

Abstract
We have analyzed the ability of three molecular clones of feline immunodeficiency virus (FIV) and an ex vivo variant to infect nine distinct specific-pathogen-free feline cell lines in tissue culture. The purpose of these studies was to elucidate mechanisms by which host cells regulate the level of virus infection and expression and to assess host cell cytokine responses to virus infection. Cells used for the analyzes included four IL-2-dependent continuous T-cell lines (104-C1, 104-C7, MCH5-4 and DB FeTs) which arose from long-term passage, followed by limiting dilution cloning of peripheral blood mononuclear cells (PBMCs); two IL-2-independent T-cell lines (104-C1DL and MCH5-4DL) which originated from two of the IL-2-dependent lines, 104-C1 and MCH5-4; respectively; Crandell feline kidney cells (CrFK); G355-5 brain-derived glial cells; and the T-cell lymphoma line, 3201. Cells were infected with FIV-PPR, FIV-34TF10, FIV 34TF10orf2rep, and a variant arising from FIV-PPR during ex vivo passage on 104-C1DL cells, termed FIV-PPRglial. Infection of the IL-2-dependent T-cell line, 104-C1, by FIV-PPR resulted in the specific and distinct upregulation of cytokine expression. In particular, these cells doubled their expression of the pleiotropic cytokines, interleukin-4 and interleukin-12 after FIV infection. Interferon-γ production also increased after infection with FIV whereas, TNFα expression remained constant. Also, a marked upregulation of MHC class II expression was noted post infection of MCH5-4 and 104-C1 cells with FIV-PPR. Similar results were obtained after infection with FIV-34TF10orf2rep, indicating that the upregulation of cytokine expression is not an isolate-specific phenomenon. Changes in cytokine and class II expression are similar to various reports for the in vivo cytokine alterations in FIV, SIV and HIV infections. The ex vivo infection of these cell lines offers a manipulable system to examine the mechanism(s) by which lentiviruses alter cytokine expression.
Keywords: FIV, Cytokine upregulation, MHC class II, Chemokine
1. Introduction
Feline immunodeficiency virus (FIV) is a lentivirus of domestic cats found associated with an AIDS-like syndrome in infected animals. As with HIV in humans, the cat virus primarily infects T-cells of the CD4+ lineage (Ackley et al., 1990; Novotney et al., 1990; Brown et al., 1991; Torten et al., 1991; Willett et al., 1991; Hoffmann-Fezer et al., 1992), but also infects CD8+ T-cells, monocytes/macrophages, and a subset of IgG-bearing B cells in vivo (Brunner and Pedersen, 1989; Brown et al., 1991; English et al., 1993; Dean et al., 1996) and in vitro (Brunner and Pedersen, 1989). Additionally, certain FIVs will productively infect some continuous feline cell lines, such as Crandell feline kidney cells (CrFK) and a glial cell line derived from cat brain, G355-5 (Phillips et al., 1990; Waters et al., 1996). The extent to which non-lymphoid cells are infected in vivo has yet to be satisfactorily determined.
There are several levels at which the cell may control virus expression. In the simplest case, the lack of a receptor or low affinity interaction with that receptor on a given cell could limit virus infection and spread. The receptor for FIV has yet to be defined, although recent reports indicate that at least certain FIVs interact with the chemokine receptor CXCR4 (Willett et al., 1997) to facilitate fusion events. Whether CXCR4 is a primary or secondary receptor remains to be determined. A molecule previously thought to be a possible receptor, CD9 (Hosie et al., 1993; Willett et al., 1994), has recently been shown to be involved in virus release from the cell, rather than entry (de Parseval et al., 1997). Relative to receptor binding affinity, it has been shown that changes in the V3 region of Env can impart the ability of certain FIVs to productively infect CrFK cells, whereas the parental molecularly cloned FIV could only productively infect PBMCs (Verschoor et al., 1995). By analogy with HIV-1, this may mean that the V3 region is involved in chemokine receptor interactions (Picard et al., 1997; Speck et al., 1997).
Down-regulation of transcription by a negative regulatory element in the retroviral long terminal repeat (LTR) is another possible control on virus expression (Thompson et al., 1994) and sequences corresponding to a negative regulatory element (NRE) are present in the FIV-LTR (Thompson et al., 1994). The influence of these sequences on host cell range has yet to be defined. We have recently shown that an intact ORF2 gene, a putative FIV transactivator (Waters et al., 1996), is necessary for productive growth of FIV-34TF10 on primary T-cells and macrophages (Waters et al., 1996). Also, a functional deoxyuridine triphosphatase (DU) is necessary for the efficient growth of FIV on primary macrophages, but is not required for growth in actively dividing T-cell lines (Lerner et al., 1995). Tests in vivo have revealed that DU− FIV-PPR is still able to infect the cat, but the distribution of infection is distinct from wild-type FIV-PPR (Lerner et al., 1995). Also, there is a five-fold increase in mutations in viral DNA isolated from primary macrophages of cats infected with DU− FIV, consistent with mutations arising from mis-incorporation of uracil into viral DNA (Lerner et al., 1995). Although productive viral infection relies on several additional factors, all the above are examples of genotypes that influence growth in one cell environment, but are dispensable in another. Such factors influence the cellular distribution of the infection and this may potentially influence the timecourse, severity, and specificity of the resultant pathology.
As an approach for examining and defining controls on virus expression, we have assembled a panel of cell lines derived from primary cells as well as transformed and continuous cell lines. We have assessed these cells for surface marker, cytokine, chemokine, and MHC class II expression, and for relative infection by a well-characterized panel of FIVs. The results show that FIV can specifically upregulate distinct cytokines in the panel of cells herein described, as well as MHC class II. These findings are consistent with the interpretation that the observed upregulations are a direct result of FIV infection, rather than a bystander effect.
2. Materials and methods
2.1. Cell lines and viruses and propagation methods
CrFK cells were obtained from the ATCC and a feline glial cell line (G355-5), was kindly provided by Don Blair (NIH, Bethesda, MD). PBMCs were prepared from heparinized whole feline blood by Ficoll-paque gradient purification (Lerner et al., 1995) and cultured in the presence of IL-2 and Concanavalin-A (Con-A). MCH5 cell lines were isolated by limiting dilution cloning of PBMCs from an FIV positive cat. The T-cell lines 104-C1 and 104-C7 were isolated from an FIV negative cat. The lymphoma cell line, 3201, (Tochikura et al., 1990) was obtained from Dr. William Hardy. The two continuous IL-2-independent cell lines, MCH5-4DL and 104-C1DL were isolated from their parental lines, MCH5-4 and 104-C1, respectively, after undergoing crisis in the absence of interleukin-2 and Con-A.
T-cell lines were cultured in RPMI-1640 medium (Mediatech) containing: 10% heat-inactivated FBS (Gemini Bioproducts), 200 μM, l-glutamine (Sigma), 1X non-essential amino acids (Sigma), 1X sodium pyruvate (Sigma), 1X MEM-vitamins (Sigma), 5.5×10−5 M β-mercaptoethanol (Gibco-BRL) and 50 μg ml−1 gentamicin sulfate (Gemini BioProducts). The lines that required mitogenic stimulation also received 50 U ml−1 human recombinant IL-2 (a gift of Hoffmann–LaRoche) and 7.5 μg ml−1 Con-A (Boehringer Mannheim).
The CrFK cells were cultured in DMEM (BioWhittaker) with the same supplements as described above for RPMI 1640 medium, without addition of IL-2 or Con-A. The G355-5 cells were cultured in McCoys-5A medium (Mediatech), also with the same supplements without Con-A and IL-2.
