

Abstract
A reverse transcription nested PCR (RT-PCR) sequencing methodology was developed and used to generate sequence data from the spike genes of three geographically and chronologically distinct human coronaviruses 229E. These three coronaviruses were isolated originally from the USA in the 1960s (human coronavirus 229E strain ATCC VR-74), the UK in the 1990s (human coronavirus 229E LRI 281) and Ghana (human coronavirus 229E A162). Upon translation and alignment with the published spike protein sequence of human coronavirus 229E ‘LP’ (isolated in the UK in the 1970s), it was found that variation within the translated protein sequences was rather limited. In particular, minimal variation was observed between the translated spike protein sequence of human coronaviruses 229E LP and ATCC VR-74 (1/1012 amino acid differences), whilst most variation was observed between the translated spike protein sequence of human coronaviruses 229E LP and A162 (47/1012 amino acid changes). Further, the translated spike protein sequence of human coronavirus 229E A162 showed three clusters of amino acid changes, situated within the 5′ half of the translated spike protein sequence.
Keywords: PCR, Human coronaviruses 229E, Spike genes
1. Introduction
Coronaviruses were first described as aetiological agents of human disease in the mid-1960s when isolated from natural common colds (Tyrrell and Bynoe, 1965, Hamre and Procknow, 1966). They derive their name from their characteristic ‘crown-like’ appearance in electron micrographs imbued by a fringe of club shaped spike (or peplomer) proteins inserted into the viral envelope. Virions are lipid enveloped and are approximately 80–120 nm in diameter; there is a single stranded genome of positive sense RNA approximately 30 kb in length. They also have a characteristic replication strategy, in that the positive sense genomic RNA is first transcribed into negative sense intermediate RNAs (by a virus encoded transcriptase) from which a nested 3′ co-terminus set of five to eight subgenomic mRNAs (six subgenomic RNAs for human coronaviruses) are transcribed. The subgenomic mRNAs have identical 3′ ends but extend for different lengths in the 5′ direction (Lai, 1990).
Antigenically, coronaviruses may be divided into two major serogroups and one minor serogroup. The two major antigenic serogroups are designated coronavirus serogroup 1 (including human coronavirus 229E) and coronavirus serogroup 2 (which includes human coronavirus OC43). The minor antigenic serogroup (coronavirus serogroup 3) currently only contains a single member, avian infectious bronchitis virus (Siddell, 1995).
All coronaviruses possess three major proteins: the nucleocapsid (N), membrane (M) and spike (S); a minor protein (sM); with some coronaviruses also possessing a haemagglutinin-esterase (HE) glycoprotein (Siddell, 1995). The spike glycoprotein is of particular importance in the infectious process because: (a) it is the site for the virus anti-receptor (Collins et al., 1982); (b) it has fusion activity (De Groot et al., 1989); and (c) it contains sites against which major neutralising antibodies are directed (Jimenez et al., 1986). The composition of the spike glycoprotein is therefore very relevant to the ability of the virus to evade the hosts' immune system (La Monica et al., 1991).
Human coronaviruses have a world-wide distribution (Hruskova et al., 1990, Matsumoto and Kawana, 1992) and infect all age groups (Gwaltney, 1980). There is evidence to suggest a role for human coronaviruses in the aetiology of enteric (Payne et al., 1986), neurological (Stewart et al., 1992) but, primarily, respiratory disease (Myint, 1995). Indeed, human coronaviruses are though to be responsible for approximately 20% of common colds (McIntosh et al., 1970), as well as lower respiratory tract infections in infants (McIntosh et al., 1974) and the exacerbation of asthma (Johnston et al., 1995). Prospective studies have indicated that such human coronavirus induced respiratory infections tend to occur in cycles, with a periodicity of approximately 3 years (Monto and Lim, 1974).
Respiratory re-infections with human coronaviruses are common (Monto and Lim, 1974). The mechanism facilitating re-infection is, however, unclear. Macnaughton (1982) indicated that coronavirus antibodies raised against human coronavirus 229E strains (serogroup 1) may not be protective against human coronavirus OC43 strains (serogroup 2) and vice versa. The existence of pre-existing coronavirus antibody directed to the same serotype is not protective against further coronavirus infection (Callow, 1985). Natural antibodies against a particular serotype of coronavirus were protective for approximately four months only, after which time re-infection by the same serotype of human coronavirus could occur.
In this study preliminary evidence was obtained that significant variation in the S protein of the virus that is unlikely to explain the basis of re-infections. A reverse transcription PCR sequencing strategy was developed which allows sequence data from the spike genes of several geographically and chronologically distinct human coronaviruses 229E to be collated and compared. By predicting the corresponding amino acid sequences of these spike genes, it has been possible to make a preliminary assessment of the degree of variation within the corresponding spike protein sequences of these isolates and those published previously.
2. Materials and methods
2.1. Viruses and cells
Human coronavirus 229E strain VR-74 was purchased from the American Type Culture Collection, MD, USA. Strain LRI 281 was isolated from nasal washings obtained in 1990 from a child with asthma at the Leicester Royal Infirmary, Leicester, UK. Strain A162 was isolated from nasal secretions obtained in 1995 from an adult presenting with the common cold at Kumasi, Ghana, West Africa. All specimens were transported to the laboratory on dry ice, aliquoted into 100 μl quantities and stored at −70°C until required.
