
PRRSV and swine β-Actin (representing swine host genomic DNA) were quantified in extracted DNA/RNA by RT-qPCR and qPCR, respectively. Equal sample volumes were used for each test to ensure comparable results. PRRSV was quantified in DNA/RNA extracted from tested samples using the commercial EZ-PRRSV™ MPX 4.0 Real Time RT-PCR kit (Tetracore, Rockville, Maryland, USA), following the manufacturer's recommendations. The β-actin quantification was done by qPCR using the SsoFast™ EvaGreen® Supermix kit (Bio-rad, Hercules, CA, USA) in order to evaluate host genome in spiked and clinical samples following each treatment. Samples were diluted in RNase free water (1:16) prior to β-actin qPCR tests. The PCR amplification program for β-actin quantification consisted of an enzyme activation step of 3 min at 98 °C followed by 40 cycles of a denaturing step (2 s at 98 °C) and an annealing/extension step (5 s at 58 °C) using the following primers: forward primer (5′-ATCTTCATGAGGTAGTCGGTCAGG-3′) and reverse primer (5′-ACCACTGGCATTGTCATGGACTCT-3′). Both primers were selected to achieve amplification efficiency between 90 and 110% (data not shown) and were designed from the NCBI GenBank mRNA sequences using web-based software primerquest from Integrated DNA technologies. All amplification steps were done on a Bio-Rad CFX 96 apparatus with results expressed as Ct values.
2.6. DNA/RNA samples amplification
Following treatment and extraction, total nucleic acid samples used for all experiments (i.e. both array detection and PMA/EMA treatment) were amplified using a modified random PCR protocol (Wang et al., 2002, Chen et al., 2011). Random-amplified samples for PVDA testing were spiked with 225 pg of purified pUC19 plasmid DNA and used both as a positive control and localization marker on array slides.
2.7. DNA array
2.7.1. DNA array development
Probe sequences targeting PRRSV American strains were selected from the PRRSV probes used on the ViroChip developed by Wang et al. (2002) and were deduced from sequences alignment of full and partial PRRSV genomic sequences gathered from the National Center for Biotechnology Information (NCBI) GenBank database using Geneious pro software, version 5.6.6 (Biomatters, Auckland, New Zealand [http://www.geneious.com/]). The PRRSV homology of candidate probes was verified with BLASTN. Each selected oligonucleotide probe (70-mers) was unique and was targeting specific PRRSV conserved regions. Reverse and forward sequences of 17 conserved regions were selected for the PRRSV genotype 2 strains. Two probes were also selected to target a specific region of pUC19 plasmid DNA as an array positioning control and one probe was selected as a negative hybridization control. A total of 37 probes were selected and were synthesized by Eurofins MWG Operon (Huntsville, AL, USA). These probes are reported in the Gene Expression Omnibus (GEO) NCBI database (accession number GSE62910). DNA array spotting was done at the National Research Council Canada, as previously described (Vuong et al., 2013).
2.7.2. DNA array hybridization and analysis
After RT and PCR random-amplification steps, PCR products were incubated with aminoallyl (aa)-dUTP (Invitrogen, Burlington, ON, Canada) in the presence of Klentaq (Clontech) as previously described (Wang et al., 2002, Chen et al., 2011). The generated aa-DNA was purified with the QIAquick PCR purification kit (QIAGEN, Toronto, ON, Canada), re-suspended in 30 μL of RNase-free water and supplemented with 3 μL of 1M sodium bicarbonate. Thereafter, the aa-DNA was incubated for 1h in the presence of 1:10 DMSO-reconstituted Cy3 Mono-Reactive Dye (GE Healthcare Life Sciences, Pittsburgh, PA, USA). Labeled DNA was then purified using a QIAquick PCR purification kit and assessed for quality using a NanoDrop 1000 spectrophotometer (Fisher Scientific, Toronto, ON, Canada).
Microarray slides were pre-hybridized at 50 °C for 1h with 85.5 μL of DIG easy Hyb buffer (Hoffmann-La Roche Limited, Mississauga, ON, Canada) supplemented with 4.5 μL of 10% (w/v) bovine serum albumin (Invitrogen, Burlington, ON, Canada) under 22 mm × 60 mm × 0.25 mm Grace Bio-Labs Hybrislip™ coverslips (Sigma–Aldrich, Oakville, ON, Canada). Subsequently, cover slips were removed by dipping the glass slides into 0.1× SSC (15 mM NaCl, 1.5mM sodium citrate) and the slides dried by a quick centrifugation. Total Cy3-labeled DNA (typically 3 μg) was dried in a Speedvac (Fisher Scientific) then suspended in 7 μL of DIG Easy Hyb buffer. Afterwards, the DNA was denatured for 5 min in a boiling water bath followed by 5 min of incubation on ice. Samples were hybridized overnight in a water bath at 50 °C under 22 mm × 22 mm Grace Bio-Labs Hybrislip™ (Sigma–Aldrich). Finally, coverslips were removed in 0.1× SSC, 0.1% (V/V) sodium dodecyl sulfate (SDS) and the slides washed three times in 0.1× SSC, 0.1% (v/v) SDS and once in 0.1× SSC for 5 min per wash. Hybridized arrays were imaged using a fluorescence scanner (ScanArray; Perkin Elmer, Mississauga, ON, Canada) and ScanArray software version 1.1. Fluorescent spot intensities were scanned at a laser fluorescent intensity of 80–100 and quantified using ScanArray software version 1.1. DNA array fluorescent intensity results were analyzed with Microsoft Excel™. The intensity of each spotted probe was compared to the average intensity of the two negative control spots. For a probe to be considered positive, the average of signal-to-noise fluorescence ratios of their duplicate spots had to be ≥2.0.