FIV strains used in this study were: FIV-34TF10, a molecular clone derived from the Petaluma isolate (Talbott et al., 1989); FIV-34TF10orf2rep (Waters et al., 1996), which contains an open Orf2 reading frame; FIV-PPR, a molecular clone derived from the San Diego isolate (Phillips et al., 1990); and FIV-PPRglial, a strain with expanded host cell range, obtained after ex vivo passage in the 104-C1DL cell line (see below).
T-cells were infected at a density of 5×106 ml−1. Virus-positive tissue culture supernatants with a known reverse transcriptase (RT) unit value were used as infectious input virus. Typically, cells were infected with 400 000 cpm RT activity, defined as assayed (below). Cells were washed in Hanks-buffered saline solution (HBSS) prior to the addition of virus. The cells were then resuspended in 1 ml of viral supernatant and incubated for 1 h at 37°C followed by a 5 min spin at 1000 rpm in a Beckman-J6B centrifuge. The virus was aspirated and the cells washed prior to culture in fresh media.
Adherent cells were infected at 50% confluency in a T25 flask. Cells were washed with HBSS and 2 ml viral supernatant was pipetted onto the monolayer. The cells were incubated for 1 h at 37°C and then washed 2 times with HBSS. Fresh media was then added to each flask.
2.2. Antibodies
Anti-feline CD4-PE and anti-feline CD8-FITC were obtained from Southern Biotech. Anti-feline CD9 (vpg15) was obtained from Dr. Brian Willett. Cross-reactive anti-canine CD21 was provided by Dr. Peter Moore, University of California, Davis, CA. Anti-feline CD11b and anti-feline CD38 were obtained from Dr. Edward Hoover, Colorado State University, Fort Collins, CO. Anti-feline MHC Class-II antibodies, CAG3-8B, CAG5-3DI and PF6J-6D1 as well as PAK32-C, an anti-FIV Gag p24 were prepared from a series of fusions from mice immunized with either control or FIV-infected tissue culture cell membranes or viral pellets (C. Grant, in press). Cross-reactive anti-human CD3 was provided by David Lo, The Scripps Research Institute. FIV-infected cat serum (`VC') was provided by Christina Hutson, Animal Hospital of Redondo Beach, CA.
2.3. Gel electrophoresis and western blotting
Sonicated samples were run on 10%–20% tricine gels (Novex) and electrophoretically transferred onto nitrocellulose membranes (Schleicher & Schuell). The membranes were briefly stained with Amido Black to visualize the protein banding pattern and then blocked using BLOTTO (Johnson et al., 1984) for an hour at room temperature. Primary antibodies were used at a dilution of 1:100. Secondary antibodies (rabbit anti-mouse-HRP and goat anti-rabbit-HRP [Cappel]) were used at a dilution of 1:500. Goat anti-feline-HRP (Southern Biotech), was used at 1:1000. The blots were washed between incubations in 0.5 M LiCl/100 mM Tris–HCl, pH 8.5, with 1% NP-40 and developed using chemiluminescence (ECL, Amersham) and autoradiography (BIOMAX MR, Kodak).
2.4. Reverse transcriptase assays
Virus infection was assessed by quantitation of pelletable reverse transcriptase activity from culture supernatants as follows: 5 ml tissue culture supernatant was filtered through a 0.45 μl uniprep filter unit (Whatmann) and 4 ml of this supernatant was centrifuged at 100 000×g for 30 min in a Beckman TL ultracentrifuge with a TLA-100 rotor. The pellets were then resuspended in 100 μl of disruption buffer (20 mM DTT, 40 mM Tris–Cl pH 8.1, 0.356 M NaCl2 and 0.2% NP-40 (USB)). Following vigorous vortexing and two rounds of freeze-thawing, 25 μl of lysate was added to a reaction mixture containing 40 mM Tris–HCl pH 8.1, 60 mM NaCl2, 0.32 U Poly(rA)·p(dT)12–18 (Pharmacia), 0.1 mCi 3H-dTTP, 4 mM dTTP and 16 mM MgCl2. The samples were incubated at 37°C for an hour and then briefly placed on ice to stop the reaction. The samples were then transferred onto DE81 filter circles (Whatmann) and incubated in 0.1 M sodium pyrophosphate for 5 min, followed by three washes in 0.3 M ammonium formate pH 7.8 and one wash in 95% ethanol, followed by air-drying. Radioactivity was quantitated on a liquid scintillation counter using Eco-Scint (National Diagnostics).
2.5. Flow cytometry
Cells (1×106) were pelleted in 12×75 mm polystyrene tubes (Falcon) at 1000 rpm for 10 min in a Beckman J-6B centrifuge and then washed 2 times in ice-cold Earle's balanced salt solution (EBSS) without phenol red (GIBCO). Primary antibodies were diluted in EBSS and incubated with target cells for 30 min at 4°C, followed by two washes with ice-cold EBSS. Cells were incubated with either FITC or PE-labeled secondary antibodies (Southern Biotech) at 4°C in the dark for 30 min. The cells were then washed 2 times with EBSS, fixed by resuspension in 500 μl 1% formaldehyde in EBSS, and kept at 4°C in the dark until sorted.
2.6. Cytokine, cell marker and viral oligonucleotides
Oligonucleotides for polymerase chain reactions (PCRs) were synthesized on a Beckman Oligo 1000 M DNA synthesizer. The sequences are as follows:
IL-1β Fwd: CAGGTTTCTGAAGCCGCCATGGCA
IL-1β Rev: GTCTCTTTAGGAAGCGCTTTCCAT
IL-2 Fwd: GCACCTACTTCAAGCTCTAC
IL-2 Rev: GATGCTTTGACAAAAGGTAATC
IL-4 Fwd: ATGGGTCTCACCTCCCAACTG
IL-4 Rev: TCGTCTTTAGCCTTTCCAAGAAG
IL-6 Fwd: CAGCTATGAACTCCCTCTCCAC
IL-6 Rev: CTCAGGCTGAACTGCAGGAA
IL-10 Fwd: TACTTGGGTTGCCAAGCCTT
IL-10 Rev: TTCACAGAGAGCTCAGTAAAT
IL-12 Fwd: GGAGGCGAGGTTCTGAGCCAT
IL-12 Rev: GTCCACCACGACTTCAATGGG
CD4 Fwd: TTCCGGAAGGTCTCTAACACGGT
CD4 Rev: CACAAGGCCTAGGACCCCACC
CD8 Fwd: ATGGCCTCTCCGGTGACTGCCC
CD8 Rev: ACTGTCCTCATAAAGAGAGTTTTAT
CD9 Fwd: CACCATGCCGGTCAAAGGAGG
CD9 Rev: GTACCTTTCATTGCAGGATTTCTG
CD20 Fwd: TCTTCACTGGTGGGCCCCAC
CD20 Rev: CTTCATTCTTTGGTTGGGAAGAT
CD56 Fwd: CTAAGGATCTCATCTGGACTTTG
CD56 Rev: TGCAAAGACCTTGAGGTGGATGG
βA Fwd: GACGAGGCCCAGAGCAAGAG-AGG
βA Rev: GATCCACATCTGCTGGAAGGTGGAC
IFNγ Fwd: AACTCTCCGAAACGATGAATTAC
IFNγ Rev: CAAATATTGCAGGCAGGACAACCA
TNFα Fwd: GCATGATCCGGGACGTGGAG
TNFα Rev: CCTCTGGGGGCCGATCACTC
TGFβ Fwd: ACCTGCAAGACCATCGACATGGAGC
TGFβ Rev: AGGAGCAGGAAGGGCCGGTTCATG
CXCR4 Fwd: ATGGAGGGGATCAGTATATACACTTCAGAT
CXCR4Rev: ACATCTGTGTTAGCTGGAGTGAAAACTTGAAGACTCAG
CCR5 Fwd: ATGGACGGGTTTCGTATATACCCTTCAGAT
CCR5 Rev: TTAGCTGGAGTGAAAACTTGAAGACT
FIV-RT Fwd: GAAGGGAAAGTAAAAAGAGCAGA
FIV-RT Rev: AGTTTCAAATCCCCACCATAATAG
FIV Gag-p24 Fwd: GCCTTCTCTGCAAATTTAACACC
FIV Gag-p24 Rev: CCTTAGCTCCTTGTCTTAACTGCA
2.7. Polymerase chain reaction (PCR)
Each 100 μl reaction contained 200 μM dNTPs (Promega), 1 mM MgCl2, 50 mM KCl, 10 mM Tris–Cl pH 9.0, 1% Triton X-100, 700 ng 3′ and 5′ primers and 5U Taq Polymerase (Promega). The thermocycler (Perkin-Elmer Cetus) was set to the following parameters: 2 min pre-soak 94°C followed by 40 cycles of: 94°C 15 s, 58°C 30 s, 72°C 45 s, followed by a final 10 min soak at 72°C. Bands were visualized on 1.2% agarose gels.