2.2. Primers
Spike gene reverse transcription and PCR primers were designed from consensus regions of the spike genes of several coronaviruses 229E utilising published data (Wesseling et al., 1994). Spike gene sequencing primers were designed by a ‘primer walking’ method utilising human coronavirus 229E strain ATCC VR-74 as template. All primers were prepared using β-cyanoethyl phosphoramidite (CEP) chemistry at the Protein and Nucleic Acid Laboratory at the University of Leicester, Leicester, UK.
2.3. Extraction of human coronavirus 229E RNA
The extraction of human coronavirus 229E RNA was based on guanidium isothiocyanate methodology (Chomczynski and Sacchi, 1987) using RNAzol B (Biogenesis Ltd, Poole, UK). Once extracted, the total RNA pellet was allowed to dry for approximately 25 min at room temperature and then resuspended in 30 μl of RNAse free ultra-high quality (UHQ) water containing 20 U/μl of RNAse inhibitor (Promega).
2.4. Reverse transcription of human coronavirus 229E RNA
All reverse transcription reactions were carried out in a final volume of 20 μl. Negative controls comprised RNAse free UHQ water which had undergone RNA extraction.
Initially, for each RNA extraction to be reverse transcribed, a reverse transcription supermix containing 2 μl of 10×MMLV reverse transcription (RT) buffer (Stratagene, Cambridge, UK), 2 μl of a 5 mM mix of deoxynucleotide triphosphates (dNTPs), 0.5 μl of 100 mM dithiothreitol (Sigma, Poole, UK), 1 μl of 10 μg/ml gelatin, 3 μl of UHQ RNAse free water and 1 μl of downstream primer LPS2 (see Table 1 ) at a stock concentration of 25 pmoles was prepared. Of this RT-super mix, 9.5 μl was then transferred to a labelled 0.5 ml sterile RNAse free Eppendorf and overlaid with sterile mineral oil (Sigma, Poole, UK). RNA extract (10 μl) was then added to its respective Eppendorf and the resultant RT/RNA mixes heated to 70°C for 5 min. After this time, the RT/RNA mixes were placed immediately on ice for 5 min and 0.5 μl of MMLV (Stratagene, Cambridge, UK) reverse transcriptase then added to each reaction mix. The reaction mixes were then placed in a pre-heated Trio-block thermocycler (Biometra, Maidstone, UK) at 37°C for 1 h. After 1 h the reverse transcription/RNA mixes were heated to 95°C for 5 min and then cooled to 4°C prior to use in the human coronavirus 229E nested spike gene PCR.
Table 1.
Primers utilised in the human coronavirus 229E spike gene RT-nested PCR and subsequent cycle sequencing reactions
| Primer pair | Fragment generated | Primer sequences |
| (1) Reverse transcription primer | 5′ GCCACAGCAACCAGTAGA 3′ | |
| (LPS2) | ||
| (2) Spike gene nested PCR primers | ||
| (LPS1) | NA | 5′ AATAATTGGTTCCTTCTAAC 3′ |
| (LPS2) | NA | 5′ GCCACAGCAACCAGTAGA 3′ |
| (JH1) | F1 | 5′ TTTGTTGCTTAATTGCTTATGG 3′ |
| (JH2) | F1 | 5′ TTTGCCAAAAGAAAAAGGGC 3′ |
| (JH3) | F2 | 5′ CCTTTTTCTTTTGGCAAAG 3′ |
| (JH4) | F2 | 5′ CCATTATAATATTGAGCAC 3′ |
| (JH5) | F3 | 5′ TGCTCAATATTATAATGG 3′ |
| (JH6) | F3 | 5′ ACAACATAATAGCA 3′ |
| (3)Cycle sequencing primers | ||
| (JH1) | F1 | 5′ TTTGTTGCTTAATTGCTTATGG 3′ |
| (JH2) | F1 | 5′ TTTGCCAAAAGAAAAAGGGC 3′ |
| (JH3) | F2 | 5′ CCTTTTTCTTTTGGCAAAG 3′ |
| (JH4) | F2 | 5′ CCATTATAATATTGAGCAC 3′ |
| (JH5) | F3 | 5′ GTGCTCAATATTATAATGG 3′ |
| (JH6) | F3 | 5′ ACAACAACATAATAGCA 3′ |
| (JH7) | F1 | 5′ TCTGATGTCATACGTTACAACC 3′ |
| (JH8) | F2 | 5′ GTAAGTACTATACTATAGG 3′ |
| (JH9) | F3 | 5′ TCTCATTAGCAATTCAGGC 3′ |
| (JH10) | F1 | 5′ TTCAGGTGATGCTCACAT 3′ |
| (JH11) | F2 | 5′ ACGTACACATCAACTTCAGG 3′ |
| (JH12) | F3 | 5′ GGATGTTGTTCATCAACAAG 3′ |
| (JH13) | F1 | 5′ CACTTTAGGTAATGTAGAAGC 3′ |
| (JH14) | F5 | 5′ CTATAATTGCTGTTCAACCACG 3′ |
| (JH15) | F3 | 5′ TGAGTGTGTCAAATCCCAG 3′ |
| (JH16) | F4 | 5′ TGACCAGTTGTCCTTTGATGTA 3′ |
| (JH17) | F5 | 5′ AGACGCCTTAAGAAATAGCG 3′ |
| (JH18) | F4 | 5′ CGTTTATTGTGTTGTACGTTG 3′ |
| (JH19) | F1 | 5′ ATGTGAGCATCACCTGAA 3′ |