2.8. High-throughput sequencing
The random amplified samples were end-repaired and A-tailed using KAPA High Throughput Library Preparation Kit with SPRI solution and Standard PCR Library Amplification/Illumina series (Kapa Biosystems, Wilmington, MA, USA). Illumina TruSeq HT dual indexed adapters (Illumina, SanDiego, CA, USA) were ligated to the amplified samples and the libraries were amplified with the KAPA kit. After the final cleanup, the quality of the libraries were assessed on High Sensitivity DNA Chips (Agilent, Santa Clara, CA, USA) using a 2100 BioAnalyzer (Agilent). Equal amounts of each library were pooled and sequenced on an Illumina MiSeq (2 × 300 paired-end reads, dual-indexed) at the Plateforme d’Analyses Génomiques de l’institut de Biologie Intégrative et des Systèmes de l’Université Laval (Quebec, QC, Canada). Raw sequencing reads were trimmed for the random amplification primers and mapped to the Sus scrofa genome v10.2 and the PRRSV IAF-Klop viral genome sequence using the gsMapper application of Newbler v2.9.
2.9. Statistical analysis
A parametric one-way ANOVA model, followed by Tukey's Multiple Comparison tests (GraphPad PRISM Version 5.03 software) was used to determine if a statistically significant difference exists between the quantification of targeted genes (β-actin or PRRSV) for each treated and untreated samples, as evaluated by qPCR and RT-qPCR. A non-parametric one-way ANOVA model, followed by Dunn's Multiple Comparison Test (GraphPad PRISM software), was used to determine if a statistically significant difference exists between the mean relative fluorescent intensity of each PRRSV detection probe on the DNA array for each treated and untreated samples. The ratio of positive PRRSV reads compared to the total amount of reads following each treatment was analyzed with a mixed linear model, with trial number as a random factor and treatment as a fixed factor, followed by Tukey's Multiple Comparison tests (SAS version 9.3 software, Cary, NC, USA). HTS results were also evaluated individually using chi-square tests in order to determine if the odds to obtained positive PRRSV reads were different following each selected treatments, compared to the total amount of reads not related to PRRSV (GraphPad PRISM software). Finally, a Spearman's non-parametric correlation was also used to evaluate the relation between the presence of DNA contaminant reads and positive PRRSV reads as well as the ratio of PRRSV reads and the PRRSV percent of coverage in HTS results (SAS version 9.3 software). Differences were considered statistically significant with a P < 0.05, with the exception of chi-square tests results where only P < 0.003 (P < 0.05/15) were considered significant.
3. Results
3.1. EMA or PMA treatment effects on host-genomic DNA and PRRSV detection in spiked samples
In order to select an effective concentration for EMA and PMA treatment, a preliminary experiment was done with PRRSV spiked lung samples (5000 TCID50/mL; data not shown) and a final EMA and PMA concentration of 100 μM was selected for the realization of subsequent experiments. Using this concentration, the effect of EMA or PMA treatments on virus genome's presence and host genomic DNA was further evaluated in tissue samples spiked with known quantities of PRRSV (5000 TCID50/mL or 50,000 TCID50/mL) but also in serum samples spiked with the same PRRSV quantities. Results with lung homogenates spiked with both concentration of PRRSV indicate that EMA and PMA treatments significantly lowered PRRSV detection compared to non-treated samples (Fig. 1A; P < 0.001 and P < 0.05 respectively). However, PRRSV detection was more negatively affected following EMA treatment (Fig. 1A; P < 0.01), indicating that PMA treatments have less negative impact on RT-qPCR PRRSV detection as observed in the preliminary experiment. Ultracentrifugation had a significant positive impact on PRRSV detection in lung tissue homogenates spiked with either concentration of PRRSV following EMA treatment (Fig. 1A; P < 0.001). In lung tissue homogenates spiked only with the highest concentration of PRRSV, ultracentrifugation had also increased significantly PRRSV detection following no treatment and following PMA treatment (Fig. 1A; P < 0.01 and P < 0.05 respectively). In spiked serum samples, only EMA treatment significantly decreased PRRSV detection (Fig. 1A; P < 0.05). Ultracentrifugation significantly improved PRRSV detection in non-treated spiked sera samples (Fig. 1A; P < 0.001) and significantly decreased PRRSV RT-qPCR detection in EMA treated spiked sera samples (Fig. 1A; P < 0.001).
Fig. 1.
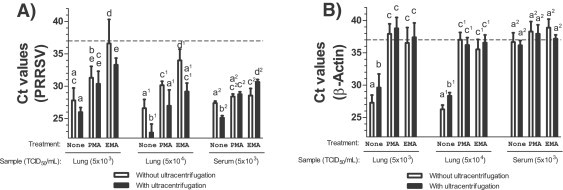
PRRSV and host genomic detection in spiked tissues following EMA or PMA treatment using qPCR. Effect of EMA and PMA treatments, with or without ultracentrifugation, on (A) PRRSV quantification and (B) host genomic DNA (β-Actin) in lung tissue homogenates spiked with PRRSV (5000 TCID50/mL or 50,000 TCID50/mL) or in serum spiked with PRRSV (5000 TCID50/mL). Results are expressed as Ct and were obtained from two to seven independent experiments. The results of each independent experiment (trial) are illustrated in Supplemental Fig. 1. Sample (TCID50/mL) represents the type of tissue spiked with PRRSV. Numbers in brackets represent the PRRSV concentration for each spiked sample expressed in TCID50/mL. Open bars represent results obtained from samples processed without an ultracentrifugation step while filled bars represent results obtained from samples treated with an ultracentrifugation step. A Ct value of 37 (dashed line) represents the limit of detection of each qPCR test. Labeling of two sets of data with different letters indicates that these two sets of data are statistically different (P < 0.05). Sets of data using letters with the same superscript number must be compared only together.
Both EMA and PMA treatments, combined or not with ultracentrifugation, were equally efficient in lowering β-actin amplification in PRRSV spiked lung tissue homogenates (Fig. 1B; P < 0.001). Ultracentrifugation of non-treated spiked lung tissue samples also slightly lowered β-actin amount (Fig. 1B; P < 0.05). In spiked sera samples, β-actin was already at the limit of detection in untreated samples, which indicates that hg/cont is much lower in sera compared to lung tissue homogenates (Fig. 1B).