2.8. Mimic constructs for quantitative PCR
Mimic constructs were prepared to serve as PCR standards for quantitation of cell markers and cytokines. The approach was similar to that previously described for performing quantitative PCR (QC-PCR) (Siebert and Larrik, 1992, Siebert and Larrik, 1993).
Oligonucleotides encoding cytokine and cell surface marker-specific sequences were synthesized and used as templates for the preparation of the arms on two separate mimic constructs. The forward and reverse arms were generated in a PCR essentially as described in Fig. 1 (steps 1 and 2). Oligonucleotide template sequences are as follows:
Cytokine mimic forward arm: GCACCTACTTCAAGCTCTACATGGGTCTCACCTCCCAACTGCAGCTATGAACTCCCTCTCCACTACTTGGGTTGCCAAGCCTTGGAGGCGAGGTTCTGAGCCATGGATCCAAA (IL-2, IL-4, IL-6, IL-10, IL-12 and BamHI, respectively).
Cytokine mimic reverse arm: AAAGATATCCCCATTGAAGTCGTGGTGGACATTTACT GAGCTCTCTGTGAATTCCTGCAGTTCAGCCTGAGCTTC TTGGAAAGGCTAAAGACGAGATTACCTTTTGTCAAA GCATC (EcoRV, IL-12, IL-10, IL-6, IL-4 and IL-2, respectively).
Cell surface marker mimic forward arm: GACGAGGCCCAGAGCAAGAGAGGTTCCGGAAGGTCTCTAACACGGTATGGCCTCTCCGGTGACTGCCCCACCATGCCGGTCAAAGGAGGAACTCTCCGAAACGATGAATTACGCATGATCCGGGACGTGGAGGGATCCAAAAA (βA, CD4, CD8, CD9, IFNγ, TNFα and BamHI, respectively).
Cell surface marker mimic reverse arm: TTTTTGATATCGAGTGATCGGCCCCCAGAGGTGGTTGTCCTGCCTGCAATATTTGCAGAAATCCTGCAATGAAAGGTACATAAAACTCTCTTTATGAGGACAGTGGTGGGGTCCTAGGCCTTGTGGTCCACCTTCCAGCAGATGTGGATC (EcoRV,TNFα,IFNγ,CD9,CD8,CD4 and βA, respectively)
Fig. 1.
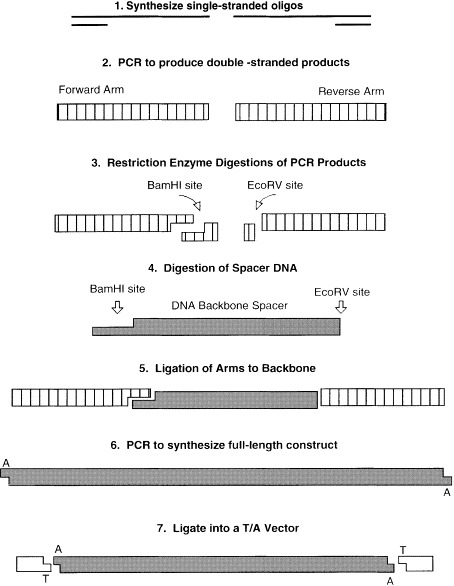
Scheme for the creation of mimic constructs for use in QC-PCR. In steps 1 and 2, double-stranded arms were amplified from oligonucleotides encoding feline cytokine- and cell marker-specific sequences. In steps 3 and 4, PCR products and backbone DNA were digested with the restriction enzymes Bam HI and Eco RV. In step 5, the DNA fragments were ligated, and then used as a template in a final PCR to create a full-length construct (step 6). Finally, in step 7, the construct was ligated into the TA cloning vector and transformed into E. coli for DNA production. DNA was purified and quantitated as detailed in Section 2.
Double-stranded forward and reverse arms were generated during PCR using sequence-specific oligonucleotide primers (bold) corresponding to the 5′ (+ strand) and 3′ (complementary) ends of the templates. Restriction enzyme sites were engineered into the templates and primers to provide the necessary directional cloning sites in the resultant products. BamHI was engineered into the 3′ ends of the forward arms and EcoRV into the 5′ ends of the reverse arms (Fig. 1, step 3)
The backbone onto which the arms were ligated was a 300 bp DNA fragment from the Env of FIV-PPR digested out of the TA vector, pCR2.1 (Invitrogen, La Jolla, CA), using the BamHI and EcoRV sites in the vector's polycloning site (Fig. 1, step 4). The fragment was gel and column-purified (Wizard PCR prep, Promega). The digested arms and backbone were set up in a three-way ligation overnight at 14°C (Fig. 1, step 5). The resulting product was used in a final PCR in which 1 μl of the ligation mixture was amplified using the IL-2 forward and reverse oliognucleotides for the Cytokine mimic and the βA forward and reverse oligonucleotides for the cell surface marker mimic (Fig. 1, step 6). The resulting full-length products were cloned into pCR2.1 (Fig. 1, step 7) and transformed into one-shot cells (Invitrogen). Utilizing blue/white selection, appropriate colonies were screened by colony PCR with each of the cytokine and cell surface marker primer pairs to verify sequence fidelity.
2.9. Quantitative PCR
Reactions were carried out as described above, with the exception of the addition of a defined amount of mimic plasmid as a dilution series. Assays ranged from 5 to 0.1 ng mimic plasmid in the quantitation of IFNγ and 3.5 to 0.035 ng for quantitation of IL-2, IL-6, IL-10 and IL-12. All cDNAs were used at a concentration in which, when amplified, the product was molar with 100 pg of β-actin mimic plasmid. PCR-based assessments of virus binding and infectivity were performed as previously described (de Parseval et al., 1997).
2.10. Colony PCR
A bacterial colony was picked with a pipet tip and resuspended in 100 μl dH20. Ten μl of this suspension was used as template in PCR, as described above.
2.11. Preparation of RNA and cDNA
Total RNA was extracted from 5×106 cells using either the RNeasy Kit (Qiagen) or the TRIzol Reagent (Gibco-BRL). Total RNA was treated with RNAse-free DNAse (Stratagene) in the presence of an RNAse inhibitor (Stratagene) and extracted with phenol/chloroform. cDNA was synthesized using an RT-PCR kit (Stratagene).