| (JH20) | F1 | 5′ GCTTCTACATTACCTAAAGTG 3′ |
| (JH21) | F1 | 5′ TACATCAAAGGACAACTGGTCA 3′ |
| (JH22) | F1 | 5′ CAACGTACAACACAATAAACG 3′ |
| (JH23) | F4 | 5′ CCTATAGTATAGTACTTAC 3′ |
| (JH24) | F4 | 5′ CCTGAAGTTGATGTGTACGT 3′ |
| (JH25) | F2 | 5′ CGTGGTTGAACAGCAATTATAG 3′ |
| (JH26) | F2 | 5′ CGCTATTTCTTAAGGCGTCT 3′ |
| (JH27) | F5 | 5′ TGCCTGAATTGCTAATGAGA 3′ |
| (JH28) | F5 | 5′ CTTGTTGATGAACAACATCC 3′ |
| (JH29) | F3 | 5′ CTGGGATTTGACACACTCA 3′ |
| (JH30) | F3 | 5′ GAACCACGTATTCCTACCAT 3′ |
| (JH31) | F3 | 5′ TTGACCAGTGAAATTAGCACCC 3′ |
| (JH32) | F3 | 5′ ATGGTAGGAATACGTGGTTC 3′ |
| (JH33) | F3 | 5′ GGGTGCTAATTTCACTGGTCAA 3′ |
2.5. Human coronavirus 229E spike gene nested PCR
All first and second round PCR reactions were carried out in a final volume of 50 μl. All stock primers had a concentration of 25 pmoles.
2.5.1. First round PCR protocol
Initially, a first round PCR supermix was prepared containing 31.6 μl of sterile UHQ water, 5 μl of 10×Thermus icelandicus PCR buffer (Advanced Biotechnologies, Leatherhead, UK), 6 μl of 25 mM magnesium chloride, 0.4 μl of a 5 mM mix of dNTPs, 1 μl of primer LPS1 (see Table 1) and 1 μl of primer LPS2 (see Table 1) per reverse transcribed specimen to be PCR amplified. This PCR supermix (44.8 μl) was then pipetted into a labelled 0.5-ml Eppendorf and overlaid with sterile mineral oil. Reverse transcribed human coronavirus 229E spike gene cDNA (or negative control cDNA) (5 μl) was then added to its respective Eppendorf and the first round PCR reaction mixes transferred to a pre-heated (95°C) Trioblock thermocycler and subjected to a ‘hot start’ and ‘touchdown’ PCR protocol with 0.2 μl ‘Red Hot’ Thermus icelandicus DNA polymerase (Advanced Biotechnologies, Leatherhead, UK). The initial phase consisted of 20 cycles of 92°C for 30 s, thermal ramp to 65°C for 1 min, thermal ramp to 72°C for 4 min then thermal ramp to 92°C. This was followed by ten cycles of 92°C for 30 s, thermal ramp to 55°C for 1 min, thermal ramp to 72°C for 4 min, then a thermal ramp to 92°C. PCR products were then cooled to 4°C and stored until second round reaction mixes had been prepared.
2.5.2. Second round PCR protocol
In the second round of the human coronavirus 229E spike gene PCR, 5 ‘second round reaction mixes’ were prepared for each of the first round PCR amplification products to be re-amplified. Initially, a second round supermix containing 33 μl of sterile UHQ water, 5 μl of 10×Thermus icelandicus PCR buffer (Advanced Biotechnologies), 6μl of 25 mM magnesium chloride and 0.4 μl of a 5 mM mix of dNTPs was prepared for each first round PCR amplification undertaken. This second round supermix (44.4 μl) was then aliquoted into labelled sterile 0.5-ml Eppendorfs. Next, five separate ‘primer pair mixes’ were prepared containing either (i) 0.2 μl of primer JH1 and 0.2 μl of primer JH2; (ii) 0.2 μl of primer JH3 and 0.2 μl of primer JH4; (iii) 0.2 μl of primer JH5 and 0.2 μl of primer JH6; (iv) 0.2 μl of primer JH16 and 0.2 μl of primer JH24; and (v) 0.2 μl of primer JH14 and 0.2 μl of primer JH28 (see Table 1) per first round PCR amplification undertaken. Each of these primer pair mixes (0.4 μl) was then added to their respective second round supermix aliquot and the resulting ‘complete mixes’ overlaid with sterile mineral oil. A 1:10 (v/v) dilution of the first round amplification products was then prepared in sterile UHQ water and 5 μl of the resultant PCR product dilution added to each of its five respective complete second round reaction mixes. Round 2 PCR reaction mixes were then transferred to a pre-heated (95°C) Trioblock thermocycler and subjected to a ‘hot start’ and ‘touchdown’ PCR protocol as already described for the first round. After completion of this second round PCR cycling regime, PCR products were cooled to 4°C and amplified PCR products observed by gel electrophoresis and ethidium bromide staining.