3.2. DNA array sensitivity with EMA or PMA treated PRRSV spiked tissues
After nucleic acid extraction, random amplification and DNA labeling were done on each spiked samples and the fluorescence of each PRRSV probe was measured following the hybridization of labeled samples on DNA arrays and compared to the negative probe fluorescence intensity (see accession number GSE62910 in the GEO NCBI database for raw data). Surprisingly, probes signals intensity and positivity were varying between experiments. For lung samples spiked with PRRSV (5000 TCID50/mL), a low number of slightly positive probes (probes relative signal intensity <5) were detected in untreated samples (Fig. 2A). Interestingly, multiple probes with relative high fluorescence intensity (>5) were detected following PMA treatment when combined with ultracentrifugation and were associated with a significantly higher number of positive probes compared to untreated samples with ultracentrifugation and the PMA treated samples without ultracentrifugation (Fig. 2A; P < 0.001). In contrast, the fluorescence intensity of detected positive probes of EMA-treated lung homogenates spiked with PRRSV (5000 TCID50/mL) were significantly lower than all other experimental groups, indicating a lower chance to detect positive probes following treatment with EMA (P < 0.05; Fig. 2A).
Fig. 2.
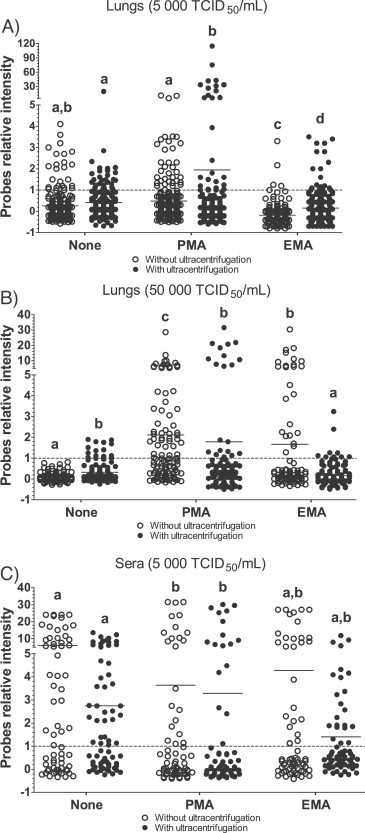
PRRSV detection in spiked samples by DNA array following EMA or PMA treatments. DNA array probes relative intensity from A) lung tissue homogenates spiked with PRRSV (5000 TCID50/mL), B) lung tissue homogenates spiked with PRRSV (50,000 TCID50/mL) and C) serum samples spiked with PRRSV (5000 TCID50/mL). Dots are relative fluorescence intensity mean values of two identical probes gathered from two to seven independent experiments (each experiment consisting of a duplicate of 34 PRRSV specific probes) and was calculated as followed: [(PFL − BFL)/BFL] where PFL represents a PRRSV probe fluorescence intensity and BFL represents the basal fluorescence level (negative control probe fluorescence). The results of each independent experiment (trial) are illustrated in Supplemental Fig. 2. The line represents the fluorescence mean value of all probes. Open dot circles represent results obtained from samples processed without an ultracentrifugation step while filled dot circles represent results obtained from samples treated with an ultracentrifugation step. Labeling of two sets of data with different letters indicates that these two sets of data are statistically different (P < 0.05). A probe relative intensity of 1 (dashed line) represents the lowest limit of DNA array positive results.
In lung samples spiked with a higher concentration of PRRSV (50,000 TCID50/mL), a small number of low intensity positive probes (fluorescence relative signal <5) or no positive probes (fluorescence relative signal <1) were detected in untreated samples (Fig. 2B). A significant increase in the number of high intensity positive probes (fluorescence relative signal >5) were found in samples treated with PMA, with or without ultracentrifugation, when compared to untreated samples without ultracentrifugation (Fig. 2B; P < 0.001). Surprisingly, only a few positive probes were found in samples treated with ultracentrifugation and EMA, similar to untreated samples. However, the number and intensity of probes in samples treated with EMA without ultracentrifugation were significantly higher compared to untreated samples without ultracentrifugation (P < 0.001).
In spiked sera samples, it was interesting to observe that all experimental groups had large numbers of probes with high intensity fluorescence signal against PRRSV, including untreated samples (Fig. 2C), indicating overall that all treatments (ultracentrifugation versus PMA/EMA) did not improve DNA array sensitivity when used with sera samples. Interestingly, PMA treatment compared to untreated sera reduced the PVDA sensitivity even if a high number of high intensity positive probes were observed in PMA treated sera (Fig. 2C; P < 0.05).
3.3. HTS efficiency with EMA or PMA treated PRRSV spiked tissues.
DNA sequences obtained from sequencing experiments were compared to the swine mitochondrial (GenBank accession number NC_000845) and chromosomal genomic DNA sequences (GenBank accession numbers NC_010443 to NC_010462) as well as the full genetic sequence of the PRRSV strain used in this study. All HTS reads associated with PRRSV sequence were also considered to evaluate the virus coverage obtained following each treatment combination.
In two out of three experiments, PMA and EMA treated PRRSV spiked lung tissues (5000 TCID50/mL) had a significant increase in the number of PRRSV reads when compared to untreated samples, as revealed by the chi-square analysis (Fig. 3A; P < 0.001). A strong correlation was found between PRRSV percent of coverage and the higher number of reads following treatment (r = 0.81, P < 0.001), indicating that the increase in PRRSV coverage is related to the augmentation in PRRSV reads. However, when all three HTS experiments were combined together in the statistical analyses, no significant differences were obtained between the amount of PRRSV specific reads against the total amount of reads (Fig. 3A; P = 0.4358) or the PRRSV percentage of coverage (Fig. 3B; P = 0.5585). Noteworthy, the experiment showing no improvement in the amount of PRRSV reads was associated with a higher level of hg/cont as revealed in Fig. 3C. In fact, a strong negative correlation was found between the ratio of PRRSV reads (number of PRRSV reads/total number of reads) and the ratio of host genomic reads (number of genomic reads/total number of reads) (r = −0.66, P < 0.005) in these three experiments. The reason why this experiment indicates a low effect of treatments on the percentage of host genomic DNA and possibly its transcripts is currently unknown but might be caused by a higher rate of host genome released during the tissue preparation.
Fig. 3.