3. Results
3.1. Cell marker, cytokines and virus susceptibility of cell lines
Table 1 summarizes the analyzes of cell lines employed in this study. The 104-C1, 104-C7, MCH5-4, and DB FeT-cells are uninfected IL-2-dependent cell lines established from long-term culture of PBMCs. The 104-C1 and -C7 cells were isolated from the same cat. The 104-C1 cells are fully infectable by most FIVs analyzed, whereas the 104-C7 cells are refractory to productive infection by all FIVs tested (Table 2 ). The DB FeT-cell line is very similar to the 104-C1 in morphology (not shown) and virus susceptibility (Table 2), but differs in the lack of expression of cytokines IL-4 and IL-6, and lack of CD3 expression (Table 1). The MCH5 cell lines were derived from an FIV-infected cat. All but the MCH5-4 line are either positive for virus expression (Fig. 2 (A)) or are FIV positive by PCR (Fig. 2(B)). It is interesting that the MCH5-4 cells remained virus negative prior to limiting dilution cloning from the other cells, in spite of the fact that they are fully infectable by most FIVs (Table 2). The results imply that MCH5-4 cells differentiated from a non-infectable to infectable phenotype some time after single cell cloning. MCH5-4 cells are similar to 104-C1 in morphology (not shown), and differ only by the lack of expression of IL-4 among the markers and cytokines measured (Table 1). Important to the present discussion, the overall virus susceptibility of all the three cell lines was similar, in spite of the distinctions noted in cell surface/cytokine expression. In contrast, the 104-C7 cell line, which was completely refractory to infection by all FIVs tested, differed from the susceptible 104-C1 cells only by the lack of expression of CD4 and CD38 among markers examined (Table 1). However, the lack of these markers did not correlate with failure to infect with FIV, when all cell lines were compared (Table 2). For example, the virus-producing MCH5-5 cell line lacks both these markers. Thus, the marker analyses described here serve to delineate these cells, but do not explain virus susceptibility per se.
Table 1.
Characterization of cell lines by RT-PCR and flow cytometry
| TNFa† | IFNy† | TGFb† | CXCR4† | CCR5† | IL 1b† | IL2† | IL4† | IL6† | IL 10† | IL12† | CD3§ | CD4†§ | CD8†§ | CD9†§ | CD20§ | CD38§ | CD56§ | MHC Class II§ | |
| CrFK | + | + | + | + | + | − | − | − | + | − | + | − | − | − | + | − | − | + | − |
| G355-5 | + | + | + | + | + | − | − | − | + | − | + | − | − | − | + | − | − | + | − |
| MCH5-1 | + | + | + | + | + | − | − | − | + | − | + | − | − | − | + | − | + | + | + |
| MCH5-2 | + | + | + | + | + | − | − | + | + | − | + | + | − | − | + | − | + | + | + |
| MCH5-4 | + | + | + | + | + | − | − | − | + | − | + | + | + | + | + | − | + | + | + |
| MCH5-4DL | + | + | + | + | + | − | − | − | + | − | + | + | + | + | − | − | + | + | − |
| MCH5-5 | + | + | + | + | + | − | − | − | + | − | + | − | − | + | + | − | − | + | + |
| MCH5-10 | + | + | + | + | + | − | − | − | + | − | + | + | − | + | + | − | − | + | + |
| 104-C1 | + | + | + | + | + | − | − | + | + | − | + | + | + | + | + | − | + | + | + |
| 104-C1DL | + | + | + | + | + | − | − | − | + | − | + | + | + | + | − | − | + | + | + |
| 104-C7 | + | + | + | + | + | − | − | + | + | − | + | + | − | + | + | − | − | + | + |
| DB FeTs | + | + | + | + | + | − | − | − | − | − | + | − | + | + | + | − | + | + | + |
| 3201 | + | + | + | + | + | − | + | − | + | − | + | − | + | + | − | − | + | − | + |
RT-PCR.
§Flow cytometry.
Table 2.
Relative quantitation of FIV infection on feline cell lines
| PPR | PPRglial | 34TF10 | 34TF10orf2rep | |
| 104-C1 | ++++ | ++++ | +/− | ++++ |
| 104-C1DL | * | ++++ | +/− | + |
| 104-C7 | − | − | − | − |
| MCH5-4 | ++++ | ++++ | +/− | ++++ |
| MCH5-4DL | * | ++++ | +++ | + |
| DB FeTs | ++++ | ++++ | − | ++++ |
| 3201 | * | ++++ | ++++ | ++++ |
| CrFK | − | ++++ | ++++ | ++++ |
| G355-5 | − | ++++ | ++++ | ++++ |
*Requires 3 week incubation period prior to productive infection
+/−, ≤10K; +, ≤50K; ++, ≥100K; +++, ≥200K; ++++, ≥400K.
Fig. 2.
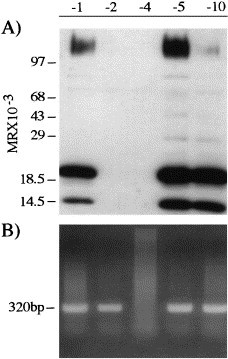
Virus expression by MCH5 cell lines. Cells were assessed for viral protein expression by Western blotting (A), and for the presence of viral DNA by PCR analyzes (B). MCH5-1, MCH5-5, and MCH5-10 were virus positive. MCH5-4 cells were virus negative. MCH5-2 cells did not produce virus, although they were positive for integrated FIV.
Experiments were performed to try to determine the level at which 104-C7 cells were blocked for FIV infection (Fig. 3 ). A PCR-based assay (de Parseval et al., 1997) was used to assess virus binding to the cell, penetration, reverse transcription, integration, and transcription, as a function of time post acute infection. As shown in Fig. 3(A), virus bound specifically not only to the fully infectable MCH5-4 and 104-C1 cells, but also to the refractory 104-C7 cells. Furthermore, as shown in Fig. 3(B), Gag-specific viral RNA and DNA could be detected in both infectable 104-C1 cells and refractory 104-C7 cells within 3 h post infection, consistent with the notion that the block was not at the level of virus binding or initial penetration events. However, from 24–72 h post infection, both unspliced and multiply spliced viral RNAs accumulated in the productively infected 104-C1 cells, whereas unspliced RNA decreased in the 104-C7 cells and multiply spliced mRNAs were barely detected (Fig. 3(B)). Additional studies indicated a reduction in closed circular DNA in the 104-C7 cells compared to the 104-C1 cells (not shown), suggestive of a failure of the viral DNA to enter the nucleus. Further studies to delineate this apparent block to virus integration are in progress.
Fig. 3.
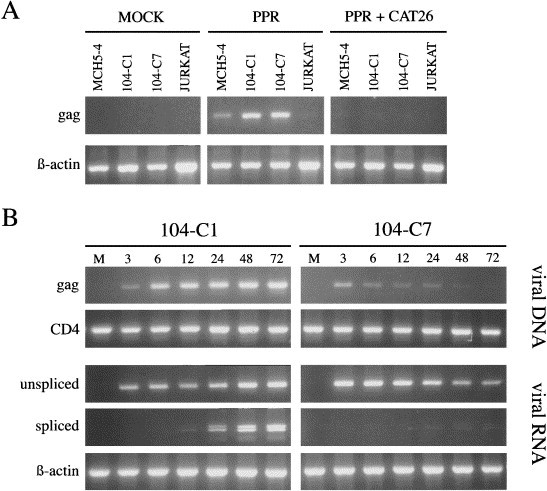
Inhibition of virus infection in 104-C7 cells. (A) Virus binding assay. Cells were incubated with medium alone, or FIV-PPR at 4°C for 30 min. An FIV-infected cat serum (CAT 26) was used as a positive control to neutralize the binding of the virus, and the human Jurkat cell line was used as a negative control. After extensive washes to remove unbound virus, total cell-associated viral RNA was detected by RT-PCR as previously described (de Parseval et al., 1997). (B) Kinetics of viral DNA and RNA synthesis in 104-C1 versus 104-C7 cells. The presence of viral DNA and RNA was monitored at the indicated times as previously described (de Parseval et al., 1997). CD4 and β-actin were used as an internal control for DNA and RNA extraction, respectively. Lane M, mock-infected control.