Fig. 1 indicates schematically the region of the human coronavirus 229E spike gene amplified by this reverse transcription and nested PCR protocol.
Fig. 1.
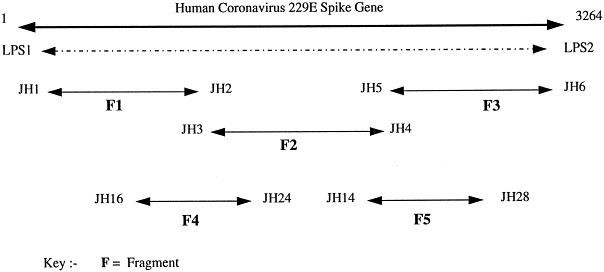
Schematic representation of the amplification products generated using the human coronavirus 229E spike gene nested PCR.
2.6. Cycle sequencing protocol
Human coronavirus 229E spike gene PCR product sequencing was undertaken using the PRISM™ di-deoxy terminator cycle sequencing kit (Applied Biosystems, Foster City, USA). Sequencing of both the sense and antisense strands of the human coronavirus 229E spike gene PCR DNA was undertaken, with some sequencing primers being used more than once to increase the accuracy of generated sequence data at a particular locus.
2.6.1. Cleaning second round PCR products
Prior to sequencing, amplified human coronavirus 229E PCR products were cleaned using ‘Qiaquick’ spin columns as detailed by the manufacturer (Qiagen, Hilden, Germany).
2.6.2. Cycle sequencing
Each individual cycle sequencing mix contained 8 μl of pre-prepared dye-terminator mix, 0.05–0.1 μg cleaned human coronavirus 229E second round PCR product, 3.2 pmol of the relevant sequencing primer and the correct volume of UHQ water to make a total cycle sequencing reaction mix volume of 20 μl. Once prepared, the cycle sequencing mix was overlaid with mineral oil and placed in a pre-heated (96°C) Trioblock thermocycler. A cycle sequencing temperature regime was then undertaken with 25 cycles of 96°C for 10 s, thermal ramp to 50°C in 64 s for 5 s, thermal ramp to 55°C in 30 s for 241 s, then thermal ramp to 94°C in 54 s, followed by cooling to 4°C.
2.6.3. Cleaning cycle sequencing products
The removal of unincorporated nucleotides and enzymes from cycle sequencing products was achieved using a standard phenol/chloroform/isoamyl alcohol extraction and sodium acetate (pH 4.5)/ethanol precipitation methodology.
2.6.4. Assimilation of sequence data and generation of consensus sequences
Cleaned cycle sequencing products were run on an ABI 373 DNA sequencer (Applied Biosystems, Foster City, USA). Resultant chromatograms were examined using Sequence Editor™ software (Applied Biosystems) and a library of text only sequences generated. Each individual human coronavirus 229E spike gene library was then assembled using AutoAssembler™ software (Applied Biosystems) to generate a human coronavirus 229E spike gene consensus sequence for that particular human coronavirus 229E isolate.
3. Results
In total, 33 sequencing primers were designed and used to sequence approximately 90% of the spike genes of human coronavirus 229E isolates ATCC VR-74, LRI 281 and A162 (when compared to the published human coronavirus 229E ‘LP’ spike gene sequence Raabe et al., 1990). Six of these 33 sequencing primers (i.e. JH1, JH2, JH3, JH4, JH5 and JH6) were also used in the initial human coronavirus 229E spike gene PCR protocol, whilst the remaining 27 primers were used as sequencing primers alone. Sequence data was collected from both the sense and antisense strands of human coronavirus 229E spike gene PCR products.
3.1. Human coronavirus 229E strain ATCC VR-74
Forty one individual primer sequences were used to construct a human coronavirus 229E strain ATCC VR-74 consensus sequence (AutoAssembler software) comprising 3122 nucleotides. This 3122-nucleotide consensus sequence was assembled from a total library of 13 021 individual nucleotides. Forty nine of these 13 021 nucleotides were deemed to have been included via mis-incorporation errors by the Thermus icelandicus and MMLV reverse transcriptase enzymes (mis-incorporation errors deemed to have occurred when the nucleotide at a particular locus within the total spike gene assemblage differed from that of the same locus in the spike gene consensus sequence and where this nucleotide difference occurred in either the sense or antisense strand only). Similarly, 24 nucleotides within the 13 021 total nucleotide assemblage were deemed to contain nucleotide additions and 16 loci nucleotide deletions (data not shown). From this data an overall mis-incorporation rate for the human coronavirus 229E reverse transcription nested spike gene PCR of 0.7% was calculated. Fig. 2 . shows the amino acid sequence obtained upon translation of the human coronavirus 229E strain ATCC VR-74 spike gene consensus sequence.
Fig. 2.