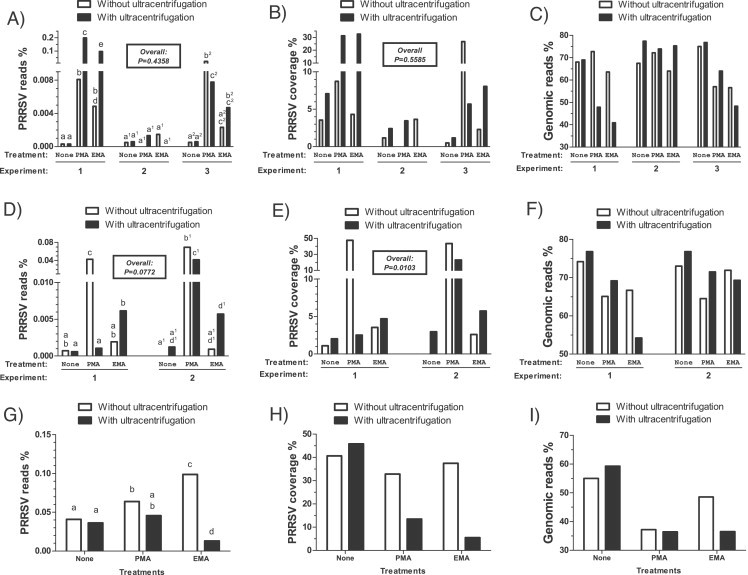
PRRSV and host genomic detection efficiency in spiked tissue samples following EMA or PMA treatment by high-throughput sequencing. HTS results gathered from (A–C) lung tissue homogenates spiked with PRRSV (5000 TCID50/mL); from (D–F) lung tissue homogenates spiked with PRRSV (50,000 TCID50/mL); and from (G–I) serum samples spiked with PRRSV (5000 TCID50/mL). The amounts of PRRSV specific reads compared to the total number of reads gathered from each HTS run (expressed as %) are reported in panels (A), (D) and (G) while the percentage coverage of PRRSV recovered from the total number of PRRSV specific reads are reported in panel (B), (E) and (H). The host genomic specific reads compared to the total number of reads gathered from each HTS run (expressed as %) are reported in panels (C), (F) and (I). Open bars represent results obtained from samples processed without an ultracentrifugation step while filled bars represent results obtained from samples treated with an ultracentrifugation step. The results from each experiment are expressed separately in each graphic. Labeling of two sets of data with different letters indicates that these two sets of data are statistically different (P < 0.05). Sets of data using letters with the same superscript number must be compared only together. The overall P-values shown in the boxes represent the statistical analysis of treatments effects taking into account all the experimental groups.
In tissue homogenates spiked with higher amounts of PRRSV (50,000 TCID50/mL), although not statistically significant, statistical analyses revealed a tendency for an increase of the ratio of PRRSV reads when the two HTS experiments were taken into account (Fig. 3D; P = 0.0772). Moreover, a significant variation was found in PRRSV coverage in those samples (Fig. 3E; P = 0.0103) and a significant increase in PRRSV coverage in PMA treated samples without ultracentrifugation was observed (Fig. 3F; P = 0.0082). Ultracentrifugation of samples containing the higher amount of PRRSV did not improve either the PRRSV number of reads or its coverage (Fig. 3D and E).
In PRRSV spiked sera, there was an important increase in the ratio of PRRSV reads and coverage in untreated samples compared to results obtained with lung homogenates (Fig. 3). This was in accordance with the lower amount of host genomic DNA detected in sera of untreated samples (Figs. 1B, and 3I and 5F). According to the chi-square analysis, a slight increase in PRRSV ratio of reads was observed following PMA and EMA treatments. However, ultracentrifugation lowered the ratio of PRRSV reads when combined with EMA treatment (Fig. 3G) and was associated with a lower PRRSV coverage (Fig. 3H).
Fig. 5.
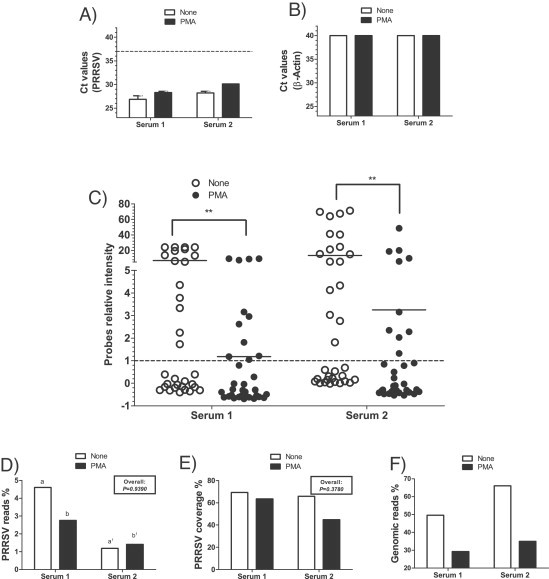
Detection of PRRSV and of host genomic DNA in clinical sera samples by RT-qPCR, DNA array and high-throughput sequencing following PMA treatment. Results obtained from two clinical serum samples by (A) PRRSV RT-qPCR; (B) swine host genomic quantification (β-Actin) qPCR; (C) DNA array; (D) HTS PRRSV specific reads compared to the total amount of reads (expressed as %); (E) HTS PRRSV percentage coverage recovered from the total number of PRRSV specific reads; and (F) host genomic specific reads compared to the total amount of reads (expressed as %). Open bars or open circles represent results obtained from untreated samples while filled bars or filled circles represent results obtained from samples treated with PMA. A Ct value of 37 (dashed line) represents the limit of detection of each qPCR test. Dots are relative fluorescence intensity mean values of two identical probes gathered from two independent experiments (each experiment consisting of a duplicate of 34 PRRSV specific probes) and was calculated as followed: [(PFL − BFL)/BFL] where PFL represents a PRRSV probe fluorescence intensity and BFL represents the basal fluorescence level (negative control probe fluorescence). The line represents the fluorescence mean value of all probes. A probe relative intensity of 1 (dashed line) represents the lowest limit of DNA array positive results. The results from each clinical case are expressed separately in each panel. The overall P values shown in boxes represent the statistical analysis of treatments effects taking into account all the experimental groups. When two sets of data or group of data are labeled with an asterisk, it indicates that these two sets of data and group are statistically different (***P < 0.001; **P < 0.01). Labeling of two sets of data with different letters indicates that these two sets of data are statistically different (P < 0.05). Only sets of data using letters with the same superscript number should be compared together.