The 3201 cell line showed several distinctions from the lymphocyte lines derived from primary cells (Table 1 and Table 2). These cells expressed IL-2, lacking from all of the other cell lines, and failed to express CD56, which was expressed by all other cells, including the non-lymphocyte lines, CrFK and G355-5. The 3201 cells lacked CD3, similar to DB FeTs, but also lacked CD9 expression, a character shared with two IL-2-independent primary cell lines 104-C1DL and MCH5-4DL (see below). Also shared with the IL-2-independent lines was an inability to be infected to high titer by FIV-PPR (Table 2). However, passage of FIV-PPR on 3201 cells as well as on IL-2- independent primary cell lines (see below) resulted in the production of FIVs with expanded host cell range (not shown). This included the non-lymphoid cell lines CrFK and G355-5, previously reported as refractory to productive infection by the parental FIV-PPR (Phillips et al., 1990; Waters et al., 1996).
3.2. Phenotypic changes associated with IL-2 independence
Two of the cell lines, 104-C1DL and MCH5-4DL, were weaned from requirement for exogenous IL-2 (Table 1). Of the cell marker/cytokine expressions examined, only the loss of CD9 expression was noted as a correlate with IL-2 independence. Interestingly, the loss of IL-2 dependence changed the susceptibility of the cells to infection with certain FIVs compared to their respective parental lines (Table 2). In particular, both IL-2-independent cell lines failed to grow FIV-PPR to high titers after acute infection, in contrast to results with the parental 104-C1 and MCH5-4 cells. FIV-34TF10orf2rep also showed a marked reduction in ability to acutely infect the IL-2-independent cells (Table 2). The simple addition of IL-2 back to the IL-2-independent cultures was insufficient to facilitate productive infection by FIV-PPR (Fig. 4 ) and did not cause reconversion of the cells to IL-2 dependence. However, continuous passage of FIV-PPR on either of the IL-2-independent cells for a period of approximately three weeks resulted in the production of virus (Fig. 4(B), only data for 104-C1DL shown). The progeny virus, termed FIV-PPRglial could be immediately propagated on 104-C1DL cells and 3201 cells (Fig. 4(A)). Additionally, FIV-PPRglial, could productively infect CrFK and G355-5 cells (Table 2). This result indicates that low-level propagation of FIV-PPR did occur on the IL-2-independent cells, which facilitated sufficient replication to allow adaptation. FIV-PPRglial was also found to be more cytopathic than it's precursor, FIV-PPR on naive cells (Fig. 4(A)). This phenomenon has also been observed when FIV-Petaluma was passaged through 3201 cells (Tochikura et al., 1990), though expanded host-cell range was not described.
Fig. 4.
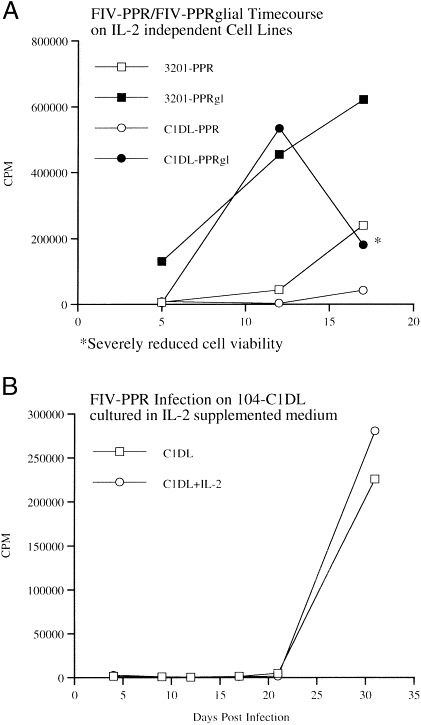
Kinetics of infection of IL-2- independent cell lines by wild type and adapted FIV-PPR. Infections were performed and monitored for the presence of reverse transcriptase activity as detailed in Section 2. (A) Both 3201 cells and 104-C1DL cells are resistant to productive infection with FIV-PPR. However, FIV-PPRglial, which arose from long-term culture of FIV-PPR on 104-C1DL cells, will productively infect both 3201 cells and IL-2 independent 104-C1DL cells. (B) Addition of exogenous IL-2 to the IL-2 independent cells does not render them susceptible to productive infection by FIV-PPR and adaptation occurs at equivalent rates with and without IL-2.
One possibility was that the above changes correlated with altered chemokine receptor expression, which might change the relative susceptibility to FIV infection, given the potential for involvement of these receptors in infection of the cell (Willett et al., 1997). However, we were unable to show any change in RNA expression of the chemokine receptor CXCR4 (Table 1; Fig. 5 ) as a function of IL-2 independence or exposure. Interestingly, the expression of CCR5 on the IL-2-independent MCH5-4DL cell line was very low in the absence of IL-2. As previously found using primate lymphocytes (Xu et al., 1996; Bleul et al., 1997), higher levels of CCR5 expression could be induced by the addition of exogenous IL-2 (Fig. 5). However, the increased expression of CCR5 did not result in imparting the ability of FIV-PPR to rapidly infect these cells (Fig. 4(B)), indicating that some other factor or factors are involved in dictating distinct virus expression as a function of IL-2 dependence.
Fig. 5.
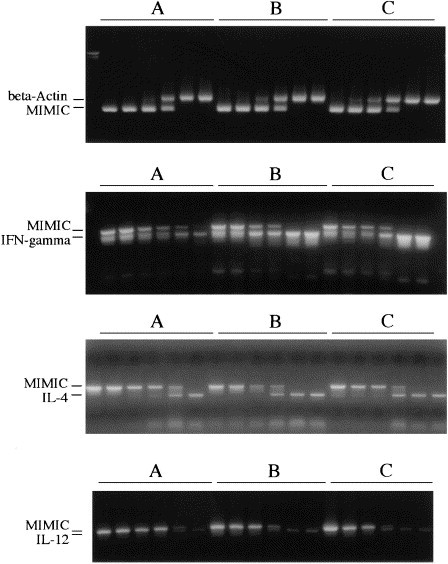
Quantitative PCR analyzes of cytokines expressed by 104-C1 cells with and without FIV infection. (A), uninfected 104-C1; (B), 104-C1 infected with FIV-PPR; (C), 104-C1 infected with FIV-34TF10orf2rep. A constant amount of a dilution series of internal standard mimic construct (see Fig. 1) was added vs. a constant amount of cellular cDNA. PCR reactions were performed and quantitated as described in Section 2. Quantitation of β-actin mRNA was performed to standardize the amount of cDNA target in each case (top panel). All the three cytokines doubled in expression as a consequence of infection with either FIV isolate.
3.3. Cytokine upregulation by FIV
We analyzed the effects of FIV infection on cytokine expression in the panel of cells described herein and quantitated the relative expression using QC-PCR (Fig. 6 ). The results showed a distinct upregulation of the pleiotropic cytokines IL-4 and IL-12 as a function of FIV infection of 104-C1 cells. Specifically, the expression of IL-4 and IL-12 increased from 350 pg in the uninfected cells to 700 pg in the FIV-PPR and FIV-34TF10orf2rep 104-C1 cells (Fig. 5, panels 3 and 4). Expression of IFNγ also increased (Fig. 5, panel 2) from 50 to 100 pg, under the same conditions. All the cell lines were negative for IL-10 (Table 1) and no upregulation was observed as a consequence of virus infection in this panel of cells (not shown). Also noted was a striking upregulation of MHC class II expression (Fig. 7 ). All of these altered expressions are consistent with the notion that upregulation of IL-4, IL-12, IFNγ, and MHC class II is a direct consequence of virus infection, based on the clonal nature of the cells.