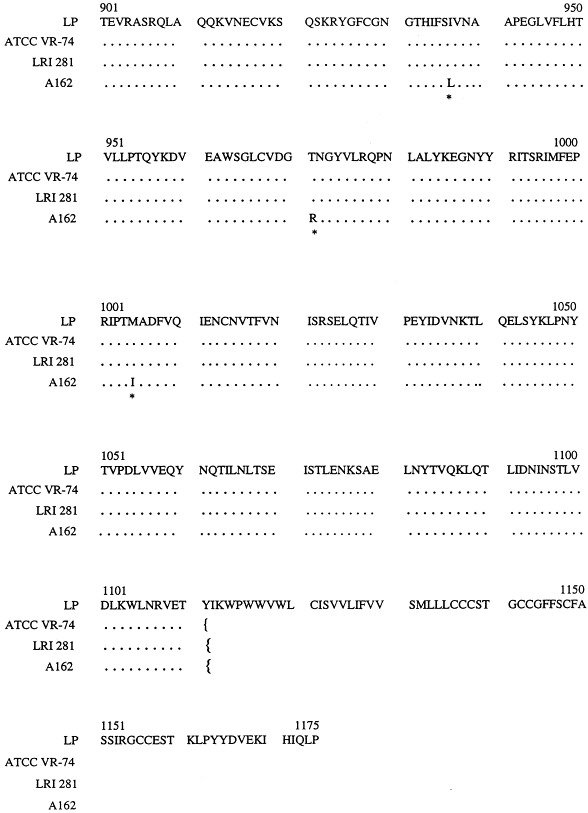
Comparison of the predicted amino acid sequence for he spike proteins of several human coronaviruses 229E. LP, human coronavirus 229E isolate LP (Raabe et al., 1990); ATCC VR-74, human coronavirus 229E ATCC VR-74 (EMBL Accession No. Y09923); LRI 281, human coronavirus 229E isolate LRI 281 (EMBL Accession No. Y10052); A162, human coronavirus 229E isolate A162 (EMBL Accession No. Y10051); ···, regions of homology between translated spike protein sequences; *, spike protein loci with an absence of conservation; }{, spike gene region sequences.
3.2. Human coronavirus 229E LRI 281
Forty six individual primer sequences were employed to construct a human coronavirus 229E LRI S gene 281 consensus sequence. A different number of primers were used for this strain because the read length from individual sequences varied with some reactions not generating the required number of bases. The resultant 3139 nucleotide consensus sequence was assembled from a total library of 14 946 individual nucleotides. Misincorporation errors were deemed to have occurred at 25 positions within this 14 946 total nucleotide assemblage, with 29 positions deemed to contain nucleotide additions and 29 positions nucleotide deletions (data not shown). Fig. 2 shows the amino acid sequence obtained upon translation of the human coronavirus 229E LRI 281 spike gene consensus sequence.
3.3. Human Coronavirus 229E A162
Forty one individual primer sequences were employed to construct a human coronavirus 229E A162 consensus sequence some 3046 nucleotides in length. This 3046 nucleotide consensus sequence was constructed from 13 974 individual nucleotides. Mis-incorporation errors were deemed to have occurred at 31 positions within this 13 974 total nucleotide assemblage, with 21 positions deemed to contain nucleotide additions and 36 positions nucleotide deletions (data not shown). Fig. 2. shows the amino acid sequence obtained upon translation of the human coronavirus 229E A162 spike gene consensus sequence.
During the sequencing of the human coronavirus 229E A162 spike gene it was found that sequence data could not be obtained from three of the 33 sequencing primers used successfully in the human coronavirus 229E strain ATCC VR-74 and human coronavirus 229E LRI 281 spike gene sequencing protocols. Upon assembly of the human coronavirus 229E A162 spike gene consensus sequence, however, it was determined that nucleotide differences within the human coronavirus 229E A162 spike gene consensus sequence (as compared to the spike gene consensus sequences of human coronavirus 229E strain ATCC VR-74 and human coronavirus 229E LRI 281) may have affected the primer binding sites for these particular sequencing primers. Fig. 3 shows the effect of human coronavirus 229E A162 spike gene sequence changes on the primer binding capacity of these three failed sequencing primers.
Fig. 3.


Failed sequencing primers and primer binding regions on the human coronavirus 229E A162 spike gene.
4. Discussion
In this study touchdown PCR methodology was used due to the fact that sequentially decreasing the annealing temperature (from a preset maximum to a preset minimum) allows PCR amplification reactions to be attempted using a wide range of PCR primers even if these primers are somewhat mis-matched with regard to their predicted annealing temperatures (Roux, 1994). Also, it was envisaged that the use of a touchdown PCR methodology would help to circumvent any non-specific priming (Don et al., 1991); this was considered particularly important because the human coronavirus 229E spike gene PCR primer design protocol utilised limited the number of possible primer sequences available for spike gene amplification. Touchdown PCR methodology did not inhibit, however, the production of extraneous PCR products during the first round of the human coronavirus 229E spike gene PCR. In effect, this meant that a nested PCR methodology had to be employed.
Extraneous first round PCR products were produced in such quantities that after addition of ‘neat’ first round PCR product to second round reaction mixes and subsequent second round PCR cycling, the extraneous first round PCR products could still be detected in the background of second round PCR products. More importantly, this background of extraneous first round PCR products tended to interfere with subsequent sequencing reactions, with the result that sequence data could not be obtained from second round PCR mixes to which neat first round PCR products had been added. By diluting the first round PCR products 1:10 (v/v) in sterile UHQ water, this problem was overcome and sequence data could be readily obtained.