3.4. High-throughput sequencing efficiency with EMA or PMA treated PRRSV positive clinical samples.
In order to confirm the effectiveness of EMA and PMA treatments to increase the sensitivity of PVDA and HTS to detect viruses, both techniques were evaluated with different PRRSV positive clinical samples gathered from PRRSV experimentally infected piglets and clinical samples submitted to the MDL. All clinical samples were ultracentrifuged and treated with PMA or untreated. PMA was selected because overall, our previous findings indicated that PMA treatment was more efficient than EMA with regards to PRRSV RT-qPCR, PVDA and HTS detection.
PRRSV and the amplifiable host genomic DNA copy numbers were significantly reduced in PMA treated lung tissues (Fig. 4 A and B; P < 0.001). The important PRRSV decrease in PMA treated samples was probably caused by the presence of damaged virions and/or non-encapsidated viral genomes within clinical samples. PRRSV was undetectable in all untreated samples by DNA array (Fig. 4C). Interestingly, several PRRSV probes were found positive for lung #1 in PMA treated samples (Fig. 4C; P < 0.001). However, PRRSV was not detected by DNA array in lung #3 and few PRRSV probes were found positive with lung #2 following PMA treatment (Fig. 4C). For HTS results, when the three clinical cases are taken into account together, results revealed that PMA tends to increase the number of reads obtained from lung tissues compared to untreated samples (Fig. 4D; P = 0.0529). Nonetheless, PMA treatment significantly improved the percentage of PRRSV HTS reads in two out of three clinical cases tested (Fig. 4D, P < 0.001). Although these experiments showed an important increase in PRRSV genomic coverage (3–6-fold), no overall significant differences were detected (Fig. 4E; P = 0.1117). No increase in PRRSV coverage was detected for lung #1 (Fig. 4E). Although lung #3 showed no increase in the percentage of PRRSV reads, an important increase (6-fold) of PRRSV coverage was detected. However, a higher host genomic DNA content is observed in this sample as suggested by the lower Ct values detected in treated and untreated sample (Fig. 4B) and the higher genomic percentage of reads (Fig. 4F), compared to lungs #1 and #2.
Fig. 4.
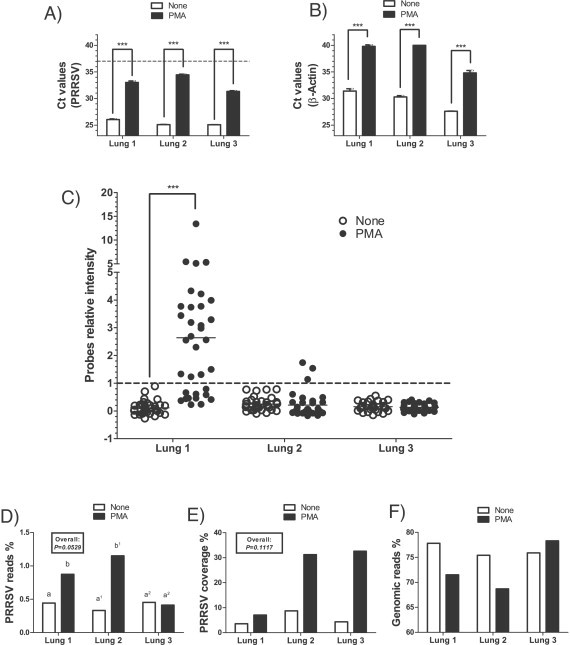
Detection of PRRSV and of host genomic DNA in clinical lung samples by RT-qPCR, DNA array and high-throughput sequencing following PMA treatment. Results obtained from three clinical lung samples by (A) PRRSV RT-qPCR; (B) swine host genomic quantification (β-Actin) qPCR; (C) DNA array; D) HTS PRRSV specific reads compared to the total amount of reads (expressed as %); (E) HTS PRRSV percentage coverage recovered from the total number of PRRSV specific reads; and (F) host genomic specific reads compared to the total amount of reads (expressed as %). Open bars or open circles represent results obtained from untreated samples while filled bars or filled circles represent results obtained from samples treated with PMA. A Ct value of 37 (dashed line) represents the limit of detection of each qPCR test. Dots are relative fluorescence intensity mean values of two identical probes gathered from three independent experiments (each experiment consisting of a duplicate of 34 PRRSV specific probes) and was calculated as followed: [(PFL − BFL)/BFL] where PFL represents a PRRSV probe fluorescence intensity and BFL represents the basal fluorescence level (negative control probe fluorescence). The line represents the fluorescence mean value of all probes. A probe relative intensity of 1 (dashed line) represents the lowest limit of DNA array positive results. Results obtained from each clinical case are expressed separately in each panel. The overall P values shown in boxes represents the statistical analysis of treatments effects taking into account all the experimental groups. When two sets of data or group of data are labeled with an asterisk, it indicates that these two sets of data and group are statistically different (***P < 0.001). Labeling of two sets of data with different letters indicates that these two sets of data are statistically different (P < 0.05). Only sets of data using letters with the same superscript number should be compared together.
PMA treatment of clinical sera had no significant effect on amplifiable PRRSV (Fig. 5A) or host genomeic DNA levels (Fig. 5B, Ct values >40). In agreement with spiked sera samples, PRRSV was strongly detected with all experimental conditions tested from both clinical sera samples by PVDA and HTS (Fig. 5C, D and E). However, the number of PRRSV positive probes was significantly lower in PMA treated samples for both sera samples (Fig. 5C; P < 0.01). HTS results revealed that the PMA treatment also lowers the ratio of PRRSV reads in one out of two cases (Fig. 5D; P < 0.001). PRRSV coverage was high in all cases (over 60%), except for serum 2 treated with PMA where the PRRSV percent of coverage was slightly higher than 40%.
4. Discussion and conclusion
Although generally preferable, direct virus isolation and identification from clinical samples are not always possible and can sometimes lead to mis-identification of the etiological agent (29). Consequently direct identification within clinical samples, using newly available genomic technologies, is desirable. During the past decade, PVDA and HTS technologies have led to the discovery of new viruses (Mishra et al., 2014, Yozwiak et al., 2012, Nakamura et al., 2009, Blomstrom et al., 2009, Mihindukulasuriya et al., 2008, Victoria et al., 2008, Hang et al., 2012, Rosseel et al., 2011). However, the sensitivity of these technologies can suffer from excessive amounts of contaminating host DNA and RNA. Any methodology to decrease the masking effect that these contaminants have on viral detection is important.