Fig. 6.
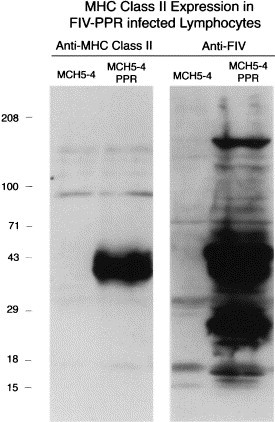
Upregulation of MHC class II expression after infection of MCH5-4 cells with FIV-PPR. Cell lysates from 1×107 cells, uninfected or infected with FIV-PPR, were prepared and analyzed by Western blot analysis using the anti-feline MHC class II monoclonal antibody CAG5-3DI (Hunt and McConnell, 1995), and with convalescent cat serum, as detailed in Section 2.
Fig. 7.
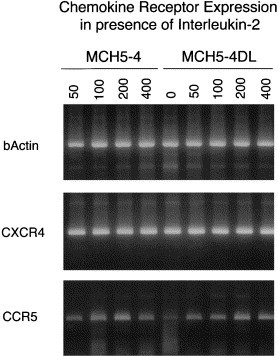
PCR analysis of CXCR4 and CCR5 chemokine receptor expression as a function of IL-2. RNA and subsequently, cDNA was prepared from either IL-2 dependent MCH5-4 cells or IL-2 independent MCH5-4DL cells. PCR was then performed as detailed in Section 2, to quantitate the expression of CXCR4 and CCR5 vs. a concentration gradient of exogenous IL-2. Quantitation of β-actin was used as a control for standardizing target cDNA (upper panel). The amount of CXCR4 mRNA was constant, regardless of cell type or level of exogenous IL-2 (middle panel). In the absence of exogenous IL-2, the expression of CCR5 was markedly reduced in MCH5-4 cells, but was upregulated in the presence of IL-2.
4. Discussion
The cell lines described above offer venues for a detailed study of the numerous aspects of virus infection, both from the standpoint of defining controls on virus entry, replication, and egress, as well as on understanding the cellular consequences of infection. Although one must apply caution in extrapolating ex vivo findings to an in vivo setting, the current findings were consistent with in vivo and ex vivo results reported for lentiviral infections including FIV, HIV-1 and SIV. Specifically, it has been reported that IL-4 (Zou et al., 1997; Benveniste et al., 1996) and IFNγ (Zou et al., 1997; Benveniste et al., 1996; Cheret et al., 1996; Rosenberg et al., 1997) are upregulated in macaques experimentally infected with SIV and additionally, IFNγ expression is upregulated in humans infected with HIV-1 (Breen et al., 1997). Interleukin-12 is upregulated in both FIV-infected cats (Dean et al., 1997) and in HIV-1-infected humans (Fantuzzi et al., 1996). Interestingly, IL-4, IL-12 and IFNγ were each found to be upregulated in cats upon infection with the coronavirus, feline infectious peritonitis virus, FIP (Gunn-Moore et al., 1997). An upregulation of IL-10 has also been a hallmark of FIV infection in vivo. However, IL-10 expression is generally considered a property of monocyte/macrophage and B lymphocytes, which are not represented in our panel of cells. The findings are consistent with a direct effect on upregulation of these cytokines and cell markers as a consequence of FIV infection, rather than an indirect bystander recruitment effect, which may occur in a heterogeneous cell population. However, the findings do not rule out that these proteins may be upregulated sequentially, as a consequence of a cascade initiated by upregulation of one of the cytokines. For example, IL-12 has been shown to upregulate the expression of IFNγ, which in turn can upregulate MHC class II. More rigorous examination of the time course of upregulation of each cytokine will determine the order in which upregulation occurs. The well-characterized cell lines described here will facilitate these studies, with a potential to perform both subtraction library and differential display analyzes to identify other upregulated species that may exist as a consequence of FIV infection.
The upregulation of MHC class II noted in these ex vivo studies has also been reported with in vivo infection by FIV (Ohno et al., 1992; Rideout et al., 1992; Hunt and McConnell, 1995). Similar observations have also been noted with infections by HIV-1 (Cantin et al., 1997a, Cantin et al., 1997b) and SIV (Kannagi et al., 1987). At first glance, this would seem to be a counterproductive move for the virus in that it would, via class II-bound virus peptide, signal the immune system that the cell was infected and target it for clearing. However, (Cantin et al., 1997a, Cantin et al., 1997b) offer a plausible explanation with regard to upregulation of class II as observed both in vitro and in vivo following infection with HIV-1. Since CD4 is the natural ligand for class II antigen, its expression on the surface of the virus in conjunction with the expression of the viral envelope protein gp120 increases the number of potential binding sites between the cell and the virus. This scenario fits well with the FIV model in that FIV preferentially infects cells of the CD4 lineage (Ackley et al., 1990; Novotney et al., 1990; Torten et al., 1991; Willett et al., 1991; Hoffmann-Fezer et al., 1992). Class II upregulation may also be a means of recruitment of CD4+ target cells for virus infection.
Of the cell lines examined, the only one that proved refractory to infection by any of the FIVs tested was the 104-C7 cell line. Virus binding studies, followed by assessment of internalized viral RNA, viral DNA, and spliced viral mRNAs revealed that the block to the infection was not at the level of virus entry. FIV-PPR was able to bind to and infect the 104-C7 cells with essentially the same kinetics as observed with the productively infected 104-C1 cell line. However, analysis of viral DNA synthesis, integration, and appearance of spliced mRNAs revealed a marked reduction in the ability of the virus to integrate into the 104-C7 genome. This, in turn, resulted in a drastic reduction in the production of a new virus and resultant spread in the culture. The reason(s) the virus fails to integrate efficiently in these cells are unclear. One possible explanation is that the majority of viral replication complex fails to enter the nucleus, perhaps because it is shunted into lysosomal vesicles or because of lack of a transport protein. Another possibility is that the 104-C7 cells are deficient in some component needed to facilitate the integration event. Importantly, these cells offer a paradigm for study of cellular control on lentiviral replication, independent of control of virus entry.
The process of adaptation to infection of some of these cell lines by FIV is also of interest. In particular, we noted that FIV-PPR did not immediately grow to high titer on the 3201 cell line. However, after several weeks in culture, a virus emerged that possessed the ability not only to rapidly and productively infect 3201 cells, but also the non-lymphoid CrFK and G355-5 cell lines which were refractory to infection with wild type FIV-PPR. Similar results were noted with FIV-PPR infection of the two IL-2-independent primary cell lines, 104-C1DL and MCH5-4DL. No significant virus growth was noted for several weeks, after which virus (FIV-PPRglial) emerged with expanded host cell range. A simple explanation would be that IL-2 independence corresponded with the loss of a primary receptor used by wild type FIV-PPR. The emerging virus adapted to utilization of a new receptor found on the IL-2-independent cells. Clearly, sufficient infection and replication by wild type FIV-PPR occurred to facilitate the process of adaptation. However, the initial affinity of interaction may have been weak, so that the level of virus replication and spread was very low until mutations occurred to increase the receptor affinity. However, it could also be due to a phenomenon that has nothing to do with receptor binding, such as an internal downregulation of transcription or the lack of components needed for efficient virus release. It is interesting to note that 3201, 104-C1DL, and MCH5-4DL cells share two features: (1) all three cell lines are IL-2-independent; and (2) all lack the surface marker CD9. Presumably 3201 cells are IL-2 `independent' because they express IL-2 albeit at low levels (Table 1). However, neither of the IL-2-independent primary cell lines express IL-2, so they are not independent simply because they possess an endogenous source of cytokine. The lack of CD9 expression is particularly interesting, given the previous findings that antibodies to this cell marker can block the propagation of FIV in tissue culture (Hosie et al., 1993; Willett et al., 1994; de Parseval et al., 1997). However, we have recently shown that this is due to a block in egress, rather than to blocking of a CD9 interaction at the point of entry (de Parseval et al., 1997). One possibility is that the lack of CD9 on the three cell lines requires that the virus adapt to an alternative pathway for egress. As with the cytokine responses noted above, the use of subtraction libraries and/or differential display on the IL-2-dependent and independent versions of 104-C1 and MCH5-4 will prove invaluable to defining the cellular distinctions that dictate each unique infection phenotype.