An automated sequencing methodology was chosen over manual methods due to the relatively large number of sequencing reactions required to generate human coronavirus 229E spike gene consensus sequences and the relatively high throughput rates achievable with automated sequencing.
The use of automated dye-terminator sequencing chemistry per se was favoured over the use of automated dye-primer sequencing chemistry due to the fact that: (a) unadulterated PCR (or sequencing) primers can be utilised in dye-terminator automated sequencing chemistry without the need to label the primers with fluorescent dye tags (greatly reducing costs); (b) dye-terminator sequencing reactions are carried out in a single tube whilst dye-primer sequencing reactions are carried out in four separate reaction tubes (again reducing sequencing costs); (c) false termination products are not detected using dye-terminator chemistry as a labelled dye-terminator must be incorporated into the DNA chain in order for the DNA to be detected; and (d) dye-terminator sequencing chemistry requires lower concentrations of template DNA allowing several sets of sequence data to be generated with several different sequencing primers from a single human coronavirus 229E PCR reaction (DNA Sequencing: Chemistry Guide, 1995).
Automated dye-terminator sequencing does however have its drawbacks. In particular, both a reduced sequencing accuracy rate (Naeve et al., 1995) and the generation of artifactual chromatogram peaks (Parker et al., 1995) have been associated with the use of such chemistry. Indeed, Parker et al. (1995) indicated that the use of automated dye-terminator sequencing chemistry resulted in chromatogram artifactual peaks whose presence was determined by the nucleotide sequence immediately 5′ (i.e. downstream) to the artefact itself. Interestingly, it was noted that the nucleotide sequences immediately 5′ to chromatogram artefactual peaks in this project did not correspond with the 5′ nucleotide sequences generating artefactual peaks indicated by Parker et al. (1995) (even though automated dye-terminator sequencing chemistry was utilised in both projects). This apparent discrepancy was brought about by the fact that Parker et al. utilised Amplitaq enzyme (Perkin Elmer) in their dye-terminator sequencing reactions, whereas in this project Amplitaq FS (FS, fluorescent sequencing) enzyme (Perkin Elmer) was utilised. The conclusion therefore being that the pattern of chromatogram artefactual peaks generated in dye-terminator sequencing chemistry reactions by these two enzymes is somewhat different.
Taking the comments made about the methodology into account, the results from the sequencing of the spike genes of the human coronavirus 229E isolates presented in this paper suggest that only minor spike protein variation exists between these chronologically and geographically distinct isolates. This appears to contrast with another member of the same coronavirus serogroup as human coronaviruses 229E, canine coronavirus (Horsburgh and Brown, 1995) in which heterogeneity was distributed throughout the spike gene sequences of two geographically distinct (one British and one American) isolates. Also, a high degree of genome recombination events has been observed during mixed infections with different murine coronavirus strains (Lai et al., 1985) and different avian infectious bronchitis virus strains (Kusters et al., 1990). Moreover, there is evidence to suggest that in the absence of selection pressure these recombination events occur in a random manner (Banner and Lai, 1990). As only minor changes are seen in the translated spike protein sequences obtained in this project, it may be possible that during human coronavirus 229E infections selection pressure plays a dominant role in limiting the degree of spike gene variation (alternatively, multiple human coronavirus 229E infections may be a relatively rare event).
One possible explanation for the degree of homology observed between the translated spike protein sequences of human coronavirus 229E ‘LP’ and human coronavirus 229E strain ATCC VR-74 relies upon the fact that both of these viruses were adapted to tissue culture by serial passaging. In particular, MRC5 (Medical Research Council No. 5) human embryonic lung fibroblasts were the final replicative host for human coronavirus 229E strain ATCC VR-74, whilst Clone 16 human embryonic lung fibroblasts (the 16th clone of heteroploid MRC-c cells Philpotts, 1983) were the final replicative host for human coronavirus 229E ‘LP’ (Raabe et al., 1990). As MRC5 and MRC-c cells are closely related, it may feasible that adaptation of human coronaviruses 229E to these similar cell lines may facilitate similar spike protein conformations. These similar spike protein conformations would be expected to have similar protein sequences and by reverse translation, similar spike gene sequences. Further, if such ‘convergent evolution’ does indeed occur, then it is possible that the spike gene sequences obtained from serially passaged, tissue culture adapted human coronaviruses 229E may differ from the spike gene sequences of the original isolates. In this case, the accuracy of spike gene sequence data from human coronaviruses 229E which had been isolated by serial passage in tissue culture may be called into question. In this project, spike gene PCR amplification of human coronavirus 229E isolates LRI 281 and A162 was undertaken directly from clinical specimens.