A new approach to lower the host genomic DNA within clinical samples is presented in this report. Our results support an increase in sensitivity of PVDA and HTS in lung tissue samples treated with PMA and, to a lesser extent, with EMA. This treatment can be done as a standalone treatment or in combination with other treatments like ultracentrifugation. Surprisingly, adding an ultracentrifugation step before the EMA or PMA treatment sometimes lowers the sensitivity of both PVDA and HTS. This is probably caused by a physical degradation of viral particles during ultracentrifugation, making these particles more sensitive to EMA or PMA treatment.
Interestingly, our results indicate a higher sensitivity of PVDA and HTS with sera samples, compared to tissue homogenates. This may be explained by two phenomena. Firstly, there is a lower concentration of hg/cont in sera samples compared to tissue homogenates, as demonstrated by β-actin DNA measurements for both untreated sera samples and tissue homogenates. Secondly, the random amplification method used to increase the amount of DNA for both HTS and PVDA techniques amplify all nucleic DNA and RNA sequences found in a sample, including hg/cont. Thus, the sensitivity of both PVDA and HTS will be highly affected by the ratio of the viral genetic material of interest over the total amount of hg/cont found in samples. This indicates that the initial amount of hg/cont in clinical samples has a deep impact on PVDA and HTS sensitivity for the identification of viruses. The variation in hg/cont content within nucleic acid extraction from clinical samples may explain the high standard deviation obtained within the results of PVDA and HTS with lung tissue homogenates spiked with the lowest PRRSV concentration. This variation can be explained by multiple factors like the extraction method, the tissue quality and the homogenization process. Also, the virus integrity in clinical samples will have to be taken into account to avoid virus particle degradation during the tissue manipulation, storage and homogenization, which can sensitize viral particles to EMA or PMA treatment. This is especially true for clinical samples where the virus type, the integrity of tissue (dead animal, sample conservation at room temperature, etc.), the amount of hg/cont or the presence of PCR inhibitors will have an critical impact on PVDA and HTS sensitivity. In addition, the efficiency of the random amplification process could be affected by the viral genome itself (its sequence and its secondary structures), making some viral genomes less compatible with the use of random amplification prior to HTS and PVDA (Codony et al., 2012b). It is important to note that our work utilized a unique virus, and does not take into account the efficiency of EMA and PMA treatments on other types of viruses. However, the outer structure of the virions of several viruses can protect the viral genetic material from PMA treatment (Elizaquivel et al., 2014), suggesting that this treatment should reduce the influence of hg/cont when used for clinical samples with other virus types.
Multiple reports have revealed differential PVDA and HTS sensitivities for viral detection and identification in clinical samples following the use of different treatment combinations, including ultracentrifugation and nuclease treatment (Mishra et al., 2014, Djikeng et al., 2008). The lowest viral load in spiked serum samples was shown by Nicholson and collaborators (2011) where they determined the limit of PVDA detection for PRRSV in spiked serum samples, not subjected to nuclease treatment, to be 10,000 TCID50/mL (Nicholson et al., 2011). Similar results were obtained in the present study but with lower virus concentration (5000 TCID50/mL).
Previous studies have reported the ratio of viral sequencing reads to total number of reads ranged between 0.00019% and 2.8% from non-nuclease treated serum and nasopharyngeal aspirates (Yozwiak et al., 2012, Nakamura et al., 2009). However, Mishra and collaborators (2014) showed a ratio (viral reads/total number of reads) of 0.00012% by HTS from muscle tissue samples (Mishra et al., 2014). In this study, rRNA was depleted and a DNase treatment was done following the RNA extraction, in order to lower hg/cont in that clinical sample (Mishra et al., 2014). These results are in accordance with our work since the percentage of PRRSV specific reads ranged between 0% and 0.00059% in spiked untreated samples and between 0% and 0.19785% in EMA and PMA-treated spiked samples. In contrast, Djikeng and collaborators (2008) have reported much higher ratios of host genomic content to isolated virus ranging between 3 and 40% (Djikeng et al., 2008) while Nakamura and collaborators (2009) have reported higher genomic content ranging between 90.0% and 94.6% (Nakamura et al., 2009). The variation of the host genomic DNA content between those studies is probably explained by the difference in purification and treatment methods used for each virus, like the use of gradient density centrifugation to obtained highly purified viruses with lower hg/cont (Djikeng et al., 2008). The ratio of host genomic DNA content reported in our study varied between 67.50% and 77.40% in untreated samples and between 40.91% and 72.76% in EMA and PMA-treated samples. This represents an improvement over the results reported by Nakamura and collaborators (2009) where no purification methods were used, and less than the ratio of hg/cont reported by Djikeng and collaborators (2008). However, in the last case, the viruses were isolated from cell culture and the virions were subsequently purified.
Thus, our results indicate that EMA and PMA treatments improve the sensitivity of PVDA and HTS to detect viruses in clinical samples contaminated with hg/cont. Furthermore, EMA/PMA treatments are faster and easier to perform than a nuclease treatment. Firstly, they require shorter incubation times compared to nuclease treatment which would result in faster processing of clinical samples in a diagnostic laboratory, and should increase the robustness of the method by exerting less stress on temperature sensitive viral particles. Secondly, EMA and PMA are easy to inactivate through light exposure, subsequently leaving EMA/PMA molecules unable to destroy newly exposed viral genomic material following nucleic acid extraction, unlike the case when residual nucleases may still be present after nucleic acid purification steps. This is especially true if RNase or DNase treatment is being used in the presence of RNA or DNA viruses, respectively. PMA/EMA treatments were more effective in tissue samples compared to sera samples which was presumably due to lower hg/cont in the latter. PMA was more effective than EMA in improving HTS and PVDA sensitivity for viral detection in clinical samples. This would be explained by the fact that EMA can leak through phospholipid bilayer membrane (Nocker et al., 2006). Moreover, ultracentrifugation appears to lower the sensitivity of HTS and PVDA following some treatments, possibly because of direct physical degradation of the viral particle. In conclusion, pre-treatment of clinical samples with EMA, and especially PMA, represents an interesting novel approach that improves PVDA and HTS sensitivity for the identification of viruses from clinical samples.