5. Conclusions
We have described the partial characterization of the cytokine and cell surface marker expression of a panel of nine feline cell lines. Furthermore, we have examined the relative ability of these cells to be productively infected by a panel of FIVs. The results show that all of these cell lines are unique and that they are differentially infectable by various isolates of FIV. FIV infection caused a marked upregulation of the pleiotropic cytokines IL-4, IL-12, and IFNγ and of MHC class II, all of which have been noted as consequences of lentiviral infection in vivo. In addition, we found that some of the primary T-cell lines could become IL-2-independent, and that commensurate with IL-2 independence, came a change in susceptibility to infection with certain FIVs. Adaptation to growth on these cells was observed after several weeks in culture, with resultant expansion of the host cell range and cytopathogenicity of the progeny virus. The cell lines described here offer a manipulable venue to further define the molecular mechanisms that control both the host-cell responses to viral infection and to identify factors that control the level and range of the virus expression.
Acknowledgements
The authors gratefully acknowledge Laurie McPherson and Michael Ingram, for their valued assistance in oligonucleotide synthesis, Pamela C. Wagaman, Ph.D. for her careful critique of the manuscript, and also, Hoffmann-La Roche for their generous contribution of recombinant human IL-2. This research was supported in part by grants from the National Institute of Mental Health (MH48870) and the National Allergy and Infectious Agents and Disease Institute (AI25825) of the National Institutes of Health.
References
- Ackley C.D., Yamamoto J.K., Levy N., Pedersen N.C., Cooper M.D. Immunologic abnormalities in pathogen-free cats experimentally infected with feline immunodeficiency virus. J. Virol. 1990;64:5652–5655. doi: 10.1128/jvi.64.11.5652-5655.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste O., Vaslin B., Le Grand R., Cheret A., Matheux F., Theodoro F., Cranage M.P., Dormont D. Comparative interleukin (IL-2)/interferon IFN-gamma and IL-4/IL-10 responses during acute infection of macaques inoculated with attenuated nef-truncated or pathogenic SICmac251 virus. Proc. Natl. Acad. Sci. USA. 1996;93:3658–3663. doi: 10.1073/pnas.93.8.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleul C.C., Wu L., Hoxie J.A., Springer T.A., Mackay C.R. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T-lymphocytes. Proc. Natl. Acad. Sci, USA. 1997;94:1925–1930. doi: 10.1073/pnas.94.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen E.C., Salazar-Gonzalez J.F., Shen L.P., Kolberg J.A., Urdea M.S., Martinez-Maza O., Fahey J.L. Circularing CD8 T-cells show increased interferon-gamma mRNA expression in HIV infection. Cell. Immunol. 1997;178:91–98. doi: 10.1006/cimm.1997.1115. [DOI] [PubMed] [Google Scholar]
- Brown W.C., Bissey L., Logan K.S., Pedersen N.C., Elder J.H., Collisson E.W. Feline immunodeficiency virus infects both CD4+ and CD8+ T-lymphocytes. J. Virol. 1991;65:3359–3364. doi: 10.1128/jvi.65.6.3359-3364.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner D., Pedersen N.C. Infection of peritoneal macrophages in vitro and in vivo with feline immunodeficiency virus. J. Virol. 1989;63:5483–5488. doi: 10.1128/jvi.63.12.5483-5488.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin, R.C., Fortin, J.-F., Lamontagne, G., Tremblay, M., 1997a. The presence of host-derived HLA-DR1 on human immunodeficiency virus type 1 increases viral infectivity. J. Virol. 71, 1922–1930 [DOI] [PMC free article] [PubMed]
- Cantin, R.C., Fortin, J.-F., Lamontagne, G., Tremblay, M., 1997b. The acquisition of host-derived major histocompatibility complex class II glycoproteins by human immunodeficiency virus type 1 accelerates the process of virus entry and infection in human T-lymphoid cells, Blood 90, 1091–1100 [PubMed]
- Cheret A., Le Grand R., Caufour P., Dereuddre-Bosquet N., Matheux F., Neildez O., Theodoro F., Maestrali N., Benveniste O., Vaslin B., Dormont D. Cytokine mRNA expression in mononuclear cells from different tissues during acute SICmac251 infection of macaques. AIDS Res. Hum. Retroviruses. 1996;12:1263–1272. doi: 10.1089/aid.1996.12.1263. [DOI] [PubMed] [Google Scholar]
- de Parseval A., Lerner D.L., Borrow P., Willett B.J., Elder J.H. Blocking of FIV infection by a monoclonal antibody to CD9 is via inhibition of virus release, rather than interference with receptor binding. J. Virol. 1997;71:5742–5749. doi: 10.1128/jvi.71.8.5742-5749.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean G.A., Reubel G.H., Moore P.F., Pedersen N.C. Proviral burden and infection kinetics of feline immunodeficiency virus in lymphocyte subsets of blood and lymph node. J. Virol. 1996;70:5165–5169. doi: 10.1128/jvi.70.8.5165-5169.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean, G., Woo, J., LaVoy, A., Moore, P., Pedersen, N., 1997. Cytokine expression in feline lymphoid tissues in health and disease. (abstract), The 2nd International Feline Immunology Workshop, University of California, Davis, CA, section 2-2
- English R.V., Johnson C.M., Gebhard D.H., Tompkins M.B. In vivo lymphocyte tropism of feline immunodeficiency virus. J. Virol. 1993;67:5175–5186. doi: 10.1128/jvi.67.9.5175-5186.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantuzzi L., Gessani S., Borghi P., Varano B., Conti L., Puddu P., Belardelli A. Induction of interleukin-12 (IL-12) by recombinant glycoprotein gp120 of human immunodeficiency virus type 1 in human monocytes/macropahges: Requirement of gamma interferon for IL-12 secretion. J. Virol. 1996;70:4121–4124. doi: 10.1128/jvi.70.6.4121-4124.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn-Moore, D.A., Caney, S.M.A., Gruffyd-Jones, T.J., Harbour, D.A., 1997. FIP- associated changed in cytokine mRNA associated changes in cytokine mRNA levels in vivo. (abstract), The 2nd International Feline Immunology Workshop, U.C., Davis, CA, section 2-13
- Hoffmann-Fezer G., Thum J., Ackley C., Herbold M., Mysliwietz J., Thefeld S., Hartmann K., Kraft W. Decline in CD4+ cell numbers in cats with naturally acquired feline immunodeficiency virus infection. J. Virol. 1992;66:1484–1488. doi: 10.1128/jvi.66.3.1484-1488.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie M.J., Willett B.J., Dunsford T.H., Jarrett O., Neil J.C. A monoclonal antibody which blocks infection with feline immunodeficiency virus identifies a possible non-CD4 receptor. J. Virol. 1993;67:1667–1671. doi: 10.1128/jvi.67.3.1667-1671.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt P., McConnell I. Variable expression of major histocompatibility complex class II in the domestic cat. Res. Vet. Sci. 1995;59:195–200. doi: 10.1016/0034-5288(95)90001-2. [DOI] [PubMed] [Google Scholar]