Though comparatively few changes were observed between the human coronavirus 229E translated spike proteins compared in this project, the majority of such changes were observed in the 5′ half of the spike protein sequence. This point is illustrated most obviously upon examination of the translated spike protein sequence of human coronavirus 229E A162, where three apparent clusters of nucleotide variation were observed, all in the 5′ half of the translated spike protein sequence. Interestingly, research by Banner et al. (1990), working with mouse hepatitis virus, indicated that the 5′ end of the murine coronavirus spike gene may be the preferred site for RNA recombination events for this particular coronavirus. Moreover, comparisons of the spike protein sequences of avian infectious bronchitis virus, feline infectious peritonitis virus, murine hepatitis virus and transmissible gastro-enteritis virus have indicated that the 3′ (C-terminal) portion of the spike protein is rather more conserved than the corresponding 5′ (amino-terminal portion) portion of the spike gene (Cavanagh, 1995).
In theory, it is possible that minor variations in the translated sequences of human coronavirus 229E spike proteins may facilitate a relatively major change to the antigenicity of the spike protein. However, comparison of the predicted antigenic indices of the translated spike proteins of human coronaviruses 229E strain ATCC VR-74, LRI 281 and A162 (data not shown) using the ‘peptidestructure’ and ‘plotstructure’ computer applications (Genetics Computer Group Inc. v8.0., Madison WI, USA) showed that no major changes in predicted antigenic indexes were observed between the translated spike protein sequences of the aforementioned human coronavirus 229E isolates.
Taken as a whole, these results indicate that variation in the spike proteins of chronologically and geographically distinct human coronaviruses 229E may be rather limited. Such an interpretation would tend to suggest that spike protein variation does not play a major role in the aetiology of human coronavirus 229E re-infection. However, in order to assess fully the role that spike protein variation has upon human coronavirus 229E re-infection, further work is required. In particular, sequencing of the spike genes from other chronologically and geographically distinct human coronaviruses 229E should be undertaken. Further, by cloning and expressing the spike proteins of human coronaviruses 229E in vitro, it may be possible to assess the role of the immune system in the aetiology of human coronavirus 229E re-infection.
Acknowledgements
JPH was supported by a studentship from the UK Medical Research Council. This work was supported in part by grants from the National Asthma Campaign and Royal Society. Automated sequencing was undertaken in the Protein and Nucleic Acid Chemistry Laboratory of the University of Leicester.
References
- Banner L.R., Keck J.G., Lai M.C. A clustering of RNA recombination sites adjacent to a hypervariable region of the peplomer gene of murine coronavirus. Virology. 1990;175:548–555. doi: 10.1016/0042-6822(90)90439-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banner L.R., Lai M.M. Random nature of coronavirus recombination in the absence of selection pressure. Virology. 1990;185:441. doi: 10.1016/0042-6822(91)90795-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callow K.A. Effect of specific humoral immunity and some non-specific factors on resistance of volunteers to respiratory coronavirus infection. J. Hyg. 1985;95:173. doi: 10.1017/s0022172400062410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. The coronavirus surface glycoprotein. In: Siddell S.G., editor. The Coronaviridae. Plenum Press; London: 1995. [Google Scholar]
- Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate phenol chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Collins A.R., Knobler R.L., Powell H., Buchmeier M.J. Monoclonal antibodies to murine hepatitis virus-4 (strain JHM) define the viral glycoprotein responsible for attachment and cell-cell fusion. Virology. 1982;119:358. doi: 10.1016/0042-6822(82)90095-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot R.J., Van Leen R.W., Dalderup M.J.M., Vennema H., Horzinek M.C., Spaan W.J. Stably expressed FIPV peplomer protein induces cell fusion and elicits neutralising antibodies in mice. Virology. 1989;171:493. doi: 10.1016/0042-6822(89)90619-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DNA Sequencing: Chemistry Guide, 1995. Part Number 903563, Perkin Elmer, Warrington, Cheshire, UK.
- Don R.H., Cox P.T., Wainwright B.J., Baker K., Mattick J.S. Touchdown PCR to circumvent spurious priming during gene amplification. Nucl. Acids. Res. 1991;19:4008. doi: 10.1093/nar/19.14.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwaltney J.M. Epidemiology of the common cold. Ann. N.Y. Acad. Sci. 1980;353:54–690. doi: 10.1111/j.1749-6632.1980.tb18905.x. [DOI] [PubMed] [Google Scholar]
- Hamre D., Procknow J.J. A new virus isolated from the human respiratory tract. Proc. Soc. Exp. Biol. 1966;121:190–193. doi: 10.3181/00379727-121-30734. [DOI] [PubMed] [Google Scholar]
- Horsburgh B.C., Brown T.D. Cloning, sequencing and expression of the S protein from two geographically distinct strains of canine coronavirus. Virus Res. 1995;39:63–74. doi: 10.1016/S0168-1702(95)00068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruskova J., Heinz F., Svandova E., Pennigerova S. Antibodies to human coronaviruses 229E and OC43 in the population of C.R. Acta. Virologica. 1990;34:346–352. [PubMed] [Google Scholar]