Acknowledgements
This manuscript was financially supported by the Canadian Swine Health Board (CSHB). C.A. Gagnon was financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC). The authors wish to thank Guy Beauchamp and Philippe Garneau for scientific support, and Miria Elias for technical support. The authors have no conflict of interest.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jviromet.2015.06.014.
Appendix A. Supplementary data
The following are the supplementary data to this article:
Colour coded individual trial results of PRRSV and host genome detection in spiked tissues following EMA or PMA treatment using qPCR. Effect of EMA and PMA treatments, with or without ultracentrifugation (UC), on PRRSV quantification in lung tissue homogenates spiked with (A) PRRSV (5000 TCID50/mL) or with (C) PRRSV (50,000 TCID50/mL) and (E) serum spiked with PRRSV (5000 TCID50/mL); and on host genomic DNA quantification (β-actin) in lung tissue homogenates spiked with (B) PRRSV (5000 TCID50/mL) or with (D) PRRSV (50,000 TCID50/mL) and (F) serum spiked with PRRSV (5000 TCID50/mL). Each trial identification number represents a unique experiment. All trials are the same than those illustrated in Supplemental Fig. 2. Results are expressed as Ct. A Ct value of 37 (dashed line) represents the limit of detection of each the PRRSV qPCR assay. See Fig. 1 for the combined statistical analyses f all trials. N.D., not determined.
Colour coded individual trial results of PRRSV detection in spiked samples by DNA array following EMA or PMA treatments. DNA array probes relative intensity from (A) lung tissue homogenates spiked with PRRSV (5000 TCID50/mL), (B) lung tissue homogenates spiked with PRRSV (50,000 TCID50/mL) and (C) serum samples spiked with PRRSV (5000 TCID50/mL). Dots are relative fluorescence intensity mean values of two identical probes from each trials (each experiment consisting of a duplicate of 34 PRRSV specific probes) and was calculated as followed: [(PFL − BFL)/BFL] where PFL represents a PRRSV probe fluorescence intensity and BFL represents the basal fluorescence level (negative control probe fluorescence). Each trial identification number represents a unique experiment. All trials are the same than those illustrated in Supplemental Fig. 1. A probe relative intensity of 1 (dashed line) represents the lowest limit of DNA array positive results. See Fig. 2 for combined statistical analyses of all trials.
References
- Barzon L., Lavezzo E., Costanzi G., Franchin E. Next-generation sequencing technologies in diagnostic virology. J. Clin. Virol. 2013;58:346–350. doi: 10.1016/j.jcv.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Bellehumeur C., Boyle B., Mandeville I., Gagnon C.A. High-throughput sequencing revealed the presence of an unforeseen parvovirus species in Canadian swine: the porcine partetravirus. Can. Vet. J. 2013;54:787–789. [PMC free article] [PubMed] [Google Scholar]
- Beura L.K., Sarkar S.N., Kwon B., Subramaniam S. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J. Virol. 2010;84:1574–1584. doi: 10.1128/JVI.01326-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomstrom A.L., Belak S., Fossum C., McKillen J. Detection of a novel porcine boca-like virus in the background of porcine circovirus type 2 induced postweaning multisystemic wasting syndrome. Virus Res. 2009;146:125–129. doi: 10.1016/j.virusres.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Chen E.C., Miller S.A., DeRisi J.L., Chiu C.Y. Using a pan-viral microarray assay (Virochip) to screen clinical samples for viral pathogens. J. Vis. Exp. 2011;50 doi: 10.3791/2536. pii: 2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem A.L., Sims J., Telang S., Eaton J.W., Chesney J. Virus detection and identification using random multiplex (RT)-PCR with 3′-locked random primers. Virol. J. 2007;4:65. doi: 10.1186/1743-422X-4-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codony F., Perez L.M., Adrados B., Agusti G. Amoeba-related health risk in drinking water systems: could monitoring of amoebae be a complementary approach to current quality control strategies? Future Microbiol. 2012;7:25–31. doi: 10.2217/fmb.11.139. [DOI] [PubMed] [Google Scholar]
- Codony F., Fittipaldi M., Lopez E., Morato J., Agusti G. Well water as a possible source of Waddlia chondrophila infections. Microbes Environ. 2012;27:529–532. doi: 10.1264/jsme2.ME12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwart E. Animal virus discovery: improving animal health, understanding zoonoses, and opportunities for vaccine development. Curr. Opin. Virol. 2012;2:344–352. doi: 10.1016/j.coviro.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A., Halpin R., Kuzmickas R., Depasse J. Viral genome sequencing by random priming methods. BMC Genomics. 2008;9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elizaquivel P., Aznar R., Sanchez G. Recent developments in the use of viability dyes and quantitative PCR in the food microbiology field. J. Appl. Microbiol. 2014;116:1–13. doi: 10.1111/jam.12365. [DOI] [PubMed] [Google Scholar]
- Firth C., Lipkin W.I. The genomics of emerging pathogens. Annu. Rev. Genomics Hum. Genet. 2013;14:281–300. doi: 10.1146/annurev-genom-091212-153446. [DOI] [PubMed] [Google Scholar]
- Gagnon C.A., Lachapelle G., Langelier Y., Massie B., Dea S. Adenoviral-expressed GP5 of porcine respiratory and reproductive syndrome virus differs in its cellular maturation from the authentic viral protein but maintains known biological functions. Arch. Virol. 2003;148:951–972. doi: 10.1007/s00705-002-0943-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon C.A., del Castillo J.R., Music N., Fontaine G. Development and use of a multiplex real-time quantitative polymerase chain reaction assay for detection and differentiation of Porcine circovirus-2 genotypes 2a and 2b in an epidemiological survey. J. Vet. Diagn. Invest. 2008;20:545–558. doi: 10.1177/104063870802000503. [DOI] [PubMed] [Google Scholar]
- Gatteschi D., Fittipaldi M., Sangregorio C., Sorace L. Exploring the no-man's land between molecular nanomagnets and magnetic nanoparticles. Angew. Chem. Int. Ed. 2012;51:4792–4800. doi: 10.1002/anie.201105428. [DOI] [PubMed] [Google Scholar]