- Johnson D.A., Gautsch J.W., Sportsman R., Elder J.H. Improved technique utilizing non-fat dry milk for analysis of protein and nucleic acids transferred to nitrocellulose. Gene Analysis Techniques. 1984;1:3–8. [Google Scholar]
- Kannagi M., Kiyotaki M., King N.W., Lord C.I., Letvin N.L. Simian immunodeficiency virus induces expression of class II major histocompatibility complex structures on infected target cells in vitro. J. Virol. 1987;61:1421–1426. doi: 10.1128/jvi.61.5.1421-1426.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner D.L., Wagaman P.C., Phillips T.R., Prospero-Garcia O., Henriksen S.J., Fox H.S., Bloom F.E., Elder J.H. Increased mutation frequency of FIV lacking functional deoxyuridine triphosphatase. Proc. Natl. Acad. Sci. USA. 1995;92:7480–7484. doi: 10.1073/pnas.92.16.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotney C., English R.V., Housman J., Davidson M.G., Nasisse M.P., Jeng C.R., Davis W.C., Tompkins M.B. Lymphocyte population changes in cats naturally infected with feline immunodeficiency virus. AIDS. 1990;4:1213–1218. doi: 10.1097/00002030-199012000-00005. [DOI] [PubMed] [Google Scholar]
- Ohno K., Watari T., Goitsuka R., Tsujimoto H., Hasegawa A. Altered surface antigen expression on peripheral blood mononuclear cells in cats infected with feline immunodeficiency virus. J. Vet. Med. Sci. 1992;54:517–522. doi: 10.1292/jvms.54.517. [DOI] [PubMed] [Google Scholar]
- Phillips T.R., Talbott R.L., Lamont C., Muir S., Lovelace K., Elder J.H. Comparison of two host cell range variants of feline immunodeficiency virus. J. Virol. 1990;64:4605–4613. doi: 10.1128/jvi.64.10.4605-4613.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard L., Simmons G., Power C.A., Meyer A., Weiss R.A., Clapham P.R. Multiple extracellular domains of CCR-5 contribute to human immunodeficiency virus type 1 entry and fusion. J. Virol. 1997;71:5003–5011. doi: 10.1128/jvi.71.7.5003-5011.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout B.A., Moore P.F., Pedersen N.C. Persistent upregulation of MHC class II antigen expression on T-lymphocytes from cats experimentally infected with feline immunodeficiency virus. Vet. Immunol. Immunopathol. 1992;35:71–81. doi: 10.1016/0165-2427(92)90122-7. [DOI] [PubMed] [Google Scholar]
- Rosenberg Y.J., Cafaro A., Brennen T., Greenhouse J.G., Villinger F., Ansari A.A., Brown C., McKinnon K., Bellah S., Yalley-Ogunro J., Elkins W.R., Gartner S., Lewis M.G. Virus-induced cytokines regulate circulating lymphocyte levels during primary SIV infections. Int. Immunol. 1997;9:703–712. doi: 10.1093/intimm/9.5.703. [DOI] [PubMed] [Google Scholar]
- Siebert P.D., Larrik J.W. Competitive PCR. Nature. 1992;359:557–558. doi: 10.1038/359557a0. [DOI] [PubMed] [Google Scholar]
- Siebert P.D., Larrik J.W. PCR mimics: Competitive DNA fragments for use as internal standards in quantitive PCR. Biotechniques. 1993;14:244–249. [PubMed] [Google Scholar]
- Speck R.F., Wehrly K., Platt E.J., Atchison R.E., Charo I.F., Kabat D., Chesebro B., Goldsmith M.A. Selective employment of chemokine receptors as human immunodeficiency virus type 21 co-receptors determined by individual amino acids within the envelope V3 loop. J. Virol. 1997;71:7136–7139. doi: 10.1128/jvi.71.9.7136-7139.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbott R.L., Sparger E.E., Lovelace K.M., Fitch W.M., Pedersen N.C., Luciw P.A., Elder J.H. Nucleotide sequence and genomic organization of feline immunodeficiency virus. Proc. Natl. Acad. Sci. USA. 1989;86:5743–5747. doi: 10.1073/pnas.86.15.5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochikura T.S., Hayes K.A., Cheney C.M., Tanabe-Tochikura A., Rojko J.L., Mathes L.E., Olsen R.G. In vitro replication and cytopathogenicity of the feline immunodeficiency virus for feline T4 thymic lymphoma 3201 cells. Virology. 1990;179:492–497. doi: 10.1016/0042-6822(90)90323-j. [DOI] [PubMed] [Google Scholar]
- Torten M., Franchini M., Barlough J.E., George J.W., Mozes E., Lutz H., Pedersen N.C. Progressive immune dysfunction in cats experimentally infected with feline immunodeficiency virus. J. Virol. 1991;65:2225–2230. doi: 10.1128/jvi.65.5.2225-2230.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson F.J., Elder J.H., Neil J.C. Cis- and trans-regulation of feline immunodeficiency virus: Identification of functional binding sites in the long terminal repeat. J. Gen. Virol. 1994;75:545–554. doi: 10.1099/0022-1317-75-3-545. [DOI] [PubMed] [Google Scholar]
- Verschoor E.J., Boven L.A., Blaak H., van Vliet A.L., Horzinek M.C., de Ronde A. A single mutation within the V3 envelope neutralization doma of feline immunodeficiency virus determines its tropism for CRFK cells. J. Virol. 1995;69:4752–4757. doi: 10.1128/jvi.69.8.4752-4757.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters A.K., de Parseval A.P., Lerner D.L., Elder J.H. The role of ORF2 in determining the host cell tropism of feline immunodeficiency virus. Virology. 1996;215:10–16. doi: 10.1006/viro.1996.0002. [DOI] [PubMed] [Google Scholar]
- Willett B.J., Hosie M.J., Dunsford T.H., Neil J.C., Jarrett O. Productive infection of T-helper lymphocytes with feline immunodeficiency virus is accompanied by reduced expression of CD4. AIDS. 1991;5:1469–1475. doi: 10.1097/00002030-199112000-00009. [DOI] [PubMed] [Google Scholar]
- Willett B.J., Hosie M.J., Jarrett O., Neil J.C. Identification of a putative cellular receptor for feline immunodeficiency virus as the feline homologue of CD9. Immunology. 1994;81:228–233. [PMC free article] [PubMed] [Google Scholar]
- Willett B.J., Picard L., Hosie M.J., Turner J.D., Adema K., Clapham P.R. Shared usage of the chemokine receptor CXCR4 by the feline and human immunodeficiency viruses. J. Virol. 1997;71:6407–6415. doi: 10.1128/jvi.71.9.6407-6415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W., Charo, I.F., Greene, W.C., Goldsmith, M.A., 1996. Interleukin-2 activiates expression of the HIV co-receptor, CKR5. (abstract) The Third West Coast Retrovirus Meeting, p. 15
- Zou W., Lackner A.A., Simon M., Durand-Gasselin I., Galanaud P., Desrosiers R.C., Emilie D. Early cytokine and chemokine gene expression in lymph-nodes of macaques infected with simian immunodeficiency virus is predictive of disease outcome and vaccine efficacy. J. Virol. 1997;71:1227–1236. doi: 10.1128/jvi.71.2.1227-1236.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]