- Jimenez G., Correa I., Melgosa M.P., Bullido M.J., Enjuanes L. Critical epitopes in transmissible gatroenteritis virus neutralization. J. Virol. 1986;60:131. doi: 10.1128/jvi.60.1.131-139.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S.L., Pattemore P.K., Sanderson G. Community study of the role of viral infections in exacerbations of asthma in 9–11 year old children. Br. Med. J. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusters J.G., Jager E.J., Neisters H.G., van der Zeijst B.A. Sequence evidence for RNA recombination in field isolates of avian coronavirus infectious bronchitis virus. Vaccine. 1990;8:605. doi: 10.1016/0264-410X(90)90018-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C. Coronavirus-organization, replication and expression of genome. Ann. Rev. Microbiol. 1990;44:303. doi: 10.1146/annurev.mi.44.100190.001511. [DOI] [PubMed] [Google Scholar]
- Lai M.M., Baric R.S., Makino S., Keck J.G., Egbert J., Leibowitz J.L., Stohlman S.A. Recombination between nonsegmented RNA genomes of murine coronaviruses. J. Virol. 1985;56:449. doi: 10.1128/jvi.56.2.449-456.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Monica N., Banner L.R., Morris V.L., Lai M.M.C. Localisation of extensive deletions in the structural genes of two neurotropic variants of murine coronavirus JHM. Virology. 1991;182:838. doi: 10.1016/0042-6822(91)90635-O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNaughton M.R. Occurrence and frequency of coronavirus infections in humans as determined by enzyme linked immunosorbant assay. Infect. Immunol. 1982;38:419. doi: 10.1128/iai.38.2.419-423.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto I., Kawana R. Virological surveillance of acute respiratory tract illnesses of children in Morioka, Japan. III. Human respiratory coronavirus. J. Jpn. Assoc. Infect. Dis. 1992;66:319–326. doi: 10.11150/kansenshogakuzasshi1970.66.319. [DOI] [PubMed] [Google Scholar]
- McIntosh K., Kapikian A.Z., Turner H.C., hartley J.W., Parrott R.H., Chanock R.M. seroepidemologic studies of coronavirus infection in adults and children. Am J. Epidemiol. 1970;91:585. doi: 10.1093/oxfordjournals.aje.a121171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh K., Chao R.K., Krause H.E., Wasil R., Mocega H.E., Mufson M.A. Copronavirus infections in lower respiratory tract disease of infants. J. Infect. Dis. 1974;130:502. doi: 10.1093/infdis/130.5.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monto A.S., Lim S.K. The Tecumseh study of respiratory illness. VI. Frequency and relationship between outbreaks of coronavirus infection. J. Infect. Dis. 1974;129:271–276. doi: 10.1093/infdis/129.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myint, S.H., 1995. Human coronavirus infections. In: Siddell, S.G. (Eds.), The Coronaviridae. Plenum Press, London. Book in the series, The Viruses. Fraenkel-Conrat, H., Wagner, R.R. (series Eds.), Plenum Press, New York.
- Naeve C.W., Buck G.A., Nieces R.L., Pon R.T., Robertson M. Accuracy of automated DNA sequencing-a multilaboratory comparison of sequencing results. Biotechniques. 1995;19:48–453. [PubMed] [Google Scholar]
- Parker L.T., Deng Q., Zakeri H., Carlson C., Nickeroson D.A., Kwok D.Y. Peak height variations in automated sequencing of PCR products using Taq dye-terminator chemistry. Biotechniques. 1995;19:116–121. [PubMed] [Google Scholar]
- Payne C.M., Ray C.G., Borduin V., Minnich L.L., Lebowitz M.D. An eight year study of the viral agents of acute gastroenteritis in humans: ultrastructural observations and seasonal distribution with a major emphasis on coronavirus-like particles. Diagn. Microbiol. Infect. 1986;5:39–54. doi: 10.1016/0732-8893(86)90090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpotts R. Clones of MRC-c cells may be superior to the parent line for the culture of 229E-like strains of human respiratory coronavirus. J. Virol. Methods. 1983;6:267–269. doi: 10.1016/0166-0934(83)90041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raabe T., Schelle P.B., Siddell S.G. Nucleotide sequence of the gene encoding the spike glycoprotein of human coronavirus HCV 229E. J. Gen. Virol. 1990;71:1065–1073. doi: 10.1099/0022-1317-71-5-1065. [DOI] [PubMed] [Google Scholar]
- Roux K.H. Using mis-matched primer pairs in touchdown PCR. Biotechniques. 1994;16:812. [PubMed] [Google Scholar]
- Siddell, S.G., 1995. The Coronaviridae: An Introduction in the Coronaviridae. Plenum Press, London. Book in the series, The Viruses. Fraenkel-Conrat, H., Wagner, R.R. (series Eds.), Plenum Press, New York.
- Stewart J.N., Mounir S., Talbot P.J. Human coronavirus gene expression in the brain of a multiple sclerosis patients. Virology. 1992;191:502. doi: 10.1016/0042-6822(92)90220-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell D.A.J., Bynoe M.L. Cultivation of a novel type of common cold virus in organ culture. Br. Med. J. 1965;1:1467. doi: 10.1136/bmj.1.5448.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling J.G., Vennema H., Godeke G-J., Horzinek M.C., Rottier P.J.M. Nucleotide sequence and expression of the spike (S) gene of canine coronavirus and comparison with the S proteins of feline and porcine coronaviruses. J. Gen. Virol. 1994;75:1789–1794. doi: 10.1099/0022-1317-75-7-1789. [DOI] [PubMed] [Google Scholar]