- Hall R.J., Wang J., Todd A.K., Bissielo A.B. Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. J. Virol. Methods. 2014;195:194–204. doi: 10.1016/j.jviromet.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang J., Forshey B.M., Kochel T.J., Li T. Random amplification and pyrosequencing for identification of novel viral genome sequences. J. Biomol. Tech. 2012;23:4–10. doi: 10.7171/jbt.12-2301-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang X., Qin C., Li Y., Liu H. Improvement of the specificity of a pan-viral microarray by using genus-specific oligonucleotides and reduction of interference by host genomes. J. Med. Virol. 2011;83:1624–1630. doi: 10.1002/jmv.22157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin W.I., Firth C. Viral surveillance and discovery. Curr. Opin. Virol. 2013;3:199–204. doi: 10.1016/j.coviro.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massart S., Olmos A., Jijakli H., Candresse T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014;188C:90–96. doi: 10.1016/j.virusres.2014.03.029. [DOI] [PubMed] [Google Scholar]
- Mihindukulasuriya K.A., Wu G., St Leger J., Nordhausen R.W., Wang D. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. J. Virol. 2008;82:5084–5088. doi: 10.1128/JVI.02722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N., Pereira M., Rhodes R.H., An P. Identification of a novel polyomavirus in a pancreatic transplant recipient with retinal blindness and vasculitic myopathy. J. Infect. Dis. 2014;210:1595–1599. doi: 10.1093/infdis/jiu250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S., Yang C.S., Sakon N., Ueda M. Direct metagenomic detection of viral pathogens in nasal and fecal specimens using an unbiased high-throughput sequencing approach. PLoS ONE. 2009;4:e4219. doi: 10.1371/journal.pone.0004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson T.L., Kukielka D., Vincent A.L., Brockmeier S.L. Utility of a panviral microarray for detection of swine respiratory viruses in clinical samples. J. Clin. Microbiol. 2011;49:1542–1548. doi: 10.1128/JCM.01876-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocker A., Cheung C.Y., Camper A.K. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J. Microbiol. Methods. 2006;67:310–320. doi: 10.1016/j.mimet.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Nocker A., Sossa-Fernandez P., Burr M.D., Camper A.K. Use of propidium monoazide for live/dead distinction in microbial ecology. Appl. Environ. Microbiol. 2007;73:5111–5117. doi: 10.1128/AEM.02987-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocker A., Mazza A., Masson L., Camper A.K., Brousseau R. Selective detection of live bacteria combining propidium monoazide sample treatment with microarray technology. J. Microbiol. Methods. 2009;76:253–261. doi: 10.1016/j.mimet.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Quinones-Mateu M.E., Avila S., Reyes-Teran G., Martinez M.A. Deep sequencing: becoming a critical tool in clinical virology. J. Clin. Virol. 2014;61:9–19. doi: 10.1016/j.jcv.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosseel T., Lambrecht B., Vandenbussche F., van den Berg T., Van Borm S. Identification and complete genome sequencing of paramyxoviruses in mallard ducks (Anas platyrhynchos) using random access amplification and next generation sequencing technologies. Virol. J. 2011;8:463. doi: 10.1186/1743-422X-8-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoria J.G., Kapoor A., Dupuis K., Schnurr D.P., Delwart E.L. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathog. 2008;4:e1000163. doi: 10.1371/journal.ppat.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong N.M., Villemur R., Payment P., Brousseau R. Fecal source tracking in water using a mitochondrial DNA microarray. Water Res. 2013;47:16–30. doi: 10.1016/j.watres.2012.09.011. [DOI] [PubMed] [Google Scholar]
- Wang D., Coscoy L., Zylberberg M., Avila P.C. Microarray-based detection and genotyping of viral pathogens. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15687–15692. doi: 10.1073/pnas.242579699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Urisman A., Liu Y.T., Springer M. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 2003;1:E2. doi: 10.1371/journal.pbio.0000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Yang F., Ren L., Xiong Z. Unbiased parallel detection of viral pathogens in clinical samples by use of a metagenomic approach. J. Clin. Microbiol. 2011;49:3463–3469. doi: 10.1128/JCM.00273-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yozwiak N.L., Skewes-Cox P., Stenglein M.D., Balmaseda A. Virus identification in unknown tropical febrile illness cases using deep sequencing. PLoS Negl. Trop. Dis. 2012;6:e1485. doi: 10.1371/journal.pntd.0001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Colour coded individual trial results of PRRSV and host genome detection in spiked tissues following EMA or PMA treatment using qPCR. Effect of EMA and PMA treatments, with or without ultracentrifugation (UC), on PRRSV quantification in lung tissue homogenates spiked with (A) PRRSV (5000 TCID50/mL) or with (C) PRRSV (50,000 TCID50/mL) and (E) serum spiked with PRRSV (5000 TCID50/mL); and on host genomic DNA quantification (β-actin) in lung tissue homogenates spiked with (B) PRRSV (5000 TCID50/mL) or with (D) PRRSV (50,000 TCID50/mL) and (F) serum spiked with PRRSV (5000 TCID50/mL). Each trial identification number represents a unique experiment. All trials are the same than those illustrated in Supplemental Fig. 2. Results are expressed as Ct. A Ct value of 37 (dashed line) represents the limit of detection of each the PRRSV qPCR assay. See Fig. 1 for the combined statistical analyses f all trials. N.D., not determined.
Colour coded individual trial results of PRRSV detection in spiked samples by DNA array following EMA or PMA treatments. DNA array probes relative intensity from (A) lung tissue homogenates spiked with PRRSV (5000 TCID50/mL), (B) lung tissue homogenates spiked with PRRSV (50,000 TCID50/mL) and (C) serum samples spiked with PRRSV (5000 TCID50/mL). Dots are relative fluorescence intensity mean values of two identical probes from each trials (each experiment consisting of a duplicate of 34 PRRSV specific probes) and was calculated as followed: [(PFL − BFL)/BFL] where PFL represents a PRRSV probe fluorescence intensity and BFL represents the basal fluorescence level (negative control probe fluorescence). Each trial identification number represents a unique experiment. All trials are the same than those illustrated in Supplemental Fig. 1. A probe relative intensity of 1 (dashed line) represents the lowest limit of DNA array positive results. See Fig. 2 for combined statistical analyses of all trials.